
Synthesis of the ZnTPyP/WO3 nanorod-on-nanorod heterojunction direct Z-scheme with spatial charge separation ability for enhanced photocatalytic hydrogen generation†
Shuanghong
Liu
a,
Siyu
Xia
a,
Jiefei
Wang
b,
Xitong
Ren
a,
Sudi
Chen
a,
Yong
Zhong
*a and
Feng
Bai
 *a
*a
aKey Laboratory for Special Functional Materials of Ministry of Education, National & Local Joint Engineering Research Center for High-efficiency Display and Lighting Technology, School of Materials Science and Engineering, Collaborative Innovation Center of Nano Functional Materials and Applications, Henan University, Kaifeng 475004, P. R. China. E-mail: zhywy8521115@163.com; baifengsun@126.com
bInternational Joint Centre for Biomedical Innovation, School of Life Sciences, Henan University, Kaifeng 475004, P. R. China
First published on 10th January 2023
Abstract
The direct Z-scheme photocatalytic system can effectively improve the separation efficiency of photogenerated carriers through the photosynthesis-based photocarrier transport model. In this study, zinc porphyrin-assembled nanorods (ZnTPyP) and WO3 nanorods’ nanorod-on-nanorod heterojunctions (ZnTPyP/WO3) were successfully prepared through a simple modified acid–base neutralization micelle-confined assembly method using WO3 nanorods as the nucleation template and ZnTPyP as building blocks. ZnTPyP achieved a controllable assembly onto WO3 nanorods through N–W coordination. ZnTPyP/WO3 nanorod-on-nanorod heterojunctions exhibited a structure-dependent photocatalytic performance for hydrogen production. The ZnTPyP/WO3 nanorod-on-nanorod heterojunctions exhibited a optimal hydrogen production rate (74.53 mmol g−1 h−1) using Pt as the co-catalyst, which was 2.64 times that of the ZnTPyP self-assembled nanorods. The improvement in the photocatalytic hydrogen production efficiency could be mainly attributed to the direct Z-scheme electron-transfer mechanism from WO3 to ZnTPyP. This is the first report of an approach using porphyrin-assembled nanostructures to construct organic–inorganic Z-scheme photocatalysts. This study offers valuable information for preparing new efficient photocatalysts based on organic supramolecular orderly aggregate materials.
1. Introduction
Hydrogen energy, which is a clean and high specific enthalpy energy source, has received tremendous attention.1–5 Photocatalytic water-splitting for hydrogen-production technology can efficiently utilize solar energy to produce hydrogen, thus minimizing environmental pollution and energy crisis.6–8 The photosynthesis-based Z-scheme photocatalytic system has recently been a cutting-edge research area due to the advantages of promoting the spatial separation of photogenerated charges to efficiently photoinduce water splitting into H2.9–14To date, traditional solution Z-scheme photocatalysts with shuttle redox couples and all-solid-state Z-scheme photocatalysts have been constructed and applied to solar H2 generation.15–18 However, the former are confined to the solution phase and need shuttle redox ion pairs to assist the transfer of photogenerated charge carriers,19,20 while the latter are disadvantageous because their charge transfer relies on conductors.21–23 Moreover, the use of precious metals as electron-transport media is limited by the high cost of metals and the complicated preparation process of composite catalysts.24,25 Direct Z-scheme photocatalysts without any solid electron mediators have evolved.26,27 Wang et al. reported a free electron-transport media of a ZnO/CdS photocatalytic hydrogen production system with a Z-scheme electron-transport mechanism, initiating the exploration of a direct Z-scheme photocatalytic system.20
The direct Z-scheme photocatalytic system allows photogenerated carriers to directly cross the composite interface without the presence of electron-transport media, thus reducing the preparation cost and improving the electron-transport efficiency.19,28,29 Hu et al. supported MoS2 quantum dots and MoS2 nanosheets on a CdS nanorods substrate to construct a MoS2/CdS composite system and achieved high photocatalytic hydrogen-production efficiency. These composites could effectively improve the photocatalytic performance.30,31 However, most of the existing direct Z-scheme photocatalysts are inorganic semiconductor materials, and they are characterized by a low visible-light utilization rate, poor hydrogen-production efficiency, high toxicity, and poor stability, thus limiting their practical applications.32,33 Furthermore, theoretical studies have shown that the match energy level plays an important role in constructing the direct Z-scheme photocatalytic system.34 Thus, this study mainly focused on the construction of a new and efficient direct Z-scheme photocatalyst.
Compared with inorganic photocatalysts, photoactive organic nanostructures are promising photocatalysts because of their easily tunable structure and function by molecular design.35–37 Porphyrins are a class of macromolecular heterocyclic organic compounds,38–40 and they exhibit good optical and photosensitizing (as light-harvesting antennas) properties owing to their highly conjugated molecular structure.41,42 Thus, porphyrins are widely used as photosensitizers in organic catalysis, solar cells, photocatalysis, nanodevices, and other fields.43–45 Porphyrin monomers form long-range orderly self-assembled H/J-aggregates, which are driven by weak forces, such as π–π stacking, hydrogen bonds, coordination, and hydrophilic/hydrophobic interactions.46–48 Based on the inheritance characteristics of the porphyrin monomer, the porphyrin assemblies can greatly expand the spectral response range (400–700 nm), thereby exhibiting an excellent electron buffering effect, photocatalytic activity, and chemical stability.49 Porphyrin assemblies, such as ZnTPyP,50 THPP,51 and InTPP,52 Pd(II) tetra(4-carboxylphenyl)porphyrin (PdTCPP),49 and Pt(II) meso-tetra (4-carboxyphenyl) porphine (PtTCPP)53 assembled nanostructures, exhibit extremely high photocatalytic hydrogen-production efficiency under visible-light irradiation. As an excellent photocatalyst, WO3 has no biological toxicity and is widely used in photocatalytic water-splitting, the photocatalytic reduction of CO2, the photocatalytic degradation of organic pollutants, and other fields.54,55 The more negative band position of WO3 (∼0.2 eV) makes the construction of a Z-scheme photocatalytic system by coupling with other semiconductor materials easier.32 However, the self-assembly process of porphyrin molecules is a complex process and is affected by numerous interacting forces, thus making the preparation of homogeneous porphyrin assemblies/inorganic semiconductor heterojunctions difficult because of the self-assembly of porphyrin molecules.56 Few studies on porphyrin-assembled heterojunction photocatalysts have been reported. Therefore, the construction of porphyrin assemblies and the WO3 direct Z-scheme photocatalytic system is a major problem to solve.
In this study, zinc porphyrin-assembled nanorods and WO3 nanorod heterojunctions (ZnTPyP/WO3) were first prepared through a simple modified acid–base neutralization micelle-confined assembly method using WO3 nanorods as a nucleation template and ZnTPyP as building blocks. ZnTPyP achieved controllable assembly onto the WO3 nanorods through N–W coordination. Then, the close structure–activity relationship between the ZnTPyP assemblies and WO3 nanorods is discussed. Finally, the improvement of the photocatalytic hydrogen-production performance and the direct Z-scheme photocatalytic system was investigated. This work will provide valuable information for designing efficient Z-scheme photocatalysts based on organic supramolecular orderly aggregate materials.
2. Experimental section
2.1. Materials and methods
Zinc meso-5, 10, 15, 20-tetra(4-pyridyl) porphyrin (ZnTPyP) was purchased from Frontier Scientific, Inc. All the surfactants, including cetryltrimethyl ammonium bromide (CTAB), sodium dodecyl sulfate (SDS), and myristyltrimethyl ammonium bromide (MTAB), were purchased from Sigma-Aldrich and used without further purification. A standard solution of sodium hydroxide (NaOH, 1 M) and a standard solution of hydrochloric acid (HCl, 1 M) were purchased from Acros Organics. Ascorbic acid (AA), sodium tungstate dihydrate (Na2WO4·2H2O), and sodium chloride (NaCl) were purchased from Aladdin Ltd. Potassium tetrachloroplatinate(II) (K2PtCl4) was purchased from J&K Chemical. Nanopure water (18.2 MΩ cm) was used in solution preparation. All the reagents were analytically pure and used without further purification.2.2. Preparation of WO3 nanorods (NRs)
WO3 nanorods were synthesized via a hydrothermal method following the previously reported procedure.57 Typically, 2.0615 g of Na2WO4·2H2O and 1.45 g of NaCl were added to 50 mL of deionized water. The solution pH values were adjusted to 2.0 with 1 M HCl solution and stirred for 1 h. Then, the solution was transferred into a Teflon-lined autoclave (100 mL). The autoclave was maintained at 180 °C for 24 h. The obtained product was filtered and washed three times with deionized water and absolute ethanol and dried at 60 °C for 8 h.2.3. Synthesis of ZnTPyP/WO3 nanorod-on-nanorod heterojunctions
The ZnTPyP/WO3 heterojunctions were synthesized using a modified encapsulation confined acid–base neutralization self-assembly method.58,59 Typically, 2.5 mg of WO3 nanorods were dispersed in 9.5 mL of CTAB aqueous solution (10.5 mM), and added into 41 μL NaOH solution (1 N). Then, 200 μL of 0.01 M ZnTPyP/HCl (0.2 M HCl) was quickly injected into the mixed solution under stirring, and the solution pH value was close to 8.61. The solution was continuously stirred for 72 h at room temperature, and centrifuged at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 min, and the obtained precipitation was washed with ultrapure water three times. Similarly, the heterojunction catalysts with different contents of WO3 nanorods were obtained by only adjusting the amount of WO3 (1, 5, and 10 mg). The experiments were performed at different pH values (3.06, 10.96, and 11.82) to optimize the experimental conditions. For comparison, pure ZnTPyP-assembled nanorods were prepared at pH 8.6 as a blank control without the addition of WO3 nanorods.
000 rpm for 10 min, and the obtained precipitation was washed with ultrapure water three times. Similarly, the heterojunction catalysts with different contents of WO3 nanorods were obtained by only adjusting the amount of WO3 (1, 5, and 10 mg). The experiments were performed at different pH values (3.06, 10.96, and 11.82) to optimize the experimental conditions. For comparison, pure ZnTPyP-assembled nanorods were prepared at pH 8.6 as a blank control without the addition of WO3 nanorods.
2.4. Photocatalytic hydrogen-production experiments
Photocatalytic experiments were performed on a glass-closed gas system (Labsolar-6A, Beijing Perfectlight Technology Co., Ltd). ZnTPyP/WO3 heterojunctions (1 mg) with 0.2 M of AA aqueous solution (50 mL) were added to the quartz reactor. The reactor with a water-cooling jacket (6 °C) was vacuumed for 20–30 min and then irradiated under a 300 W Xenon lamp with a cutoff filter (λ > 400 nm). The amount of hydrogen was analyzed by an online gas chromatography instrument (GC-7920) equipped with a thermal conductivity detector (TCD).2.5. Photoelectrochemical tests
The Mott–Schottky plots and photocurrent response were conducted on an electrochemical workstation (Autolab, Holland) equipped with a standard three-electrode system. The prepared samples were coated on fluorine-doped tin oxide (FTO) substrate as the working electrode, while Ag/AgCl electrode and a platinum plate were used as the reference and counter electrodes, respectively, and the electrode potential was +0.20 V vs. NHE potential. The experiments were performed in 0.2 M Na2SO4 electrolyte solution purged with N2 to remove O2 before the measurements. The photocurrent density was recorded under visible-light switching on and off modes (λ > 400 nm, 300 W Xenon lamp).2.6. Characterization
The morphology of the samples was characterized by field emission scanning electron microscopy (FESEM, Nova Nano SEM 450JS and M-7001F) and transmission electron microscopy (TEM, JEOL JEM-2100 and Titan G260-300). The chemical valences were measured via X-ray photoelectron spectroscopy (XPS, Thermo Scientific K-Alpha). The absorption spectra were determined by ultraviolet–visible spectrophotometry (UV–vis, Agilent Cary60). Fourier-transform infrared spectrometry (FT-IR, Bruker VERTEX70) was used to determine the functional group spectra. The crystal characteristics were determined by X-ray powder diffraction (XRD, Bruker D8 Advance, Germany) with Cu Kα radiation. Deionized water was produced by an ultrapure water meter (Thermo Scientific GenPure Pro UV-TOC, American). The photocatalytic experiments were performed on a Labsolar-6A photocatalytic system (Beijing Perfectlight Co. Ltd, China) with the light power provided by a 300 W Xenon lamp with a UVCUT 400 nm filter (PLS-SXE300D, Beijing Perfectlight Co. Ltd, China). The Mott–Schottky plots and photocurrent response were conducted on an electrochemical workstation (Autolab, Holland) in a standard three-electrode system.3. Results and discussion
3.1. Structural characteristics of the ZnTPyP/WO3 nanorod-on-nanorod heterojunctions
The ZnTPyP/WO3 nanorod-on-nanorod heterojunctions were prepared using a modified acid–base neutralization micelle-confined self-assembly method. The WO3 nanorods were first synthesized via a hydrothermal method using NaCl as a capping agent. ZnTPyP nanorods were then in situ grown onto WO3 nanorods using a modified acid–base neutralization self-assembly method according to our previous report (Fig. 1).58,59 Typically, the as-prepared WO3 nanorods were dispersed in CTAB aqueous solution under stirring to make their surfaces evenly coated with emulsifier micelles. Then, NaOH solution was injected to make the surface of the WO3 nanorods form a large number of negative charges in an alkaline environment. After that, the protonated ZnTPyP/HCl solution was injected into the mixture, and ZnTPyP–H44+ was adsorbed on the surface of the WO3 nanorods under positive–negative charges attraction. ZnTPyP achieved a controllable assembly onto the WO3 nanorods using WO3 nanorods as the nucleation template driven by non-covalent interactions, such as hydrophobic–hydrophobic, π–π stacking, Zn–N axial coordination interactions, and so on. Under the self-assembly growth aging, the ZnTPyP/WO3 nanorod-on-nanorod heterojunctions were finally formed. The preparation details are presented in the Experimental section. Moreover, the morphological structures of the ZnTPyP/WO3 heterojunctions could be easily adjusted by changing the content of WO3 nanorods, the pH value, and the emulsifier types.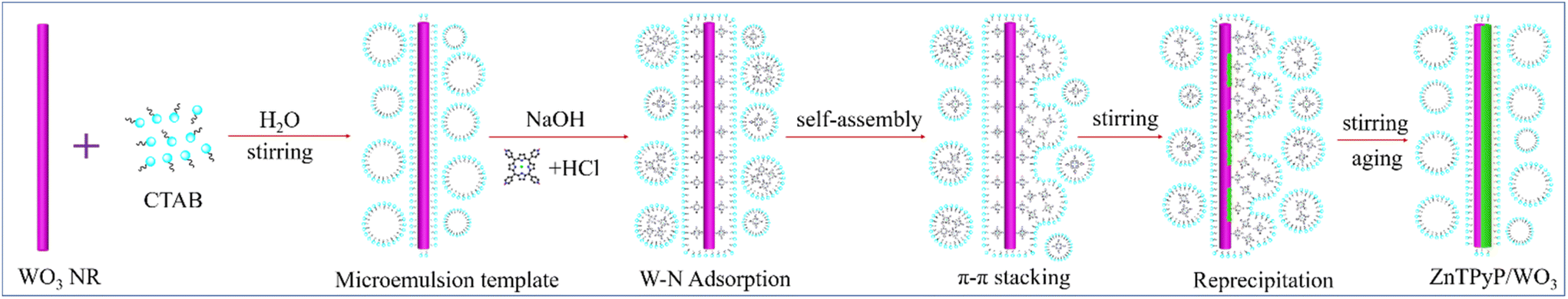 | ||
| Fig. 1 Schematic illustration of the fabrication of ZnTPyP/WO3 nanorod-on-nanorod heterojunctions. | ||
A series of ZnTPyP/WO3 nanorod-on-nanorod heterojunctions with various WO3 loadings were prepared in parallel. Pure WO3 nanorods and ZnTPyP nanorods were prepared as a control. The SEM images showed that the obtained pure ZnTPyP nanorods (with a diameter of 200 nm and length of ∼2 μm, Fig. S1a†) and the obtained pure WO3 nanorods (with a diameter of 100 nm and length of ∼1 μm, Fig. S1b†) had a regular morphology and uniform size. The morphology of the WO3 nanorods and structure evolution of the ZnTPyP/WO3 composites were further investigated. When the input amount of WO3 NRs reached 10 mg (the ZnTPyP retained 2 mM), no sign of the ZnTPyP assemblies nanorods growth was observed in the TEM image. The ZnTPyP molecules only adsorbed and started to nucleate on the surface of WO3 NRs (Fig. 2a). As the content of WO3 gradually decreased (2.5–5 mg), ZnTPyP assemblies and WO3 nanorods leaned together and a formed nanorod-on-nanorod heterostructure (Fig. 2b), and then ZnTPyP nanorods grew to several micrometers (Fig. 2c). When the input amount of WO3 was reduced to 1 mg, numerous ZnTPyP assemblies were wrapped around the surface of WO3, forming a ZnTPyP/WO3 core–shell structure (Fig. 2d). Collectively, the formation of the ZnTPyP/WO3 heterostructure was performed in two consecutive stages: the formation of ZnTPyP nanorods via a homogeneous nucleation with the assistance of surfactants, followed by the in situ growth of ZnTPyP on the nanorod surface via a heterogeneous nucleation. The TEM images showed that ZnTPyP and WO3 were effectively combined and retained their original nanorod morphology. Moreover, the TEM results showed that with decreasing the WO3 loading in the samples, the growth of the ZnTPyP and template for the nucleation sites increased and decreased, respectively, resulting in a significant change in the morphology of ZnTPyP assemblies, gradually increasing the length further along the WO3 NRs. Therefore, ZnTPyP/WO3 exhibited different morphologies and structures by controlling the WO3 loading, which further affected the separation of the photogenerated carriers.
 | ||
| Fig. 2 TEM of the ZnTPyP/WO3 heterojunction with various ZnTPyP nanorods loading: (a) 8.1, (b) 15.0, (c) 26.1, (d) 46.9 wt%, vs. ZnTPyP wt%, where CTAB was used as an emulsifier, and the pH value was approximately 8.60. | ||
In order to unveil the formation process of the ZnTPyP/WO3 heterostructure, a series of pH-dependent experiments were performed with CTAB used as an emulsifier (Fig. S2†). WO3 is an acidic oxide and cannot exist in an alkaline environment for a long time, while ZnTPyP molecules form assemblies only in an environment of pH > 3, and the hexagonal nanorods have the best hydrogen-production performance and are formed at pH 11.5.60 Thus, the pH of the assembly reaction solution can affect the nucleation of zinc porphyrin molecules on WO3 nanorods, thereby affecting the structure of the heterojunctions. At pH 3.06, ZnTPyP was assembled into rough and irregular islands on the surface of the composites (Fig. S2a†) because ZnTPyP tended to form nanodisks in an acidic environment, and the protonated ZnTPyP adsorbed and assembled on the surface of WO3 nanorods in the absence of NaOH adding during the assembly process. At pH 8.83, the ZnTPyP in the heterojunctions were assembled into nanorods with a smooth surface (Fig. S2b†). At pH 10.96, a mixture of ZnTPyP assemblies and ZnTPyP/WO3 heterojunctions were formed (Fig. S2c†). When the pH value was raised to 11.82, the WO3 nanorods disappeared, and only the ZnTPyP assemblies could be observed (Fig. S2d†). These transitions could be attributed to the decomposition of the WO3 nanorod templates in the strongly alkaline solution during the self-assembly process. Thus, the optimal pH of the assembly solution for the synthesis of ZnTPyP/WO3 was between 8 and 9. Besides, the effects of different emulsifier types on the assembly behavior of ZnTPyP molecules on the surface of WO3 nanorods are also discussed. When SDS (Fig. S3a†) and MTAB (Fig. S3b†) were used as emulsifiers, ZnTPyP/WO3 nanorod-on-nanorod heterostructures were also obtained.
The typical morphology of the obtained ZnTPyP/WO3 nanorod-on-nanorod heterostructure consisting of 26.1 wt% ZnTPyP is shown in Fig. 3a. The morphology of ZnTPyP nanorods attached on WO3 nanorods was largely maintained. The contrast difference and lattice fringes in the high-resolution TEM image confirmed the effective composite of the two materials. The HRTEM images (Fig. 3b and c) showed that the ZnTPyP/WO3 heterojunctions exhibited a well-resolved lattice fringe with the spacing of 0.38 nm, corresponding to the interplanar spacing of (010) lattice plane of the WO3 nanorod (Fig. 3d). The corresponding HAADF image showed the existence of the two materials (Fig. 3e), and the intimate interfacial contact formed between WO3 and ZnTPyP. The EDS elemental mapping distribution of C, N, Zn, O, and W showed that C, N, and Zn were distributed on the surface of the entire heterojunction material. However, C, N, and Zn in the ZnTPyP assemblies were concentrated on one side of the entire heterojunction material surface. This phenomenon could be attributed to the ZnTPyP monomer adsorbed on the surface of the WO3 NRs, and a large amount of ZnTPyP that preferentially grew into assemblies along one side on the surface of WO3 NRs, further indicating the effective combination of ZnTPyP assemblies with WO3 nanorods to a form nanorod-on-nanorod heterostructure (Fig. 3f–k).
 | ||
| Fig. 3 (a) TEM images, (b–d) HRTEM, and (e–k) STEM image and EDS elemental mapping of ZnTPyP/WO3 nanorod-on-nanorod heterostructure (26.1 wt%). | ||
3.2. Chemical structure analysis
The crystal structure of the organic supramolecular self-assemblies reflected the packing mode of the internal molecules and determined the photocatalytic performance of the ZnTPyP assemblies. The powder XRD pattern of the ZnTPyP nanorods showed that diffraction peaks appeared at 5.4°, 9.4°, 10.8°, 11.5°, 14.8°, and 18.9°, corresponding to the crystal planes of (110), (300), (220), (201), (131), and (241), respectively, suggesting hexagonal stacking (Fig. 4a). The XRD diffraction peaks of WO3 nanorods appeared at 13.95°, 23.20°, 24.33°, 28.11°, 33.58°, and 36.58°, corresponding to the hexagonal WO3 crystal planes of (100), (001), (110), (200), (111), and (201), respectively, consistent with the standard card (JCPDS 85-2460).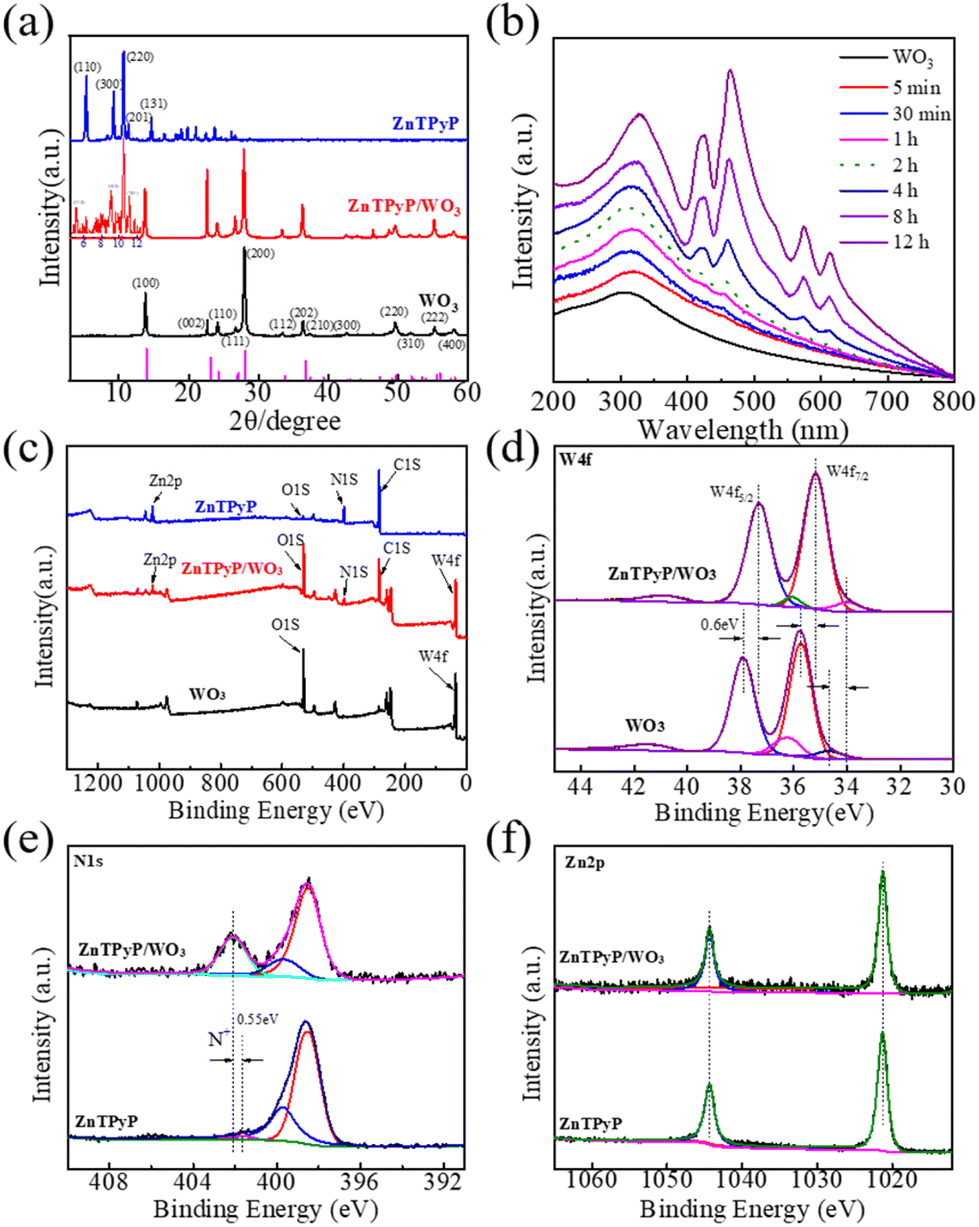 | ||
| Fig. 4 (a) XRD patterns; insert is the corresponding amplified area in (a). (b) Time progression of ZnTPyP monomer aggregation on WO3 nanorods monitored by UV–vis absorption spectroscopy. (c) XPS survey spectrum of the ZnTPyP/WO3 nanorod-on-nanorod heterostructure. High-resolution XPS spectra of (d) W 4f, (e) N 1s, and (f) Zn 2p in pure WO3 nanorods and ZnTPyP/WO3 (26 wt%). | ||
The XRD patterns of the ZnTPyP/WO3 heterojunction were mainly dominated by WO3 nanorods, indicating that the crystal structure of the WO3 nanorods did not significantly change during the self-assembly process. The insert corresponding enlarged view of Fig. 4a shows that the ZnTPyP-assembled nanorods in the heterojunction had hexagonal stacking, according to our previous report.50,60 Nitrogen adsorption–desorption isotherms were performed to detect the specific surface area of the nanorod-on-nanorod heterojunctions. The ZnTPyP/WO3 nanorod-on-nanorod heterojunctions showed a maximum surface area of 21.5 m2 g−1 (Fig. S4†). The thermogravimetric analysis (TGA) results showed that the thermal stability of the heterojunctions was significantly increased with the introduction of WO3 nanorods (Fig. S5†).
Greater insights into the ZnTPyP/WO3 nanorod-on-nanorod heterostructure growth process were obtained by UV–vis spectroscopy (Fig. 4b). For reaction times below 2 h, there shows no apparent ZnTPyP-assembled nanostructures formed. After 4 h, the characteristic absorption peak of the ZnTPyP assemblies could be observed. The corresponding UV–vis diffuse reflectance spectra (DRS) were also characterized, as shown in Fig. S6.† The WO3 NRs showed intensive absorption with an absorption edge at ∼410 nm and a weak absorption coverage of the whole visible region. The wide absorption peak was mainly due to the micrometer-scale size of the WO3 NRs, resulting from the abundant oxygen vacancies existing on the surface of the WO3 NRs. The ZnTPyP-assembled nanorods had B-band absorption peaks at 420 and 460 nm, and Q-band absorption peaks at 578 and 605 nm. ZnTPyP/WO3 exhibited the optical absorption feature of WO3 combined with that of ZnTPyP, and four new peaks of ZnTPyP were observed, with the peaks at 417 and 455 nm corresponding to Soret bands, and the peaks at 572 and 611 nm corresponding to Q bands. Compared with the ZnTPyP powder, the ZnTPyP/WO3 exhibited a broadened and red-shift for the Q bands, contributing to the strong interaction interface. Larger aggregates were formed via further assembling driven by the axial coordination interactions, hydrogen bonding, and hydrophobic interactions, which further indicated that the heterojunction material effectively broadened the photoresponse region, which would be useful for photocatalytic applications.
The elemental composition and chemical states of the ZnTPyP/WO3 heterojunction were characterized via XPS. The XPS survey spectra revealed that WO3 was mainly comprised of W and O, while ZnTPyP was comprised of C, N, and Zn. The ZnTPyP/WO3 heterojunction material was comprised of both elements (Fig. 4c). The fine spectrum of W 4f showed that both the WO3 and the ZnTPyP/WO3 heterojunction exhibited two double W 4f states. From the spectrum, the diffraction peak at the binding energy of 35.75 eV corresponded to W 4f7/2, and 37.90 eV corresponded to W 4f5/2, and both diffraction peaks corresponded to W6+. The second doublet peaks were at 34.70 and 36.25 eV with a weaker binding energy, corresponding to W 4f7/2 and W 4f5/2 of W5+, respectively. While in the ZnTPyP/WO3 spectrum, the positions of the W 4f7/2 orbital and W 4f5/2 orbital peaks of W6+ were 35.15 and 37.30 eV, respectively, and the positions of the W 4f7/2 orbital and W 4f5/2 orbital peaks of W5+ were 34.00 and 36.25 eV, respectively, which implied that the electron density of the W element increased. The peak position of the W element was red-shifted at 0.60 eV after recombining with ZnTPyP (Fig. 4d). From the N element spectrum, the binding energies of N 1s in the pyridine group were 401.65 and 402.10 eV before and after being compounded with WO3, respectively, and the binding energy was increased by 0.45 eV (Fig. 4e), indicating that the electron density around the N atom had decreased, attributed to the N of pyridyl (in ZnTPyP) coordinated with WO3 through the W–N coordination bond. The peak position of the Zn element in the center of porphyrin did not change, mainly because the Zn atom was in the center of the porphyrin ring, and the surrounding atomic environment did not change during the recombination process (Fig. 4f). The binding energy of W decreased, and the increase in the binding energy of N confirmed the strong N–W coordination between WO3 and ZnTPyP, which in turn contributed to the formation of the ZnTPyP/WO3 heterojunction.
To further dissect the binding of WO3 and ZnTPyP, ZnTPyP/WO3 heterostructures were treated with acid or alkali to corrode the WO3 NRs and ZnTPyP NRs (Fig. S7†). The optical photo color of the ZnTPyP/WO3 heterojunction was claybank and similar to ZnTPyP nanorods. After the ZnTPyP/WO3 heterojunction was washed with hydrochloric acid (HCl), the color became a light green and close to that of the WO3 NRs (Fig. S7a†). The ZnTPyP components showed nanorods with hollow grooves after the WO3@ZnTPyP core–shell nanostructures were treated with NaOH (Fig. S7b†), which confirmed that the WO3 nanorods were wrapped within the ZnTPyP. The ZnTPyP/WO3 heterojunctions retained their nanorod structures after washing with HCl (Fig. S7c†). The XRD diffraction peak of the ZnTPyP/WO3 heterojunction after acid treatment was the same as that of pure WO3, indicating that the WO3 structure was maintained during the co-assembly process. After the ZnTPyP/WO3 heterojunction was treated with alkali, the XRD diffraction peaks were identical to those of pure ZnTPyP (Fig. S7d†). These results indicated that the ZnTPyP assemblies in the heterojunction existed in the form of hexagonal stacking.50,60 The residual ZnTPyP was further examined via Fourier-transform infrared spectroscopy (FT-IR). The corresponding absorption peak of the ZnTPyP monomer appeared at 1490 cm−1, mainly corresponding to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CN vibrations of the pyridine ring. A new absorption peak of the ZnTPyP/WO3 heterojunction treated with HCl appeared at 1464 cm−1, indicating that the ZnTPyP molecules were adsorbed on WO3 NRs because of the size effect. Compared with the ZnTPyP monomer, the absorption peak of the pyridine ring was red-shifted by 26 cm−1, which was mainly caused by the coordination between the N–W (Fig. S7e†).
C and CN vibrations of the pyridine ring. A new absorption peak of the ZnTPyP/WO3 heterojunction treated with HCl appeared at 1464 cm−1, indicating that the ZnTPyP molecules were adsorbed on WO3 NRs because of the size effect. Compared with the ZnTPyP monomer, the absorption peak of the pyridine ring was red-shifted by 26 cm−1, which was mainly caused by the coordination between the N–W (Fig. S7e†).
Given the above characterization data, we assumed the possible formation process of the heterojunctions (Fig. 5). In the alkaline emulsifier solution, the surface of the WO3 nanorods was gradually etched to form negatively charged WO42−. After adding the acidic porphyrin protonation solution, a layer of porphyrin molecules was adsorbed through electrostatic interaction and W–N coordination. Then, the absorbed layer was taken as the core of the assembly under the π–π interaction between the porphyrin molecules and gradually assembled on the surface of the WO3 nanorods to form porphyrin assemblies. With the change in the content of WO3 nanorods, heterojunction materials with different binding modes were gradually formed, which further affected the transmission path of photogenerated electrons. Therefore, the formation of ZnTPyP mainly included the following two processes: first, numerous ZnTPyP monomers were adsorbed on the WO3 surface through the force of W–N coordination. Then, the ZnTPyP monomer in solution took the WO3 nanorods as the nucleation site and preferentially continuously formed long-range orderly nanorods combined with WO3 NRs driven by π–π stacking and formed nanorod-to-nanorod nanostructures. Based on the WO3 loading amount, the different combined models of ZnTPyP/WO3 heterojunction could be obtained, which realized the controllable assembly of ZnTPyP on WO3 NRs. The close contact between the ZnTPyP NRs and WO3 NRs favored the transportation of the assembly of ZnTPyP on WO3 NRs. The close contact between ZnTPyP and WO3 NRs favored the transportation of photogenerated charges.
 | ||
| Fig. 5 Schematic of the formation process of ZnTPyP/WO3 heterojunctions by various interactions. | ||
3.3. Photocatalytic performance and mechanism analysis
The steady-state photoluminescence (PL) spectra measurements of ZnTPyP NRs and ZnTPyP/WO3 nanorod-on-nanorod heterojunctions revealed an emission peak centered at ∼660 nm (Fig. S8†). Then the time-resolved fluorescence decay profiles of the ZnTPyP/WO3 nanorod-to-nanorod nanostructures were tested (Fig. 6a). The fluorescence lifetimes τ of ZnTPyP and ZnTPyP/WO3 under 650 nm laser excitation were 1.44 and 1.71 ns, respectively, while their non-radiative transition lifetimes τ1 were 0.25 and 0.17 ns, and their spontaneous emission transition lifetimes τ2 were 1.19 and 1.54 ns, respectively. The decrease in the non-radiative transition lifetime τ1 by 0.08 ns and the increase in the radiative transition lifetime τ2 by 0.35 ns of the photogenerated electrons of the ZnTPyP/WO3 heterojunctions were mainly because the photogenerated electrons in WO3 recombined with the photogenerated holes in ZnTPyP through the N–W configuration, that is, the photogenerated electrons recombined along the “Z” path for migration, thereby effectively improving the photogenerated electron lifetime. The photocurrents of the different samples under visible-light irradiation (UVCUT > λ400 nm) are shown in Fig. 6b. The ZnTPyP/WO3 nanorod-to-nanorod nanostructures exhibited the highest photocurrent response intensity compared to the physical mixture of ZnTPyP nanorods and WO3 nanorods (ZnTPyP, WO3) and ZnTPyP nanorods, while the WO3 NRs and ZnTPyP nanorods showed comparatively low photoelectric responses, indicating that the ZnTPyP/WO3 nanorod-to-nanorod nanostructures had the highest electron-transfer rate and separation efficiency, mainly because of the N–W configuration, while the (ZnTPyP, WO3) mixture only exhibited a physical contact surface between the ZnTPyP and WO3. N–W bonds were more conducive to the migration and transfer of photogenerated electrons.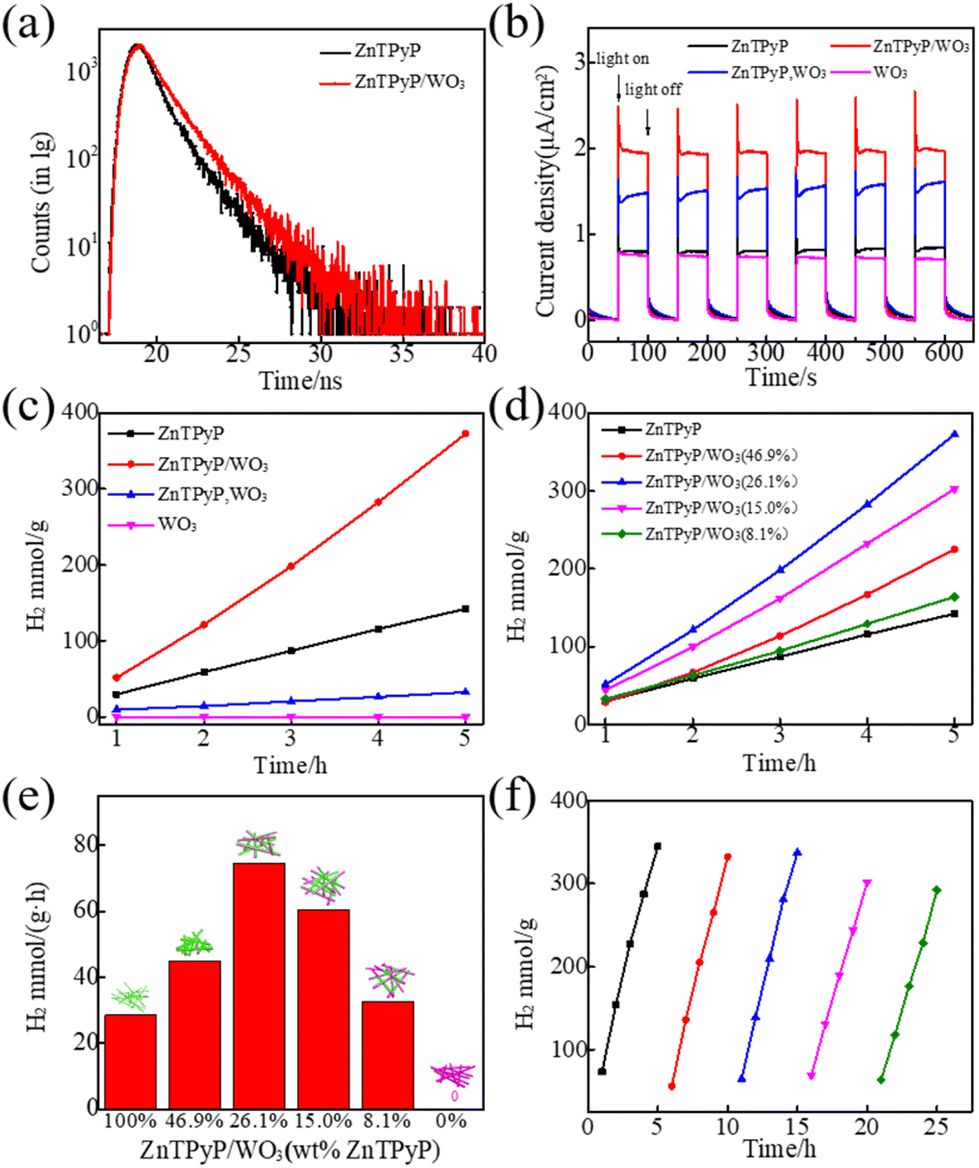 | ||
| Fig. 6 (a) Time-resolved fluorescence decay profiles of pure ZnTPyP nanorods and ZnTPyP/WO3 nanorods. (b) Photocurrent responses of pure ZnTPyP nanorods, pure WO3 nanorods, a mixture of ZnTPyP, WO3, and ZnTPyP/WO3 nanorods. (c) Comparison of the photocatalytic H2 production rate of pure ZnTPyP, pure WO3, and a mixture of ZnTPyP, WO3 and ZnTPyP/WO3. (d) Comparison of the photocatalytic H2-production rate of various ZnTPyP wt%. (e) Average H2-generation rate of ZnTPyP/WO3 with various ZnTPyP wt%. (f) Photocatalytic cycles of H2-production rates; the reaction conditions were as follows: 3 mg of ZnTPyP/WO3 and 0.2 M of AA aqueous solution (100 mL), 1 N NaOH used to adjust the solution pH to 3.5, and a 300 W Xe lamp with a 400 nm CUT filter. | ||
The photocatalytic H2-production activity of the prepared samples was evaluated with AA as a sacrificial electron donor and 1 wt% Pt co-catalyst loading (Fig. 6c). The hydrogen-production rates of the ZnTPyP nanorods and (ZnTPyP, WO3) physical mixture were 28.44 mmol g−1 h−1 and 6.47 mmol h−1, respectively. The ZnTPyP/WO3 nanorod-to-nanorod nanostructures exhibited the highest photocatalytic activity for H2 evolution (74.53 mmol g−1 h−1), which was 2.62 times that of the ZnTPyP nanorods and 11.5 times that of the (ZnTPyP, WO3) mixture. As a control, neither triethanolamine (TEOA) aqueous solution (Fig. S9a†), CH3OH aqueous solution (Fig. S9b†), or Na2S/Na2SO3 aqueous solution (Fig. S9c†) as sacrificial electron donors could evolve a detectable amount of H2 under these conditions. This was because the acidic sacrificial agent systems (AA) provided a large amount of proton source, which proved that the protonation of pyridine group of ZnTPyP facilitated photocatalytic HER, which could not occur in the neutral CH3OH sacrificial agent system, or in the alkaline TEOA and Na2S/Na2SO3 systems, which were consistent with other previous reports.61,62 Pristine WO3 nanorods failed to show any appreciable photocatalytic H2-production activity as the control. The dramatic enhancement of the photocatalytic activity of the ZnTPyP/WO3 nanorod-to-nanorod nanostructures was because of the embedding of WO3 in ZnTPyP, whereby the intimate interfacial contact between them effectively promoted charge separation.
The ZnTPyP/WO3 heterojunction exhibited a morphological and structure-dependent photocatalytic performance (Fig. 6d and e). Upon the enhancement of the ZnTPyP wt%-loading content from 0 to 26.1%, the H2-production rates exhibited an increasing trend and then a decreasing trend as the ZnTPyP-loaded increased continuously. The H2-production rates exhibited an increasing trend (from 28.44 mmol g−1 h−1 to 74.53 mmol g−1 h−1) when the ZnTPyP/WO3 was transformed from the core–shell structure to the nanorod-on-nanorod heterostructure. This increase was because the porphyrin-assembled shell affected the absorption of light energy by WO3 and led to a large accumulation of holes in WO3 in the core–shell ZnTPyP@WO3. The system was in a dynamic equilibrium between the light-harvesting and active sites. When the material formed a nanorod-on-nanorod heterostructure, both WO3 and ZnTPyP in the heterojunctions were exposed to visible light, thus reducing the optical shielding effect. However, the photogenerated electrons of WO3 NRs could rapidly recombine with the photogenerated holes of the ZnTPyP assemblies through the heterojunction interface to improve the separation efficiency of the photogenerated electron–hole pairs in ZnTPyP, thereby improving the efficiency of ZnTPyP. The structural stability of the heterojunction material played a crucial role in the photocatalytic activity of the material. After 5 cycles of photocatalytic hydrogen production, the catalytic rate did not significantly decrease (Fig. 6f).
After a 5 h hydrogen production test, the morphology and structure of ZnTPyP/WO3 remained intact, and no considerable change was observed compared with before the photocatalytic reaction (Fig. S10†). The heterojunction exhibited a certain structural stability during the photocatalytic process. Interestingly, potassium tetrachloroplatinate (K2PtCl4) was added to the solution of ZnTPyP/WO3 to reduce Pt2+ into Pt nanoparticles (Pt NPs) as a co-catalyst. The Pt NPs were all deposited on one side of the porphyrin assemblies, indicating that Pt2+ was preferentially reduced and deposited by photogenerated electrons generated in ZnTPyP (Fig. S10†). Moreover, the effect remained the same at 5 wt% Pt loading (Fig. S11†). The ZnTPyP assemblies were at the photogenerated electron-rich end, which confirmed that the photocatalytic hydrogen-production reaction mainly occurred in the ZnTPyP region, indicating that ZnTPyP/WO3 was a direct Z-scheme photocatalytic system.
To further explore the photogenerated electron-transport process of ZnTPyP/WO3 in the photocatalytic process, the band energies of WO3 NRs and ZnTPyP NRs were characterized. According to the UV diffuse reflectance spectra, the energy gaps (Eg) of the ZnTPyP assemblies and WO3 NRs were calculated to be 1.97 and 2.80 eV, respectively (Fig. 7a). The Mott–Schottky curve showed that the conduction band (ECB) positions of the ZnTPyP assemblies and WO3 NRs were −0.83 and 0.23 eV, respectively (Fig. 7b). According to the equation Eg = EVB − ECB, the EVB values of the ZnTPyP assemblies and WO3 were calculated to be 1.14 and 3.03 eV, respectively. From the XPS valence-band spectra, the energy gap between the valence-band and the Fermi level (Evf) of the ZnTPyP NRs and WO3 NRs were 1.71 and 2.70 eV, respectively (Fig. 7c). Thus, the Fermi energy levels (Ef) of the ZnTPyP assemblies and WO3 were calculated to be −0.57 and 0.33 eV, respectively (Fig. 7d). The Ef of WO3 was higher than that of ZnTPyP. When the ZnTPyP NRs and WO3 NRs came in contact, a new steady-state built-in electric field was spontaneously formed on the interface of the heterojunction material,34 and the energy band was bent at the heterojunction interface (Fig. 7d). Under visible-light radiation, both ZnTPyP and WO3 generated photogenerated electron holes. The photogenerated electrons in the conduction band of WO3 passed through the interface and recombined with the photogenerated holes in the valence band of ZnTPyP with the participation of the built-in electric field; that is, the photogenerated carriers of the heterojunction material were transferred via a direct Z-scheme pathway. The transfer of the photogenerated carriers effectively improved the separation rate of photogenerated electron–hole pairs of ZnTPyP, resulting in the more negative conduction band energy of ZnTPyP and generating more photogenerated electrons for photocatalytic hydrogen production (Fig. 7e).
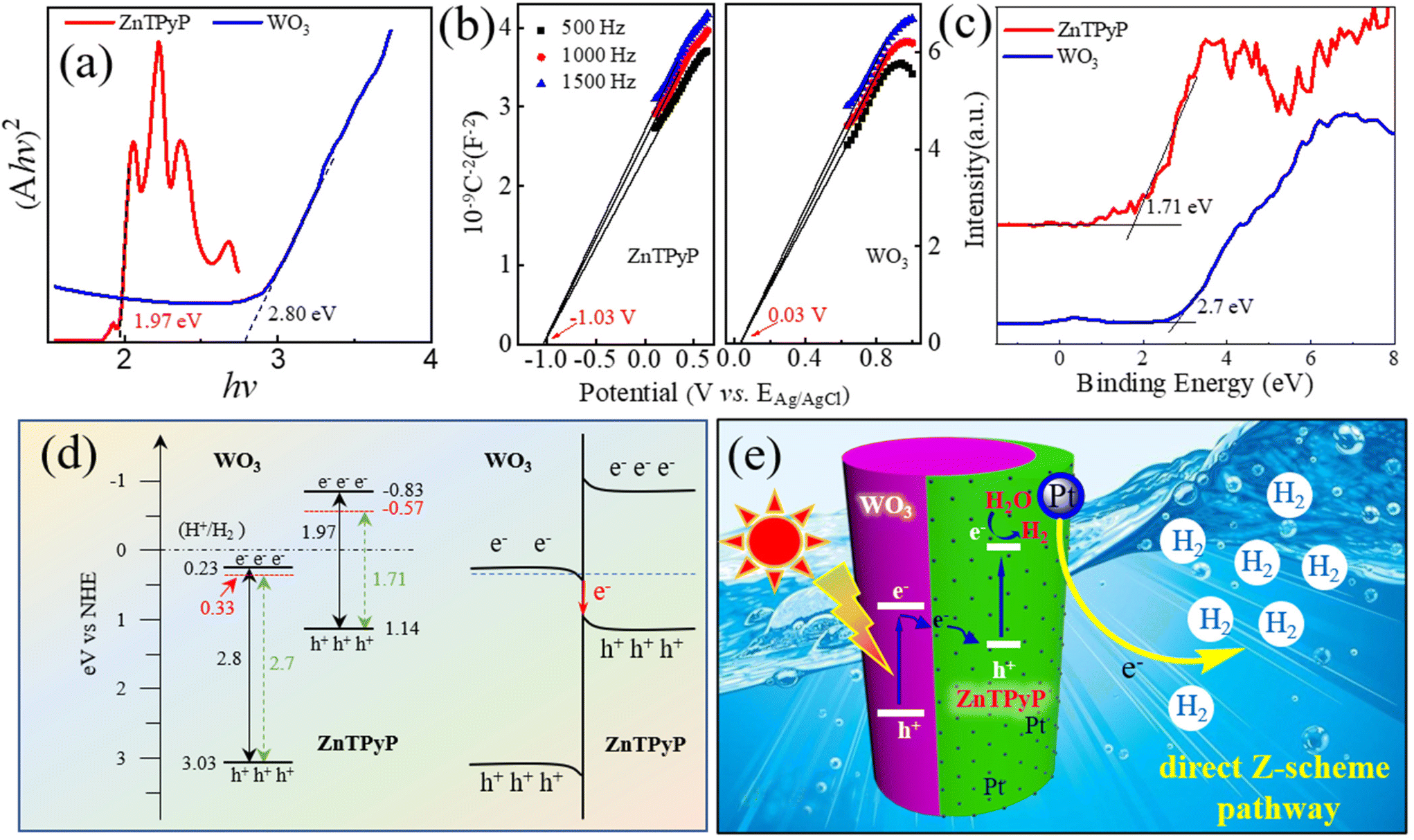 | ||
| Fig. 7 (a) Tauc plots of (αhv)2vs. Eg, (b) Mott–Schottky plots, and (c) valence-band XPS spectra of pure ZnTPyP NRs and pure WO3 NRs. (d) Energy level and (e) photocatalytic mechanism scheme and the possible charge separation of direct Z-scheme ZnTPyP/WO3. | ||
4. Conclusions
In summary, a ZnTPyP/WO3 nanorod-on-nanorod direct Z-scheme photocatalytic heterojunction was prepared via a modified acid–base neutralization micelle confinement assembly method using WO3 NRs as the confined template and ZnTPyP monomer as the building blocks. A series of ZnTPyP/WO3 heterojunctions with different morphological structures were prepared through N–W coordination by controlling the process factors, such as the pH value of the assembly solution and the amount of WO3 NRs. The hydrogen-production performance test results showed that the hydrogen-production rate of the ZnTPyP/WO3 nanorod-to-nanorod heterojunction was as high as 74.53 mmol g−1 h−1, which was 2.62 times that of ZnTPyP assemblies. The electrons of the ZnTPyP/WO3 nanorod-to-nanorod heterojunction nanomaterial were transported via a direct Z-scheme pathway during the photocatalytic process, which effectively improved the photocatalytic performance. This study provides a new strategy for preparing Z-scheme photocatalyst based on organic supramolecular orderly aggregate materials.Author contributions
S. Liu: Investigation, methodology, data curation, formal analysis, writing-original draft. S. Xia: Investigation, methodology, data curation. X. Ren: Data curation, formal analysis. J. Wang: Formal analysis, methodology, visualization, writing – review & editing. S. Chen: Formal analysis. Y. Zhong: Conceptualization, data curation, funding acquisition, supervision, writing – review & editing. F. Bai: Conceptualization, data curation, formal analysis, funding acquisition, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21802032 and U21A2085), China Postdoctoral Science Foundation (2019TQ0081) and Zhongyuan high level talents special support plan (No. 204200510009).References
- Y. Song, K. Ji, H. Duan and M. Shao, Exploration, 2021, 1, 20210050 CrossRef
.
- D. Wei, X. Shi, R. Qu, K. Junge, H. Junge and M. Beller, ACS Energy Lett., 2022, 3734–3752 CrossRef CAS
- X. Zhang, T. Peng and S. Song, J. Mater. Chem. A, 2016, 4, 2365–2402 RSC
- M. Brodt, K. Müller, J. Kerres, I. Katsounaros, K. Mayrhofer, P. Preuster, P. Wasserscheid and S. Thiele, Energy Technol., 2021, 9, 2100164 CrossRef CAS
- T. Yamamura, T. Nakanishi, J. Lee, S. Yamate and J. Otomo, Energy Fuels, 2022, 36, 9745–9756 CrossRef CAS
- Z. Wang, T. Hisatomi, R. Li, K. Sayama, G. Liu, K. Domen, C. Li and L. Wang, Joule, 2021, 5, 344–359 CrossRef CAS
- L. Wang, H. Fan and F. Bai, MRS Bull., 2020, 45, 49–56 CrossRef
- Y. Bai, Z. Hu, J. X. Jiang and F. Huang, Chem. – Asian J., 2020, 15, 1780–1790 CrossRef CAS PubMed
- Y. Wang, X. Shang, J. Shen, Z. Zhang, D. Wang, J. Lin, J. C. S. Wu, X. Fu, X. Wang and C. Li, Nat. Commun., 2020, 11, 3043–3054 CrossRef CAS PubMed
- S. Liu, S. Xia, S. Liu, M. Li, J. Sun, Y. Zhong, F. Zhang and F. Bai, Chem. J. Chin. Univ., 2022, 20220512 Search PubMed
- E. A. Reyes Cruz, D. Nishiori, B. L. Wadsworth, N. P. Nguyen, L. K. Hensleigh, D. Khusnutdinova, A. M. Beiler and G. F. Moore, Chem. Rev., 2022, 122, 16051–16109 CrossRef CAS PubMed
- S. Nishioka, K. Hojo, L. Xiao, T. Gao, Y. Miseki, S. Yasuda, T. Yokoi, K. Sayama, T. E. Mallouk and K. Maeda, Sci. Adv., 2022, 8, eadc9115 CrossRef CAS PubMed
- D. Zhao, Y. Wang, C.-L. Dong, Y.-C. Huang, J. Chen, F. Xue, S. Shen and L. Guo, Nat. Energy, 2021, 6, 388–397 CrossRef CAS
- Y. Tachibana, L. Vayssieres and J. R. Durrant, Nat. Photonics, 2012, 6, 511–518 CrossRef CAS
- A. Iwase, Y. H. Ng, Y. Ishiguro, A. Kudo and R. Amal, J. Am. Chem. Soc., 2011, 133, 11054–11057 CrossRef CAS PubMed
- H. J. Yun, H. Lee, N. D. Kim, D. M. Lee, S. Yu and J. Yi, ACS Nano, 2011, 5, 4084–4090 CrossRef CAS PubMed
- J. E. Yourey, J. B. Kurtz and B. M. Bartlett, J. Phys. Chem. C, 2012, 116, 3200–3205 CrossRef CAS
- K. Maeda, D. Lu and K. Domen, ACS Catal., 2013, 3, 1026–1033 CrossRef CAS
- K.-Y. Jiang, X.-C. Dai, Y. Yu, Q.-L. Mo and F.-X. Xiao, J. Phys. Chem. C, 2018, 122, 12291–12306 CrossRef CAS
- X. Wang, G. Liu, Z. G. Chen, F. Li, L. Wang, G. Q. Lu and H. M. Cheng, Chem. Commun., 2009, 3452–3454 RSC
- H. Li, Y. Gao, Y. Zhou, F. Fan, Q. Han, Q. Xu, X. Wang, M. Xiao, C. Li and Z. Zou, Nano Lett., 2016, 16, 5547–5552 CrossRef CAS PubMed
- H. Li, H. Yu, X. Quan, S. Chen and Y. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 2111–2119 CrossRef CAS PubMed
- Y. Zou, J.-W. Shi, D. Ma, Z. Fan, C. Niu and L. Wang, ChemCatChem, 2017, 9, 3752–3761 CrossRef CAS
- T. H. Jeon, D. Monllor-Satoca, G. H. Moon, W. Kim, H. I. Kim, D. W. Bahnemann, H. Park and W. Choi, Nat. Commun., 2020, 11, 967–976 CrossRef CAS PubMed
- B. Niu, D. Wu, J. Wang, L. Wang and W. Zhang, Appl. Surf. Sci., 2020, 528, 146965 CrossRef CAS
- Q. Xu, L. Zhang, B. Cheng, J. Fan and J. Yu, Chem, 2020, 6, 1543–1559 CAS
- Z. Sun, W. Wang, Q. Chen, Y. Pu, H. He, W. Zhuang, J. He and L. Huang, J. Mater. Chem. A, 2020, 8, 3160–3167 RSC
- L. Jiang, X. Yuan, G. Zeng, J. Liang, X. Chen, H. Yu, H. Wang, Z. Wu, J. Zhang and T. Xiong, Appl. Catal., B, 2018, 227, 376–385 CrossRef CAS
- W. Yu, J. Chen, T. Shang, L. Chen, L. Gu and T. Peng, Appl. Catal., B, 2017, 219, 693–704 CrossRef CAS
- X. L. Yin, L. L. Li, W. J. Jiang, Y. Zhang, X. Zhang, L. J. Wan and J. S. Hu, ACS Appl. Mater. Interfaces, 2016, 8, 15258–15266 CrossRef CAS PubMed
- X.-L. Yin, G.-Y. He, B. Sun, W.-J. Jiang, D.-J. Xue, A.-D. Xia, L.-J. Wan and J.-S. Hu, Nano Energy, 2016, 28, 319–329 CrossRef CAS
- R. Abe, K. Shinmei, N. Koumura, K. Hara and B. Ohtani, J. Am. Chem. Soc., 2013, 135, 16872–16884 CrossRef CAS PubMed
- J. Zheng, X. Liu and L. Zhang, Chem. Eng. J., 2020, 389, 124339 CrossRef CAS
- C. F. Fu, X. Wu and J. Yang, Adv. Mater., 2018, 30, e1802106 CrossRef PubMed
- Y. Chen, C. Yan, J. Dong, W. Zhou, F. Rosei, Y. Feng and L. N. Wang, Adv. Funct. Mater., 2021, 31, 2104099 CrossRef CAS
- O. Dumele, J. Chen, J. V. Passarelli and S. I. Stupp, Adv. Mater., 2020, 32, e1907247 CrossRef PubMed
- J. Xu, W. Li, W. Liu, J. Jing, K. Zhang, J. Yang, E. Zhu, J. Li and Y. Zhu, Angew. Chem., Int. Ed., 2022, 61, e202212243 CAS
- W. Wei, J. Sun and H. Fan, MRS Bull., 2019, 44, 178–182 CrossRef
- F. Bai, K. Bian, B. Li, C. Karler, A. Bowman and H. Fan, MRS Bull., 2020, 45, 135–141 CrossRef
- Q. Li, Y. Bao and F. Bai, MRS Bull., 2020, 45, 569–573 CrossRef
- Y. Zhong, S. Liu, J. Wang, W. Zhang, T. Tian, J. Sun and F. Bai, APL Mater., 2020, 8, 120706 CrossRef CAS
- J. Yang, J. Jing and Y. Zhu, Adv. Mater., 2021, 33, e2101026 CrossRef PubMed
- S. Tian, S. Chen, X. Ren, Y. Hu, H. Hu, J. Sun and F. Bai, Nano Res., 2020, 13, 2665–2672 CrossRef CAS
- S. Tian, S. Chen, X. Ren, R. Cao, H. Hu and F. Bai, Nano Res., 2019, 12, 3109–3115 CrossRef CAS
- T. E. Karam, N. Siraj, J. C. Ranasinghe, P. E. Kolic, B. P. Regmi, I. M. Warner and L. H. Haber, J. Phys. Chem. C, 2020, 124, 24533–24541 CrossRef CAS
- Y. Zhong, J. Wang and Y. Tian, MRS Bull., 2019, 44, 183–188 CrossRef
- S. Chen, X. Ren, S. Tian, J. Sun and F. Bai, MRS Adv., 2020, 5, 2147–2155 CrossRef CAS
- J. Lu, Z. Li, W. An, L. Liu and W. Cui, Nanomaterials, 2019, 9, 1321–1336 CrossRef CAS PubMed
- R. Cao, G. Wang, X. Ren, P. C. Duan, L. Wang, Y. Li, X. Chen, R. Zhu, Y. Jia and F. Bai, Nano Lett., 2022, 22, 157–163 CrossRef CAS PubMed
- J. Wang, Y. Zhong, L. Wang, N. Zhang, R. Cao, K. Bian, L. Alarid, R. E. Haddad, F. Bai and H. Fan, Nano Lett., 2016, 16, 6523–6528 CrossRef CAS PubMed
- P. Zhang, B. Y. Guan, L. Yu and X. W. Lou, Chem, 2018, 4, 162–173 CAS
- M. Xiao, Z. Wang, M. Lyu, B. Luo, S. Wang, G. Liu, H.-M. Cheng and L. Wang, Adv. Mater., 2019, 31, 1801369 CrossRef PubMed
- R. Cao, J. Wang, Y. Li, J. Sun and F. Bai, Nano Res., 2022, 15, 5719–5725 CrossRef CAS
- R. Lin, J. Wan, Y. Xiong, K. Wu, W. C. Cheong, G. Zhou, D. Wang, Q. Peng, C. Chen and Y. Li, J. Am. Chem. Soc., 2018, 140, 9078–9082 CrossRef CAS PubMed
- L. Liang, X. Li, Y. Sun, Y. Tan, X. Jiao, H. Ju, Z. Qi, J. Zhu and Y. Xie, Joule, 2018, 2, 1004–1016 CrossRef CAS
- T. Hasobe, H. Sakai, K. Mase, K. Ohkubo and S. Fukuzumi, J. Phys. Chem. C, 2013, 117, 4441–4449 CrossRef CAS
- J. Wang, E. Khoo, P. S. Lee and J. Ma, J. Phys. Chem. C, 2008, 112, 14306–14312 CrossRef CAS
- F. Bai, Z. Sun, H. Wu, R. E. Haddad, E. N. Coker, J. Y. Huang, M. A. Rodriguez and H. Fan, Nano Lett., 2011, 11, 5196–5200 CrossRef CAS PubMed
- F. Bai, H. Wu, R. E. Haddad, Z. Sun, S. K. Schmitt, V. R. Skocypec and H. Fan, Chem. Commun., 2010, 46, 4941–4943 RSC
- Y. Zhong, J. Wang, R. Zhang, W. Wei, H. Wang, X. Lu, F. Bai, H. Wu, R. Haddad and H. Fan, Nano Lett., 2014, 14, 7175–7179 CrossRef CAS PubMed
- J. Yang, A. Acharjya, M.-Y. Ye, J. Rabeah, S. Li, Z. Kochovski, S. Youk, J. Roeser, J. Grüneberg, C. Penschke, M. Schwarze, T. Wang, Y. Lu, R. Krol, M. Oschatz, R. Schomäcker, P. Saalfrank and A. Thomas, Angew. Chem., Int. Ed., 2021, 60, 19797–19803 CrossRef CAS PubMed
- J. Yang, S. Ghosh, J. Roeser, A. Acharjya, C. Penschke, Y. Tsutsui, J. Rabeah, T. Wang, S. Y. D. Tameu, M.-Y. Ye, J. Grüneberg, S. Li, C. Li, R. Schomäcker, R. V. D. Krol, S. Seki, P. Saalfrank and A. Thomas, Nat. Commun., 2022, 13, 6317–6327 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr05777h |
| This journal is © The Royal Society of Chemistry 2023 |