
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning the electrical properties of graphene oxide through low-temperature thermal annealing†
Cataldo
Valentini
 a,
Verónica
Montes-García
a,
Pietro Antonio
Livio
a,
Tomasz
Chudziak
bc,
Jésus
Raya
d,
Artur
Ciesielski
*ab and
Paolo
Samorì
*a
a,
Verónica
Montes-García
a,
Pietro Antonio
Livio
a,
Tomasz
Chudziak
bc,
Jésus
Raya
d,
Artur
Ciesielski
*ab and
Paolo
Samorì
*a
aUniversité de Strasbourg and CNRS, ISIS, 8 allée Gaspard Monge, 67000 Strasbourg, France. E-mail: ciesielski@unistra.fr; samori@unistra.fr
bCentre for Advanced Technologies, Adam Mickiewicz University, Uniwersytetu Poznańskiego 10, 61-614 Poznań, Poland
cFaculty of Chemistry, Adam Mickiewicz University, Uniwersytetu Poznańskiego 8, 61-614 Poznań, Poland
dUniversité de Strasbourg, CNRS, Membrane Biophysics and NMR, Institute of Chemistry, 1 Rue Blaise Pascal, 67000 Strasbourg, France
First published on 16th February 2023
Abstract
During the last fifteen years, the reduction of electrically insulating graphene oxide (GO) through the elimination of oxygen containing functional groups and the restoration of sp2 conjugation yielding its conducting form, known as reduced graphene oxide (rGO), has been widely investigated as a scalable and low-cost method to produce materials featuring graphene-like characteristics. Among various protocols, thermal annealing represents an attractive green approach compatible with industrial processes. However, the high temperatures typically required to accomplish this process are energetically demanding and are incompatible with the use of plastic substrates often desired for flexible electronics applications. Here, we report a systematic study on the low-temperature annealing of GO by optimizing different annealing conditions, i.e., temperature, time, and reduction atmosphere. We show that the reduction is accompanied by structural changes of GO, which affect its electrochemical performance when used as an electrode material in supercapacitors. We demonstrate that thermally-reduced GO (TrGO) obtained under air or inert atmosphere at relatively low temperatures (<300 °C) exhibits low film resistivities (10−2–10−4 Ω m) combined with unaltered resistance after 2000 bending cycles when supported on plastic substrates. Moreover, it exhibits enhanced electrochemical characteristics with a specific capacitance of 208 F g−1 and a capacitance retention of >99% after 2000 cycles. The reported strategy is an important step forward toward the development of environmentally friendly TrGO for future electrical or electrochemical applications.
1. Introduction
Graphene oxide (GO) is a unique multifunctional two-dimensional material (2DM) composed of carbon, oxygen, and hydrogen atoms in variable ratios. Firstly synthesized almost 150 years ago,1 nowadays GO is mainly produced by treating natural graphite flakes with strong oxidants, primarily by making use of the modified Hummers’ method.2 The presence of different oxygenated functional groups (OFGs), such as epoxy, hydroxyl and carbonyl,3,4 on the basal plane and the edges provides GO with a unique set of mechanical and optical properties along with a good dispersibility and colloidal stability in many solvents, particularly in water.5–7 In addition, by using well-known chemistry strategies, these OFGs can serve as sites for chemical modification, endowing GO with potential applicability in chemical sensing,8 solar cells,9 drug delivery,10 water desalination11 or as an active material in energy storage systems (ESSs)12 among others. Unfortunately, because most of the GO production methods use strong oxidants, such as potassium permanganate, GO possesses a significant number of defects in its crystalline structure. This is concomitant with the loss in electrical conductivity, which limits the direct application of GO in electronics.13,14 Tremendous efforts have been devoted toward removing OFGs from GO via reduction processes with the goal of enhancing the degree of conjugation in the carbon network through the formation of sp2 species, ultimately boosting the electrical properties of the material aiming at approaching the characteristics of graphene.15–17 GO reduction yielding electroactive reduced graphene oxide (rGO) can be achieved through a variety of (electro)chemical (CrGO) or thermal (TrGO) methods.18,19 Chemical reduction is currently the most common and efficient approach for reducing GO using a plethora of reducing agents including hydrazine, sodium borohydride (NaBH4), hydrogen iodide, or L-ascorbic acid.20–26 However, the price of chemicals, together with the high quantities of generated chemical waste, makes this methodology industrially undesirable.27 Chemical reagents can be avoided when employing the electrochemical method. In this case, the reduction process relies on the GO-electrode electron exchange, and it can be performed inside an electrochemical cell in the presence of an aqueous buffer solution. Yet, in electrochemical reduction, the deposition and reduction of GO is limited to substrates that can act as electrodes. Even though chemical reduction is one of the most efficient and therefore the most used solution-processed protocols, it is inconvenient as it yields a stoichiometric amount of chemical waste. Therefore, thermal reduction represents one of the most attractive reduction methods due to its low environmental impact. Thermal reduction comprises the annealing of GO under a controlled atmosphere, leading to H2O, CO2, and CO as desorption products. It is now well established that under vacuum and annealing temperature ranging between 100 and 185 °C, interlamellar H2O molecules are desorbed, while epoxide and carbonyl groups are removed between 185–300 °C and 500–700 °C, respectively. In the ranges 700–900 °C and 900–1200 °C ether and hydroxyl groups are removed, leading to a material with a high C/O ratio.28,29 However, such high temperatures are energetically very demanding and incompatible with GO thermal reduction on plastic substrates for flexible electronics. Typical substrates employed in flexible and wearable electronics, such as polyethyleneterephthalate (PET)30 and polyimide (PI),31 retain their mechanical properties at temperatures up to 175 °C and 232 °C, respectively.30,31 For these reasons, the thermal reduction of GO at relatively low temperatures is highly sought after.During the last few years, a few works have been reported where the thermal reduction of GO was achieved at relatively low temperatures (<300 °C) (Table S1, ESI†). For instance, it has been found that the reduction temperature can be decreased to 150 °C at atmospheric pressure when GO is dispersed in propylene carbonate as well as in a mixture of water and organic solvents.32 Grossman et al. measured the sheet resistance of GO films annealed at 50 and 80 °C in air. After five days of annealing at 80 °C, the sheet resistance dropped from 1010 Ω sq−1 of pristine GO to 105 Ω sq−1 mainly due to the interlamellar H2O desorption.33 Chhowalla et al. reported that GO films reduced under ultra-high vacuum (UHV) for 15 minutes at 150 and 200 °C have a resistivity of about 10 and 10−2 Ω m, respectively, and when the reduction is performed in Ar/H2 for 15 minutes at 200 °C, a resistivity of 10−2 Ω m was obtained.34
Thermal reduction in air has been investigated by Nazarov et al. in a range of temperatures from 130 to 200 °C with an annealing time of 15 minutes, showing a decrease in resistivity from 4 × 105 Ω m to 4 × 10−2 Ω m. The latter was ascribed to a mechanism comprising first H2O desorption, followed by OFG desorption releasing CO and CO2.35 These last two examples highlight that independently from the reduction atmosphere, when annealing is performed at 200 °C for 15 minutes, the resistivity drops always to ∼10−2 Ω m. Sun et al. studied the kinetics and thermodynamics of GO deoxygenation under argon by thermogravimetric analysis coupled with a mass detector. The CO and CO2 signals dramatically increased with a temperature near 180 °C, yet traces of CO2 were already detected at 130 °C.36 Although these examples prove that GO thermal reduction can be accomplished at relatively low temperatures, the relationship between changes in conductivity with other parameters such as the chemical composition, surface area or crystallinity of TrGO has not yet been elucidated. To the best of our knowledge, there is no systematic study that explores and optimizes the annealing conditions and correlates them with the changes in the GO structure and functions. For instance, as can be seen in Table S1† the reported annealing time has been varied between 15 minutes and 9 days depending on the temperature used. To fill such an important gap, here we report the thermal reduction of GO at different low temperatures (i.e., 100, 130, 150, 200, 250 and 300 °C) under air and inert atmospheres. We investigated the effect of annealing conditions on the chemical structure, various physico-chemical parameters, and the electrochemical performance of the resulting TrGO used as an electrode in a symmetric supercapacitor with a two-electrode system using KOH 6 M as an aqueous electrolyte. Graphene-based materials, such as GO, rGO or high-level oxygen-functionalized GO, have been widely used as active materials in ESS.37–41 In particular, OFGs endow these materials with a pseudocapacitive nature, making them ideal candidates for supercapacitors. It has been reported that the pseudocapacitance of GO probably comes from electrochemical reactions such as >C–OH ↔ >C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O + H+ + e− at the electrode interfaces.37 For instance, Li et al. reported that high-level oxygen-functionalized GO exhibits not only an exceptional supercapacitor performance, reaching a value of specific capacitance as high as 285 F g−1 at 1 A g−1, but also a high surfaces area of 512 m2 g−1.40 Interestingly, a similar electrochemical performance (capacitance of 264 F g−1 at 0.1 A g−1) was achieved for exfoliated graphene, having a much lower density of OGFs.42 Therefore, not only the presence and density of OFGs are key to achieve a state-of-the-art electrochemical performance, but also other parameters should be carefully controlled. In this work, we investigated the fine tuning of the reduction degree of GO by varying the annealing temperature, time and atmosphere to allow the modulation of the surface area, conductivity and density of electrochemically-active groups, boosting the electrochemical performance of GO. To demonstrate the full compatibility of the reduction strategy with the use of plastic substrates, we fabricated proof-of-concept devices consisting of ca. 66 nm thick TrGO films supported on flexible polyethylene terephthalate (PET) substrates and monitored their resistance during 2000 bending cycles.
O + H+ + e− at the electrode interfaces.37 For instance, Li et al. reported that high-level oxygen-functionalized GO exhibits not only an exceptional supercapacitor performance, reaching a value of specific capacitance as high as 285 F g−1 at 1 A g−1, but also a high surfaces area of 512 m2 g−1.40 Interestingly, a similar electrochemical performance (capacitance of 264 F g−1 at 0.1 A g−1) was achieved for exfoliated graphene, having a much lower density of OGFs.42 Therefore, not only the presence and density of OFGs are key to achieve a state-of-the-art electrochemical performance, but also other parameters should be carefully controlled. In this work, we investigated the fine tuning of the reduction degree of GO by varying the annealing temperature, time and atmosphere to allow the modulation of the surface area, conductivity and density of electrochemically-active groups, boosting the electrochemical performance of GO. To demonstrate the full compatibility of the reduction strategy with the use of plastic substrates, we fabricated proof-of-concept devices consisting of ca. 66 nm thick TrGO films supported on flexible polyethylene terephthalate (PET) substrates and monitored their resistance during 2000 bending cycles.
2. Results and discussion
2.1. Annealing conditions
The thermal reduction of GO under air and N2 atmospheres was first followed by thermogravimetric analysis (TGA) (Fig. S1, ESI†). The two diagrams show a similar trend, with a first mass loss at 75 °C attributed to interlayer H2O desorption and a second mass loss at around 200 °C due to CO and CO2 desorption (Fig. S1, ESI†), achieving a total mass loss of ∼45% of the initial weight at 300 °C in agreement with the literature.33 We then monitored the kinetics of the reduction process by means of TGA with a ramp of 10 °C min−1 and an isotherm of 4 hours at 100, 130, 150, 200, 250 and 300 °C (Fig. 1 and S2, ESI†). Interestingly, to the best of our knowledge, TGA kinetic analysis of GO thermal reduction is still unexplored. Under both atmospheres, for 100, 130 and 150 °C, the TrGO weight constantly decreases for 4 hours with a mass loss of about 20, 28 and 35%, respectively. At higher temperatures, the plateau of mass loss (ca. 45%) is reached after ∼30 minutes. The kinetic analysis at 150 °C is followed up to 24 hours (Fig. S3, ESI†) and the plateau of ∼45% mass loss was achieved after 12 hours for both atmospheres. TGA kinetic analysis reveals that 150 and 200 °C are the key threshold temperatures to produce TrGO with a good degree of reduction. However, to gain greater insight into the reduction process, the electrical resistivity is used as an internal gauge by performing thin film conductivity measurements. The films of GO (thickness of 66 ± 11 nm, Fig. S4 and Table S2, ESI†) were annealed at 150 and 200 °C under air and under argon, and the film resistivity was measured at different annealing times with a four-point probe (FPP) to follow the kinetics of the reduction process (Fig. 1b and S5, ESI†). Importantly, N2 and argon are tested as inert atmospheres (see the Experimental section for details) and no substantial differences in the electrical characteristics are observed on the resulting TrGO. Under air and inert atmospheres (Ar and N2), the film resistivity gradually decreases upon annealing at 200 °C with the lowest resistivity being achieved after 4 hours (∼3 × 10−3 Ω m). At 150 °C under air, the onset of conductivity in TrGO is observed after 4 hours of reduction with the resistivity continuously decreasing even after 24 hours (∼3 × 10−2 Ω m). Conversely, when the annealing is performed under argon, the onset of TrGO's conductivity (∼2 × 10−1 Ω m) is observed only after 24 hours. To demonstrate the full compatibility of our reduction process with substrates employed in flexible electronics, the annealing of a GO film deposited on polyethylene terephthalate (PET) was performed at 150 °C for 24 h. Then, the mechanical stability of the films was tested by performing 2000 bending cycles (Fig. S6, ESI†). The resistance of the film is constant for the 2000 bending cycles performed with a subtle increase below 1%. Although 24 hours are needed to produce conductive TrGO when the annealing is executed at 150 °C, the reduction time can be reduced to 4 hours when the annealing is performed at temperatures ≥200 °C. Therefore, our subsequent characterization studies were carried out after annealing the samples for 4 hours since the TGA analyses show the plateau achievement for most of the reduction temperatures investigated (Fig. 1a). Fig. 1c shows the room-temperature resistivity of 66 nm thick films of TrGO prepared under air and argon at different annealing temperatures. The resistivity measured for TrGO is above the detection limit of the employed instrument (i.e. 107 Ω sq−1) for temperatures below 150 °C under argon and below 100 °C under air. Under an argon atmosphere, from 200 °C to 300 °C, the resistivity of TrGO decreases from 3 × 10−3 to 4 × 10−4 Ω m, thereby approaching the resistivity of graphite (10−5 Ω m)43–45 and two orders of magnitude lower than that of rGO annealed at 200 °C in Ar/H2 or UHV (10−2 Ω m).34 In contrast, the thermal reduction of GO under air shows a constant decrease in film resistivity from 1 Ω m at 130 °C to 10−3 Ω m at 250 °C. Conversely, at 300 °C, an increase in the resistivity of TrGO is observed, which could be ascribed to the partial oxidation of the GO layers since its ignition temperature is 317 °C.46 The differences in the kinetics obtained from TGA and the film resistivity measurements may suggest structural reorganization of Csp3–O to Csp2–O taking place, which is not accompanied by any weight changes, yet, it determines a transition of the electrical nature of GO from insulating to conductive.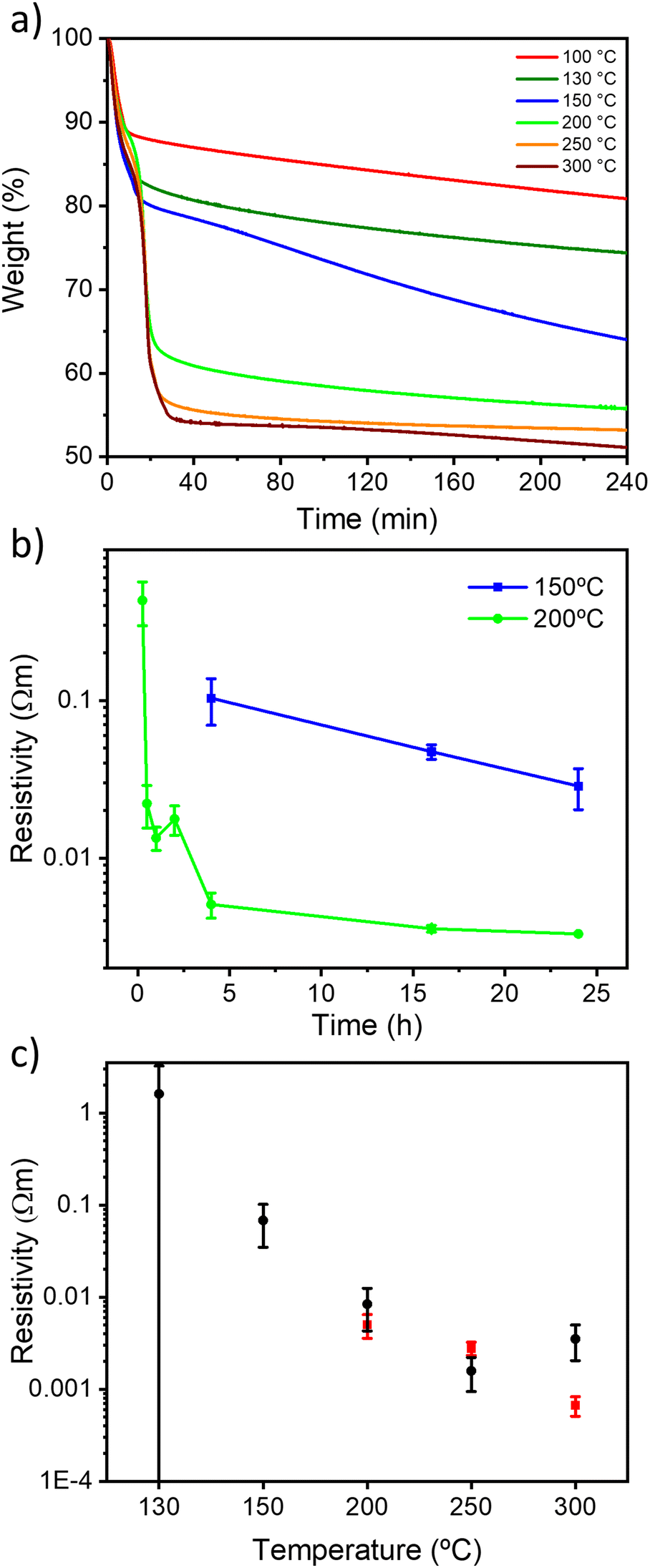 | ||
| Fig. 1 (a) TGA measurements of weight loss versus time for the annealing of GO in air at different temperatures for 4 h, (b) kinetic resistivity measurements for the annealing of GO in air at 150 °C and 200 °C for 24 hours, and (c) resistivity measurements for TrGO in air (black dots) and argon (red dots) for 4 hours. | ||
2.2. Compositional, chemical and structural characterization
To gain insight into the chemical composition of TrGO, X-ray photoelectron spectroscopy (XPS) analyses were performed on the GO powders annealed for 4 hours (Fig. 2 and S7–S9, ESI†). From the survey spectra, a C/O ratio of 2.5 is estimated for pristine GO (Fig. S7, ESI†). During the annealing, H2O, CO and CO2 are released and, consequently, the C/O ratio increases, indicating the reduction process. The C/O ratio increases between 130 and 200 °C for the samples annealed either under air or nitrogen; however, while the C/O ratio increases gradually when the annealing is performed under N2, more rapid changes are monitored when the annealing is performed in air (Fig. S7, ESI†). For instance, at 150 °C the C/O ratio is 5.05 versus 3.40 for TrGO under air and N2, respectively, indicating that the reduction process at lower temperatures occurs more efficiently in air when compared to the N2 environment. The high-resolution C 1s spectrum is fitted using 5 Gaussian–Lorentzian curves for the 5 chemical environments: 284.4 eV C–C (Csp2–Csp2), 285.15 eV C–O (including Csp2–O–Csp2, Csp3–OH and Csp2–OH), 286.48 eV C–O–C (Csp3–O–Csp3), 287.38 eV CO, and 288.50 eV COOR (including COOH and lactone) (Fig. 2a and b and S8a, ESI†).47Fig. 2c and d show the evolution of the area of each component as a function of annealing temperature. As expected, the area of the C–C peak increases with the annealing temperature under both atmospheres due to the decreasing amount of oxygen in the sample. On the other hand, the area of the C–O–C peak decreases abruptly at 150 °C when annealing is performed under air and at 200 °C when it is performed under N2. However, the area of the other three peaks remains roughly constant. To gain a greater understanding of the nature of the OFGs, the high-resolution XPS of O 1s spectrum is fitted with 3 Gaussian–Lorentzian curves: 531.08 eV CO, 532.03 eV Csp3–O (including Csp3–O–Csp3, and Csp3–OH), and 533.43 eV Csp2–O (including Csp2–O–Csp2 and Csp2–OH) (Fig. S8b and S9, ESI†).47Fig. 2e and f show how the relative area of each component varies with the annealing temperature. The area of the Csp3–OH peak decreases starting from 150 °C when the reduction is performed under air and from 200 °C under N2, following the same trend as the C–O–C peak. In parallel, the relative area decrement of the Csp3–O peak is accompanied by a drastic increase in the area of the Csp2–O peak, indicating the inclusion of oxygen atoms in the conjugated carbon network. From XPS analysis, we can conclude that the threshold temperatures for the thermal reduction of GO under air and N2 amount to 150 °C and 200 °C, respectively. Further insight into the chemistry of the reduction process was obtained by solid-state NMR magic angle spinning (ssNMR-MAS) analyses (Fig. 3). As previously reported,48–51 the ssNMR spectrum of GO is deconvoluted in eight curves (Fig. S10, ESI†) corresponding to 60.4 ppm (13Csp3–O–13Csp3), 70.6 ppm (13Csp3–OH), 78.9 ppm (13Csp3–OH, close to defects), 100.2 ppm (13C–OOR), 126.7 ppm (13Csp2–13Csp2), 134.7 ppm (13Csp2–13Csp2 close to defects), 162.4 ppm (Csp2–O (including Csp2–O–Csp2 and Csp2–OH)) and 187.9 ppm (13CO).48 In agreement with the XPS analysis, the intensity of peaks related to sp3 carbons decreases with the increasing annealing temperature, accompanied by the increase of the intensity of sp2 carbon peaks (Fig. 3c and d). At 150 °C under argon (Fig. 3b and d), a small decrease in the area of 13Csp3–O–13Csp3 and 13Csp3–O peaks is observed with respect to the pristine GO spectra. However, at 150 °C under air (Fig. 3a and c), these peaks are almost suppressed and the 13Csp2–13Csp2 peak arises significantly. The spectra of TrGO at 300 °C under both atmospheres show almost the same profile, the 13Csp2–13Csp2 peaks being predominant (Fig. 3a and b). As observed from the XPS analysis, the only OFG formed during the annealing is 13Csp2–O and a quantitative evaluation is done by plotting the relative areas of the fitted ssNMR spectra as a function of the reduction temperature (Fig. 3c and d). The 13Csp2–O peak increases drastically from 4.7% to 31.2% after the reduction under air at 150 °C and is kept almost constant at 300 °C (27%). However, when the reduction is performed under argon, only at 300 °C the 13Csp2–O peak increases sharply to 37%. The generation of 13Csp2–O species may be the confirmation of our initial statement where we observed no changes in the weight in the TGA analysis, yet the film resistivity continues to decrease for a certain period of time. In full agreement with the high-resolution XPS, the ssNMR experiments confirm that the reduction of GO can occur at a lower temperature under air when compared to the inert atmosphere.
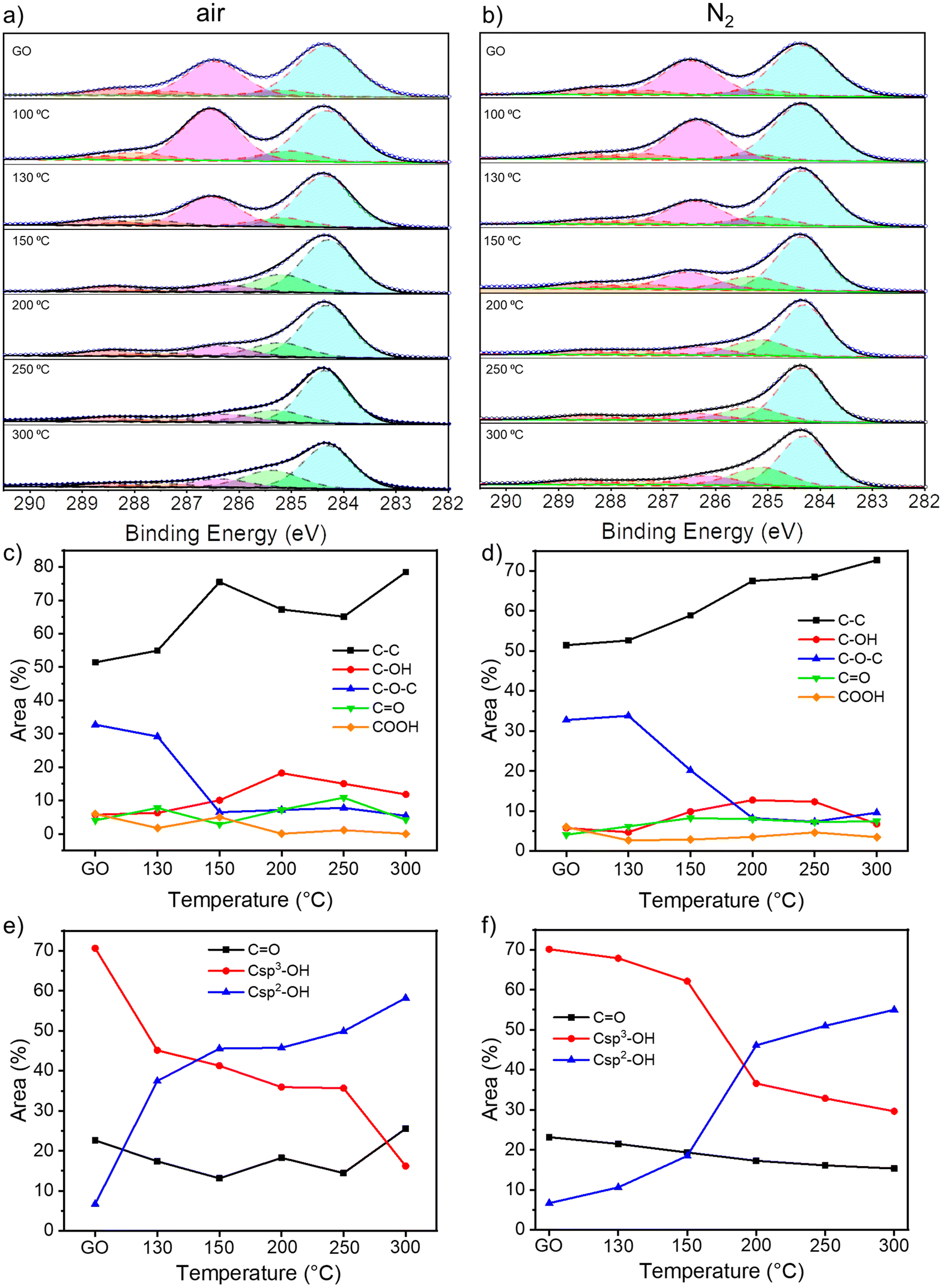 | ||
| Fig. 2 High-resolution XPS C 1s peaks of thermally reduced GOs under (a) air and (b) N2; temperature dependence of the relative contribution of C 1s peak components estimated by dividing the area under each component by the whole C 1s peak area for (c) air and (d) N2; temperature dependence of the relative contribution of O 1s peak components estimated by dividing the area under each component by the whole O 1s peak area for (e) air and (f) N2. | ||
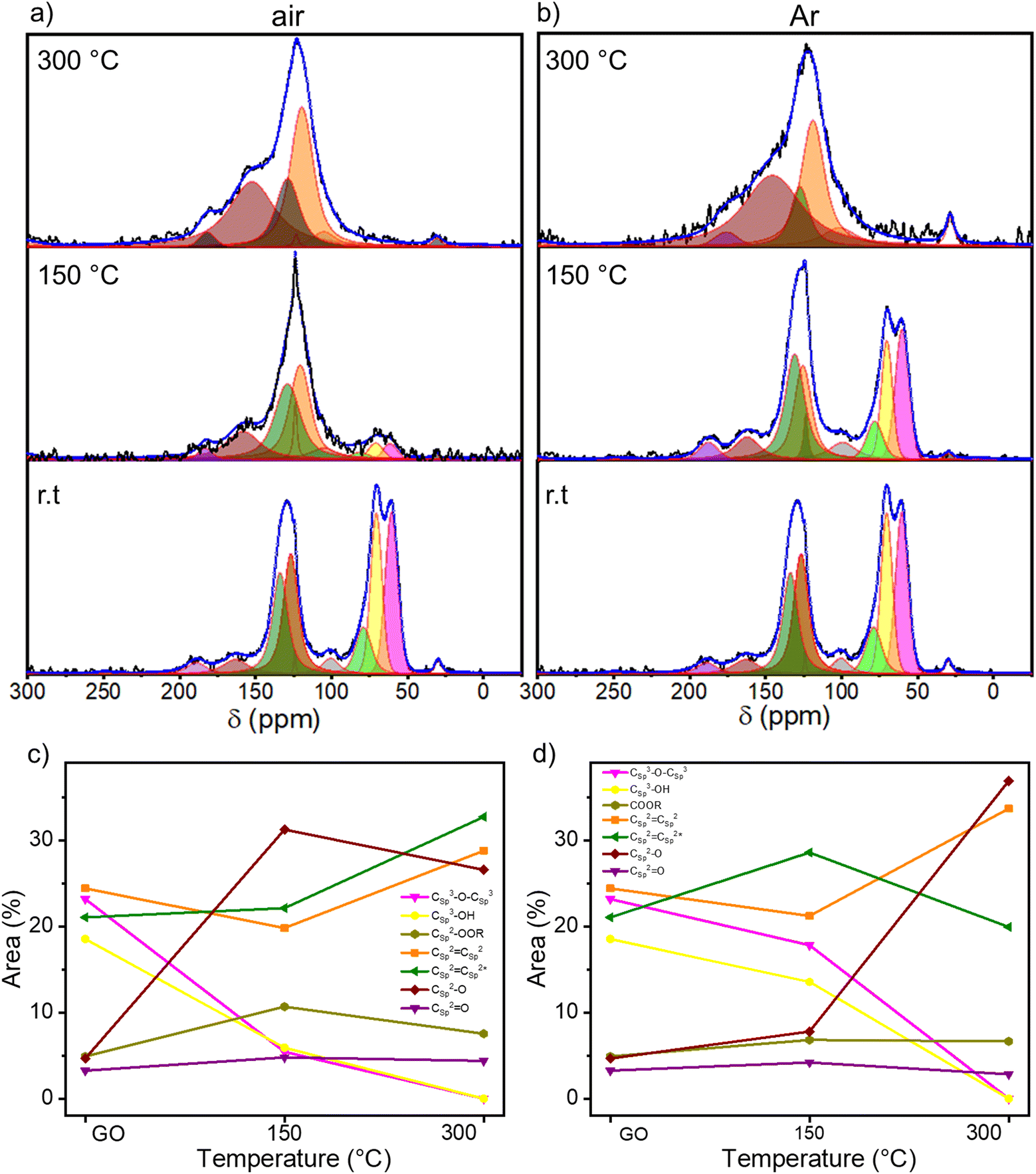 | ||
| Fig. 3 Solid-state MAS-NMR C 1s spectra of thermally rGO under (a) air and (b) argon; temperature dependence of the relative contribution of NMR spectral components estimated by dividing the area under each component by the whole peak area for (c) air and (d) argon. | ||
As previously reported, the features in the Raman spectra of GO and rGO (i.e., band position, intensity ratio, and width) can be related to structural properties such as the oxygen content, crystallinity, and reduction degree of GO.52–55 The Raman spectra of TrGO are deconvoluted using four Lorentzian curves, which consist of the first-order Raman modes, namely: D, G, D′′, and D′ bands (Fig. S11–S14 and Tables S3 and S4, ESI†). The D band, located at 1330–1355 cm−1, arises from the A1g breathing modes of the six-membered rings that are activated by defects such as vacancies, grain boundaries, and disorders in the carbon lattice and the double resonant processes near the K point of the Brillouin zone (BZ) boundary.55 The G band at 1580–1600 cm−1 corresponds to the first-order allowed Raman mode E2g.52 The additional bands (D′ and D′′) arise from the defects present in the graphitic structure of the carbon material. The physical origin of the D′ band at 1610–1620 cm−1 is still not fully understood. Some authors have attributed it to the disorder-induced phonon mode due to crystal defects, whereas other authors have attributed this band to the double vacancy corresponding to pentagonal and octagonal rings, usually referred to as 5–8–5 defects.52 The D′′ band at 1500–1550 cm−1 is related to the amorphous phase and its intensity is inversely related to the crystallinity.56 It is well established that the relative intensity of the D band with respect to the G band (ID/IG ratio) is an insightful parameter to estimate the degree of defects in the GO derivatives and it has been related to the inverse of the crystallite size on basal planes (1/La) through the Tuinstra–Koenig model.57 Upon GO thermal reduction, the intensity of the G peak increases and hence the ID/IG ratio decreases with the annealing temperature (Fig. S13, ESI†). The oxygen atomic percentage obtained by XPS is also included in Fig. S13, ESI† for a better understanding of the degree of GO reduction. Fig. S13a, ESI† shows that upon thermal reduction under air, an abrupt decrease of both the ID/IG ratio and oxygen atomic percentage at temperatures ≥150 °C is observed, with both parameters becoming stable (ID/IG = ∼1.42 and ∼15% oxygen atoms). However, when the thermal reduction is performed under N2, a more gradual decrease in both parameters is observed, reaching similar minimum values to the reduction under air above 250 °C (Fig. S13b, ESI†). The intensities of the D′ and D′′ bands also decrease with the degree of reduction, as the number of defects on the basal plane of GO is lowered. Fig. S14, ESI† shows that the ID′/IG ratio and the ID′′/IG ratio decrease with the annealing temperature both under air and N2. It is also important to note that while the ID′/IG ratio decreased to ∼20% of its initial value, the ID′′/IG ratio decreased ∼45% (under air) and ∼35% (under N2) of their respective initial values. From this result two conclusions can be drawn: (i) thermal annealing reduces mainly the defects related to the D′′ band and (ii) this process is more pronounced for annealing under air, leading to a more crystalline structure.52,53 The effect of temperature on the crystallinity of TrGO is investigated by powder X-Ray diffraction (PXRD) (Fig. 4 and S14–S16†). Pristine GO diffraction pattern displays one characteristic peak at 2θ = ∼11° (peak I) with a full-width half maximum (FWHM) of ∼ 1.2° related to the (002) family of planes (Fig. 4a and b).
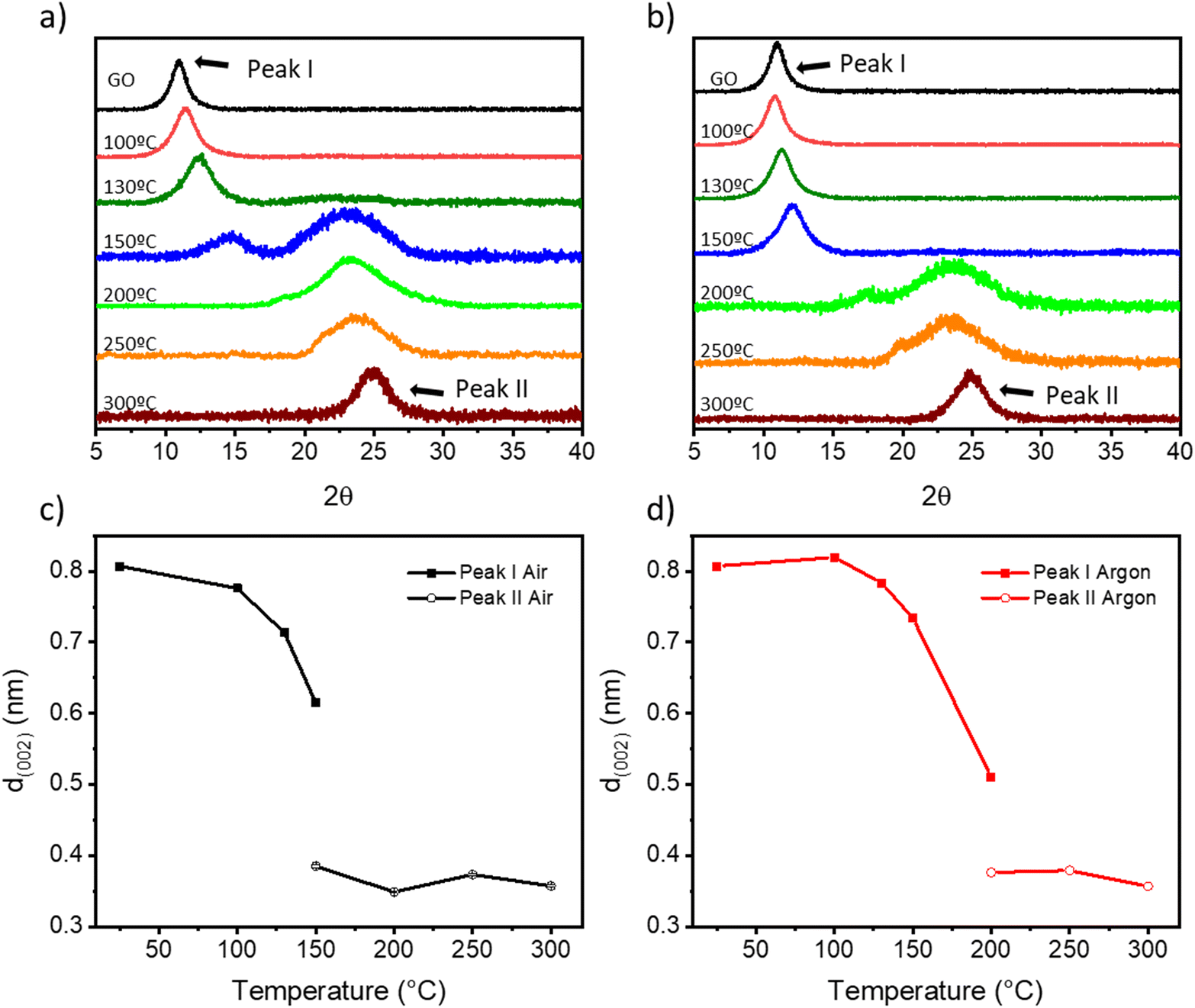 | ||
| Fig. 4 (a and b) Diffraction patterns of TrGO under (a) air and (b) argon at different temperatures; (c and d) calculated d-spacing for peak I (full squares) and peak II (empty circles) for the samples annealed under (c) air and (d) argon. | ||
On the other hand, rGO exhibits one characteristic peak at 2θ = ∼25° (peak II) with a larger FWHM of ∼5°, five times larger compared to peak I, which indicates a smaller crystallite size.58Fig. 4a and b show a gradual disappearance of peak I and a gradual increase of peak II upon increasing the annealing temperature. In agreement with previous characterization studies, such as TGA, XPS and Raman spectroscopy, when the annealing is performed under air, peak II appears at a lower temperature (i.e., 150 °C) compared to annealing performed under argon (i.e., 200 °C), and the full reduction is achieved at 200 °C under both atmospheres. From the PXRD patterns different parameters can be determined (e.g., interlayer distance and the number of GO layers) (see the Experimental section for details). First, from the scattering angle (2θ) of each peak (Fig. S15, ESI†), we can quantify the d-spacing parameter (Fig. 4c and d). At 150 °C in air and 200 °C in argon, there is the coexistence of peaks I and II, hence two d-values are determined. The estimated d-spacing of GO is ∼0.8 nm, in agreement with the reported values.52,59 When GO is annealed under air from 100 to 150 °C, peak I shifts toward higher scattering angles, indicating a contraction of the interlayer spacing. This effect can be attributed to the evaporation of the entrapped H2O. Conversely, when GO is annealed under argon from 100 to 150 °C, only a slight shift of peak I from 11° to 12° is recorded (Fig. 4b). Above 150 °C (under air) and 200 °C (under argon), peak I intensity drops significantly and peak II becomes the major feature in the diffractogram with a calculated d-spacing of TrGO of ∼0.3 nm.52,59 The further shrinking of the interlayer distance is related to the partial removal of the OFGs from the GO sheets. Furthermore, considering the crystallite size (Lc) (Fig. S16, ESI†) and the interlayer spacing d (002) (Fig. 4c and d), we estimated the theoretical number of (r)GO sheets per crystallite domain (Fig. S17, ESI†). Our results clearly show that for GO annealed in air few layer-thick rGO sheets (4–6 layers) can be produced already at 150 °C. Remarkably, the low-temperature annealing under both atmospheres (i.e., air and argon) allows us to produce high-quality few-layer rGO, starting from commercially available GO with a high percentage of monolayers (>95%) with promising characteristics for integration into electronic devices. The porosity of (Tr)GO annealed at different temperatures under air or argon is evaluated by recording N2 adsorption–desorption isotherms at 77 K (Fig. S18 and Table S5, ESI†). The adsorption isotherms of TrGO exhibited type-I sorption isotherms, with steep rises appearing at a low relative pressure and type-IV sorption features with adsorption/desorption hysteresis at higher pressure. The calculated Brunauer–Emmett–Teller (BET) surface area of TrGO increases gradually with the rising annealing temperature up to 300 °C, from 12.61 m2 g−1 for the as-prepared GO to 480.44 m2 g−1, when the annealing is performed under air. In contrast, when the annealing is performed under argon the surface area abruptly increases at 200 °C to 439.92 m2 g−1, and then remains roughly constant. The average pore size increases gradually with thermal treatment temperature, following the same tendency as the specific surface area (Table S5, ESI†). Fig. S18c and d, ESI† show the differential distribution of pore volumes versus pore sizes for TrGO under (S18c†) air and (S18d†) argon. It reveals that all materials have a predominant mesopore distribution with sizes in the range between 10 and 100 nm. The work function of (Tr)GO was calculated by photoelectron yield spectroscopy in air (PYSA) (Fig. S19, ESI†). The work function of GO (5.59 ± 0.03 eV) decreases gradually with the increase in the annealing temperature under both atmospheres until reaching a stable value of ∼5.14 eV.
2.3. Electrochemical characterization
The electrochemical properties of all the GO samples were evaluated in a symmetrical two-electrode cell by cyclic voltammetry (CV), galvanostatic charge/discharge (GCD) and electrochemical impedance spectroscopy (EIS). Fig. 5a and d show the cyclic voltammograms of GO thermally reduced under (a) air and under (d) argon using a 6 M KOH aqueous electrolyte between 0 and 0.6 V at a scan rate of 50 mV s−1. The CV curves of TrGO show a quasi-rectangular shape, indicating the pseudocapacitive nature of (Tr)GO.37 When the reduction is performed under air, the transition from GO to TrGO is gradual, starting at 130 °C but reaching an actual quasi-rectangular shape from 150 °C. The area of the CV curve increases with the temperature of annealing up to 200 °C. Above 250 °C, the voltammogram starts to contract slightly, which is attributed to the initiation of the combustion of the material, as demonstrated by TGA and film resistivity measurements. Interestingly, when the reduction is performed under argon there are only two clear states of reduction. At 200 °C the area of the CV curves abruptly increases, and above this temperature, the CV curves became stable. Rate capability is also an important feature for energy storage devices. The CV curves for GO (Fig. S20, ESI†) and TrGO under air (Fig. S21, ESI†) and under argon (Fig. S22, ESI†) at different scan rates were obtained. When GO is annealed above 150 °C or 200 °C under air or argon, respectively, the cyclic voltammograms remain in box-like shapes with a little deviation at lower potentials, even when the scan rate increases to 200 mV s−1, implying a quick charge propagation and hence a good rate capability.42 On the other hand, the CV curves of GO and GO annealed below 150 °C or 200 °C under air or argon respectively deviate from the ideal box-shape, indicating that the device is being overcharged, as a result of the poor material conductivity as well as the occurrence of parasitic side reactions, such as decomposition of the electrode material, the electrolyte, or a combination of both.60 This finding is in full agreement with the film resistivity analysis.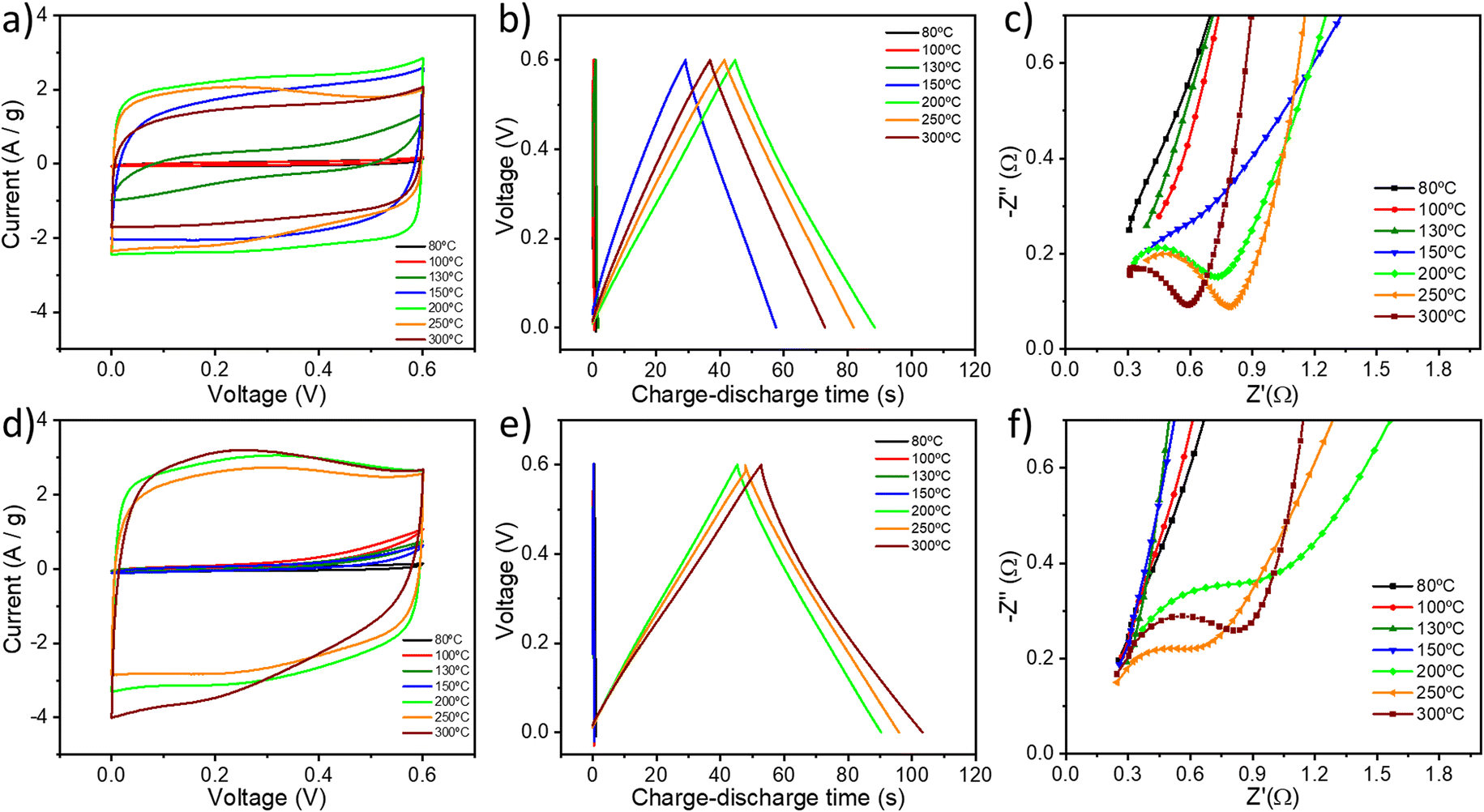 | ||
| Fig. 5 Electrochemical characterization of thermally reduced GO at different temperatures under (a–c) air and under (d–f) argon. (a and d) CV curves at a scan rate of 50 mV s−1. (b and e) GCD curves at a current density of 1 A g−1. (c and f) Magnification of the high-frequency region of the Nyquist plots. | ||
The pseudocapacitive behaviour of TrGO under two different atmospheres is confirmed by galvanostatic charge/discharge curves, as shown in Fig. 5b and e. The voltage–time curve exhibits a quasi-linear shape under both atmospheres and similar trends to those obtained by CV. The specific capacitances of TrGO were calculated from GCD curves at different current densities, as can be seen in Fig. S23 and S24, ESI† (see the Experimental section for calculation details). With an increase of the current density, the capacitances of GO and TrGO decrease very slowly under both atmospheres, indicating, in agreement with CV at different scan rates, that (Tr)GO has excellent rate capability. The maximum specific capacitance for TrGO reduced under air amounts to 154.8 ± 5.4 F g−1 at a current density of 0.1 A g−1 (Fig. S24, ESI†) for samples reduced at 200 °C. On the other hand, the maximum value observed under argon is slightly higher 173.5 ± 12.6 F g−1 at a current density of 0.1 A g−1 (Fig. S24, ESI†) at 300 °C. These values represent an outstanding 60-fold increase when compared to the initial capacitance of GO (∼2.5 F g−1). The values of specific capacitance measured for TrGO are similar using 1 M H2SO4 and 6 M KOH as aqueous electrolytes (Fig. S25, ESI†).61
The evolution of the specific capacitance values as a function of the annealing temperature can result from three different contributions. First, the increase in surface area with the annealing temperature (Fig. S26a and c, ESI†) is directly proportional to the electrochemical performance. The surface area of pristine GO (∼13 m2 g−1) increases up to 40 times (∼480 m2 g−1) upon thermal annealing under both atmospheres due to the gradual removal of OFGs. However, it has been reported that the GO annealing at higher temperatures (≥500–800 °C) would lead to a decrease in surface area, which is due to the partial coalescence and overlap of graphene sheets.62–64 In the present case, we observed this phenomenon starting at a temperature of 300 °C, with the crystallite dimension obtained from XRD. Therefore, the largest surface area is achieved at low annealing temperatures (<300 °C). Secondly, it is also important to highlight that the presence of heteroatoms and functional groups is advantageous, as they can provide a large additional pseudocapacitance.37 Interestingly, Zhao et al. showed that the annealing of GO at temperatures between 200 and 500 °C can lead to materials with higher surface area and the specific capacitance exhibited a decrease from 200 °C due to the high loss of OFGs.62 As evidenced by XPS and ssNMR analyses, GO samples annealed at a temperature <300 °C feature small amounts of OFGs, in particular Csp2–O, which seem to be the key for the enhanced electrochemical performance of TrGO. Lastly, film conductivity also plays a key role in the electrochemical performance as low resistivity guarantees a quick charge propagation through the electrode material. In the present case, as we are removing OFGs, the mechanism is governed by the increase in the surface area of the GO and the increase in film conductivity (Fig. S26b and d, ESI†), yielding capacitance values comparable to the state of the art of TrGO (Table S6, ESI†).42,62,63,65–68 When the reduction is performed under argon, the increase in specific capacitance with the annealing temperature is followed by the increase in both surface area and film conductivity. Differently, when the reduction is carried out under air the same trend also occurs for the specific capacitance and film conductivity but not for the surface area at 150 and 200 °C. We can conclude that the increase in film conductivity has a higher effect on the electrochemical performance of TrGO under air, while under argon the effect of surface area and film conductivity cannot be distinguished. Importantly, a high annealing temperature (>300 °C, independently from the reduction atmosphere) is detrimental for the electrochemical performance of TrGO. The TrGO samples prepared under such conditions feature a smaller surface area and decreased pseudocapacitance due to the reduction of OFGs on GO's surface. Furthermore, an increase in the interface electrical resistance arises from the overlap of graphene sheets. By employing the mildest and optimized annealing conditions, our TrGO exhibits state of the art capacitance (Table S6, ESI†) compared to other TrGO prepared at low temperatures. The device's electrochemical performance depends on its individual components, which are the electrode, electrolyte, separator and current collector.69 Although the charges are mainly stored in the electrode material (i.e., TrGO), the other components also play key roles in the overall electrochemical performance.70 The influence of the current collector is tested by comparing carbon paper with Ni foam. As can be seen in Fig. S27, ESI† the specific capacitance of thermally reduced GO at 200 °C under air and 6 M KOH as an electrolyte increases from 154 F g−1 to 208 F g−1 at 0.1 A g−1 current density using a Ni foam current collector. This increase originates from the higher electrical conductivity of the Ni foam current collector as well as the stability of the whole system at higher voltages (up to 1 V), as can be seen in Fig. S27b and c, ESI.† Furthermore, the electrochemical impedance spectroscopy data are analysed using Nyquist and Bode plots (Fig. 5c and f and S28, ESI†). The Nyquist plot of an ideal double-layer capacitor should be a vertical line, parallel to the imaginary axis.71 The presence of a semicircle at high frequencies is related to the different OFGs on the GO surface endowing it with a pseudocapacitive behavior. For GO and TrGO under air up to 130 °C and up to 150 °C under argon, no semicircle can be observed (Fig. 5c and f and S28a and d, ESI†) due to the intrinsic internal resistance of the electrode materials being too high as has been shown in the film resistivity analysis (Fig. 1c). The experimental results are well fitted with the two indicated circuits (Scheme S1†). Rs is the combined internal resistance, including the interfacial contact resistance of the material with the current collector, the ohmic resistance of the electrolyte and the intrinsic resistance of the current collector, which can be obtained from the high-frequency region of the Nyquist plot at the intersection on the real axis.72Rct is the interfacial charge transfer resistance, corresponding to the diameter of the semicircle, which represents the resistance of electrochemical reactions at the electrode surface.73 Furthermore, the semicircle can also reflect a constant phase element (CPE) due to the double-layer behaviour. In the middle frequency region, the sloping transmission line corresponds to the Warburg element ZW, describing the transfer and diffusion of the electrons and electrolyte ions in the pores of the electrode materials. The nearly vertical line in the low frequency region reflects an excellent capacitive behaviour of the electrodes. The fitting parameters can be seen in Table S7, ESI.† The low Rct values are consistent with the fact that at a scanning rate as high as 200 mV s−1, the CV curve still shows a rectangular shape with small distortion, indicating that the TrGO samples show a good rate capability.
From the Nyquist and Bode plots an important parameter can be calculated, the conductance of the (Tr)GO film (Gp) (see the Experimental section for details), which can be converted to conductivity when normalized by the film thickness. Fig. S29a, ESI† shows the Gp of (Tr)GO under air at different temperatures in the frequency range of 0.1–106 Hz. From 0.1–10 Hz, there is no dependence of the annealing temperature and Gp, and from 104–106 Hz Gp is not stable. Therefore, for a better comparison, the frequency range of 10–104 Hz is selected. Fig. S29b and c, ESI† show the Gp of (Tr)GO under air (Fig. S29b†) and under argon (Fig. S29c†) at different temperatures. The same trend, in full agreement with previous characterization techniques, is obtained. When the annealing is performed under air, Gp gradually increases from 150 °C with the annealing temperature. Differently, when the annealing is performed under argon, Gp increases abruptly at 200 °C and then becomes constant at higher annealing temperatures. In all cases, the film thickness is ∼1 mm and the conductivity plot (Fig. S30, ESI†) exhibits similar values compared to the ones obtained from film resistance measurements (Fig. 1c, S26b and d, ESI†). The energy and power densities of TrGO annealed at 200 °C under air are plotted in Fig. S31, ESI† with: (1) the highest energy density of 7.35 W h kg−1 achieved at a power density of 0.057 kW kg−1 and (2) the highest power density of 2.86 kW kg−1 achieved at an energy density of 6.62 W h kg−1. These values of energy and power densities are well suited for energy storage applications74 Finally, to investigate the electrochemical stability of TrGO at 200 °C under air, GCD cycling was performed at a current density of 3 A g−1 (Fig. S32, ESI†). After 2000 cycles, the capacitance decay is only 0.4%, indicating that TrGO has excellent cycle durability.
3. Conclusions
In summary, we presented an optimized, scalable, easily controllable, and low-temperature (<300 °C) annealing procedure without chemical treatments for the production of reduced graphene oxide with electrical properties at will. The use of multiple techniques made it possible to gain an unprecedented insight and control over the compositional, structural, morphological and (electro)chemical characteristics of the materials. The oxygen atoms’ reorganization on the GO surface to promote H2O and CO2 release is favoured in a range of temperatures between 130 and 200 °C as shown from the TGA, XPS and ssNMR analyses. Therefore, it is possible to achieve the elimination of OFGs without promoting the ignition of the carbon framework when the reduction is performed in air. The main OFG formed during the annealing is Csp2–O, which does not result in any mass loss but it determines a variation of the electrical nature of GO from insulating to conductive. We have demonstrated that film resistivities of ∼10−2–10−4 Ω m can be achieved by annealing under air between 150 and 200 °C for 24 and 4 hours, respectively, and under an inert atmosphere from 200 °C for 4 hours. Such results indicate the viability of GO reduction upon annealing at temperatures as low as 150 °C, compatible with the use of plastic substrates. Interestingly, films supported on plastic substrates showed unaltered resistance when subjected to 2000 bending cycles. This represents an important step forward for application in wearable and flexible electronics. Thermally-reduced GO at 200 °C under air exhibited an enhanced electrochemical performance (a specific capacitance of 208 F g−1 at a current density of 0.1 A g−1 and a capacitance retention >99% after 2000 cycles). Although the loss of OFGs is detrimental to the electrochemical performance, it is compensated with a high gain in the surface area and film conductivity, yielding ∼450 m2 g−1 and 103 S m−1, respectively. Our protocol can be performed in any lab and it is scalable for industrial applications.4. Experimental section
4.1. Materials
Graphene oxide (GO, 4 mg mL−1, monolayer content >95%, Graphenea), polytetrafluoroethylene (PTFE) binder (Sigma Aldrich), N-methyl-2-pyrrolidone (NMP) (Sigma Aldrich), potassium hydroxide (KOH) (Fisher), sulfuric acid (H2SO4) (Sigma Aldrich), tetraethylammonium tetrafluoroborate ((C2H5)4NBF4) (Acros Organics), and acetonitrile (Carlo Erba reagents).4.2. Methods
4.3. Solid-state MAS NMR
All experiments were performed at room temperature on an AVANCE 500 MHz wide bore spectrometer (Bruker™) operating at a frequency of 500.12 MHz for 1H NMR and 188.5 MHz for 13C NMR. As GO is generally barely protonated the classical cross-polarization magic angle spinning (CP/MAS) proves ineffective in obtaining 13C quantitatively,75 Then a DP/MAS (direct polarization with magic angle spinning) pulse scheme was preferred by using a speed synchronized spin echo sequence with proton decoupling during acquisition.76 The latter allows us to obtain undistorted line shapes and filter out background probe signals, giving integrable spectra providing recycling delays that fulfill quantitative rules, i.e. equal to ca. 3 to 5 times that of the longitudinal relaxation time (13C T1). Under our conditions (speed and fields) T1s were measured to be less than 300 ms (saturation/recovery method, data not shown). 13C spectra were recorded using a triple resonance MAS probe (Bruker™), allowing the samples to be spun at 22.5 kHz after packing them inside 3.2 mm o.d. zirconia rotors (closed with Vespel caps). Recycle-time delays were set to 1.5 seconds and 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 240 transients over 8192 time-domain points separated by 2 μs dwell time were added, leading to a 61.035156 Hz pt−1 spectral resolution (spectral width = 250 kHz and total acquisition time per spectrum = 4 hours 30 min). 13C NMR and 1H NMR RF-fields were 60 kHz and 95 kHz, respectively. For the 300 °C thermally treated samples, the conductivity was such that it was very difficult to tune the probe. The concern was fixed by dispersing these powders in silica.77 Raw data were processed with a 150 Hz Lorentzian filter followed by Fourier transformation without zero filling. Chemical shifts are referenced to TMS by the substitution method using adamantane as an external reference. Spectral deconvolution was performed within Topspin™ software suite from Bruker™ using a CSA (chemical shift anisotropy) model.
240 transients over 8192 time-domain points separated by 2 μs dwell time were added, leading to a 61.035156 Hz pt−1 spectral resolution (spectral width = 250 kHz and total acquisition time per spectrum = 4 hours 30 min). 13C NMR and 1H NMR RF-fields were 60 kHz and 95 kHz, respectively. For the 300 °C thermally treated samples, the conductivity was such that it was very difficult to tune the probe. The concern was fixed by dispersing these powders in silica.77 Raw data were processed with a 150 Hz Lorentzian filter followed by Fourier transformation without zero filling. Chemical shifts are referenced to TMS by the substitution method using adamantane as an external reference. Spectral deconvolution was performed within Topspin™ software suite from Bruker™ using a CSA (chemical shift anisotropy) model.
4.4. Four-point probe measurements
Film resistivities were measured with a Jandel probe, Model RM3000, and the limit of detection is 107 Ω sq−1. The resistivity (ρ) was obtained using the following equation:| ρ = Rs·l | (1) |
4.5. Atomic force microscopy (AFM)
AFM characterization was carried out to determine the GO film thickness using a Bruker Dimension Icon microscope in intermittent contact mode using a TESPA-V2 silicon tip with a force constant (k) = 42 N m−1.4.6. Bending characterization
GO water solution (0.4 mg mL−1) was spray coated onto PET substrates at 100 °C (2.5 × 1.2 cm2, 125 μm thick) and annealed at 150 °C for 24 h in air. The reduced film of GO on PET was allowed to contact electrically with conductive copper tape. The sheet resistance of the film was 40 kΩ sq−1. The stability test of the TrGO conductive film to fatigue bending was carried out by performing 2000 bending cycles with a bending radius of 6 mm using a digital force gauge (Mark-10, M7-025E, ∼25 N) equipped with a motorized test stand (Mark-10, ESM-S-8 303E). All the above-mentioned tests were performed by applying a bias voltage of 1 V by means of a Keithley 2635B.4.7. Fabrication of supercapacitors
The electrochemical performance of thermally-reduced graphene oxide was measured in a two-electrode symmetric supercapacitor system. Two electrodes were assembled in CR2032 stainless steel coin-type cells with a porous cellulose membrane as a separator and 6 M KOH or 1 M H2SO4 aqueous solution or 1 M tetraethylammonium tetrafluoroborate ((C2H5)4NBF4) in acetonitrile as an electrolyte. The preparation procedure of the electrodes is shown as follows. First, a paste was prepared by fully mixing 90 wt% of sample (18 mg), 10 wt% polytetrafluoroethylene (PTFE) binder (2 mg) and a certain amount of N-methyl-2-pyrrolidone (NMP) using an agate mortar and pestle. The paste was coated over the carbon paper or Ni foam electrode, which was then subjected to a quick dry at 80 °C. The electrodes were completely dried at 80 °C in a vacuum oven for 16 h. Finally, the Ni foam electrodes were pressed at 10 MPa for 1 minute. The mass loading of (thermally reduced) graphene oxide was ∼0.5 mg in each electrode.The devices were electrically characterized by cyclic voltammetry (CV), galvanostatic charge/discharge (GCD), and electrochemical impedance spectroscopy (EIS) using a Metrohm Autolab PGSTAT204 potentiostat/galvanostat and the Autolab DuoCoin cell holder (Metrohm AG). The frequency range for the impedance spectra was from 0.1 Hz to 100 kHz with a sine-wave voltage signal amplitude of 50 mV (root mean square, RMS). CV and GCD tests were carried out between 0 and 0.6 V. Stability tests were performed using a battery testing system (Neware).
4.8. Calculation of the specific capacitances, energy densities and power densities
From charge–discharge measurements, the specific capacitances (Cp), energy densities (E), and power densities (P) of (thermally reduced) graphene oxide were obtained from the acquired data using the following equations:78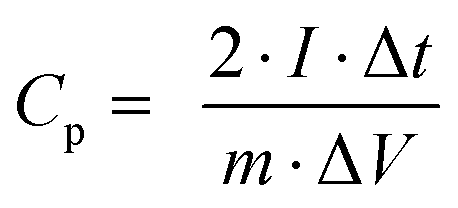 | (2) |
| E = ½·Cp·ΔV2 | (3) |
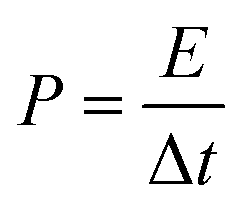 | (4) |
4.9. Calculation of conductance (Gp)
As previously reported,79 we can express the sample Gp in terms of the measurable, fundamental Z-related parameters: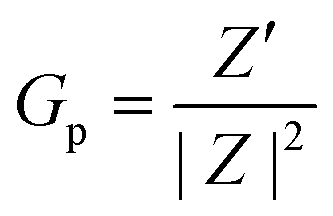 | (5) |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the funding from the European Commission through the ERC project SUPRA2DMAT (GA-833707), the Graphene Flagship Core 3 project (GA-881603) and the Marie Sklodowska Curie ETN project BORGES (GA-813863), as well as the Agence Nationale de la Recherche through the Interdisciplinary Thematic Institute SysChem via the IdEx Unistra (ANR-10-IDEX-0002) within the program Investissement d'Avenir, the International Center for Frontier Research in Chemistry (icFRC), the Institut Universitaire de France (IUF), and the National Science Centre, Poland (grant no. 2019/35/B/ST5/01568 and 2021/41/N/ST5/01112).References
- A. K. Geim, Phys. Scr., 2012, T146, 014003 CrossRef.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339–1339 CrossRef CAS.
- J. Guerrero-Contreras and F. Caballero-Briones, Mater. Chem. Phys., 2015, 153, 209–220 CrossRef CAS.
- N. I. Zaaba, K. L. Foo, U. Hashim, S. J. Tan, W.-W. Liu and C. H. Voon, Procedia Eng., 2017, 184, 469–477 CrossRef CAS.
- D. Chen, H. Feng and J. Li, Chem. Rev., 2012, 112, 6027–6053 CrossRef CAS PubMed.
- V. Agarwal and P. B. Zetterlund, Chem. Eng. J., 2021, 405, 127018 CrossRef CAS.
- J. W. Suk, R. D. Piner, J. An and R. S. Ruoff, ACS Nano, 2010, 4, 6557–6564 CrossRef CAS PubMed.
- C. Anichini, W. Czepa, D. Pakulski, A. Aliprandi, A. Ciesielski and P. Samorì, Chem. Soc. Rev., 2018, 47, 4860–4908 RSC.
- Y. Song, X. Li, C. Mackin, X. Zhang, W. Fang, T. Palacios, H. Zhu and J. Kong, Nano Lett., 2015, 15, 2104–2110 CrossRef CAS PubMed.
- J. Liu, L. Cui and D. Losic, Acta Biomater., 2013, 9, 9243–9257 CrossRef CAS PubMed.
- D. Pakulski, A. Gorczyński, D. Marcinkowski, W. Czepa, T. Chudziak, S. Witomska, Y. Nishina, V. Patroniak, A. Ciesielski and P. Samorì, Nanoscale, 2021, 13, 10490–10499 RSC.
- S. Witomska, Z. Liu, W. Czepa, A. Aliprandi, D. Pakulski, P. Pawluć, A. Ciesielski and P. Samorì, J. Am. Chem. Soc., 2019, 141, 482–487 CrossRef CAS PubMed.
- Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22, 3906–3924 CrossRef CAS PubMed.
- G. Eda and M. Chhowalla, Adv. Mater., 2010, 22, 2392–2415 CrossRef CAS PubMed.
- A. Ciesielski and P. Samorì, Chem. Soc. Rev., 2014, 43, 381–398 RSC.
- X. Zhang, L. Hou, A. Ciesielski and P. Samorì, Adv. Energy Mater., 2016, 6, 1600671 CrossRef.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed.
- J. J. Xingfa Gao and S. Nagase, J. Phys. Chem. C, 2010, 114, 832–842 CrossRef.
- Y. Shao, J. Wang, M. Engelhard, C. Wang and Y. Lin, J. Mater. Chem., 2010, 20, 743–748 RSC.
- L. G. Guex, B. Sacchi, K. F. Peuvot, R. L. Andersson, A. M. Pourrahimi, V. Strom, S. Farris and R. T. Olsson, Nanoscale, 2017, 9, 9562–9571 RSC.
- S. Abdolhosseinzadeh, H. Asgharzadeh and H. Seop Kim, Sci. Rep., 2015, 5, 10160 CrossRef CAS PubMed.
- J. Zhang, H. Yang, G. Shen, P. Cheng, J. Zhang and S. Guo, Chem. Commun., 2010, 46, 1112–1114 RSC.
- K. K. H. De Silva, H. H. Huang and M. Yoshimura, Appl. Surf. Sci., 2018, 447, 338–346 CrossRef CAS.
- C. Mattevi, G. Eda, S. Agnoli, S. Miller, K. A. Mkhoyan, O. Celik, D. Mastrogiovanni, G. Granozzi, E. Garfunkel and M. Chhowalla, Adv. Funct. Mater., 2009, 19, 2577–2583 CrossRef CAS.
- B. L. Kyu Hyung Lee, S.-J. Hwang, J.-U. Lee, H. Cheong, K. S. Oh-Sun Kwon and N. H. Hur, Carbon, 2014, 69, 327–335 CrossRef.
- S. P. Lee, G. A. M. Ali, H. H. Hegazy, H. N. Lim and K. F. Chong, Energy Fuels, 2021, 35, 4559–4569 CrossRef CAS.
- C. J. Li and B. M. Trost, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 13197–13202 CrossRef CAS PubMed.
- N. D. K. Tu, J. Choi, C. R. Park and H. Kim, Chem. Mater., 2015, 27, 7362–7369 CrossRef.
- M. Pelaez-Fernandez, A. Bermejo, A. M. Benito, W. K. Maser and R. Arenal, Carbon, 2021, 178, 477–487 CrossRef CAS.
- J. P. Jog, J. Macromol. Sci., Part A: Pure Appl.Chem., 1995, 35, 531–553 CrossRef.
- V. Ratta, E. J. Stancik, A. Ayambem, H. Pavatareddy, J. E. McGrath and G. L. Wilkes, Polymer, 1999, 40, 1889–1902 CrossRef CAS.
- W. Chen and L. Yan, Nanoscale, 2010, 2, 559–563 RSC.
- P. V. Kumar, N. M. Bardhan, S. Tongay, J. Wu, A. M. Belcher and J. C. Grossman, Nat. Chem., 2014, 6, 151–158 CrossRef CAS PubMed.
- C. Mattevi, G. Eda, S. Agnoli, S. Miller, K. A. Mkhoyan, O. Celik, D. Mastrogiovanni, G. Granozzi, E. Garfunkel and M. Chhowalla, Adv. Funct. Mater., 2009, 19, 2577–2583 CrossRef CAS.
- O. M. Slobodian, P. M. Lytvyn, A. S. Nikolenko, V. M. Naseka, O. Y. Khyzhun, A. V. Vasin, S. V. Sevostianov and A. N. Nazarov, Nanoscale Res. Lett., 2018, 13, 139 CrossRef PubMed.
- H. L. Kuibo Yin, Y. Xia, H. Bi, J. Sun, Z. Liu and L. Sun, Nano-Micro Lett., 2011, 3, 51–55 CrossRef.
- B. Xu, S. Yue, Z. Sui, X. Zhang, S. Hou, G. Cao and Y. Yang, Energy Environ. Sci., 2011, 4, 2826–2830 RSC.
- B. C. Kim, W. J. Cho, W. G. Lee, S. J. Kim, R. Jalili, S. Y. Park, G. G. Wallace, K. H. Yu and S. J. Chang, Synth. Met., 2014, 193, 110–116 CrossRef CAS.
- Y. Tian, Z. Yu, L. Cao, X. L. Zhang, C. Sun and D.-W. Wang, J. Energy Chem., 2021, 55, 323–344 CrossRef CAS.
- Z. Li, S. Gadipelli, Y. Yang, G. He, J. Guo, J. Li, Y. Lu, C. A. Howard, D. J. L. Brett, I. P. Parkin, F. Li and Z. Guo, Energy Storage Mater., 2019, 17, 12–21 CrossRef.
- P. L. B. Zhaoa, Y. Jianga, D. Pana, H. Taob, J. Songa, T. Fanga and W. Xua, J. Power Sources, 2012, 198, 423–427 CrossRef.
- W. Lv, D. M. Tang, Y. B. He, C.-H. You, Z. Q. Shi, X.-C. Chen, C.-M. Chen, P. X. Hou, C. Liu and Q. H. Yang, ACS Nano, 2009, 3, 3730–3736 CrossRef CAS PubMed.
- C. G. Powell, American institute of physics handbook, McGraw-Hill, Dallas, 3rd edn, 1972 Search PubMed.
- W. W. Tyler and A. C. Wilson, Phys. Rev., 1953, 89, 870–875 CrossRef CAS.
- H. O. Pierson, Handbook of Carbon, Graphite, Diamonds and Fullerenes, 1993 Search PubMed.
- K. Woll, T. Neuhauser, C. Acuña, D. Diaz-Droguett and A. Rosenkranz, Lubricants, 2019, 7, 96 CrossRef.
- A. Ganguly, S. Sharma, P. Papakonstantinou and J. Hamilton, J. Phys. Chem. C, 2011, 115, 17009–17019 CrossRef CAS.
- I. A. Vacchi, C. Spinato, J. Raya, A. Bianco and C. Ménard-Moyon, Nanoscale, 2016, 8, 13714–13721 RSC.
- S. Guo, J. Raya, D. Ji, Y. Nishina, C. Ménard-Moyon and A. Bianco, Nanoscale Adv., 2020, 2, 4085–4092 RSC.
- M. A. Lucherelli, J. Raya, K. F. Edelthalhammer, F. Hauke, A. Hirsch, G. Abellán and A. Bianco, Chem. – Eur. J., 2019, 25, 13218–13223 CrossRef CAS PubMed.
- I. A. Vacchi, S. Guo, J. Raya, A. Bianco and C. Ménard-Moyon, Chem. – Eur. J., 2020, 26, 6591–6598 CrossRef CAS PubMed.
- S. Claramunt, A. Varea, D. López-Díaz, M. M. Velázquez, A. Cornet and A. Cirera, J. Phys. Chem. C, 2015, 119, 10123–10129 CrossRef CAS.
- A. A. K. King, B. R. Davies, N. Noorbehesht, P. Newman, T. L. Church, A. T. Harris, J. M. Razal and A. I. Minett, Sci. Rep., 2016, 6, 19491 CrossRef CAS PubMed.
- J. B. Wu, M. L. Lin, X. Cong, H. N. Liu and P. H. Tan, Chem. Soc. Rev., 2018, 47, 1822–1873 RSC.
- A. Y. Lee, K. Yang, N. D. Anh, C. Park, S. M. Lee, T. G. Lee and M. S. Jeong, Appl. Surf. Sci., 2021, 536, 147990 CrossRef CAS.
- S. Vollebregt, R. Ishihara, F. D. Tichelaar, Y. Hou and C. I. M. Beenakker, Carbon, 2012, 50, 3542–3554 CrossRef CAS.
- F. Tuinstra and J. L. Koenig, Chem. Phys., 1970, 53, 1126–1130 CAS.
- S. Hun, in In Physics and Applications of Graphene - Experiments, ed. S. Mikhailov, InTech, 2011, ch. 5, DOI:10.5772/14156.
- I. Sengupta, S. Chakraborty, M. Talukdar, S. K. Pal and S. Chakraborty, J. Mater. Res., 2018, 33, 4113–4122 CrossRef CAS.
- T. S. Mathis, N. Kurra, X. Wang, D. Pinto, P. Simon and Y. Gogotsi, Adv. Energy Mater., 2019, 9, 1902007 CrossRef CAS.
- C. Zhong, Y. Deng, W. Hu, J. Qiao, L. Zhang and J. Zhang, Chem. Soc. Rev., 2015, 44, 7484–7539 RSC.
- B. Zhao, P. Liu, Y. Jiang, D. Pan, H. Tao, J. Song, T. Fang and W. Xu, J. Power Sources, 2012, 198, 423–427 CrossRef CAS.
- H. Zhang, V. V. Bhat, N. C. Gallego and C. I. Contescu, ACS Appl. Mater. Interfaces, 2012, 4, 3239–3246 CrossRef CAS PubMed.
- Y. Wang, Z. Shi, Y. Huang, Y. Ma, C. Wang, M. Chen and Y. Chen, J. Phys. Chem. C, 2009, 113, 13103–13107 CrossRef CAS.
- L. T. Le, M. H. Ervin, H. Qiu, B. E. Fuchs and W. Y. Lee, Electrochem. Commun., 2011, 13, 355–358 CrossRef CAS.
- H. Cao, X. Peng, M. Zhao, P. Liu, B. Xu and J. Guo, RSC Adv., 2018, 8, 2858–2865 RSC.
- Z. Li, S. Gadipelli, Y. Yang and Z. Guo, Small, 2017, 13, 1702474 CrossRef PubMed.
- L. P. Bakos, L. Sárvári, K. László, J. Mizsei, Z. Kónya, G. Halasi, K. Hernádi, A. Szabó, D. Berkesi, I. Bakos and I. M. Szilágyi, Nanomaterials, 2020, 10, 2313 CrossRef CAS PubMed.
- J. Zhao and A. F. Burke, J. Energy Chem., 2021, 59, 276–291 CrossRef CAS.
- P. Sinha and K. K. Kar, in Handbook of Nanocomposite Supercapacitor Materials II: Performance, ed. K. K. Kar, Springer International Publishing, Cham, 2020, pp. 71–87, DOI:10.1007/978-3-030-52359-6_3.
- M. D. Stoller, S. Park, Y. Zhu, J. An and R. S. Ruoff, Nano Lett., 2008, 8, 3498–3502 CrossRef CAS PubMed.
- E. Frackowiak and F. Béguin, Carbon, 2001, 39, 937–950 CrossRef CAS.
- D. Pakulski, V. Montes-García, A. Gorczyński, W. Czepa, T. Chudziak, P. Samorì and A. Ciesielski, J. Mater. Chem. A, 2022, 10, 16685–16696 RSC.
- C. Xu, B. Xu, Y. Gu, Z. Xiong, J. Sun and X. S. Zhao, Energy Environ. Sci., 2013, 6, 1388–1414 RSC.
- J. Raya, A. Bianco and J. Hirschinger, Phys. Chem. Chem. Phys., 2020, 22, 12209–12227 RSC.
- E. L. Hahn, Phys. Rev., 1950, 80, 580–594 CrossRef.
- J. C. C. Freitas, F. G. Emmerich, G. R. C. Cernicchiaro, L. C. Sampaio and T. J. Bonagamba, Solid State Nucl. Magn. Reson., 2001, 20, 61–73 CrossRef CAS PubMed.
- M. D. Stoller and R. S. Ruoff, Energy Environ. Sci., 2010, 3, 1294–1301 RSC.
- V. Montes-García, R. F. de Oliveira, Y. Wang, A. Berezin, P. Fanjul-Bolado, M. B. González García, T. M. Hermans, D. Bonifazi, S. Casalini and P. Samorì, Adv. Funct. Mater., 2021, 31, 2008554 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr06091d |
| This journal is © The Royal Society of Chemistry 2023 |