
Atomic layer deposition of SnO2 using hydrogen peroxide improves the efficiency and stability of perovskite solar cells†
 bc
and
Nam-Gyu
Park
*ac
bc
and
Nam-Gyu
Park
*ac
Abstract
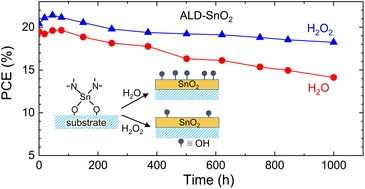
Low-temperature processed SnO2 is a promising electron transporting layer in perovskite solar cells (PSCs) due to its optoelectronic advantage. Atomic layer deposition (ALD) is suitable for forming a conformal SnO2 layer on a high-haze substrate. However, oxygen vacancy formed by the conventional ALD process using H2O might have a detrimental effect on the efficiency and stability of PSCs. Here, we report on the photovoltaic performance and stability of PSCs based on the ALD-SnO2 layer with low oxygen vacancies fabricated via H2O2. Compared to the ALD-SnO2 layer formed using H2O vapors, the ALD-SnO2 layer prepared via H2O2 shows better electron extraction due to a reduced oxygen vacancy associated with the highly oxidizing nature of H2O2. As a result, the power conversion efficiency (PCE) is enhanced from 21.42% for H2O to 22.34% for H2O2 mainly due to an enhanced open-circuit voltage. Operational stability is simultaneously improved, where 89.3% of the initial PCE is maintained after 1000 h under an ambient condition for the H2O2-derived ALD SnO2 as compared to the control device maintaining 72.5% of the initial PCE.
- This article is part of the themed collections: Nanoscale Most Popular 2023 Articles, 2024 Lunar New Year Collection and Halide Perovskite Optoelectronics
 Please wait while we load your content...
Please wait while we load your content...

