
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Correlative radioimaging and mass spectrometry imaging: a powerful combination to study 14C-graphene oxide in vivo biodistribution†
Hélène
Cazier
a,
Carole
Malgorn
b,
Dominique
Georgin
c,
Nathalie
Fresneau
cd,
Fabrice
Beau
b,
Kostas
Kostarelos
 efg,
Cyrill
Bussy
ef,
Stéphane
Campidelli
d,
Mathieu
Pinault
h,
Martine
Mayne-L'Hermite
h,
Frédéric
Taran
c,
Christophe
Junot
a,
François
Fenaille
a,
Antoine
Sallustrau
*c and
Benoit
Colsch
*a
efg,
Cyrill
Bussy
ef,
Stéphane
Campidelli
d,
Mathieu
Pinault
h,
Martine
Mayne-L'Hermite
h,
Frédéric
Taran
c,
Christophe
Junot
a,
François
Fenaille
a,
Antoine
Sallustrau
*c and
Benoit
Colsch
*a
aUniversité Paris Saclay, CEA, INRAE, Département Médicaments et Technologies pour la Santé (DMTS), SPI, 91191 Gif-sur-Yvette, France
bUniversité Paris Saclay, CEA, INRAE, Département Médicaments et Technologies pour la Santé (DMTS), SiMos, 91191 Gif-sur-Yvette, France
cUniversité Paris Saclay, CEA, INRAE, Département Médicaments et Technologies pour la Santé (DMTS), SCBM, 91191 Gif-sur-Yvette, France. E-mail: antoine.sallustrau@cea.fr
dUniversité Paris Saclay, CEA, CNRS, NIMBE, LICSEN, 91191 Gif-sur-Yvette, France
eNanomedicine Lab, Faculty of Biology, Medicine and Health, AV Hill Building, University of Manchester, Manchester M13 9PL, UK
fNational Graphene Institute, University of Manchester, Manchester M13 9PL, UK
gCatalan Institute of Nanoscience and Nanotechnology (ICN2), UAB Campus Bellaterra, Barcelona 08193, Spain
hUniversité Paris-Saclay, CEA, CNRS, NIMBE, LEDNA, 91191 Gif-sur-Yvette, France
First published on 20th February 2023
Abstract
Research on graphene based nanomaterials has flourished in the last decade due their unique properties and emerging socio-economic impact. In the context of their potential exploitation for biomedical applications, there is a growing need for the development of more efficient imaging techniques to track the fate of these materials. Herein we propose the first correlative imaging approach based on the combination of radioimaging and mass spectrometry imaging for the detection of Graphene Oxide (GO) labelled with carbon-14 in mice. In this study, 14C-graphene oxide nanoribbons were produced from the oxidative opening of 14C-carbon nanotubes, and were then intensively sonicated to provide nano-size 14C-GO flakes. After Intravenous administration in mice, 14C-GO distribution was quantified by radioimaging performed on tissue slices. On the same slices, MS-imaging provided a highly resolved distribution map of the nanomaterial based on the detection of specific radical anionic carbon clusters ranging from C2˙− to C9˙− with a base peak at m/z 72 (12C) and 74 (14C) under negative laser desorption ionization mass spectrometry (LDI-MS) conditions. This proof of concept approach synergizes the strength of each technique and could be advantageous in the pre-clinical development of future Graphene-based biomedical applications.
Introduction
Since Geim and Novoselov's Nobel prize winning discovery in 2004,1 research on Graphene Based Nanomaterials (GBNs) has steadily expanded over the years.2,3 GBNs have demonstrated unique properties that could be exploited in diverse industrial sectors (e.g. batteries, touch screens, anti-corrosion primers)4,5 and have important societal impacts.6 Progress in methodology development for higher quality synthesis and versatile surface functionalisation of GBNs has also paved the way towards their medical and biomedical applications.7–9 However, the commercial exploitation of GBN properties depends critically on their biocompatibility and toxicity profile. In fact, there is a growing concern about the risk of GBN materials towards human health.10 Graphene Oxide (GO) is recognised as one of the most preferential GBN materials for biomedical applications11–13 due to its distinctive physico-chemical properties (large surface area, thinness, high conductivity) and ease to be chemically functionalised.14 In such kind of applications, a direct and long-term interaction of the nanomaterial with the human body can be expected, which imply a careful evaluation of their biodistribution for health and safety considerations. Intravenous (i.v.) route is one of the most common routes of administration for these specific applications. Several studies using i.v. as a mode of administration evidenced an extensive accumulation in the liver, the lungs, spleen or in the kidneys over time.15,16 In a recent study, reduced GO was also found in the brain which support the possibility for such nanomaterials to cross biological barriers.17 The development of new methods for the long-term detection and absolute quantification of these materials and more specifically GO, is undoubtedly an important challenge to better understand the possible risks of long-term biopersistence of such materials and in order to make them safer-by-design.Raman spectroscopy imaging,18 photoacoustic spectroscopy19 or intrinsic fluorescence properties have proven to be useful techniques to detect GO in biological samples. However, they present limitations such as slow imaging speed, weak photoluminescence signals, low sensitivity, and do not allow absolute quantification. Recently, Mass Spectrometry Imaging (MSI) has emerged as a powerful tool, providing structural information with an excellent specificity and the capacity to map quantitatively multiple molecular classes such as drugs,20,21 lipids,22 peptides,23 proteins,24 as well as metabolites25,26 or carbon-based nanomaterials.27,28 In 2015, Chen et al. reported the use of laser desorption/ionization mass spectrometry (LDI-MS) for the detection, mapping, and quantification of Carbon Nanotubes (CNTs), Carbon Dots (CDTs) and GO in mouse tissue sections using a specific MS signature consisting of anionic carbon clusters.29 Inspired by these results, we recently published an optimised MSI method to assess the biodistribution of GO and rGO in rodent tissues.30 However, one of the main limitation of MSI is its lower sensitivity than radioimaging,31 despite its better spatial resolution and the fact that it allows identification.
Radiolabelling methods (using 188Re, 125I, 99mTc, 198Au, 111In)32–38 combined with highly sensitive radioimaging techniques have already been used to track labelled GO in vitro or in vivo. However, these approaches involve a chemical modification of the surface of the nanomaterials. This may in return alter the physicochemical properties of the nanoparticle or the labels may be cleaved from the GBN surface during in vivo circulation, hence affecting their reliable localisation and quantification. Nevertheless, the direct incorporation of carbon-14 (14C) into the backbone of GBNs has proven to be a method of choice to circumvent these drawbacks. In 2013, a graphitized material was labelled with 14C to assess and quantify its uptake and excretion in Daphnia magna.39 A year later, Czarny et al. labelled CNTs with 14C and demonstrated their translocation through the air-blood barrier towards distant organs supporting human health risk assessment of CNTs.31 Quantitative measurement of radioactivity using 14C performed by Mao et al. revealed the biodistribution and excretion mechanisms of 14C-labelled carbon-based nanoparticles in mice after intratracheal instillation.40 Moreover, the degradation of the 14C-labelled nanoparticles by liver cells was shown by the capture and quantification of 14CO2 breathed out by mice after intravenous injection.41
Considering the need for new analytical methods to ensure a reliable detection, localisation and quantification of GBN in complex biological environments, we propose here an original correlative imaging approach consisting in combining the strengths of both MSI and radio-imaging to provide absolute quantification, spatial resolution and specific signature information, for the in vivo tracking of GO in mice (Fig. 1).
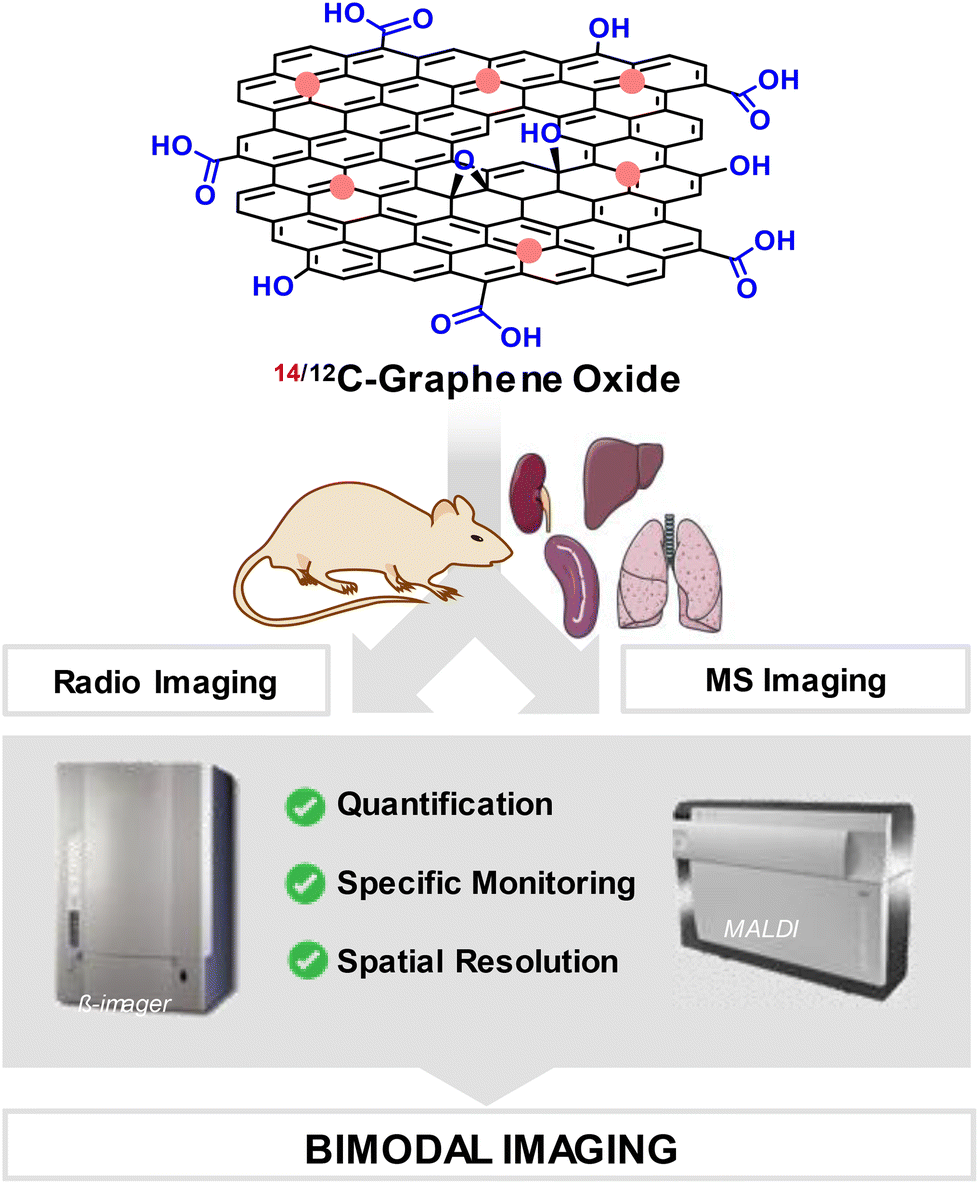 | ||
| Fig. 1 Combinaison of radio-imaging and mass spectrometry imaging techniques for the detection of 14/12C-graphene oxide. | ||
Experimental
Materials
Organic solvents (Sigma-Aldrich) were used without further purification. 14C-benzene (0.5 mL, 4.63 GBq mmol−1, 58 MBq mg−1) was purchased from Quotient Bioresearch Radiochemicals Ltd (UK) with a radiochemical and chemical purity > 99.9%. The scintillation fluid (Gold Star, Meridian Biotechnology Ltd, (UK) was purchased from PerkinElmer (USA). The deionized water used in the experiment was prepared by a Millipore Milli-Q ultra-purification system (resistance > 18.2 MΩ cm).Material characterisation
Graphene oxide nanoribbons and graphene oxide were analysed by transmission electron microscopy (TEM) (Philips CM 30; CEA Saclay, DEN-LM2E, France and LVEM5 Low-voltage Transmission Electron Microscope, CORDOUAN Technologies, Pessac, France) at an accelerating voltage of 5 kV and a magnification from 25 to 200 kV according to samples. Raman spectra were recorded on a Horiba Jobin–Yvon LabRam ARAMIS equipped with a laser excitation at 532 nm. The chemical composition of the non-radiolabelled graphene oxide nanoribbons and graphene oxide was evaluated by XPS (X-ray induced photoelectron spectroscopy) analysis using a Kratos Analytical Axis Ultra DLD spectrometer (UK) with monochromatic Al Kα X-ray radiation (hν = 1486.6 eV). Graphene oxide was imaged by Atomic Force Microscopy (AFM) (Multimode 8 atomic force microscope, Bruker, UK) in tapping mode using OTESPA tips (Bruker, UK) with a curvature radius of 10 nm, resonant frequency of 300 kHz, and a spring constant of 42 N m−1. A high-performance autoradiography imager (β-imager TM 2000, Biospace Lab, Paris, France) was used for μ-particle counting and absolute radioactivity quantification (detection threshold of 0.01 cpm mm−2 for 14C) allowing real-time radioactive imaging. Mass Spectrometry Imaging experiments were performed on an UltraFleXtreme™ Matrix-assisted Laser Desorption Ionization – tandem Time-of-Flight mass spectrometer (MALDI-TOF/TOF, Bruker Daltonics, Bremen, Germany).Graphene oxide synthesis
Graphene oxide was produced in a multistep procedure (Fig. 2A) from Carbon Nanotubes synthetized by a catalytic chemical vapour deposition process specifically developed for the use of benzene as source of carbon, as previously reported.31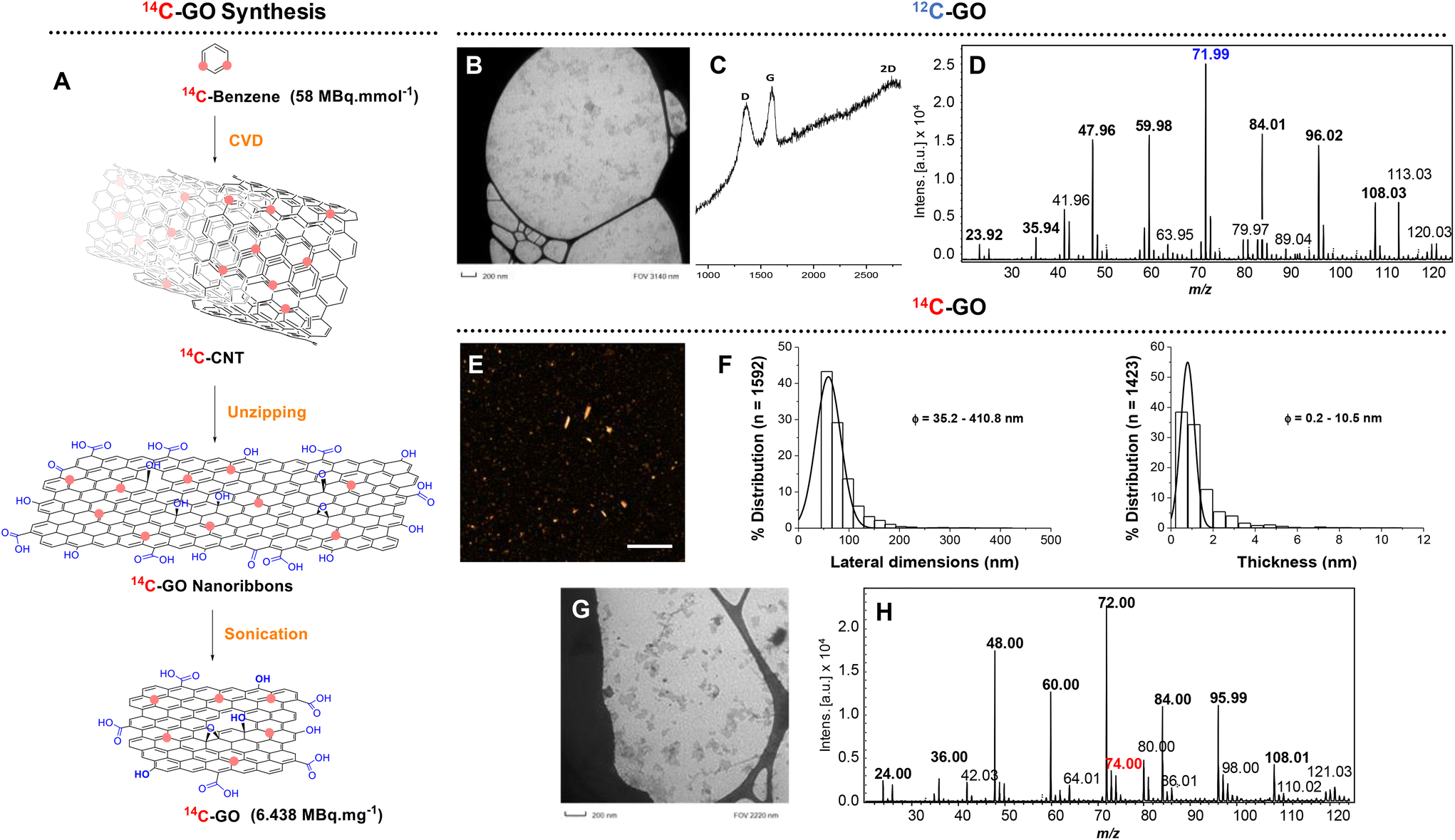 | ||
| Fig. 2 (A) Strategy for the synthesis of 14C-GO; (B) TEM image of 12C-GO; (C) Raman spectrum of 12C-GO; (D) LDI mass spectrum of 12C-GO; (E) AFM heigh image of 14C-GO (scale bar 1 μm); (F) size and thickness distribution of 14C-GO from the AFM heigh image. (G) and (E) TEM image of 14C-GO; (H) LDI mass spectrum of 12/14C-GO. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 15 min (Beckman Coulter, rotor F630), the supernatant was removed and 20 mL of MilliQ water was added in each tube. A second centrifugation step (25000 rpm, 15 min) was performed before the supernatant was removed and the pellet dispersed in water (30 mL). After 5 min stirring, the dispersion was sonicated for 10 min. Then HCl (4 mL, 20%) was added. After 5 min stirring, the solution was centrifuged (25000 rpm, 15 min). Four additional steps of centrifugation were performed for which the supernatant was systematically removed and replaced by 20 mL of Milli-Q water. After the last centrifugation step, the pellet was collected and dispersed in Milli-Q water (12 mL). The recovered nanoribbons (17.5 mg, 1.46 mg mL−1) were characterized before the synthesis of GO (ESI†).
000 rpm for 15 min (Beckman Coulter, rotor F630), the supernatant was removed and 20 mL of MilliQ water was added in each tube. A second centrifugation step (25000 rpm, 15 min) was performed before the supernatant was removed and the pellet dispersed in water (30 mL). After 5 min stirring, the dispersion was sonicated for 10 min. Then HCl (4 mL, 20%) was added. After 5 min stirring, the solution was centrifuged (25000 rpm, 15 min). Four additional steps of centrifugation were performed for which the supernatant was systematically removed and replaced by 20 mL of Milli-Q water. After the last centrifugation step, the pellet was collected and dispersed in Milli-Q water (12 mL). The recovered nanoribbons (17.5 mg, 1.46 mg mL−1) were characterized before the synthesis of GO (ESI†).
In vivo experiments
Radioimaging
A high-performance autoradiography imager was used for the quantitative determination of radioactivity in dried tissue sections. Correct calibration was verified between each sample analysis by counting the radioactivity of a reference sample (blood with a known quantity of 14C-GO). Organs from animals exposed to 14/12C-GO were processed and analysed as described in Table S2.†Mass spectrometry imaging
Methods for 14C-GO and 12C-GO detection were developed on an UltraFleXtreme™ MALDI-TOF/TOF mass spectrometer as described before.30 The MALDI-TOF instrument operated in the negative ionization reflectron mode, using an optimized extraction delay set at 10 ns. MS analyses were first performed at a 2 kHz laser repetition rate with a Smartbeam II laser ((Bruker Daltonics, Germany), which delivered a gross flat top beam profile with small laser beam focusing at 90% of fluence. Source and TOF pressure were below 1 × 10−7 mbar and voltages were adjusted as follows: IS1 = 20.02 kV, IS2 = 18.02 kV, lens = 6.99 kV, reflectron = 21.03 kV, and reflectron 2 = 10.93 kV. MS measurements were performed from m/z 0 to 200 with 1000 accumulated shots per spectrum. Images were acquired with a spatial resolution set at 25 μm for biodistribution studies and 100 μm for quantification assay. Data processing was achieved using FlexImaging 4.0 or SCiLS Lab 2023a Pro (Bruker Daltonics, Bremen, Germany). The molecular images were reconstituted based on the average intensities of the C6˙− anionic radical ion at m/z 72, which proved to be the most intense radical anion for the 14C-GO signature.30Results and discussion
Preparation and characterisation of graphene oxide
First developed and optimized with carbon-12 materials, the following strategy was applied to synthesize 14C-GO. It started with the longitudinal opening or “unzipping” of carbon nanotubes synthesized by Catalytic Chemical Vapor Deposition (CCVD)42–44 from 14C-benzene, as previously reported.31 The 14C-CNT were then oxidized in presence of KMnO4 and H2SO4 leading to the formation of 14C-graphene oxide nanoribbons. After being washed and centrifuged several times, the nanoribbons were sonicated for 12 hours using a Misonix Ultrasonic Convertor in an aqueous solution of SDBS leading to a suspension of 14C-GO (3.4 mg, 22 MBq, 6.438 MBq mg−1). As expected, the specific activity of 14C-GO measured by β-imager remained identical to the starting CNTs (6.438 MBq mg−1).The physicochemical characterization of 12C-GO and 14C-GO was performed to confirm the consistency of the production method and quality of the engineered materials. Raman analysis performed on 12C-GO showed peaks at 1363 cm−1 (D band), 1611 cm−1 (G band) and at 2730 cm−1 (2D band), all of which are characteristic of oxidized graphenic materials. The D band stands for the presence of defects (Csp3 and structural defects) in the sp2 structure and the G band corresponds to the remaining the graphitic mode of graphene. The defects in the structure were evaluated by the ID/IG ratio that equals to ∼1 here. Transmission electron microscopy (TEM) analysis of 12C-GO showed nanometre scale graphene oxide particles (<100 nm) (Fig. S8†) with a size distribution similar to 14C-GO and a narrow distribution under 100 nm (Fig. 2) corresponding to 75% of all particles. The morphology of 14C-GO was determined by AFM. It showed that the lateral dimension ranged from 50 to 400 nm, with a main size distribution centred below 100 nm, in agreement with TEM imaging. The average thickness was about 1 nm, indicating that 14C-GO nanoparticles were mainly made of 1 to 2 layers of graphene oxide sheets, given that GO interlayer distance is 0.86 nm.45 XPS analyses were performed only on 12C-GO nanoribbons and 12C-GO to avoid any radioactive contamination of the equipment. The deconvolution C1s spectrum of 12C-GO nanoribbons exhibits 3 major peaks at 284.5, 287, and 288 eV. The peak at 284.5 eV is assigned to C–C and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C. The peak at 287 eV is assigned to C–O species (C-OH and epoxy groups), and the remaining shoulder at 288 eV corresponds to the CO binding energy. A similar peak distribution was observed for 12C-GO. However the contribution of the C–C/CC peak and the C–O peak changes from the GO nanoribbons to GO (Fig. S3 and S11†) with a decrease of the C–O peak. However, there is no major change observed in the CO species.
C. The peak at 287 eV is assigned to C–O species (C-OH and epoxy groups), and the remaining shoulder at 288 eV corresponds to the CO binding energy. A similar peak distribution was observed for 12C-GO. However the contribution of the C–C/CC peak and the C–O peak changes from the GO nanoribbons to GO (Fig. S3 and S11†) with a decrease of the C–O peak. However, there is no major change observed in the CO species.
Additionally, XPS of 12C-GO nanoribbons indicates that the carbon content was the dominant feature (ca. 67.5%) as expected, and the oxygen content was found to reach almost a third of the total amount (ca. 31.5%), confirming the oxidized nature of the nanoribbons (Fig. S3 and S4†). Quantitative analysis of 12C-GO also indicated that carbon was predominant (ca. 69.0%) with a significant proportion of oxygen (ca. 30.3%) (Fig. S11 and S12†).
Furthermore, the C/O ratio increases from 2.14 in GO nanoribbons to 2.28 in GO, revealing the elimination of some oxygen functional groups after the ultrasonication step. This was confirmed by the work of Le et al.46 where they described the effect of sonication on GO leading to the removal of various oxygen groups and leading to increased C/O ratios.
Combining radio and MS imaging
Before assessing the biodistribution of GO in tissues by MSI and radio-imaging, adequate sample preparation and experimental conditions have been optimized as mentioned below.An isotopically diluted graphene oxide dispersion of 14C-GO and 12C-GO in SDBS referred as 14/12C-GO dispersion (3.2 MBq mg−1, 500 μg mL−1, 1.6 MBq mL−1) was prepared in order to increase the MSI signal of 14C-GO and avoid saturation of the radio-imaging signal. Mice were then exposed to 50 μg (161 kBq) and 75 μg (241 kBq) corresponding respectively to the injection of 100 μl and 150 μl of the 14/12C-GO dispersion by intravenous administration. Mice were sacrificed 15 min after injection. Lungs, liver, spleen and kidneys were collected, snap-frozen, cryo-sectioned and mounted on either ITO or Superfrost® slides for further analysis.
To perform optimal GO detection in tissue sections by MSI, the methodology previously developed for the detection of unlabelled GO and rGO30 was used for the radiolabelled GO. In this case, ITO slides have been used to provide both radioimaging and MSI analysis of the same tissue slice. LDI-MS parameters were thus optimised directly on the 14/12C-GO exposed tissues. For that, an extraction delay of 10 nanoseconds, a fluence of 90% in reflectron acquisition and in negative ionisation mode were used. In respect to the MSI laser, the parameters were fixed with the “minimum” focus, a random movement of the laser within the “raster”, and a spatial resolution of either 100 μm or 25 μm for biodistribution studies. Finally, an internal calibration was performed on the expected theoretical signature before acquisition of the MS images in order to guarantee mass measurement accuracy.
Following these first steps, we then investigated the LDI-MS signature of 14C-GO and ability to produce specific MSI images (Fig. 3). To do so, a comparative analysis of 14C-GO and 12C-GO was carried out on lung tissues exposed to 14/12C-GO. LDI-MS profiles of 12C-GO (Fig. 2D) and 14/12C-GO (Fig. 3C) indicated similar radical anionic carbon clusters ranging from C2˙− to C9˙−, which was consistent with our previous results.27 The most intense peak at m/z 71.99 was detected for both labelled and unlabelled nanoparticles. Interestingly, a slightly more intense peak was observed at m/z 74, related to 14C, due to the incorporation of the carbon isotope (one 14C for 35 12C) into the structure of GO. The overlay of the two MS images of a lung section, namely the m/z 72 (blue; Fig. 3A) and the m/z 74 (red; Fig. 3D), together with the corresponding H&E staining of an adjacent lung section (Fig. 3B) revealed an even distribution of 14/12C-GO materials across the lungs (Fig. 3E).
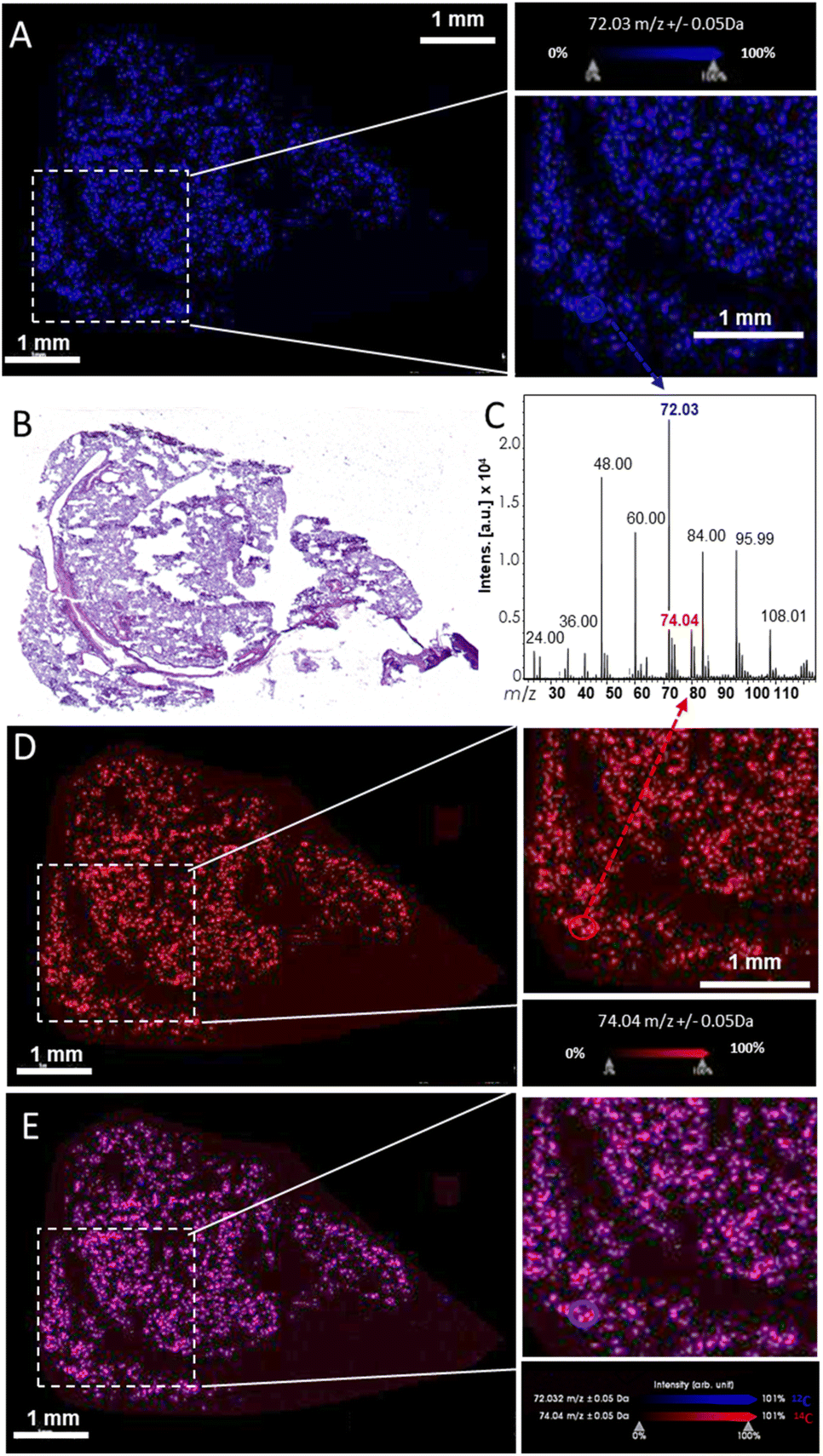 | ||
| Fig. 3 MSI analysis of a lung section from a mouse exposed to 50 μg of 14C/12C-GO with a spatial resolution of 25 μm. The MS image was acquired with an accumulation of 1000 shots per spectrum and per pixel at 2 kHz with a DIE of 10 ns, small laser focus and 90% laser fluency. (A) Molecular image of 12C-GO was represented using m/z 72 ion. (B) H&E staining of the same lung section. (C) MS profile of 14/12C-GO. (D) MS images of the m/z 74 corresponding to 14C-GO in the same section. (E) Overlay (purple) of the two MS images, namely the m/z 72 (blue) and the m/z 74 (red), corresponding to the 14/12C-GO (14C/12C6˙−). | ||
After 14/12C-GO sample preparation and the validation of a specific LDI-MS signal for 14/12C-GO, radio-imaging and MSI were successively performed on the same slice to investigate 14/12C-GO fate in vivo. Here, ITO slides used specifically for MSI analyses, was also tested and validated for radioimaging acquisition allowing both radioimaging and MSI analysis of the same tissue slice. Since MSI is a destructive analytical technique, radioimaging analysis was thus performed first providing a sensitive assessment of the GO biodistribution and allowing to select the most relevant organs or area onto which MSI will be carried out.
β-Imager analysis has been performed based on the average measurements of three adjacent organ sections. The GO amount found per section was then normalised to the total organ weight and expressed in percentages. Cumulating the four organs analysed, a total of 96.8% and 97.1% of the total injected doses was recovered for the 50 μg and 75 μg doses, respectively (Table S3†). Biodistribution images (Fig. 4B and Fig. S18†) show the predominant accumulation of the labelled GO in the liver for both injected doses (i.e., 50 and 75 μg) (n = 3 slices from the same mouse) corresponding respectively to 51% and 57% of the total injected dose. A strong signal was also found in lungs with 45% and 39% for 50 μg and 75 μg injected doses. Substantial accumulation in lungs is consistent with previous short-term distribution studies after GO intravenous administration reported in the literature.47,48 Following iv injection, GO particles are large enough to be entrapped in the first capillary networks they encountered (i.e., lungs and liver), which in turn reduces their transport and accumulation in other organs. Indeed, much less radioactivity was found in the spleen and kidneys where less than 1% of the total injected dose was detected, irrespective of the injected dose. Despite this quantitative assessment of the radioactivity distribution in the different organs, the low spatial resolution of β-imaging prevented a more precise evaluation of the GO distribution in the different organs of interest. MSI was therefore used as a complementary technique providing higher resolution to circumvent this drawback. MSI was thus performed on liver and lungs for both injected doses as they correspond to the main targeted organs.
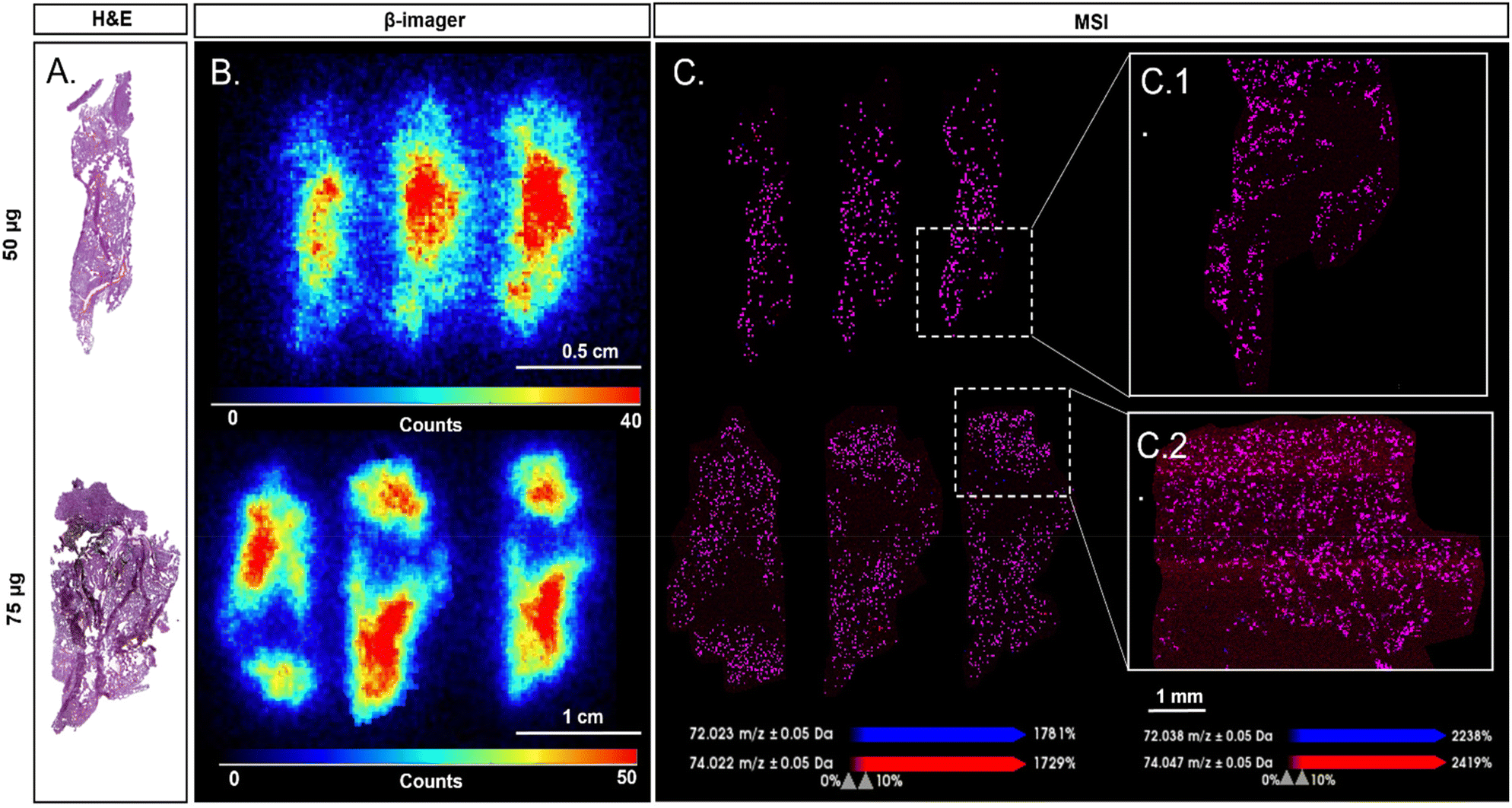 | ||
| Fig. 4 Comparison between H&E, radio and MS imaging of lung sections from mice exposed to 50 μg and 75 μg of 14/12C-GO. (A) H&E staining of a successive slice of lung section. (B) β-Imager acquisition of 50 μg and 75 μg injection dose with a spatial resolution of 150 μm. (C) MSI analysis of the same lung section from mice exposed to 50 μg and 75 μg of 14/12C-GO with a spatial resolution of 25 (insert C.1 and C.2) and 100 μm. The MS images were acquired with an accumulation of 1000 shots per spectrum and per pixel at 2 kHz with an extraction delay of 10 ns, small laser focus and 90% laser fluency. Molecular images of GO were represented using the overlay of maps (purple) obtained for m/z 72 (blue) and 74 (red) ions. | ||
Since MSI is mainly used for molecular mapping, the acquisition of mass spectra at different coordinates allows the representation of several molecules or nanoparticles with an average spatial resolution of 20 μm or less.49,50 This spatial resolution corresponds to the distance between two acquisitions on the same tissue slice. MSI was first performed with a spatial resolution of 100 μm providing a preliminary assessment of the distribution of GO in the different organs. While sensitivity of MSI did not allow the detection of the lower amount of labelled GO present in the spleen and kidneys due to the estimated limit of detection (LOD) of our LDI-MSI method (above 5 ng mg−1 of GO),3014/12C-GO distribution in both lungs and liver was spatially resolved by this method (Fig. S19 and S20†). However, MSI images of the liver sections showed fewer GO specific signals compared to the lungs, which might be counterintuitive considering the distribution results provided by radioimaging. This can be explained by the physiological differences between liver and lungs, and by the fact that MSI images only consider the biodistribution of GO on the slice surface of each organ, while the radioimaging quantification is calculated according to the weight of the whole organ. MSI was thus performed on lungs with a spatial resolution of 25 μm (Fig. 4C and Fig. S21†) providing higher spatial resolution than β-imager, which has an average resolution of 150 μm (Fig. 4B).51
We decided then to compare radioimaging and MSI on lung tissue images. Here, 14/12C-GO images of lungs obtained by LDI-MSI overlapped with radioactivity areas revealed by β-imaging and thus confirmed the presence of labelled GO nanoparticles in exposed mice (Fig. 4). Furthermore, MSI provided additional information on the biodistribution of 14/12C-GO in tissues by highlighting the alveolar aspect of lung tissue (Fig. 4C1 and C2). Areas without radioactivity were also found in MSI images with a finer resolution, allowing an accurate superposition between MSI, β-imaging and H&E staining images. Interestingly, due to the spatial resolution gap between MSI and β-imaging, radio-imaging seems to show a decreasing intensity of the 14/12C-GO from the centre to the edges of the section; this was not found in MSI.
Conclusions
In the present study, we demonstrated how the combination of radioimaging and MS imaging can be used as a correlative imaging approach to provide absolute quantification and high resolution spatial distribution of 14C-labelled graphene oxide in selected organs of accumulation after intravenous administration in mice.Labelled GO nanoparticles dispersed in 0.05% SDBS in water, were detectable by LDI-MS without addition of MALDI matrix, owing to GO aromatic structure favouring desorption/ionisation phenomena. Moreover, a profile of carbon and radical clusters was detected with structures from C2˙− to C9˙−. MS imaging parameters were then optimised to enable the successful detection of GO signature in tissue sections. Biodistribution studies with both β-imager and MSI were finally performed on the same tissue section for each organ tested. Quantitative assessment performed with β-imager showed greater amounts of GO in the liver than in the lungs, spleen and kidneys. Biodistribution within each organ was obtained with a higher spatial resolution for MSI (25 μm) than for β-imager (150 μm) even though the best signal was obtain for the lungs.
Combining the strength of radioimaging quantification and MS imaging spatial resolution, the present work highlights the benefit of the proposed bimodal approach to provide simultaneously absolute quantification and high resolution biodistribution of GBN accumulated within biological tissues. Application of this proof of concept based on advanced quantification and analytical methods for the detection and quantification of GO could be extended to other GBN for the safe and sustainable development of future GBN-based industrial biomedical applications.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Ethical statements
All animal procedures were performed in accordance for Care and Use of Laboratory Animals of the Directive 2010/63/EU of the European Parliament, which had been approved by the Ministry of Agriculture, France. The project was approved by the French Ethics Committee CEEA (Comité d'Ethique en Expérimentation Animale). The French Ethics Committee CEEA (Comité d'Ethique en Expérimentation Animale) is responsible for overseeing the provisions for the care and well-being of animals used for research and educational purposes at CEA-Saclay and serves the public by ensuring compliance with all legal and ethical standards regarding the use of vertebrate animals in research and teaching at CEA-Saclay.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Leon Newman and Dr Artur F. Rodrigues for the AFM analysis, which was performed at the Bio-AFM Facility of the University of Manchester, Faculty of Biology Medicine and Health. This work was supported by the Commissariat à l'Energie Atomique et aux Energies Alternatives (PhD fellowship to H. Cazier) and by the MetaboHUB infrastructure (ANR-11-INBS-0010 grant). This project also received funding of the GrapheneCore3 project from the European Union's Horizon 2020 research and innovation program under grant agreement no. 785219.References
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666 CrossRef CAS PubMed.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183 CrossRef CAS PubMed.
- K. S. Novoselov, V. I. Fal'ko, L. Colombo, P. R. Gellert, M. G. Schwab and K. A. Kim, Nature, 2012, 490, 192 CrossRef CAS PubMed.
- B. G. Nassef, G. A. Nassef and M. A. Daha, Int. J. Mater. Eng., 2020, 10, 1–12 Search PubMed.
- Y. Zhu, H. Ji, H.-M. Cheng and R. S. Ruoff, Natl. Sci. Rev., 2017, 5, 90 CrossRef.
- C. Alvial-Palavicino and K. Konrada, Futures, 2019, 109, 192 CrossRef.
- A. Paul, A. Hasan, H. A. Kindi, A. K. Gaharwar, V. T. Rao, M. Nikkhah, S. R. Shin, D. Krafft, M. R. Dokmeci, D. Shum-Tim and A. Khademhosseini, ACS Nano, 2014, 8, 8050 CrossRef CAS PubMed.
- J. Wu, A. Chen, M. Qin, R. Huang, G. Zhang, B. Xue, J. Wei, Y. Li, Y. Cao and W. Wang, Nanoscale, 2015, 7, 1655 RSC.
- T. Nezakati, A. Seifalian, A. Tan and A.M. Seifalian, Chem. Rev., 2018, 118, 6766 CrossRef CAS PubMed.
- A. B. Seabra, A. J. Paula, R. de Lima, O. L. Alves and N. Duran, Chem. Res. Toxicol., 2014, 27, 159 Search PubMed.
- L. Ou, B. Song, H. Liang, J. Liu, X. Feng, B. Deng, T. Sun and L. Shao, Part. Fibre Toxicol., 2016, 13, 57 CrossRef PubMed.
- K. V. Krishna, C. Menard-Moyon, S. Verma and A. Bianco, Nanomedicine, 2013, 8, 1669 CrossRef CAS PubMed.
- C. Chung, Y.-K. Kim, D. Shin, S.-R. Ryoo, B. H. Hong and D.-H. Min, Acc. Chem. Res., 2013, 46, 2211 CrossRef CAS PubMed.
- M. Nurunnabi, K. Parvez, M. Nafiujjaman, V. Revuri, H. A. Khan, X. Feng and Y. Lee, RSC Adv., 2015, 5, 42141 RSC.
- B. Fadeel, C. Bussy, S. Merino, E. Vázquez, E. Flahaut, F. Mouchet, L. Evariste, L. Gauthier, A. J. Koivisto, U. Vogel, C. Martín, L. G. Delogu, T. Buerki-Thurnherr, P. Wick, D. Beloin-Saint-Pierre, R. Hischier, M. Pelin, F. Candotto Carniel, M. Tretiach, F. Cesca, F. Benfenati, D. Scaini, L. Ballerini, K. Kostarelos, M. Prato and A. Bianco, ACS Nano, 2018, 12, 10582 CrossRef CAS PubMed.
- T. Fan, L. Yan, S. He, Q. Hong, F. Ai, S. He, T. Ji, X. Hu, E. Ha, B. Zhang, Z. Li, H. Zhang, X. Chen and J. Hu, Chem. Soc. Rev., 2022, 51, 7732 RSC.
- S. Syama, W. Paul, A. Sabareeswaran and P. V. Mohanan, Biomaterials, 2017, 131, 121 CrossRef CAS PubMed.
- D. Guarnieri, P. Sánchez-Moreno, A. E. Del Rio Castillo, F. Bonaccorso, F. Gatto, G. Bardi, C. Martín, E. Vázquez, T. Catelani, S. Sabella and P. P. Pompa, Small, 2018, 14, 1800227 CrossRef PubMed.
- H. F. Zhang, K. Maslov, G. Stoica and L. V. Wang, Nat. Biotechnol., 2006, 24, 848 CrossRef CAS PubMed.
- S. Schulz, M. Becker, M. R. Groseclose, S. Schadt and C. Hopf, Curr. Opin. Biotechnol., 2019, 55, 51 CrossRef CAS PubMed.
- J. Xue, H. Liu, S. Chen, C. Xiong, L. Zhan, J. Sun and Z. Nie, Sci. Adv., 2018, 4, eaat9039 CrossRef CAS PubMed.
- A. P. Bowman, R. M. A. Heeren and S. R. Ellis, Trends Anal. Chem., 2019, 120, 115197 CrossRef.
- A. R. Buchberger, N. Q. Vu, J. Johnson, K. DeLaney and L. A. Li, J. Am. Soc. Mass Spectrom., 2020, 31, 1058 CrossRef CAS PubMed.
- D. J. Van, J. M. Spraggins and R. M. Caprioli, Curr. Opin. Chem. Biol., 2019, 48, 64 CrossRef PubMed.
- J. Huang, S. Gao, K. Wang, J. Zhang, X. Pang, J. Shi and J. He, Chin. Chem. Lett., 2022, 107865 CrossRef.
- Y. Lv, T. Li, C. Guo, C. Sun, F. Tang, L. Huang, Z. Luo, X. Li, R. Zhang, Q. Zang, J. He and Z. Abliz, Chin. Chem. Lett., 2019, 30, 461 CrossRef CAS.
- Z. Jing, W. Hao-Yang and G. Yin-Long, Chin. J. Chem., 2005, 23, 185 CrossRef.
- M. C. P. Mendonça, E. S. Soares, M. B. de Jesus, H. J. Ceragioli, M. S. Ferreira, R. R. Catharino and M. A. da Cruz-Höfling, J. Nanobiotechnol., 2015, 13, 78 CrossRef PubMed.
- S. Chen, C. Xiong, H. Liu, Q. Wan, J. Hou, Q. He, A. Badu-Tawiah and Z. Nie, Nat. Nanotechnol., 2015, 10, 176 CrossRef CAS PubMed.
- H. Cazier, C. Malgorn, N. Fresneau, D. Georgin, A. Sallustrau, C. Chollet, J.-C. Tabet, S. Campidelli, M. Pinault, M. Mayne, F. Taran, V. Dive, C. Junot, F. Fenaille and B. Colsch, J. Am. Soc. Mass Spectrom., 2020, 31, 1025 CrossRef CAS PubMed.
- B. Czarny, D. Georgin, F. Berthon, G. Plastow, M. Pinault, G. Patriarche, A. Thuleau, M. Mayne-L'Hermite, F. Taran and V. Dive, ACS Nano, 2014, 8, 5715 CrossRef CAS PubMed.
- X. Zhang, J. Yin, C. Peng, W. Hu, Z. Zhu, W. Li, C. Fan and Q. Huang, Carbon, 2011, 49, 986 CrossRef CAS.
- B. Li, J. Yang, Q. Huang, Y. Zhang, C. Peng, Y. Zhang, Y. He, J. Shi, W. Li, J. Hu and C. Fan, NPG Asia Mater., 2013, 5, e44 CrossRef CAS.
- K. Yang, H. Gong, X. Shi, J. Wan, Y. Zhang and Z. Liu, Biomaterials, 2013, 34, 2787 CrossRef CAS PubMed.
- Y.-J. Lu, C.-W. Lin, H.-W. Yang, K.-J. Lin, S.-P. Wey, C.-L. Sun, K.-C. Wei, T.-C. Yen, C.-I. Lin, C.-C. M. Ma and J.-P. Chen, Carbon, 2014, 74, 83 CrossRef CAS.
- Y. Fazaeli, O. Akhavan, R. Rahighi, M. R. Aboudzadeh, E. Karimi and H. Afarideh, Mater. Sci. Eng., C, 2014, 45, 196 CrossRef CAS PubMed.
- S.-C. Jang, S.-M. Kang, J. Y. Lee, S. Y. Oh, A. E. Vilian, I. Lee, Y.-K. Han, J. H. Park, W.-S. Cho, C. Roh and Y. S. Huh, Int. J. Nanomed., 2018, 13, 221 CrossRef CAS PubMed.
- R. Singh, D. Pantarotto, L. Lacerda, G. Pastorin, C. Klumpp, M. Prato, A. Bianco and K. Kostarelos, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 3357 CrossRef CAS PubMed.
- X. Guo, S. Dong, E. J. Petersen, S. Gao, Q. Huang and L. Mao, Environ. Sci. Technol., 2013, 47, 12524 CrossRef CAS PubMed.
- L. Mao, M. Hu, B. Pan, Y. Xie and E. J. Petersen, Part. Fibre Toxicol., 2016, 13, 57 CrossRef PubMed.
- K. Lu, S. Dong, T. Xia and L. Mao, ACS Nano, 2021, 15, 396 CrossRef CAS PubMed.
- M. Pinault, M. Mayne-L'Hermite, C. Reynaud, V. Pichot, P. Launois and D. Ballutaud, Carbon, 2005, 43, 2968 CrossRef CAS.
- D. Georgin, B. Czarny, M. Botquin, M. Mayne-L'Hermite, M. Pinault, B. Bouchet-Fabre, M. Carriere, J.-L. Poncy, Q. Chau, R. Maximilien, V. Dive and F. Taran, J. Am. Chem. Soc., 2009, 131, 14658 CrossRef CAS PubMed.
- C. Castro, M. Pinault, D. Porterat, C. Reynaud and M. Mayne-L'Hermite, Carbon, 2013, 61, 585 CrossRef CAS.
- B. Lian, J. Deng, G. Leslie, H. Bustamante, V. Sahajwalla, Y. Nishina and R. K. Joshi, Carbon, 2017, 116, 240 CrossRef CAS.
- G. T. T. Le, N. Chanlek, J. Manyam, P. Opaprakasit, N. Grisdanurak and P. Sreearunothai, Chem. Eng. J., 2019, 373, 1212 CrossRef CAS.
- J. H. Liu, S. T. Yang, H. Wang, Y. Chang, A. Cao and Y. Liu, Nanomedicine, 2012, 7, 1801 CrossRef CAS PubMed.
- D. A. Jasim, L. Newman, A. F. Rodrigues, I. A. Vacchi, M. A. Lucherelli, N. Lozano, C. Ménard-Moyon, A. Bianco and K. Kostarelos, J. Controlled Release, 2021, 338, 330 CrossRef CAS PubMed.
- A. D. Feenstra, M. E. Dueñas and Y. J. Lee, J. Am. Soc. Mass Spectrom., 2017, 3, 434 CrossRef PubMed.
- M. Kompauer, S. Heiles and B. Spengler, Nat. Methods, 2017, 14, 90 CrossRef CAS PubMed.
- N. Barthe, K. Chatti, P. Coulon, S. Maîtrejean and B. Basse-Cathalinat, Nucl. Instrum. Methods Phys. Res., Sect. A, 2007, 527, 41 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr06753f |
| This journal is © The Royal Society of Chemistry 2023 |