
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn situ quantitative single-molecule study of site-specific photocatalytic activity and dynamics on ultrathin g-C3N4 nanosheets†
Shuyang
Wu
,
Jenica Marie L.
Madridejos
,
Jinn-Kye
Lee
 ,
Yunpeng
Lu
,
Rong
Xu
and
Zhengyang
Zhang
*
,
Yunpeng
Lu
,
Rong
Xu
and
Zhengyang
Zhang
*
School of Chemistry, Chemical Engineering and Biotechnology, Nanyang Technological University, 21 Nanyang Link, Singapore 637371. E-mail: zhang.zy@ntu.edu.sg
First published on 26th January 2023
Abstract
Graphitic carbon nitride (g-C3N4) has attracted extensive research attention in recent years due to its unique layered structure, facile synthetic route, visible-light-responsive nature, and excellent photocatalytic performance. However, an insightful investigation of site-specific catalytic activities and kinetics on g-C3N4 is still warranted. Here, we fabricated ultrathin g-C3N4 nanosheets through thermal exfoliation. The optimized sample exhibits a high specific surface area of 307.35 m2 g−1 and a remarkable H2 generation activity of 2008 μmol h−1 g−1 with an apparent quantum efficiency of 4.62% at λ = 420 nm. Single-molecule fluorescence microscopy was applied for the first time to spatially resolve the reaction heterogeneities with nanometer precision (∼10 nm). The catalytic kinetics (i.e., reactant adsorption, conversion, and product dissociation) and temporal activity fluctuations were in situ quantified at individual structural features (i.e., wrinkles, edges, and basal planes) of g-C3N4. It was found that the wrinkle and edge exhibited superior photocatalytic activity due to the intrinsic band modulation, which are 20 times and 14.8 times that of the basal plane, respectively. Moreover, due to the steric effect, the basal plane showed the highest adsorption constant and the lowest direct dissociation constant. Density functional theory (DFT) simulations unveiled the adsorption energies of reactant and product molecules on each structure of g-C3N4, which support our experimental results. Such investigation would shed more light on the fundamental understanding of site-specific catalytic dynamics on g-C3N4, which benefits the rational design of 2D layered materials for efficient solar-to-chemical energy conversion.
1. Introduction
Photocatalytic energy conversion is a promising way to generate renewable green sources of energy (e.g., H2, CO, CH4, methanol, ethanol, etc.) and photosynthesize chemical feedstocks (e.g., NH3, nitrites, fertilizers, etc.). It provides an energy-saving and environmentally benign approach to convert solar energy to chemical energy under ambient and mild conditions. Among various photocatalyst materials, laminar g-C3N4 nanosheets have attracted great research attention in photocatalytic applications due to their unique structural features and electronic band properties. The layered structure with a large lateral size provides many active sites for reactant adsorption and chemical conversion. In particular, when decorating with functional groups, the modified surface has enormous potential in anchoring and accommodating the guest species, which forms multifunctional heterostructures with remarkable catalytic activity. The moderate bandgap of ∼2.7 eV makes g-C3N4 a visible-light-responsive photocatalyst that absorbs the light spectrum up to 460 nm.1 The relatively negative conduction band position (−1.13 eV) provides sufficient reduction potential for photoreduction of water, CO2, N2 and other compounds.2 Besides, the high stability, low cost, nontoxicity and simple preparation routes make g-C3N4 a promising candidate for a variety of applications. Despite these numerous advantages, the photocatalytic activity of g-C3N4 is still hindered by the high charge recombination rate, lack of surface-active sites and limited visible light utilization. Therefore, there is a need to further investigate the fundamental catalytic properties of g-C3N4 and explore effective methods (e.g., surface engineering, morphology design, electronic band modulation, etc.) to improve its photoactivity.Until now, numerous efforts have been devoted to investigating the physicochemical properties and photocatalytic performance of g-C3N4. For example, high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM)3,4 and scanning tunnelling spectroscopy (STS)5,6 were used to characterize the structural features and electronic properties of g-C3N4 down to atomic resolution, respectively. However, a direct visualization of single-turnover events on g-C3N4 and a fundamental understanding of catalytic activities and dynamics at the individual sites of g-C3N4 are still missing. The dynamic steps in a chemical reaction include the adsorption, activation and conversion of reactants and the dissociation of products. All these kinetic steps determine the turnover frequency of a catalytic event. Hence, our work intends to apply the single-molecule fluorescence (SMF) imaging to fill the gaps and construct a bridge between the catalyst structure and activity at the nanoscale. Traditional ensemble methods (e.g., X-ray diffraction, UV-vis spectroscopy, transient photocurrent, etc.) are usually combined with bulk performance measurements (i.e., photocatalytic H2 generation, CO2 reduction, N2 fixation, etc.) to reveal the “structure–activity” correlations at the ensemble level.7 However, it is challenging to unveil spatially single-particle and temporally varying catalytic properties using the ensemble techniques owing to the inherent heterogeneity of g-C3N4 in morphology, structural features, and surface and interface properties. To achieve an in-depth understanding of structure–activity correlations with nanometer precision, there is a great need to in situ monitor surface catalytic dynamics at the single-molecule level with a high spatiotemporal resolution. Therefore, our work with SMF microscopy would provide such information in these blank areas and help address the heterogeneity challenge in the catalytic study of g-C3N4.
SMF microscopy has achieved great momentum in recent years in investigating chemical reaction heterogeneities and kinetics at the nanoscale. Compared with traditional methods, it shows great advantages in localizing catalytic active species and reactive sites and unveiling underlying reaction mechanisms on various structures (e.g., 1D nanowires/nanorods, 2D nanoplates/nanosheets, 3D interconnected and hierarchical structures).8–10 Huang et al. applied SMF imaging to quantitatively study the photocatalytic activities and dynamics at the individual defects of 2D layered InSe.11 It was found that the vacancy showed 30 times higher photoactivity, 7 times smaller adsorption constant and 26 times smaller dissociation constant compared to the basal plane. Tachikawa et al. used SMF microscopy to obtain the nanoscale mapping of adsorption sites on TiO2 nanoplates and investigated the effect of crystal facets on the adsorption preference of a dye.12 It was discovered that the adsorption ability was (101) > (001) ≈ (100) in neutral water but the order was reversed in an aprotic solvent, suggesting the significant impact of solvents on the adsorption of organic molecules. These findings prove that SMF microscopy is an effective technique to study the site-specific reactivities and kinetics of 2D materials. However, such important nature and catalytic properties of g-C3N4 have not been studied and remain unclear. Hence, it is highly desirable to utilize SMF microscopy to spatially resolve the reaction heterogeneities and in situ quantify the catalytic kinetics at the individual structures of g-C3N4.
Herein, 2D laminar g-C3N4 nanosheets were synthesized via thermal exfoliation. The optimized sample exhibits an ultrathin thickness of 9.8 nm and a high specific surface area (SSA) of 307.35 m2 g−1. The larger SSA and better charge separation efficiency lead to an outstanding photocatalytic H2 generation rate of 2008 μmol h−1 g−1. Due to the remarkable spatial resolution (∼10 nm) and the ability to study the real-time dynamic process, SMF microscopy was applied to in situ reveal the surface active-site distributions, site-specific catalytic kinetics, and dynamic activity fluctuations on g-C3N4. The catalytic turnover rate, chemical conversion, adsorption and direct/indirect dissociation rate constants were quantitively determined at each structure. The wrinkle shows the highest photoactivity, fluctuation rate, and direct dissociation rate constant, but the lowest adsorption rate constant among all structures. Such microscopic information on the catalytic process would help establish a structure–dynamics relationship for g-C3N4 at the nanoscale and in turn provide fundamental guidance on the catalyst design.
2. Experimental section
2.1. Synthesis of photocatalysts
Bulk g-C3N4, hereafter referred to as CNB, was synthesized by the thermal polymerization of melamine under an air atmosphere.13 In a typical synthesis, 5 g of melamine was calcined in a crucible with a cover at 500 °C for 2 h at a heating rate of 2 °C min−1. Then, the sample was subject to further thermal treatment at 520 °C for 2 h. The yellow product was ground into powder for further use. g-C3N4 nanosheets, hereafter referred to as CNS, were prepared by further thermal treatment of CNB. First, 1 g of CNB was spread into an ark to form a thin layer of the material with optimal contact with air. Then, it was heated to 520 °C at a heating rate of 2 °C min−1 and maintained at this temperature. The samples after 2 h and 6 h calcination are named CNS-2 h and CNS-6 h, respectively.2.2. Materials characterization
The phase information of g-C3N4 was analyzed by X-ray diffraction (XRD) using a Bruker D2 Phaser diffractometer. The incident beam is Cu Kα irradiation (λ = 1.54184 Å) at 10 mA and 30 kV. Field emission scanning electron microscopy (FESEM) images were taken using a JEOL JSM 6701F microscope. Transmission electron microscopy (TEM) images were obtained using a JEOL JEM-2100Plus microscope. Atomic-force microscopy (AFM) images were taken using a Bruker Dimension XR Icon AFM. Fourier transform infrared (FTIR) spectra were recorded using a PerkinElmer spectrophotometer (Spectrum 100). Photoluminescence (PL) spectra were recorded using a Cary Eclipse fluorescence spectrophotometer. UV-vis diffuse reflection spectroscopy (DRS) was performed using a Shimadzu UV-2450 spectrophotometer. The Brunauer–Emmett–Teller (BET) surface area, isotherms and pore size distribution were measured by N2 adsorption and desorption at 77 K using a Micromeritics 3Flex system.2.3. Photoelectrochemical measurement
A conventional electrochemical system with three electrodes was used to measure the photocurrent and electrochemical impedance. The system consists of the working electrode, Pt counter electrode and Ag/AgCl reference electrode, which were controlled by a CHI 660E electrochemical workstation. A catalyst solution was prepared by dispersing 2 mg of photocatalyst in a 1 mL solution containing 20 μL of 5 wt% Nafion and 980 μL of ethanol. After 20 min ultrasonication, 30 μL of catalyst solution was deposited on conductive ITO glass (0.196 cm2). Then, the photocatalyst-deposited ITO glass was dried at 80 °C overnight. A 300 W Xe lamp (Newport) with a cut-off filter (λ > 400 nm) was used as the light source. The photocurrent was recorded under on/off illumination cycles in 0.5 M Na2SO4 solution with a bias voltage of 0.6 V. EIS spectra were obtained with a bias voltage of 10 mV in the frequency range of 0.1 Hz–105 Hz.2.4. Photocatalytic measurement
The photocatalytic activity of H2 generation was measured in a 300 mL glass reactor, which was connected to a gas evacuation and circulation system. In a typical experiment, 20 mg of photocatalyst was dispersed into 100 mL of 15 vol% TEOA solution. 200 μL of 1 mg Pt mL−1 H2PtCl6 solution (equivalent to 1 wt% Pt loading) was added as a cocatalyst. After 15 min ultrasonication, the reactor was placed under light irradiation (λ > 400 nm), which was a 300 W Xe lamp (Newport) with a cut-off filter. Through evacuating the reactor and purging with Ar several times, the air in the system was removed. The reactor was finally purged with Ar at around 30 Torr. The temperature of the solution was maintained at 18 °C using a circulatory cooling water system. The H2 generated was measured with an online gas chromatograph (Agilent 6890N). Stability test was conducted under the same conditions with four consecutive runs and each run lasted for 6 h. The evolved H2 after each run was removed before the next run started.2.5. Single-molecule fluorescence imaging
SMF imaging was conducted using a prism-type total internal reflection fluorescence (TIRF) microscope (Nikon Eclipse Ti-U). To prepare a photocatalyst-deposited coverslip, several drops of catalyst suspension (2 mg mL−1) were deposited on a glass coverslip and fully dried in an 80 °C oven. The coverslip with the photocatalyst was assembled with a microflow chamber (75.5 mm × 25.5 mm × 0.6 mm) using a double-side tape and sealed tightly. Then, it was transferred to the microscope stage. The probe solutions were deoxygenated by bubbling N2 gas for 30 min before introducing into the microflow chamber. Then, a steady stream of probe solution containing different concentrations of resazurin (i.e., 0.02, 0.05, 0.1, 0.2, 0.5 and 1 μM) was pumped into the chamber at a flowrate of 20 μL min−1. A circularly polarized 405 nm laser (2 mW, L6CC Oxxius) and a 532 nm laser (50 mW) were used as light sources to photoexcite the catalyst and resorufin molecules, respectively. A band-pass filter (ET605/70 m, Chroma) was used to minimize the photocatalyst PL signal. Before irradiation, the brightfield images were collected. Upon illumination, the emission signal passed through an oil-immersion objective lens (Nikon Plan Apo λ 100×, NA 1.45) and collected using an electron multiplying charge coupled device (EMCCD) camera (Andor iXon3) at 60 Hz. For each run, a movie with more than 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 frames was collected and then processed with software.
000 frames was collected and then processed with software.
2.6. Computational details
Adsorption energy calculations were performed with the Vienna Ab initio Simulation package (VASP).14–17 In our simulations, carbon nitride nanosheets (g-C3N4) have been studied by density functional theory (DFT) via the generalized gradient approximation (GGA) with the Perdew–Burke–Ernzerhof (PBE)18,19 functional with a plane-wave cut-off energy of 450 eV.20,21 The basal plane and wrinkle surfaces were based on a previous work22 on g-C3N4 while the edge was generated by terminating basal plane's N-atoms with hydrogen and 10 Å vacuum was added to signify the edge of the material. Optimization calculations were performed using energy and force convergence limits of 10–4 eV per atom and 0.01 eV Å−1 for different surface slabs with explicit dispersion corrections using D3 Grimme's method (DFT-D3).23,24 Geometric constraints were applied to both the basal plane and edge g-C3N4 surfaces to avoid corrugation to wrinkle structures. Brillouin zone integration was performed with a 3 × 3 × 1 Monkhorst–Pack grid for the different surface slabs. A vacuum height of 20 Å between different layers was set to prevent interactions between layers. To optimize the gas-phase adsorbates (resazurin and resorufin), the unit cell is set to 20 Å × 20 Å × 20 Å to avoid interactions between neighboring molecules with a 1 × 1 × 1 Monkhorst–Pack grid. A series of initial locations and orientations of resazurin and resorufin on all three surfaces was considered based on the symmetry and possible essential atomic interactions. Geometry optimization was performed on all cases and the final adsorption geometry was determined by the most stable case in energy. The adsorption energy (Eads) was calculated according to eqn (1), where Etotal is the total energy calculated for the substrate with the adsorbed reactant or product, Esubstrate is the calculated energy of the clean substrate, and Egas-phase adsorbate is the calculated energy of the adsorbed gas phase molecule.| Eads = − (Etotal − Esubstrate − Egas-phase adsorbate) | (1) |
3. Results and discussion
3.1. Physicochemical properties of the as-prepared photocatalysts
FESEM and TEM were used to investigate the morphology and structure of the as-prepared g-C3N4. As shown in Fig. 1a and d, the CNB sample exhibits a thick laminar structure and consists of bulk solid aggregates with a lateral size of several micrometers. After thermal exfoliation for 2 h, the thickness of g-C3N4 is significantly reduced, which is found to be 26.1 nm by AFM (Fig. S1a and S1b†). During the heating process, through overcoming the weak interlayer van der Waals force, the thick CNB is thermally exfoliated to thin g-C3N4 nanosheets (Fig. 1b and e). CNS-2 h shows a soft and loose structure of agglomerates and exhibits great flexibility and tunability. Moreover, the lateral size also decreases due to thermal oxidation. After 6 h thermal treatment, the thickness of g-C3N4 is further reduced to 9.8 nm (Fig. S1c and S1d†). The CNS-6 h sample shows an ultrathin and graphene-like morphology with a rough surface (Fig. 1c and f). This is due to the gradual degradation of planar polymeric melon units in the g-C3N4 layers during the thermal etching process.25 The basal planes tend to bend and fold and the edges are ragged, which is caused by minimizing the surface energy of g-C3N4 sheets. Therefore, several wrinkle structures can be clearly identified on CNS-6 h in Fig. S2a and S2b.† Besides, as shown in the photograph of the samples (Fig. S3†), it can be observed that CNS-6 h shows a macroscopic foam-like structure that is loose and porous. Consequently, CNS-6 h has a much lower stacking density (18.6 mg cm−3) compared to CNB (360.4 mg cm−3). Such an ultralight nature indicates that CNS-6 h possesses a high SSA and outstanding dispersion properties in an aqueous solution. | ||
| Fig. 1 Morphology of the as-prepared samples. FESEM and TEM images of (a and d) CNB, (b and e) CNS-2 h, and (c and f) CNS-6 h. | ||
The BET surface areas and pore structures of the as-prepared samples were studied by N2 adsorption–desorption measurements at 77.4 K. As shown in Fig. 2a, CNS-2 h and CNS-6 h exhibit typical IV isotherms that have considerable adsorption in the high relative pressure range (P/P0 > 0.8). This indicates the existence of meso- and macropores in the structure.26 The hysteresis loops can be classified into type H3 with no limiting adsorption at high P/P0, suggesting the presence of slit-shaped pores in the nanosheets. The SSAs of CNS-6 h and CNS-2 h are found to be 307.35 and 234.27 m2 g−1, which are 57 and 43 times that of CNB, respectively (Table S1†). This proves that thermal oxidation successfully exfoliates the bulk g-C3N4 into ultrathin nanosheets, consistent with the TEM and AFM results. In addition, CNS-6 h exhibits a larger average pore size (49.5 nm) than that of CNS-2 h (34.7 nm), while CNB has no obvious pore size distribution peak (Fig. S4†). This is probably because the thermal etching process destroyed some tri-s-triazine units in the plane, which forms large pores and holes.27,28 The pore volumes of CNS-6 h (1.330 cm3 g−1) and CNS-2 h (1.097 cm3 g−1) are 38 and 31 times that of CNB (0.035 cm3 g−1) respectively, confirming the porous nature of exfoliated g-C3N4. It is well known that a higher SSA and the porous structure provide more active sites for surface reactions and facilitate the mass transfer, leading to superior photocatalytic activity. The XRD patterns of the as-prepared samples are shown in Fig. 2b. The low-angle peak at 13.1° is ascribed to the interplanar structural packing of tri-s-triazine motifs indexed as (100) planes. The peak becomes less prominent with a prolonged thermal oxidation time, which can be attributed to the simultaneous decrease of planar size when CNB is etched by calcination. The diffraction peak at around 27.5° arises from the (002) plane reflections of interlayer periodic stacking. It was found that the peak is shifted from 27.31° for CNB to 28.01° for CNS-6 h, which suggests that the interlayer gallery distance is reduced after exfoliation.25 Previous work has revealed that the monolayers in bulk g-C3N4 tend to be undulated but could be planarized after thermal treatment, leading to a denser packing.29 Therefore, the thermal oxidation in our synthesis results in a denser stacking of g-C3N4 layers thus decreasing the interlayer gallery distance.
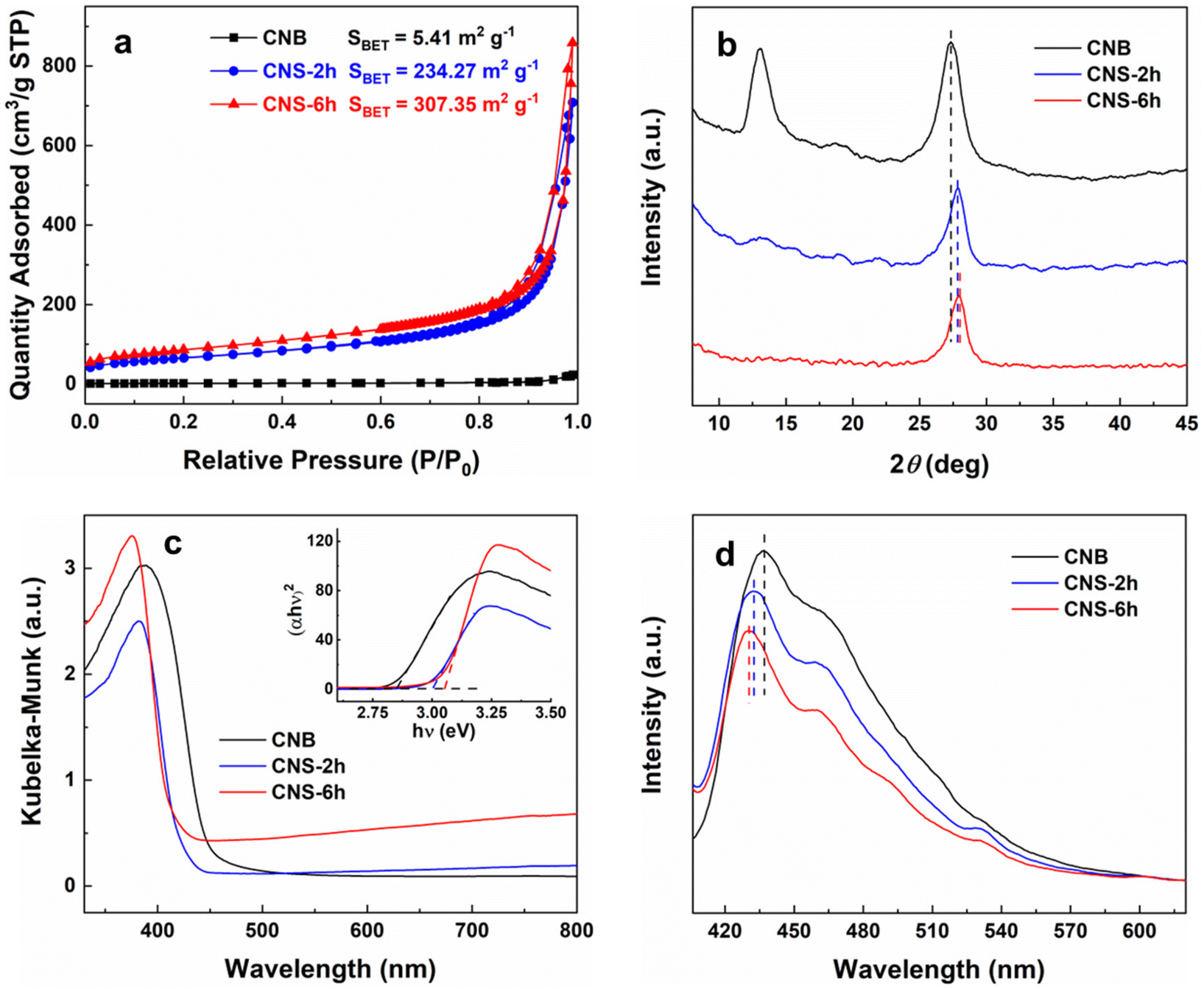 | ||
| Fig. 2 Physicochemical properties of the as-prepared samples. (a) BET adsorption–desorption isotherms, (b) XRD patterns, (c) UV-vis DRS spectra, and (d) the steady-state PL spectra of the as-prepared samples. | ||
FTIR spectroscopy was conducted to further verify the structure of g-C3N4 before and after exfoliation. In Fig. S5,† all the samples possess typical characteristic IR peaks of g-C3N4. The characteristic band at 808 cm−1 is assigned to the vibrational bending of the triazine ring system.30 The absorption bands in the range of 1000–1700 cm−1 arise from the stretching mode of aromatic heterocycles.31 The broad peaks at 3000–3600 cm−1 correspond to the N–H bond stretching. The shape of these peaks becomes sharper with increasing exfoliation time. This is most likely caused by more ordered packing and an array of polymeric melon units in the nanosheets after long-time thermal treatment.25 The optical properties and electronic band structures of the as-prepared samples were studied by UV-vis DRS and PL spectra. As shown in Fig. 2c, the absorption edges of CNS-6 h and CNS-2 h have an obvious blue shift compared to CNB. The bandgap energies of the samples were calculated by transforming the DRS data into the Tauc plot (inset of Fig. 2c), and are found to be 2.85, 3 and 3.05 eV for CNS-6 h, CNS-2 h and CNB, respectively. The blue shift of the absorption edge with an enlarged bandgap energy for CNS-6 h can be ascribed to the quantum confinement effect by shifting the conduction band (CB) and valence band (VB) positions in the opposite direction,32 and the reduction of the in-plane conjugation length stemmed from thermal etching.33 Similar phenomena were also observed for CNS with other exfoliation times (Fig. S6a†). It can be observed that CNS-6 h has an enhanced visible light absorption compared with CNS-2 h and CNB. Such a phenomenon is related to the multiple scattering effect originating from its porous structure.34 Furthermore, steady-state PL spectroscopy validates the band structure of the samples (Fig. 2d). The blue-shift of the emission peak is in agreement with the UV-vis DRS results, which was attributed to the reduced thickness and larger bandgap of CNS-6 h. Moreover, the decreased intensity of the emission peak suggests a better charge separation efficiency of CNS-6 h, leading to a superior photocatalytic activity.
3.2. Photocatalytic activity
The charge separation and migration properties of the photocatalysts were studied by transient photocurrent and EIS spectra. In Fig. 3a, CNS-6 h shows the highest photocurrent density, around 1.7 and 6.5 times those of CNS-2 h and CNB, respectively. This demonstrates that the ultrathin nanosheets of CNS-6 h facilitate the separation of photogenerated charges. An EIS study was conducted to investigate the charge transfer kinetics at the interface between the photocatalyst and the electrolyte. As shown in Fig. 3b, CNS-6 h has the smallest arc radius among the samples, indicating the lowest interfacial charge transfer resistance and the fastest charge migration rate. The reduced thickness of CNS-6 h also favors the mass transfer in the photocatalyst by shortening the transportation distance, which improves the charge mobility.34 The photocatalytic performance of the samples was evaluated by water splitting under visible light irradiation (λ > 400 nm). As shown in Fig. 3c and S6b,† CNS-6 h shows the highest photocatalytic H2 generation rate of 2008 μmol h−1 g−1 among all the exfoliated samples, which is 1.4 times that of CNS-2 h and 15.4 times that of CNB. The apparent quantum efficiency (AQE) of CNS-6 h was calculated to be 4.62% at λ = 420 ± 10 nm and the turnover number (TON) was estimated to be over 235 after 6 h. Besides, CNS-6 h exhibits superior or comparable photocatalytic activity for H2 generation compared to some of the best Pt-loaded g-C3N4 catalysts (Table S2†). The trend of the photocatalytic performance is in good agreement with those of PL, photocurrent and EIS results. Therefore, the remarkable H2 generation ability of CNS-6 h can be ascribed to its superior charge separation efficiency and faster charge diffusion rate. The stability test of H2 generation is shown in Fig. 3d. After four consecutive runs, the activity of CNS-6 h is decreased by 9.4%. This indicates a good photocatalytic stability of CNS-6 h, arising from the outstanding stability of the g-C3N4 inherent structure. | ||
| Fig. 3 Photophysical properties and photocatalytic activities of the samples. (a) Transient photocurrent density, (b) EIS spectra and (c) photocatalytic activity of the as-prepared samples (20 mg of photocatalysts, 1 wt% Pt, 15 vol% TEOA, 300 W Xe lamp, >400 nm). (d) Stability test of CNS-6 h under the same conditions. | ||
3.3. SMF study of site-specific catalytic activities, dynamics and fluctuations
To study the photocatalytic activities and dynamics at individual structural features (i.e., wrinkles, edges and basal planes) of g-C3N4, SMF imaging was used to visualize the real-time mapping of photoreactions and unveil the catalytic heterogeneities at each structure with nanometer resolution. The fluorogenic reaction was designed to characterize the site-specific reactivity of photo-induced e− and quantify the dynamic process for the photoreaction on g-C3N4. Among various probe molecules, non-fluorescent resazurin can be photoreduced by e− to generate highly fluorescent resorufin (Fig. 4a). In turn, the mapping of photoactive species (i.e., e−) could be visualized and the catalytic process including the reactant adsorption, activation and conversion, and product dissociation would be quantified at different sites (Fig. 4b). During imaging, CNS-6 h is deposited and immobilized on a quartz slide with the resazurin solution (0.02–1 μM) continuously flowing over the sample. As shown in Fig. 4a, CNS-6 h is photoexcited by a 405 nm laser to produce e− and h+, followed by the subsequent photoreduction of resazurin to resorufin. Another 532 nm laser was used to excite the resorufin molecules and the fluorescent signal was collected using a highly sensitive and high-speed camera. In the SRM image shown in Fig. 5a, each bright dot represents a fluorescent burst stemmed from the formation of a resorufin molecule at one reactive site. In the intensity trajectory (Fig. 5b), each stochastic on–off cycle with a one-step intensity change represents a single photocatalytic turnover event. The product formation appeared as a sudden increase in the signal while the product dissociation signified as a sudden signal decrease. The fluorescence intensity of a typical fluorescent burst (inset of Fig. 5c) distributed within a few pixels as a point spread function (PSF). The center position of the fluorescent burst is localized by fitting the signal intensity using a 2D elliptical Gaussian function (Fig. 5c and eqn (S1)†). The localization precision can be improved with less background noise and more photons collected from each frame. In our experiment, TIRF microcopy effectively reduces the background noise, and the high numerical aperture of the objective lens allows more light to be collected. Consequently, the single-molecule images with high signal to noise ratios were collected with the spatial resolution calculated to be 10 nm (eqn (S1)–(S3)†).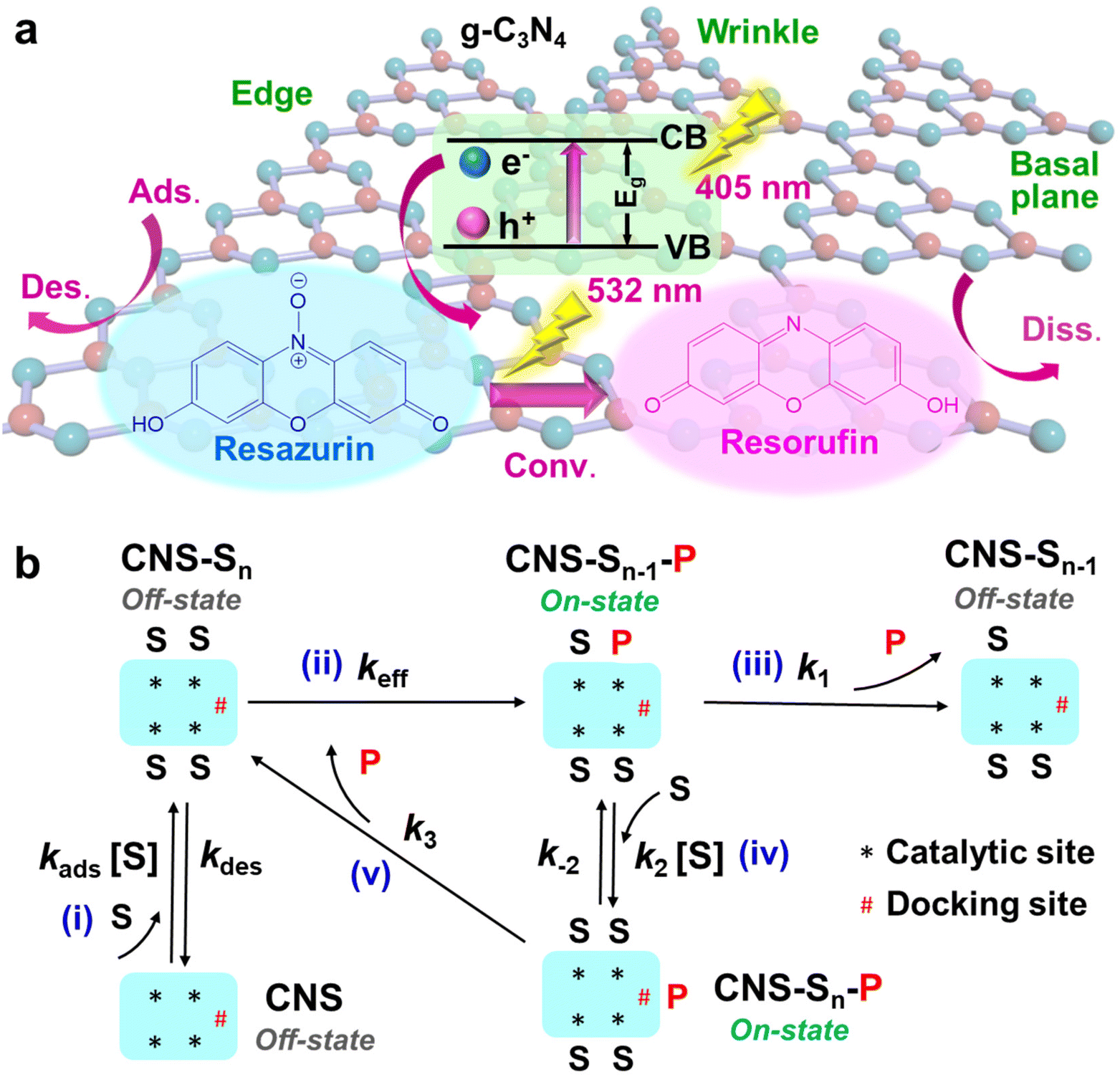 | ||
| Fig. 4 SMF imaging of the photocatalytic process on CNS-6 h. (a) Schematic of the photocatalytic reduction of resazurin to resorufin. (b) Schematic diagram of reactant adsorption, chemical conversion and product dissociation. S and P denote the substrate and product, respectively. | ||
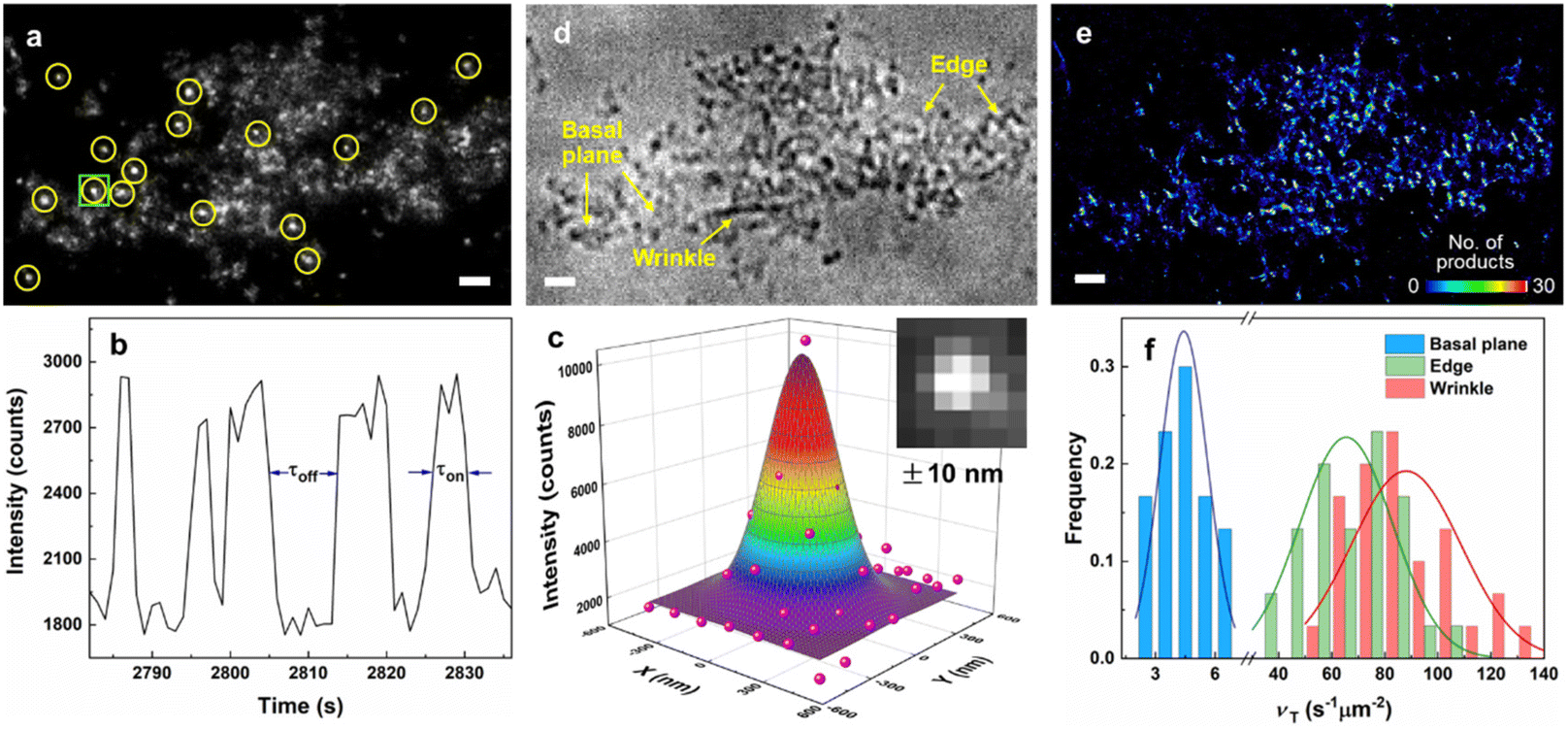 | ||
| Fig. 5 SMF imaging of photocatalytic events and activities on CNS-6 h. (a) A typical image of fluorescent resorufin molecules (in yellow circles) in a single frame. (b) A segment of fluorescence intensity trajectory from a 480 nm × 480 nm area in the green square in (a), bin time: 30 ms. (c) 2D Gaussian fitting of the intensity profile of a fluorescent burst in the inset (from the area in the green square in (a)). (d) The bright-field image and (e) density map (bin size: 20 nm × 20 nm) of CNS-6 h with resazurin. Scale bar: 2 μm. (f) Histogram distribution of catalytic turnover rates (νT) at wrinkles, edges and basal planes (based on more than 20 positions from each structure). | ||
The brightfield image of CNS-6 h is shown in Fig. 5d, wherein the wrinkle, edge and basal plane are clearly identified. It is generally known that the edge and basal plane are commonly present in 2D materials. On the other hand, the wrinkle can be formed by the folding or bending (wrinkling) of basal planes (Fig. S7a†) during the exfoliation process.11,35Fig. 5e shows the corresponding colored density map, representing the number of catalytic events at different locations of CNS-6 h. The distribution of photoactive species and catalytic heterogeneity at each structural feature are clearly revealed. The wrinkle and edge possess a higher photoactivity with more catalytic events compared to the basal plane. In order to quantify the site-specific photoactivities and dynamics, image segmentation was carried out to determine the size of reaction subregions at each structure (Fig. S8†). Counting the single turnover events at these subregions enables us to calculate the specific catalytic turnover rate, νT, which is the number of catalytic events over time and the area. The averaged specific catalytic turnover rate (〈νT〉, where 〈〉 denotes averaging) was obtained by fitting νT with its frequency (Fig. 5f). 〈νT〉 is correlated with the photocatalytic activity at individual structural features. As shown in the histogram distribution of νT (Fig. 5f) for each structure, the 〈νT〉 of wrinkles, edges and basal planes are determined to be 88.0, 65.5 and 4.4 s−1 μm−2, respectively. Therefore, the wrinkle and edge exhibit a superior photocatalytic activity, which are 20 times and 14.8 times those of the basal plane, respectively. Such phenomena are presumably due to the presence of tensile strain (TS) and compressive strain (CS) in the wrinkle structure and the low-coordination environment at the edge position. This would change the electronic band structure and charge separation properties of g-C3N4, which will be explained in the later parts.
To further explore the site-specific activities and dynamics on g-C3N4, we deconvoluted the kinetic steps in the photoreduction of resazurin and quantitatively determine the rate constants of resazurin adsorption, conversion and resorufin dissociation. In the fluorescence trajectories (Fig. 5b), τoff is the waiting time before the formation of resorufin molecules and τon is the residence time before resorufin dissociates from the g-C3N4 surface.36 Therefore, calculating 〈τoff〉−1 and 〈τon〉−1 enables us to determine the reaction rate (νRZ = 〈τoff〉−1/area) of resazurin and the dissociation rate (νd = 〈τon〉−1) of the product resorufin. As shown in Fig. 6a, different structural features of g-C3N4 (the same g-C3N4 in Fig. 5d and e) exhibit distinct photocatalytic reaction kinetics. The kinetic data for all structures are fitted into the Langmuir–Hinshelwood (LH) kinetic model.37,38 At a lower [RZ], the reaction rate instantly increases but gradually saturates at a higher [RZ]. In the LH kinetic model, the reaction kinetics can be quantitatively described as  , where keff is the chemical conversion rate constant (Fig. 4b, step (ii)) and KRZ (i.e., kads/kdes) is the adsorption equilibrium constant (Fig. 4b, step (i)). In Fig. 6b, wrinkles exhibit the highest conversion rate constant (80.1 s−1 μm−2), which is 1.5 and 2.2 times those of edges and basal planes, respectively. This could be probably ascribed to the intrinsic band modulation and local build-in electrical field arising from the presence of TS, CS and defects. In Fig. S7b,† the increasing TS could downshift the CB and upshift the VB,39,40 which forms type-I band alignment with the original g-C3N4 (Fig. S7c†). The reduced bandgap energy enables the generation of more photo-induced e− and h+. Besides, with the larger CS, the CB and VB are upshifted simultaneously, which forms type-II alignment with the pristine band and thus improves the charge separation (Fig. S7d†). It is well-known that edges with large coordination unsaturation contain disordered structures and defects.41,42 Such defects would create intermediate mid-gap states above the VB or shift the VB upward (Fig. S7e†), which improves the light absorption and spatial separation of charges. Thus, the photocatalytic activity is enhanced at these unsaturated sites compared to the basal plane. In Fig. 6c, the basal plane shows the highest adsorption equilibrium constant (KRZ), which is 13.6% and 15.6% higher than those of edges and wrinkles, respectively. This is probably due to the steric effect on the basal plane, in which the large planar π–π resazurin molecule preferably lays down on the flat basal plane via the van der Waals (vdW) force.43,44 Therefore, the basal plane exhibits the largest KRZ with the strongest vdW force with resazurin. However, wrinkles with a folding or bending configuration and the sharp edges would obstruct the flat adsorption of aromatic resazurin and weaken the vdW force, which reduces the KRZ of these structures. Besides, we performed DFT simulations to calculate the adsorption energies of resazurin and resorufin on each structural feature of g-C3N4 at various starting configurations (Fig. S9–S13†). Fig. 7 presents the most stable structures for each adsorbate–surface interaction and both resazurin and resorufin maintain the same configuration for each surface. The DFT results reveal that the adsorption energies of resazurin on basal planes, edges and wrinkles are calculated to be 1.35, 1.23 and 1.15 eV, respectively. It has been previously reported that corrugation in the wrinkle sites of g-C3N4 weakens the π delocalization over triazine moieties,22 thus the interactions with resazurin and resorufin are weakened. The termination of the edge with hydrogen most likely decreased the electron localization in the nearby structural holes, which could have led to reduce the adsorption of the adsorbates. The trend of the energies is consistent with that in our experimental data, which further confirms the adsorption properties of each structural feature of g-C3N4.
, where keff is the chemical conversion rate constant (Fig. 4b, step (ii)) and KRZ (i.e., kads/kdes) is the adsorption equilibrium constant (Fig. 4b, step (i)). In Fig. 6b, wrinkles exhibit the highest conversion rate constant (80.1 s−1 μm−2), which is 1.5 and 2.2 times those of edges and basal planes, respectively. This could be probably ascribed to the intrinsic band modulation and local build-in electrical field arising from the presence of TS, CS and defects. In Fig. S7b,† the increasing TS could downshift the CB and upshift the VB,39,40 which forms type-I band alignment with the original g-C3N4 (Fig. S7c†). The reduced bandgap energy enables the generation of more photo-induced e− and h+. Besides, with the larger CS, the CB and VB are upshifted simultaneously, which forms type-II alignment with the pristine band and thus improves the charge separation (Fig. S7d†). It is well-known that edges with large coordination unsaturation contain disordered structures and defects.41,42 Such defects would create intermediate mid-gap states above the VB or shift the VB upward (Fig. S7e†), which improves the light absorption and spatial separation of charges. Thus, the photocatalytic activity is enhanced at these unsaturated sites compared to the basal plane. In Fig. 6c, the basal plane shows the highest adsorption equilibrium constant (KRZ), which is 13.6% and 15.6% higher than those of edges and wrinkles, respectively. This is probably due to the steric effect on the basal plane, in which the large planar π–π resazurin molecule preferably lays down on the flat basal plane via the van der Waals (vdW) force.43,44 Therefore, the basal plane exhibits the largest KRZ with the strongest vdW force with resazurin. However, wrinkles with a folding or bending configuration and the sharp edges would obstruct the flat adsorption of aromatic resazurin and weaken the vdW force, which reduces the KRZ of these structures. Besides, we performed DFT simulations to calculate the adsorption energies of resazurin and resorufin on each structural feature of g-C3N4 at various starting configurations (Fig. S9–S13†). Fig. 7 presents the most stable structures for each adsorbate–surface interaction and both resazurin and resorufin maintain the same configuration for each surface. The DFT results reveal that the adsorption energies of resazurin on basal planes, edges and wrinkles are calculated to be 1.35, 1.23 and 1.15 eV, respectively. It has been previously reported that corrugation in the wrinkle sites of g-C3N4 weakens the π delocalization over triazine moieties,22 thus the interactions with resazurin and resorufin are weakened. The termination of the edge with hydrogen most likely decreased the electron localization in the nearby structural holes, which could have led to reduce the adsorption of the adsorbates. The trend of the energies is consistent with that in our experimental data, which further confirms the adsorption properties of each structural feature of g-C3N4.
 | ||
| Fig. 6 Photocatalytic kinetics on different structural features of CNS-6 h in Fig. 5. The dependence of (a) resazurin reaction rate (νRZ) and (d) resorufin dissociation rate (νd) on resazurin concentration. The error bars are the SDs of the Gaussian fittings of νRZ and νd distributions from many subregions (≥20) at each structure. (b) Reaction rate constants (keff), (c) adsorption equilibrium constants (KRZ) and dissociation rate constants of (e) direct (k1) and (f) indirect (k3) dissociation at each structure of CNS-6 h. | ||
 | ||
| Fig. 7 Optimized geometries of the most stable adsorption of resazurin and resorufin on the (a and d) basal plane, (b and e) edge, and (c and f) wrinkle of g-C3N4. The rest of configurations are presented in the ESI.† BP, RZ and RF denote the basal plane, resazurin and resorufin, respectively. | ||
The dissociation behavior of fluorescent resorufin on each structure of g-C3N4 is shown in Fig. 6d. The trend exhibits an initial rapid dissociation rate at a lower [RZ] and a decreased rate with increasing [RZ] followed by a saturated stable rate at a higher [RZ]. Such results indicate that the dissociation behavior of resorufin is not likely a simple direct dissociation process which is independent of the resazurin concentration, but a complex process including the reactant-assisted dissociation pathway. Based on previous works,38,45 two possible parallel pathways may exist in the resorufin dissociation process during the τon period: the direct (Fig. 4b, step (iii)) and indirect (Fig. 4b, step (iv) and (v)) dissociation pathways. The indirect pathway involves the participation of reactants, which is also considered as a reactant-assisted pathway. The resorufin dissociation process can be fitted by the equation of  , where
, where  . k1 and k3 represent the dissociation rate constants of direct and indirect dissociation pathways, which dominate at the low and high [RZ], respectively. In the k1 step, the conversion and dissociation take place at the catalytic site. However, in the k3 process, the product is replaced by the substrate at the catalytic site and transferred to the docking site, where the subsequent dissociation occurs (Fig. 4b).38 The adsorption and desorption rate constants of resazurin in the indirect dissociation are expressed as k2 and k−2. Through fitting the dissociation kinetics, the direct dissociation rate constants k1 for wrinkles, edges, and basal planes are calculated to be 68.9, 37.4 and 33.0 s−1 (Fig. 6e). This could be explained by the steric effect, where the flat resorufin molecules tend to preferably adsorb on the planar basal plane via an intense vdW interaction. DFT calculations reveal that resorufin has the strongest binding strength to basal planes, followed by edges and wrinkles (Fig. 7d–f). Thus, the direct dissociation rate constant shows the opposite trend to the adsorption energy, where wrinkles possess the largest k1. In addition, k1 is larger than k3 for all the structures of g-C3N4 (Fig. 6e and f), which explains why the total dissociation rate reduces with increasing [RZ]. Generally, reactant-assisted (indirect) dissociation dominates at a high concentration, while direct dissociation prevails at a low concentration. In Fig. 6f, the indirect dissociation rate constants k3 for wrinkles, edges, and basal planes are determined to be 18.5, 22.7 and 26.5 s−1. Based on the DFT results (Fig. 7), by comparing the adsorption energies of resazurin and resorufin at the same surface, we discovered that the adsorption energies of resazurin are larger than those of resorufin at all three slabs: (a) basal planes with 0.11 eV difference, then (b) edges with 0.08 eV difference, and (c) wrinkles with 0.05 eV difference. This suggests that resazurin is more likely to replace the resorufin molecules at these catalytic sites, i.e., preference for the reactant-assisted dissociation pathway. The adsorption energy differences explain why basal planes have the highest k3 while wrinkles possess the lowest k3. Besides, the opposite trend of k1 and k3 further suggests that the two dissociation behaviors of products on g-C3N4 are in competition. Hence, we deconvoluted the kinetic steps in a photocatalytic reaction and quantitatively determined the rate constants for reactant adsorption, chemical conversion and product dissociation at the individual structural features of g-C3N4. Such investigation would provide more insight into the fundamental understanding of site-specific activities and dynamics on 2D materials.
. k1 and k3 represent the dissociation rate constants of direct and indirect dissociation pathways, which dominate at the low and high [RZ], respectively. In the k1 step, the conversion and dissociation take place at the catalytic site. However, in the k3 process, the product is replaced by the substrate at the catalytic site and transferred to the docking site, where the subsequent dissociation occurs (Fig. 4b).38 The adsorption and desorption rate constants of resazurin in the indirect dissociation are expressed as k2 and k−2. Through fitting the dissociation kinetics, the direct dissociation rate constants k1 for wrinkles, edges, and basal planes are calculated to be 68.9, 37.4 and 33.0 s−1 (Fig. 6e). This could be explained by the steric effect, where the flat resorufin molecules tend to preferably adsorb on the planar basal plane via an intense vdW interaction. DFT calculations reveal that resorufin has the strongest binding strength to basal planes, followed by edges and wrinkles (Fig. 7d–f). Thus, the direct dissociation rate constant shows the opposite trend to the adsorption energy, where wrinkles possess the largest k1. In addition, k1 is larger than k3 for all the structures of g-C3N4 (Fig. 6e and f), which explains why the total dissociation rate reduces with increasing [RZ]. Generally, reactant-assisted (indirect) dissociation dominates at a high concentration, while direct dissociation prevails at a low concentration. In Fig. 6f, the indirect dissociation rate constants k3 for wrinkles, edges, and basal planes are determined to be 18.5, 22.7 and 26.5 s−1. Based on the DFT results (Fig. 7), by comparing the adsorption energies of resazurin and resorufin at the same surface, we discovered that the adsorption energies of resazurin are larger than those of resorufin at all three slabs: (a) basal planes with 0.11 eV difference, then (b) edges with 0.08 eV difference, and (c) wrinkles with 0.05 eV difference. This suggests that resazurin is more likely to replace the resorufin molecules at these catalytic sites, i.e., preference for the reactant-assisted dissociation pathway. The adsorption energy differences explain why basal planes have the highest k3 while wrinkles possess the lowest k3. Besides, the opposite trend of k1 and k3 further suggests that the two dissociation behaviors of products on g-C3N4 are in competition. Hence, we deconvoluted the kinetic steps in a photocatalytic reaction and quantitatively determined the rate constants for reactant adsorption, chemical conversion and product dissociation at the individual structural features of g-C3N4. Such investigation would provide more insight into the fundamental understanding of site-specific activities and dynamics on 2D materials.
To gain more insight into the temporal fluctuation of reaction rates, we further analyzed the single-turnover trajectories to obtain the time dependence of the rate of turnovers (Fig. 8a), which is the quantity of off–on cycles per unit time. This perturbation with time reflects the dynamic disorders in chemical kinetics, which usually occurs at the high concentration of substrates [S], known as catalysis-induced activity fluctuations. It arises from the rate changes in catalytic conversion (τoff reaction), product dissociation (τon reaction) or both.46 The contribution of these two processes can be analyzed separately by calculating their autocorrelation functions, Cτ(m) = 〈Δτ(0)Δτ(m)〉/〈Δτ(0)2〉.37,47 Here, Δτ(m) = τ(m) − 〈τ〉, τ refers to either τoff or τon and m is the event index number in sequence. In Fig. 8b and c, Cτoff and Cτon(Cτoff, Cτon ≥ 0) exhibit an exponential decay with m, which proves the presence of fluctuations in both τoff and τon reactions. The fitted decay constants for Cτoff − m (Fig. 8b) and Cτon− m (Fig. 8c) are determined to be moff = 2.7 and mon = 4.2 turnovers, respectively. The average turnover time for this trajectory is 0.8 s. Thus, the fluctuation correlation times for τoff and τon reactions are calculated to be 2.2 s and 3.4 s. The two correlation times demonstrate the fluctuation timescales for the catalytic reaction (keff) and indirect product dissociation (k3) which dominates at the saturated [S] (1 μM). The insets of Fig. 8b and c show the histogram of correlation times for the catalytic reaction (τoff) and dissociation (τon) based on numerous locations (≥20). The wide distribution of the histogram suggests the different timescales of activity fluctuations at each structure. As reported by previous literature studies, this temporal activity variation could be relevant to small-scale dynamic surface restructuring with nanometer dimension.38,48,49 It can alter the conversion rate constant (keff = knT) by changing the reactivity per catalytic site and the quantity of catalytic sites for τoff reactions. It also changes the dissociation rate constants (i.e., k1, k2, and k3) for τon reactions. Both will contribute to the temporal variations of activities and oscillatory kinetics. Therefore, the fluctuation correlation times are also the timescales of dynamic surface restructuring. The inverse of the correlation time, that is the fluctuation rate represents the surface restructuring rate. Such a phenomenon is triggered by the distinct adsorbate–surface interactions at each site of the surface structure and this catalysis-induced nature should be affected by [S].46 As shown in Fig. 8d–f, the fluctuation rates of τoff and τon reactions at each structure increase with faster rates of catalytic turnovers, which follows a positive linear correlation. With saturated [RZ] (1 μM), the effect of resazurin migration among different sites causing activity fluctuation is minimal, because the substrates tend to occupy all surface sites at high [RZ]. Moreover, it was found that the slopes (k) of τoff for wrinkles (k = 12.9) and edges (k = 9.7) are much higher than those of basal planes (k = 0.245). Similar results were also obtained for τon reactions with wrinkles and edges having larger slopes. This could be explained by the discrepancy between local mechanical and energy properties at each structure. Distortions such as CS and TS are present in wrinkles, while edges contain unsaturated coordination. Such a configuration would increase the surface energy and reduce the restructuring activation energy as compared to the basal plane.48 Thus, the dynamic activity fluctuations at wrinkles and edges are more evident than those at basal planes.
 | ||
| Fig. 8 Dynamic activity fluctuations at different structural features of CNS-6 h. (a) A typical trajectory of the rate of turnovers on the basal plane with 1 μM resazurin. Each data point is calculated based on every 10 turnovers. Autocorrelation functions of (b) τoff and (c) τon reactions from the single-turnover trajectory in (a). Insets: histograms of the correlation times of τoff and τon reactions calculated from numerous subregions (≥20). Dependence of fluctuation rates on the rate of turnovers on (d) wrinkles, (e) edges and (f) basal planes. Each data point is an average of two trajectories. | ||
In addition, it can be noted that the fluctuation rates for τoff and τon deviate a lot at higher turnover rates for both wrinkles and edges. The difference in the fluctuation rates of τoff and τon demonstrates that the catalytic conversion and dissociation exhibit distinct temporal fluctuation dynamics. No cross-correlation was observed between τoff and τoff reactions (Fig. S14†). Thus, due to the different temporal perturbation behaviors, we propose that at higher turnover rates, the sites where catalytic reactions proceed (i.e., catalytic sites) are different from the sites where dissociation occurs (i.e., docking sites). In other words, under high [RZ] with high turnover rates, the dissociation process on wrinkles and edges tends to undergo indirectly (Fig. 4b, step (iv) and (v)), while the basal plane has no such feature under this condition. This is most likely caused by active sites fully occupied by resazurin molecules due to its higher adsorption on the basal plane. Moreover, the replacement of resazurin by resorufin is unfavored as previously discussed above (Fig. 7). Based on the above discussions, we discovered that the different structural features of g-C3N4 exhibit distinct temporal fluctuation behaviors, which provide hints on the study of activity difference at the saturated [S]. Such investigation would benefit the catalyst design and system optimization for real practical applications, usually performed under the high [S].
4. Conclusion
In conclusion, we have successfully fabricated ultrathin g-C3N4 nanosheets with a remarkable photocatalytic activity for H2 generation. The superior performance is ascribed to the reduced thickness, larger SSA and better charge separation properties, which are verified by TEM, AFM, BET, PL, EIS and transient photocurrent results. Besides the measurement at the ensemble level, we applied SMF imaging to spatially resolve the catalytic heterogeneities at nanometer resolution. The reaction kinetics and temporal activity fluctuations are in situ quantified at the individual structural features of g-C3N4. It was found that the wrinkle has the highest photoactivity, direct dissociation rate constant and activity fluctuation rate, followed by the edge and basal plane. However, regarding the adsorption rate constants, the order is reversed. The trends of catalytic activity and kinetics on the different structures of g-C3N4 are attributed to the electronic band modulation and steric effect. The experimental data are also supported by our DFT simulation results, which further confirms the catalytic properties of each g-C3N4 structure. Therefore, our work constructs a bridge between the catalyst structure and reaction dynamics at the nanoscale, which helps address the heterogeneity challenge in nanocatalysis. Such work can be extended to investigate the catalytic activities and kinetics at other structures (e.g., vacancies, boundaries, porous networks, etc.) of a variety of 2D layered materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support from the Ministry of Education, Singapore, under its Academic Research Fund Tier 1 (No. RG10/20, RG60/21 and RG83/20), and the Singapore Agency for Science, Technology and Research (A*STAR) AME YIRG grant (No. A2084c0065) and MTC IRG grant (No. M21K2c0110).References
- J. Fu, J. Yu, C. Jiang and B. Cheng, Adv. Energy Mater., 2018, 8, 1701503 CrossRef.
- Q. Hao, Y. Huang, D. Chen, Y. Liu, W. Wei and B.-J. Ni, Chin. J. Catal., 2020, 41, 249–258 CrossRef CAS.
- S. An, G. Zhang, T. Wang, W. Zhang, K. Li, C. Song, J. T. Miller, S. Miao, J. Wang and X. Guo, ACS Nano, 2018, 12, 9441–9450 CrossRef CAS PubMed.
- G. Liu, Y. Huang, H. Lv, H. Wang, Y. Zeng, M. Yuan, Q. Meng and C. Wang, Appl. Catal., B, 2021, 284, 119683 CrossRef CAS.
- T. Jeong, H. Piao, S. Park, J.-H. Yang, G. Choi, Q. Wu, H. Kang, H. J. Woo, S. J. Jung and H. Kim, Appl. Catal., B, 2019, 256, 117850 CrossRef CAS.
- K. Chen, Z. Chai, C. Li, L. Shi, M. Liu, Q. Xie, Y. Zhang, D. Xu, A. Manivannan and Z. Liu, ACS Nano, 2016, 10, 3665–3673 CrossRef CAS PubMed.
- Q. Hao, G. Jia, W. Wei, A. Vinu, Y. Wang, H. Arandiyan and B.-J. Ni, Nano Res., 2020, 13, 18–37 CrossRef CAS.
- M. J. K. Ow, J. J. Ng, J. X. Yong, B. Y. L. Quek, E. K. Yeow and Z. Zhang, ACS Appl. Nano Mater., 2020, 3, 3163–3167 CrossRef CAS.
- N. M. Andoy, X. Zhou, E. Choudhary, H. Shen, G. Liu and P. Chen, J. Am. Chem. Soc., 2013, 135, 1845–1852 CrossRef CAS PubMed.
- Z. Ristanović, J. P. Hofmann, G. De Cremer, A. V. Kubarev, M. Rohnke, F. Meirer, J. Hofkens, M. B. Roeffaers and B. M. Weckhuysen, J. Am. Chem. Soc., 2015, 137, 6559–6568 CrossRef PubMed.
- T.-X. Huang, B. Dong, S. L. Filbrun, A. A. Okmi, X. Cheng, M. Yang, N. Mansour, S. Lei and N. Fang, Sci. Adv., 2021, 7, eabj4452 CrossRef CAS PubMed.
- T. Tachikawa, T. Ohsaka, Z. Bian and T. Majima, J. Phys. Chem. C, 2013, 117, 11219–11228 CrossRef CAS.
- S. Yan, Z. Li and Z. Zou, Langmuir, 2009, 25, 10397–10401 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- L. M. Azofra, D. R. MacFarlane and C. Sun, Phys. Chem. Chem. Phys., 2016, 18, 18507–18514 RSC.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- P. Niu, L. Zhang, G. Liu and H. M. Cheng, Adv. Funct. Mater., 2012, 22, 4763–4770 CrossRef CAS.
- Y. Li, R. Jin, Y. Xing, J. Li, S. Song, X. Liu, M. Li and R. Jin, Adv. Energy Mater., 2016, 6, 1601273 CrossRef.
- Q. Han, B. Wang, J. Gao, Z. Cheng, Y. Zhao, Z. Zhang and L. Qu, ACS Nano, 2016, 10, 2745–2751 CrossRef CAS PubMed.
- S. Guo, Y. Zhu, Y. Yan, Y. Min, J. Fan and Q. Xu, Appl. Catal., B, 2016, 185, 315–321 CrossRef CAS.
- M. Groenewolt and M. Antonietti, Adv. Mater., 2005, 17, 1789–1792 CrossRef CAS.
- C. Wu, S. Xue, Z. Qin, M. Nazari, G. Yang, S. Yue, T. Tong, H. Ghasemi, F. C. R. Hernandez and S. Xue, Appl. Catal., B, 2021, 282, 119557 CrossRef CAS.
- S. Wu, J.-K. Lee, P. C. Lim, R. Xu and Z. Zhang, Nanoscale, 2022, 14, 5612–5624 RSC.
- A. P. Alivisatos, Science, 1996, 271, 933–937 CrossRef CAS.
- J. Xu, L. Zhang, R. Shi and Y. Zhu, J. Mater. Chem. A, 2013, 1, 14766–14772 RSC.
- Z. Tong, D. Yang, Y. Sun, Y. Nan and Z. Jiang, Small, 2016, 12, 4093–4101 CrossRef CAS PubMed.
- J. Pető, T. Ollár, P. Vancsó, Z. I. Popov, G. Z. Magda, G. Dobrik, C. Hwang, P. B. Sorokin and L. Tapasztó, Nat. Chem., 2018, 10, 1246–1251 CrossRef PubMed.
- B. Dong, Y. Pei, F. Zhao, T. W. Goh, Z. Qi, C. Xiao, K. Chen, W. Huang and N. Fang, Nat. Catal., 2018, 1, 135–140 CrossRef.
- K. S. Han, G. Liu, X. Zhou, R. E. Medina and P. Chen, Nano Lett., 2012, 12, 1253–1259 CrossRef CAS PubMed.
- W. Xu, J. S. Kong, Y.-T. E. Yeh and P. Chen, Nat. Mater., 2008, 7, 992–996 CrossRef CAS PubMed.
- Y. Liang, C. Long, J. Li, H. Jin, B. Huang and Y. Dai, ACS Appl. Energy Mater., 2018, 1, 5394–5401 CAS.
- D. J. Trainer, Y. Zhang, F. Bobba, X. Xi, S.-W. Hla and M. Iavarone, ACS Nano, 2019, 13, 8284–8291 CrossRef CAS PubMed.
- C. Mondelli, B. Puértolas, M. Ackermann, Z. Chen and J. Pérez-Ramírez, ChemSusChem, 2018, 11, 2859–2869 CrossRef CAS PubMed.
- X. Yu, S. F. Ng, L. K. Putri, L. L. Tan, A. R. Mohamed and W. J. Ong, Small, 2021, 17, 2006851 CrossRef CAS PubMed.
- T. Chen, S. Chen, Y. Zhang, Y. Qi, Y. Zhao, W. Xu and J. Zeng, Angew. Chem., 2016, 128, 1871–1875 CrossRef.
- G. Chen, N. Zou, B. Chen, J. B. Sambur, E. Choudhary and P. Chen, ACS Cent. Sci., 2017, 3, 1189–1197 CrossRef CAS PubMed.
- W. Xu, J. S. Kong and P. Chen, J. Phys. Chem. C, 2009, 113, 2393–2404 CrossRef CAS.
- X. Zhou, W. Xu, G. Liu, D. Panda and P. Chen, J. Am. Chem. Soc., 2010, 132, 138–146 CrossRef CAS PubMed.
- H. P. Lu, L. Xun and X. S. Xie, Science, 1998, 282, 1877–1882 CrossRef CAS PubMed.
- P. Chen, X. Zhou, N. M. Andoy, K.-S. Han, E. Choudhary, N. Zou, G. Chen and H. Shen, Chem. Soc. Rev., 2014, 43, 1107–1117 RSC.
- W. Xu, J. S. Kong and P. Chen, Phys. Chem. Chem. Phys., 2009, 11, 2767–2778 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr06077a |
| This journal is © The Royal Society of Chemistry 2023 |