
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceScalemic natural products
Sarah
Mazzotta
a,
Vincenzo
Rositano
ab,
Luca
Senaldi
b,
Anna
Bernardi
 a,
Pietro
Allegrini
b and
Giovanni
Appendino
*c
a,
Pietro
Allegrini
b and
Giovanni
Appendino
*c
aDipartimento di Chimica, Università degli Studi di Milano, Via Golgi 19, 20133 Milano, Italy
bIndena SpA, Via Don Minzoni 6, 20049 Settala, MI, Italy
cDipartimento di Scienze del Farmaco, Largo Donegani 2, 28100 Novara, Italy. E-mail: giovanni.appendino@uniupo.it
First published on 13th July 2023
Abstract
Covering: up to the end of 2022
The area of scalemic natural products is often enigmatic from a mechanistic standpoint, since low optical purity is observed in compounds having multiple contiguous stereogenic centers resulting from mechanistically distinct biogenetic steps. A scalemic state is rarely the result of a sloppy enzymatic activity, rather resulting from the expression of antipodal enzymes/directing proteins or from the erosion of optical purity by enzymatic or spontaneous reactions. Evidence for these processes is critically reviewed, identifying the mechanisms most often associated to the enzymatic generation of scalemic natural products and also discussing analytical exploitations of natural products' scalemicity.
 Sarah Mazzotta | Sarah Mazzotta received her Master's degree from the University of Calabria in 2016 and she obtained her PhD at University of Seville (2020). Her PhD research activity was focused on the design and synthesis of novel antimicrobial small molecules, especially against adenovirus infections. She was also involved in the preparation of new bioactive compounds from natural sources. Since 2020, she is a postdoctoral research fellow at the University of Milan and she works on the generation of novel glycomimetics as bacterial lectin ligands useful in anti-adhesion therapy for microbial diseases. |
 Vincenzo Rositano | Vincenzo Rositano received his Bachelor's (2017) and Master's degree (2019) from the University of Pisa under the supervision of Prof. Anna Iuliano, working on asymmetric catalysis. In 2021 he moved to Milan where is now pursuing his doctoral study at University of Milan, in partnership with Indena. His research is focused on the development of processes for industrial preparation of active pharmaceutical ingredients, from plant and microbial origin. |
 Luca Senaldi | Luca Senaldi received his Bachelor's (2014) and Master's degree (2016) from the University of Milan under the supervision of Prof. Anna Bernardi. In 2020 he received his PhD degree from the University of Milan at the end of the high-level apprenticeship period at Indena, a company specialized in the preparation of pharmaceutical and food ingredients. He is currently working at Indena Research and Development and his research focuses on the design and development of processes for the industrial preparation of active pharmaceutical ingredients. |
 Anna Bernardi | Anna Bernardi obtained her PhD at the Università degli Studi di Milano in 1986. After a post-doctoral period at Columbia University (NY), she returned to Milano in 1989 as a research associate and became full professor of Organic Chemistry in 2005. She has worked on the development of stereoselective methodologies in organic synthesis, the design of new reagents and, for the past 20 years, on the design and synthesis of carbohydrate mimics (glycomimetics). She received the Quilico Medal of the Società Chimica Italiana in 2021. |
 Pietro Allegrini | Pietro Allegrini received his BS/MS degree from the University of Milan in 1987 and completed the specialization course in Chemical Synthesis at the University of Milan in 1991. He is currently the Research and Development Director at Indena SpA. His research activity has been devoted to process chemistry applied to the industrial synthesis of fine chemicals and active pharmaceutical ingredients as well as natural products. He is co-founder of ISPROCHEM, an international school of process chemistry. In 2018 he has been awarded with the Industrial Research Prize of the Italian Chemical Society – Organic Chemistry division. |
 Giovanni Appendino | Giovanni Appendino is Emeritus Professor at the University del Piemonte Orientale, Department of Pharmaceutical Sciences, Novara (Italy). His research activity has focused on the isolation, chemical modification, and total synthesis of bioactive natural products of plant origin. His studies were recognized by the Italian Chemical Society (Quilico- and Piria medals), by the Phytochemical Society of Europe (Rhone-Poulenc Rorer award and Bruker prize) and by GA (Honorary membership). |
1 Introduction
The occurrence of different enantiomeric forms of natural products can be traced back to the early 19th century observation of an opposite optical rotation in French and American turpentines, with French turpentine being laevorotatory and American turpentine dextrorotatory.1 Turpentine was one of the first organic substances discovered to show optical activity, and enantiomeric forms of α-pinene (1, Fig. 1) occur indeed in its source plants (Pinus maritima L. for French turpentine and mainly P. palustris Mill for American turpentine).1 The existence of racemic natural products, that is of an equimolar mixture of both enantiomers in a single producer, was actually first reported with compounds better referred to as racemized rather than racemic, since loss of enantiomeric purity had occurred during extraction and/or isolation, as in paratartaric (racemic) acid ((±)-2)2 and atropine ((±)-3),3 but truly racemic natural products were then shown to exist, and certain biogenetic pathways are now recognized to be significantly associated to the generation of racemic natural products.4 More puzzling is the occurrence of scalemic (enantiomerically enriched) natural products, not only simple monoterpenes like α-pinene (1), known to occur as (+)-, (−)-, (±)-α-pinene as well as in various ratios of its enantiomers,5 but also more complex and polycyclic compounds like the meroterpene cis-Δ9-tetrahydrocannabinol (4)6 and the alkaloid cephalotaxine (5).7 The number of these occurrences increased with the advent of chiral HPLC as routine analytical tool for natural product mixture. The occurrence of natural scalemates is apparently ambiguous, since chiral natural products are either made by enzymes, and are therefore expected to be enantiomerically pure, or spontaneously self-assemble from reactive species generated by enzymes, and are therefore expected to be racemic [or a mixture of diastereomers in case of pre-existing stereogenic element(s)].4 Scalemates are a bump in the road, a deviance from a binary logic (enantiomeric purity or racemic state) also plagued by semantic problems. Thus, racemic refers to an equimolar mixture of two enantiomers, and has therefore an associated numeric value, while scalemic refers to an uneven ratio of two enantiomers, and is only qualitative, covering a whole spectrum of values. Methodologic limits also exist, and absolute enantiomeric purity and absolute lack of enantiomeric purity are pure abstractions, the ends of a continuous line, with the meaning of “high enantiomeric purity” being context-dependent. In the analysis of terpene hydrocarbons, a class of compounds generally highly scalemic or even racemic, a 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 enantiomeric ratio could be reported as a noteworthy enantiomeric purity, while in a pharmaceutical context a ratio lower than 99:1 could cause concern and require additional purification. A similar situation exists in the literature on enantioselective synthesis, where the bar of enantiomeric purity has been constantly raised. The first enantioselective reaction was reported by Bredig and Fiske in 1912,8 and produced, under quinine catalysis, mandelonitrile (6) from two achiral precursors (benzaldehyde and hydrocyanic acid) in ca 8% enantiomeric excess (ee). Conversely, the first disclosure of the sharpless asymmetric dihydroxylation in 1988 reported crude ee >95% for trans-stilbene as a substrate,9 and ee > 99% are nowadays not uncommon in the literature on asymmetric synthesis.
:10 enantiomeric ratio could be reported as a noteworthy enantiomeric purity, while in a pharmaceutical context a ratio lower than 99:1 could cause concern and require additional purification. A similar situation exists in the literature on enantioselective synthesis, where the bar of enantiomeric purity has been constantly raised. The first enantioselective reaction was reported by Bredig and Fiske in 1912,8 and produced, under quinine catalysis, mandelonitrile (6) from two achiral precursors (benzaldehyde and hydrocyanic acid) in ca 8% enantiomeric excess (ee). Conversely, the first disclosure of the sharpless asymmetric dihydroxylation in 1988 reported crude ee >95% for trans-stilbene as a substrate,9 and ee > 99% are nowadays not uncommon in the literature on asymmetric synthesis.
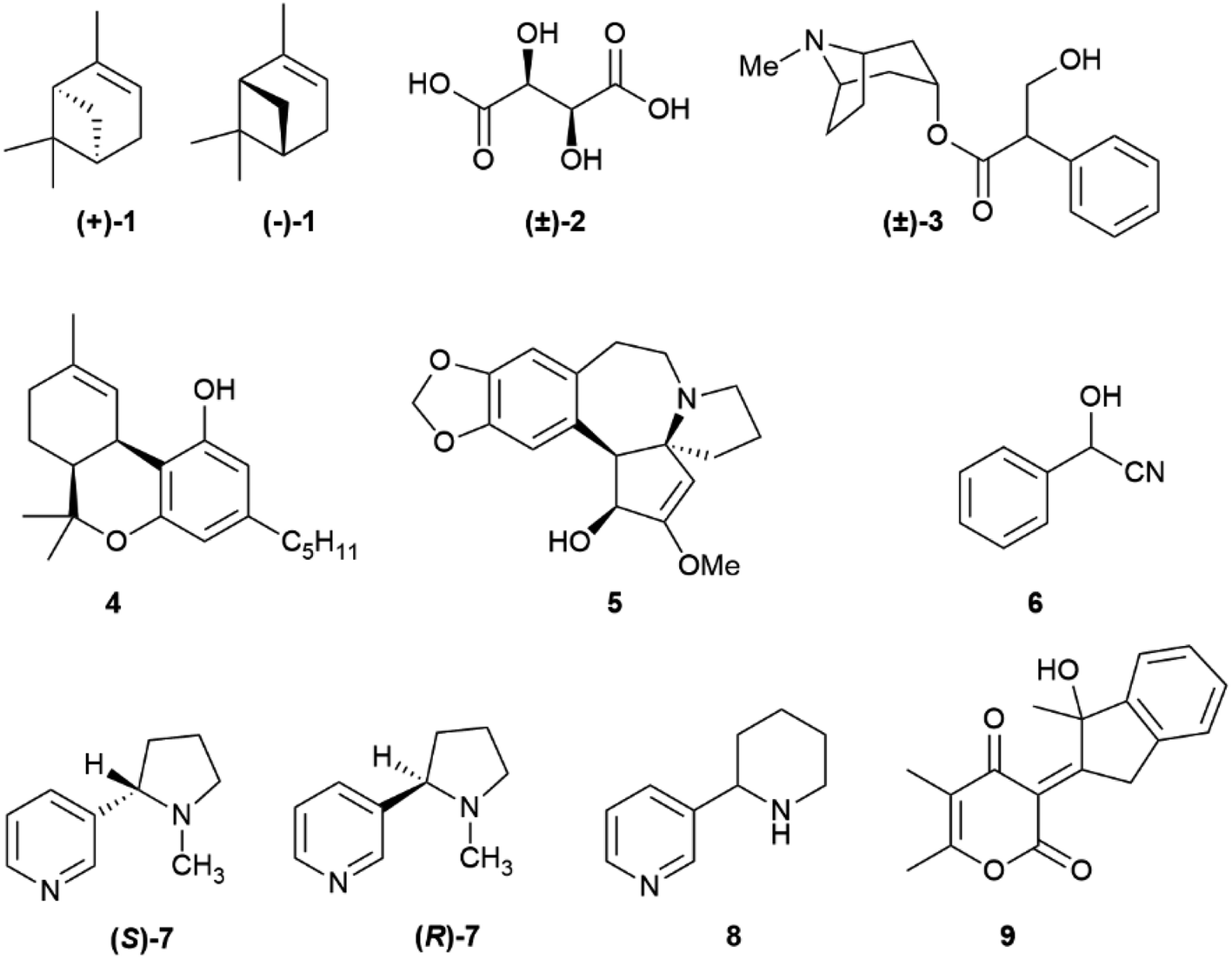 | ||
| Fig. 1 Chemical structures of compounds 1–9. | ||
A notation problem also exists. While enantiomerically pure compounds can be drawn according to their absolute configuration (when known) and racemates can be indicated by (±) or “rac”, there is not accepted notation for scalemates. In this review, for compounds with a single stereogenic element (center or axis) we do not draw configurations for scalemic forms. For compounds with multiple stereocenters, we draw the configuration of the predominant isomer. Information about its enantiomeric composition must be found in the text.
The occurrence of racemic natural products has been the subject of several recent reviews,10–15 one comprehensively covering the years 2012–2019.15 Conversely, the grey area of natural products' optical “impurity” (scalemicity) has so far been largely overlooked, and the inventory of scalemic natural products is probably underestimated for various reasons. The first one is that the optical purity of novel natural products is rarely reported, and novel natural products are assumed, when chiral and optically active, to be also enantiomerically pure. Thus, out of the 286 articles reporting novel chiral natural products published by the Journal of Natural Products in 2017, enantiomeric purity was evaluated only in 31 articles (<15%).16 The second reason is that, as discussed in the next section, whenever a highly scalemic mixture is subjected to a physicochemical process that involves a phase change (evaporation, crystallization, achiral chromatography), self-disproportionation of enantiomers (SDE) can occur, with a redistribution between the two phases. As a result, for highly scalemic compounds, optical purity is generally upgraded in the mother liquors of crystallization and in the faster moving chromatographic fractions, and downgraded in the complementary phase or fractions.17 Since the physical and spectroscopic properties of novel natural products are preferentially reported on compounds carefully purified by chromatography and/or crystallization, the possibility exists that scalemates could potentially be reported as racemates or quasi-racemates. Finally, a compound can be produced as a scalemate and then upgraded to enantiomeric purity by enzymatic activity, nicotine being a remarkable example. Nicotine is produced in the roots of the tobacco plant (Nicotiana tabacum L.) as a scalemate, with (S)-nicotine ((S)-7) as the dominant enantiomer by a factor of ca 95:5. However, during xylemic translocation to the leaves, (R)-nicotine ((R)-7) is enantioselectively N-demethylated, resulting in high optical purity (>99%) for the alkaloid isolated from the leaves.18 Interestingly, anabasine (8), the teratogenic piperidine homologue of nicotine, is not significantly accumulated in tobacco leaves, and only occurs, in a highly scalemic form, in the stalks,19 an observation highlighting the relevance of the N-methyl group, present in nicotine but not in anabasine, as a “handle” for the enzymatic “resolution” of nicotinoid alkaloids. Incidentally, (R)-nicotine is significantly more toxic than (S)-nicotine, but has a lower affinity for cholinergic receptors and almost negligible recreational activity. The presence of synthetic racemic nicotine in “tobacco-free” e-cigarettes liquids is therefore potentially dangerous, since consumers, by dosing their puffs with (S)-nicotine ((S)-7), also inhale significant amounts of the more toxic (R)-enantiomer ((R)-7).20
The purpose of this review is to foster attention on scalemate natural products, highlighting that the optical purity of novel compounds cannot be taken for granted, especially for some classes of natural products, and should be validated by chiral analysis, either spectroscopic or chromatographic. We do not aim to provide a comprehensive list of natural scalemates, inevitably incomplete for the reasons discussed above, but rather to identify biogenetic pathways and mechanisms associated to the generation of scalemate natural products, highlighting the areas most in need of investigation and discussing the opportunity that scalemic natural products offer to biomedical research.
2 Self-disproportionation of enantiomers (SDE) during the purification of scalemates
Any type of physicochemical operation which involves phase separation (crystallization, solubilization, chromatography, distillation, sublimation) has the potential to alter the composition of scalemates, differently redistributing them into enantio-enriched and enantio-depleted phases.17,20 For compounds with significant volatility, even trivial operations like solvent removal by rotary evaporation21,22 or vacuum drying23 have been reported to produce scalemates enantioenriched by the loss of the faster evaporating/sublimating racemate. In asymmetric synthesis, it is generally possible to avoid fractionation and directly measure ee on crude reaction mixtures by chiral chromatography or by NMR. Conversely, the complexity of biomass extracts makes extensive chromatographic steps, often associated to crystallization, mandatory to reach a purity level sufficient for the structure elucidation and the determination of enantiomeric purity of isolated natural products. Thus, SDE is unavoidable, at least to some degree, and especially in compounds highly functionalized and prone to intermolecular interactions.17,24 For this reason, it is difficult to go beyond the identification of the dominant enantiomer and to make a quantitative comparison between the ee of natural scalemates from different sources and/or different purification processes.SDE has so far received scarce attention within the natural products community, even though the first systematic studies in the area can be traced back to a series of seminal observations by Otto Wallach on crystal terpene derivatives.25 Working with brominated carvones, Wallach reported in 1895 that racemic crystals tend to be denser and have a higher melting point and a lower solubility than their homochiral analogues (Wallach rule).26 Although exceptions are known, racemic crystals, representing more than 90% of the crystalline racemic mixtures, are generally more tightly packed and thermodynamically more stable than their enantiopure counterparts.26 A chiral packing can be accommodated only in a limited set of the crystallographic space groups, which are, conversely, all available for racemic crystals. Crystallization of a scalemate can therefore afford racemic or quasi-racemic crystals even from highly biased scalemic mixtures.26 A remarkable example was reported by Sharpless in Organic Synthesis, describing the upgrade of the ee of a diol obtained by asymmetric dihydroxylation from 84% to 97% by selective crystallization of a quasi-racemate of the minor enantiomer.27 Mixtures of racemic and enantiopure crystals can, in principle, be separated based on their different density, and ultracentrifugation has indeed be reported to efficiently separate mechanical mixtures of racemic and enantiopure crystals of alanine.28 The situation is well summarized by ternary diagrams of solubility phase (Fig. 2).29 The racemate is characterized by two eutectic points (E and E′) that define five different regions. The upper region (A-1) corresponds to the non-saturated solution, and the two lateral triangles (A-2) to pure crystals of each enantiomers. Adjacent to the A-2 phases, two triangular regions (A-3) represent mechanical mixtures of racemate and enantiopure crystals, while the central conical phase corresponds to the racemate (A-4). A scalemate whose composition falls in the E–E′ projection at the base of the triangle will produce a racemate, while scalemates whose composition correspond to the AE and A′E projections will afford pure enantiomers. A decrease of temperature and the presence of groups that favour intermolecular interactions will raise the vertex of the solubility curve, enlarging the E–E′ projection, and favouring the crystallization of a racemate. The phase diagram clearly shows that if the composition of the scalemic mixture has high content of one of the enantiomers, it is probale to ends in the pure D and pure L solids regions and hence the enantiomeric composition can be further enriched. On the other hand, when the starting composition of the mixtures is close to racemic ratios (R), the obtained crystals are often racemic. Anyway, in both the cases the starting scalemic mixture is altered by crystallization.
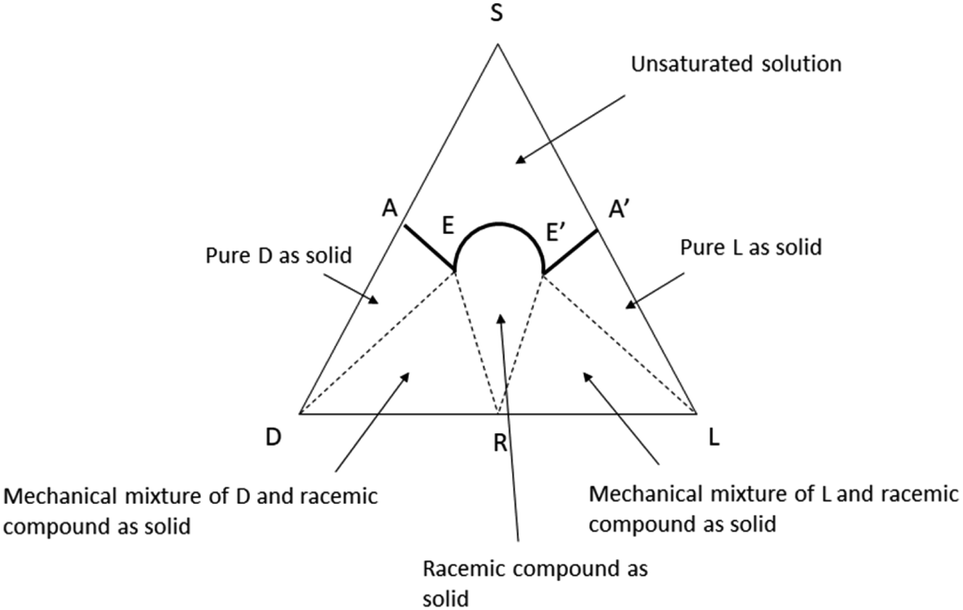 | ||
| Fig. 2 Ternary solubility diagram of true racemic compounds. | ||
“Optical purification” by crystallization makes it possible that natural products reported as crystalline racemates could actually be scalemates, with loss of optical rotation being associated not to the removal of impurities, but to self-disproportionation. This was shown for the fungal ketide colletopyrandione (9, Fig. 1), which occurs as a scalemate in extracts but crystallizes as a racemate,30 and SDE tests should be applied to assess its contribution to the isolation of a crystalline racemate natural product. In this context, it should be remarked that the measurement of optical purity is sensitive to the presence of impurities, especially when a compound has low optical rotation, and the same holds for electronic circular dichroism (ECD) when the impurity has UV absorption similar to the one of the main product. When the presence of a scalemate is suspected, it is therefore advisable to rely on a chromatographic determination of enantiomeric composition or on NMR derivatization with chiral reagents, rather than simply on measurement of optical rotation.
The considerations discussed for crystallization hold also for other preparative processes of purification, like chromatography, distillation, and sublimation. The last technique is not of general relevance for natural products purification, but chromatography is also the largest area of SDE manifestation, since applicable to all scalemates independently on their physical state. Dreiding was one of the first to warn that achiral chromatography should be used with great care for the purification of scalemates, since the enantiomeric composition of chromatographically pure fractions is affected by the different solubility of racemic and enantiopure crystals in the eluent.31 Similar observations were also reported by Kagan,32 but, despite the gigantic state of Dreidig and Kagan in stereochemical studies, the research community has so far largely failed to receipt these warning notes. The issue of scalemate chromatography is particularly challenging for natural products, since crude extracts contain a host of optically pure small molecules, whose solubilization renders de facto non-achiral the chromatographic eluant. For these reasons, it is not unconceivable that the racemic state of some natural products might be dubious and in need of re-evaluation, while early measurements of ee, especially with chiral NMR or chiral chromatography, should be used to investigate differences in the stereochemical state of native and isolated natural products.
3 The origin of scalemic natural products
3.1 Partial racemization of enantiopure compounds
Partial racemization during isolation and/or purification is the most obvious reason for the isolation of a natural product in a scalemic form, but post-synthetic erosion as well as upgrade of optical purity are also documented. Two major types of mechanisms underlie the spontaneous erosion of optical purity, namely enolization and pericyclic reactivity.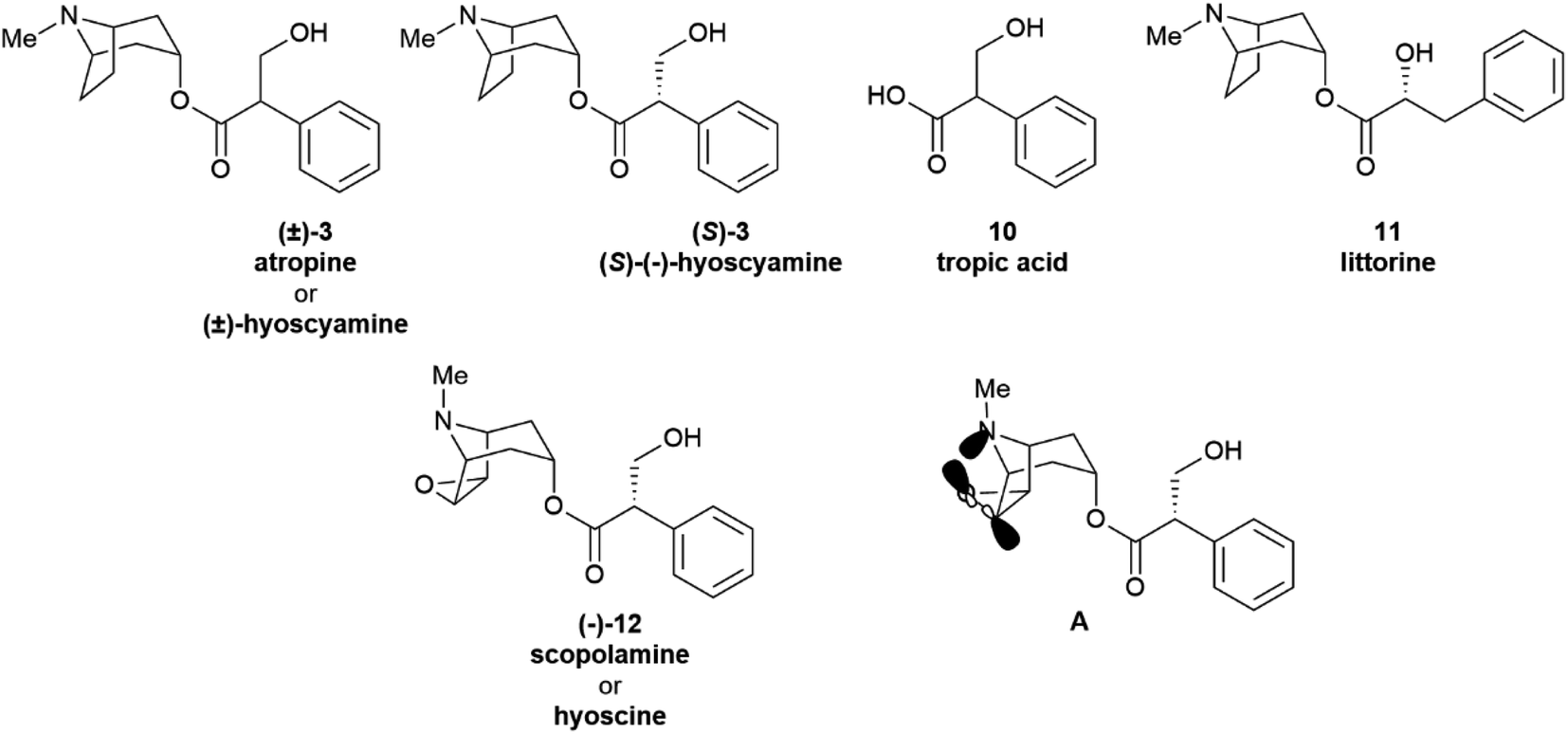 | ||
| Fig. 3 Tropane alkaloids and related compounds. | ||
The stereogenic center of tropic acid is established at the level of the phenyllactate ester of tropine (littorine, 11, Fig. 3) via a process mediated by an oxidative rearrangement induced by an iron-based oxygenase (CYP80F1). After initial homolyses of the benzylic C–H bond from the high-valent oxo-iron species (Scheme 1), rearrangement of the carbon-centered benzyl radical to the oxygen-centered cyclopropanol radical 13 occurs, with hydroxyl radical rebound eventually taking place at the homobenzyl carbon and not at the one where the original radical was generated (Scheme 1).34 This process is strictly stereoregulated, and the side-chain stereogenic center of the tropic acid moiety of 11 is generated with high selectivity for the S configuration. The two enantiomers of hyoscyamine have distinct biological profiles. Thus, most cholinergic activity resides in the natural S-(−)-enantiomer, but (+)-hyoscyamine has, surprisingly, more potent excitatory effects on the central nervous system.35 Because of this, and to secure standardization of dosages, the European Pharmacopoeia requires that the optical purity of atropine (3), expressed as (−)-hyoscyamine ((−)-3), should not exceed 9%.36
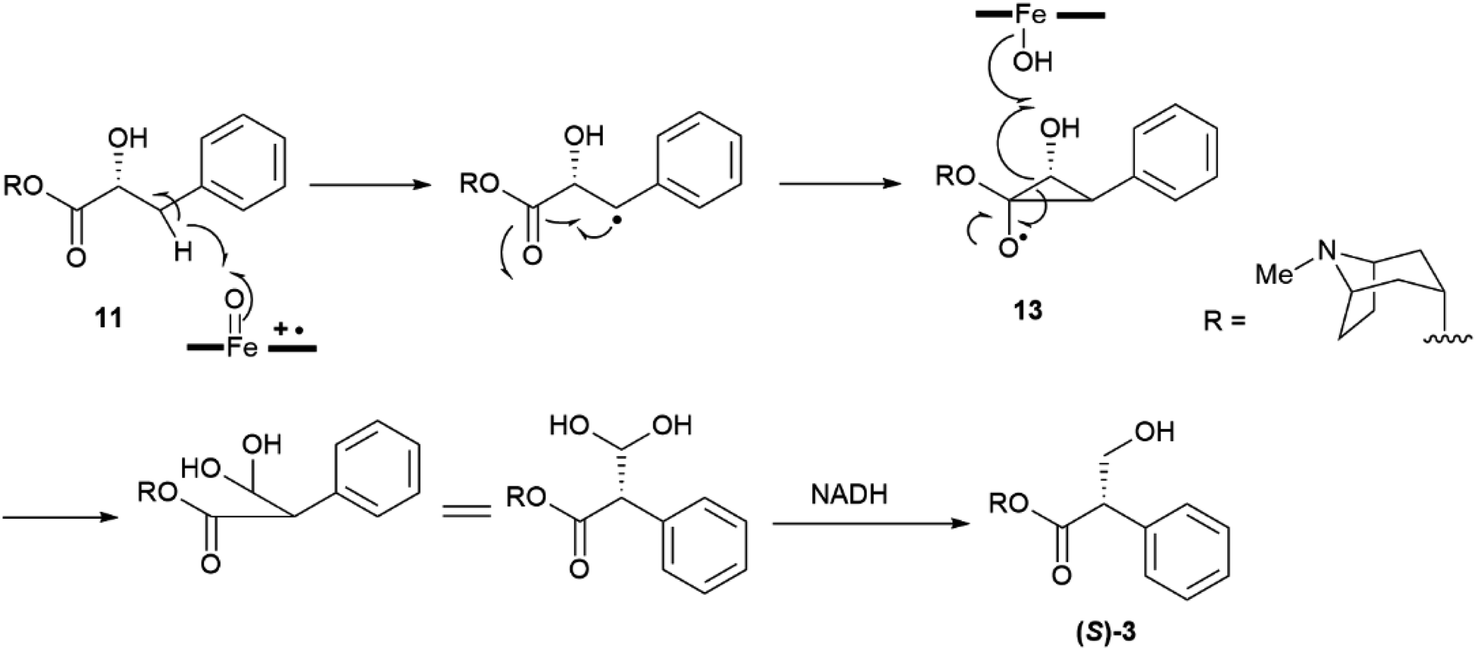 | ||
| Scheme 1 Oxidative rearrangement of the hydroxyphenyllactic acid ester 11 to (−)-hyoscyamine ((S)-3) via the cyclopropanol 13. | ||
Remarkably, the self-catalysed racemization of the closely related alkaloid scopolamine (hyoscine, (−)-12, Fig. 3) to atroscine ((±)-12) is slower than the one of hyoscyamine to atropine, since, despite the structural similarity between the two alkaloids, scopolamine is significantly less basic than hyoscyamine (pKa 7.60 vs. 9.85).37 This difference is presumably due to a combination of the inductive effects associated to the oxyrane oxygen as well as to homoanomeric donation of the nitrogen lone pair on the 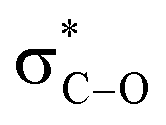 bonds of the epoxide ring (A, Fig. 3). In contrast to hyoscyamine, where it occupies a pseudo-axial orientation, in scopolamine the nitrogen lone-pair is pseudo-equatorially orientated, which allows orbital interaction with the carbon–oxygen bonds of the oxirane.38 Despite the structural similarity of atropine (3) and scopolamine ((−)-12) the European Pharmacopoeia requires tests of optical “impurity” for the former and of optical “purity” for the latter ([α]D −33 and −39).36
bonds of the epoxide ring (A, Fig. 3). In contrast to hyoscyamine, where it occupies a pseudo-axial orientation, in scopolamine the nitrogen lone-pair is pseudo-equatorially orientated, which allows orbital interaction with the carbon–oxygen bonds of the oxirane.38 Despite the structural similarity of atropine (3) and scopolamine ((−)-12) the European Pharmacopoeia requires tests of optical “impurity” for the former and of optical “purity” for the latter ([α]D −33 and −39).36
Partial self-racemization also underlies the long controversies on the structure of pelletierine (14), the major alkaloid from pomegranate root bark (Fig. 4). This piperidine alkaloid was first isolated in 1878 by Tanret in France, and named after Joseph Pelletier (1788–1842), one of the founding fathers of alkaloid research.39 After salification, pelletierine (14) could be crystallized both in an optically active laevorotatory form and a racemate. The latter was assumed by Tanret to be an isomer of the laevorotatory compound and named isopelletierine, while a related, optically inactive, alkaloid was referred to as pseudopelletierine (15).39 Pseudopelletierine (15), an achiral compound, holds a special position in the history of chemistry, since it was the starting material for Wilstaetter's synthesis of cyclooctatetraene, a compound that fostered interest in the nature of aromaticity.40
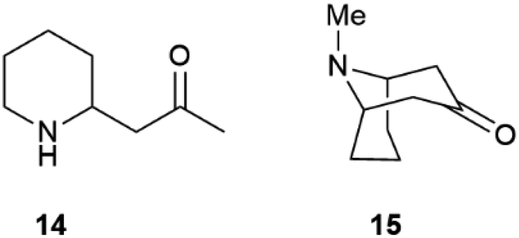 | ||
| Fig. 4 Pelletierine 14 and pseudopelletierine 15. | ||
Pelletierine and isopelletierine were considered constitutional isomers, the former being an aldehyde and the latter a ketone. In a further bold development to reconciliate the failure to correlate isopelletierine to the hemlock alkaloid conidrine (see infra), pelletierine and isopelletierine were considered no longer constitutional isomers but diastereomers, assuming the presence of a stereogenic non-inverting nitrogen in both compounds.41 The structure of isopelletierine was eventually unambiguously solved by Meissenheimer, who synthesized it from 2-methylpyridine, while the discovery that pelletierine is configurationally labile in the presence of bases finally clarified its relationship with isopelletierin.41 The configurational lability of pelletierine is due to retro-aza-Michael equilibration with an open form (Scheme 2), a process base-catalyzed and almost unavoidable in the standard workflow of alkaloid isolation, which involves extraction with acids of the plant material and then basification of the extract to recover its basic constituents.41 In contrast, crystalline salts of pelletierine are remarkably stable toward racemization, and an analysis of the original Tanret sample of pellettierine sulfate carried out 80 years after its preparation showed substantial retention of optical purity.42
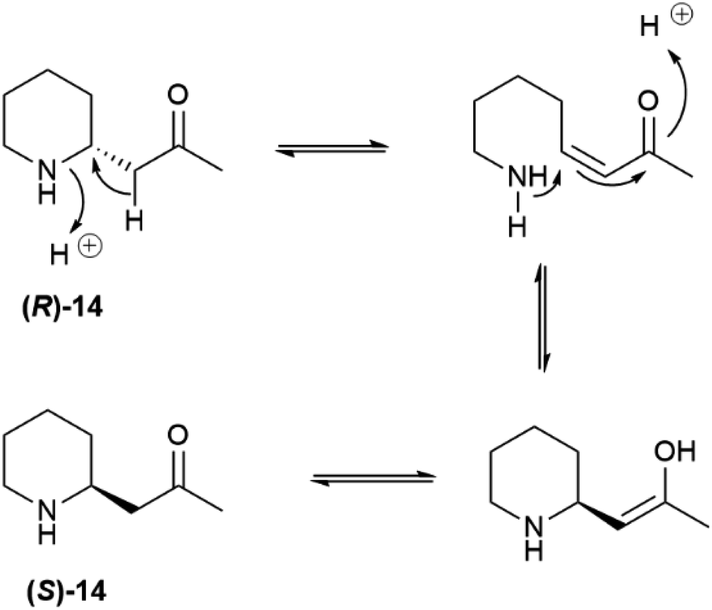 | ||
| Scheme 2 Racemization of pelletierine (14) via retro-aza-Michael fragmentation. | ||
In conclusion, the major pomegranate alkaloid is synthesized in the plant in optically active form as pelletierine ((R)-14), which then partially isomerizes to isopelletierine ((S)-(+)-14). To avoid confusion, the name pelletierine should be used for the optically active native compound, and rac-pelletierine ((±)-14) for its racemized version. In sharp contrast with nicotine-related alkaloids, tropane and tropane-related alkaloids are therefore produced in an enantioselective way, and their isolation as scalemates or racemates is due to spontaneous racemization induced by the presence of an easily enolized carbonyl.
A complex set of tautomeric and rotameric equilibria is also responsible for enantiopurity erosion of the phenanthroperylenequinone hypericin (16, Fig. 5).43 This phototoxic red pigment has been isolated from plants, insects and protozoa, but its best known source is St. John's worth (Hypericum perforatum L.), in whose flowering toppings it can occur in concentrations as high as 0.3% on dry weight basis.44 Hypericin is responsible for hypericism, a potentially deadly sun-dependent intoxication of farm animals. It has been suggested that hypericin derives from emodin (17) or emodin anthrone (18) by oxidative dimerization associated to a single phenol oxidase coupling protein (Hyp-1).44 Hypericin shows a remarkable structural complexity, not evident from a superficial inspection of its formula, and associated to torsional and tautomeric equilibria.45
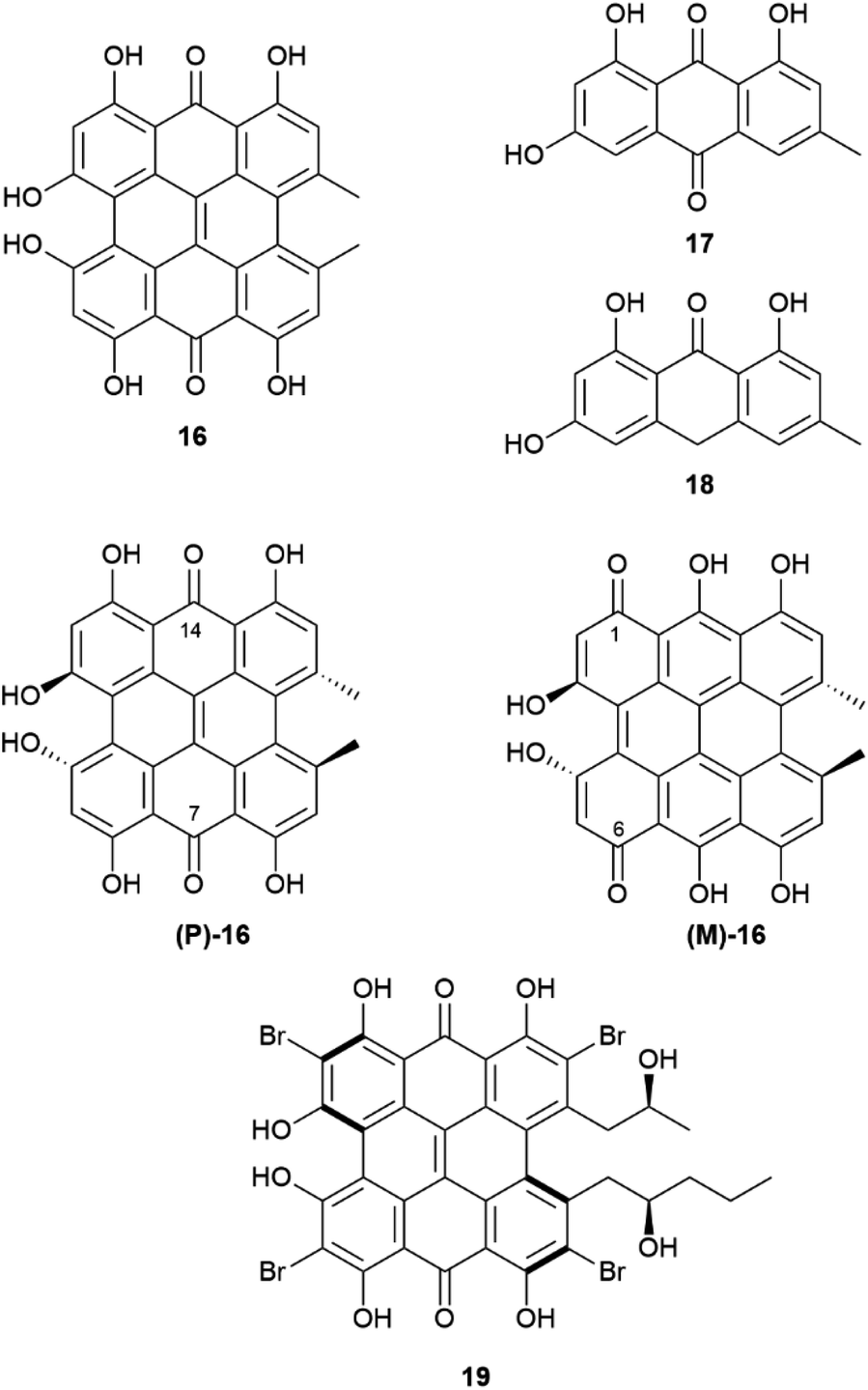 | ||
| Fig. 5 Hypericine 16, its biosynthetical precursors 17 and 18 and its two chiral tautomers (P)-16 and (M)-16. Structure of the hypericin-related pigment gymnochrome A (19). | ||
In short, two sets of axially chiral tautomers exist ((P)-16 and (M)-16), differing for the location of the quinone moiety on the central (7,14-tautomer (P)-16) or on the peripheral (1,6-tautomer (M)-16) biphenyl systems. In the solid state, the least stable 1,6-peripheral quinone system ((M)-16) prevails in salts, while the more stable 7,14-tautomer ((P)-16) prevails in the non-ionized compounds.46,47 All tautomers have a propeller C2-symmetric and chiral shape, and racemization occurs via interconversion of P (aS) and M (aR)-tautomers. The racemization barrier is low, ca 20 kcal mole−1, with a half-life of around 30 minutes at 35 °C. Therefore, hypericin is obtained by isolation under normal room temperature conditions as a racemic compound.45 On the other hand, in cell environment, hypericin could be present in an enantiopure P-configuration, stabilized by chiral interaction with other chiral molecules, and evidence has been obtained by investigating the association of hypericin with albumin.46 This enantiomer has, indeed, a CAS number (548-04-9), and is, apparently, commercialized, although the synthetic origin of this material implies confusion with rac-hypericin, having CAS number 1372719-41-9. In principle, by working at low temperature (4 °C), it could be possible to isolate or resolve hypericin, since its enantiomers show a reasonable configurational stability at 15 °C. On the other hand, substituted hypericins like the marine pigment gymnochrome A (18) and related compounds, were isolated in optically active form. Regrettably, optical purity was not reported.46
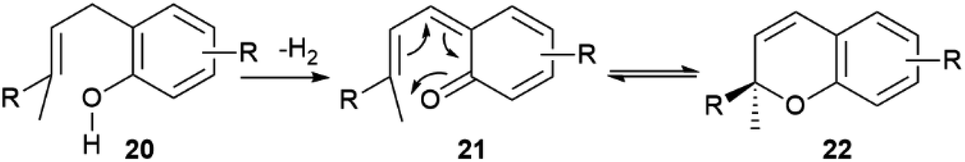 | ||
| Scheme 3 Generation of the chromene system 22 by tandem dehydrogenation-electrocyclization of o-isoprenylphenols (R = prenyl, geranyl). | ||
When the isoprenyl group is higher than a prenyl (geranyl, farnesyl), a stereogenic carbon is generated in the pericyclic process, and the resulting chromenes are chiral. Since the electrocyclization is rapid and the intermediate vinylogous quinonmethides are generated in the chiral environment of the dehydrogenase enzyme,48 the stereogenic carbon is seemingly generated in an enantiopure form, and many chromenes are, indeed, isolated in high enantiomeric purity (e.g. daurichromenic acid (23) from, various Rhododendron species).50 However, cycloreversion during biomass storage followed by spontaneous electrocyclization can erode this purity, with formation of highly scalemic or almost racemic compounds, as exemplified by cannabichromene (CBC, 24a) from Cannabis sativa L. and by rhodonoids, a class of compounds derived from its tetra-nor analogue 24b and occurring in Rhododendron species.51–53
Cycloreversion is induced by light and, to a lesser extent by heating, and is not significantly affected by treatment with acids and bases.53 Reversibility of the electrocyclization critically depends on the substitution pattern of the aromatic ring. The presence of a carboxylic group para to the chromenic oxygen significantly slows cycloreversion, an effect presumably due to stabilization by resonance of the aromatic ground-state, which leads to an increased activation energy for dearomatization (Scheme 3, conversion of 22 to 21).48 Cannabichromene (CBC, 24a) and its analogues with a modified alkyl group54 are the starting material for the generation of other phytocannabinoids, which are therefore expected to be scalemic. However, cannabicyclol (25), obtained from cannabichromene (CBC, 24a) by photochemical or acidic cyclization,49 was always isolated as a racemic compound, even though the stereogenic carbon of the starting chromene (24a) is not involved in the process (Fig. 6).
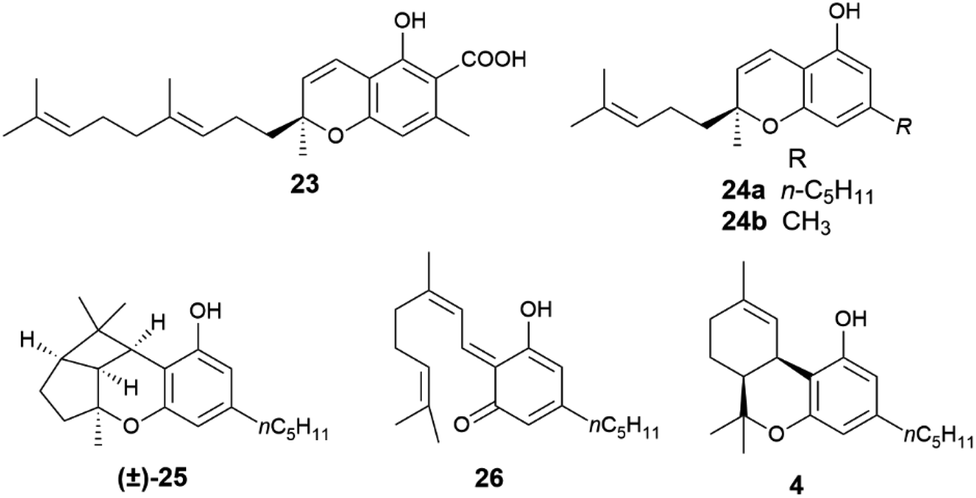 | ||
| Fig. 6 Formulae of compounds 23–26 and 4. | ||
Unlike its trans-isomer, a highly enantiopure compound,51cis-Δ9-tetrahydrocannabinol (cis-Δ9-THC, 4) is scalemic,6 and could derive from an intramolecular hetero-Diels–Alder cycloaddition of the vinylogous quinonmethide 26 where the ω-, and not the proximal, trisubstituted double bond is involved in the pericyclic process (Scheme 3, R = prenyl).
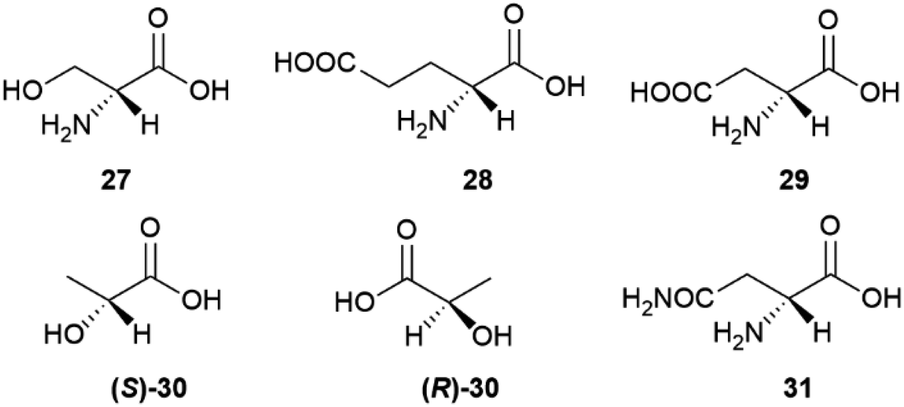 | ||
| Fig. 7 Structures of the proteinogenic amino acids 27–29 and the hydroxyacids 30–31. | ||
D-amino acids play a relevant role also in mammalian metabolism, resulting into an overall scalemic state in some organs and at specific development stages. Thus, significant amounts of D-serine (27) occur in rat brain from birth until seven months of age.57 The production of this amino acid then decreases and remains associated to the function of glutamate receptors of the N-methyl-D-aspartate (NMDA) type, where D-serine behaves as an endogenous agonist, acting at the strychnine-insensitive glycine site.57 While D-serine only occurs in brain, D-aspartic acid (29) also occurs in other organs, undergoing development-dependent changes with age.58 In the prefrontal cortex of human brain, aspartic acid was found to be scalemic, with dominance of the D-enantiomer by a 6:4 ratio at week 14 of gestation, and a decrease to trace level by week 41.59 Just like D-serine (27), also D-aspartic acid binds to NMDA receptors at the strychnine-insensitive glycine site.58S-[L, (+)]. Lactic acid ((S)-30) is produced enantioselectively in glycolysis, but bacteria can invert its configuration, resulting in a generally scalemic or even racemic state for lactic acid of microbial origin.60 The formation of (R)-D-lactic acid ((R)-30) is involved in the mechanism of bacterial resistence to vancomycin.60 This glycopeptide antibiotic inhibits peptidoglycan synthesis by complexing the terminal D-Ala-D-Ala sequence of the peptidoglycan precursor. Replacement of the terminal D-alanine with D-lactic acid confers resistance to vancomycin, as does the replacement with D-serine (27).60
For unclear reasons, some plants produce and accumulate significant amounts of D-amino acids in free form along with their proteinogenic version.55 A remarkable case is represented by asparagine, which is accumulated as a scalemate in the initial phases of seed germination by various leguminous plants. The non-proteinogenic D-enantiomer (31) is then metabolized, eventually resulting in the selective accumulation of the natural L-form in adult tissues.61 The scalemate state of asparagine in leguminous seeds was discovered by Arnaldo Piutti in 1886 in vetches (Vicia sativa L.) seedlings, and his observation of the distinct taste of the two enantiomers of this amino acid was the first report of different physiological effects associated to a pair of enantiomers.61,62 In the wake of the studies by Guareschi on asparagine,63 Piutti crystallized 20 kg of the L-enantiomer from an aqueous extract of 6.5 metric tons of germinated vetches, obtaining then a second crop of crystals from the mother liquors. Much to his surprise, the second crystal crop was a conglomerate of hemihedral crystals, from which, by manual (!!) separation, he eventually obtained 100 g of intensely sweet D-asparagine.60,61 A sample of Piutti's asparagina destrogira dolce (dextrorotatory sweet asparagine) was recently discovered in the Schiff collection at the Florence Chemical Department.62 Despite the popularity of this historical milestone of molecular chirality, the rationale for the production of sweet D-asparagine during germination of some legumes is unknown and puzzling, since seedlings do not tend to accumulate attractive, but rather deterrent (bitter of pungent) compounds. Also unknown are the metabolic fate of the non-proteinogenic version of asparagine and how the human sweet receptor, a T1R2 and T1R3 heterodimer which cannot distinguish between the enantiomeric forms of most sugars, can do so with the enantiomeric forms of asparagine.64
In most cases where the origin of the D-form of proteinogenic amino acids was investigated, specific racemases were shown to isomerize the corresponding L-isomer.56 Most of these enzymes require pyridoxal 5′ phosphate as cofactor, and isomerization occurs via reversible formation of a Schiff base with the 4-formyl group of pyridoxal and formation of an aza-quinoid intermediate.56 However, other racemases, and specifically the bacterial version of glutamate and aspartate racemase, do not require a cofactor, and exploit cysteine residues for the deprotonation–riprotonation of the glutamate α-position.56
Configurational inversion of proteinogenic amino acid generally precedes their incorporation in secondary metabolites, but partial racemization of the amino acid cysteine occurs during the biosynthesis of firefly luciferin. Bioluminescence is associated to the enzymatic production of luciferins, a class of compounds capable of light emission upon oxidation.65 Firefly luciferin is a benzothiazole derivative [(S)-2-(6′-hydroxy-2′-benzothiazolyl)-2-thiazoline-4-carboxylic acid, (S)-32, D-luciferin, Scheme 4], which emits light with loss of CO2 when oxidized by the enzyme firefly luciferase to form oxyluciferin 33.66
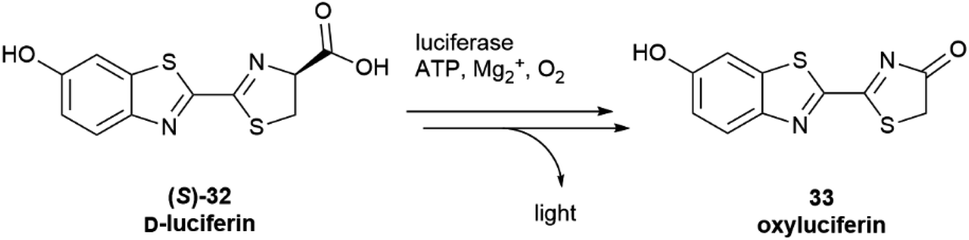 | ||
| Scheme 4 The oxidation of D-luciferin (S)-32 by luciferase is the basis of luciferin bioluminescence. | ||
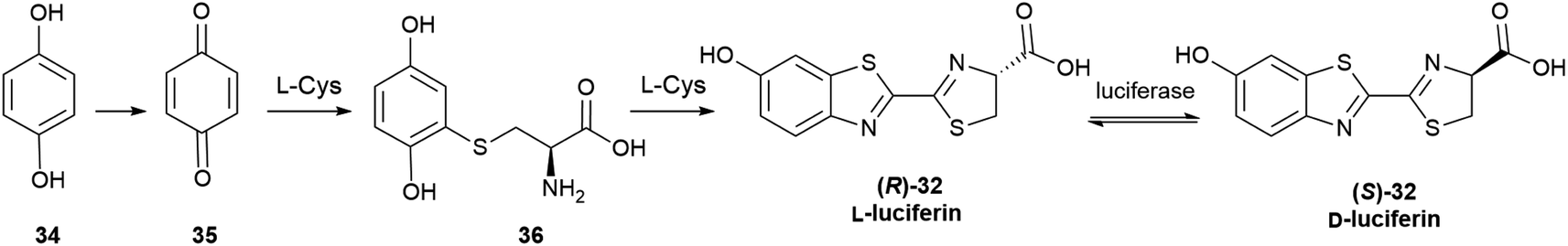
Only D-luciferin is recognized by the enzyme66 and its oxidation is associated to the emission of light of pH-dependent maxima, but luciferin isolated from various insects is a scalemate, with the D-enantiomer dominating by a factor of ca 4:1.67L-Luciferin (R)-32 (Scheme 5) is the biosynthetic precursor of D-luciferin (S)-32, and is a strong inhibitor of firefly luciferase, interfering with the light-emitting reaction.
 | ||
| Scheme 5 Biosynthesis of D-luciferin (S)-32 in fireflies. | ||
The first biosynthesis studies on luciferin from fireflies and beetles identified hydroquinone 34 and L-cysteine as its precursors, and later studies clarified the stereochemical aspects of the process (Scheme 5).68 Hydroquinone 34 is oxidized to p-quinone 35, which undergoes thia-Michael addition of L-cysteine affording L-cysteinylhydroquinone (36), next turned into L-luciferin (R)-32 by reaction with a second molecule of L-cysteine in a yet mechanistically unclarified step.
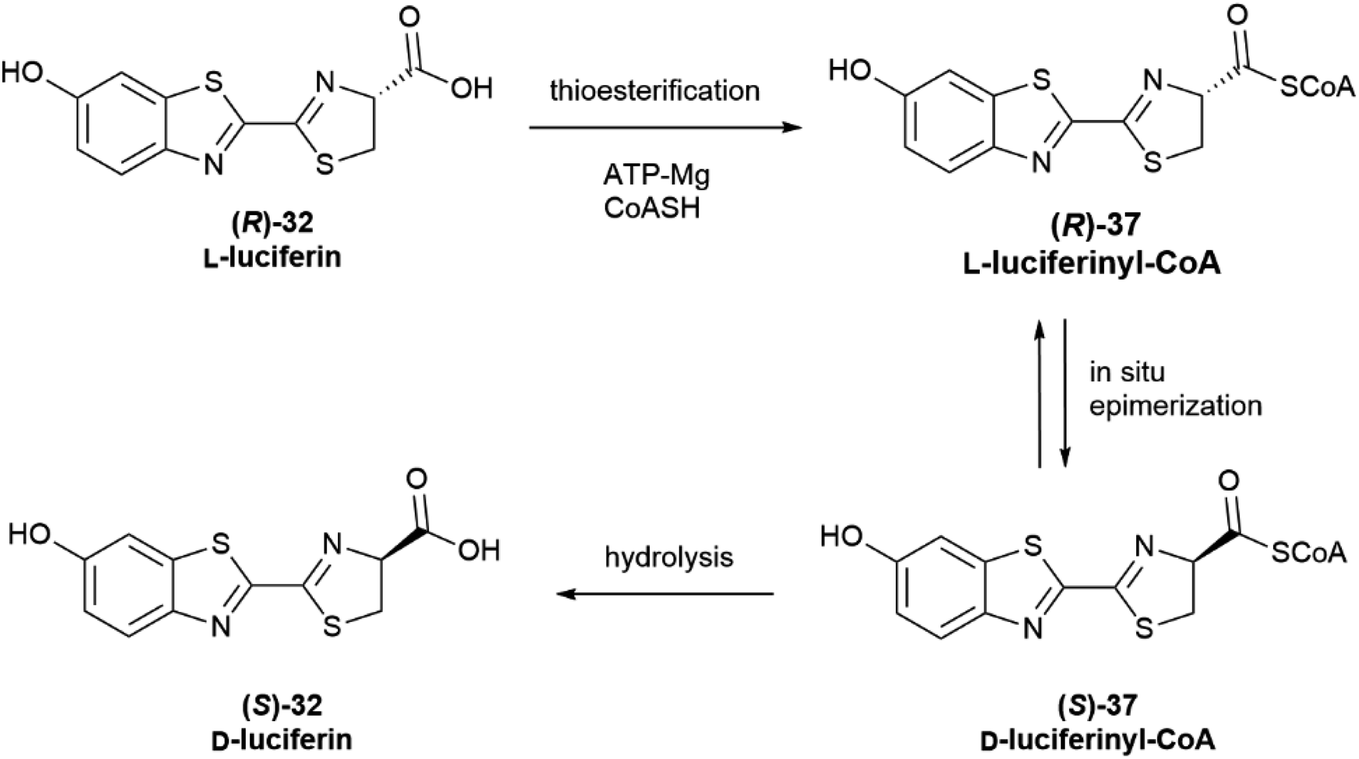
The configurational inversion has been shown to occur in three steps (Scheme 6). The carboxylic group of L-luciferin (R)-32 is first enzymatically activated to the corresponding CoA thioester (R)-37. This compound is configurationally unstable due to the increased acidity of the α-carbon, and spontaneously enolizes, affording by re-protonation D-luciferyl-CoA (S)-37, next hydrolized to D-luciferin (S)-32.68,69
 | ||
| Scheme 6 Epimerization of luciferin in firefly and beetle. | ||
Noteworthy, luciferase appears to show different enzymatic activity for the two enantiomers, behaving as a thioesterase towards L-luciferin (Scheme 6) and as an oxidase towards D-luciferin (Scheme 4).65 Moreover, D-luciferin inhibits the synthesis of L-luciferyl-CoA, and therefore self-limits its production, while an increased concentration of free L-luciferin inhibits the oxidation reaction, hindering the bioluminescence process.65
3.2 Co-expression of antipodal enzymes or directing proteins
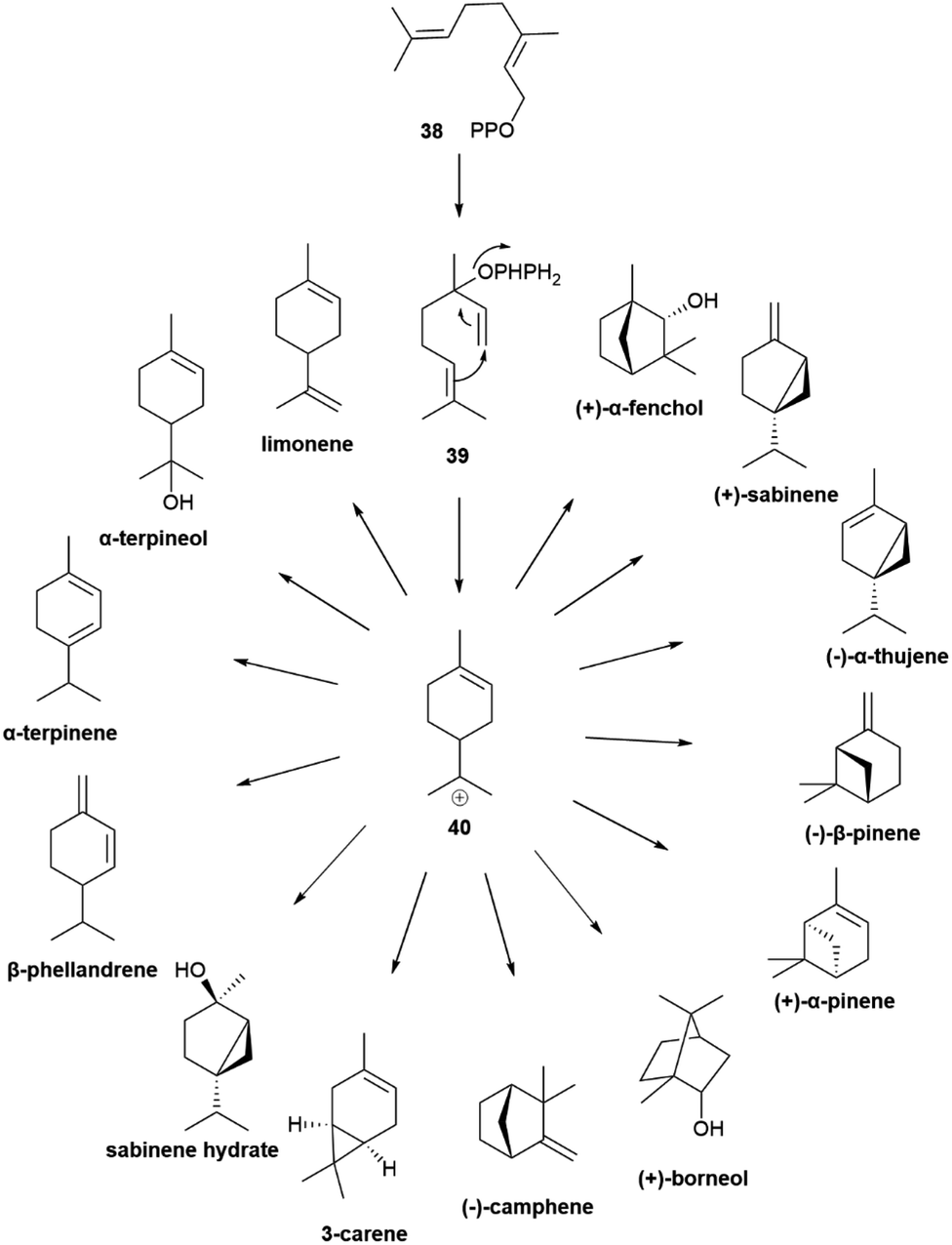 | ||
| Scheme 7 Monoterpenes derived from the para-menthane cation 40. For compounds having multiple stereocenters, the configuration of the more common enantiomer was depicted (ROMPP Lexicon, Thieme Verlag, 1997). | ||
The cyclic cationic 40 can then undergo distinct rearrangements, and is eventually quenched by proton loss or water trapping (Scheme 7). In the oxidase phase, the primary skeleta are then decorated with oxygen functionalities which, in turn, can trigger additional ring rearrangements and closures that significantly expand skeletal diversity. The oxidase phase relies on cytochrome P450 monooxygenases that show high substrate enantiospecificity,73 and effectively upgrade the enantiomeric purity of their hydrocarbon substrates. With the notable exception of alcohols, like α-terpineol (41, Scheme 7 and Fig. 8), directly generated form a cyclase primary cation by water trapping, the cyclic monoterpene alcohols show very high optical purity, as exemplified by menthol (42, Fig. 8).70 The cyclase phase in the biosynthesis of triterpenoids is of the cascade-type, and is triggered by protonation of a double bond or an epoxide rather than by ionization of a pyrophosphate. However, this difference alone does not explain the enantiomeric purity of these compounds, since the cascade cyclization mode can also operate in diterpenoids, where it can generate enantiomeric forms.72 The occurrence of triterpenoids in a single enantiomeric form could perhaps be traced to the high genomic burden associated to building enantiomorphic active sites for large substrates. Thus, it may not be by chance that antipodal monoterpene synthases recognize enantiomeric substrate folds,74 while enantiodivergence associated to antipodal sesquiterpene synthases exploits differences in reaction mechanism (see infra).75
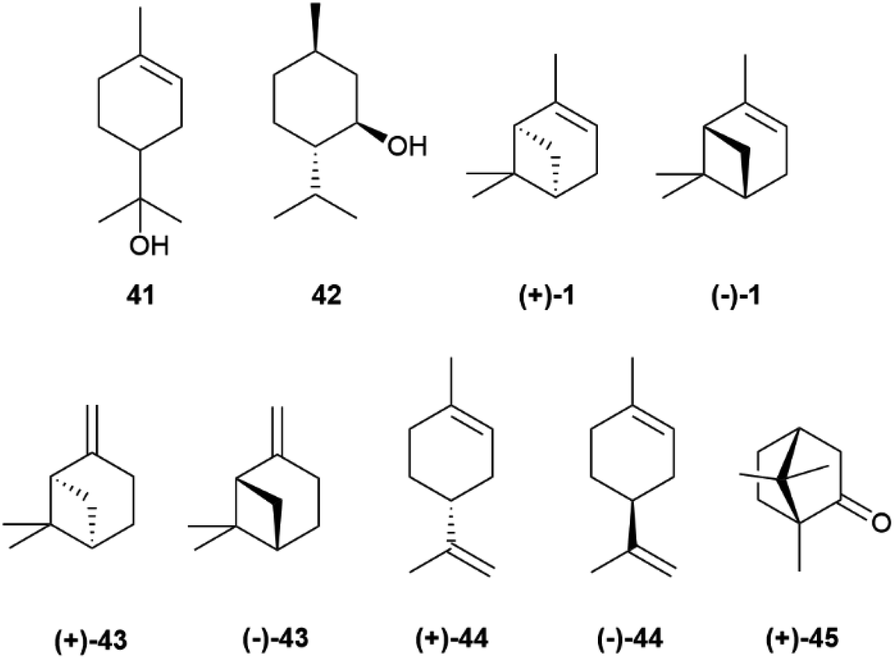 | ||
| Fig. 8 Structure of chiral monoterpenes 41–45 and 1. | ||
The enantiomeric composition trait of monoterpene hydrocarbons differs greatly even between closely related compounds. Thus, while (+)-α-pinene ((+)-1, Fig. 8), (−)-β-pinene ((−)-43) and (+)-limonene ((+)-44), can be isolated in large amounts and with ee around or even higher than 90%, their enantiomers ((−)-1, (+)-43 and (−)-44, respectively) are only available in lower amounts and optical purity.70 The optical purity of pinenes can be upgraded (see Section 4), and they can serve as starting material for the synthesis of other enantiopure terpene hydrocarbons, including (−)-limonene ((−)-44), scarcely available by isolation (Fig. 8). Since many oxygenated monoterpenes are crystalline, the cheap scalemic form of the “big three” can be used to prepare oxygenated terpenes. Indeed, most camphor (45) on the market is nowadays obtained not by isolation but by synthesis from natural α-pinene.70
Many monoterpene synthases have been cloned and heterologously expressed in bacteria and yeasts.75 However, yields have so far remained significantly lower than those achieved for hemiterpenes and sesquiterpenes, some of which can be obtained by fermentation in transgenic organisms in two- or even three digits mg yields per liter.76
The scalemic state of monoterpene hydrocarbons is generally due to the co-expression of antipodal synthases,74 and more rarely to the generation of uneven ratios of both enantiomers by the same cyclase.77 All monoterpene synthases share a mechanism based on the ionization of a linear isoprenyl diphosphate by a trinuclear magnesium(II) cluster bound to an aspartate-rich motif (DDXD) in the active site, a feature generally conserved in plant, microbial and fungal isoprenoid synthases.77 The cation is then trapped, in a head to tail fashion, by the distal double bond, generating a cationic species that can be directly quenched by proton loss or water trapping (Scheme 7). Alternatively, the cation can undergo one of more sequences of Wagner–Meerwein rearrangement or of hydride shift before the termination step.77 Cyclases are not generally highly selective in the termination step and, therefore, at directing the biosynthetic flux down one specific reaction manyfold. Thus, half of the known terpene cyclases generate, in addition to a main reaction product, a constellation of additional products which generally makes up >10% of the reaction mixture.77
Geranyl pyrophosphate (38, Scheme 8) cannot cyclize directly, since the interaction of its cationic head with the nucleophilic terminal double bond of the substrate would generated an E-cyclohexene. Therefore, isomerization to linalyl pyrophosphate (39) and next to a linaly cation 40 must take place in the course of the reaction, leading to two enantiomeric helical conformations A and B (Scheme 8) for the achiral linalyl cation precursor. Alternatively, but much more rarely, the cyclase can directly recognize a neryl pyrophosphate substrate, like in tomato glandular trichomes.76
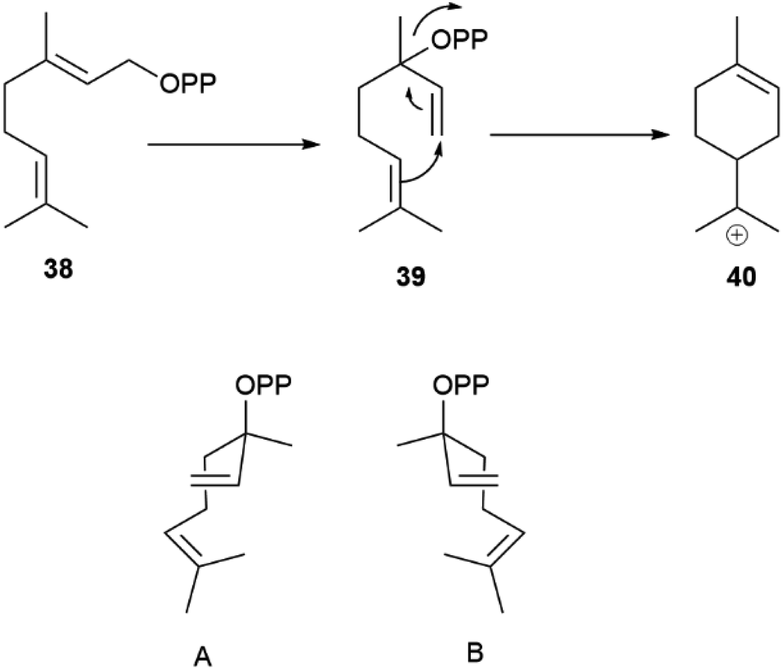 | ||
| Scheme 8 Head-to-tail cyclization of geranyl diphosphate (38) via enantiomeric folds (A and B, 3aR and 3aS, respectively) of linalyl diphosphate (39). | ||
Generally, only a single cissoid anti-endo fold is recognized by the cyclase (Scheme 8). Therefore, when more than one constitutional isomer is generated by distinct termination steps, only monoterpenes belonging to the same configurational series are formed.76 Thus, the (−) limonene synthase from Abies grandis (Douglas ex D. Don) Lindl. in addition to (−)-limonene ((−)-44) also produces (−)-α- and (−)-β-pinenes ((−)-1 and (−)-43, respectively), all three compounds deriving from the same (S)-menthenyl cation ((S)-40).78 Exceptions are, however known, and some terpene cyclases produce scalemic cyclic terpenes or mixtures of cyclic terpenes derived from different folds of the linalyl precursor.77 Remarkably, terpene synthases with different enantioselectivity can be co-expressed. Thus, from the loblolly pine (Pinus taeda L.), a major pulpwood species and the most common tree species in US after red maple,79 two enantioselective (+) and (−)-α-pinene synthases were cloned, while the α-terpineol synthase from the same source plant produced a scalemate, with only ca 80% enantiomeric excess.74 The two antipodal α-pinene synthases from the loblolly pine show relatively low level (75%) of sequence homology, suggesting that they diverged from a common ancestor long ago.74 Modifications of chemoselectivity in terpene synthases require the change of only a few amino acids, but the sequence differences between the two antipodal α-pinene synthases shows that the alteration of substrate enantioselectivity has a much higher genomic cost.74
Germacrene D synthase is the best characterized case of differently co-expressed antipodal and enantioselective terpene synthases.75,80 Germacrene D (46, Fig. 9) is an important intermediate in sesquiterpene biosynthesis, and occurs across a wide range of organisms, from bacteria to liverworts and plants. The (+)-enantiomer ((+)-46) is typical of lower organisms, while most plant produce the (−)-enantiomer ((−)-46).80
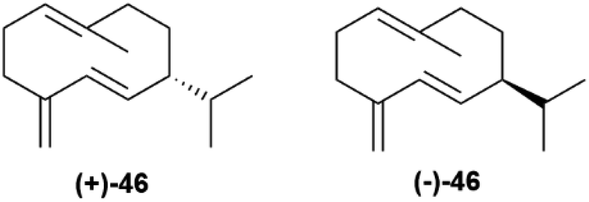 | ||
| Fig. 9 Structure of the two germacrene D-enantiomers. | ||
Asteraceous species from the genus Solidago are an exception, since they produce both enantiomeric forms of germacrene D in a ratio which depends on season, location and plant part investigated, with an almost racemic state detected in most accessions analyzed.81 Two antipodal germacrene D synthases were characterized from S. altissima L. (tall goldenrod), and, by using deuterium-labelled substrates, it was shown that they differ in the mechanism of charge propagating step.75,80 Both enzymes generate an 11-germacradienyl cation (48, Scheme 9) by intramolecular interaction between the electrophilic diphosphate site and the nucleophilic ω-double bond of farnesyl pyrophosphate (47, Scheme 9), and both enzymes form the Δ5(15) exomethylene by proton loss from the allylic C-15 methyl of a C-6 germacra-Δ1(10),5-dienyl cation (48, Scheme 9). However, the propagation step in the (−)-germacrene D synthase involves a 1,3 anionic shift of the β-C-6 hydrogen to C-11 (Scheme 9b), while the formation (+)-germacrene D (+)-46 involves a sequence of two 1,2-hydride shifts, the first one from the α-hydrogen at C-7 to C-11, and the second one of the 6β-hydrogen to C-7 (Scheme 9a). As a result of the different shift of the 6β-hydrogen, the two enantiomeric germacrene D are formed. Both Solidago germacrene D synthases were cloned, and showed a remarkable sequence identity, as expected for a mechanism of enantio-divergency based on a reaction course rather than on the recognition of enantiomeric chiral substrate folds.80
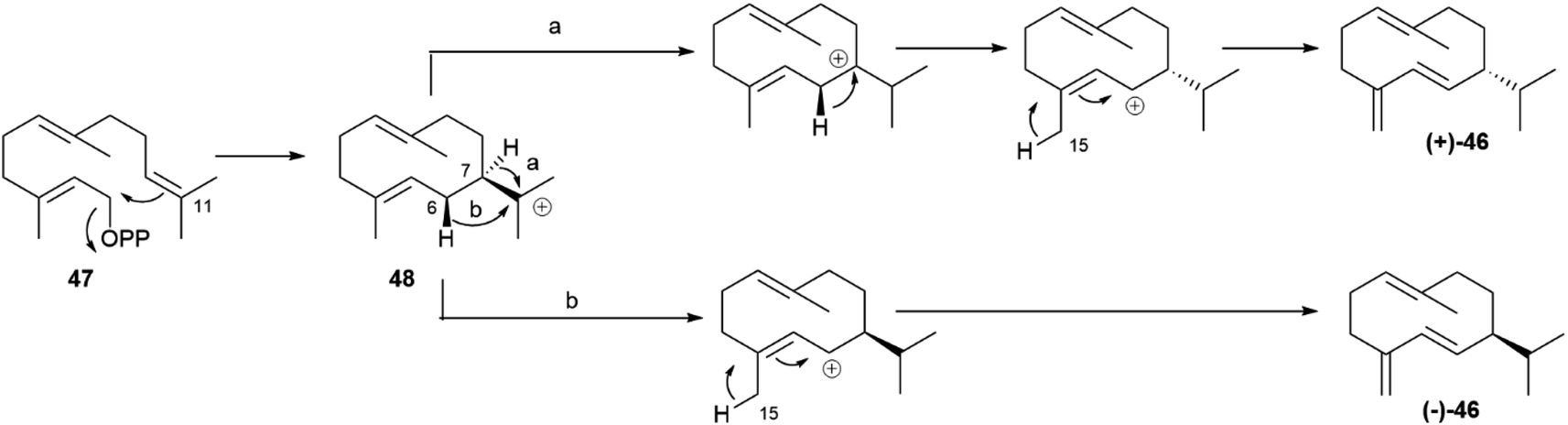 | ||
| Scheme 9 Mechanism for the enantioselective formation of (+)- and (−)-germacrene D ((+)-46 and (−)-46, respectively) by antipodal germacrene D synthases. | ||
 | ||
| Scheme 10 Biogenetic derivation of pyrrolidine (n = 1) and piperidine (n = 2) alkaloids. | ||
Some of these alkaloids and pseudoalkaloids are of significant relevance for toxicology and our cultural history. Coniine, the poisonous principle of spotted hemlock (Conium maculatum L.), was the first “alkaloid” to be structurally elucidated (Hofmann, 1881)83 and synthesized (Ladenburg, 1886).84 Coniine 49 and its N-methylderivative (50), that actually are pseudoalkaloids, are scalemic, with a prevalence of the (S)-(+)-enantiomer ((S)-49) (Fig. 10).85 Coniine binds to the nicotinic acethylcholine receptor (nAChR), activating and eventually blocking it, with depolarization of post synaptic neuromuscular junctions and flaccid paralysis.85 The pharmacological potency of coniine enantiomers is different, with the (R)-(−) enantiomer ((R)-49) outperforming the (S)-(+) enantiomer ((S)-49) in terms of both cholinergic activity (one order of magnitude) and overall acute toxicity (murine LD50 7 mg vs. 12 mg kg−1 by IV administration in mice).85 Chronic administration of sub-lethal dosages of coniine is associated to teratogenic effects, and it has been suggested that the major toxicity of (R)-(−) coniine ((R)-49) is retained also in terms of chronic effects.85 The toxicological relevance of coniine is not only historical (Socrates's death sentence was carried out with hemlock juice), since it is still associated to cattle loss and to human fatalities, both by direct consumption of the plant86 and via the food chain. Thus, coniine is not toxic for quails (Cortunix corturnix coturnix), and possibly also some other birds, which can impudently eat its seeds and store the alkaloid in their flesh.87 The consumption of quails can be associated to severe toxicity (coturnism) with death and rhabdomyolysis in severe cases.87 Quails also feed on other toxic plants like henbane and water hemlock, and thus coniine could be only one of the toxic cocktail accumulated by these birds. The involvement of a hereditary enzyme deficiency has also been suggested to play a role, but no specific enzyme has yet been identified.87 Coniine originates from the NADPH-dependent reduction of achiral γ-conicein (51) by a specific reductase.88 In turn, γ-conicein originates from 52, the reductive transamination product of 5-oxooctanal, a polyketide construct.89 The transcriptome sequencing of coniin biosynthesis in C. maculatum has been reported, and eight transcripts for γ-coniceine reductase were identified.89 However, additional studies to validate the candidates' function and confirm their role in planta have not yet been carried out, and therefore the presence of functionally distinct forms of the enzyme needs confirmation. Coniine-type alkaloids also occur in plants unrelated to hemlock: seven carnivorous species of the genus Sarracenia and twelve Aloes have been reported to contain these alkaloids.90 It has been speculated that S. flava uses coniine to paralyze its insect prays, but the enantiomeric composition of coniin in these plants is not known.
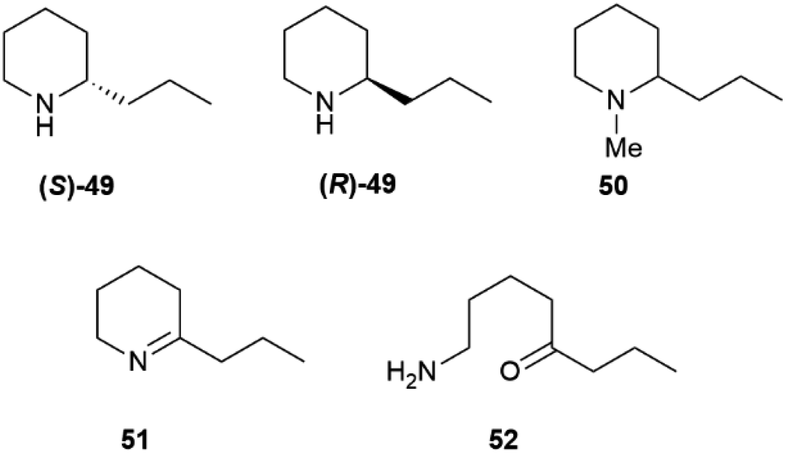 | ||
| Fig. 10 Structures of piperidine alkaloids 49–51 and their precursor 52. | ||
Nicotine (S)-7, at least when isolated from tobacco leaves, can be considered almost optically pure due to the selective demethylation of the R-enantiomer ((R)-7,Fig. 1) described in the introduction. On the other hand, its related minor tobacco alkaloids are scalemic [nornicotine (53) and anatabine (54)] or almost racemic [anabasine, 8] (Fig. 11).91,92
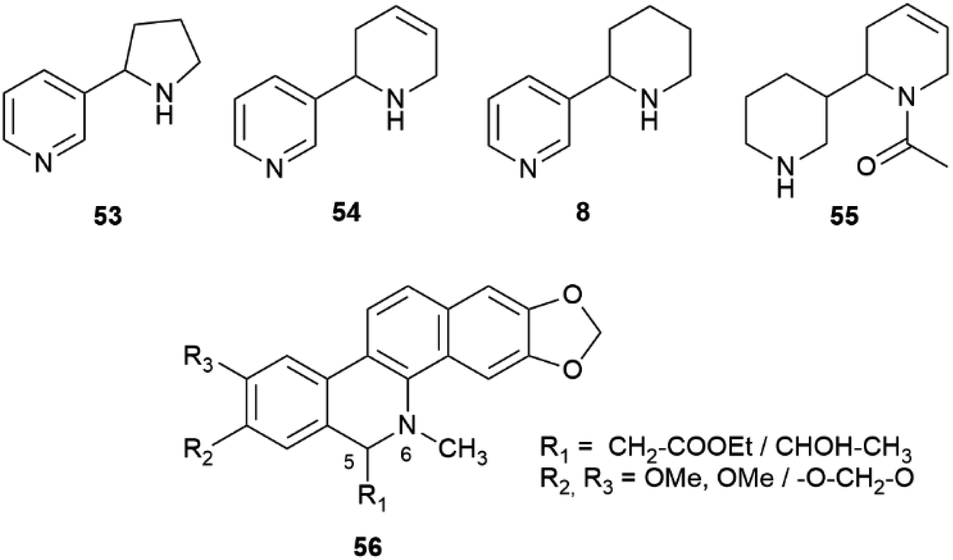 | ||
| Fig. 11 Structures of “minor” tobacco alkaloids (53, 54, 8, 55) and of the benzophenanthridine alkaloids of general formula 56. | ||
Overall, the issue of chirality seems to have been largely overlooked also in the study of the genomic and enzymology of pyridine alkaloid biosynthesis, one contributing reason being the contrasting literature data on their enantiomeric composition, possibly related to SDE associated to the volatily of these alkaloids. Tobacco alkaloids are all structurally similar, but can have different biogenetic origin.91,92 Nicotine (S)-7 and anabasine 8 derive from the convergence of the diamine and the NADPH pathways (Scheme 11) and only differ for the starting amino acid precursor.
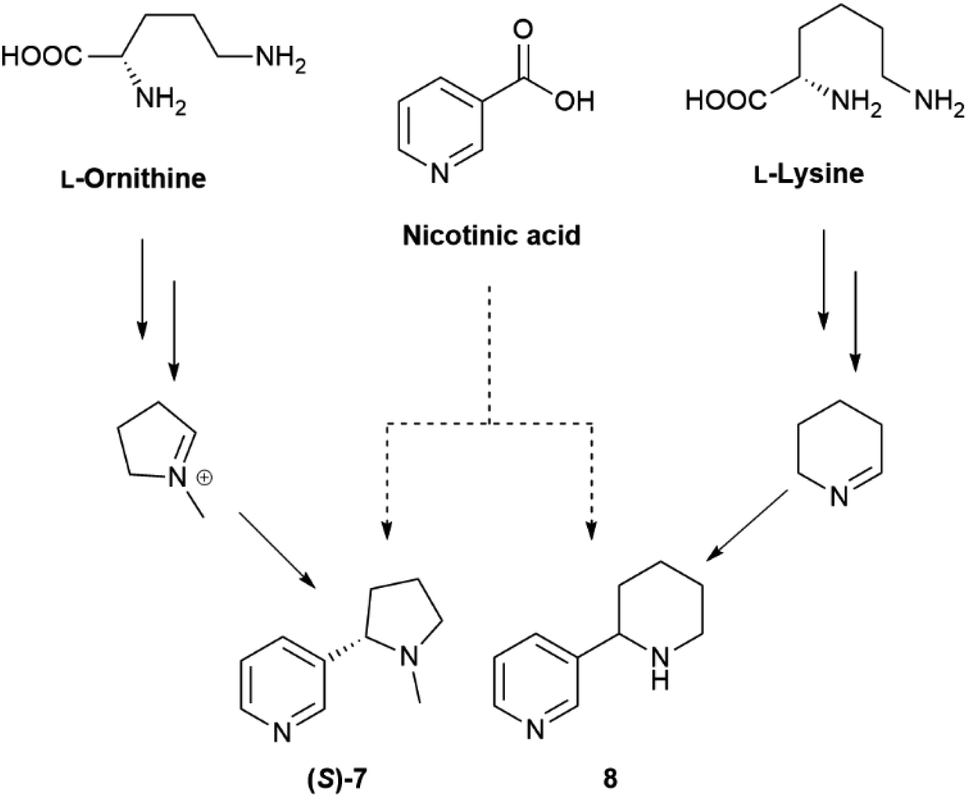 | ||
| Scheme 11 Biosynthesis of nicotine (S)-7 and anabasine 8. | ||
On the other hand, anatabine (54) derives from the dimerization of two nicotinic acid-derived dihydropyridine intermediates, and nornicotine (53) from the demethylation of nicotine.91 Chirality is associated to the formation of the dinuclear system, and different versions of the coupling enzyme seemingly exist. Also puzzling is the racemic state of anabasine (8) in the leaves, which sharply contrasts with the optical purity of nicotine. Anabasine is a potent teratogen, and is the major alkaloid of Nicotiana glauca L., where it occurs as a scalemate.82 The teratogenic alkaloid ammodendrine (55, Fig. 11) from varius lupin (Lupinus spp. vv.) species is biogenetically related to anatabine, and is also scalemic.82
Intermolecular iminium ion trapping can also be at the origin of scalemate formation, as exemplified by a series of dihydrobenzophenathridine alkaloids from celandine (Chelidonium majus L.) of general formula 56 (Scheme 12), derived from nucleophilic addition to a 5,6-iminium ion 57. The nucleophilc species is derived from an acetate unit 58via enolization or from a thiamine-associated umpolung synthon 59.93
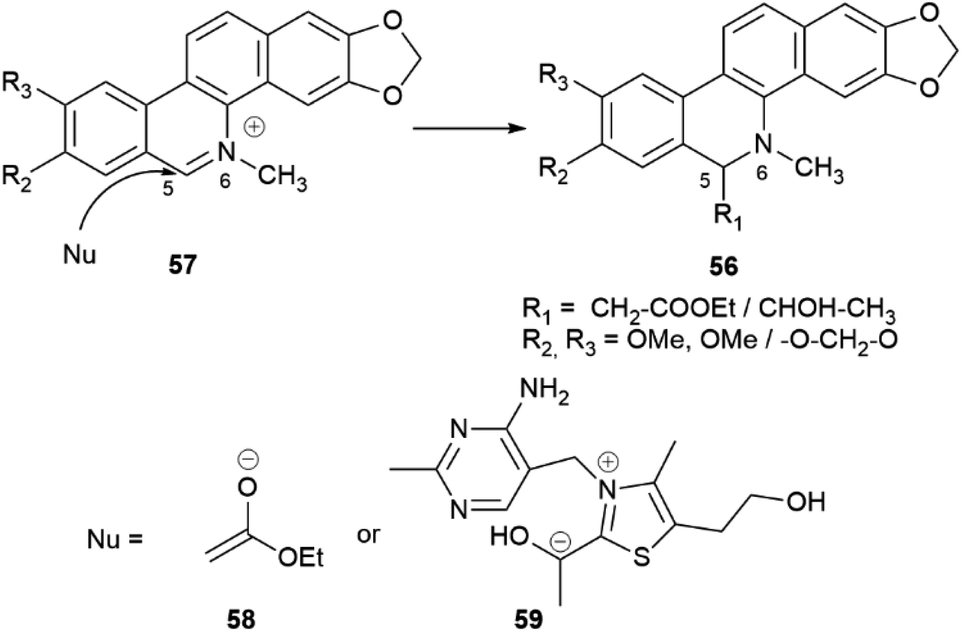 | ||
| Scheme 12 Non-enantioselective formation of alkaloids of general formula 56. | ||
3.2.3.1 Alkyl coupling. The monoelectronic oxidation of alkenylphenols by laccases and related oxygenases generates stabilized radicals that can couple with different regio- and stereo-chemical results. Of particular relevance is coupling at the distal alkyl carbon, since it is involved in the generation of the complex polymer lignin as well as of lignans, a class of C6–C3 dimers of wide distribution in plants. While lignin is racemic, lignans originating from this coupling, as well as their reductive cleavage products, are generally obtained as scalemates by isolation, and only more rarely as optically pure or racemic species. The oxidative dimerization of the monolignol E-coniferyl alcohol (60, Scheme 13) to the furofuran pinoresinol (61), and the subsequent stepwise reduction to the furan lariciresinol (62) and the dibenzylbutane secoisolariciresinol (63) exemplify the early steps of lignan biosynthesis.94,95 The principal isomers isolated from Forsythia intermedia are depicted in Scheme 13. However, the compounds are generated in variable enantiomeric composition, depending on several factors.
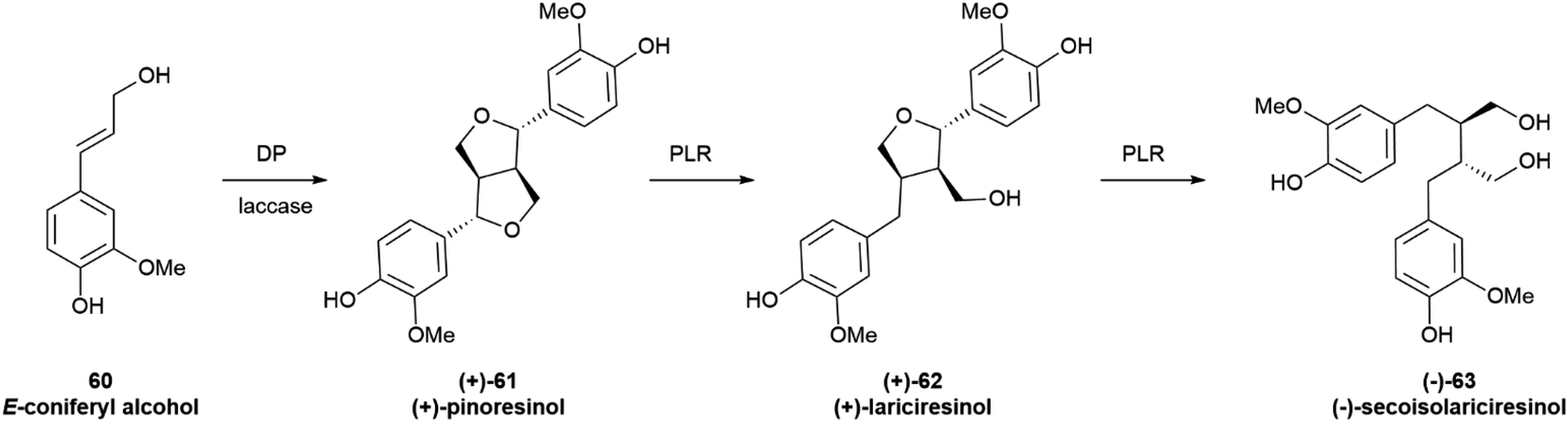 | ||
| Scheme 13 Oxidative coupling of E-coniferyl alcohol (60) to the furofuran lignan pinoresinol (61) is followed by stepwise reduction to lariciresinol (62) and to secoisolariciresinol (63). DP = Dirigent protein, PLR = Pinoresinol-lariciresinol reductases. The major isomers isolated from Forsythia intermedia are depicted. However, the compounds are generated in variable enantiomeric composition depending on several factors detailed in the text. | ||
The products of further downstream biogenetic oxidative elaboration are generally enantiomerically pure, as exemplified by podophyllotoxin (64). Thus, while pinoresinol (61) occurs in scalemic forms of opposite enantiodominance even within same plant, the same enantiomeric form of podophyllotoxin (64, Fig. 12) and its analogues have been isolated in enantiopure form in not less than nine distinct and taxonomically unrelated plant families.96
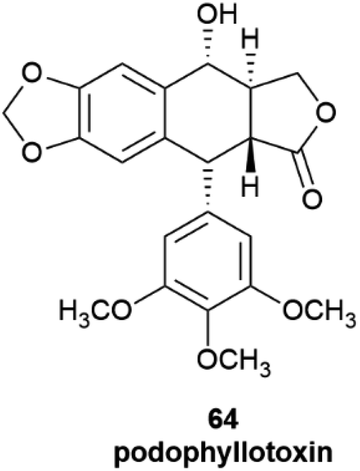 | ||
| Fig. 12 Podophillotoxin 64. | ||
The different enantiopurity of lignin and lignans is related to the cellular milieu where the monoelectronic oxidation takes place. The dimerization of monolignol, as exemplified by coniferyl alcohol (60) is initiated by a single-electron transfer catalysed by a laccase or a peroxidase (Scheme 14). The resulting delocalized radical has three distinct coupling modes, and can generate 8-O-4′-, 8–8′- or 8–5′- linked dimers.97 The bis-quinonmethide generated in the 8–8′ coupling step next undergoes a double intramolecular oxa-Michael addition, eventually generating a diaryl furofuran system (61). Coupling outside the cell membrane is regio- and stereo-random (Scheme 14, left), generating an achiral polymer (lignin) where all three types of coupling are represented. Conversely, intracellular coupling is chemoselective, and leads regioselectively to dimers (monolignols) that originate only from 8–8′ coupling and have a variable degree of enantiopurity (Scheme 14, right). This remarkable difference has been related to the association of the oxidizing enzyme(s) with specific non-enzymatically active cytoplasmatic proteins, that have been named Directing Proteins (DP).98 These ubiquitous proteins bind the reactive radicals generated by the oxidase enzymes, fluxing them into distinct coupling manifolds.
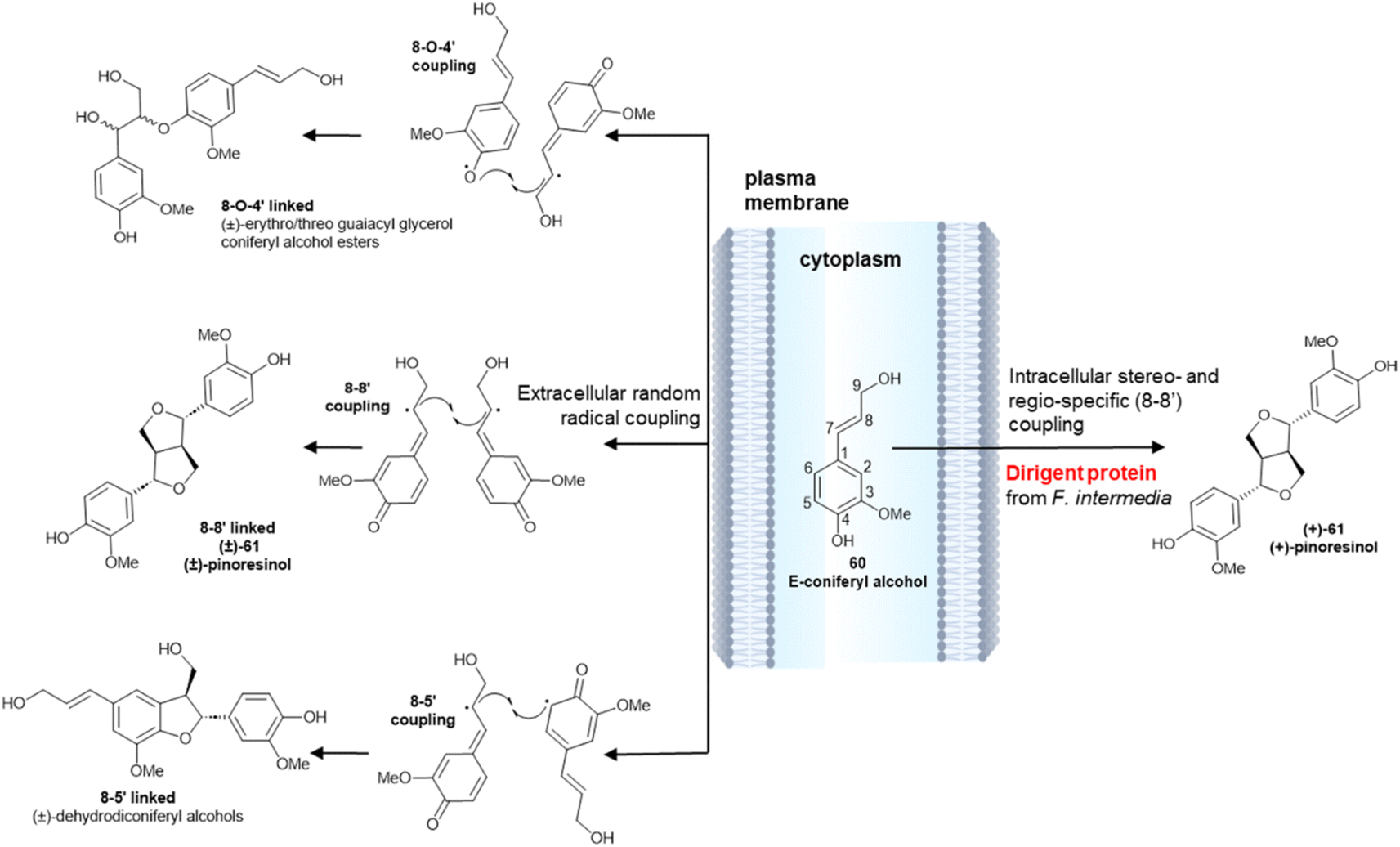 | ||
| Scheme 14 Random radical coupling of E-coniferyl alcohol 60vs. stereo- and regio-specific intracellular coupling in the presence of a Dirigent Protein from Forsythia intermedia (ref. 98). | ||
The seminal discovery of DP was reported in 1997 as a result of extensive studies on the biosynthesis of lignans in plants from the genus Forsythia, a prolific producer of 8,8′-lignans. Using in vivo tracer labelling studies, the formation of (+) pinoresinol ((+)-61, Scheme 15) was investigated in cell cultures and in cell-free extracts of the plant, using hydrogen peroxide as a cofactor. The formation of (+)-61 involved the cooperation of two proteins, only one of which, a laccase, endowed with enzymatic activity. The second protein did not affect the rate of the one-electron oxidation step, rather it appeared to bind the laccase generated radical to secure regio- and enantioselectivity of the coupling step.98,99 When the reaction was carried out with the laccase alone, a complex mixture of racemic isomers was generated, highlighting the key role of DP to steer in a regio- and enantioselective fashion the coupling reaction.
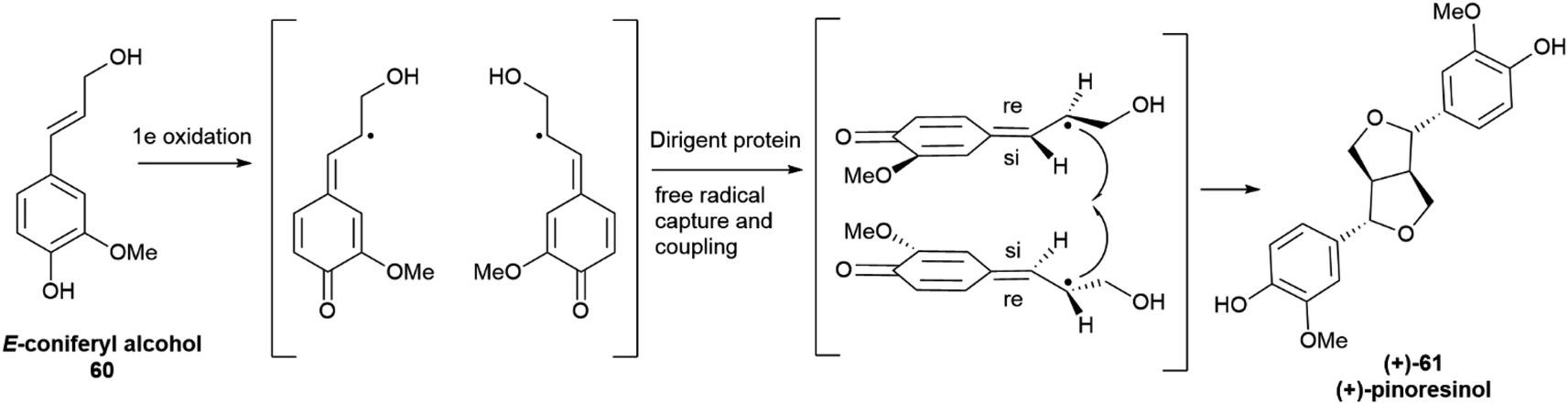 | ||
| Scheme 15 Directing protein-mediated oxidative coupling of the monolignol 60 (coniferyl alcohol) into (+)-pinoresinol (+)-61 in Forsythia intermedia. | ||
DP showing opposite enantioselectivity relative to the one described in Forsythia were characterized from other plants.100 Crystal structures were obtained for the DPs associated to the generation of (+) pinoresinol ((+)-61) in P. sativum (PsDRR20) and of (−)-pinoresinol ((−)-61) in Arabidopsis thaliana L. (PsDRR20 and AtDIR6, respectively).101 Both proteins are eight-stranded antiparallel β-barrel trimers, and present two-lobed cavities with adjacent binding sites to accommodate a pair of coniferyl alcohol radicals in an orientation suitable for the 8–8′ coupling. One DP favours coupling at the si–si faces, and the other one at the re–re face of the interacting radicals, leading to (R,R)- or (S,S)-bisquinonmethides, and next to (+) or (−)-pinoresinol.102
Despite the existence of enantioselective DPs, pinoresinol occurs in many plants as a scalemate,94 presumably because of the presence of antipodal DPs, or because of a suboptimal production of a single DP and a kinetic mismatch between the rate of radical generation by the laccase and the rate of radical trapping by the DP. Different organs of the same plant can also contain different enantiomers of pinoresinol, due to the expression of distinct DPs. Thus, burdock (Arctium lappa L.) contains the (+)-enantiomer (+)-61 in petioles and the (−)-enantiomer (−)-61 in the seeds.103 Environmental factors also play a role, since DPs are thermolabile, and are inactivated by temperatures >33 °C.104 The erosion of enantiomeric purity due to a suboptimal concentration of DP is also associated to a reduction of regioselectivity, with generation of other types of couplings (8–5′-and 8-O-4′).104
The furofuran system of the primary coupling product pinoresinol next undergoes reductive cleavage, with formation of monocyclic and then acyclic derivatives (62 and 63 in Scheme 13, respectively). In this way, the furan-type lignan lariciresinol (62) and the dibenzyl-type lignan secoisolariciresinol (63) are generated from the furofuran lignan pinoresinol (61) through the agency of the NADPH-dependent pinoresinol/lariciresinol reductase (PLR). Two isoforms of this enzyme have been characterized, displaying opposite enantioselectivity.105 The enantiomeric ratio of the starting pinoresinol is significantly affected by the agency of these two reductases, which show various degrees of enantioselectivity toward their substrates and different levels of expression in different plant tissues. Thus, pinoresinol reductase kinetically resolves its scalemic (−)-substrate in the seeds of A. lappa L., but significantly erodes the enantiomeric excess of scalemic (−) pinoresiol in Wikstroemia sikokiana Franch. & Sav. and in Forsythia koreana (Rehder) Nakai.106 Similarly, lariciresinol reductase from F. koreana upgrades the enantiomeric excess of its substrate, generating (−)-secoisolariciresinol (−)-63 in >99% ee from a highly scalemic (−) lariciresinol (ee ca 35%). In contrast, the lariciresinol reductase in the seeds of A. lappa L. significantly erodes the optical purity of its substrate, generating (−)-secoisolariciresinol of 38% ee from optically pure (−) lariciresinol.106
In summary, furofuran lignans and their products of reductive cleavage are a major class of scalemic natural products. The scalemic state is primarily associated to a suboptimal involvement of Dirigent proteins in the oxidative coupling of the monolignol precursor, but the enantiomeric composition can be significantly affected by the activity of reductases en route to monofuran and dibenzylbutane lignans. The further oxidative modification of these compounds is instead associated to the activity of highly enantioselective oxidizing enzymes, and aryltetralin lignans and other biogenetically downstream lignans are generally isolated in enantiomerically pure form.
Little systematic attention has been given to the enantiomeric composition of neolignans, the result of 8–5′ coupling of the quinonmethide radicals (Scheme 14), but it has been suggested that the scalemic state discovered in gardenifolin A (65, Fig. 13) and related compounds from Gardenia ternifolia could be widespread, or even the norm within this class of compounds.107
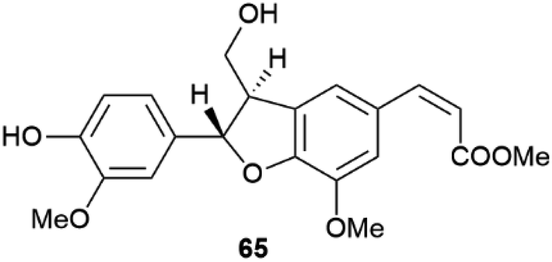 | ||
| Fig. 13 Structure of gardenifolin A 65. | ||
3.2.3.2 Aryl coupling. Biaryl coupling is involved in the biosynthesis of compounds from different classes, and one could cogently argue that the presence of an electron-rich aryl moiety is an irresistible invitation for Nature to expand the diversity of secondary metabolites. In the presence of a directing stereocenter, coupling can be highly regio- and enantioselective. Thus, under the agency of specific P450 oxidases,108 the two aromatic moieties of the benzylisoquinoline alkaloid (R)-reticuline ((R)-66) couple in an ortho–para fashion to salutaridine (67) en route to morphine (68), while in (S)-reticuline ((S)-66) they couple in an ortho–ortho fashion to corytuberine (69), en route to magnoflorine (70) (Scheme 16).108
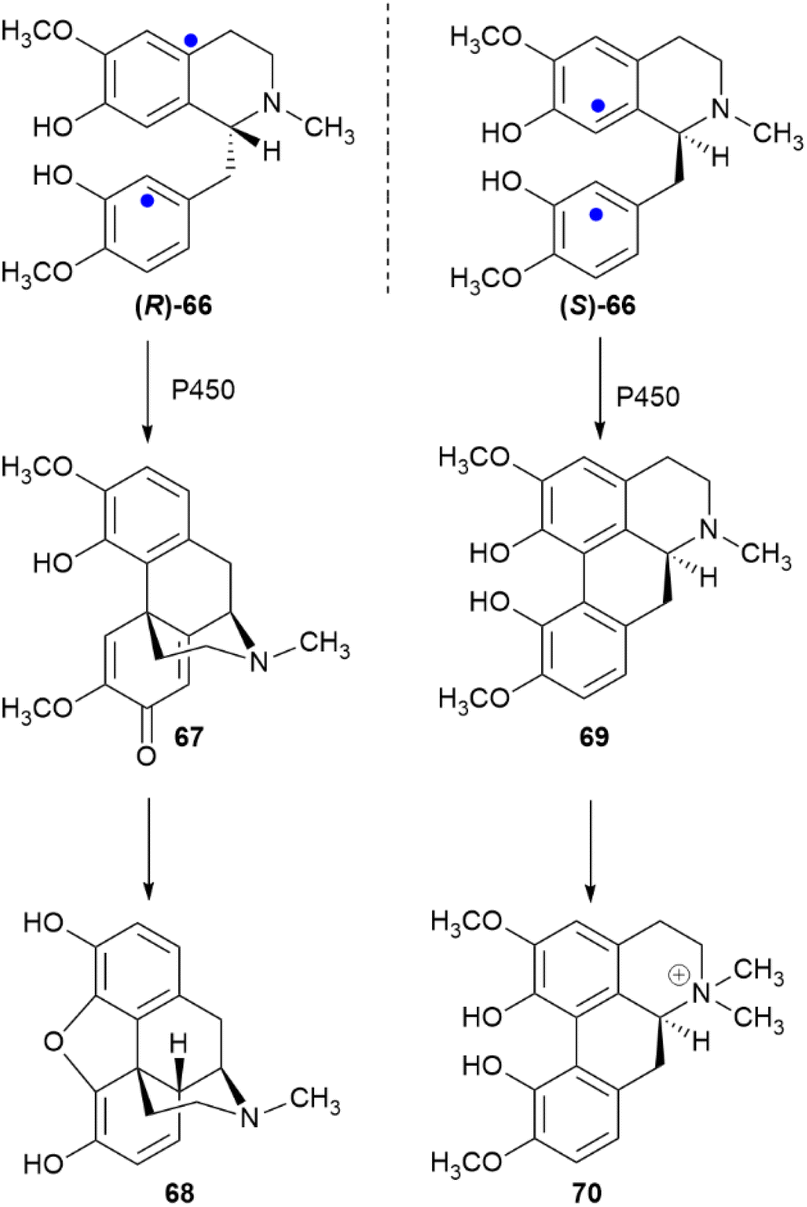 | ||
| Scheme 16 Enantiodivergent metabolic role of reticuline (66). The sites of aryl coupling are indicated by blue dots. | ||
In the absence of directing stereocenter(s), aryl coupling is more “plastic” and can generate enantiomerically pure compounds, racemates or scalemates, depending on the specificity of the oxidase involved and/or the involvement of dirigent proteins. The enantiomeric state of chiral biphenyls and their derivatives is, in any case, complex, since a low enantioselectivity in the coupling step leading to a scalemic mixtures of atropoisomers can be followed by enantioselective reactions (oxidations or glycosidations), while the presence of aromatic rings facilitates racemization reactions associated to the configuration of benzylic/vinylic positions. Chiral neolignans containing a biphenyl system are generally racemic or scalemic, but the involvement of directing proteins in their biosynthesis is unclear. Remarkably, both atropoisomers are retained even after further elaboration, including not only hydroxylation but also combination with monoterpenes with stereogenic centers, like in magmentanes (e.g. magmentane A, 71, Fig. 14) from Magnolia officinalis L.109 In compounds derived from the degradation of axially chiral phenolics, biogenetically closely related and co-occurring compounds can show a surprisingly different optical purity, as shown by the chebulic acid derivatives of Euphorbia hirta L.110 In this plant, various enantiopure esters of the degraded biphenyl chebulic acid (e.g. euphorhirtin F, 72, Fig. 14) occur along with their racemic coumarinic analogues (e.g. euphorhirtin H [(±)-73, Fig. 14]) from which, however, the scalemic rearranged cyclization product 75 (euphorhirtin L) is derived by an intramolecular Claisen condensation (Scheme 17). The racemic state of the coumarin derivatives might be associated to the increased acidity of C-3, a vinylogous α-carbonyl position, while the scalemic state of the rearranged compound could result from the activity of enantio-discriminating enzyme(s). Interestingly, the scalemic state of 75 was suggested by a crystal analysis, which showed a spatial group (P1) typical of a scalemate.110 Chiral phase HPLC next confirmed the presence of a 1:3 mixture of enantiomers in the optically active natural product,110 and one wonders how many chiral “biaryloids” assumed to be enantiopure actually deserve this status.
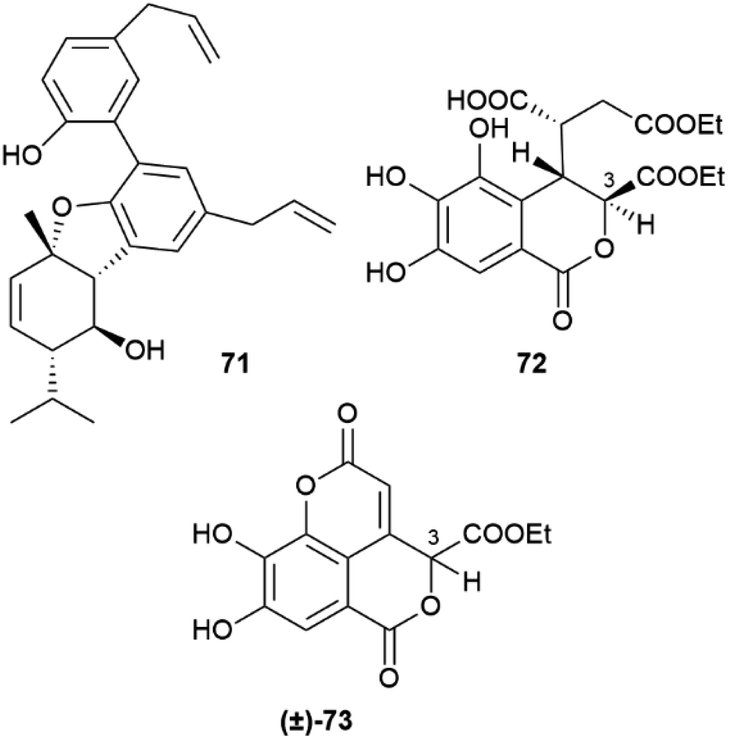 | ||
| Fig. 14 Structure of magmentane A 71, euphorhirtin F 72, euphorhirtin H 73. | ||
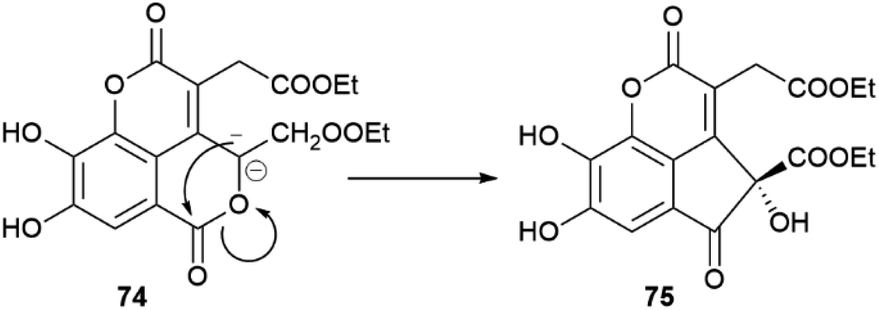 | ||
| Scheme 17 Enzymatic conversion of a racemic coumarin-type euphoritin 74 into scalemic euphorhirtin L (75) by intramolecular Claisen condensation. | ||
Another remarkable example of the plasticity of aryl coupling is usnic acid (76, Fig. 15), the archetypal lichen constituent. This compound can occur in both enantiopure forms, as a scalemate, or as a racemate depending on the lichen species.111 In addition, some lichens also contain iso-usnic acid (77), a signature of a non-regioselective coupling.111 Usnic acid used to be a popular ingredient in weight-loss products, but was later banned by FDA because of its association to severe liver damage.112 Remarkably, the huge toxicological literature on the hepatotoxicity of usnic acid often ignores its stereochemical diversity. Since the biological profile of the two enantiomers is not overlapping,113 one wonders if some controversial aspects of the toxicity profile of usnic acid could actually be related to the study of samples having a different enantiomeric composition.
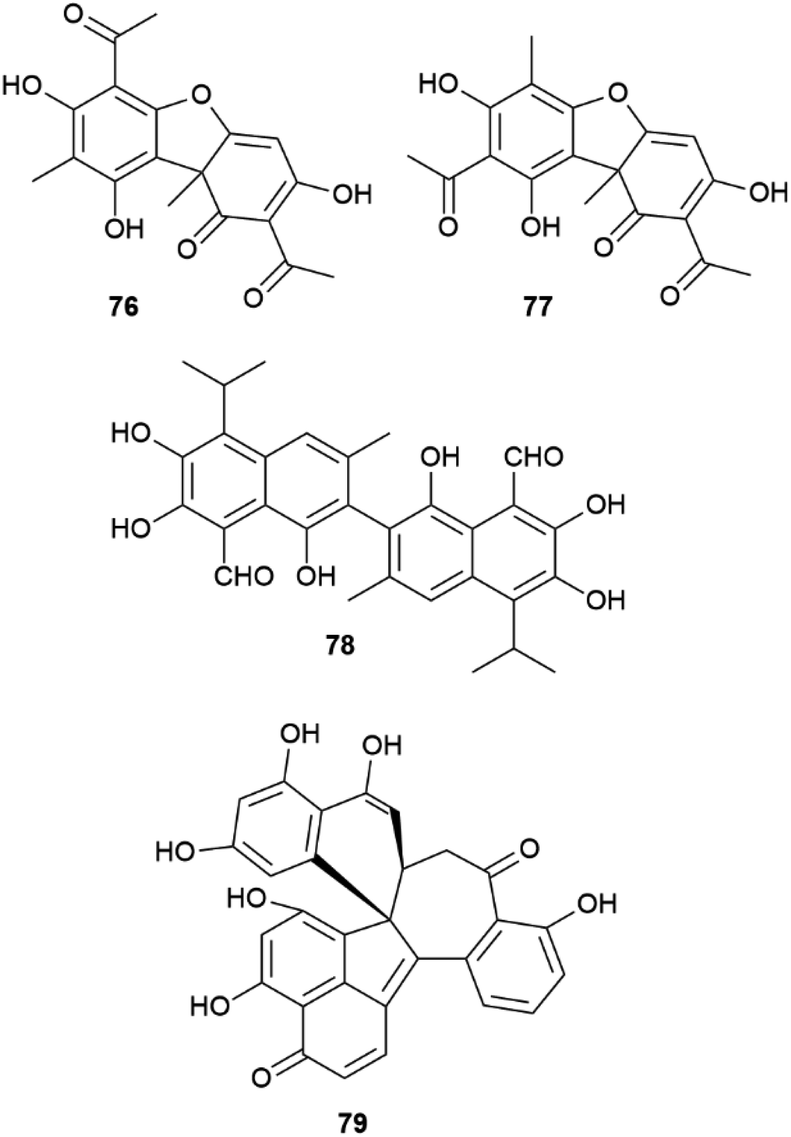 | ||
| Fig. 15 Structure of usnic acid (76), iso-usnic acid (77), gossypol (78), dalesconol C (79). | ||
Also the binaphthyl gossypol (78) shows a different distribution in the cotton plant in terms of enantiopurity. Depending on the cotton variety or of the Gossypium species, scalemic forms with different dominant enantiomers have been described, with the (+) atropoisomeric form generally prevailing.114 A specific dirigent protein (GhDIR4) has been described to confer atroposelectivity to hemigossypol coupling in the presence of laccase and oxygen.115 GhDIR4 was the second dirigent protein to be discovered and cloned, providing support to the general involvement of these agents in oxidative coupling reactions, not only in lignans biosynthesis.115 The laccase-promoted dimerization of hemigossypol to gossypol in the presence of GhDIR4 p reduces (+)-gossypol in more than 80% ee. The (−) enantiomer prevails in some Gossypium species, but characterization of the directing protein from these plants could not be carried out.115
For complex phenolics, the generation of scalemic products of aryl coupling does not depend on directing proteins, but could be related to conformational preferences of the intermediate radicals, as suggested for dalesconol C (79, Fig. 15) and related tri-naphtyls.116 Different helicities of the reacting radical(s) could be recognized differently in terms of π–π interactions by the active site of laccases, affording a scalemate whose composition depends, assuming a kinetic control, on the relative conformational stability of the radical substrate(s). This concept is, in principle, of general relevance, and worth of a systematic investigation.
Within compounds whose chirality is associated to biaryl coupling, the anticancer alkaloid cephalotaxine (5, Scheme 18) presents an additional interesting case. Depending on the Cephalotaxus species, cephalotaxine can be isolated both as enantiopure or scalemic, a surprising observation since this compound features three stereogenic carbons.7 A possible explanation can be found in the stereocontrol exerted by the stereocenter C-1 of the isoquinoline 80 over the phenol coupling that ultimately generates 81 and the subsequent cascade of events leading to cephalotaxine. Thus, after ortho–para coupling and cleavage of the 1–8a bond, the chiral dibenzo azacyclodecane 81 could stereospecifically undergo hydride abstraction coupled to nitrogen trapping (82) and afford the spirane 83, which is next converted into cephalotaxine 5 by a decarboxylative benzylic acid rearrangement and alkylative modification of the phenolic hydroxyls.117 If this entire process is ultimately controlled by the configuration of carbon C-1 in 80, the different enantiomeric purity of cephalotaxine from different sources relates back to stereocontrol in the generation of this stereocenter.
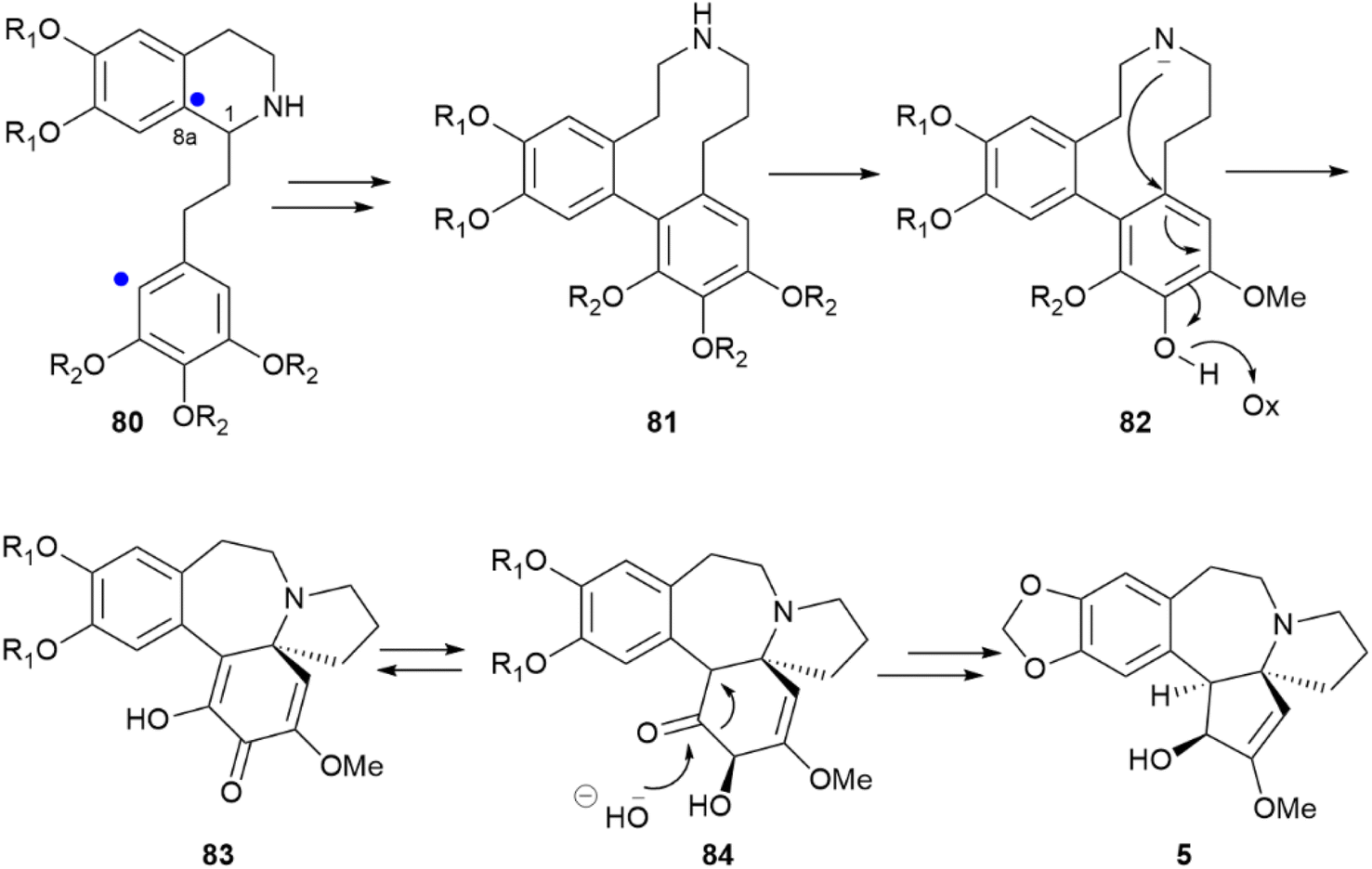 | ||
| Scheme 18 Biosynthesis of the alkaloid cephalotaxine (5) from a phenethylisoquinoline precursor (80). R1 = OH or –CH2–; R2 = OH or OMe. The sites of aryl coupling are indicated by a blue dot. | ||
Calanolides are another remarkable example of substrate-controlled enantiospecific amplification of a scalemic stereocenter. These non-nucleoside reverse transcriptase inhibitors, exemplified by calanolides A and B (85 and 86, Fig. 16), have been isolated in a range of scalemic states as well as in more biologically active enantiopure form from various trees of the Calophyllum genus.118,119 Calanolides are a veritable apax in the literature of anti-HIV drugs, not only for their non-nucleoside nature, but also for ability to alternatively bind two distinct sites of reverse transcriptase. In calanolide-producing plants, from an achiral gem-dimethyl substituted pyranocoumarin (simplified in Scheme 19 as A), a chiral compound is generated in scalemic form by hydratation (Scheme 19B), Wagner-Meerwein rearrangement of one of the gem-methyl groups (Scheme 19C), and hydride migration from C-11 to C-10 (Scheme 19D). The resulting vic-dimethyl substituted derivative [4-propyl dipetalolactone (87)] is then hydrated regioselectively to scalemic calanolides. The configuration at C-10 of 87 controls the final oxidative elaboration of the chromene double bond by specific oxygenases, affording in scalemic form all possible relative configurations at the three stereocenters. The conversion of chromene 87 into scalemic calanolides is, apparently, under genetic control, since different Calophyllum species contain calanolides of different optical purity.120
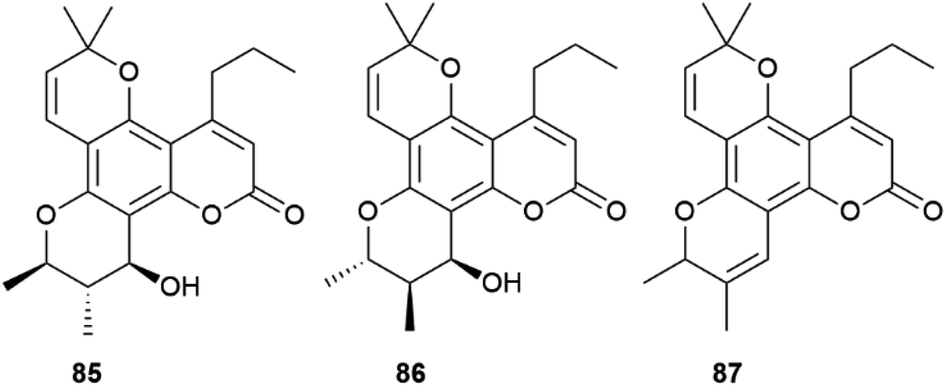 | ||
| Fig. 16 Structure of calanolides A (85), calanolides B (86) and 4-propyldipetalolactone (87). | ||
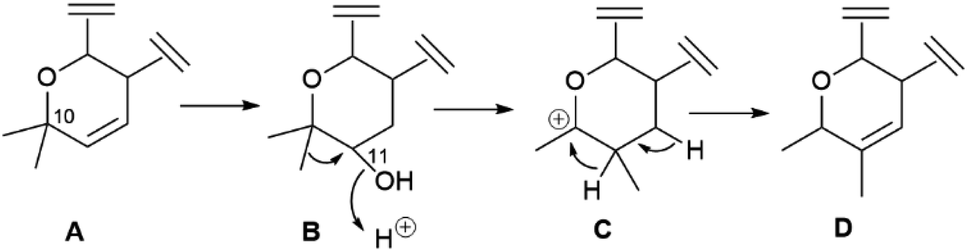 | ||
| Scheme 19 Possible biogenetic derivation of the calanolide precursor 87 from a gem-dimethylated precursor (only the dihydropyrane moiety is shown for clarity). | ||
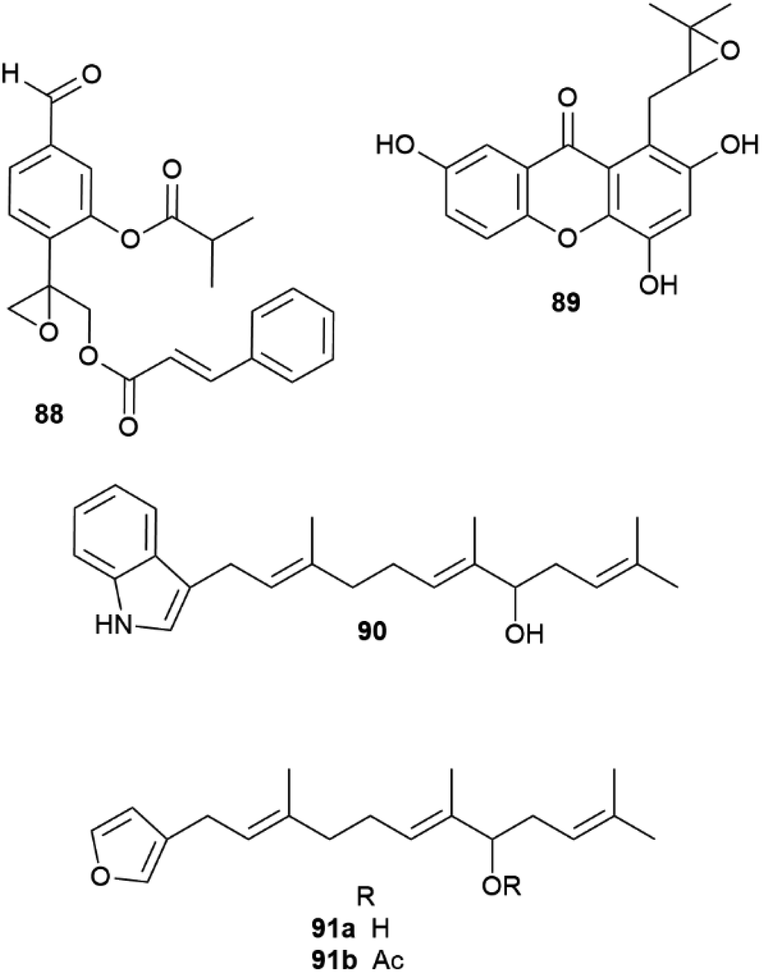 | ||
| Fig. 17 Structure of epoxythymols (88), mangoxanthone B (89), 8-farnesylindole (90), 12-hydroxyambliofuran (91a) and its acetate (91b). | ||
The allylic hydroxylation of isoprenoid moieties can also be associated to the generation of scalemates and racemates. Thus, 3-farnesylindole (90, Fig. 17) was isolated from Anomianthus dulcis in 33% ee in favour of the R-enantiomer,124 and the marine furanoditerpene 12-hydroxyambliofuran (91a) and its acetate (91b) were obtained from a Spongia species as a ca. 3:1 mixture of enantiomers.125
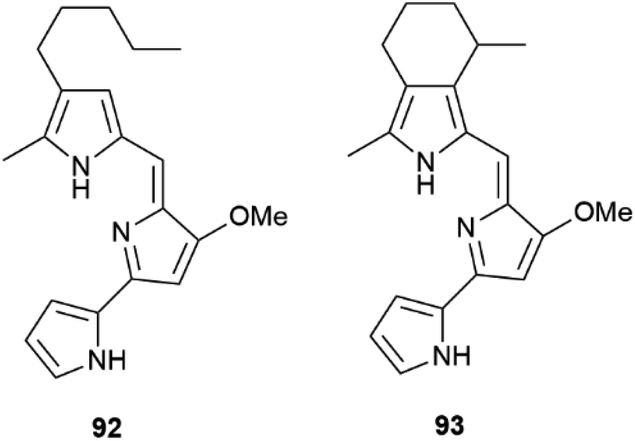 | ||
| Fig. 18 Structure of prodigiosin (92) ancycloprodigiosin (93). | ||
Many insect pheromones are also generated as scalemates by nonenantioselective hydroxylation of a non-activated carbon (see Section 6).127
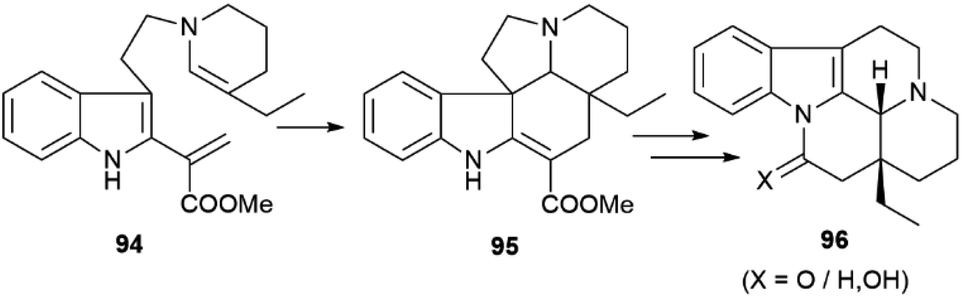 | ||
| Scheme 20 Intramolecular cycloaddition in eburnane alkaloids from the genus Kopsia. | ||
A related regular demand Diels–Alder cycloaddition from the secodine precursor (97, Scheme 21) is involved in the formation of andranginine (98), a scalemic Craspidospermum alkaloid.129
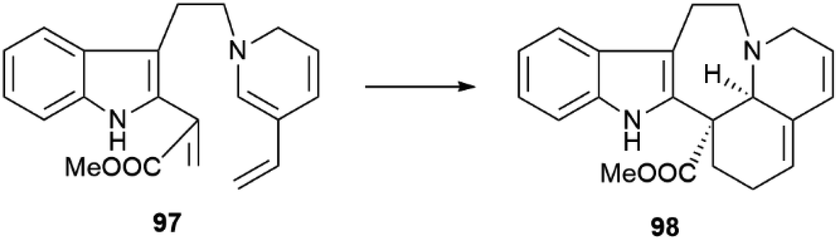 | ||
| Scheme 21 Diels–Alder reaction in the formation of andranginine (98). | ||
4 Strategies to upgrade the optical purity of scalemic natural products
The occurrence of a chiral natural product as a scalemate or a racemate rather than in an enantiopure form has interesting implications, the most obvious being that both enantiomers are available by isolation. This is an important asset not only for bioactivity studies on structurally complex natural products, but also for all synthetic applications, both as chirons (chiral building blocks) and as chiral auxiliaries, catalysts and reagents in asymmetric synthesis. The flip side of the coin is, however, that the racemic and the scalemic states imply “optical impurity”, and resolution or an ee upgrade will be necessary. In terms of synthetic applications, the most important scalemic natural products is α-pinene (1, Fig. 1). This monoterpene olefin and its derivatives have been extensively used as building blocks for the synthesis of natural and synthetic compounds and in asymmetric synthesis. For both uses, an upgrade of optical purity is necessary, since the ee of commercial α-pinene 1 does not exceed 92% for the (+)-enantiomer ((+)-1) and 81% for the more expensive (−)-enantiomer ((−)-1). More optically pure (−)-α-pinene can be obtained by treatment of (−)-β-pinene ((−)-43, Scheme 22), available by isolation in up to 92% ee, with the potassium salt of 1,3-propandiamine (potassium 3-aminopropanamide, KAPA, Scheme 22 left). The superbase isomerizes the exocyclic double bond to the thermodynamically more stable endocyclic position, with deprotonation occurring selectively at the allylic methylene since the one at the allylic methine would generate an anti-Bredt allyl anion. In this way, also (−)-pinene becomes available in >90% optical purity.130 Further upgrading of α-pinenes capitalizes on the preparation of a homodimeric borane by reaction with diborane and equilibration with an excess of the starting scalemic α-pinene (Scheme 22, right).130 Due to steric hindrance, hydroboration stops at the dialkylborane (diisopinoamphenylborane, Ipc2BH, 99) stage. This, after evaporation of the solvent, is suspended in THF and equilibrated with an excess of α-pinene. Statistic considerations favour the formation of the homodimeric borane, which precipitates from the reaction mixture. Enantiomerically upgraded α-pinenes 1 (>99% ee) could then be obtained by decomposition of the borane. The resulting diisopinocampheylborane Ipc2BH 99 has multiple uses in asymmetric synthesis.131,132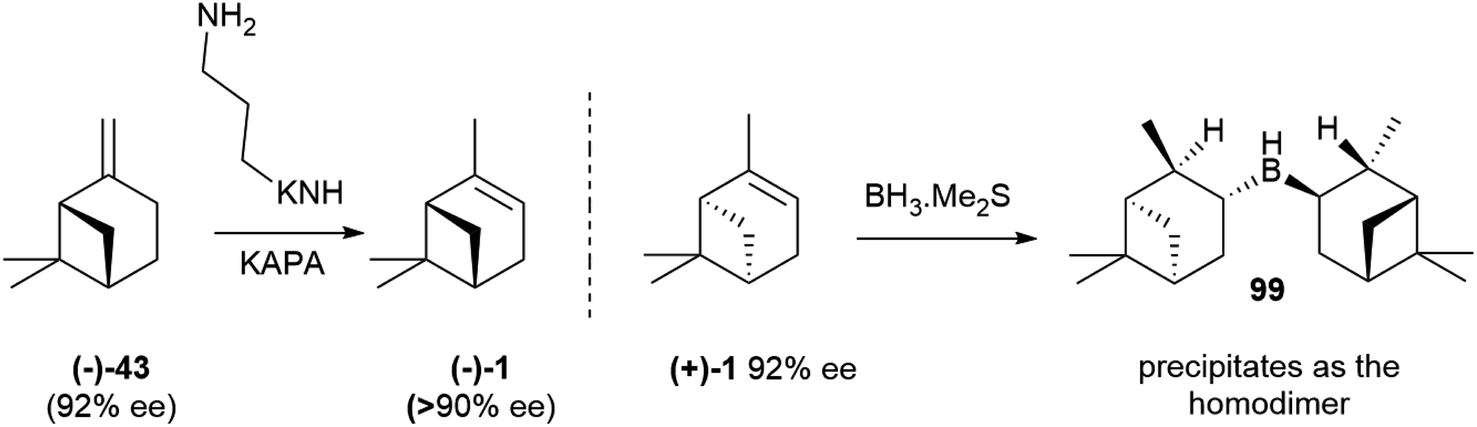 | ||
| Scheme 22 Upgrading the optical purity of α-pinenes (from ref. 130). | ||
5 Analytical applications of scalemic natural products
The determination of the enantiomeric excess of scalemic constituents provides an important asset for quality control, with interesting applications in the realm of essential oil authentication and food chemistry. Volatile scalemic monoterpenoids have proven useful to fight adulteration in the market of essential oils.133Due to a high natural variability of composition, the definition of a reference profile for an essential oil requires the analysis of a large number of certified genuine samples, and is expressed, at least for those having an official monograph like the European Pharmacopoeia, into specific ranges of concentration, or of concentration ratios, for key markers.133 Even so, the analytical landscape can be complicated by the presence of multiple botanical sources, by additional manipulation like rectification to remove sesquiterpenes or dilution with turpentine, as well as by intended or unintended exchange of raw material. In this scenario, the determination of the dominant enantiomer of a scalemic constituent can be a precious asset for authentication, as exemplified by the analysis of “pine oil”. Scalemic α-pinene is the major constituents of pine oils of different botanical origin and commercial value.134 In the essential oils from Scots pine (Pinus sylvestris L.), P. cembra L., and P. sibirica Du Tour, the (+) enantiomer (1, Fig. 19) prevails, while in those from P. nigra J. F. Arnold and P. mugo Turra (−)-α-pinene (1) dominates. The oil from P. sylvestris is the one of highest market value, but an analysis of several commercial samples labelled as P. sylvestris and complying with the European Pharmacopoeia criteria for this oil, actually showed a predominance of the (−) rather than the (+) enantiomer.134 This, coupled with the overall profile of the oils, identified P. nigra, a major turpentine source, as the probable adulterant.134
 | ||
| Fig. 19 Monoterpene markers of Scots pine, citrus, lime and tea tree essential oils. | ||
A similar situation exists for “citrus oils”, where the determination of linalool enantiomeric ratio is useful to distinguish between bitter- and sweet orange oils: in the more valuable bitter orange oil, (−)-linalol ((−)-100, Fig. 19) is the most abundant enantiomer (60–89%), while in sweet orange (+)-linalol ((+)-100) dominates.135 A lower optical purity for linalool and its acetate is diagnostic of adulteration for the oil of bergamot and lavender.133 The determination of enantiomeric ratios is also useful to identify the preparation process of an oil, as well as the geographical origin of the starting biomass. A combination of the enantiomeric ratios of linalool and α-terpineol (41, Fig. 19) can distinguish between lime oils prepared with different contact time of the peel and the pulp of the fruit, with an increased scalemicity trait being diagnostic of a longer contact time,135 while the composition of scalemic terpinen-4-ol 101 can distinguish between tea tree [Melaleuca alternifolia (Maiden & Betche) Cheel] oils of Australian and Chinese origin.133,136 In the more valuable Australian oil, the S-enantiomer of 101 prevails, with a ca 55:45 enantiomeric ratio, while in the cheaper Chinese oil the R-enantiomer is more abundant, with an R/S enantiomer ratio of ca. 58:42. The terpinen-4-ol enantiomeric ratio is now included in the ISO 4730 monograph of tea tree oil.137
Milk contains only traces of proteinogenic amino acids from the D-series, a profile not significantly affected by pasteurization or sterilization, and associated to the presence of lactic bacteria. During storage, however, bacterial enzymatic activity and/or bacterial cell lysis increase the concentration of proteinogenic amino acids from the D-series, and especially D-alanine 102 (Fig. 20), a bacterial cell wall constituent.138 The presence of D-amino acids increases steadily during cheese ripening, and in aged cheeses like Parmigiano Reggiano, the scalemicity of aspartic and glutamic acid can be used as a ripening marker.139
 | ||
| Fig. 20 Structure of proteinogenic amino acids used as biomarkers. | ||
Interestingly, the scalemicity of certain proteinogenic amino acids can also be used to determine the age of fossilized human bones and teeth too old to be dated by 14C-based isotopic analysis.140 Thus, the content of D-aspartic acid in post-mortem dental enamel increases by ca. 0.1% every year due to spontaneous racemization and this, as well as the conversion of L-isoleucine (103) to D-alloisoleucine (104, Fig. 20) has also proved useful to age estimation up to one million year of fossilized human dental remains.141
6 The importance of being scalemic: when optical impurity is advantageous
The biological profile of enantiomeric natural products is rarely overlapping, although remarkable examples of lack of enantioselective activity are known, as exemplified by the nicotinergic frog alkaloid epibatidine (105, Fig. 21).142 In many cases, the activity of a scalemate can be approximated by a “weighed” combination of the one of its constituents, but significant deviations from this linear analysis have been reported with insects pheromones, where it is not uncommon that both enantiomers are recognized, but elicit distinct activity.127 Chirality is a critical aspect of insect communication, but it is still a poorly known process involving both olfactory receptors and their associated odorant-binding proteins, which often show promiscuous binding toward enantiomers.143 On the other hand, many insect pheromones are linear compounds whose structure show low conformational constraint to assist recognition by a macromolecule.143 Using enantiopure synthetic versions of chiral pheromones, it was surprisingly demonstrated that in some cases a well-defined scalemic version is necessary for optimal activity. Thus, the macrolide pheromone 106 of the grain beetle Cryptolestes turcicus showed activity only as a scalemic mixture, with an optimal ratio between the R- and S-enantiomers of 85:15, while both pure enantiomers were inactive.144,145 In other cases, a scalemate simply outperformed its enantiomeric constituents as well as its corresponding racemic mixture. Studies on Myrmica scabrinodis demonstrated that the 9:1 scalemate of octan-3-ol 107 was more potent than the R-enantiomer, while the S-form was inactive.146 Also interesting is the observation that the most potent stereochemical version of tribolure 108, the aggregation pheromone of the red flour beetle (Tribolium castaneum), a worldwide pest of stored products, is a 4:1 mixture of the (4R,8R)- and (4R,8S)-diastereomers, and none of the single diastereo- and enantiomerically pure forms is found.147 The combined activation of distinct receptors with different affinity for their enantiomeric (diastereomeric) ligands could underlie the need of a specific combination of isomers for bioactivity, but the precise details remain unknown. Insect olfactory receptors are ion channels, while the mammal olfactory receptors are GPCR, and their regulation is presumably distinct, while the ability of insect olfactory receptors to discriminate isotopomers is still highly debated for its implication on the theory of olfaction.148
 | ||
| Fig. 21 Structure of some chiral pheromones. | ||
7 Conclusions
The scalemic state of natural products seems to have been largely underestimated in terms of both inventory and biogenetic implications. It is generally associated, either directly or via directing protein, with enzymatic activity, and is common in specific classes of chiral compounds, like monoterpene hydrocarbons and biaryls. Also the intramolecular trapping of iminium ions can be associated with a non-negligibile production of scalemates. However, additional enzymatic activity can lead to kinetic resolution via the enantioselective elaboration of only one enantiomer, as shown for nicotine in tobacco leaves. The enantiomer not recognized by downstream enzymes can be significantly accumulated, like ent-oxidosqualene (109, Fig. 22), not a substrate for oxydosqualene cyclases and a major constituent of jasmine absolute,71 or, alternatively, can be diverted to other metabolic pathways, although few systematic studies exist in this area. | ||
| Fig. 22 3R-2,3-oxidosqualene (ent-squalene oxide), the enantiomeric triterpenoid from jasmine absolute. | ||
Historically, optical activity was one of the first physico-chemical properties associated to natural products and, indeed, chiral natural products are generally optically active, but not necessarily enantiopure. The production of racemic versions of specific chiral secondary metabolites is associated to the presence of non-enzymatic biogenetic steps or to spontaneous racemization, and can be viewed as a strategy of bioactivity expansion, since enantiomers rarely show an overlapping biological profile. A similar consideration applies to the production of scalemates, which, however, are generally the result of enzymatic activity, either direct or mediated by directing proteins, and therefore more “expensive” in genomic terms. This genomic profligacy could be viewed as a manoeuvre to increase the diversity of a natural products inventory and better adapted it to a specific environment, a view supported by the strong activity on insects of many terpenes and sesquiterpenes synthesized as scalemates (see Section 6). Compared to the production of racemic compounds, the production of scalemates is plastic and environmentally tunable, as shown by the spectacular gradient of α-pinene configuration and scalemicity in the Amazon rainforest emission,149 a chiral circadian show affected by altitude, temperature and herbivore activity, and for which we appreciate the quid, but do not understand the quia.
8 Author contributions
All authors equally contributed to the review, with the corresponding author coordinating their activity and conceptualizing the topic of this review.9 Conflicts of interest
There are no conflicts to declare.10 Acknowledgements
We would like to dedicate this article to the memory of Dr Daniele Ciceri, too prematurely taken from the affection of his family and from our friendship.11 Notes and references
- C. H. Herty, J. Am. Chem. Soc., 1908, 30, 863–867 CrossRef CAS.
- A. Findlay, Nature, 1937, 140, 1937 Search PubMed.
- S. Scholtz, L. MacMorris, F. Krogmann and G. U. Auffarth, J. Eye Study Treat., 2019, 1, 51–58 Search PubMed.
- L. M. Bouthillette, V. Aniebok, D. A. Colosimo, D. Brumley and J. B. Macmillan, Chem. Rev., 2022, 122, 14815–14841 CrossRef CAS PubMed.
- R. J. Nyamwihura and I. V. Ogungbe, RSC Adv., 2022, 12, 11346–11375 RSC.
- M. A. Schafroth, G. Mazzoccanti, I. Reynoso-Moreno, R. Erni, F. Pollastro, D. Caprioglio, B. Botta, G. Allegrone, G. Grassi, A. Chicca, F. Gasparrini, J. Gertsch, E. M. Carreira and G. Appendino, J. Nat. Prod., 2021, 84, 2502–2510 CrossRef CAS PubMed.
- W. Huang and Y. X. Li, Sci. Sin., 1980, 23, 835 CAS.
- G. Bredig and P. S. Fiske, Biochem Z., 1912, 46, 7–23 Search PubMed.
- E. N. Jacobsen, I. Markó, W. S. Mungall, G. Schroder and K. B. Sharpless, J. Am. Chem. Soc., 1988, 110, 1968–1970 CrossRef CAS.
- J. M. Finefield, D. H. Sherman, M. Kreitman and R. M. Williams, Angew. Chem., Int. Ed., 2012, 51, 4802–4836 CrossRef CAS PubMed.
- A. Zask and G. Ellestad, Chirality, 2018, 30, 157–164 CrossRef CAS PubMed.
- A. J. E. Novak and D. Trauner, Trends Chem., 2020, 2, 1052–1065 CrossRef CAS.
- A. Zask and G. A. Ellestad, Chirality, 2021, 33, 915–930 CrossRef CAS PubMed.
- G. T. M. Bitchagno, V. A. Nchiozem-Ngnitedem, D. Melchert and S. A. Fobofou, Nat. Rev. Chem., 2022, 6, 806–822 CrossRef PubMed.
- J.-H. Yu, Z.-P. Yu, R. J. Capon and H. Zhang, Molecules, 2022, 27, 1279 CrossRef CAS PubMed.
- J. M. Batista, E. W. Blanch and V. Da Silva Bolzani, Nat. Prod. Rep., 2015, 32, 1280–1302 RSC.
- J. Han, A. Wzorek, K. D. Klika and V. A. Soloshonok, Molecules, 2021, 26, 2757 CrossRef CAS PubMed.
- B. Cai, A. M. Jack, R. S. Lewis, R. E. Dewey and L. P. Bush, Phytochemistry, 2013, 95, 188–196 CrossRef CAS PubMed.
- S. T. Lee, K. Wildeboer, K. E. Panter, W. R. Kem, D. R. Gardner, R. J. Molyneux, C. W. T. Chang, F. Soti and J. A. Pfister, Neurotoxicol. Teratol., 2006, 28, 220–228 CrossRef CAS PubMed.
- A. K. Duell, P. J. Kerber, W. Luo and D. H. Peyton, Chem. Res. Toxicol., 2021, 34, 1718–1720 Search PubMed.
- J. Han, O. Kitagawa, A. Wzorek, K. D. Klika and V. A. Soloshonok, Chem. Sci., 2018, 9, 1718–1739 RSC.
- S. Abás, C. Arróniz, E. Molins and C. Escolano, Tetrahedron, 2018, 74, 867–871 CrossRef.
- H. Doucet, E. Fernandez, T. P. Layzell and J. M. Brown, Chem.–Eur. J., 1999, 5, 1320–1330 CrossRef CAS.
- S. Tsuzuki, H. Orita, H. Ueki and V. A. Soloshonok, J. Fluorine Chem., 2010, 131, 461–466 CrossRef CAS.
- O. Wallach, Justus Liebigs Ann. Chem., 1895, 286, 119–143 CrossRef CAS.
- C. P. Brock, W. B. Schweizer and J. D. Dunitz, J. Am. Chem. Soc., 1991, 113, 9811–9820 CrossRef CAS.
- O. Ryu and K. B. Sharpless, Org. Synth., 1998, 9, 251 Search PubMed.
- Y. Mastai, A. Völkel and H. Cölfen, J. Am. Chem. Soc., 2008, 130, 2426–2427 CrossRef CAS PubMed.
- H. Lorenz, A. Perlberg, D. Sapoundjiev, M. P. Elsner and A. Seidel-Morgenstern, Chem. Eng. Process.: Process Intensif., 2006, 45, 863–873 CrossRef CAS.
- M. Masi, A. Cimmino, A. Boari, M. Chiara, G. Marcin, G. Pescitelli, A. Tuzi, M. Vurro and A. Evidente, Tetrahedron, 2017, 73, 6644–6650 CrossRef CAS.
- W. -L. Tsai, K. Hermann, E. Hug, B. Rohde and A. S. Dreiding, Helv. Chim. Acta, 1985, 68, 2238–2243 CrossRef CAS.
- P. Diter, S. Taudien, O. Samuel and H. B. Kagan, J. Org. Chem., 1994, 59, 370–373 CrossRef CAS.
- F. J. Muhtadi and M. M. A. Hassan, in Analytical Profiles of Drug Substances, ed. Florey K., 1990, pp. 477–551 Search PubMed.
- K. L. Kohnen-Johannsen and O. Kayser, Molecules, 2019, 24, 796 CrossRef PubMed.
- A. R. Cushny, J. Pharmacol. Exp. Ther., 1920, 15, 105–127 CAS.
- Scopolamine e atropine Polarim. test; Eur. Pharmacopoeia, http://https//pheur.edqm.eu/home Search PubMed.
- W. D. Dettbarn, E. Heilbronn, F. C. G. Hoskin and R. Kitz, Neuropharmacology, 1972, 11, 727–732 CrossRef CAS PubMed.
- I. V. Alabugin, in Stereoelectronic Effects: a Bridge Between Structure and Reactivity, Wiley, 2016, pp. 8–36 Search PubMed.
- C. Tanret, Hebd. Scéances Acad. Sci., 1878, 86, 1270–1272 Search PubMed.
- R. Willstätter and E. Waser, Ber. Dtsch. Chem. Ges., 1911, 44, 3423–3445 CrossRef.
- D. Sicker, K.-P. Zeller, H.-U. Siehl and S. Berger, in Natural Products. Isolation, Structure Elucidation and Synthesis, Wiley-VCH, 2020, Supporting Information to Chapter 1 Search PubMed.
- R. E. Gilman and L. Marion, Bull. Soc. Chim. Fr., 1961, 1993–1995 CAS.
- A. Kubin, F. Wierrani, U. Burner, G. Alth and W. Grunberger, Curr. Pharm. Des., 2005, 11, 233–253 CrossRef CAS PubMed.
- H. P. Bais, R. Vepachedu, C. B. Lawrence, F. R. Stermitz and J. M. Vivanco, J. Biol. Chem., 2003, 278, 32413–32422 CrossRef CAS PubMed.
- D. Skalkos, E. Tatsis, I. P. Gerothanassis and A. Troganis, Tetrahedron, 2002, 58, 1925–4929 CrossRef.
- R. Altmann, C. Etzlstorfer and H. Falk, Monatsh. Chem., 1997, 128, 785–793 CrossRef CAS.
- S. P. B. Vemulapalli, J. C. Fuentes-Monteverde, N. Karschin, T. Oji, C. Griesinger and K. Wolkenstein, Mar. Drugs, 2021, 19, 445 CrossRef CAS PubMed.
- M. Iijima, R. Munakata, H. Takahashi, H. Kenmoku, R. Nakagawa, T. Kodama, Y. Asakawa, I. Abe, K. Yazaki, F. Kurosaki and F. Taura, Plant Physiol., 2017, 174, 2213–2230 CrossRef CAS PubMed.
- D. Caprioglio, D. Mattoteia, A. Minassi, F. Pollastro, A. Lopatriello, E. Muňoz, O. Taglialatela-Scafati and G. Appendino, Org. Lett., 2019, 21, 6122–6125 CrossRef CAS PubMed.
- Y. Kashiwada, K. Yamazaki, Y. Ikeshiro, T. Yamagishi, T. Fujioka, K. Mihashi, K. Mizuki, L. M. Cosentino, K. Fowke, S. L. Morris-Natschke and K. H. Lee, Tetrahedron, 2001, 57, 1559–1563 CrossRef CAS.
- G. Mazzoccanti, O. H. Ismail, I. D'Acquarica, C. Villani, C. Manzo, M. Wilcox, A. Cavazzini and F. Gasparrini, Chem. Commun., 2017, 53, 12262–12265 RSC.
- F. Pollastro, D. Caprioglio, D. Del Prete, F. Rogati, A. Minassi, O. Taglialatela-Scafati, E. Munoz and G. Appendino, Nat. Prod. Commun., 2018, 13, 1189–1194 CrossRef.
- H. Jeon, G. Kang, M. J. Kim, J. S. Shin, S. Han and H. Y. Lee, Org. Lett., 2022, 24, 2181–2185 CrossRef CAS PubMed.
- L. O. Hanuš, S. M. Meyer, E. Muñoz, O. Taglialatela-Scafati and G. Appendino, Nat. Prod. Rep., 2016, 33, 1357–1392 RSC.
- G. Genchi, Amino Acids, 2017, 49, 1521–1533 CrossRef CAS PubMed.
- T. Yoshimura and N. Esaki, J. Biosci. Bioeng., 2003, 96, 103–109 CrossRef CAS PubMed.
- A. Hashimoto, T. Nishikawa, T. Oka and K. Takahashi, J. Neurochem., 1993, 60, 783–786 CrossRef CAS PubMed.
- D. S. Dunlop, A. Neidle, D. McHale, D. M. Dunlop and A. Lajtha, Biochem. Biophys. Res. Commun., 1986, 141, 27–32 CrossRef CAS PubMed.
- A. Hashimoto, S. Kumashiro, T. Nishikawa, T. Oka, K. Takahashi, T. Mito, S. Takashima, N. Doi, Y. Mizutani, T. Yamazaki, T. Kaneko and E. Ootomo, J. Neurochem., 1993, 61, 348–351 CrossRef CAS PubMed.
- A. P. Kuzin, T. Sun, J. Jorczak-Baillass, V. L. Healy, C. T. Walsh and J. R. Knox, Structure, 2000, 8, 463–470 CrossRef CAS PubMed.
- J. Gal, Chirality, 2012, 24, 959–976 CrossRef CAS PubMed.
- L. Colli and A. Guarna, Substantia, 2018, 2, 125–130 Search PubMed.
- G. C. Tron, A. Minassi, G. Sorba, M. Fausone and G. Appendino, Beilstein J. Org. Chem., 2021, 17, 1335–1351 CrossRef CAS PubMed.
- J. Gal, Helv. Chim. Acta, 2013, 96, 1617–1657 CrossRef CAS.
- A. S. Tsarkova, Front. Ecol. Evol., 2021, 9, 667829 CrossRef.
- Y. Oba, N. Yoshida, S. Kanie, M. Ojika and S. Inouye, PLoS One, 2013, 8, e84023 CrossRef PubMed.
- J. Maeda, D. Kato, M. Okuda, M. Takeo, S. Negoro, K. Arima, Y. Ito and K. Niwa, Biochim. Biophys. Acta Gen. Subj., 2017, 1861, 2112–2118 CrossRef CAS PubMed.
- S. Kanie, T. Nishikawa, M. Ojika and Y. Oba, Sci. Rep., 2016, 6, 24794 CrossRef CAS PubMed.
- R. Zhang, J. He, Z. Dong, G. Liu, Y. Yin, X. Zhang, Q. Li, Y. Ren, Y. Yang, W. Liu, X. Chen, W. Xia, K. Duan, F. Hao, Z. Lin, J. Yang, Z. Chang, R. Zhao, W. Wan, S. Lu, Y. Peng, S. Ge, W. Wang and X. Li, Sci. Rep., 2020, 10, 15882 CrossRef CAS PubMed.
- W. Liu, in Handbook of Chiral Chemicals, ed. D. J. Ager, 1999, pp. 83–102 Search PubMed.
- D. Joulain, Flavour Fragr. J., 2021, 36, 526–553 CrossRef.
- J. F. Biellmann, Chem. Rev., 2003, 103, 2019–2033 CrossRef CAS PubMed.
- U. Bathe and A. Tissier, Phytochemistry, 2019, 161, 149–162 CrossRef CAS PubMed.
- M. A. Phillips, M. R. Wildung, D. C. Williams, D. C. Hyatt and R. Croteau, Arch. Biochem. Biophys., 2003, 411, 267–276 CrossRef CAS PubMed.
- I. Prosser, I. G. Altug, A. L. Phillips, W. A. König, H. J. Bouwmeester and M. H. Beale, Arch. Biochem. Biophys., 2004, 432, 136–144 CrossRef CAS PubMed.
- S. Sarria, B. Wong, H. G. Martín, J. D. Keasling and P. Peralta-Yahya, ACS Synth. Biol., 2014, 3, 466–475 CrossRef CAS PubMed.
- J. Degenhardt, T. G. Köllner and J. Gershenzon, Phytochemistry, 2009, 70, 1621–1637 CrossRef CAS PubMed.
- J. Bohlmann, C. L. Steele and R. Croteau, J. Biol. Chem., 1997, 272, 21784–21792 CrossRef CAS PubMed.
- S. Nix, Ten most common trees in the United States, https://www.treehugger.com/ten-most-common-trees-united-states-3971258 Search PubMed.
- C. O. Schmidt, H. J. Bouwmeester, S. Franke and W. A. König, Chirality, 1999, 11, 353–362 CrossRef CAS.
- C. S. Chanotiya and A. Yadav, Nat. Prod. Commun., 2008, 3, 263–266 CrossRef CAS.
- S. T. Lee, D. R. Gardner, C. W. T. Chang, K. E. Panter and R. J. Molyneux, Phytochem. Anal., 2008, 19, 395–402 CrossRef CAS PubMed.
- A. W. Hofmann, Ber. Dtsch. Chem. Ges., 1881, 14, 705–713 CrossRef.
- A. Ladenburg, Ber. Dtsch. Chem. Ges., 1886, 19, 439–441 CrossRef.
- S. T. Lee, B. T. Green, K. D. Welch, G. T. Jordan, Q. Zhang, K. E. Panter, D. Hughes, C. W. T. Chang, J. A. Pfister and D. R. Gardner, Chem. Res. Toxicol., 2008, 21, 2061–2064 Search PubMed.
- L. M. Labay, A. Chan-Hosokawa, J. W. Homan, M. M. McMullin, F. X. Diamond, M. M. Annand, S. M. Marco and J. M. Hollenbach, Forensic Sci. Int., 2022, 341, 111500 CrossRef CAS PubMed.
- K. A. Yeung, P. R. Chai, B. L. Russell and T. B. Erickson, J. Med. Toxicol., 2022, 18, 321–333 CrossRef CAS PubMed.
- M. F. Roberts, Phytochemistry, 1975, 14, 2393–2397 CrossRef CAS.
- G. Peddinti, H. Hotti, T. H. Teeri and H. Rischer, Sci. Rep., 2022, 12, 17562 CrossRef CAS PubMed.
- N. V. Mody, R. Henson, P. A. Hedin, U. Kokpol and D. H. Miles, Experientia, 1976, 32, 829–830 CrossRef CAS.
- D. W. Armstrong, X. Wang, J. T. Lee and Y. S. Liu, Chirality, 1999, 11, 82–84 CrossRef CAS.
- M. Kajikawa, N. Sierro, H. Kawaguchi, N. Bakaher, N. V. Ivanov, T. Hashimoto and T. Shoji, Plant Physiol., 2017, 174, 999–1011 CrossRef CAS PubMed.
- A. J. Deng, H. J. Zhang, Q. Li, Z. H. Li, Z. H. Zhang, L. Q. Wu, L. Li and H. L. Qin, Phytochemistry, 2017, 144, 159–170 CrossRef CAS PubMed.
- S. Suzuki, Wood Res., 2002, 89, 52–60 CAS.
- T. Umezawa, Phytochem. Rev., 2003, 2, 371–390 CrossRef CAS.
- L. Jin, Z. Song, F. Cai, L. Ruan and R. Jiang, Molecules, 2023, 28, 302 CrossRef CAS PubMed.
- C. T. Walsh and B. S. Moore, Angew. Chem., Int. Ed., 2019, 58, 6846–6879 CrossRef CAS PubMed.
- L. B. Davin, H. Bin Wang, A. L. Crowell, D. L. Bedgar, D. M. Martin, S. Sarkanen and N. G. Lewis, Science, 1997, 275, 362–366 CrossRef CAS PubMed.
- L. B. Davin and N. G. Lewis, Plant Physiol., 2000, 123, 453–461 CrossRef CAS PubMed.
- K. W. Kim, C. A. Smith, M. D. Daily, J. R. Cort, L. B. Davin and N. G. Lewis, J. Biol. Chem., 2015, 290, 1308–1318 CrossRef PubMed.
- B. Pickel, M. A. Constantin, J. Pfannstiel, J. Conrad, U. Beifuss and A. Schaller, Angew. Chem., Int. Ed., 2010, 49, 202–204 CrossRef CAS PubMed.
- R. Gasper, I. Effenberger, P. Kolesinski, B. Terlecka, E. Hofmann and A. Schaller, Plant Physiol., 2016, 172, 2165–2175 CrossRef CAS PubMed.
- S. Suzuki, T. Umezawa and M. Shimada, Biosci., Biotechnol., Biochem., 2002, 66, 1262–1269 CrossRef CAS PubMed.
- S. C. Halls, L. B. Davin, D. M. Kramer and N. G. Lewis, Biochemistry, 2004, 43, 2587–2595 CrossRef CAS PubMed.
- A. T. Dinkova-Kostova, D. R. Gang, L. B. Davin, D. L. Bedgar, A. Chu and N. G. Lewis, J. Biol. Chem., 1996, 271, 29473–29482 CrossRef CAS PubMed.
- T. Okunishi, T. Umezawa and M. Shimada, J. Wood Sci., 2000, 46, 234–242 CrossRef CAS.
- D. T. Tshitenge, D. Feineis, S. Awale and G. Bringmann, J. Nat. Prod., 2017, 80, 1604–1614 CrossRef CAS PubMed.
- N. Ikezawa, K. Iwasa and F. Sato, J. Biol. Chem., 2008, 283, 8810–8821 CrossRef CAS PubMed.
- C. Li, C. J. Li, J. Ma, J. W. Huang, X. Y. Wang, X. L. Wang, F. Ye and D. M. Zhang, Bioorg. Chem., 2019, 88, 102948 CrossRef CAS PubMed.
- Z. Yang, B. Su, Y. Wang, H. Liao, Z. Chen and D. Liang, J. Nat. Prod., 2020, 83, 985–995 CrossRef CAS PubMed.
- Y. Kinoshita, Y. Yamamoto, I. Yoshimura, T. Kurokawa and S. Huneck, J. Hattori Bot. Lab., 1997, 83, 173–178 Search PubMed.
- S. Chitturi and G. C. Farrell, J. Gastroenterol. Hepatol., 2008, 23, 366–373 CrossRef CAS PubMed.
- A. Galanty, P. Paśko and I. Podolak, Phytochem. Rev., 2019, 18, 527–548 CrossRef CAS.
- Y. Liu, L. Wang, L. Zhao and Y. Zhang, Nat. Prod. Rep., 2022, 39, 1282–1304 RSC.
- I. Effenberger, B. Zhang, L. Li, Q. Wang, Y. Liu, I. Klaiber, J. Pfannstiel, Q. Wang and A. Schaller, Angew. Chem., Int. Ed., 2015, 54, 14660–14663 CrossRef CAS PubMed.
- W. Fang, S. Ji, N. Jiang, W. Wang, G. Y. Zhao, S. Zhang, H. M. Ge, Q. Xu, A. H. Zhang, Y. L. Zhang, Y. C. Song, J. Zhang and R. X. Tan, Nat. Commun., 2012, 3, 1039 CrossRef PubMed.
- P. Allegrini and G. Appendino, Manuscr. Prep.
- L. Nahar, A. Das Talukdar, D. Nath, S. Nath, A. Mehan, F. M. D. Ismail and S. D. Sarker, Molecules, 2020, 25, 4983 CrossRef CAS PubMed.
- J. H. Cardellina, H. R. Bokesch, T. C. McKee and M. R. Boyd, Bioorganic Med. Chem. Lett., 1995, 5, 1011–1014 CrossRef CAS.
- M. Mariner, B. Mcmahon and R. Boyd, J. Pharmacol. Exp. Ther., 1996, 279, 652–661 Search PubMed.
- M. Arreaga-Gonz, A. J. Oliveros-Ortiz, E. Rosa, G. Rodr, J. M. Torres-Valencia, C. M. Cerda-Garc, P. Joseph-Nathan and A. G. Mario, J. Nat. Prod., 2021, 84, 707–712 CrossRef PubMed.
- H. M. Arreaga-González, G. Rodríguez-García, R. E. Del Río, J. A. Ferreira-Sereno, H. A. García-Gutiérrez, C. M. Cerda-García-Rojas, P. Joseph-Nathan and M. A. Gómez-Hurtado, J. Nat. Prod., 2019, 82, 3394–3400 CrossRef PubMed.
- L. Yang, D. Zhang, J. B. Lia, X. Zhang, N. Zhou, W. Y. Zhang and H. Lu, J. Asian Nat. Prod. Res., 2022, 24, 624–633 CrossRef CAS PubMed.
- T. Promchai, T. Thaima, R. Rattanajak, S. Kamchonwongpaisan, S. G. Pyne and T. Limtharakul, Nat. Prod. Res., 2021, 35, 2476–2481 CrossRef CAS PubMed.
- P. A. Searle and F. Molinzki, Tetrahedron, 1994, 50, 9893–9908 CrossRef CAS.
- R. E. Johnson, T. De Rond, V. N. G. Lindsay, J. D. Keasling and R. Sarpong, Org. Lett., 2015, 17, 3474–3477 CrossRef CAS PubMed.
- K. Mori, Bioorg. Med. Chem., 2007, 15, 7505–7523 CrossRef CAS PubMed.
- K. W. Chong, J. S. Y. Yeap, S. H. Lim, J. F. F. Weber, Y. Y. Low and T. S. Kam, J. Nat. Prod., 2017, 80, 3014–3024 CrossRef CAS PubMed.
- C. Kan-Fan, G. Massiot, A. Ahond, B. C. Das, H. P. Husson, P. Potier, A. I. Scott and C. C. Wei, J. Chem. Soc., Chem. Commun., 1974, 5, 164–165 RSC.
- H. C. Brown and P. K. Jadhav, J. Am. Chem. Soc., 1983, 105, 2092–2093 CrossRef CAS.
- H. C. Brown and P. Veeraraghavan Ramachandran, J. Organomet. Chem., 1995, 500, 1–19 CrossRef CAS.
- I. Paterson, J. M. Goodman, M. Anne Lister, R. C. Schumann, C. K. McClure and R. D. Norcross, Tetrahedron, 1990, 46, 4663–4684 CrossRef CAS.
- F. Capetti, A. Marengo, C. Cagliero, E. Liberto, C. Bicchi, P. Rubiolo and B. Sgorbini, Molecules, 2021, 5610 CrossRef CAS PubMed.
- M. Allenspach, C. Valder, D. Flamm and C. Steuer, Sci. Rep., 2021, 11, 16923 CrossRef CAS PubMed.
- I. Bonaccorsi, D. Sciarrone, A. Cotroneo, L. Mondello, P. Dugo and G. Dugo, Rev. Bras. Farmacogn., 2011, 21, 841–849 CrossRef CAS.
- N. W. Davies, T. Larkman, P. J. Marriott and I. A. Khan, J. Agric. Food Chem., 2016, 64, 4817–4819 CrossRef CAS PubMed.
- Essential Oil of Melaleuca, Terpinen-4-ol Type (Tea Tree Oil), http://www.iso.org/ Search PubMed.
- I. Gandolfi, G. Palla, L. Delprato, F. D. E. Nisco and R. Marchelli, J. Food Sci., 1992, 57, 377–379 CrossRef CAS.
- F. Bellesia, A. Pinetti, L. Simon-sarkadi, C. Zucchi, J. Csapó, B. Weimer, L. Caglioti and G. Pályi, in Advances in Asymmetric Autocatalysis and Related Topics, 2017, pp. 357–367 Search PubMed.
- P. M. Helfman and J. L. Bada, Nature, 1976, 262, 279–281 CrossRef CAS PubMed.
- G. Rainer, Am. J. Phys. Anthropol., 2006, 48, 2–48 Search PubMed.
- M. I. Damaj, K. R. Creasy, A. D. Grove, J. A. Rosecrans and B. R. Martin, Brain Res., 1994, 664, 34–40 CrossRef CAS PubMed.
- C. Sims, M. A. Birkett and D. M. Withall, Insects, 2022, 13, 368 CrossRef PubMed.
- A. C. Oehlschlager, G. G. S. King, H. D. Pierce, A. M. Pierce, K. N. Slessor, J. G. Millar and J. H. Borden, J. Chem. Ecol., 1987, 13, 1543–1554 CrossRef CAS PubMed.
- J. G. Millar, H. D. Pierce, A. M. Pierce, A. C. Oehlschlager and J. H. Borden, J. Chem. Ecol., 1985, 11, 1071–1081 CrossRef CAS PubMed.
- M. C. Cammaerts and K. Mori, Physiol. Entomol., 1987, 12, 381–385 CrossRef CAS.
- T. Suzuki, J. Kozaki, R. Sugawara and K. Mori, Chem. Pharm. Bull., 1984, 19, 15–20 CAS.
- M. Paoli, A. Anesi, R. Antolini, G. Guella, G. Vallortigara and A. Haase, Sci. Rep., 2016, 6, 21893 CrossRef CAS PubMed.
- N. Zanoni, D. Leppla, P. I. Lembo Silveira de Assis, T. Hoffmann, M. Sá, A. Araújo and J. Williams, Commun. Earth Environ., 2020, 1, 1–11 CrossRef.
| This journal is © The Royal Society of Chemistry 2023 |