
Block copolymer self-assembly to pattern gold nanodots for site-specific placement of DNA origami and attachment of nanomaterials†

Abstract
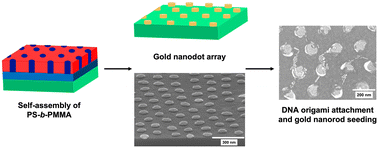
Directed placement of DNA origami could play a key role in future integrated nanoelectronic devices. Here we demonstrated the site-selective attachment of DNA origami on gold dots formed using a pattern transfer method through block copolymer self-assembly. First, a random copolymer brush layer is grafted on the Si surface and then poly (styrene-b-methylmethacrylate) block copolymer is spin-coated to give a hexagonal nanoarray after annealing. UV irradiation followed by acetic acid etching is used to remove the PMMA, creating cylindrical holes and then oxygen plasma etching removes the random copolymer layer inside those holes. Next, metal evaporation, followed by lift-off creates a gold dot array. We evaluated different ligand functionalization of Au dots, as well as DNA hybridization to attach DNA origami to the nanodots. DNA-coated Au nanorods are assembled on the DNA origami as a step towards creating nanowires and to facilitate electron microscopy characterization of the attachment of DNA origami on these Au nanodots. The DNA hybridization approach showed better DNA attachment to Au nanodots than localization by electrostatic interaction. This work contributes to the understanding of DNA-templated assembly, nanomaterials, and block copolymer nanolithography. Furthermore, the work shows potential for creating DNA-templated nanodevices and their placement in ordered arrays in future nanoelectronics.
- This article is part of the themed collection: Emerging concepts in nucleic acids: structures, functions and applications
 Please wait while we load your content...
Please wait while we load your content...

