
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAssemblies of poly(N-vinyl-2-pyrrolidone)-based double hydrophilic block copolymers triggered by lanthanide ions: characterization and evaluation of their properties as MRI contrast agents†
Marjorie
Yon
a,
Laure
Gibot
 a,
Stéphane
Gineste
a,
Pascale
Laborie
b,
Christian
Bijani
c,
Christophe
Mingotaud
a,
Olivier
Coutelier
a,
Franck
Desmoulin
de,
Carine
Pestourie
e,
Mathias
Destarac
a,
Diana
Ciuculescu-Pradines
*a and
Jean-Daniel
Marty
*a
a,
Stéphane
Gineste
a,
Pascale
Laborie
b,
Christian
Bijani
c,
Christophe
Mingotaud
a,
Olivier
Coutelier
a,
Franck
Desmoulin
de,
Carine
Pestourie
e,
Mathias
Destarac
a,
Diana
Ciuculescu-Pradines
*a and
Jean-Daniel
Marty
*a
aLaboratoire des IMRCP, Université de Toulouse, CNRS UMR 5623, Université Toulouse III – Paul Sabatier, France. E-mail: eliza.ciuculescu-pradines@univ-tlse3.fr; jean-daniel.marty@univ-tlse3.fr
bPlateforme scientifique et technique Institut de Chimie de Toulouse ICT – UAR 2599, Université de Toulouse, CNRS, Toulouse, France
cLCC-CNRS, Université de Toulouse, CNRS, UPS, Toulouse, France
dToulouse NeuroImaging Center (ToNIC), Inserm, University of Toulouse—Paul Sabatier, Toulouse, France
eCREFRE-Anexplo, Université de Toulouse, Inserm, UT3, ENVT, Toulouse, France
First published on 24th January 2023
Abstract
Because of the formation of specific antibodies to poly(ethylene glycol) (PEG) leading to life-threatening side effects, there is an increasing need to develop alternatives to treatments and diagnostic methods based on PEGylated copolymers. Block copolymers comprising a poly(N-vinyl-2-pyrrolidone) (PVP) segment can be used for the design of such vectors without any PEG block. As an example, a poly(acrylic acid)-block-poly(N-vinyl-2-pyrrolidone) (PAA-b-PVP) copolymer with controlled composition and molar mass is synthesized by reversible addition–fragmentation chain transfer (RAFT) polymerization. Mixing this copolymer with lanthanide cations (Gd3+, Eu3+, Y3+) leads to the formation of hybrid polyion complexes with increased stability, preventing the lanthanide cytotoxicity and in vitro cell penetration. These new nanocarriers exhibit enhanced T1 MRI contrast, when intravenously administered into mice. No leaching of gadolinium ions is detected from such hybrid complexes.
Introduction
Lanthanide based nanomaterials are a very important class of imaging probes in the biomedical field.1,2 These include, for example, the most successful contrast agents for magnetic resonance imaging (MRI), which benefit from the excellent paramagnetic properties of gadolinium ions, or fluorescent labels which use the luminescent properties of Europium. However, beyond the attractive properties of lanthanide ions, the ligands which escort them play a key role in controlling the performances of the contrast agent: (i) regarding the relationship with the captive lanthanide ion, ligands should be capable of strong and multiple interactions allowing one to reach suitable and efficient concentrations for an optimized signal, to prevent them from leaching and to tune the parameters which influence the desired properties; (ii) regarding the ligand/biological interface, ligands should act as a shield, keeping the toxic lanthanide ions out of contact with the biological media and ensure biocompatibility and transport of the payload with long blood-circulation time and possibly specific recognition.Double hydrophilic block copolymers (DHBCs)3–6 have unique advantages for the design of vectors incorporating lanthanide ions.7–9 Typically, one of the blocks which is ionizable can interact with polyvalent metal ions, spontaneously inducing self-assembly of the copolymer into micelle structures and encapsulation of metal ions into the core, through multiple electrostatic interactions, while the second non-ionizable block forms the shell of micelles in contact with the exterior medium. The negatively ionizable groups of poly(acrylic acid) (PAA), in combination with the obvious advantages of poly(ethylene glycol) (PEG) such as hydrophilicity, biocompatibility, Food and Drug Administration (FDA) approval, stealth behaviour etc. make PAA-b-PEG block copolymers of choice for loading polyvalent metallic ions.7–15 These copolymers can be synthesized by reversible deactivation radical polymerization (RDRP) from functional PEG.16 This requires first the modification of one of the end groups of PEG in one or two synthetic steps.17,18 These modified PEGs then serve either as macro-chain transfer agents for the copolymerization of acrylic acid (AA) by reversible addition–fragmentation chain transfer (RAFT) polymerization19 or as macroinitiators for the copolymerization of acrylic esters by atom transfer radical polymerization (ATRP) followed by ester cleavage.17 Although PEG synthesis requires specific conditions in the laboratory, its highly industrialized polymerization allows the access to medical grade PEGs with a wide variety of molar masses, and their easy implementation into block copolymers. However, the extensive use of PEGs in biomedical field shows that it may have possible drawbacks, thus there is increasing evidence for unexpected immune responses occurring against PEGylated nanocarriers upon repeated administration, an issue becoming more relevant considering the currently running SARS-CoV2 vaccination campaigns.20–22 Therefore, there is an urgent need to develop alternatives to not run out of treatments or diagnostic methods20–22 using PEG-based copolymers. Also, colloids based on the association of PEG-based polymers have limitations related to possible aggregation of PEG-coated NPs during lyophilization.23
The substitution of the PEG block with another block offers new perspectives for the preparation of original hybrid polyion complexes (HPICs). These DHBCs would provide new opportunities to enrich the knowledge related to their micellization in the presence of polyvalent metallic ions and to the functionalities of the resulting objects in regard to biomedical applications. In this context, the choice of poly(N-vinyl-2-pyrrolidone), PVP, seems especially relevant: PVP is a highly water soluble, biocompatible polymer, with an ability to prevent nanoparticles opsonization.24–26 It has been used as a plasma expander27 and it is used as carrier of iodine (Inadine, Aerodine, Betadine) for antiseptic usage.28 As a food additive, PVP is used as a stabilizer and has the E number E1201.29
In addition, the polymerization of N-vinyl-2-pyrrolidone (VP) can be controlled by RDRP methods.30,31 Lastly, the use of hydrophilic PVP in colloid formulations has led to colloids with improved stability thanks to the cryoprotective properties of PVP that prevent aggregation during lyophilization.32 In association with PAA homopolymer, PVP has been mainly used in the composition of interpolymer complexes, based on cooperative hydrogen bonds.33–36
Surprisingly, there are only few examples describing the synthesis of PAA-b-PVP diblock copolymers: one by consecutive Cu-mediated ATRP of VP and t-butyl acrylate followed by t-butyl ester cleavage,37 and one by direct aqueous RAFT polymerization of AA and VP.38 Moreover, to our knowledge, only one example has reported the use of PAA-b-PVP block copolymer for encapsulation of zinc phthalocyanine and doxorubicine into supramolecular assemblies to achieve chemo-photodynamic therapy but no attempt to assemble them in the presence of polyvalent metallic ions has been reported so far.37
We report here a more effective and metal free RAFT synthetic strategy to gain access to a PAA-b-PVP block copolymer with controlled composition and molar mass. The formation of HPICs through the interaction of this copolymer with different cations (Gd3+, Eu3+, Y3+) was then studied by scattering techniques, NMR and ATR-FTIR spectroscopy and fluorescence experiments (Scheme 1). The stability of these HPICs in culture medium was addressed, together with their toxicity and interactions with cells, in vitro. Finally, their MRI relaxivity in comparison to the one of HPICs based on PAA-b-PEG copolymer, was evaluated both in vitro and in vivo in mice model.
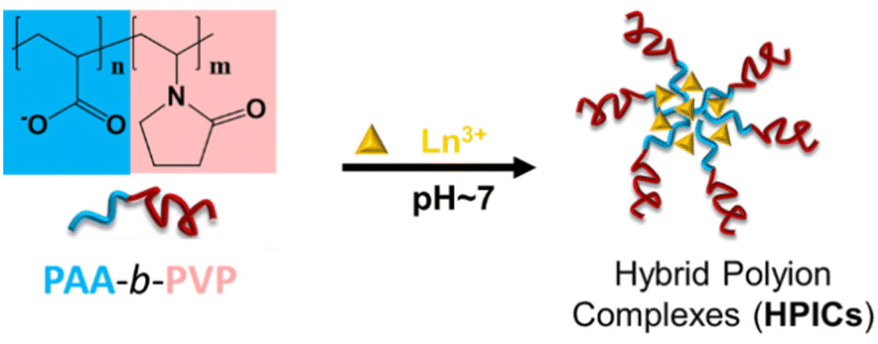 | ||
| Scheme 1 Strategy of formation of hybrid polyion complexes (HPICs). Lanthanides cations Ln3+ were either Gd3+, Eu3+ or Y3+. | ||
Results and discussion
Synthesis of PAA-b-PVP block copolymer
The synthesis of a PAA-b-PVP block copolymer with number-average molar masses (Mn) of respective blocks similar to the ones of PAA3k-b-PEG6k which was previously used for the formation of Gd3+-HPICs, was targeted.7 We did not opt for the direct synthesis of PAA-b-PVP by xanthate-mediated aqueous RAFT polymerization38 as previously reported due to encountered side reactions (acidity-induced formation of N-(α-hydroxyethyl)pyrrolidone from NVP) for the Mn and composition of interest. Instead, a PAA-b-PVP diblock copolymer was obtained through consecutive RAFT polymerization of methyl acrylate (MA) and VP mediated by O-ethyl-S-(1-methoxycarbonyl) ethyldithiocarbonate (xanthate XA1) followed by cleavage of the methyl ester of MA units under basic conditions (Fig. 1A). Unlike direct aqueous synthesis of PAA-b-PVP for which average molar masses could not be determined by SEC, this indirect approach allows us to determine the Mn and dispersity of first block and diblock before deprotection by organic SEC: this represents a signification advantage. Polymerization of MA was initiated by azobisisobutyronitrile (AIBN) in the presence of XA1 at 70 °C in dioxane for 6 h until complete monomer conversion as determined by 1H NMR. A PMA with a Mn,NMR of 2750 g mol−1 in agreement with the theoretical value was determined by 1H NMR (Table 1 and Fig. S1†). Size exclusion chromatography (SEC) analysis of the PMA showed a monomodal molar mass distribution, with Mn,SEC = 2530 g mol−1 also in good agreement with the expected Mn and corresponding to a number-average degree of polymerization (DPn) equal to 32. A dispersity (Đ) equal to 1.4 was determined, which is in excellent agreement with the chain transfer constant to XA1 (2 < Ctr,XA1 < 3) for acrylate monomers.39 Block copolymerization of VP from the PMA32-XA1 precursor was successfully carried out in dioxane at 35 °C with 2,2′-azobis(4-methoxy-2,4-dimethylvaleronitrile (V-70) as evidenced by SEC and 1H NMR analysis (Fig. 1B and Fig. S2†). The molar ratio of PMA to PVP was determined by 1H NMR: PMA/PVP = 0.54 (0.6 expected) leading to a DPn equal to 59 for the PVP block (see Fig. S2†). Finally, cleavage of ester functions of the PMA block was performed in basic conditions. It could be remarked in the 1H-NMR spectra of the hydrolyzed copolymer (Fig. S3†), the absence of the methyl signal of the ester group at 3.7 ppm. In Fig. 1C, ATR-FTIR spectra also evidenced the disappearance of the carbonyl stretch band of the ester group at 1736 cm−1 and the appearance of the carboxylate (OCO) asymmetric and symmetric stretching modes at 1564 cm−1 and 1402 cm−1, respectively. | ||
| Fig. 1 Synthesis and characterization of PAA-b-PVP copolymer. (A) Schematic description of the different steps of the polymer synthesis. (B) Size exclusion chromatograms (RI traces) in DMF/LiBr 10 mM of PMA32-b-PVP59 (red line) and PMA32 (black line). (C) ATR-FTIR spectra of PMA32 (black line), PMA32-b-PVP59 (red line) and PAA32-b-PVP59 (blue line). | ||
| Polymer |
M
n,th![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a (g mol−1) a (g mol−1) |
M
n,NMRb (g mol−1) |
M
n,SECc (g mol−1) (Đ) |
DPn,PVP/DPn,PMAd (theoretical value) |
|---|---|---|---|---|
| a M n,th = [Monomer]0/[RAFTagent]0 × Conversion (Monomer) × MW (Monomer) + MW (RAFT agent). b Calculated from 1H NMR (see details in Fig. S1†). c Determined by SEC-RI-MALS in DMF/LiBr (10 mM). d Calculated from 1H NMR (see Fig. S2,† for details). | ||||
| PMA | 2750 | 2750 | 2530 (1.4) | 0/32 (0/32) |
| PMA-b-PVP | 8300 | 9400 | 8630 (1.1) | 59/32 (50/30) |
The 1H NMR spectrum of the targeted PAA32-b-PVP59 block copolymer confirmed its composition (Fig. S3†).
Formation and characterization of HPICs
HPICs were formed by addition of trivalent lanthanide cations (Gd3+, Eu3+, Y3+) to aqueous solutions of the PAA32-b-PVP59 copolymer followed by the adjustment of the pH to 7. The effect of the addition of increasing amounts of trivalent lanthanide cations was monitored, by dynamic light scattering (DLS) measurements. The ratio, R, between the positive charges arising from the trivalent lanthanide cations and the potentially available negative charges from the ionized or ionizable carboxylic acid monomer units of the polymer was used to define the HPICs: R = 3·[Ln3+]/[AA]. As an example, mono-angle DLS results for Gd3+/PAA32-b-PVP59 HPICs solutions are given in Fig. 2A. Similar results obtained for Eu3+ and Y3+ are given in Fig. S4.† For R values lower than 0.8, the measured light scattering intensity was low. Consequently, the precision and the significance of Z-average values obtained from the correlograms are poor and not significant of the presence of colloidal objects. For R larger than 0.8 and up to 1, an increase of scattered intensity was measured associated with a decrease of Z-average size. As previously observed in the case of PAA-b-PEG copolymer,8,40 these results suggest the formation of colloids, triggered by the addition of gadolinium ions and achieved for R = 1. Around R = 1, these colloids exhibit a diameter of ca. 25 nm (in term of Z-average) with a PDI of 0.27 suggesting a large monomodal distribution or a multimodal distribution. A non-negative least-squares (NNLS) analysis (see Fig. S5†) confirmed the presence of large objects (diameter larger than 100 nm) but at a very small concentration (estimated less than 10−2 %). Further multi-angle DLS analysis (Fig. S6†) confirmed that the solution contains mainly Brownian colloids with a radius of 12.1 ± 1.3 nm. Similar sizes were obtained with Y3+ and Eu3+ ions (respectively 14.3 ± 1.5 nm and 12.2 ± 1.4 nm, Fig. S6†). These values are comparable to the 11.5 ± 1.2 nm radius obtained for HPICs formed by Gd3+ and PAA3k-b-PEG6k copolymer.7 Zeta potential was measured as a function of R in order to evaluate the surrounding electric potential of the colloidal objects (Fig. S7†). Pristine PAA32-b-PVP59 polymer solutions were found with an overall negative zeta potential value of about ζ = −32 ± 2 mV at pH 7, which is due to the negatively charged carboxylate groups of the PAA block in solution. Addition of Gd3+ ions then induces the decrease of the zeta potential until R = 1 where a plateau value at about ζ ≈ 0 mV is reached for R ≥ 1. The objects corresponding to a ratio R = 1 were observed by conventional transmission electron microscopy (TEM) (see Fig. S8A†). Even if it was possible to visualize them, the contrast was poor and got worse with the magnification, typical of weakly scattering materials. This is probably due to the relative low density of the gadolinium ions in the polymer matrix sample (very roughly estimated from the PAA density around few gadolinium per nm3), to the small size of the nanoparticles and to the drying process which could disorganize the HPICs. However, as shown in Fig. S8A,† a mean diameter of the objects of 11.5 nm with a standard deviation σ = 1.5 nm, obtained by a Gaussian fit could be calculated from such TEM pictures. Additionally, the sample was mapped by scanning transmission electron microscopy using high angle annular dark-field detector (HAADF-STEM) in order to obtain imaging only from incoherently scattered electrons (Rutherford scattering from the nucleus of the atoms) and thus to obtain a better contrast due to variations in atomic number Z of the elements in the sample.41,42 In Fig. 2B and Fig. S8† the regions containing gadolinium ions appear white in contrast to the polymer. The diameter of the white dots is of 6.4 nm (σ = 0.8 nm) slightly smaller than the one observed in conventional TEM in agreement with the sequestering of the Gd3+ ions in the polymer objects. This estimation of the core size is close to the one measured by ASAXS on other types of HPICs.15 | ||
| Fig. 2 Characterization of Ln3+/PAA32-b-PVP59 HPICs. (A) Typical evolutions of the scattered light intensity (black squares) and Z-average diameter (red dots), measured by a mono-angle DLS instrument for Gd3+/PAA32-b-PVP59 HPICs versus the R ratio. The lines are just guide for the eyes. Because of self-association of the polymer, the Z-average size obtained for R below ca. 0.8 corresponds to polydispersed nano-objects. Above R equals to ca. 0.8, the size distribution is quasi mono-modal. *At the highest ratio (R = 2.2) the formation of hydroxide species were evidenced by ATR-FTIR experiments, so corresponding values do not correspond solely to HPICs formation. (B) Scanning transmission electron microscopy images using high-angle annular dark-field detector (HAADF-STEM) and inset: the respective size distribution histogram. (C) Emission spectra (λex = 256 nm) of Eu3+/PAA32-b-PVP59 HPICs at different R ratios. (D) Normalized intensities of the 7D0 → 7F2 transition at 615 nm in respect to the Eu3+ concentration as a function of R ratios. (E) ATR-FTIR spectra of PAA32-b-PVP59 (blue line) and of Gd3+/PAA32-b-PVP59 HPICs for different R ratios. (F) 1H NMR spectra of Y3+/PAA32-b-PVP59 HPICs at different R ratios. (G) Evolution of the integral ratio of the protons associated to the carboxylate block, to the ones of PVP block with the R ratio. | ||
Further proofs of the interaction of lanthanides ions with the PAA32-b-PVP59 copolymer, leading to the formation of well-defined objects, were obtained by investigating the luminescence properties of Eu3+ ions and by infrared spectroscopy, respectively. The emission spectra (λex = 256 nm) of Eu3+/PAA32-b-PVP59 HPICs at different R ratios, in aqueous solutions at pH 7 are shown in Fig. 2C.
Strong red emission was detected at 590 nm and 615 nm, corresponding respectively to the 7D0 → 7F1 and 7D0 → 7F2 transitions of Eu3+,43 while no significant intensity was detected in the emission spectrum of an aqueous solution of Eu(NO3)3 with a concentration of Eu3+ similar to the one in HPICs with R = 1. Moreover, the number of water molecules in the first coordination sphere of Eu3+ ions, estimated using fluorescence lifetime measurements and the equation proposed by R. M. Supkowski et al.44 was found around 3.7 ± 0.2 water molecules for all R ratios. This is significantly lower than the 9 expected for the fully hydrated europium salt in water45 (see Table S1†). As the emission of Eu3+ is highly dependent on its coordinated water molecules, due to non-radiatively deactivation of the 7D0 excited state by energy transfer to the OH vibrational modes of coordinated water molecules,43 the enhanced emission of Eu3+/PAA32-b-PVP59 HPICs illustrates that the Eu3+ environment is less hydrated in the presence of the copolymer.46 Coordinated water molecules are replaced by carboxylates units of the copolymer which electrostatically interact with the positively charged Eu3+ ions. Additionally up to R = 1, Eu3+ ions in the HPICs have close coordination environments as shown by the constant values of normalized intensities of the 7D0 → 7F2 transition at 615 nm in respect to the Eu3+ concentration (see Fig. 2D). For R > 1, the decrease of the normalized intensity should correspond to the presence of free (and poorly emitting) europium ions in solution.
The ATR-FTIR spectra of Gd3+/PAA32-b-PVP59 HPICs, measured as powder solids obtained by lyophilization of the solutions, at different R ratios are shown in Fig. 2E. For a better visualization, all spectra were normalized to the absorbance of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O amide stretching band (at 1665 cm−1) of the pyrrolidone ring of the PAA32-b-PVP59 polymer. In the polymer at pH 7, the carboxylate (OCO) asymmetric and symmetric stretching modes appeared at 1562 cm−1 (νas) and 1402 cm−1 (νsym), respectively. Addition of Gd3+ ions shifted the wavenumbers of the two carboxylate νas and νsym bands at 1555 cm−1 and 1363 cm−1, respectively. The progressive increase in intensity of these two bands with increasing amount of Gd3+ ions until R = 1 is especially noteworthy. For higher ratio these intensities remained constant (Fig. S9A†). This behavior is consistent with the progressive neutralization of the negatively charged carboxylate groups of the polymer by the Gd3+ ions until R = 1. For R exceeding the neutralization ratio (R > 1), no additional interactions with carboxylate functions were formed. The difference (noted Δ between the wavenumbers of carboxylate symmetric and asymmetric peaks) is frequently used to determine the type of coordination with the metal center.47,48 For a monodentate bonding, Δ increases compared to the free carboxylate anion; for bidentate chelating coordination Δ decreases and for bidentate bridging, Δ remains similar to the free anion.47 The higher value of Δ = 190 cm−1 in the presence of Gd3+ ions and for all R ratios, compared to the one of the free carboxylate (Δ = 161 cm−1) suggests a monodentate mode coordination of carboxylates of the polymer to the Gd3+ ions. This type of coordination is consistent with the hard acid character of the Gd3+ ions, favoring electrostatic interactions with lack of strong directionality in binding donor groups, in the gadolinium first coordination sphere.49 The complexation between the Gd3+ ions and the carboxylate groups of the PAA block until R = 1, is also evidenced by the progressive shift to lower wavelength, of the characteristic stretching band of the amide CO group of the pyrrolidone ring initially at 1665 cm−1 up to 1658 cm−1. This complexation releases the NVP units of the PVP block from the self-association with the AA units of the PAA block, and thus the characteristic stretching band of the amide CO group shifts to lower wavelength, characteristic of a NVP environment in pure PVP, as observed by comparing the ATR-FTIR spectra of the PAA-b-PVP copolymer and the PVP homopolymer (Fig. S9C†).
O amide stretching band (at 1665 cm−1) of the pyrrolidone ring of the PAA32-b-PVP59 polymer. In the polymer at pH 7, the carboxylate (OCO) asymmetric and symmetric stretching modes appeared at 1562 cm−1 (νas) and 1402 cm−1 (νsym), respectively. Addition of Gd3+ ions shifted the wavenumbers of the two carboxylate νas and νsym bands at 1555 cm−1 and 1363 cm−1, respectively. The progressive increase in intensity of these two bands with increasing amount of Gd3+ ions until R = 1 is especially noteworthy. For higher ratio these intensities remained constant (Fig. S9A†). This behavior is consistent with the progressive neutralization of the negatively charged carboxylate groups of the polymer by the Gd3+ ions until R = 1. For R exceeding the neutralization ratio (R > 1), no additional interactions with carboxylate functions were formed. The difference (noted Δ between the wavenumbers of carboxylate symmetric and asymmetric peaks) is frequently used to determine the type of coordination with the metal center.47,48 For a monodentate bonding, Δ increases compared to the free carboxylate anion; for bidentate chelating coordination Δ decreases and for bidentate bridging, Δ remains similar to the free anion.47 The higher value of Δ = 190 cm−1 in the presence of Gd3+ ions and for all R ratios, compared to the one of the free carboxylate (Δ = 161 cm−1) suggests a monodentate mode coordination of carboxylates of the polymer to the Gd3+ ions. This type of coordination is consistent with the hard acid character of the Gd3+ ions, favoring electrostatic interactions with lack of strong directionality in binding donor groups, in the gadolinium first coordination sphere.49 The complexation between the Gd3+ ions and the carboxylate groups of the PAA block until R = 1, is also evidenced by the progressive shift to lower wavelength, of the characteristic stretching band of the amide CO group of the pyrrolidone ring initially at 1665 cm−1 up to 1658 cm−1. This complexation releases the NVP units of the PVP block from the self-association with the AA units of the PAA block, and thus the characteristic stretching band of the amide CO group shifts to lower wavelength, characteristic of a NVP environment in pure PVP, as observed by comparing the ATR-FTIR spectra of the PAA-b-PVP copolymer and the PVP homopolymer (Fig. S9C†).
To get more information on the formation of HPICs, 1H NMR experiments were performed using Y3+. Indeed, yttrium presents coordination chemistry similar to the one of lanthanide ions and it is not paramagnetic.50 The longitudinal relaxation time (T1) of the protons which can be associated with the mobility of molecular chains was measured (Fig. S10†). The decreasing of the mobility of the PAA chain of the copolymer, due to complexation with the Gd3+ ions was evidenced by the increasing of the relaxation time (T1) of the corresponding protons. The T1 values were further taken into account to acquire 1H NMR spectra in quantitative conditions and to determine the evolution of the integral ratio of the protons associated to the carboxylate block, to the ones of PVP block. As can be seen on Fig. 2F and Fig. S11 and S12,† while the peaks related to PVP block around 3.22, 2.36, 2.22, 1.94 and 1.63 ppm remained roughly constant, the ones associated to the carboxylate block at 1.5 and 2.1 ppm (respectively due to the hydrogen in β and α of the carboxylate function) progressively disappeared when R increased. Using trioxane as a reference, it was evidenced that both PVP and PAA signals decreased with increasing R ratio (Fig. S12A†). However, the protons relative to PAA block were more impacted than the ones of the PVP block (Fig. S12B†). The selective suppression of these peaks suggests the preferential Y3+ binding on PAA residues as expected from the intrinsic chelating capability of acrylates and the rigidification of the PAA block. It confirms the results issued from ATR-FTIR and fluorescence. This might be ascribed to the formation of HPICs with a core–shell structure with a Y3+/PAA core surrounded by a PVP shell. As expected, Eu3+ based HPICs presented similar 1H NMR spectra (Fig. S13†).
The amount of free ions as a function of R was assessed by using inductively coupled plasma – mass spectroscopy (ICP-MS) analysis. Free ions were separated from HPICs by an ultra-filtration process. In the case of Gd3+ based system, free Gd3+ ions were evidenced in solution only for molar ratios R higher than 1. These observations confirm the strong interaction of Gd3+ ions with carboxylate functions and are consistent with the former observations on the interaction of Gd3+ ions with carboxylate groups in PAA3k-b-PEG6k type polymers.7
Data from ATR-FTIR, NMR, DLS, TEM and zeta potential supported the formation of HPICs with Gd3+, Eu3+, Y3+ ions interacting first with the PAA block. When the R ratio is high enough (approximatively above 0.8), these interactions led to the formation of HPICs nano-objects with a core mainly composed of PAA crosslinked by cations surrounded by a shell mainly composed of PVP as depicted in Scheme 1. The formation of hybrid inorganic/polymeric nanoparticles is complete when the R ratio is close to unity. The polymer architecture appears to be essential as the formation of HPICs could not be observed when a statistical copolymer (from commercial source) was used instead of block copolymer (Fig. S14†). The obtained structure is similar in term of architecture and average size to the one obtained with PAA3k-b-PEG6k copolymer.7 Nevertheless, it has to be noted that some differences appear for higher ratios. Indeed, for ratios larger than 2 and on the contrary to what was observed with PAA3k-b-PEG6k copolymers, a slight flocculation was observed and could be related to an undefined mixture of gadolinium hydroxide and polymers. Hence, for ratios R > 1, beyond the composition assuring the neutralization of carboxylates by Gd3+ ions, the infra-red spectra at pH 7 shown clear evidence of the appearance of gadolinium hydroxides (Fig. S9†). The bands at 745 cm−1 and 814 cm−1 are characteristic of the bending vibrations of Gd–O–H (δOH and γOH, respectively).51–56 At 1627 cm−1 the O–H stretching vibration appears as a shoulder to the CO amide band of pyrrolidone ring. Such a phenomenon was not observed for PAA3k-b-PEG6k probably due to the poor interaction of PEG moieties with hydroxides species.7 Interestingly for R < 1 and at pH 7 the complexation of ions with carboxylate prevent the formation of such hydroxides. The ratio R = 1 was chosen for the following experiments.
Colloidal stability of HPICs in biological medium
The colloidal behavior of HPICs (R = 1) was assessed in high ionic strength conditions by addition of NaCl solutions of concentration up to 2 mol L−1. No substantial aggregation was evidenced through DLS measurements up to 1 mol L−1 NaCl (Fig. 3A and Fig. S15†). A slight loss of the scattered light intensity is indicative of the presence of some aggregates beyond 1 mol L−1 NaCl. This result is in good agreement with quasi null zeta potential value indicating that the colloidal stability of these HPICs objects does not rely on electrostatic stability but rather on steric stabilization supported by hydrated PVP chains on the corona of the HPICs architecture. DLS measurements within 2 weeks demonstrated the stability of the HPICs with time, without any release of Gd3+ ions being detected. It is noteworthy the stability of the HPICs down to a pH of 5 as demonstrated by DLS and fluorescence measurements (Fig. S16†), meaning that these objects could be used to image the acidic extracellular environment of tumors. The integrity of HPICs was also maintained with high dilution (up to 50 times) (Fig. 3B and Fig. S17†). Stability over this range of dilution demonstrates that the NPs are not in equilibrium with a large quantity of free copolymers and Gd3+ ions. | ||
| Fig. 3 Assessment of HPICs colloidal stability, cytotoxicity and cell penetration. (A) Colloidal stability of Gd3+ HPICs in high ionic strength conditions as determined by mono-angle DLS measurements: the average diameter of the HPICs remains constant with the addition of the increasing concentrations of NaCl. (B) Evidence of the constant diameter of Gd3+ HPICs with the dilution as measured by mono-angle DLS. The scattered light intensity decreases due to the dilution. The lines are just guide for the eyes. (C) Time-resolved emission spectra (λex = 256 nm) of Eu3+ HPICs in different media (SfCM for serum free culture medium and CCM for complete cell culture medium). (D) Quantification of HPICs cytotoxicity on human colorectal tumor cells HCT-116 after 48 h of treatment. n = 6. Data are represented as mean ± SEM. Statistical differences were analyzed either by 1-way ANONA followed by Dunnett's multiple comparisons post-test for each condition compared to the control one “no treatment” (# symbols, p < 0.05) or by 2-way ANOVA followed by Turkey's post-test to compare conditions with each other (blue or red stars, *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001). (E) Flow cytometry analysis of PAA32-b-PVP59 levels in HCT-116 cells when incubated over 6 h with PAA32-b-PVP59 alone, Eu(NO3)3 and Eu3+ HPICs and for condition of incubation at 4 °C. Statistical analysis by 1-way ANOVA followed by Tukey's multiple comparisons test. Ns = non-significant; **** = p < 0.0001. Data are represented as a mean value ± SEM. (F) ICP-MS analysis quantifying penetration of free Eu3+ and of Eu3+ in PAA32-b-PVP59 HPICs in HCT-116 cells. Statistical analysis by unpaired-t test. * = p < 0.05, **** = p < 0.0001. Data are represented as a mean value ± SEM. | ||
A key issue concerns the stability of HPICs in complete cell culture medium containing 10% of fetal bovine serum (FBS). Nevertheless, attempts to evidence the presence of HPICs by tracking their light scattering failed because of the presence of fetal bovine serum rich in proteins and lipids with considerable sizes. (Fig. S18A†) However, in serum-free cell culture medium (without proteins but rich in inorganic salts such as: carbonate, phosphate, chloride and nitrate) the integrity of the Eu3+/PAA32-b-PVP59 HPICs could be evidenced by DLS (Fig. S18A†). In contrast, Eu(NO3)3 forms big aggregates in this medium57 (Fig. S18B†). To further investigate the HPICs stability in complete cell culture medium, luminescent measurement were performed by using Eu3+ HPICs as a valuable luminescent probe.1,55,56 In order to remove the parasite fluorescence signals mainly arising from red phenol fluorescence present in complete cell culture medium, time-resolve detection experiments were performed.58 As shown in Fig. S18C,† the luminescence intensity of Eu(NO3)3 is significantly increased both in complete cell culture medium and serum free culture medium compared to water, confirming the complexation of Eu3+ ions by the constituents present in these media.57 The luminescence intensity of Eu3+/PAA32-b-PVP59 HPICs is also modified in both media (Fig. 3C). The medium-dependent variation in intensity follows the order: serum-free culture medium, water and complete cell culture medium. Moreover, the ratio of the luminescent intensity of the transition bands 5D0 → 7F2 (615 nm) to 5D0 → 7F1 (590 nm), the first one being solely dependent on the environment, decreases by 22% from 3.2 in water to 2.5 in serum-free culture medium and by 28% in complete cell culture medium.43,56,59 Despite the nearly identical fine structure of the luminescence spectra of Eu3+/PAA32-b-PVP59 HPICs and Eu(NO3)3 in the different media, they do not match at all, demonstrating a different and specific chemical environment of Eu3+ ions in these two samples and the role of the PAA32-b-PVP59 polymer in the protection of Eu3+ ions. In addition, the luminescence intensity of Eu3+/PAA3k-b-PEG6k HPICs in complete cell culture medium is much more affected than Eu3+/PAA32-b-PVP59 HPICs one (see Fig. S18D†). Based on the spectroscopic finding, it could be concluded that the HPICs are affected by the culture medium57 but that HPICs based on PAA-b-PVP are more stable in such complex environment. However, the exact description of the nature of the interaction of HPICs with the serum protein is not trivial and is the scope of a future work.60
HPICs in vitro prevent lanthanides cytotoxicity and cell penetration
Cytotoxicity of the different elements forming the PAA32-b-PVP59 HPICs was assessed in vitro, in complete cell culture medium, on human colorectal tumor cells HCT- 116. Cell viability was measured using PrestoBlue assay. For that purpose, cells were incubated for 48 h with increasing concentrations of polymer PAA32-b-PVP59, Eu(NO3)3 or Gd(NO3)3 aqueous solutions, and Eu3+/PAA32-b-PVP59 HPICs or Gd3+/PAA32-b-PVP59 HPICs (Fig. 3D). Results concerning the polymer PAA32-b-PVP59 show a limited cytotoxicity toward HCT-116, since at the highest concentration (0.1 wt%) at least 80% of cells were viable. This is considerably more than the viability (only 50%) of the HCT-116 cells observed for PAA3k-b-PEG6k copolymer at 0.09 wt%.8 Eu3+ ions from Eu(NO3)3 significantly and statistically affect cell viability, over the whole range of concentrations used. For Gd3+ ions from Gd(NO3)3 cell viability decreases to less than 50% at 0.9 mM. In a remarkable manner, when these ions were encapsulated within the PAA32-b-PVP59 HPICs structure, their cytotoxcity undoubtedly and statistically decreased. Thus, the HPICs were better tolerated than the free Gd3+ or Eu3+ ions especially at the highest concentrations. These results point to the beneficial role of the PAA32-b-PVP59 polymer for decreasing toxicity of the lanthanide ions, even that their environment is affected by the complete cell culture medium as demonstrated above. A similar decrease in cytotoxicity was observed when lanthanide was encapsulated in PAA3k-b-PEG6k polymer.8 The proliferative effect of Eu-HPICs at certain concentrations can be noted, a phenomenon that we are not currently able to explain in terms of the biological mechanisms involved. In order to study the Eu3+/PAA32-b-PVP59 HPICs cell uptake, flow cytometry experiments were performed. For that purpose, HCT-116 cells were incubated for 6 h with 0.07 wt% PAA32-b-PVP59, 0.9 mM Eu(NO3)3 aqueous solution and the corresponding Eu3+/PAA32-b-PVP59 HPICs (Fig. 3E). As expected, no fluorescence coming from Eu3+ was detected due to technical limitations of the conventional, dedicated to biology cytometer, i.e. excitation laser and simultaneous fluorescence detection was clearly not optimal for Eu3+ excitation.61However, fluorescence signal (Excitation = 405 nm, Emission Max = 711 nm) was detected for the samples incubated with PAA32-b-PVP59 polymer, due to autofluorescence of the PVP block of the polymer.62 This represents a real advantage compared to the non autofluorescent PAA3k-b-PEG6k copolymer, thus avoiding the use of additional fluorescent dyes. It was found that the percentage of cell labelling is low. 12% of the cell population was labelled in the presence of PAA32-b-PVP59 copolymer and twice less, approximately 7% in the presence of Eu3+/PAA32-b-PVP59 HPICs.
Interestingly, when incubation with copolymer was performed at 4 °C, no fluorescent signal was observed within the cells, meaning that endocytosis is a major actor in the process of internalization of the copolymer. Complementary, for the quantification of europium inductively coupled plasma-mass spectrometry (ICP-MS) was used. The quantity of Eu3+ ions, when the cells were incubated with 0.9 mM of Eu(NO3)3, amounts to about 23.6 nmol Eu3+/20 × 106 cells after 6 h of exposure, comparable with the value reported for Eu3+ ions uptake into FaDu cells.57 (Fig. 3F) The amount of Eu3+ ions decreases at 0.5 nmol Eu3+/20 × 106 cells, when the cells were incubated with Eu3+/PAA32-b-PVP59 HPICs containing 0.9 mM of Eu3+ ions. This is in agreement with the low percentage of labelling found by flow cytometry experiments. Furthermore, these results correlate to the cell viability data. Thus our results underline that HPICs organization prevents lanthanide ions and polymer cytotoxicity and cell penetration compared to their free counterparts, mainly by increasing architecture stability of the nanocarrier.
Gd3+ HPICs demonstrate increased r1 relaxivity and enhanced MRI signal on mice
The relaxivity of both types of Gd3+ HPICs, formulated either with PAA32-b-PVP59 or PAA3k-b-PEG6k copolymers, was determined in the absence and presence of complete cell culture medium (Fig. S19†). In water, the r1 relaxivity of Gd3+/PAA32-b-PVP59 HPICs was measured to be 42 ± 0.36 mM−1 s−1 (25 °C, 0.47T) very close to the one measured for Gd3+/PAA3k-b-PEG6k HPICs in the same conditions i.e. 33 ± 0.36 mM−1 s−1 (whereas a value equal to 48 ± 2 mM−1 s−1 was measured at 1.4T).7 However, when mixed with the complete cell culture medium the r1 relaxivity falls at 12 ± 1.02 mM−1 s−1 and 11 ± 0.85 mM−1 s−1 for Gd3+/PAA32-b-PVP59 and Gd3+/PAA3k-b-PEG6k HPICs, respectively (Table 2). This behavior is consistent with the modifications of the HPICs in complete cell culture medium as suggested by luminescence experiments. But still, the values are larger than the values displayed by commercial molecular complexes which are lower than 6 mM−1 s−1 in human plasma at 37 °C and 0.47T.63| Medium | Relaxivity r1 (mM−1 s−1) | ||
|---|---|---|---|
| Gd3+/PAA32-b-PVP59 | Gd3+/PAA3k-b-PEG6k | Gd3+ | |
| Water | 42 ± 0.36 | 33 ± 0.36 | 13 ± 0.96 |
| Complete cell culture medium | 12 ± 1.02 | 11 ± 0.85 | 6.4 ± 0.31 |
Similar values of r1 relaxivity were measured on micellar systems based on Gd chelates64,65 or on compounds based on other paramagnetic ions such as Mn2+ or Fe3+.66,67 However, their fabrication is less straightforward than the system that we propose which in comparison is very simple and avoid the use of specific ligands and additional chemicals.
The insignificant cytotoxicity of the Gd3+/PAA32-b-PVP59 HPICs up to 1.3 mM Gd (0.1 wt% of polymer) allowed one to perform in vivo experiments on mice and to obtain preliminary determinations of MR contrast efficacy, pharmacokinetic properties, and tolerance. The in vivo MR contrast was assessed after intravenous (IV) bolus injection of Gd3+/PAA32-b-PVP59 HPICs and compared to the use of GdDOTA with an equivalent Gd concentration. Tissue uptake and elimination properties were evaluated using a T1-weighted dynamic sequence of coronal images centered on the abdominal cavity, acquired during 30 min after IV injection of Gd3+/PAA32-b-PVP59 HPICs or GdDOTA at 15 μmol kg−1 equivalent Gd concentration. In Fig. 4A, regions of interest are given corresponding to the renal cortex of both kidneys and the bladder. Comparatively to GdDOTA, Gd3+/PAA32-b-PVP59 HPICs significantly increased contrast in all the studied organs: an enhanced contrast of about +67% was measured in T1-weighted images of the intravascular space for Gd3+/PAA32-b-PVP59 HPICs after 30 minutes (1% for GdDOTA, Fig. 4A and B) and showed similar trends than the ones previously measured with Gd3+/PAA3k-b-PEG6k HPICs.7 In addition, elimination process for the Gd3+/PAA32-b-PVP59 HPICs was followed by monitoring signal enhancement vs. time in the intravascular space, the kidney and the bladder.(Fig. 4B) Whereas GdDOTA was quickly eliminated from the blood circulation as assessed by the high signal intensity in the bladder after 30 min, Gd3+/PAA32-b-PVP59 HPICs residence time was relatively long showing a persistent enhancement of the vascular signal. Moreover, direct injection in pelvis enabled to estimate the in vivo colloidal stability of Gd3+/PAA32-b-PVP59 HPICs (Fig. 4C). Indeed, whereas GdDOTA, slowly diffuse out of the peritoneal cavity, contrast enhancement remained confined to the intraperitoneal cavity for the Gd3+/PAA32-b-PVP59 HPICs indicating the non-penetration of the pelvic barrier of the HPICs and no leak of the Gd3+ ions out of the HPICS. Quantitative biokinetic studies are still needed, but these preliminary studies of a relatively simple HPIC, apparently not toxic to mice, showed surprisingly good stability and superior magnetic relaxivity properties in vitro and in vivo, even at high magnetic field.
 | ||
| Fig. 4 Assessment of MR contrast efficacy on mice. (A) Coronal T1-weighted images 30 min after injection of GdDOTA (middle), Gd3+/PAA32-b-PVP59 HPICs (bottom) in comparison with a control (top). The images were centered on the kidneys (left) and bladder (right). (B) Typical evolution of the MRI signal following the administration (arrow) of GdDOTA (blue) or Gd3+/PAA32-b-PVP59 HPICs (black) from the vascular space (rectangular shaped ROI taken in the slice at the level of the inferior vena cava, the kidney (ellipse shaped ROI taken in a horizontal image covering all the right kidney) and in the Bladder (circular shaped ROI taken in an horizontal image covering all the Bladder). A.U. (arbitrary unit of the MRI signal expressed as mean value ± SD measured in ROIs). (C). Horizontal slices of the abdomen acquired after 40 min following the intraperitoneal administration of Gd3+/PAA32-b-PVP59 HPICs. Cut at the kidney level on the left and at the level of the bladder on the right. | ||
Conclusions
In this work, we demonstrated the pertinence of using a double hydrophilic block copolymer including a PVP block as alternative to the frequently used PEG one, for the elaboration of lanthanide HPICs. For the first time, lanthanide ions were associated to a PAA-b-PVP block copolymer. This seldomly reported combination of blocks was successfully achieved through a straightforward methodology based on RAFT polymerization. It was demonstrated that these new HPICs showed similar well-defined architectures, size and size distribution as the analogues based on PAA-b-PEG copolymer. The PAA-b-PVP copolymer had low toxicity alone and effectively contributed to the decrease of the toxicity of the lanthanide ions when they were embedded into the HPICs. Despite clear evidence of interaction with the serum proteins from the complete cell culture medium, which partially alters the fluorescence and relaxivity properties, the composition of the HPICs seems to be still mostly unaffected which is not the case of the PAA-b-PEG analog. This is supported in vitro by a low toxicity and low cell penetration of the HPICs compared to their individual constituents and in vivo by the impossibility of these HPICs to penetrate the pelvic barrier of mice, with no leak of the Gd3+ ions as demonstrating by monitoring the magnetic resonance contrast of the peritoneal cavity.Finally, the PAA-b-PVP HPICs showed enhanced T1-MRI contrast compared to the GdDOTA (of about +67% measured in T1-weighted images of the intravascular space after 30 minutes after intravenous injection into mice) and equivalent relaxivity efficiency compared to the PAA-b-PEG analogues, thus contributing as a new interesting probe to be used in MRI.
This work paves the way to future studies aiming at developing original HPICs based on new architectures of AA/VP copolymers, supported by the versatility of xanthate-mediated RAFT polymerization.
Experimental methods
Chemicals
(2,2′-Azobis(4-methoxy-2,4-dimethylvaleronitrile) (V-70) (Wako Chemicals), 1,3,5-trioxane (≥99%) (Sigma Aldrich), diethyl ether and absolute ethanol (VWR), N,N-dimethylformamide anhydrous (99.8%) (Sigma Aldrich), potassium hydroxide (KOH) (Sigma Aldrich), Gd(NO3)3·6H2O (99.99% trace metals basis) (Sigma Aldrich), Eu(NO3)3·5H2O, (99.9% trace metals basis) (Sigma Aldrich), Y(NO3)3·6H2O (99.8% trace metals basis) (Sigma Aldrich), NaCl, ACS, (99.0% min.) (Alfa Aesar), were used as received. 1,4-Dioxane (≥99.0%) (VWR) and methyl acrylate (99%, contains ≤100 ppm monomethyl ether hydroquinone as inhibitor) (Sigma Aldrich) were filtrated on alumina. 1-Vinyl-2-pyrrolidone (NVP) (purum, ≥97.0% (GC)) (Sigma Aldrich) was distilled prior to use. 2,2′-Azobis(2-methylpropionitrile) (98%) (AIBN) (Sigma Aldrich) was recrystallized from methanol and dried at room temperature under vacuum. The methyl 2((ethoxycarbonothioyl)thio)-propanoate RAFT agent (XA1) was synthesized according to a previously published procedure.68 The statistical PAA416-r-PVP270 copolymer was purchased from Polymer Source.Polymer synthesis
![[H with combining low line]](https://www.rsc.org/images/entities/b_char_0048_0332.gif) 2–CH(CO2CH3)–S(CS)–OEt), 1.3–1.6 (3H, C3–CH2–O– end-group), 1.6–1.8 (2(n − 1)H, –C2–CH(CO2CH3)–), 2.25–2.5 (1(n − 1)H, –CH2–C(CO2CH3)–, 3.7 (3(n + 1)H, –CO2C3), 4.4 (1H, –CH2–C(CO2CH3)–S(CS)–OEt), 4.7 (2H, CH3–C2–O– end-group). The degree of polymerization (DPn) was calculated by as presented in Fig. S1.†Mn,th = 2750 g mol−1. Mn,NMR = 2750 g mol−1; ATR-FTIR spectroscopy: ν(CO) ester group = 1736 cm−1.
2–CH(CO2CH3)–S(CS)–OEt), 1.3–1.6 (3H, C3–CH2–O– end-group), 1.6–1.8 (2(n − 1)H, –C2–CH(CO2CH3)–), 2.25–2.5 (1(n − 1)H, –CH2–C(CO2CH3)–, 3.7 (3(n + 1)H, –CO2C3), 4.4 (1H, –CH2–C(CO2CH3)–S(CS)–OEt), 4.7 (2H, CH3–C2–O– end-group). The degree of polymerization (DPn) was calculated by as presented in Fig. S1.†Mn,th = 2750 g mol−1. Mn,NMR = 2750 g mol−1; ATR-FTIR spectroscopy: ν(CO) ester group = 1736 cm−1.
Step 2: Block copolymerization of 1-vinyl-2-pyrrolidone (NVP) in 1,4-dioxane. The macro-RAFT agent (PMA-XA1) from step 1 (2 g, 7 × 10−3 mol), NVP (3.8 g, 3.4 × 10−2 mol), 1,4-dioxane (3.5 g, 4 × 10−2 mol), V-70 (46 mg, 1.5 × 10−4 mol) and 1,3,5-trioxane (0.1 g, 1.1 × 10−3 mol) were introduced at room temperature in a Schlenk flask. The reaction mixture was degassed by purging with argon for 30 min, heated and stirred at 35 °C for 12 hours. The reaction mixture was then cooled down at room temperature and a sample was withdrawn to measure NVP conversion (97%) and calculate the DPn of the PVP block by 1H NMR analysis (Fig. S2†): (300 MHz in CDCl3) δ ppm: 1.20 (3H, C3–CH2–O– end-group), 1.30–1.5 (2mH, –N–CH2–C2–CH2–CO(N)), 1.6–1.8 (2nH, –C2–CH(CO2CH3)–), 2.0–2.18 (2mH, –C2–CH(NCH2CH2CH2CO(N))–), 2.18–2.25 (1nH, –CH2–C(CO2CH3)), 2.25–2.5 (2mH, –N–CH2–CH2–C2–CO(N)), 3–3.5 (2mH, –N–C2–CH2–CH2–CO(N)), 3.7 (3(n + 1)H, –CO2C3), 3.8–4.0 (1mH, –CH2–C(NCH2CH2CH2CO(N))–), 4.6 (2H, CH3–C2–O– end-group) (Mn,th = 8300 g mol−1. Mn,NMR = 9300 g mol−1. The final composition was established as PMA32-b-PVP59. ATR-FTIR spectroscopy: ν(CO) ester group = 1736 cm−1 and ν(CO) PVP ring = 1674 cm−1. For the purification, the crude reaction solution was precipitated in diethyl ether, filtrated and dried in a vacuum oven over night to remove solvent traces.
Step 3: Hydrolysis of PMA-b-PVP to form PAA-b-PVP. The PMA-b-PVP copolymer obtained in step 2 (4.5 g, 5.4 × 10−4 mol), ethanol (15.8 g, 0.34 mol) and potassium hydroxide (KOH) (9.5 g, 0.17 mol) were introduced at room temperature in a 50 ml round-bottom flask. The reaction mixture was refluxed (60 °C) for 12 hours, cooled down at room temperature and concentrated under vacuum. The residue was introduced in a dialysis membrane with a Molecular Weight Cut-Off (MWCO) of 3.5 kDa and dialyzed in pure MiliQ water in a 2L beaker for 2 days. Finally, the aqueous PAA-b-PVP solution was lyophilized to obtain PAA-b-PVP as a powder. This final product was characterized by 1H NMR (300 MHz in D2O) δ ppm (Fig. S3†): range 1.4–1.53 (2nH, –C2–CH(COOH)–), 1.6–1.75 (2mH, –N–CH2–C2–CH2–CO(N)), 1.82–2.05 (2mH, –C2–CH(NCH2CH2CH2CO(N))–), 2.07–2.19 (1nH, –CH2–C(COOH)–), 2.2–2.5 (2mH, –N–CH2–CH2–C2–CO(N)), 3.0–3.35 (2mH, –N–C2–CH2–CH2–CO(N)), 3.4–3.8 (1mH, –CH2–C(NCH2CH2CH2CO(N))–) and by ATR-FTIR spectroscopy: ν(CO) PVP ring = 1674 cm−1 and νas(OCO) = 1564 cm−1 and νsym(OCO) = 1402 cm−1.
HPICs preparation
0.5 wt% solution of PAA32-b-PVP59 were prepared by dissolution of the necessary quantity of the copolymer in Milli-Q water (18.2 mΩ). Gd3+, Eu3+ and Y3+ stock solutions (65 mM) were prepared by dissolution of Gd(NO3)3·6H2O, Eu(NO3)3·5H2O and Y(NO3)3·6H2O, respectively in Milli-Q water. HPICs were formed by mixing solutions of PAA32-b-PVP59 (0.5 wt%) with solutions of Gd3+, Eu3+ and Y3+, respectively. Lanthanide ions’ concentrations were adjusted in order to vary the molar fraction R = 3·[Ln3+]/[AA] between the positive charges of lanthanide ions and the negative ones of the polymers between 0.2 and 2.2 within the HPICs. After mixing, the pH of the solutions was adjusted to 6.8–7.Experimental characterization
1 H NMR experiments were performed on a 300 MHz Bruker spectrometer using CDCl3 and D2O as solvent at room temperature. Chemical shifts were reported in ppm.1 H NMR experiments-Y 3+-titration were performed on a Bruker Avance 600 MHz spectrometer equipped with a 5 mm triple resonance inverse Z-gradient probe (TBI 1H, 31P, BB). The sample were prepared in a mixture of 10% vol. D2O and 90% vol. H2O and the pH was adjusted to 7. The temperature was set to 25 °C.
1H spin–lattice relaxation times (T1) were measured using the excitation sculpting inversion-recovery pulse sequence t1iresgp. The relaxation delay was 6 s and the acquisition time was 1 s. For each measurement, the recovery times were from 50 ms to 6 s and 8 points were collected.
Size exclusion chromatography (SEC) measurements were performed with a system composed of an Agilent technologies guard column (PLGel20 μm, 50 × 7.5 mm) and a set of three columns (TSKgel Alpha-2500, Alpha-3000, Alpha-3500 TOSOH BIOSCIENCE). Detections were realized using a Varian Prostar UV detector (dual wavelength analysis at 290 and 254 nm), a MiniDawn TREOS multi-angle light scattering detector (Wyatt Technology Corporation) and a Wyatt Optilab® rEX refractive index detector. DMF LiBr (10 mM) was used as eluent at 50 °C (flow rate, 1 mL min−1). Prior to injections, the samples were prepared at a concentration of 10 mg mL−1 and filtered through a 0.45 μm PTFE filter. Recorded data were treated using the Wyatt Astra 7.1 software. The dn/dc value for the PMA-XA1 was found to be 0.0558 and for the block copolymer 0.0799 mL g-1. This last one was calculated by using the dn/dc value of the PVP of 0.093 mL g-1.
Mono-angle dynamic light scattering and zeta potential measurements were realized using a Zetasizer Nano-ZS (Malvern Instruments, Ltd, UK) with integrated 4 mW He–Ne laser, λ = 633 nm. Light scattering intensity (at 173°) was measured using the same recording parameters for all the samples. In order to obtain Z-average sizes of the colloidal structures, the correlation functions were analyzed via the cumulant method. For each sample, 5 series of 10 recorded signals of 10 seconds were realized for size data records and only 3 series for zeta potential data, at a controlled temperature of 25 °C.
Multi-angle dynamic light scattering experiments were conducted on a 3D LS spectrometer from LS instruments (Switzerland) at 20 °C. Working with a laser at 660 nm, this instrument recorded light scattering for scattering angles between 15 and 150°. Typical acquisition time for each angle was set to 30 s. All data were then analyzed using a laboratory made software program (named M-STORMS).69 Cumulant or NNLS method led to an estimation of the average decay rate Γ for each angle. The linear dependency of Γ versus the square of the scattering vector q (Γ = q2·D) gives the diffusion coefficient of the nano-objects and therefore their radius through the Stokes–Einstein equation:  where T is the temperature and η the viscosity of the solution (taken as the one of pure water).
where T is the temperature and η the viscosity of the solution (taken as the one of pure water).
exp(−t/τ) by using Origin curve-fitting program, where I(t) is the total luminescence intensity at time t, I(0) the luminescence intensity at t = 0 ms, and τ is the corresponding lifetime.
Biological in vitro assays
000 HCT-116 were seeded in 96-well plates. The day of experiment, cells were incubated for 48 h at 37 °C with increasing concentration of Gd3+ and Eu3+ HPICs and its individual components, respectively, PAA-b-PVP polymer and Gd(NO3)3 or Eu(NO3)3 aqueous solutions, at similar concentrations as the ones tested for HPICs. Cell viability was quantified using PrestoBlue reagent (Fisher scientific) according to the manufacturer's instructions. Briefly, after 48 h of incubation, cell culture medium was removed and cells were incubated for 30 min at 37 °C with 100 μl of 1× PrestoBlue reagent diluted in PBS, before reading absorbance on a plate reader at 570 nm and 600 nm Synergy H1 (Biotek, Winooski, VT, USA). Six biological replicates were produced and analyzed for each condition. Data analysis was performed using GraphPad Prism 8 program (GraphPad Software, Inc., La Jolla, CA, USA) and data were expressed as mean ± SEM. Statistical comparisons were performed using one-way analysis of variance (ANOVA) followed by Dunnett's post-test in comparison to the control condition. *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001.
000 HCT-116 were seeded in 12-well plates. Cells were incubated for 6 hours with cell culture medium containing each component of the HPICs, respectively PAA-b-PVP polymer alone (0.07 wt%), or Eu(NO3)3 aqueous solution (0.9 mM) or corresponding Eu-HPICs. An additional set of experiments was performed at 4 °C instead or 37 °C for PAA-b-PVP polymer alone (0.07 wt%) condition. After incubation, cells were washed once with PBS and then cells were trypsinized and transferred on ice within a U-bottom 96-well plate. Cells were analyzed with a BD LSR Fortessa flow cytometer equipped with an HTS module for plate reading, using the 405 nm laser for excitation and fluorescence emission was collected using the BV 711A filter, which allowed the detection of the maximum of signal. At least 20000 events were acquired in each well. Data were analyzed with FlowJo v10 (FlowJo). Three biological replicates were produced and analyzed for each condition. Statistical comparisons were performed using one-way analysis of variance (ANOVA) followed by Tukey's multiple comparison post-test. *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001.
In vivo experiments
All in vivo experimental procedures were approved by our institutional animal care and use committee CEEA122 (APAFIS 5192-2016041911336422 and 34703-2022011811542488) and conducted in compliance with the Ethics Committee pursuant to European legislation translated into French Law as Decret 2013-118 dated 1st of February 2013.
MR image acquisitions were performed on a Biospec 7T dedicated to small animals (Bruker, Wissenbourg, France). Acquisitions of the abdominal images were carried out with a 40 mm transmit-receive volume coil and triggered on breathing to reduce motion artifacts. T1 weighted images were acquired using Flash sequence with the following parameters: TR = 220 ms; TE = 3.5 ms; flip angle: 40°; number of average: 4; FOV: 40 × 40 mm; resolution 200 × 200 μm; 13 slices of 1 mm thickness; fat suppression; acquisition time: 2 min.
Author contributions
Marjorie Yon: Investigation and writing (supporting); Laure Gibot: investigation, conceptualization (supporting) and writing (supporting); Stéphane Gineste: investigation; Pascale Laborie: investigation; Christian Bijani: investigation; Christophe Mingotaud: validation and methodology and writing (supporting); Olivier Coutelier: validation, supervision (supporting) and writing (supporting); Franck Desmoulin: in vivo investigation (lead) and writing (supporting); Carine Pestourie: in vivo investigation (supporting); Mathias Destarac: validation and writing (supporting); Diana Ciuculescu-Pradines: conceptualization (equal), methodology (equal), project administration (equal), supervision (equal), writing (equal); Jean-Daniel Marty: conceptualization (equal), methodology (equal), project administration (equal), supervision (equal), writing (equal).Data availability statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the Imaging Core Facility TRI-IPBS, (Toulouse) and more precisely Penelope Viana for her help with the flow cytometry experiments, the CMEAB platform, (Toulouse) for TEM facilities, Laurent Weingarten for the HAADF-STEM analysis and the Raimond Castaing microcharacterization platform facilities, (Toulouse), Technopolym service and ICT (Institut de Chimie de Toulouse) (ICT-UAR 2599) https://www.ict.ups-tlse.fr for the Size Exclusion Chromatography analysis, 300 MHz NMR and IR facilities, LSPCMIB laboratory (Toulouse) for the access to the luminescent spectroscopy facilities, LCC laboratory (Toulouse) for the 600 MHz NMR facilities, Chantal Galaup and Marc Guerre for the fruitful discussions, the doctoral school Science de la Matiere de Toulouse, the Ministère de l'Enseignement Supérieur et de la Recherche and Toulouse Tech Transfer for the financial support.References
- S. S. Syamchand and G. Sony, J. Lumin., 2015, 165, 190–215 CrossRef CAS.
- J. Wahsner, E. M. Gale, A. Rodríguez-Rodríguez and P. Caravan, Chem. Rev., 2019, 119, 957–1057 CrossRef CAS PubMed.
- A. El Jundi, S. J. Buwalda, Y. Bakkour, X. Garric and B. Nottelet, Adv. Colloid Interface Sci., 2020, 283, 102213 CrossRef CAS PubMed.
- A. Harada and K. Kataoka, Polym. J., 2018, 50, 95–100 CrossRef CAS.
- B. V. K. J. Schmidt, Macromol. Chem. Phys., 2018, 219, 1700494 CrossRef.
- D. Taton, A.-Z. Wilczewska and M. Destarac, Macromol. Rapid Commun., 2001, 22, 1497–1503 CrossRef CAS.
- C. Frangville, Y. Li, C. Billotey, D. R. Talham, J. Taleb, P. Roux, J.-D. Marty and C. Mingotaud, Nano Lett., 2016, 16, 4069–4073 CrossRef CAS PubMed.
- M. Yon, S. Gineste, G. Parigi, B. Lonetti, L. Gibot, D. R. Talham, J.-D. Marty and C. Mingotaud, ACS Appl. Nano Mater., 2021, 4, 4974–4982 CrossRef CAS.
- N. M. Pinkerton, L. Behar, K. Hadri, B. Amouroux, C. Mingotaud, D. R. Talham, S. Chassaing and J.-D. Marty, Nanoscale, 2017, 9, 1403–1408 RSC.
- H.-W. Shin, H. Sohn, Y.-H. Jeong and S.-M. Lee, Langmuir, 2019, 35, 6421–6428 CrossRef CAS PubMed.
- Y.-H. Jeong, H.-W. Shin, J.-Y. Kwon and S.-M. Lee, ACS Appl. Mater. Interfaces, 2018, 10, 23617–23629 CrossRef CAS PubMed.
- E. Seo, J. Kim, Y. Hong, Y. S. Kim, D. Lee and B.-S. Kim, J. Phys. Chem. C, 2013, 117, 11686–11693 CrossRef CAS.
- H. J. Lee, S. E. Kim, I. K. Kwon, C. Park, C. Kim, J. Yang and S. C. Lee, Chem. Commun., 2010, 46, 377–379 RSC.
- S. Tanaka, J. Lin, Y. V. Kaneti, S. Yusa, Y. Jikihara, T. Nakayama, M. B. Zakaria, A. A. Alshehri, J. You, Md. S. A. Hossain and Y. Yamauchi, Nanoscale, 2018, 10, 4779–4785 RSC.
- S. Gineste, B. Lonetti, M. Yon, J. Giermanska, E. Di Cola, M. Sztucki, Y. Coppel, A. F. Mingotaud, J. P. Chapel, J. D. Marty and C. Mingotaud, J. Colloid Interface Sci., 2022, 9, 698 CrossRef.
- X. Zhang, K. Wang, M. Liu, X. Zhang, L. Tao, Y. Chen and Y. Wei, Nanoscale, 2015, 7, 11486–11508 RSC.
- K. Zhang, M. C.-L. Yeung, S. Y.-L. Leung and V. W.-W. Yam, Chem, 2017, 2, 825–839 CAS.
- G. Pound, F. Aguesse, J. B. McLeary, R. F. M. Lange and B. Klumperman, Macromolecules, 2007, 40, 8861–8871 CrossRef CAS.
- K. H. Markiewicz, L. Seiler, I. Misztalewska, K. Winkler, S. Harrisson, A. Z. Wilczewska, M. Destarac and J.-D. Marty, Polym. Chem., 2016, 7, 6391–6399 RSC.
- M. Mohamed, A. S. Abu Lila, T. Shimizu, E. Alaaeldin, A. Hussein, H. A. Sarhan, J. Szbeni and T. Ishida, Sci. Technol. Adv. Mater., 2019, 20, 710 CrossRef CAS PubMed.
- K. Knop, R. Hoogenboom, D. Fischer and U. S. Schubert, Angew. Chem., Int. Ed., 2010, 49, 6288 CrossRef CAS PubMed.
- M. C. Castells and E. J. Phillips, N. Engl. J. Med., 2021, 384(7), 643 CrossRef CAS PubMed.
- L. Luo, M. Ranger, D. G. Lessard, D. Le Garrec, S. Gori, J.-C. Leroux, S. Rimmer and D. Smith, Macromolecules, 2004, 37, 4008–4013 CrossRef CAS.
- Y. Luo, Y. Hong, L. Shen, F. Wu and X. Lin, AAPS PharmSciTech, 2021, 22, 34 CrossRef CAS PubMed.
- A. Benahmed, M. Ranger and J. Leroux, Pharm. Res., 2001, 18, 323–328 CrossRef CAS PubMed.
- T. Song, F. Gao, S. Guo, Y. Zhang, S. Li, H. You and Y. Du, Nanoscale, 2021, 13, 3895–3910 RSC.
- H. A. Ravin, A. M. Seligman and J. Fine, N. Engl. J. Med., 1952, 247, 921–929 CrossRef CAS.
- J. A. Brydson, Plast. Mater, Elsevier, 1999, pp. 466–477 Search PubMed.
- V. P. Torchilin, J. Microencapsulation, 1998, 15, 1 CrossRef CAS PubMed.
- S. Aroua, E. G. V. Tiu, M. Ayer, T. Ishikawa and Y. Yamakoshi, Polym. Chem., 2015, 6, 2616–2619 RSC.
- M. A. de Farias and M. do C. Gonçalves, Polímeros, 2016, 26, 1–10 CrossRef CAS.
- T. M. Amis, J. Renukuntla, P. K. Bolla and B. A. Clark, Pharmaceutics, 2020, 12, 892 CrossRef CAS PubMed.
- M.-K. Chun, C.-S. Cho and H.-K. Choi, J. Appl. Polym. Sci., 2004, 94, 2390–2394 CrossRef CAS.
- M.-K. Chun, C.-S. Cho and H.-K. Choi, J. Controlled Release, 2002, 81, 327–334 CrossRef CAS PubMed.
- Z. S. Nurkeeva, V. V. Khutoryanskiy, G. A. Mun, A. B. Bitekenova, S. Kadlubowski, Y. A. Shilina, P. Ulanski and J. M. Rosiak, Colloids Surf., A, 2004, 236, 141–146 CrossRef CAS.
- T. Swift, C. C. Seaton and S. Rimmer, Soft Matter, 2017, 13, 8736–8744 RSC.
- R. Liang, S. You, L. Ma, C. Li, R. Tian, M. Wei, D. Yan, M. Yin, W. Yang, D. G. Evans and X. Duan, Chem. Sci., 2015, 6, 5511–5518 RSC.
- A. Guinaudeau, O. Coutelier, A. Sandeau, S. Mazières, H. D. Nguyen Thi, V. Le Drogo, D. J. Wilson and M. Destarac, Macromolecules, 2014, 47, 41–50 CrossRef CAS.
- M. Jacquin, P. Muller, G. Lizarraga, C. Bauer, H. Cottet and O. Théodoly, Macromolecules, 2007, 40, 2672–2682 CrossRef CAS.
- M. Mestivier, J. R. Li, A. Camy, C. Frangville, C. Mingotaud, F. Benoît-Marquié and J. Marty, Chem. – Eur. J., 2020, 26, 14152–14158 CrossRef CAS PubMed.
- J. Loos, E. Sourty, K. Lu, G. de With and S. v. Bavel, Macromolecules, 2009, 42, 2581–2586 CrossRef CAS.
- S. Mourdikoudis, R. M. Pallares and N. T. K. Thanh, Nanoscale, 2018, 10, 12871–12934 RSC.
- K. Binnemans, Coord. Chem. Rev., 2015, 295, 1–45 CrossRef CAS.
- R. M. Supkowski and W. DeW. Horrocks, Inorg. Chim. Acta, 2002, 340, 44–48 CrossRef CAS.
- S. A. Cotton and P. R. Raithby, Coord. Chem. Rev., 2017, 340, 220–231 CrossRef CAS.
- J. Zhang, Y. Liu, Y. Li, H. Zhao and X. Wan, Angew. Chem., 2012, 124, 4676–4680 CrossRef.
- G. Deacon, Coord. Chem. Rev., 1980, 33, 227–250 CrossRef CAS.
- C. C. R. Sutton, G. da Silva and G. V. Franks, Chem. – Eur. J., 2015, 21, 6801–6805 CrossRef CAS PubMed.
- H. G. Brittain, F. S. Richardson and R. B. Martin, J. Am. Chem. Soc., 1976, 98, 8255–8260 CrossRef CAS PubMed.
- S. A. Cotton, in EIBC, ed. R. A. Scott, John Wiley & Sons, Ltd, Chichester, UK, 2011, p. eibc0195 Search PubMed.
- S. Majeed and S. A. Shivashankar, J. Mater. Chem. C, 2014, 2, 2965 RSC.
- F. Chen, X. H. Zhang, X. D. Hu, W. Zhang, R. Zeng, P. D. Liu and H. Q. Zhang, J. Alloys Compd., 2016, 664, 311–316 CrossRef CAS.
- J.-G. Kang, B.-K. Min and Y. Sohn, Ceram. Int., 2015, 41, 1243–1248 CrossRef CAS.
- Y. Jia, T. Luo, X.-Y. Yu, B. Sun, J.-H. Liu and X.-J. Huang, RSC Adv., 2013, 3, 15805 RSC.
- J. Zhang, L. Dai, A. M. Webster, W. T. K. Chan, L. E. Mackenzie, R. Pal, S. L. Cobb and G. Law, Angew. Chem., Int. Ed., 2021, 60, 1004–1010 CrossRef CAS PubMed.
- J. Yu, D. Parker, R. Pal, R. A. Poole and M. J. Cann, J. Am. Chem. Soc., 2006, 128, 2294–2299 CrossRef CAS PubMed.
- S. Sachs, A. Heller, S. Weiss, F. Bok and G. Bernhard, Toxicol. In Vitro, 2015, 29(7), 1555 CrossRef CAS PubMed.
- M. Sy, A. Nonat, N. Hildebrandt and L. J. Charbonnière, Chem. Commun., 2016, 52, 5080–5095 RSC.
- D. Parker and J. Yu, Chem. Commun., 2005, 3141 RSC.
- C. De Rosa, A. Melchior, M. Sanadar, M. Tolazzi, A. Giorgetti, R. P. Ribeiro, C. Nardon and F. Piccinelli, Inorg. Chem., 2020, 59, 12564–12577 CrossRef CAS PubMed.
- M. A. Condrau, R. A. Schwendener, P. Niederer and M. Anliker, Cytometry, 1994, 16, 187–194 CrossRef CAS PubMed.
- G. Song, Y. Lin, Z. Zhu, H. Zheng, J. Qiao, C. He and H. Wang, Macromol. Rapid Commun., 2015, 36, 278–285 CrossRef CAS PubMed.
- M. Rohrer, H. Bauer, J. Mintorovitch, M. Requardt and H.-J. Weinmann, Invest. Radiol., 2005, 40, 715 CrossRef PubMed.
- G. Zhang, R. Zhang, X. Wen, L. Li and C. Li, Biomacromolecules, 2008, 9, 36–42 CrossRef CAS PubMed.
- P. Mi, H. Cabral, D. Kokuryo, M. Rafi, Y. Terada, I. Aoki, T. Saga, I. Takehiko, N. Nishiyama and K. Kataoka, Biomaterials, 2013, 34, 492–500 CrossRef CAS PubMed.
- Z. Mo, Q. Li, K. Zhao, Q. Xu, H. Hu, X. Chen, Y. Luo, B. Chi, L. Liu, X. Fang, G. Liao, Z. Xu, J. Wang and S. Yang, ACS Appl. Mater. Interfaces, 2022, 14(8), 10001–10014 CrossRef CAS PubMed.
- Z. Mo, M. Qiu, K. Zhao, H. Hu, Q. Xu, J. Cao, Y. Luo, L. Liu, Z. Xu, C. Yi, Z. Xiong, G. Liao and S. Yang, J. Colloid Interface Sci., 2022, 611, 193–204 CrossRef CAS PubMed.
- X. Liu, O. Coutelier, S. Harrison, T. Tassaing, J.-D. Marty and M. Destarac, ACS Macro Lett., 2015, 4, 89 CrossRef CAS PubMed.
- M. Dionzou, A. Morère, C. Roux, B. Lonetti, J. D. Marty, C. Mingotaud, P. Joseph, D. Goudounèche, B. Payré, M. Léonetti and A. F. Mingotaud, Soft Mater., 2016, 12, 2166–2176 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Complementary results. See DOI: https://doi.org/10.1039/d2nr04691a |
| This journal is © The Royal Society of Chemistry 2023 |