
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolar driven CO2 reduction with a molecularly engineered periodic mesoporous organosilica containing cobalt phthalocyanine†
M.
Angeles Navarro
ab,
Sunanda
Sain
 b,
Maximilian
Wünschek
c,
Christian M.
Pichler
cd,
Francisco J.
Romero-Salguero
a,
Dolores
Esquivel
*a and
Souvik
Roy
*b
b,
Maximilian
Wünschek
c,
Christian M.
Pichler
cd,
Francisco J.
Romero-Salguero
a,
Dolores
Esquivel
*a and
Souvik
Roy
*b
aDepartamento de Química Orgánica, Instituto Químico para la Energía y el Medioambiente (IQUEMA), Facultad de Ciencias, Universidad de Córdoba, Campus de Rabanales, 14071 Córdoba, Spain. E-mail: q12esmem@uco.es
bSchool of Chemistry, The University of Lincoln, Green Lane, Lincoln LN6 7TS, UK. E-mail: sroy@lincoln.ac.uk
cInstitute of applied Physics, TU Vienna, Wiedner Hauptstraße 8-10, 1040 Vienna, Austria
dCentre of electrochemical and surface technology, Viktor Kaplan Straße 2, 2700 Wiener Neustadt, Austria
First published on 11th January 2023
Abstract
A molecular cobalt phthalocyanine (CoPc) catalyst has been integrated in an ethylene-bridged periodic mesoporous organosilica (PMO) to fabricate a hybrid material, CoPc-PMO, that catalyses CO2 reduction to CO in a photocatalytic system using [Ru(bpy)3]2+ (bpy = 2,2′-bipyridine) as a photosensitizer and 1,3-dimethyl-2-phenyl-2,3-dihydro-1H-benzo[d]imidazole (BIH) as an electron donor. CoPc-PMO displays a Co-based turnover number (TONCO) of >6000 for CO evolution with >70% CO-selectivity after 4 h irradiation with UV-filtered simulated solar light, and a quantum yield of 1.95% at 467 nm towards CO. This system demonstrates a benchmark TONCO for immobilised CoPc-based catalysts towards visible light-driven CO2 reduction.
 Souvik Roy | Dr Souvik Roy is Senior Lecturer in the School of Chemistry at the University of Lincoln. He completed PhD in Chemistry from Arizona State University, followed by postdoctoral work at CEA Grenoble, Uppsala University, and finally, at University of Cambridge, where he was awarded a Marie-Curie Individual Fellowship. Since 2020, he started his independent research career at University of Lincoln, where his group works at the interface of molecular and materials chemistry, focusing on developing new concepts and technologies for production of fuels and chemicals using surplus renewable electricity and solar energy. |
Introduction
Climate change induced by the increasing atmospheric CO2 levels, combined with growing global energy demand, have spurred widespread interest in developing CO2 mitigation technology that will allow CO2 upcycling into value-added products. To that goal, solar-driven CO2 conversion into carbon-based energy carriers and feedstocks represents a promising strategy towards CO2 utilisation and recycling.1 Since the pioneering work reported by Lehn and co-workers2 on photochemical reduction of CO2 using ReI complexes as photosensitizer and catalytic unit, a wide range of molecular catalysts based on transition metal complexes have been developed to mediate light-driven CO2 conversion to C1 feedstocks, CO and formic acid.3 While molecular catalysts offer distinct advantages including tuneability, high product selectivity, and low overpotential, they are typically used in solution as homogeneous catalysts, which prevents catalyst recycling and often leads to decomposition via diffusional pathways.4 Heterogenization of molecular catalysts on solid supports presents a promising strategy to circumvent these problems and combine the benefits of homogeneous and heterogeneous systems.5,6Cobalt phthalocyanine (CoPc) has been studied for electrocatalytic CO2 reduction since the 1970s,7,8 and has recently attracted renewed attention due to its excellent catalytic performance upon heterogenisation on electrodes,9–11 reticular materials,12 and semiconductors.13,14 Among them, integration of CoPc into high-surface area scaffolds with ordered micro- or meso-porosity is particularly interesting because such architecture offer high loading of accessible catalyst units. Metal organic frameworks (MOFs) and covalent organic frameworks (COFs) have been employed as scaffolds to immobilise metal phthalocyanines into their skeleton. However these systems have been targeted towards conductive frameworks for application in CO2 electroreduction reaction (eCO2R).15–17 There are relatively few reports on photocatalytic CO2 reduction using heterogenised CoPc in colloidal suspension in the presence of a separate light absorber and electron donor.13,18 A porous support is key in such photocatalyst design to allow diffusion of different components (sensitiser, donor, and substrate) within the pores and reaction with the immobilised CoPc units.
In this context, periodic mesoporous organosilicas (PMOs) present a promising family of porous materials with well-ordered structures that have attracted great interest as a scaffold for mounting molecular catalysts.19 These hybrid materials, synthesized from organo-bridged alkoxysilane precursors in presence of a structure-directing agent, possess ordered mesostructures with high surfaces areas, tailored hydrophobicity/hydrophilicity and tuneable pore sizes, making them a versatile platform for introducing molecular metal complexes for CO2 reduction. However, their insulating character has limited their application exclusively to photocatalytic applications. Until now, the few examples of PMOs reported in literature as heterogenous catalysts for photochemical CO2 reduction are based on anchored metal bipyridine complexes on the mesochannels or into pore walls of these materials.20–24 First studies developed PMOs with chromophores in the framework, such as biphenyl20 and acridone groups,21 and Re-bipyridine complexes anchored in their mesochannels. The PMO support was used as light-harvesting antenna to enhance the photocatalytic CO2 reduction of the ReI complex. More recently, bipyridine-bridged PMOs that allow immobilisation of metal complexes have been developed. The first pioneering study on bipyridine-PMOs for CO2 photoreduction was reported by Inagaki et al.,22 who successfully integrated molecular Ru- and Re-bipyridine complexes as photosensitizer and catalytic units in the same framework. Following a similar approach, a precious-metal-free, Mn carbonyl bipyridine-PMO catalyst was synthesised through the immobilisation of Mn-complexes on the appended bipyridine ligands on PMO.23 The Mn-bpy-PMO material displayed photocatalytic CO2 reduction activity in the presence of Ru(bpy)32+ sensitiser, albeit with poor product selectivity. Both CO and formate were produced from CO2 with Mn-based turnover numbers of 168 and 292, respectively.
Herein, we aim to integrate cobalt-phthalocyanine molecular catalysts into the pore walls of a periodic mesoporous organosilica for photocatalytic reduction of CO2 to CO. For this, we firstly prepared a novel cobalt-phthalocyanine bridged precursor bearing four alkoxysilane groups tethered from cobalt-phthalocyanine skeleton. Then, this precursor was successfully incorporated into the ordered mesostructure of an ethylene-bridged periodic mesoporous organosilica using a one-pot synthesis via co-condensation method in the presence of octadecyltrimethylammonium bromide (OTAB) as structure-directing agent.
Experimental
Synthesis of cobalt phthalocyanine bridged periodic mesoporous organosilica material (CoPc-PMO)
CoPc-PMO material was prepared via self-assembly assisted co-condensation of the cobalt phthalocyanine bridged alkoxysilane precursor, CoPc(NCO), and conventional bis-silane precursor, BTEE (1,2-bis(triethoxysilyl)ethane) (Fig. 1) (see ESI for Experimental details, Scheme S1†). In a general synthesis,25 octadecyltrimethylammonium bromide (OTAB) (0.85 g, 2.1 mmol) was dissolved in a basic solution of deionised water (53 mL) and NaOH (6 M, 0.89 mL). After stirring this solution overnight at 40 °C, the mixture of organosilanes (2.04 mmol) (3 mol% CoPc(NCO) in 5 mL of ethanol and 97 mol% BTEE) was added dropwise to the reaction mixture under vigorous stirring. The resulting mixture was stirred at room temperature for 24 h and was further aged at 97 °C for 4 days under static conditions. A greenish blue powder was collected by filtration and thoroughly washed with distilled water. The as-synthesized material (1 g) was refluxed in a solution containing 50 mL EtOH and 1 mL HCl solution (32% wt) for 12 h to remove the surfactants from the pores. After repeating this extraction process twice, the residual solid was filtered, washed with EtOH and dried under vacuum at 80 °C to give CoPc-PMO (0.44 g). | ||
| Fig. 1 Schematic illustration of the synthesis of CoPc-PMO using a molecular CoPc building block containing four alkoxysilane anchors. | ||
Photocatalytic CO2 reduction experiments
Photocatalytic experiments were carried out in a borosilicate vial (total volume 8 mL) with constant stirring under visible light irradiation using a solar light simulator (SciSun-LP-150) equipped with AM 1.5G filter and an UV cut-off filter (>400 nm). Typically, CoPc-PMO catalyst (1–3 mg) was suspended into 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 acetonitrile/triethanolamine mixture (4 mL) containing Ru(bpy)3(PF6)2 (0.5 mM) and BIH (20 mM). When BIH was not present, TEOA served as the electron donor. The solution was sonicated in an ultrasonic bath for 15 min, and purged with CO2 for 15 min. The head space above the reaction solution was sampled with a gas-tight syringe at different time intervals for product analysis using an SRI gas chromatograph. Control experiments were performed under identical conditions without one of the components: visible light, sacrificial donor (BIH and TEOA), photosensitizer, catalyst, and CO2. Turnover number for CO (TONCO) was determined using the following equation, TONCO = nCO/nCoPc (nCO = moles of CO produced per mg catalyst, nCoPc = moles of CoPc present per mg catalyst). CO selectivity was calculated from the following equation, %CO = [nCO/(nCO + nH2)] × 100%.
:1 acetonitrile/triethanolamine mixture (4 mL) containing Ru(bpy)3(PF6)2 (0.5 mM) and BIH (20 mM). When BIH was not present, TEOA served as the electron donor. The solution was sonicated in an ultrasonic bath for 15 min, and purged with CO2 for 15 min. The head space above the reaction solution was sampled with a gas-tight syringe at different time intervals for product analysis using an SRI gas chromatograph. Control experiments were performed under identical conditions without one of the components: visible light, sacrificial donor (BIH and TEOA), photosensitizer, catalyst, and CO2. Turnover number for CO (TONCO) was determined using the following equation, TONCO = nCO/nCoPc (nCO = moles of CO produced per mg catalyst, nCoPc = moles of CoPc present per mg catalyst). CO selectivity was calculated from the following equation, %CO = [nCO/(nCO + nH2)] × 100%.
Recycling tests
For the recycling experiments, the photocatalytic reaction mixture was centrifuged after 1 h irradiation, and the catalyst was washed three times with acetonitrile to remove adsorbed TEOA and Ru(bpy)3(PF6)2. The catalyst was redispersed in fresh MeCN/TEOA solution containing Ru(bpy)3(PF6)2 (0.5 mM) and BIH (20 mM) and irradiated under identical conditions.In situ ICP analysis
Measurements were performed using an Agilent 7900 ICP-MS. The ICP-MS uses a collision cell with 5 mL min−1 flow of helium as cell gas. External calibration was performed with multielement standard solutions provided by Agilent and Inorganic Ventures. Downstream of the electrochemical cell the analyte was mixed with an internal standard solution containing gallium having a similar mass as cobalt. The in-house built electrochemical flow cell was made from PTFE with an opening for introducing a blue LED (465 nm, 3500 mcd light intensity). 250 μl of a CoPc-PMO suspension in acetone (concentration 20 mg mL−1) was deposited on carbon paper on an area of approx. 3 mm2, which was corresponding to irradiation area of the blue LED. As electrolyte 10% TEOA, 90% H2O and 320 mg L−1 Ru(bpy)3Cl2, purged with CO2 was used and pumped with a flow of 2.4 mg s−1 through the measurement cell. Flow measurements were conducted for a duration of 1 h with alternating light on–off cycles of 2 min.Results and discussion
Synthesis and characterisation
The novel PMO material with cobalt phthalocyanine (CoPc) moieties integrated on the pore walls of the silica framework was synthesised by co-condensation of CoPc-bridged alkoxysilane precursor (CoPc(NCO)) and a large excess of a conventional bis-silane precursor, 1,2-bis(triethoxysilyl)ethane (BTEE) (97%), in the presence of a cationic surfactant (Fig. 1). The surfactant was removed from the as-synthesized CoPc-PMO by solvent extraction using a mixture of aqueous HCl and ethanol to produce a pale greenish blue solid. The cobalt loading in the CoPc-PMO material was determined by ICP-MS after acid-digestion of the sample. Two materials were synthesised with Co loading of 4.6 and 2.7 μmol g−1. All physical characterisation discussed below and the photocatalysis experiments were carried out using CoPc-PMO with 4.6 μmol Co g−1, unless noted otherwise.Powder X-ray diffraction (PXRD) pattern of CoPc-PMO catalyst is depicted in Fig. 2a, which displayed an intense diffraction peak at 2θ = 1.8° with a d-spacing of 49.5 Å and two broad peaks at higher incidence angles. These peaks can be indexed as (100), (110) and (200) reflections, indicative of materials with 2D-hexagonal (P6 mm) mesostructures.26 These findings were further confirmed by transmission electron microscopy of CoPc-PMO which showed a highly ordered structure with parallel channel pores of uniform diameter (Fig. 2b).
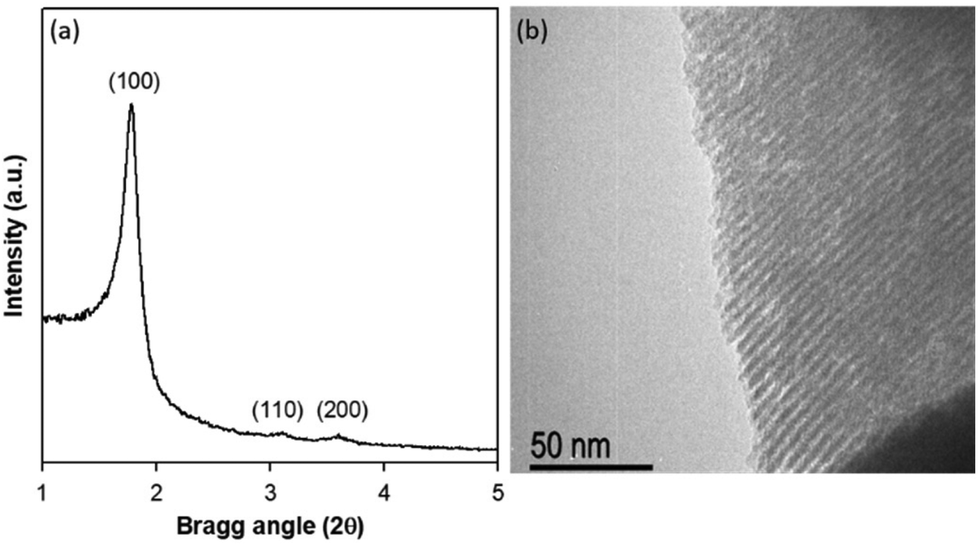 | ||
| Fig. 2 Powder X-ray diffraction pattern (a) and TEM image (b) of CoPc-PMO catalyst. | ||
Nitrogen adsorption–desorption isotherm of CoPc-PMO displayed a type-IV isotherm with a capillary step at P/P0 = 0.4–0.8 (Fig. S1†). The isotherm pattern is consistent with the mesoporous structure of CoPc-PMO with relatively broad pore size distributions ranging from 2–10 nm (Fig. S1 inset†). Similar results were obtained for PMOs containing heterocyclic rings of tris[3-(trimethoxysilyl)propyl]isocyanurate in the pore walls.27,28 The BET specific surface area of CoCp-PMO was 949 m2 g−1 with BJH average pore diameter around 3.4 nm and total pore volume of 1.1 cm3 g−1.
IR analyses was used to confirm the formation of CoPc(NCO) from hydroxylated CoPc-precursor [CoPc(OH)4] as well as to demonstrate the successful incorporation of the CoPc building blocks into the pore walls of PMO. ATR-FTIR monitoring of the reaction between CoPc(OH)4 and 3-(triethoxysilyl)propyl isocyanate showed disappearance of the –N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CO stretching band at 2265 cm−1 and concurrent appearance of a new stretching vibration at 1710 cm−1, consistent with formation of the carbamate linkage (Fig. S2†).29,30
CO stretching band at 2265 cm−1 and concurrent appearance of a new stretching vibration at 1710 cm−1, consistent with formation of the carbamate linkage (Fig. S2†).29,30
Additionally, the FT-IR spectrum of CoPc-NCO precursor (Fig. 3a) displayed the characteristic vibrational bands of the phthalocyanine skeleton at 1605, 1549, 1390 and 1093 cm−1 and the Co–N bond at 910 and 804 cm−1.31,32 Retention of the CoPc fingerprint IR bands in the FT-IR spectrum of CoPc-PMO confirmed the immobilisation of CoPc. Additional vibrations at ∼2900 cm−1 in CoPc-PMO can be assigned to the C–H stretching of propyl chains of the silane precursor and the ethylene bridges.33 The presence of molecular CoPc units in the material was further confirmed by UV-vis spectroscopy. The transmission UV-vis spectrum of CoPc(OH)4 in DMF consisted of the characteristic B-band (or Soret band) at 280 nm, associated to the metal to ligand charge transfer (S0 → S2), and the Q band at 674 nm, associated with the transitions from the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO) (S0 → S1) (Fig. 3b).34 Once this cobalt phthalocyanine compound was incorporated in the PMO through the corresponding CoPc(NCO) silane precursor, the resulting PMO material preserved both adsorption bands. Interestingly, a small red-shift of the Q-band maximum to ∼695 nm was observed alongside an additional shoulder peak at ∼640 nm, indicative of a face-to-face aggregation. The spectrum also showed peak broadening in CoPc-PMO, which have been reported in aggregated phthalocyanines and cofacially-aligned phthalocyanine aggregates on covalent organic frameworks and PMOs.35,36
 | ||
| Fig. 3 (a) FT-IR spectra of CoPc(NCO) and CoPc-PMO catalyst. (b) UV-vis spectrum of CoPc(OH)4 in solution (CH3OH) (black line) and UV-vis DRS of CoPc-PMO (red line). | ||
X-ray photoelectron spectroscopy (XPS) measurements were performed to confirm the incorporation of the phthalocyanine moieties on the material as well as the oxidation state of cobalt species on the samples. The survey spectra of CoPc(OH)4 and CoPc-PMO showed the peaks of Si, C, N, O and Co (Fig. 4a). The high resolution C1s spectrum of CoPc(OH)4 was fitted with four components at 284.7, 285.6, 286.5 and 288.4 eV, assigned to the carbon atoms of the aromatic rings (Car), carbon linked to hydroxyl groups (C–O), pyrrole carbons linked to nitrogen (CN) and π–π* excitations, respectively.37 These characteristic peaks from phthalocyanine skeleton were also observed on the C1s XPS spectrum of CoPc-PMO along with two new contributions at 285.2 and 287.5 eV for C–N and CO bonds from urethane groups (Fig. 4b).
 | ||
| Fig. 4 (a) Survey spectra, (b) C1s, (c) N1s and (d) Co2p high-resolution XPS spectra of CoPc(OH)4 and CoPc-PMO. | ||
In the N1s region (Fig. 4c), the existence of two peaks at 398.8 and 400.2 eV for CoPc(OH)4 are associated to the two types of characteristic nitrogen atoms for phthalocyanines: the central N atoms coordinated to the Co+2 and the aza nitrogen atoms on the macrocycle, respectively.38 For CoPc-PMO catalyst, these contributions were accompanied by two additional peaks for –NHCO– and C–N+ with binding energies at 400.9 and 402.8 eV, respectively. The latter peak indicates the existence of quaternary nitrogen, which could be attributed to the remaining OTAB surfactants in pores after extraction processes.39 The Co2p spectrum for CoPc(OH)4 showed two intense peaks at 780.9 and 796.4 eV associated to electron transitions of Co2p3/2 and Co2p1/2, respectively, along with their shake-up satellite bands at 786.5 and 803.1 eV (Fig. 4d). These observations are consistent with the CoII oxidation state. These typical characteristic peaks were also observed for CoPc-PMO sample supporting the integration of molecular CoPc units in the PMO.
Photocatalysis studies
In a typical experiment 1 mg of CoPc-PMO was dispersed in a CH3CN:TEOA (4:1) mixture containing Ru(bpy)3(PF6)2 (0.5 mM), purged with CO2 to saturate the colloidal suspension, and irradiated under non-filtered simulated solar light (100 mW cm−2, AM 1.5G). Evolution of the gaseous products was monitored by analysing the headspace gas by gas chromatography. The photocatalytic reaction produced syngas with 34.1% CO after 2 h irradiation (entry 1, Table 1). The amount of CO evolved after 2 h was 0.58 ± 0.08 μmol per mg of catalyst, corresponding to a turnover number (TONCO) of 126 ± 18 based on the initial CoPc loading. The syngas ratio dropped to ∼25% CO after 24 h, which was presumably caused by photodegradation of catalyst (CoPc-PMO) and/or photosensitiser [Ru(bpy)3]2+. The photocatalytic reaction was optimised by increasing the amount of CoPc-PMO from 1 mg to 3 mg, which showed that higher CoPc-PMO concentration in photoreactor is detrimental to the catalysis, with the TONCO dropping from 126 (1 mg catalyst), to 70 (2 mg catalyst) and 60 (3 mg catalyst), albeit with slight gain in CO selectivity from 34% (1 mg catalyst) to 43% (3 mg catalyst) (Fig. S3†). While increasing the amount of catalyst increases the number of active sites, high loading on solid catalyst can make the suspension turbid and block the light penetration.40
| Entry | PS | Catalyst | e− donor | λ range (nm) | CO (μmol mg−1) | H2 (μmol mg−1) | TONCO |
|---|---|---|---|---|---|---|---|
|
a Condition: 1 mg CoPc-PMO (4.6 μmol Co g−1), 4 mL MeCN/TEOA (4:1), 0.5 mM Ru(bpy)32+, 2 h irradiation under UV-visible light.
b [BIH] = 10 mM.
c CoPc-PMO with a Co loading of 2.7 μmol g−1.
d Total H2/CO in the headspace after 2 h.
|
|||||||
| 1 | Ru(bpy)32+ | CoPc-PMO | TEOA | >300 | 0.58 | 1.12 | 126 |
| 2 | Ru(bpy)32+ | CoPc-PMO | TEOA | Dark | 0 | 0 | — |
| 3 | Ru(bpy)32+ | CoPc-PMO | — | >300 | 0 | 0 | — |
| 4 | — | CoPc-PMO | TEOA | >300 | 0 | 0 | — |
| 5 | Ru(bpy)32+ | — | TEOA | >300 | 0 | 0.75d | — |
| 6 | Ru(bpy)32+ | CoPc-PMO | TEOA | >400 | 0.51 | 0.55 | 111 |
| 7b | Ru(bpy)32+ | CoPc-PMO | BIH | >400 | 6.35 | 3.20 | 1377 |
| 8c | Ru(bpy)32+ | CoPc-PMO | BIH | >400 | 5.32 | 1.48 | 1972 |
| 9 | Ru(bpy)32+ | — | BIH | >400 | 0.10d | 0.92d | — |
| 10 | Eosin Y | CoPc-PMO | TEOA | >400 | 0 | 0 | — |
| 11 | 4CzIPN | CoPc-PMO | TEOA | >400 | 0 | 0 | — |
The CO-selectivity of the CoPc-PMO/Ru(bpy)32+ combination was relatively low (30–40%) in comparison to electrocatalytic CO2R by CoPc, which is well known for its high CO selectivity. This could be attributed to UV-light mediated photodegradation of Ru(bpy)32+,41 which is enhanced by the presence of CO2 to yield chemical species that are active towards H2 production.42,43 Control experiment in the absence of CoPc-PMO supports this hypothesis since a comparable amount of H2 (1.12 μmol and 0.75 μmol H2 in the presence and absence of CoPc-PMO, respectively, under identical conditions) and negligible CO are produced (entry 5, Table 1). To minimise photodegradation of the sensitiser, an UV cut-off filter (λ > 400 nm) was employed and the photocatalysis was performed under visible light irradiation. A similar amount of CO was generated after 2 h, while the H2 evolution was significantly suppressed leading to an improved CO selectivity of 48.1% and a TONCO of 111 (entry 6, Table 1). As shown in Fig. 5b, CO evolution started to plateau around 4–6 h and a total of 1.10 ± 0.02 μmol CO was produced per mg of CoCp-PMO after 6 h, corresponding to a TONCO of 233 ± 6. However, H2 evolution continued at a nearly constant rate during 6 h irradiation. Interestingly, under non-filtered irradiation, the CO evolution activity levelled off at ∼2 h, further supporting photodegradation of the sensitiser and/or catalyst by the UV light (Fig. S3†).
 | ||
| Fig. 5 (a) Proposed reaction scheme for photocatalytic CO2 reduction. (b) Time course for photocatalytic CO and H2 evolution by CoPc-PMO in the presence of Ru(bpy)32+ as sensitiser and TEOA as donor. Condition: CO2-saturated 4:1 MeCN/TEOA, ∼1 mg CoPc-PMO (Co loading 4.6 μmol mg−1), 0.5 mM [Ru(bpy)3]2+, and visible light irradiation (100 mW cm−2, AM 1.5G, λ > 400 nm. (c) CO and H2 evolution trace when BIH (10 mM) was used as the donor. The CO selectivity is shown as a bar plot. Reaction conditions are same as (b). (d) CO evolved during four 1-hour recycling runs with CoPc-PMO (≈4 mg) is plotted as black trace and the grey bar plot shows the CO selectivity (%). (e) Cobalt ICP-MS signal in solution during chopped irradiation of CoPc-PMO under continuous flow of CO2 saturated photocatalysis solution. Condition: 5 mg CoPc-PMO deposited on carbon paper, 0.5 mM [Ru(bpy)3]2+ solution in aqueous 10% TEOA, blue LED, 2 min light/dark cycle. | ||
The TONCO values obtained for CoPc-PMO are comparable to those reported for other supported CoPc-based materials tested under similar photocatalytic conditions.14 In control experiments without Ru(bpy)32+, TEOA, CO2, and light (Table 1), CO was not detected by GC, confirming CO2 as the source of CO. Substituting Ru(bpy)32+ with two other molecular organic sensitisers, Eosin Y and 4CzIPN, led to zero photocatalytic activity (entries 10 and 11, Table 1). This indicates that CoPc-PMO is not a stand-alone photocatalyst for CO2R and it is only active when Ru-sensitiser is used. The photoexcited states of the alternative organic sensitisers might not be sufficiently reducing to mediate the CO2 reduction catalysis. Photostability of CoPc-PMO was probed using in situ ICP MS analysis under continuous flow. CoPc-PMO was deposited on carbon paper and mounted in an in situ flow cell while the photocatalysis solution (CO2 saturated aqueous solution containing 0.5 mM Ru(bpy)3Cl2 and 10% TEOA) was constantly passed through the cell and fed into the ICP-MS at a flow rate of 2.4 mg s−1. Carbon paper is a suitable support that prevents physical detachment of the catalyst powder and ensures that the Co detected is derived only from chemical leaching and/or photocorrosion. A blue LED was mounted in the flow cell to directly irradiate the catalyst coated carbon paper, and by alternating on/off cycles of 2 minutes, it can be determined if light irradiation has any influence on the Co leaching from the material. As shown in Fig. 5e, the Co signal of the ICP-MS is low and close to the baseline, indicating that the loss of Co through leaching is minimal. More importantly, the presence of light has no influence on the degree of Co leaching. If significant photo-induced Co leaching would be occurring, it would be expected that the Co counts in the ICP-MS would rise during the “light on” phases. However, the Co signal remained steady throughout the light on/off cycles, demonstrating good photostability of CoPc-PMO material. The Co loss observed during longer measurement times can be attributed to the accumulation of the very low baseline Co leaching observed in the 1 h experiment.
After the promising results with TEOA as the sacrificial donor, we investigated whether the photocatalytic activity of CoPc-PMO could be further improved by using BIH (1,3-dimethyl-2-phenyl-2,3-dihydro-1H-benzo[d]-imidazole) as a donor to supplement TEOA (Fig. 5a). Photocatalysis was performed in CO2 saturated acetonitrile/TEOA mixture (4:1 v/v) in the presence of [Ru(bpy)3]2+ (0.5 mM) and BIH (10 mM) under visible light irradiation. As shown in Fig. 5c, addition of BIH led to much improved catalytic performance with evolution of 6.35 ± 0.22 μmol CO mg−1 after 2 h irradiation at (66.5 ± 0.7)% CO-selectivity (entry 7, Table 1). The total amount of CO produced after 6 h was 11.5 ± 0.3 μmol CO mg−1 (average rate ∼1.9 μmol CO mg−1 h−1), corresponding to a Co-based TONCO of 2478 ± 41, which is an order of magnitude higher than analogous CoPc-based photocatalysts reported in literature. For comparison, photocatalysts with CoPc-based catalysts supported on C3N4 (carbon nitride) and TiO2 have shown Co-based TONCO of 10–100 with the CO evolution rates ranging from 0.01 to 0.25 μmol CO mg−1 h−1, under similar reaction conditons.14,32 The CO-selectivity of CoPc-PMO system dropped slightly after the first hour, but it remained approximately constant afterwards at ∼65% (Fig. 5c). Control experiment without CoPc-PMO showed generation of trace amount of CO (entry 9, Table 1).
Interestingly, lowering the CoPc loading in the material from 4.6 μmol g−1 to 2.7 μmol g−1 led to a superior activity towards CO evolution with a formation of 16.32 ± 1.36 μmol CO mg−1 at 72% CO selectivity after 4 h visible light irradiation (Fig. S4,† entry 8, Table 1). This corresponds to an average TOFCO (turnover frequency) of 1511 ± 123 h−1 over the course of 4 h. After overnight irradiation (15 h), the CO evolution ceased and a TONCO of 6836 ± 112 was obtained, which represents the total CoPc turnovers for the system before complete catalyst deactivation. Notably, an excellent CO selectivity of ∼84% was observed during the first hour of photocatalysis (CO yield 1.74 ± 0.07 μmol h−1 mg−1), which gradually decreased as H2 evolution rate was enhanced from Ru-sensitiser-derived by-products. The quantum yield (QY) for CO evolution by CoPc-PMO was determined to be (1.95 ± 0.08)% at 467 nm irradiation (blue LED), by the ferrioxalate actinometer method (Fig. S5 and S6†).
Heterogenous nature of the CoPc-PMO catalyst was studied by four 1 h recycling experiments which showed a steady loss of CO evolution activity after each run (Fig. 5d). However, the CO-selectivity was maintained at ∼65% throughout all four cycles, suggesting that the lower CO evolution is caused by loss of active catalytic centres by Co2+ leaching. The degradation products in solution promotes H2 evolution and therefore, upon recycling the solid catalyst the CO selectivity remained unchanged. The loss of Co2+ was confirmed by ICP analysis which showed a Co loading of 0.0021 mmol g−1 after four recycling runs, corresponding to a loss of 53% Co. Recycling experiments performed without BIH displayed similar trend albeit with a lower yield and selectivity towards CO (Fig. S7†). Post catalysis characterisation of CoPc-PMO by PXRD and TEM showed retention of its inherent hexagonal mesostructure (Fig. S8†). FT-IR spectrum of CoPc-PMO after four catalytic cycles showed vibration bands characteristic of phthalocyanine rings (Fig. S9†). However, UV-vis spectrum of the material after photocatalysis was dominated by adsorbed Ru(bpy)32+ species which masked the potential peaks for CoPc (Fig. S10†).
From a mechanistic perspective, the reaction is initiated by photoexcitation of Ru(bpy)32+ to the triplet state which is reductively quenched by BIH to generate [Ru(bpy)2(bpy˙−)]+.44 The oxidised BIH is deprotonated by TEOA, yielding strongly reducing BI˙ species that reduces Ru(bpy)32+ to generate a second [Ru(bpy)2(bpy˙−)]+ species. Two equivalents of reduced sensitisers subsequently reduce CoPc-PMO to (CoPc2−)-PMO that can mediate CO2 to CO conversion.45
Conclusions
In summary, we have reported highly efficient and selective photocatalytic CO2 reduction by a robust Co-phthalocyanine-PMO catalyst. Under visible light irradiation and in the presence of Ru(bpy)32+ sensitiser and BIH donor, CoPc-PMO material shows a Co-based TONCO of ∼6800 for CO evolution and an average TOFCO of ∼1500 h−1 over the course of 4 h; these are among the highest values reported for supported non-precious metal-based molecular catalysts and compares favourably to previously reported PMO-based material containing molecular Mn-catalyst. Furthermore, this work provides a rare example of directly integrating a CO2 reduction catalyst into PMO by a co-condensation method and provides a versatile platform for heterogenisation of molecular catalysts on inorganic porous matrices.Author contributions
DE, FJR-S, MAN and SR designed the project. MAN carried out the synthesis and characterisation of the materials, and MAN and SS performed the photocatalysis experiments. MW and CMP performed the in situ ICP measurements. All authors contributed to drafting the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
SR acknowledges funding from the Royal Society (RGS\R2\202350), RSC (Research Enablement Grant, Award E20-3474), and University of Lincoln. MAN, FJR-S and DE acknowledge funding from the Spanish Ministry of Science and Innovation for project RTI2018-101611-B-I00 and PDC2022-133973-I00, Andalusian Regional Government (Project ProyExcel_00492 and FQM-346 group) and Feder Funds. MAN acknowledges funding from Erasmus+ programme. CMP acknowledges funding of the Austrian research promotion agency (FFG COMET program grant 46305692). CMP and MW thank Prof. Markus Valtiner and Lukas Kalchgruber for helpful discussions and suggestions.References
- K. Li, B. Peng and T. Peng, ACS Catal., 2016, 6, 7485–7527 CrossRef CAS.
- J. Hawecker, J.-M. Lehn and R. Ziessel, J. Chem. Soc., Chem. Commun., 1983, 536–538 RSC.
- E. E. Benson, C. P. Kubiak, A. J. Sathrum and J. M. Smieja, Chem. Soc. Rev., 2009, 38, 89–99 RSC.
- C. D. Windle and E. Reisner, Chimia, 2015, 69, 435 CrossRef CAS PubMed.
- A. Perazio, G. Lowe, R. Gobetto, J. Bonin and M. Robert, Coord. Chem. Rev., 2021, 443, 214018 CrossRef CAS.
- X. Liu, S. Inagaki and J. Gong, Angew. Chem., Int. Ed., 2016, 55, 14924–14950 CrossRef CAS PubMed.
- C. M. Lieber and N. S. Lewis, J. Am. Chem. Soc., 1984, 106, 5033–5034 CrossRef CAS.
- S. Meshitsuka, M. Ichikawa and K. Tamaru, J. Chem. Soc., Chem. Commun., 1974, 158 RSC.
- N. Han, Y. Wang, L. Ma, J. Wen, J. Li, H. Zheng, K. Nie, X. Wang, F. Zhao, Y. Li, J. Fan, J. Zhong, T. Wu, D. J. Miller, J. Lu, S.-T. Lee and Y. Li, Chem, 2017, 3, 652–664 CAS.
- M. Wang, K. Torbensen, D. Salvatore, S. Ren, D. Joulié, F. Dumoulin, D. Mendoza, B. Lassalle-Kaiser, U. Işci, C. P. Berlinguette and M. Robert, Nat. Commun., 2019, 10, 3602 CrossRef PubMed.
- S. Ren, D. Joulié, D. Salvatore, K. Torbensen, M. Wang, M. Robert and C. P. Berlinguette, Science, 2019, 365, 367–369 CrossRef CAS PubMed.
- N. Huang, K. H. Lee, Y. Yue, X. Xu, S. Irle, Q. Jiang and D. Jiang, Angew. Chem., Int. Ed., 2020, 59, 16587–16593 CrossRef CAS PubMed.
- S. Roy and E. Reisner, Angew. Chem., Int. Ed., 2019, 58, 12180–12184 CrossRef CAS PubMed.
- H. Li, W. Xu, J. Qian and T.-T. Li, Chem. Commun., 2021, 57, 6987–6990 RSC.
- J. Yi, D. Si, R. Xie, Q. Yin, M. Zhang, Q. Wu, G. Chai, Y. Huang and R. Cao, Angew. Chem., Int. Ed., 2021, 60, 17108–17114 CrossRef CAS PubMed.
- M. Lu, M. Zhang, C. Liu, J. Liu, L. Shang, M. Wang, J. Chang, S. Li and Y. Lan, Angew. Chem., Int. Ed., 2021, 60, 4864–4871 CrossRef CAS PubMed.
- B. Han, X. Ding, B. Yu, H. Wu, W. Zhou, W. Liu, C. Wei, B. Chen, D. Qi, H. Wang, K. Wang, Y. Chen, B. Chen and J. Jiang, J. Am. Chem. Soc., 2021, 143, 7104–7113 CrossRef CAS PubMed.
- T. Dhanasekaran, J. Grodkowski, P. Neta, P. Hambright and E. Fujita, J. Phys. Chem. A, 1999, 103, 7742–7748 CrossRef CAS.
- P. Van Der Voort, D. Esquivel, E. De Canck, F. Goethals, I. Van Driessche and F. J. Romero-Salguero, Chem. Soc. Rev., 2013, 42, 3913–3955 RSC.
- H. Takeda, M. Ohashi, T. Tani, O. Ishitani and S. Inagaki, Inorg. Chem., 2010, 49, 4554–4559 CrossRef CAS PubMed.
- Y. Ueda, H. Takeda, T. Yui, K. Koike, Y. Goto, S. Inagaki and O. Ishitani, ChemSusChem, 2015, 8, 439–442 CrossRef CAS PubMed.
- Y. Kuramochi, M. Sekine, K. Kitamura, Y. Maegawa, Y. Goto, S. Shirai, S. Inagaki and H. Ishida, Chem. – Eur. J., 2017, 23, 10301–10309 CrossRef CAS PubMed.
- X. Wang, I. Thiel, A. Fedorov, C. Copéret, V. Mougel and M. Fontecave, Chem. Sci., 2017, 8, 8204–8213 RSC.
- M. Waki, K. Yamanaka, S. Shirai, Y. Maegawa, Y. Goto, Y. Yamada and S. Inagaki, Chem. – Eur. J., 2018, 24, 3846–3853 CrossRef CAS PubMed.
- M. Waki, N. Mizoshita, T. Tani and S. Inagaki, Angew. Chem., Int. Ed., 2011, 50, 11667–11671 CrossRef CAS PubMed.
- M. Á. Navarro, D. Cosano, A. Bhunia, L. Simonelli, V. Martin-Diaconescu, F. J. Romero-Salguero and D. Esquivel, Sustainable Energy Fuels, 2022, 6, 398–407 RSC.
- M. G. Dekamin, E. Arefi and A. Yaghoubi, RSC Adv., 2016, 6, 86982–86988 RSC.
- A. Yaghoubi, M. G. Dekamin, E. Arefi and B. Karimi, J. Colloid Interface Sci., 2017, 505, 956–963 CrossRef CAS PubMed.
- A. Corma, D. Das, H. García and A. Leyva, J. Catal., 2005, 229, 322–331 CrossRef CAS.
- J. Amaro-Gahete, D. Esquivel, M. V. Pavliuk, C. Jiménez-Sanchidrián, H. Tian, S. Ott and F. J. Romero-Salguero, Catalysts, 2022, 12, 254 CrossRef CAS.
- D. Verma, R. Dash, K. S. Katti, D. L. Schulz and A. N. Caruso, Spectrochim. Acta, Part A, 2008, 70, 1180–1186 CrossRef PubMed.
- G. Liu, Y. Wang, Y. Zhou, J. Cao, M. Yuan and H. Lv, J. Colloid Interface Sci., 2021, 594, 658–668 CrossRef CAS PubMed.
- M. Á. Navarro, J. Amaro-Gahete, J. R. Ruiz, C. Jiménez-Sanchidrián, F. J. Romero-Salguero and D. Esquivel, Dalton Trans., 2022, 51, 4884–4897 RSC.
- C. G. Claessens, U. Hahn and T. Torres, Chem. Rec., 2008, 8, 75–97 CrossRef CAS PubMed.
- E. L. Spitler and W. R. Dichtel, Nat. Chem., 2010, 2, 672–677 CrossRef CAS PubMed.
- F. Auras, Y. Li, F. Löbermann, M. Döblinger, J. Schuster, L. M. Peter, D. Trauner and T. Bein, Chem. – Eur. J., 2014, 20, 14971–14975 CrossRef CAS PubMed.
- V. V. Maslyuk, V. Y. Aristov, O. V. Molodtsova, D. V. Vyalikh, V. M. Zhilin, Y. A. Ossipyan, T. Bredow, I. Mertig and M. Knupfer, Appl. Phys. A, 2009, 94, 485–489 CrossRef CAS.
- K. Müller, M. Richter, D. Friedrich, I. Paloumpa, U. I. Kramm and D. Schmeißer, Solid State Ionics, 2012, 216, 78–82 CrossRef.
- X. Li, Y. Tang, L. Liu, Y. Zhang, R. Sheng and Y. NuLi, J. Colloid Interface Sci., 2022, 608, 2455–2462 CrossRef CAS PubMed.
- A. Kumar and G. Pandey, Mater. Sci. Eng. Int. J., 2017, 1, 106–114 Search PubMed.
- A. Call, M. Cibian, K. Yamamoto, T. Nakazono, K. Yamauchi and K. Sakai, ACS Catal., 2019, 9, 4867–4874 CrossRef CAS.
- J. Hawecker, J.-M. Lehn and R. Ziessel, Nouv. J. Chim., 1983, 7, 271–277 CAS.
- J. L. Grant, K. Goswami, L. O. Spreer, J. W. Otvos and M. Calvin, J. Chem. Soc., Dalton Trans., 1987, 2105–2109 RSC.
- D. Hong, Y. Tsukakoshi, H. Kotani, T. Ishizuka and T. Kojima, J. Am. Chem. Soc., 2017, 139, 6538–6541 CrossRef CAS PubMed.
- M. Zhu, R. Ye, K. Jin, N. Lazouski and K. Manthiram, ACS Energy Lett., 2018, 3, 1381–1386 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr06026d |
| This journal is © The Royal Society of Chemistry 2023 |