
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNon-directed C–H arylation of electron-deficient arenes by synergistic silver and Pd3 cluster catalysis†
Jian
Yao
ab,
Jiahui
Bai
b,
Xi
Kang
 c,
Manzhou
Zhu
c,
Yinlong
Guo
*b and
Xiaoming
Wang
*bd
c,
Manzhou
Zhu
c,
Yinlong
Guo
*b and
Xiaoming
Wang
*bd
aShanghai Key Laboratory for Molecular Engineering of Chiral Drugs, Frontiers Science Center for Transformative Molecules, School of Chemistry and Chemical Engineering, Shanghai Jiao Tong University, 800 Dongchuan Road, Shanghai 200240, China
bState Key Laboratory of Organometallic Chemistry, Center for Excellence in Molecular Synthesis, Shanghai Institute of Organic Chemistry, University of Chinese Academy of Sciences, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: ylguo@sioc.ac.cn; xiaoming@sioc.ac.cn
cDepartment of Chemistry and Center for Atomic Engineering of Advanced Materials, Anhui University, Hefei 230601, China
dSchool of Chemistry and Materials Science, Hangzhou Institute for Advanced Study, University of Chinese Academy of Sciences, 1 Sub-lane Xiangshan, Hangzhou 310024, China
First published on 10th January 2023
Abstract
Transition-metal clusters have attracted great attention in catalysis due to their unique reactivity and electronic properties, especially for novel substrate binding and activation modes at the bridging coordination sites of metal clusters. Although palladium complexes have demonstrated outstanding catalytic performance in various transformations, the catalytic behaviors of polynuclear palladium clusters in many important synthetic methodologies remain much less explored so far. Herein, we disclose the use of an atomically defined tri-nuclear palladium (Pd3Cl) species as a catalyst precursor in Ag(I)-assisted direct C–H arylation with aryl iodides under mild conditions. This catalyst system leads to the formation of synthetically important biaryls in good yields with high site selectivities without the assistance of directing groups.
Introduction
While homogeneous catalysis with mononuclear transition metal complexes has dominated the field of catalytic organic synthesis over the past decades, there has been growing interest in metal cluster catalysis since such species provide a potential bridge from mononuclear metal complexes to nanoparticles (NPs) (Scheme 1a).1–9 For example, small-sized clusters of palladium complexes have been used as models of a Pd metal surface and can also act as reservoirs to alternative catalytically active species.10–18 Several species of tripalladium clusters have been isolated and characterized; however, to date the documented examples of catalysis using Pd3 remain relatively limited (Scheme 1b).19–23 In this context, Malacria and Maestri et al. observed highly promising activities with Pd trimers in cycloadditions and hydrogenations.24–28 Another cluster, [Pd3(C7H7)2]2+, was found to be an active catalytic species for the cycloisomerization reaction of 2-phenylethynylaniline by Liu et al.29 Notably, a serious question in the area of cross-coupling reactions is the involvement of small Pd clusters as catalytically active species. For example, Wei and Zhu et al. used the Pd3 cluster as an efficient catalyst for the Suzuki–Miyaura reaction in 2017 (Scheme 1c-i).30,31 In addition, Schoenebeck and coworkers reported an elegant study using a Pd trimer for catalytically C–I selective cross-coupling of polyhalogenated arenes with Grignard reagents in 2019 (Scheme 1c-ii).32 In 2019 and 2021, the Fairlamb group studied a Pd3 cluster-catalyzed site-selective cross-coupling of halogenated heteroarenes with organoboron reagents, and the Pd3 cluster could be generated in situ from a Pd(I) dimer species, which can enhance activity and selectivity in the cross-coupling of 2-bromo-pyridine or 2,4-dibromopyridine (Scheme 1c-iii).33,34 Pérez-Ruiz and Leyva-Pérez et al. reported ligand-free, few-atom palladium cluster-catalyzed α-selective intramolecular Mizoroki–Heck coupling of iodoaryl cinnamates and intermolecular coupling of aryl iodides with styrenes (Scheme 1c-iv).35 Due to the great potential of Pd clusters in C–C bond cross-coupling reactions, we aimed to investigate the catalytic behavior of polynuclear palladium clusters in the area of synthetically important C–H functionalization.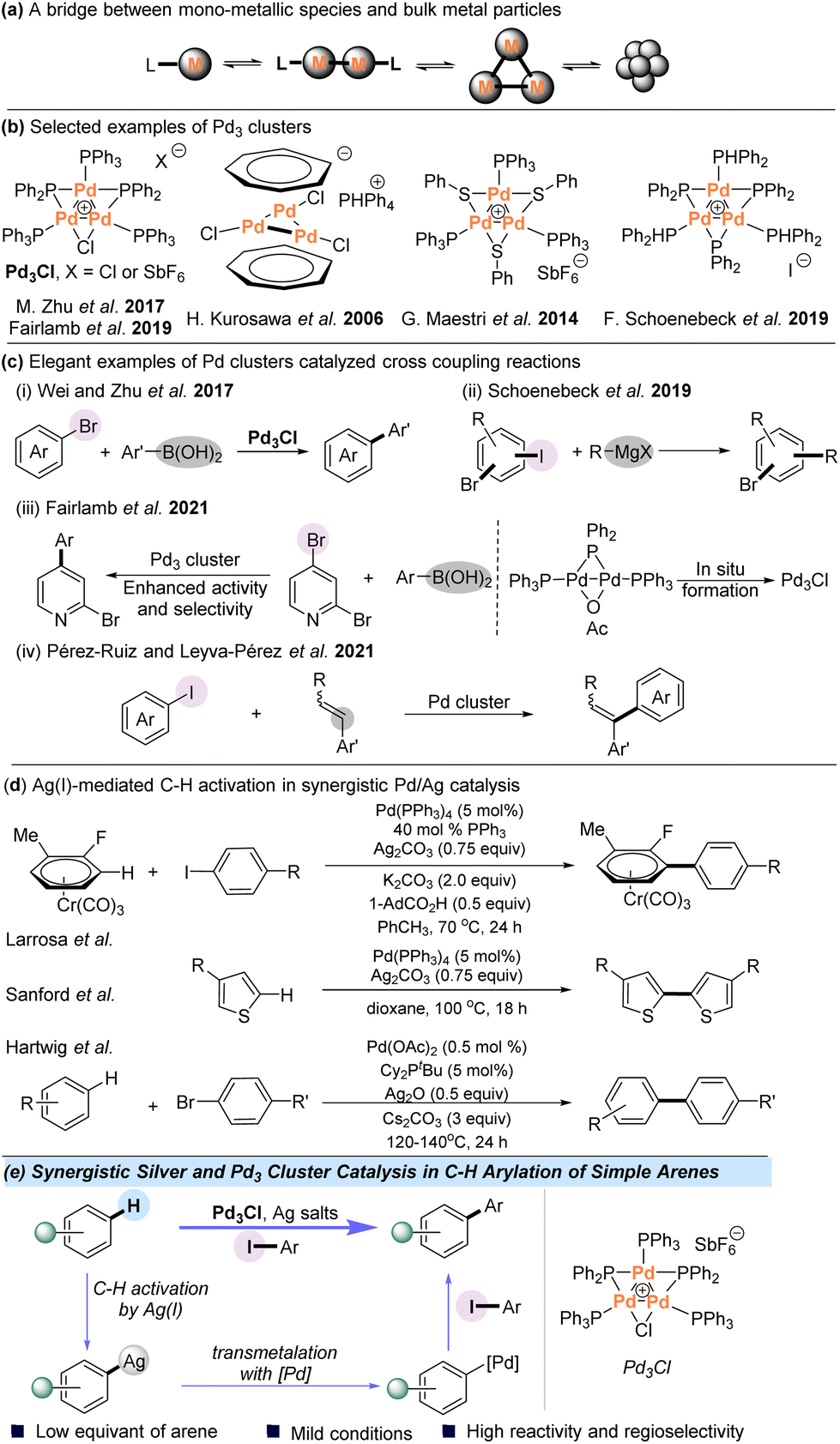 | ||
| Scheme 1 Trinuclear cationic palladium clusters and strategies for the direct C–H arylation. | ||
In recent years, C–H arylation has become a versatile tool for the construction of biaryl structures, which are common motifs in pharmaceuticals, agrochemicals, and organic materials.36–40 Despite great advances achieved in this area, the intermolecular arylation of simple arenes, which do not contain directing groups, remains a longstanding synthetic challenge.41–55 Besides the powerful Pd-catalyzed C–H activation,56–61 catalysis based on Pd/Ag cooperative bimetallic catalytic systems has been successfully developed for the C–H functionalization of simple arenes (Scheme 1d). Burés and Larrosa et al. disclosed that Ag(I) is actually responsible for the crucial C–H activation in the Pd/Ag-mediated C–H arylation system of electron-deficient arenes.62–67 The Sanford group also reported the same role of Ag(I) in the homocoupling of thiophenes at 100 °C.68 In addition, Hartwig et al. demonstrated AgOPiv-mediated C–H activation in selective allylation of aryl C–H bonds and direct arylation with aryl bromides.69,70 In these pioneering discoveries, Ag(I) cleaves the aryl C–H bond and the palladium catalyst enables the formation of the coupling products. Combined with the great potency of the Pd3 cluster in C–C bond cross-coupling reactions, we envisioned that Pd clusters might be utilized to achieve direct arylation of aryl C–H bonds with the assistance of Ag(I) under mild conditions.
All-metal palladium clusters are intriguing cyclic molecules that could present delocalized metal–metal bonds71–73 and the aromatic Pd–Pd bond of Pd3+ clusters can easily interact with Ag+ ions.74 Herein, we report the use of an atomically defined cationic [Pd3Cl(PPh2)2(PPh3)3]+[SbF6]− (Pd3Cl) cluster as a competent catalytic species for Ag(I)-assisted direct C–H arylation with aryl iodides, providing various biaryls in moderate to good yields (Scheme 1e). Mechanistic studies indicate that the phosphine-ligated silver complex is responsible for cleaving the C–H bond and the formation of Pd3I is detected in the preliminary mechanistic studies. It seems that the Pd3 core most likely remains intact throughout the reaction course.
Results and discussion
Reaction development and scope
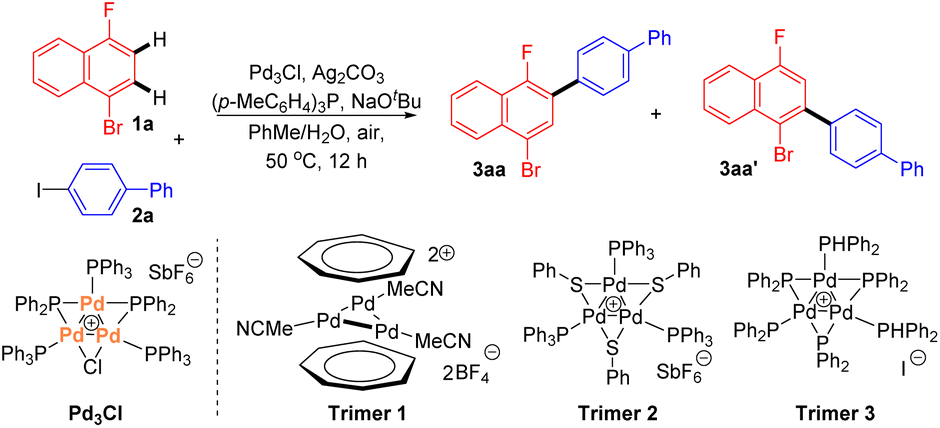
Initially, we chose 1-bromo-4-fluoronaphthalene 1a and aryl iodide 2a as the substrates to commence our investigation using Pd3Cl as the Pd catalyst. A careful survey revealed that cross-coupling product 3aa was generated in a 70% isolated yield with 5.0 mol% Pd3Cl as the catalyst in the presence of Ag2CO3, (p-MeC6H4)P and NaOtBu in the solvent (PhMe/H2O). In this case, only monoarylation product 3aa was observed. Notably, the reaction exhibited excellent regioselectivity (>20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 3aa:3aa′) (Table 1, entry 1). The effects of several reaction parameters on the catalysis are also shown in Table 1. The ligand for the Ag(I) salt was found to play an important role in the catalysis since under otherwise identical conditions, none of the reactions using PPh3, (p-OMeC6H4)3P or (p-FC6H4)3P afforded the expected product 3aa (entries 2–4 vs. 1). K2CO3 was used instead of NaOtBu and a much lower yield and regioselectivity were obtained in this case (entry 5). Other Ag salts, such as Ag2O, AgF, or AgOAc, had no beneficial effect on forming the cross-coupling product 3aa in comparison with the reaction using Ag2CO3 (entry 6). Changing the reaction solvent to trifluoroacetic acid (TFA) or hexafluoroisopropanol (HFIP) led to the complete inhibition of the catalysis (entry 7). These results suggested that a judicious choice of ligand, silver salt, and solvent is crucial for the reaction. Other palladium trimers 1–3 also showed activity in this reaction, giving slightly lower yields than that using Pd3Cl (entries 8–10). Without the silver salt or palladium catalyst, product 3aa was only detected in a trace amount (entry 11).
:1 3aa:3aa′) (Table 1, entry 1). The effects of several reaction parameters on the catalysis are also shown in Table 1. The ligand for the Ag(I) salt was found to play an important role in the catalysis since under otherwise identical conditions, none of the reactions using PPh3, (p-OMeC6H4)3P or (p-FC6H4)3P afforded the expected product 3aa (entries 2–4 vs. 1). K2CO3 was used instead of NaOtBu and a much lower yield and regioselectivity were obtained in this case (entry 5). Other Ag salts, such as Ag2O, AgF, or AgOAc, had no beneficial effect on forming the cross-coupling product 3aa in comparison with the reaction using Ag2CO3 (entry 6). Changing the reaction solvent to trifluoroacetic acid (TFA) or hexafluoroisopropanol (HFIP) led to the complete inhibition of the catalysis (entry 7). These results suggested that a judicious choice of ligand, silver salt, and solvent is crucial for the reaction. Other palladium trimers 1–3 also showed activity in this reaction, giving slightly lower yields than that using Pd3Cl (entries 8–10). Without the silver salt or palladium catalyst, product 3aa was only detected in a trace amount (entry 11).
|
|
|||
|---|---|---|---|
| Entry | Deviation from standard conditions | Yieldb (%) | Ratiob (3aa/3aa′) |
| a 1a (1.0 mmol), 2a (0.2 mmol), Ag2CO3 (0.4 mmol), ligand (0.8 mmol), NaOtBu (0.4 mmol) and Pd3Cl catalyst (5 mol%) in PhMe/H2O (0.5 mL/0.5 mL) at 50 °C for 12 h. b Determined by GC. c Isolated yield. | |||
| 1 | None | 71c (70) | >20:1 |
| 2 | PPh3 | — | — |
| 3 | (p-OMeC6H4)3P | Trace | — |
| 4 | (p-FC6H4)3P | — | — |
| 5 | K2CO3 | 23 | 3:1 |
| 6 | Other Ag sources: Ag2O, AgOAc or AgF | Trace | — |
| 7 | TFA or HFIP as solvent | — | — |
| 8 | Trimer 1 | 68 | >20:1 |
| 9 | Trimer 2 | 62 | >20:1 |
| 10 | Trimer 3 | 64 | >20:1 |
| 11 | Without [Pd] or [Ag] | Trace | — |
With the optimized reaction conditions in hand, the generality of this reaction was explored with various substituted arenes and aryl iodides. As shown in Scheme 2, a broad range of electron-deficient arenes was found to be compatible with the protocol, and the reactions gave the corresponding products in good yields and high selectivites. 1,4-Dibromonaphthalene 1b and 1-fluoronaphthalene 1c were found to be compatible with the current system, affording the corresponding single isomers in good yields (3ba and 3ca). A reaction of 1-bromo-4-fluorobenzene 1d with 2a led to the formation of 3da in high yield with good regioselectivity, and 1-bromo-3-fluorobenzene 1e also worked well, giving the single isomer of 3ea in 61% yield. The substrates of difluorobenzenes 1f, 1g and 1i were all well tolerated, and high yields and selectivities were obtained. Simple arenes without a fluorine substituent, such as 1-bromo-4-chlorobenzene 1h, also reacted smoothly under the optimal conditions to form biaryl 1he in good yield with a mixture of two regioisomers. 1,4-Dibromobenzene 1j and 1,4-dichlorobenzene 1k were also tested and afforded the corresponding biaryls 3ja and 3ka in good yields. Notably, good yields (63% and 57%) and excellent selectivities (>20/1) were found for the reactions of substrates with trifluoromethyl groups (1l and 1m). Sterically hindered arene 1,3,5-tribromobenzene 1n also proceeded smoothly to afford the corresponding arylation product 3na in 76% yield. It is noteworthy that the reaction using fluorobenzene or substituted fluorobenzenes, such as 4-methyl, 2-methoxy and 4-acetyl, also gave the corresponding products with excellent selectivities, albeit in moderate to good yields (3oa–3ra). It was found that the standard reaction conditions in Table 1 (entry 1) were not applicable to the C–H arylation of electron-rich arenes. Next, aryl iodides 2 that underwent direct arylation with 1-bromo-4-fluoronaphthalene (1a) were tested. The alkyl-substituted aryl iodides worked well under optimal reaction conditions to provide the corresponding products in high yields with good selectivities (3ab–ad). Other substrates containing an electron-donating group (2e–h), such as methoxy, phenoxy, thiomethyl or diphenylamine, were converted smoothly to the corresponding products 3ae–3ah with high selectivities. Substrates bearing reactive functionalities, such as ketone, pyridine, pyrrole or aniline moieties, were compatible with the reaction conditions, providing the corresponding biaryls (3aj–3ap) in moderate to high yields with excellent selectivities.
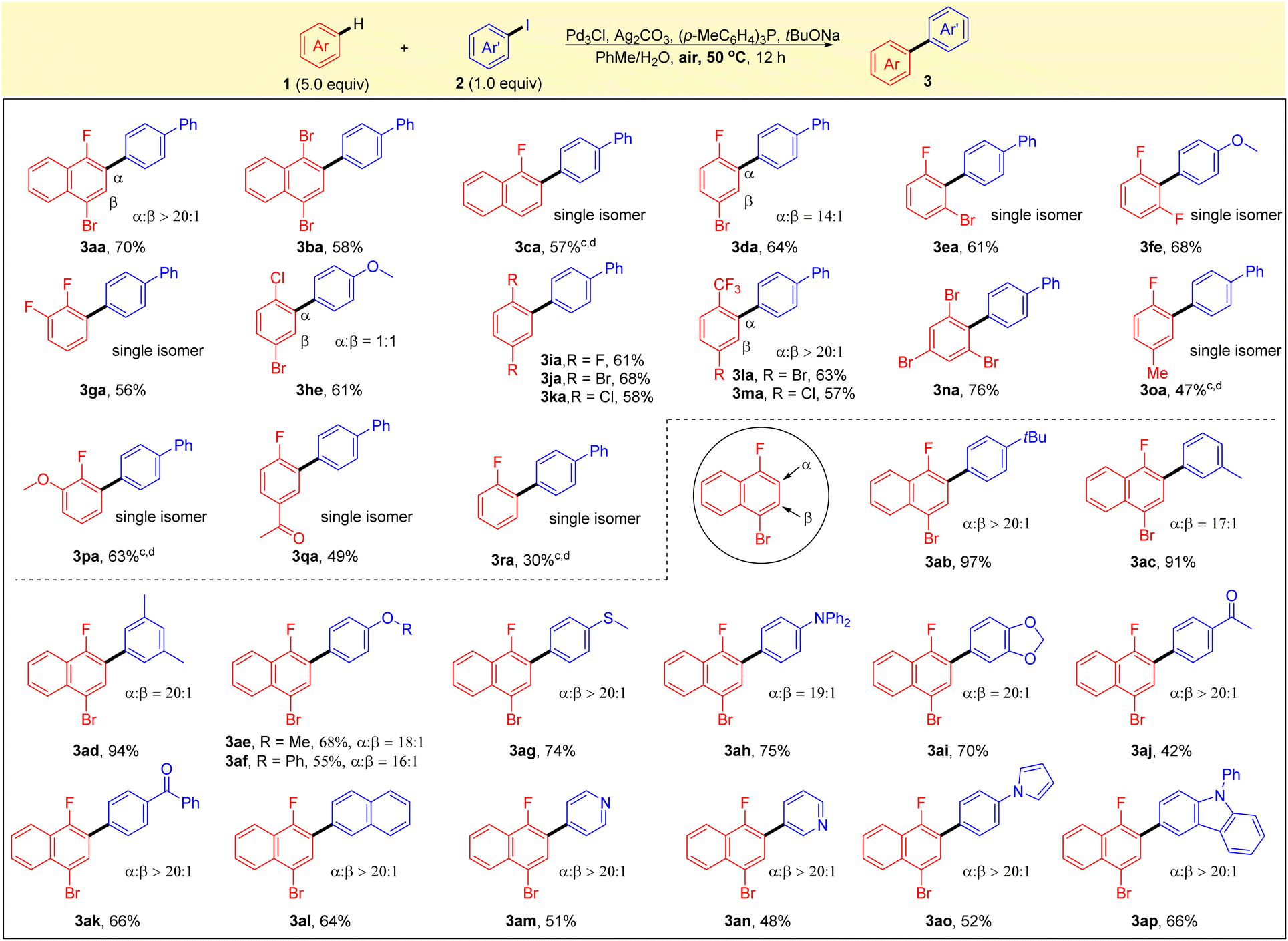 | ||
| Scheme 2 Substrate scope. aUnless otherwise stated, reactions were performed with 1 (1.0 mmol, 5 equiv.), 2 (0.2 mmol), Ag2CO3 (0.4 mmol), (p-MeC6H4)3P (0.8 mmol), NaOtBu (0.4 mmol) and 5 mol% Pd3Cl catalyst in PhMe/H2O (0.5 mL/0.5 mL) at 50 °C for 12 h. bIsolated yield. cPh2CyP was used instead of (p-MeC6H4)3P. dStirred at 80 °C. | ||
The protocols for Pd3Cl-catalyzed direct C–H arylation have been established, but the exact nature of the active species is elusive. Several attempts were undertaken to shed light on the reaction mechanism (Scheme 3). ESI-MS was used to track the palladium cluster involved in the reaction of 1-bromo-4-fluoronaphthalene 1a with aryl iodide 2a performed under standard conditions. A signal with an m/z of 1603 was detected in this case, which might be attributed to [Pd3I(PPh2)2(PPh3)3]+ (Pd3I, theoretical mass of 1602.99 Da) (Scheme 3a-i), indicating that the tripalladium core might be retained. However, we failed to isolate Pd3I after the reaction.
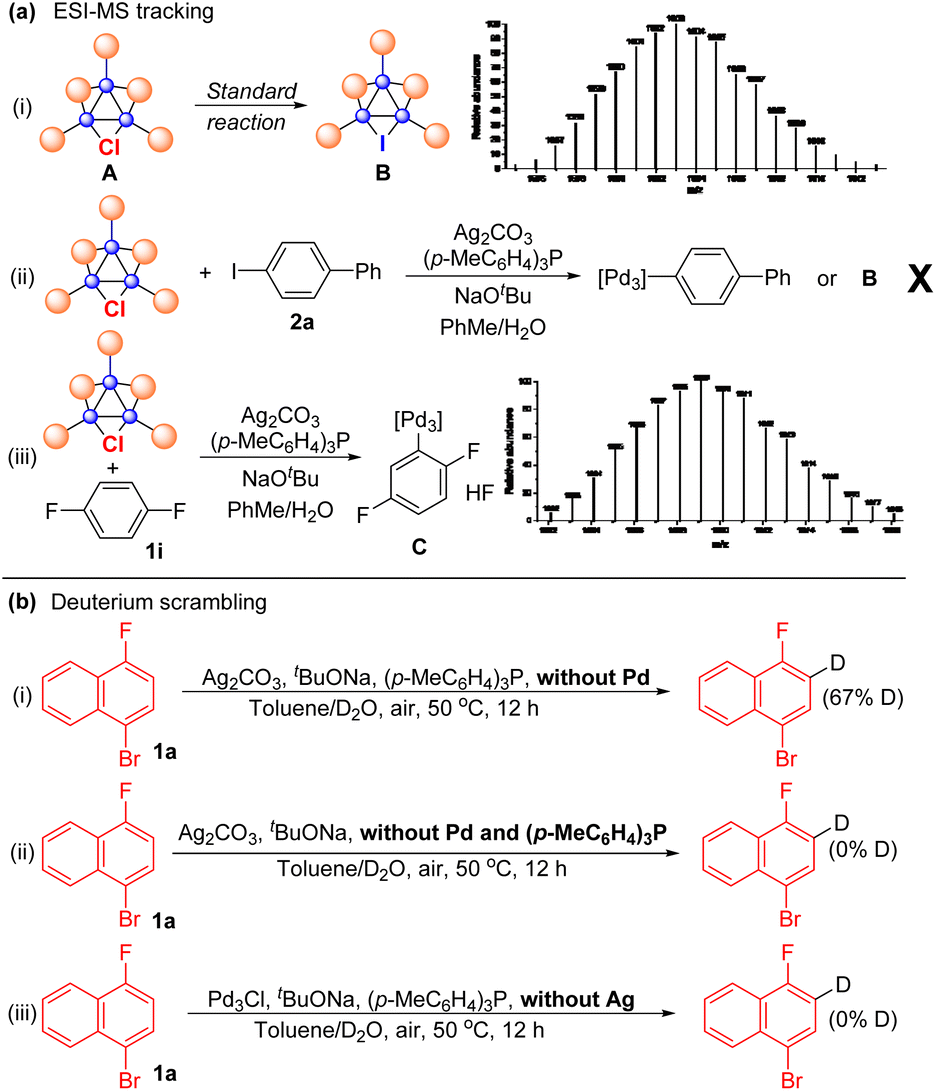 | ||
| Scheme 3 Preliminary mechanistic studies. | ||
No signal corresponding to Pd3Ar (Ar = biphenyl) or Pd3I was detected in the mass spectrum for the reaction mixture of Pd3Cl, Ag2CO3 and aryl iodide 2a, probably suggesting that Pd3Cl might not directly react with aryl iodide (Scheme 3a-ii). Notably, Pd3Ar (Ar = 2,5-difluorophenyl) was detected by ESI-MS when Pd3Cl was treated with 1,4-difluorobenzene 1i under otherwise standard conditions in the absence of an aryl iodide, indicating that the tripalladium core was retained and the aryl Pd3 species might be a key intermediate formed from Pd3Cl and the arene under the assistance of the Ag(I) salt (Scheme 3a-iii). We failed to track the signal of the formed aryl Pd3 species in the 31P NMR or 1H NMR analysis of the reaction mixture.30,34 To further probe the C–H activation step, arene 1a was treated with a mixture of toluene and D2O under standard conditions in the absence of Pd3Cl, affording 67% deuterated 1a with deuterium incorporation (Scheme 3b-i). Other parallel experiments were also conducted, though deuterium scrambling was not observed under the conditions without Pd and the (p-MeC6H4)3P ligand or without Ag salts (Scheme 3b-ii and iii), indicating that the phosphine-ligated silver complex is responsible for cleaving the C–H bond in the direct arylation reaction.61–70,75–81 Notably, 31P-NMR analysis of the solution of Pd3Cl and (p-MeC6H4)3P suggested that ligand exchange between (p-MeC6H4)3P and PPh3 in the cluster occured.82 In addition, the Pd3Cl cluster mainly remained intact in the reaction mixture between Pd3Cl and NaOtBu by 31P-NMR analysis (for details, see the ESI†), indicating that the base did not interact with Pd3Cl directly. Kinetic isotope effect (KIE) studies via the parallel reactions of deuterated 1-bromo-4-fluoronaphthalene d-1a and nondeuterated 1a were also conducted, respectively, giving the corresponding products with a primary kH/kD of 2.2, suggesting that the C–H bond cleavage is likely to be rate determining in the catalysis.83 These available mechanistic data are consistent with those of synergistic Ag-mediated C–H activation and Pd3 core-catalyzed arylation.61–70,75–81 In this case, the initial C–H bond activation of arenes using a phosphine-ligated Ag(I) salt in the reaction can result in the formation of Ag(I)–aryl species, which would undergo transmetalation with Pd3Cl. The oxidative addition and reductive elimination of Pd complexes and aryl iodides would deliver the product and regenerate the Pd3I catalyst. However, due to the similar results in the reactions using other palladium trimers and mononuclear palladium sources, the mechanism of clusters acting as a reservoir to mononuclear Pd species or aggregation to nanoparticles as the catalytically active species is also possible.
Conclusions
In summary, we have developed an efficient method for direct C(sp2)–H arylation with aryl iodides using a palladium trimer catalyst under the assistance of phosphine-ligated Ag(I). It should be highlighted that the current direct arylation of an aryl C–H bond proceeds smoothly under mild conditions which is highly appealing from the perspective of practical applications. Mechanistic experiments suggested that catalytic intermediates feature an atomically defined cationic tri-nuclear Pd cluster core that may function throughout the cycle. The present study may pave the way to a broader application of the Pd3 cluster in synthetic transformations.Author contributions
J. Y. and X. W. directed the project; J. Y., X. K., M. Z. and X. W. designed the experiments; J. Bai and Y. G. performed all the SAESI-MS studies; J. Y. performed all the experiments and analyzed all the data; J. Y. and X. W. wrote the manuscript with contributions from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from the National Key R&D Program of China (No. 2021YFA1500100) and the National Natural Science Foundation of China (21821002). The authors appreciate the valuable discussion of this work with Prof. Mian Guo and Prof. Hong Yi at Wuhan University.References
- R. H. Holm, P. Kennepohl and E. I. Solomon, Chem. Rev., 1996, 96, 2239–2314 CrossRef CAS PubMed.
- G. A. Somorjai, A. M. Contreras, M. Montano and R. M. Rioux, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10577–10583 CrossRef CAS PubMed.
- P. Buchwalter, J. Rosé and P. Braunstein, Chem. Rev., 2015, 115, 28–126 CrossRef CAS PubMed.
- S. Cao, F. Tao, Y. Tang, Y. Li and J. Yu, Chem. Soc. Rev., 2016, 45, 4747–4765 RSC.
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed.
- Y. Du, H. Sheng, D. Astruc and M. Zhu, Chem. Rev., 2020, 120, 526–622 CrossRef CAS PubMed.
- J. Tang and L. Zhao, Chem. Commun., 2020, 56, 1915–1925 RSC.
- R. Jin, G. Li, S. Sharma, Y. Li and X. Du, Chem. Rev., 2021, 121, 567–648 CrossRef CAS PubMed.
- A. D. Burrows and D. M. P. Mingos, Transition Met. Chem., 1993, 18, 129–148 CrossRef CAS.
- I. I. Moiseev, T. A. Stromnova and M. N. Vargaftik, J. Mol. Catal., 1994, 86, 71–94 CrossRef CAS.
- I. I. Moiseev and M. N. Vargaftik, New J. Chem., 1998, 22, 1217–1227 RSC.
- D. M. P. Mingos and R. Vilar, Synthesis and Reactivity of Palladium Cluster Compounds, J. Organomet. Chem., 1998, 557, 131–142 CrossRef CAS.
- T. Murahashi and H. Kurosawa, Coord. Chem. Rev., 2002, 231, 207–228 CrossRef CAS.
- K. J. Bonney and F. Schoenebeck, Chem. Soc. Rev., 2014, 43, 6609–6638 RSC.
- K. Osakada, Y. Tsuchido and M. Tanabe, Coord. Chem. Rev., 2020, 412, 213195 CrossRef CAS.
- Q. Liu and L. Zhao, Chin. J. Chem., 2020, 38, 1897–1908 CrossRef CAS.
- C. Fricke, T. Sperger, M. Mendel and F. Schoenebeck, Angew. Chem., Int. Ed., 2021, 60, 3355–3366 CrossRef CAS PubMed.
- G. W. Bushnell, K. R. Dixon, P. M. Moroney, A. D. Rattray and C. E. Wan, J. Chem. Soc., Chem. Commun., 1977, 709–710 RSC.
- S. Blanchard, L. Fensterbank, G. Gontard, E. Lacúte, G. Maestri and M. Malacria, Angew. Chem., Int. Ed., 2014, 53, 1987–1991 CrossRef CAS PubMed.
- X. Y. Deng, J. H. Lin and J. C. Xiao, Org. Lett., 2016, 18, 4384–4387 CrossRef CAS PubMed.
- R. Usui and Y. Sunada, Inorg. Chem., 2021, 60, 15101–15105 CrossRef CAS PubMed.
- K. M. Appleby, E. Dzotsi and N. W. J. Scott, Organometallics, 2021, 40, 3560–3570 CrossRef CAS.
- A. Monfredini, V. Santacroce, P. A. Deyris, R. Maggi, F. Bigi, G. Maestri and M. Malacria, Dalton Trans., 2016, 45, 15786–15790 RSC.
- A. Monfredini, V. Santacroce, L. Marchio, R. Maggi, F. Bigi, G. Maestri and M. Malacria, ACS Sustainable Chem. Eng., 2017, 5, 8205–8212 CrossRef CAS.
- M. Lanzi, T. Cañeque, L. Marchio, R. Maggi, F. Bigi, M. Malacria and G. Maestri, ACS Catal., 2018, 8, 144–147 CrossRef CAS.
- C. Cecchini, M. Lanzi, G. Cera, M. Malacria and G. Maestri, Synthesis, 2019, 51, 1216–1224 CrossRef CAS.
- A. Serafino, N. Camedda, M. Lanzi, N. D. Ca’, G. Cera and G. Maestri, J. Org. Chem., 2021, 86, 15433–15452 CrossRef CAS PubMed.
- C. L. Lv, H. Cheng, W. He, M. I. A. Shah, C. Q. Xu, X. J. Meng, L. Jiao, S. Q. Wei, J. Li, L. Liu and Y. D. Li, Nano Res., 2016, 9, 2544–2550 CrossRef CAS.
- F. Fu, J. Xiang, H. Cheng, L. Cheng, H. Chong, S. Wang, P. Li, S. Wei, M. Zhu and Y. Li, ACS Catal., 2017, 7, 1860–1867 CrossRef CAS.
- Y. Yun, H. Sheng, J. Yu, L. Bao, Y. Du, F. Xu, H. Yu, P. Li and M. Zhu, Adv. Synth. Catal., 2018, 360, 4731–4743 CrossRef CAS.
- C. J. Diehl, T. Scattolin, U. Englert and F. Schoenebeck, Angew. Chem., Int. Ed., 2019, 58, 211–215 CrossRef CAS PubMed.
- N. W. J. Scott, M. J. Ford, C. Schotes, R. R. Parker, A. C. Whitwood and I. J. S. Fairlamb, Chem. Sci., 2019, 10, 7898–7906 RSC.
- N. W. J. Scott, M. J. Ford, N. Jeddi, A. Eyles, L. Simon, A. C. Tanner, A. C. Whitwood, C. E. Willans and I. J. S. Fairlamb, J. Am. Chem. Soc., 2021, 143, 9682–9693 CrossRef CAS PubMed.
- F. Garnes-Portolés, R. Greco, J. Oliver-Meseguer, J. Castellanos-Soriano, M. C. Jiménez, M. López-Haro, J. C. Hernández-Garrido, M. Boronat, R. Pérez-Ruiz and A. Leyva-Pérez, Nat. Catal., 2021, 4, 293–303 CrossRef.
- D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174–238 CrossRef CAS PubMed.
- L. C. Campeau, D. R. Stuart and K. Fagnou, Aldrichimica Acta, 2007, 40, 35–41 CAS.
- L. Ackermann, R. Vicente and A. R. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792–9826 CrossRef CAS PubMed.
- M. Simonetti, D. M. Cannas and I. Larrosa, Adv. Organomet. Chem., 2017, 67, 299–399 CrossRef CAS.
- S. Yuan, J. B. Chang and B. Yu, Top. Curr. Chem., 2020, 378, 23 CrossRef CAS PubMed.
- N. R. Deprez, D. Kalyani, A. Krause and M. S. Sanford, J. Am. Chem. Soc., 2006, 128, 4972–4973 CrossRef CAS PubMed.
- K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2007, 129, 11904–11905 CrossRef CAS PubMed.
- N. Lebrasseur and I. Larrosa, J. Am. Chem. Soc., 2008, 130, 2926–2927 CrossRef CAS PubMed.
- L. Caron, L. C. Campeau and K. Fagnou, Org. Lett., 2008, 10, 4533–4536 CrossRef CAS PubMed.
- O. René and K. Fagnou, Org. Lett., 2010, 12, 2116–2119 CrossRef PubMed.
- T. H. Park, A. J. Hickman, K. Koh, S. Martin, A. G. Wong-Foy, M. S. Sanford and A. J. Matzger, J. Am. Chem. Soc., 2011, 133, 20138–20141 CrossRef CAS PubMed.
- Y. N. Wang, X. Q. Guo, X. H. Zhu, R. Zhong, L. H. Cai and X. F. Hou, Chem. Commun., 2012, 48, 10437–10439 RSC.
- A. M. Wagner, A. J. Hickman and M. S. Sanford, J. Am. Chem. Soc., 2013, 135, 15710–15713 CrossRef CAS PubMed.
- T. Yan, L. Zhao, M. He, J. F. Soulé, C. Bruneau and H. Doucet, Adv. Synth. Catal., 2014, 356, 1586–1596 CrossRef CAS.
- M. He, J. F. Soulé and H. Doucet, ChemCatChem, 2015, 7, 2130–2140 CrossRef CAS.
- C. Colletto, S. Islam, F. Juliá-Hernández and I. Larrosa, J. Am. Chem. Soc., 2016, 138, 1677–1683 CrossRef CAS PubMed.
- A. Hfaiedh, H. B. Ammar, J. F. Soulé and H. Doucet, Org. Biomol. Chem., 2017, 15, 7447–7455 RSC.
- J. Kim and S. H. Hong, ACS Catal., 2017, 7, 3336–3343 CrossRef CAS.
- L. Y. Liu, J. X. Qiao, K. S. Yeung, W. R. Ewing and J. Q. Yu, J. Am. Chem. Soc., 2019, 141, 14870–14877 CrossRef CAS PubMed.
- L. Y. Liu, J. X. Qiao, K. S. Yeung, W. R. Ewing and J. Q. Yu, Angew. Chem., Int. Ed., 2020, 59, 13831–13835 CrossRef CAS PubMed.
- T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS PubMed.
- H. M. L. Davies, J. Du Bois and J. Q. Yu, Chem. Soc. Rev., 2011, 40, 1855–1856 RSC.
- N. Kuhl, M. N. Hopkinson, J. Wencel-Delord and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 10236–10254 CrossRef CAS PubMed.
- J. Wencel-Delord and F. Glorius, Nat. Chem., 2014, 5, 369–375 CrossRef PubMed.
- J. F. Hartwig and M. Larsen, ACS Cent. Sci., 2016, 2, 281–292 CrossRef CAS PubMed.
- P. Wedi and M. van Gemmeren, Angew. Chem., Int. Ed., 2018, 57, 13016–13027 CrossRef CAS PubMed.
- P. Ricci, K. Kramer, X. C. Cambeiro and I. Larrosa, J. Am. Chem. Soc., 2013, 135, 13258–13261 CrossRef CAS PubMed.
- P. Ricci, K. Krämer and I. Larrosa, J. Am. Chem. Soc., 2014, 136, 18082–18086 CrossRef CAS PubMed.
- D. Whitaker, J. Burés and I. Larrosa, J. Am. Chem. Soc., 2016, 138, 8384–8387 CrossRef CAS PubMed.
- C. Colletto, A. Panigrahi, J. Fernandez-Casado and I. Larrosa, J. Am. Chem. Soc., 2018, 140, 9638–9643 CrossRef CAS PubMed.
- M. Batuecas, J. Luo, I. Gergelitsova, K. Krámer, D. Whitaker, I. J. Vitorica-Yrezabal and I. Larrosa, ACS Catal., 2019, 9, 5268–5278 CrossRef CAS PubMed.
- A. Panigrahi, D. Whitaker, I. J. Vitorica-Yrezabal and I. Larrosa, ACS Catal., 2020, 10, 2100–2107 CrossRef CAS PubMed.
- M. D. Lotz, N. M. Camasso, A. J. Canty and M. S. Sanford, Organometallics, 2017, 36, 165–171 CrossRef CAS.
- S. Y. Lee and J. F. Hartwig, J. Am. Chem. Soc., 2016, 138, 15278–15284 CrossRef CAS PubMed.
- A. Tlahuext-Aca, S. Y. Lee, S. Sakamoto and J. F. Hartwig, ACS Catal., 2021, 11, 1430–1434 CrossRef CAS PubMed.
- A. I. Boldyrev and L.-S. Wang, Chem. Rev., 2005, 105, 3716–3757 CrossRef CAS PubMed.
- J. M. Mercero, A. I. Boldyrev, G. Merino and J. M. Ugalde, Chem. Soc. Rev., 2015, 44, 6519–6534 RSC.
- I. Fernández, G. Frenking and G. Merino, Chem. Soc. Rev., 2015, 44, 6452–6463 RSC.
- Y. Wang, A. Monfredini, P. A. Deyris, F. Blanchard, E. Derat, G. Maestri and M. Malacria, Chem. Sci., 2017, 8, 7394–7402 RSC.
- K. L. Bay, Y. F. Yang and K. N. Houk, J. Organomet. Chem., 2018, 864, 19–25 CrossRef CAS.
- A. L. Mudarra and S. M. H. Perez-Temprano, Org. Biomol. Chem., 2019, 17, 1655–1667 RSC.
- T. Bhattacharya, S. Dutta and D. Maiti, ACS Catal., 2021, 11, 9702–9714 CrossRef CAS.
- G. Athavan, T. N. Tanner, A. C. Whitwood, I. J. S. Fairlamb and R. N. Perutz, Organometallics, 2022, 22, 3175–3184 CrossRef.
- L. A. Wilkinson, J. A. Pike and J. W. Walton, Organometallics, 2017, 36, 4376–4381 CrossRef CAS.
- W. P. Li, D. D. Yuan, G. Q. Wang, Y. Zhao, J. Xie, S. H. Li and C. J. Zhu, J. Am. Chem. Soc., 2019, 141, 3187–3197 CrossRef CAS PubMed.
- Y. Shimoyama, J. Kuwabara and T. Kanbara, ACS Catal., 2020, 10, 3390–3397 CrossRef CAS.
- K. R. Dixon and A. D. Rattray, Inorg. Chem., 2002, 41, 1099–1103 CrossRef PubMed.
- E. M. Simmons and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 3066–3072 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr05825a |
| This journal is © The Royal Society of Chemistry 2023 |