
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural advances toward understanding the catalytic activity and conformational dynamics of modular nonribosomal peptide synthetases
Ketan D.
Patel
 ,
Monica R.
MacDonald
,
Syed Fardin
Ahmed
,
Jitendra
Singh
and
Andrew M.
Gulick
*
,
Monica R.
MacDonald
,
Syed Fardin
Ahmed
,
Jitendra
Singh
and
Andrew M.
Gulick
*
University at Buffalo, Department of Structural Biology, Jacobs School of Medicine and Biomedical Sciences, 55 Main St. Buffalo, NY 14203, USA. E-mail: amgulick@buffalo.edu
First published on 28th April 2023
Abstract
Covering: up to fall 2022.
Nonribosomal peptide synthetases (NRPSs) are a family of modular, multidomain enzymes that catalyze the biosynthesis of important peptide natural products, including antibiotics, siderophores, and molecules with other biological activity. The NRPS architecture involves an assembly line strategy that tethers amino acid building blocks and the growing peptides to integrated carrier protein domains that migrate between different catalytic domains for peptide bond formation and other chemical modifications. Examination of the structures of individual domains and larger multidomain proteins has identified conserved conformational states within a single module that are adopted by NRPS modules to carry out a coordinated biosynthetic strategy that is shared by diverse systems. In contrast, interactions between modules are much more dynamic and do not yet suggest conserved conformational states between modules. Here we describe the structures of NRPS protein domains and modules and discuss the implications for future natural product discovery.
 (Left to Right)Syed Fardin Ahmed, Ketan D. Patel, Andrew M. Gulick, Jitendra Singh and Monica R. MacDonald | Syed Fardin Ahmed received his BS in biochemistry at Ithaca College. He is currently pursuing his PhD in structural biology with interest in NRPSs involved in siderophore production. Ketan(kumar) D. Patel received his BSc in Microbiology from Arts, Science & Commerce College, Kholwad and his MSc and PhD in Microbiology from The Maharaja Sayajirao University of Baroda, Vadodara, under the supervision of Prof. Sanjay Ingle. He performed postdoctoral research with Sankaranarayanan at the Center for Cellular & Molecular Biology. He is currently a Research Scientist, focused on delineating principles of natural product biosynthesis and the discovery of novel natural products. Professor Andrew Gulick has a long-standing interest in the structural enzymology of proteins involved in natural product biosynthesis. Jitendra Singh received his BS in biomedical informatics from New York City College of Technology. He is jointly mentored by Professors Gulick and Thomas Grant, working on free standing NRPSs from gut bacteria and developing solution scattering algorithms. Monica MacDonald received her BS in biochemistry from SUNY Stony Brook University and her MS in medicinal chemistry at Stevens Institute of Technology. Her thesis research is centered on pyrrolobenzodiaziepene biosynthesis. All authors are members of the Department of Structural Biology, Jacobs School of Medicine and Biomedical Sciences, University at Buffalo. |
1. Nonribosomal peptide synthetases
1.1. Peptide natural products produced by modular enzymes
Microorganisms produce specialized natural products that allow them to survive in diverse environments. These molecules are secreted from the cell and play multiple roles to support the producer organism, including chemical signaling and nutrient acquisition, or cause detrimental effects to other competing species or host cells. These molecules can be divided into multiple classes based on their chemical nature and biosynthetic strategy.1 Interest in these natural products has spanned more than fifty years as many molecules have been employed directly or as inspiration for the development of new pharmaceuticals.2One class of important natural products are peptides derived from the modular nonribosomal peptide synthetases (NRPSs). In contrast to ribosomally produced and post-translationally modified peptide (RiPPs) natural products,3 which are translated through the normal ribosomal machinery and heavily modified to their final active form, NRPS peptides are produced enzymatically through a modular assembly line process.4 Here, multiple catalytic domains that carry out sequential steps in the biosynthesis are joined in large multidomain proteins.5–10 This ribosome-independent approach frees the NRPS clusters from the constraints imposed by the ribosome and amino acyl-tRNA synthetases and allows for the incorporation of nonproteinogenic amino acids, fatty acids, aryl acids, and hydroxy acids that can be joined via amide and ester linkages. The diversity of NRPS peptides11 is driven not only by the use of unconventional building blocks but additionally by the use of other catalytic domains that can further modify the peptide with methylations, epimerizations, halogenations, and the formation of chemical cross-links.12 A common feature of NRPS derived peptides is the macrocyclization through a peptide bond between the N-terminus or hydroxy or amine side chains and the C-terminal carboxylate. Combined, hundreds of substrates have been observed in different NRPS peptides. As will be described below, this modular strategy of joining multiple catalytic domains results in large proteins that exhibit conserved sequence motifs making them easy to detect through genome mining,13–16 facilitating the discovery and classification of new products. This also highlights the wealth of natural product biosynthesis yet to be discovered.
1.2. Core domains of NRPS enzymes
A common feature of NRPSs is the modular architecture that encodes multiple domains on a single polypeptide that function together in a coordinated fashion to produce the peptide product. In the most dramatic cases, often seen with fungal NRPS enzymes, the entire biosynthetic pathway may exist within a single protein chain. For example, Acremonium, a fungal symbiont associated with a marine sponge, produces the hexadecapeptide acremopeptaibols A–F through the activity of a 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 165 residue NRPS that catalyzes dozens of necessary steps in the formation of the final peptide.17 More commonly, the complete NRPS biosynthetic cluster will include multiple multidomain proteins that operate in tandem through a series of intra- and intermolecular catalytic steps. In some systems, specific domains called communication (COM) or docking domains are present on the termini of proteins to facilitate the intermolecular transfer of the peptide intermediate from one protein to another.18 These domains have been explored functionally and structurally, and may offer the potential to guide novel protein interactions through engineering methods.19–22
165 residue NRPS that catalyzes dozens of necessary steps in the formation of the final peptide.17 More commonly, the complete NRPS biosynthetic cluster will include multiple multidomain proteins that operate in tandem through a series of intra- and intermolecular catalytic steps. In some systems, specific domains called communication (COM) or docking domains are present on the termini of proteins to facilitate the intermolecular transfer of the peptide intermediate from one protein to another.18 These domains have been explored functionally and structurally, and may offer the potential to guide novel protein interactions through engineering methods.19–22
This multidomain biosynthetic strategy can be compared to an assembly line in which the growing peptide – covalently attached to the carrier protein domains – is shuttled between different catalytic domains. In this approach, the protein is organized into modules, with each module generally responsible for the incorporation of a single residue. Each module contains a carrier protein to which the substrates and intermediates are covalently bound during the biosynthesis. The carrier thus visits neighboring catalytic domains for loading, chemical modification, and peptide bond formation, ultimately passing the peptide downstream to the next module for elongation. NRPSs can either operate in a linear fashion in which each module functions a single time and there is a direct correspondence between the number of modules and the number of residues in the final peptide, or can act iteratively in which modules act multiple times to produce the final product.
Ultimately, the peptide is released through the activity of a final C-terminal domain that catalyzes hydrolysis, cyclization, or reductive release.23 The NRPSs are often compared with the polyketide and fatty acid synthases (PKS and FAS, respectively) that employ a similar modular biosynthetic strategy.24,25 The obafluorin pathway (Fig. 1),26,27 a two module NRPS highlights several common and unusual features, including free-standing and modular proteins that catalyze conventional and unusual overall chemical reactions.
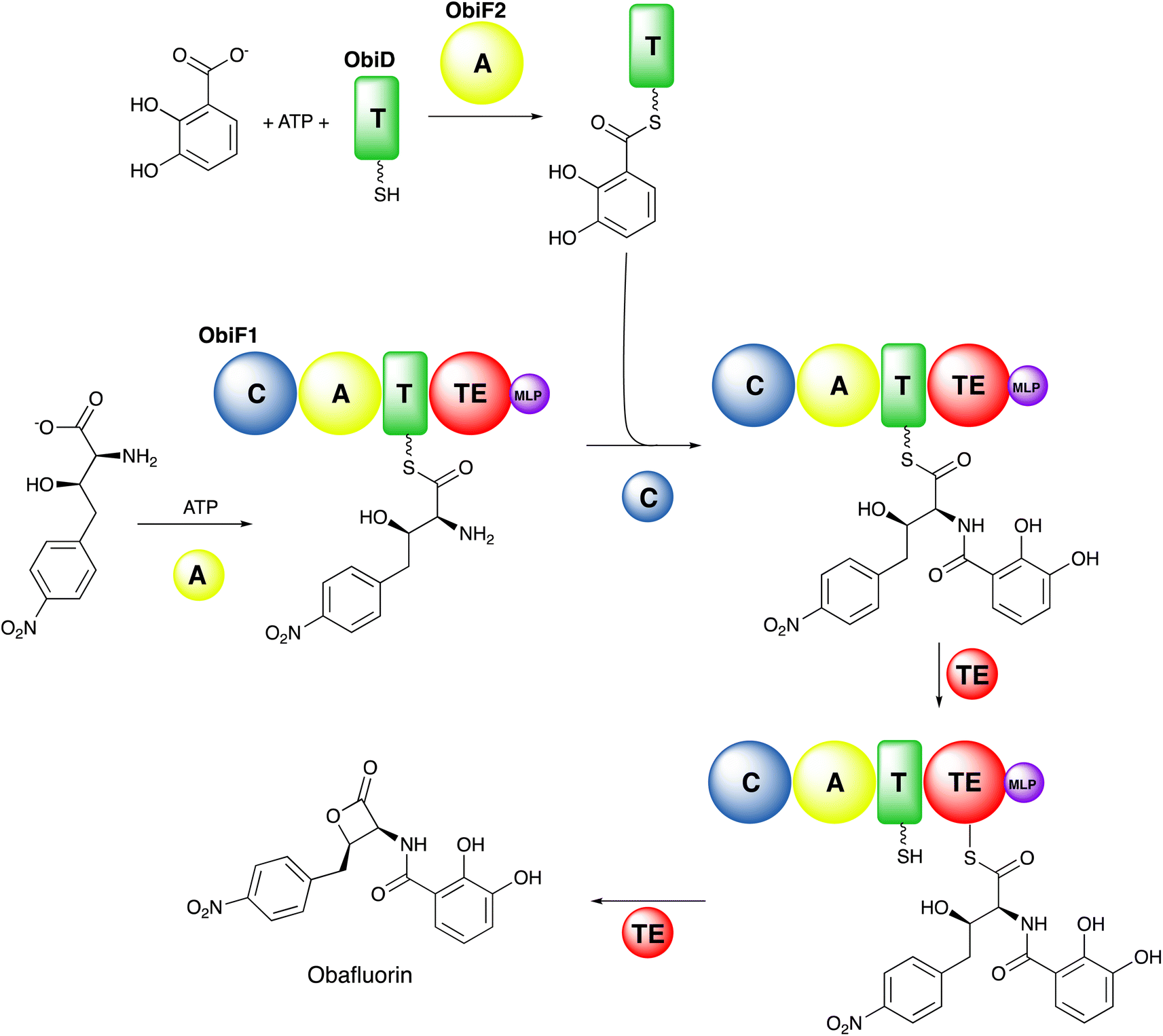 | ||
| Fig. 1 Overview of NRPS biosynthesis. The two module NRPS system for obafluorin biosynthesis is shown, highlighting the existence of free-standing (ObiD and ObiF2) and modular (ObiF1) enzymes. ObiF2 first loads a molecule of 2,3-dihydroxybenzoate onto the pantetheine cofactor of the carrier protein ObiD. After the ObiF1 adenylation domain loads the nonproteinogenic substrate onto the adjacent carrier domain, the ObiF1 condensation domain catalyzes amide bond formation. The ObiF1 thioesterase domain then catalyzes β-lactone formation, mediated by the formation of the acyl enzyme intermediate with a catalytic cysteine, releasing the obafluorin product. | ||
The adenylation domains are members of a large superfamily of enzymes that include acyl- and aryl-CoA synthetases, fatty acyl-AMP ligases,36 and beetle luciferases.37,38 A number of conserved sequence motifs have been identified,38,39 including the A3 motif (S/T)(S/T)G(S/T)TGxPK for binding the nucleotide phosphates, the A7 motif (S/T)GD that interacts with the ribose hydroxyls, and a catalytic A10 motif PxxxxGK that contains a critical lysine that is used in the adenylation step. Additionally, the adenylation domains contain a conserved A8 hinge motif Rx(D/K)xxxxxxG that is used for a dramatic conformational change that occurs between the two partial reactions. Because of their role as gatekeepers for peptide sequence, NRPS adenylation domains have been attractive targets for bioengineering to produce novel natural products that has clinical relevance.40
The second histidine of the HHxxxDG motif was initially thought to perform the proton abstraction by acting as a general base as in the homologous acetyltransferase superfamily.41,43 However, as several condensation domains retain catalytic activity upon mutation of the second histidine44,45 and the homologous cyclization domains lack it altogether,46 this histidine residue is now proposed to contribute with other residues to proper positioning of the α-amino group of the acceptor substrate.47
NRPS condensation domains can be classified as LCL and DCL domains, where the substrates for LCL are two L-amino acids, while DCL domains use a donor D-amino acid with acceptor L-amino acid.48 Interestingly, the DCL domains appear to be the evolutionary precursor to specialized condensation domains that have the ability to catalyze the formation of the β-lactam ring in nocardicin biosynthesis.49 Additionally, some condensation domains transfer a fatty acid from a fatty acyl-CoA or acyl-PCP to an acceptor amino acid and are considered lipoinitiation domains.50 Several recent reviews highlight the varied catalytic activites of NRPS domains with homology to the condensation domain.42,51
Thioesterase domains can be classified as either type I or type II. Type I domains are normally found on the C-terminus of the final module, and cleave the intermediate peptide product from the biosynthetic pathway. Unlike type I domains, type II thioesterase domains are not covalently linked to an NRPS module.55 While type II thioesterases can release intermediate or final products, they have also been observed to play an editing role to remove undesired or non-reactive substrates/moieties that are stalled on the PCP phosphopantetheine arm.55,56
1.3. Additional specialized catalytic domains
In addition to the core catalytic domains that are responsible for the activation of building blocks, formation of peptide or ester linkages, and release of the peptide product from the assembly line, many NRPS biosynthetic clusters contain embedded functional domains that carry out additional modifications to the intermediates and products. These domains may be inserted into the boundaries between the core domains or are in some cases inserted into surface loops within the catalytic domains.These diverse reactions catalyzed by the homologous domain correlate with variations in the core HHxxxDG motif.48 Epimerization domains have HHxxxD motif while dual epimerization and condensation domains have HH[I/L]xxxxGD motif. On the other hand, cyclization domains have DxxxxD motif46 and terminal condensation domains have SHxxxDx motif. Like the condensation domains, the role of some active site residues have remained unanswered in cyclization domain, although mutation of aspartate residues in DxxxxD motif abolished the activity.62,63 Epimerization activity was reduced when the second histidine and aspartate of the HHxxxD motif were mutated in GrsA-E1 indicating their role in catalysis.59 Similarly, the second histidine and glycine of the HHxxxDG motif in the terminal condensation domain TqaA, supporting a role in catalysis.64
Finally, some NRPS modules for glycopeptide antibiotics contain so-called X-domains with homology to the condensation domain that recruit cytochrome P450 oxygenases to perform side-chain crosslinking on the NRPS-bound peptide.65
The presence of NRPS modifying domains embedded within a conventional module raise questions about the timing of the modifications. A methyltransferase domain could act at any of the multiple stages in the catalytic cycle, including acting upon the free amino acid substrate, on the adenylate intermediate, or on the loaded substrate bound as a thioester. Additionally, methyl transfer could occur to the loaded amino acid prior to or after peptide bond formation. In the pyochelin NRPS system, a methyltransferase domain exists in PchF, embedded within a cysteine-activating adenylation domain that is part of an NRPS with a Cy-A-(MT)-PCP-TE architecture.84,85 Thus, methyl transfer could occur on free cysteine, the cysteine–AMP intermediate, the cysteine thioester with the PCP domain, the condensed peptide, the cyclized peptide, or, as the cyclized thiazoline is converted by the free-standing reductase PchG into a thiazolidine,86,87 to either of these oxidation states. Studies with the ethyl esters that model the pantetheine loaded intermediates of all the potential peptide precursors with full length PchF provide compelling evidence that the methyltransferase activity is the penultimate step, occurring after the PchG catalyzed reduction of the thiazoline to a thiazolidine ring.79 Once methylation has occurred, the only remaining step in pyochelin biosynthesis is the hydrolytic release catalyzed by the thioesterase domain.
1.4 Related NRPS-like proteins
In addition to conventional NRPSs harboring multiple modules, there exist standalone single modules of NRPSs that have been termed NRPS-like proteins. These modules have an N-terminal adenylation domain, a carrier domain and a C-terminal domain architecture. While these proteins do not produce a peptide, their similarity to NRPSs and the insights that may arise warrant their inclusion herein.Enzymes responsible for reduction of carboxylic acids to aldehydes were first reported in the fungus Neurospora crassa and required Mg2+, NADPH, and ATP for activity.90 Considering the broad range of carboxylic substrates reduced by these enzymes, the proteins were termed as carboxylic acid reductases (CARs). Structural and sequence analysis established CARs as similar to NRPS.91 Several other proteins including amino acid reductases LnaA, LnbA, and Nps1 (ref. 92) were reported to have similarity with CARs and also described as NRPS-like. Similar to NRPS, the adenylation domain specifically loads the substrate onto a carrier domain, which transports it to the C-terminal domain. In CARs and other modules with A-PCP-R architecture, the reductase domain catalyzes release of the product yielding an aldehyde derivative.93 A unique NRPS-like protein glycine betaine reductase in fungi has two reductase domains at C-terminus (A-PCP-R-R) to perform two sequential reductions of glycine betaine to form choline.94
Additional proteins that function as a stand-alone module (or present in cluster) have similar architecture with an N-terminal adenylation domain and a carrier domain but C-terminal has a condensation domain (A-PCP-C).95 Single modules with an A-PCP-C architecture have been found mostly in fungal natural product clusters including tryptoquialanine, alanditrypinone, cottoquinazolie and fumiquinazolines.96
Several eukaryotic proteins exist with a tridomain architecture consisting of adenylation and PCP domains joined to a variety of domains at the C-terminus. The fungal Lys2 protein contains a C-terminal reductase domain that catalyzes the reductive cleavage of the loaded α-aminoadipic acid to form α-aminoadipic-δ-semialdehyde, an intermediate in lysine biosynthesis.97,98 The Ebony protein of Drosophila melanogaster contains a C-terminal aryl-alkylamine N-acetyltransferase domain that is involved in the production of β-alanyl-dopamine and -histamine.99,100 The transferase domain contains low sequence homology to other family members and the structural classification was only possible through structure determination.101 Examination of predicted active site residues of eukaryotic members of this family has been performed, highlighting shared features and aiding in the characterization of additional structures.102
A final category of NRPS-like proteins include the cationic homopolyamino acid synthetases (CHPAS) like poly-ε-lysine synthetase.103 These CHPAS have an unusual membrane bound domain at C-terminus end which is proposed to have three small condensation domains and synthesize polymers of cationic amino acids of length up to 35-mer.103,104
2. The structural biology of nonribosomal peptide synthetase domains
The assembly line architecture of NRPSs has excited structural biologists for 25 years. Not surprisingly, the first studies focused on individual domains of NRPSs, employing either genetically truncated proteins or natural single domain enzymes, the so-called type II NRPSs.105 The earliest structures of the core catalytic domains included the phenylalanine activating adenylation domain of the gramicidin S synthetase (PDB 1AMU),106 a free-standing condensation domain from vibriobactin biosynthesis (PDB 1L5A),45 a free-standing aryl adenylating enzyme from the bacillibactin biosynthetic pathway (PDB 1MDB),107 and a truncated thioesterase domain from an NRPS responsible for the production of surfactin (PDB 1JMK).108 These studies set the stage in the early 2000s for our understanding of the respective chemical mechanisms and, particularly for the adenylation domains, provided views of the active site binding pockets that dictate substrate specificity and selectivity.Over the next decade, structures soon followed of the auxiliary domains that further diversify NRPS products. Additionally, structures of didomain complexes have been solved that illustrate that binding interface between catalytic domains and the carrier protein domains that deliver the substrates. A recent review explores the interfaces between catalytic and carrier domains.5 We describe here the structures of individual NRPS domains before moving to structural studies of full NRPS modules (Section 3). By our count, there are currently over 150 PDB entries of more than 60 different NRPS proteins (Table 1). These structures lay the foundation for the discussion of multidomain structures and the conformational dynamics that govern the NRPS structural cycle in subsequent sections. We additionally draw the reader's attention to reviews that have identified strategies that have been used to characterize structurally the challenging, dynamic NRPS proteins, and the importance of chemical tools to trap these enzymes in meaningful catalytic states.9,109
| Protein name | PDB accession code and description |
|---|---|
| a This table is mirrored and updated at: https://www.acsu.buffalo.edu/%7Eamgulick/NRPSChart.html | |
| Condensation | |
| VibH | 1L5A, free-standing domain from vibriobactin biosynthesis45 |
| CDA | CDA synthetase47 |
| 4JN3, selenomethionine | |
| 4JN5, native | |
| 5DU9, covalent substrate analog | |
| Tcp12 | 4TX2, catalytically inactive, oxygenase-recruiting X-domain from teicoplanin synthetase65 |
| BmdB | 5T3E, cyclization domain from bacilliamide E synthetase164 |
| TqaA | Fumiquinazoline F terminating condensation domain64 note that the PCP-CT is included in list multidomain proteins below |
| 5DIJ, native | |
| 5EGF, selenomethionine | |
| 5DLK, truncation of 10 residues | |
| RzmA | Lipoinitiation domain of rhizomide synthetase50 |
| 7C1H, wild-type | |
| 7C1K, R148A mutant | |
| 7C1L, R148A mutant plus C8-CoA | |
| 7C1P, H140V/R148A | |
| 7C1R, H140V/R148A + C8-CoA | |
| 7C1S, H140V/R148A + C8CoA + Leu-SNAC | |
| 7C1U, H140V/R148A | |
| HMWP2 | 7JTJ, 7JUA, second cyclization domain of yersiniabactin synthetase165 |
| AmbE | 7R9X, dehydrating condensation domain227 |
| HMWP2 | 7RY6, first cyclization domain of yersiniabactin synthetase166 |
|
|
| Adenylation | |
| PheA | 1AMU, adenylation domain of GrsA106 |
| DhbE | Aryl adenylating enzyme from bacillibactin biosynthesis107 |
| 1MD9, AMP and 2,3-dihydroxybenzoic acid | |
| 1MDB, AMP and 2,3-dihydroxybenzoic acid | |
| 1MDF, unliganded | |
| DltA | NRPS-like D-Ala-ligase involved in cell wall biosynthesis |
| B. cereus 231,232 | |
| 3DHV), D-Ala-AMP | |
| 3FCC), Mg-ATP | |
| 3FCE), ATP | |
| 4PZP), unliganded | |
| B subtilis 233 | |
| (3E7W and 3E7X, AMP) | |
| BasE | Free-standing domain from acinetobactin biosynthesis234,235 |
| 3O82, DHB-AMS | |
| 3O83, 3O84, 3U16, 3U17, alternate inhibitors | |
| SidN | 3ITE, NRPS for a fungal siderophore236 |
| VinN | Vicenistatin NRPS237 |
| 3WVN, aspartate | |
| 3WV4, unliganded | |
| 3WV5, 3-methylaspartate | |
| ApnA | Promiscuous domain from anabaenopeptin NRPS238 |
| 4D4G, AMPPNP | |
| 4D4H, unliganded | |
| 4D4I, AMPPNP + arginine | |
| 4D56, AMP + tyrosine | |
| 4D57, AMP + arginine | |
| AlmE | 4OXI, NRPS-like domain involved in glycyl transfer to lipopolysaccharide bound to glycyl-AMP239 |
| CAR | NRPS-like carboxylate reductase.91 Additional multidomain structures below |
| 5MSC, AMP | |
| 5MSD, AMP + benzoic acid | |
| 5MST, AMP + fumarate | |
| Thr1 | Free-standing NRPS-like domain involved in production of chlorothreonine240 |
| 5N9W, unliganded | |
| 5N9X, two chains bound to ATP or Thr-AMP | |
| MbtA | 5KEI, free-standing domain of mycobactin NRPS pathway241 |
| CahJ | Free-standing domain of cahuitamycin NRPS pathway242 |
| 5WM2, AMP + salicylate | |
| 5WM3, salicyl-AMP | |
| 5WM4, 6-methylsalicyl-AMP | |
| 5WM5, 5-methylsalicyl-AMP | |
| 5WM6, benzoyl-AMP | |
| 5WM7, AMP | |
| FscH | 6EA3, fuscachelin domain in complex with MLP |
| EntE | Free-standing domain of enterobactin NRPS pathway.243 Additional didomains below |
| 6IYK, 2-nitrobenzoyl-AMP | |
| 6IYL, 3-cyanobenzoyl-AMP | |
| GR01_22995 | 6OZ1, NRPS-like domain of carboxylic acid reductase bound to AMP244 |
| StsA | Structures of a keto acid activating domain.140 Additional didomain below |
| 6ULX and 6ULY, oxopentanoyl-AMP | |
| NpsA | Free-standing domain of tilimycin/tilivalline pathway.124 Additional didomain below |
| 6VHV, full-length bound to 3-hydroxybenzoyl-AMS | |
| 6VHT and 6VHU, unliganded Acore | |
| 6VHW, Acore bound to 3-hydroxybenzoyl-AMS | |
| 6VHX, Acore bound to3-hydroxyanthranilyl-AMS | |
| 6VHZ, Acore bound to anthranilyl-AMS | |
| DltA | 7VHV, Staphylococcus aureus DltA bound to ATP245 |
| PchD | Free-standing adenylation domain of pyochelin biosynthesis183 |
| 7TYB, salicyl-AMS | |
| 7TZ4, 4-cyanosalicyl-AMS | |
| CmnG | Adenylation domain of CmnG from capreomycin biosynthesis246 |
| 7XBS, unliganded | |
| 7XBT, AMP | |
| 7XBU, capreomycidine | |
| 7XBV, AMPCPP | |
|
|
| Thioesterase | |
| SrfA-C | 1JMK, surfactin pathway108 |
| FenTE | 2CB9, fengycin NRPS pathway150 |
| Vlm2 | Valinomycin NRPS-catalytic serine replaced with diaminobutyryl nucleophile151 |
| 6ECB and 6ECC, wildtype | |
| 6ECD, bound tetradepsipeptide | |
| 6ECE and 6ECF, bound dodecadepsipeptide | |
| NocB | 6OJC, bifunctional domain from nocardicin NRPS bound to a fluorophosphonate inhibitor152 |
| Skyxy | 7CRN and 7DXO, bifunctional domain from skyllamycin NRPS pathway153 |
|
|
| Epimerization | |
| TycA | 2XHG, C-terminal epimerization domain from TycA160 |
| TycB3 | 6TA8, epimerization domain from TycB3 (ref. 161) |
| Reductase | |
| Mps2 | 4DQV, 4U5Q, reductase domain from an uncharacterized NRPS67 |
| AusA | Reductase domain from aureusimine biosynthesis169 |
| 4F6C, selenomethionine | |
| 4F6L, native | |
| MxaA | Reductase domain from myxalimid biosynthesis170 |
| 4U7W, NADH | |
| 4W4T, unliganded | |
| CAR | 5MSO and 5MSU, reductase domain from NRPS-like carboxylate reductase bound to NADP + ligand91 |
|
|
| Multi-domain proteins | |
| TycC | 2JGP, PCP-condensation from tyrocidine NRPS43 |
| EntB | 2FQ1, didomain isochorismatase-ArCP domain from enterobactin biosynthesis247 |
| SrfA-C | 2VSQ, termination module from surfactin biosynthesis bound to leucine in adenylation domain175 |
| EntF | PCP-thioesterase domains from enterobactin biosynthesis |
| 2ROQ, NMR structure154 | |
| 3TEJ, crystal structure155 | |
| Complete termination module (C-A-PCP-TE)143,176 | |
| 5T3D, holo-PCP trapped with vinylsulfonamide | |
| 5JA1, holo-PCP trapped with vinylsulfonamide with YbdZ MLP | |
| 5JA2, holo-PCP trapped with vinylsulfonamide with nonnative MLP | |
| EntE-EntB | 3RG2 and 4IZ6, fusion protein between the EntE adenylation domain and the ArCP of EntB from enterobactin biosynthesis trapped with vinylsulfonamide inhibitor121,122 |
| PA1221 | Didomain Aden-PCP domain from uncharacterized Pseudomonas NRPS115 |
| 4DG8, AMP | |
| 4DG9, holo-PCP trapped with vinylsulfonamide inhibitor | |
| SlgN1 | Complex of an MLP with an adenylation domain139 |
| 4GR4, unliganded | |
| 4GR5, AMPCPP | |
| AB3403 | Termination module (C-A-PCP-TE) from uncharacterized NRPS from Acinetobacter baumannii143 |
| 4ZXH, holo-PCP | |
| 4ZXI, holo-PCP + AMP + glycine | |
| McyG | 4R0M, didomain adenylation-PCP domain from microcystin NRPS248 |
| TqaA | 5EJD, didomain PCP with condensation termination domain64 |
| LgrA | Series of multidomain structures from linear gramicidin synthetase89 |
| 5ES5, F-A, with Asub in open or adenylate-forming conformations | |
| 5ES6, F-Acore | |
| 5ES7, F-Acore with AMPCPP, valine, and 5-formyl-THF | |
| 5ES8, F-A-PCP trapped in thiolation state with vinylsulfonamide | |
| 5ES9, F-A-PCP trapped in formylation state | |
| 5JNF, F-Acore with unusual crystal packing249 | |
| 6ULZ, F-Acore mutants140 | |
| Dimodular structures from linear gramicidin synthetase144 | |
| 6MFW and 6MFX, F-A-PCP-C in peptide donation state | |
| 6MFY, F-A-PCP-C-A in peptide donation state | |
| 6MFZ, F-A-PCP-C-A-PCP in peptide forming state | |
| 6MG0, F-A-PCP-C-A in thioester-forming state within the first adenylation domain with vinylsulfonamide inhibitor | |
| DhbF | 5U89, cross-module structure A-PCP-C with MLP domain179 |
| TioS + TioT | 5WMM, methyltransferase domain interrupting an adenylation domain, along with MLP174 |
| GrsA | 5ISX, PCP and epimerization domains of gramicidin NRPS163 |
| CAR | Didomain constructs from NRPS-like carboxylate reductase91 |
| 5MSP, PCP-Re didomain bound to NAD+ | |
| 5MSS, A-PCP didomain bound to AMP | |
| 5MSV, holo-PCP-Re didomain bound to NAD+ | |
| 5MSW, A-PCP didomain bound to AMP | |
| EpoB | 5T7Z and 5T81, cyclization and docking domain20 |
| FmoA3 | Tridomain Cy-A-PCP from an NRPS producing a free-radical scavenging peptide180 |
| 6LTA, unliganded | |
| 6LTB, AMPPNP | |
| 6LTC, α-methylseryl-AMP | |
| 6LTD, cryo-EM structure bound to α-methylseryl-AMP | |
| HitB + HitD | 6M01, complex of adenylation and carrier domain achieved with pantetheine crosslinker126 |
| OxyA | 6M7L, complex of X-domain with OxyA oxidase in glycopeptide biosynthesis250 |
| BdObiF1 | 6N8E, terminating module C-A-PCP-TE-MLP of obafluorin biosynthesis, with MLP interacting upstream bound to β-hydroxy substrate128 |
| PltF + PltL | 6O6E, complex of adenylation domain with PCP in thioester-forming conformation using vinylsulfonamide inhibitor |
| Txo1 and Txo2 | Condensation-adenylation proteins from modules 1 and 2 of teixobactin NRPS177 |
| 6OYF, Txo1 CA didomain | |
| 6OZV, Txo1 C-A didomain bound to AMP | |
| 6P1J, Txo2 C-A didomain | |
| 6P3I, Txo1 C-A didomain bound to Mg2+ | |
| 6P4U, Txo1 C-A didomain bound to Mg2+ + AMP | |
| StsA | 6ULW, keto acid activating adenylation domain tridomain with A-KR as well as pseudo Asub domain |
| NpsA-ThdA | 6VHV, fused didomain adenylation domain and PCP in tilivalline biosynthesis124 |
| Mru_0351 | 6VTJ, didomain PCP-Re from archaeal NRPS168 |
| FscG | Didomain PCP-condensation. PCP interacts with neighboring asymmetric unit to model the acceptor PCP position147 |
| 7KVW, holo-PCP in acceptor state | |
| 7KW0, loaded PCP in acceptor state | |
| 7KW2, holo-PCP in acceptor state, mutant enzyme | |
| 7KW3, PCP domain alone | |
| BmdBC | Modules of BmdB (Cy-A-T) in complex with the BmdC oxidase dimer181 |
| 7LY4, cryo-EM structure bound to FMN | |
| 7LY5, crystal structure of adenylation domains bound to BmdC oxidase dimer | |
| 7LY6, crystal structure of BmdC dimer | |
| 7LY7, crystal structure of Cy-A-PCP bound to BmdC dimer with BmdB trapped in thioester-forming state | |
| PchE | Cryo-EM structures of the PchE module (PCP-Cy-A-E-PCP) dimer with epimerization domain inserted into Asub162 |
| 7EMY, PCP-Cy-A-E in thioester-forming state, PCP2 disordered | |
| 7EN1, PCP-Cy-A-E-PCP in post condensation state | |
| 7EN2, PCP-Cy-A-E-PCP in peptide bond-forming state | |
| AmbB | Didomain PCP-C complexes146 |
| 7X0E, unliganded | |
| 7X0F, holo-PCP into donor site of condensation domain | |
| 7X17, loaded-PCP bound to donor site of condensation domain | |
2.1. Carrier protein domains
Critical to the modular assembly line architecture of NRPS enzymes is the peptidyl carrier protein that is covalently bound to and transports the substrate and peptide intermediates. The small 8 kDa carrier domains contain four α-helices. The first, second, and fourth helices are of similar lengths, encompassing 3–4 turns of the helix (Fig. 2A). The third helix is shorter, adopting fewer than two complete turns.28 The N-terminus of helix α2 contains the serine residue that is post-translationally modified with a phosphopantetheine cofactor, a conversion from apo to holo that is catalyzed by a phosphopantetheinyl transferase (PPTase) that transfers the cofactor from a molecule of CoA.32 As CoA is commonly found in the cell as the acetyl-CoA thioester and the limited ability of PPTase enzymes to distinguish CoA from a CoA thioester, many NRPS clusters harbor proof-reading thioesterases that convert the acetyl-pantetheine to the free thiol.55 The full PCP domain is often 70–80 residues in length, with the critical serine residue positioned about half way through the sequence. The loop joining the first two helices is the longest and most variable. As this loop along with the N-terminus of helix α2 border the site of pantetheinylation, these regions influence the interaction with catalytic domains,5 as described in the structures discussed below.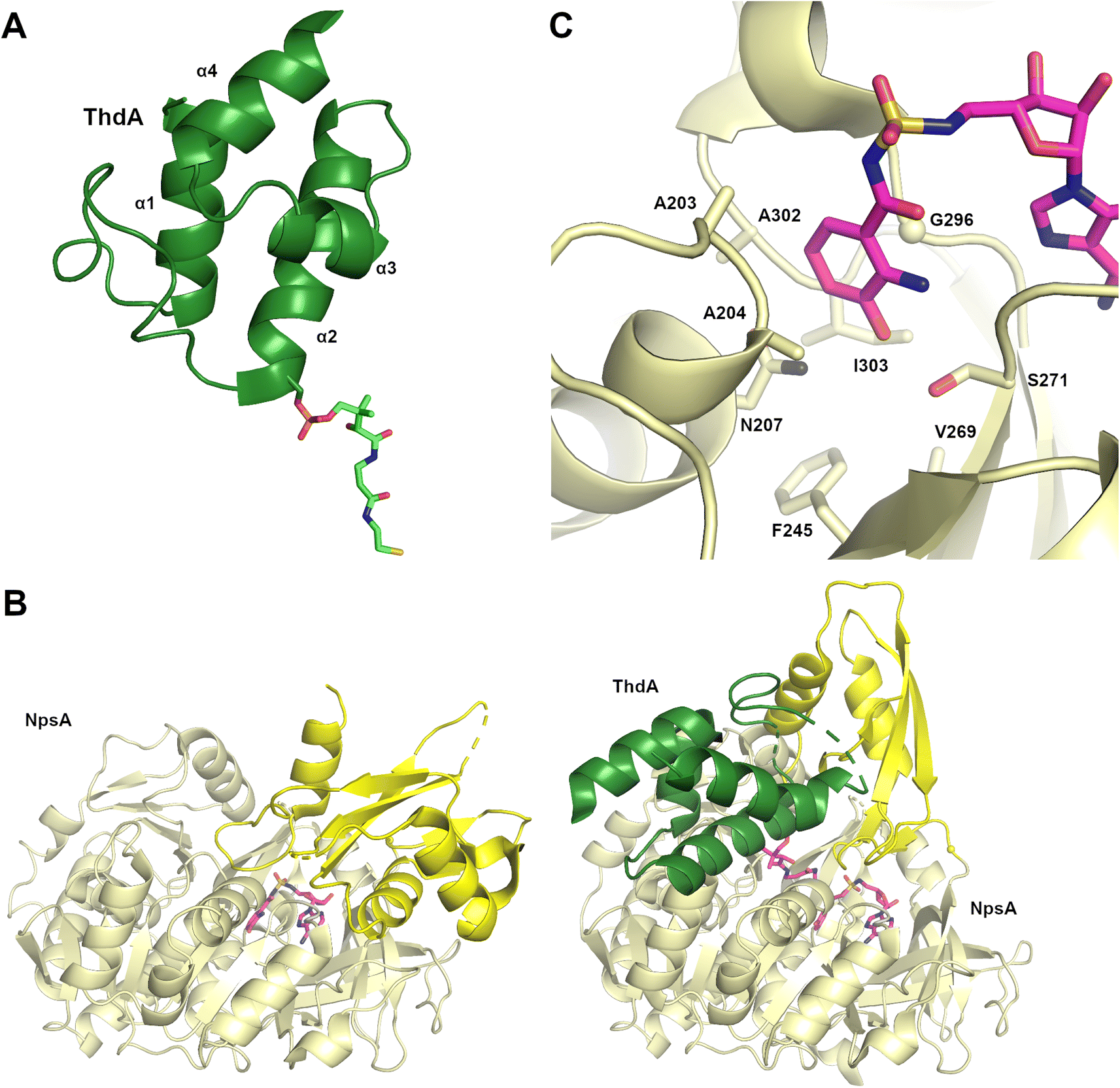 | ||
| Fig. 2 Adenylation and PCP domains from the tilivalline biosynthetic pathway. (A) The PCP domain of ThdA is shown (PDB 6VHY, chain C) illustrating four α-helices. The phosphopantetheine cofactor is attached to Ser34 (Ser542 in the NpsA-ThdA fusion protein). (B) The NpsA free-standing adenylation domain is shown in the adenylate-forming conformation (left, PDB 6VHV) and the thioester-forming conformation (right, PDB 6VHY) in complex with ThdA (green). The Asub domain is shown in brighter yellow. Bound in the active site of the adenylate-forming conformation is an adenylate mimic. On the right, the NpsA complex with ThdA highlights the rotation of the Asub domain to accommodate PCP binding. The pantetheine cofactor reacts with the vinylsulfonamide mechanism-based inhibitor. The hinge residue located between the Acore and Asub domain is shown as a small sphere. (C) The active site of NpsA highlights nine structurally conserved residues from the Acore domain that form the specificity-conferring Stachelhaus code. | ||
2.2. Adenylation domains
The A10 lysine from the Asub domain plays an important catalytic role in binding the ATP and substrate. The requirement for this residue for acyl-adenylate formation has been demonstrated experimentally in homologous protein family members110–114 and NRPS adenylation domains.115,116 In the gramicidin synthetase adenylation domain structure, this lysine interacted at the active site with oxygens from the AMP phosphate and phenylalanine substrates. Early studies with homologous acyl-CoA synthetases38,117,118 showed that binding of CoA, and presumably of the pantetheine in adenylation domains, accompanies the rotation of the Asub domain by ∼140° to present a second face to the active site. Extensive biochemical analysis confirmed the domain alternation hypothesis38 that the Asub domain adopts these two conformations, an adenylate-forming conformation and a thioester-forming conformation, for the two partial reactions. As described below, structures of NRPS adenylation domains and larger multidomain complexes confirm these two critical conformations and of the role of the thioester-forming conformation in binding the PCP and creating a pantetheine tunnel through which the cofactor approaches the active site.
The first structurally characterized adenylation–PCP interaction was derived from the first module of the enterobactin NRPS pathway, consisting of the free-standing adenylation domain EntE and the acyl-carrier protein EntB (PDB 3RG2 and 4IZ6).121,122 Two techniques facilitated crystallization. First, the two domains were genetically joined, requiring the design of a linker informed by homologous multidomain proteins. Second, the complex was trapped in the thioester-forming conformation by using an aryl-adenosine vinylsulfonamide inhibitor123 that allows a covalent bond to form between the inhibitor and pantethine group of EntB. In the complex, the Asub domain of EntE adopted the thioester-forming conformation. The EntE–EntB structure crystallized as a domain-swapped dimer as the EntB of one protein interacted with the EntE of another. The structural interface was supported through structure-guided mutagenesis that improved the ability of an EntE homolog to recognize EntB. A similar interaction was therefore expected to occur in a non-fused system including Asub rotation and key hydrophobic and hydrogen bonding interactions occurring between the adenylation domain and PCP to facilitate the passing of substrate. Critically, the structure validated the proposal38 that the Asub would adopt the thioester-forming conformation first observed with acetyl-CoA synthetase117 to load the PCP.
Additional structures, including a natural didomain PA1221 (PDB 4DG9)115 and another chimeric NpsA/ThdA (Fig. 2B, PDB 6VHY)124 displayed similar adenylation–PCP interactions relying on hydrophobic interaction occurring on helix 2 of the PCP domain and the Acore of the adenylation domain, as well as hydrogen bonding interactions and salt bridges occurring on loop 1 of the PCP domain and the Asub.
While the prior structures employed fused adenylation and PCP domains, the structure of two separate proteins have also been studied with a chemical biology approach. The type II PCP and adenylation proteins PltL and PltF were captured with a vinylsulfonamide inhibitor (PDB 6O6E),125 showing an interface that differed slightly from the previously described orientations due to less contribution of helix 2 from the PCP domain. In another approach to trap the transient interaction of free-standing adenylation and PCP domains, a bromoacetamide-modified pantetheine group was installed on HitD, a PCP from the hitachimycin NRPS.126 This reacted covalently with a cysteine residue that was engineered into the active site of the adenylating protein HitB. This structure (PDB 6M01) showed a similar overall conformation to those seen previously and highlighted the largely conserved nature of the interactions of the adenylation and carrier domains.127
Finally, several carboxylic acid reductase (CAR) enzymes have been used for structural characterization of adenylation–PCP complexes.91 In one example, a CAR (PDB 5MSS) retained similar interactions as other didomains, but did not require the common adenosine vinylsulfonamide inhibitor to trap the complex into a thiolation state. With minor variations, the complex and interface between the adenylation and carrier domains have largely been consistent across complete NRPS modules, as discussed below.
Structures of MLP proteins were determined including the PA2412 protein from the pyoverdine biosynthetic pathway (PDB 2PST) and MbtH (PDB 2KHR), illustrating a small core β-sheet with a long α-helix that packed against the sheet (Fig. 3A).133,136 The analysis of the high-resolution crystal structure of PA2412 with MLP sequences allowed for the identification of conserved sequence motifs. In particular, three conserved tryptophan residues were identified, two of which formed a shallow pocket on the face of the protein. This cavity lacked the depth or other features common to active sites, suggesting that MLPs may interact with conserved features of NRPS proteins.
 | ||
| Fig. 3 MLP domains interact with adenylation domains. (A) The structure of PA2412 (PDB 2PST), the MLP from pyoverdine biosynthesis, illustrating the conserved tryptophan residues. (B). The structure of the SlgN1 adenylation domain (PDB 4GR5), which contains an MLP natively fused at its N-terminus. Bound in the active site is a molecule of AMPCPP; that the Asub domain was disordered in the structure. (C) Close up view of SlgN1 alanine from the Acore domain inserted into the tryptophan pocket of the MLP domain. | ||
Subsequent studies with the capreomycin and viomycin didomain adenylation-PCP proteins, CmnO and VioO, showed adenylation activity only in the presence of stoichiometric amounts of their partner MLP, CmnN and VioN.137 Similar results were seen with the pacidamycin NRPS protein PacL, containing a C-A-PCP architecture, and the MLP PacJ.138 Additional clues into MLP function were provided by the ability of MLPs to co-purify with heterologously expressed NRPS modules.137 When the kinetic parameters of several adenylation domains with and without a co-expressed MLP were compared, the affinity of adenylation domains for amino acid substrate were increased by >10-fold in the presence of the MLP partner. These results suggested that MLPs physically interact with adenylation domains and enhance their activity. The role of the conserved tryptophan residues of PacJ were confirmed as mutation of both tryptophan residues on the MLP domain, eliminated the stimulation of the adenylation activity of PacL. Furthermore, this biochemical analysis recapitulated earlier cell-based studies, showing crosstalk between MLPs and adenylation domains from homologous NRPS pathways.138
Delineation of the structural interaction of an MLP with an adenylation domain was achieved with the crystal structure of SlgN1, an MLP-adenylation didomain protein involved in the biosynthesis of the antibiotic streptolydigin (PDB 4GR5).139 The SlgN1 structure (Fig. 3B) illustrated how the conserved tryptophan pocket on the surface of the MLP cradles an alanine residue of the SlgN1 adenylation domain (Fig. 3C). The mutation of this alanine to a glutamate abolished activity of the SlgN1 didomain. Subsequent structures of MLPs in complex with larger NRPS modules have all demonstrated a similar interface with an alanine, or sometimes a proline residue, projecting into the tryptophan pocket. MLPs have been observed to interact with adenylation domains in either the adenylate- or thioester-forming conformations, suggesting that MLP activity is not dependent on the adenylation domain conformation. While the structure of the MLP-adenylation domain interface is conserved, the mechanistic details of the MLP activation remains unclear.
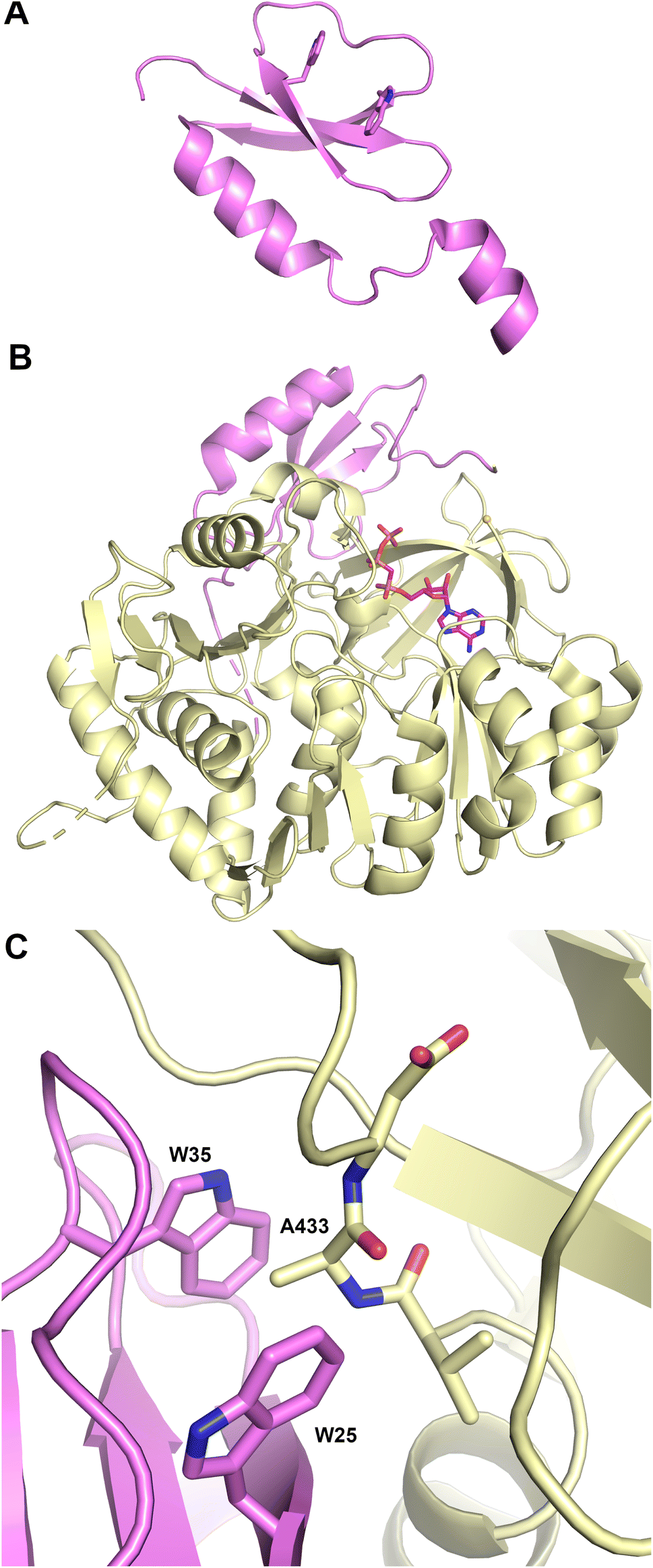
2.3. Condensation domains
The structure (Fig. 4A) revealed that this protein was a pseudo-dimer that consists of N- and C-terminal lobes that each contain a β-sheet surrounded by α-helices. The two lobes are joined by an α-helical linker and interact more closely at one side, forming an overall V-shaped domain. Between the lobes is a cleft which houses the active site, containing the conserved HHxxxDG motif that is present on the central β-strand of the N-terminal lobe. From within the C-terminal lobe, another loop that has been termed the lid or latch reaches over to the N-terminal lobe, forming one or two strands at the end of the N-terminal β-sheet.42,51 Comparisons of the relative orientation of these two lobes in different condensation domain structures illustrate that the angle between the lobes can vary, raising the possibility that the cavity between the two subdomains may open and close to facilitate interaction with partner PCPs and to adopt a catalytic conformation. However, in all cases except one, comparison of multiple structures of a single protein show that each adopts the same relative orientation. The one example of a condensation that has been structurally characterized in two states is RzmA, the lipoinitiating condensation domain from the rhizomide A NRPS system.50 Here seven structures have been solved that adopt two different states that differ by 12° rotation between the lobes, calculated using the DynDom server.142
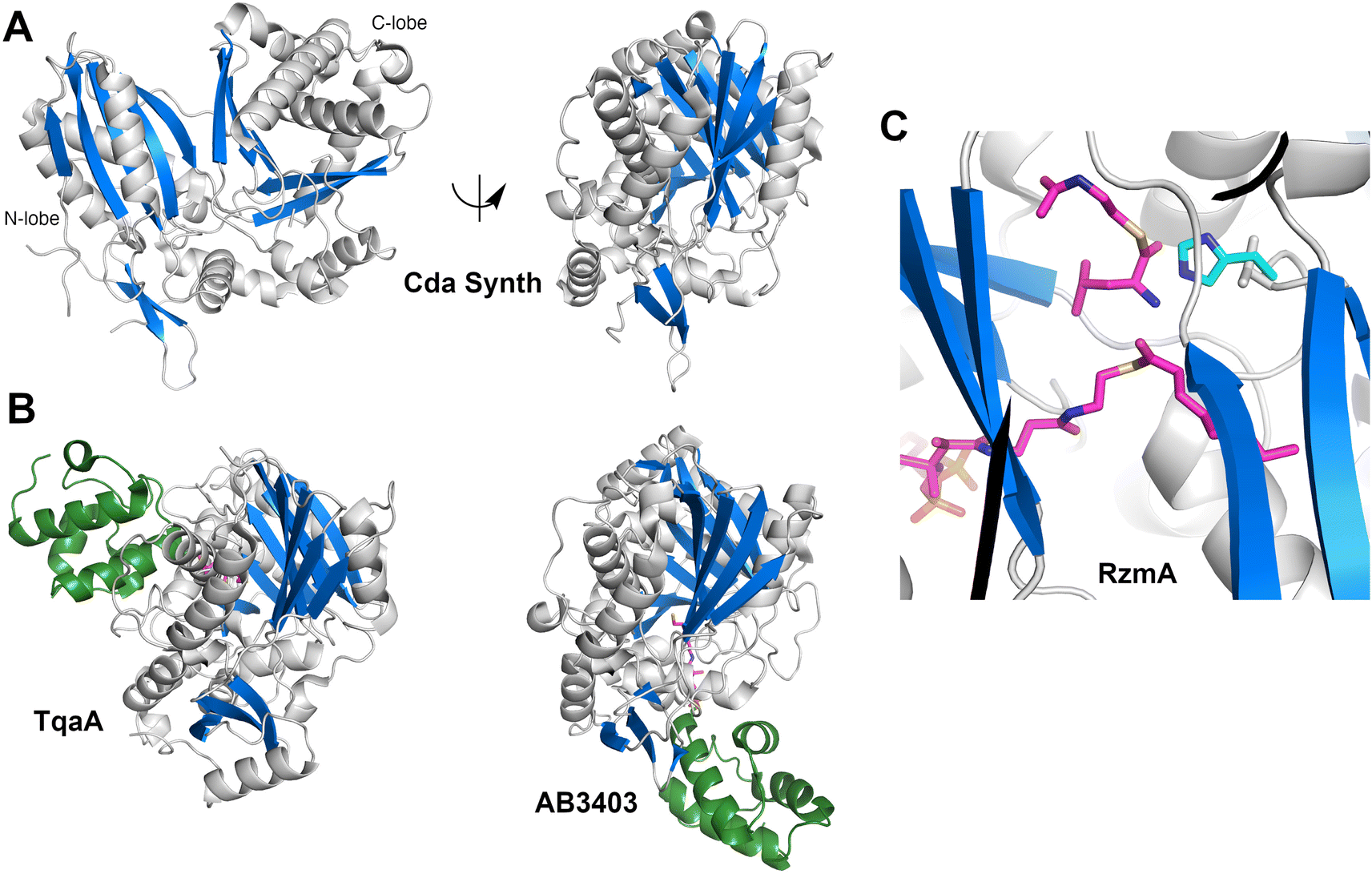 | ||
| Fig. 4 Structures of NRPS condensation domains. (A) Two views of CDA synthetase (PDB 5DU9), rotated by approximately 90° around the Y-axis. The left panel highlights the N- and C-terminal lobes. (B.) Structures of complexes of condensation domains with PCP illustrate the donor (TqaA, PDB 5EJD) and acceptor (AB3403, PDB 4ZXI) sites. Both structures are shown in the same orientation as the right panel of CDA synthetase. (C) The active site of a catalytically inert mutant of RzmA (PDB 7C1S) shows a molecule of octanoyl-CoA, representing the donor substrate, and leucyl S-(N-acetylcystamine) as a surrogate for the acceptor substrate. The catalytic His140 (cyan) superimposed from a wild-type structure was mutated to a valine to capture the two substrates. | ||
To catalyze peptide bond formation, the condensation domain must interact with two loaded PCPs that deliver an upstream donor peptide and the downstream acceptor amino acid to the active site (Fig. 4B). The structure of TqaT (PDB 5EJD), the fungal terminating condensation domain has been determined with the upstream PCP.64 This structure illustrates that the interface with the donor PCP exists primarily on the C-terminal lobe, delivering the pantetheine along the open end of the cavity. In contrast, the acceptor PCP, illustrated in the structures of the AB3403 and ObiF1 (PDBs 4ZXI and 6N8E),128,143 is positioned on the opposite face of the condensation domain with the α2 and α3 helices of the PCP interacting with the N-terminal helix of the N-lobe. The binding interfaces allow the delivery of the loaded pantetheines into the active site near the HHxxxDG motif. As described below, the structure of the dimodular LgrA protein (PDB 6MFZ) illustrates both carrier proteins interacting simultaneously at the condensation domain.144
Information about the active site was first provided through the use of biochemical probes with the initiating condensation domain of the calcium-dependent antibiotic (CDA) synthetase, one of the early condensation domain structures to have been determined.47,145 Mutation of a residue bordering the active site to a cysteine, allowed covalent modification with a N-(4-bromobutyl)alanine amide residue to form a covalent adduct to mimic the acceptor amino acid at the end of the pantetheine. The reactive α-amine of the alanine mimic interacted with the catalytic histidine (PDB 5DU9).
A recent elegant study employed the starter condensation domain of RzmA,50 a 7-module NRPS protein that initiates with a Cs-A-PCP module, where Cs represents a lipoinitation starter condensation domain. This domain loads a fatty acid from an acyl-CoA onto a leucine residue loaded by the first module. The structure was solved without ligands and bound to an octanoyl-CoA. Additionally, a catalytically compromised enzyme was examined (Fig. 4C) bound to the octanoyl-CoA donor and a leucine-SNAC (S-N-acetylcysteamine) acceptor mimic (PDB 7C1S), providing a view of the active site, with a position for the fatty acyl tail. The leucine amine overlays nicely with the amine of the tethered CDA synthetase structure. The position of the fatty acid from the donor side also provides a possible explanation for the glycine residue of the catalytic motif as the lack of a side chain provides entry of the acyl group into a mostly hydrophobic cavity in the N-terminal lobe.50
The dimodular structures of LgrA, described in greater detail below, include a structure in which a loaded PCP is bound at the donor site of the condensation domain.144 Here, the formylvaline is bound covalently to an aminopantetheine cofactor analog and adopts a position in the condensation domain active site that placed close to a reactive position. The formyl group interacts with an active site tyrosine side chain and a rotation of the amide group that mimics the thioester linkage would allow the carbonyl to adopt a proper position for attack by the acceptor amine.
A fourth view of a liganded condensation domain was provided from the PCP-C didomain from AmbB (PDB 7X0F), an NRPS responsible for the production of the antimetabolite 2-amino-4-methoxy-3-butenoic acid (AMB). The structure solved with the apo, holo, and alanine-loaded carrier protein illustrates the donor side panthethine tunnel and the positioning of the upstream amino acid near the catalytic histidine.146 Finally, a PCP-C didomain from the fuscachelin NRPS fortuitously crystallized with two molecules in the asymmetric unit, with the PCP of one protein chain interacting with the acceptor site of a neighboring molecule.147 Several structures were again determined, including apo and holo carrier proteins. Additionally, a non-hydrolyzable glycyl thioether mimic was employed (PDB 7KW0) to project into the active site, adopting a position slightly deeper into the active site cavity than in previous structures.
2.4. Thioesterase domains
These structures revealed that both type-I and type-II NRPS thioesterase domain structures have an α/β hydrolase fold consisting of a central seven-stranded β-sheet surrounded by two to three helices on either side (Fig. 5A). In the NRPS thioesterase domains, the first β-strand present in the conventional α/β hydrolase fold family is missing or forms a loop. In addition to the core α/β hydrolase fold, the thioesterase domains have a lid region inserted between β6 and β7 strands that folds over the active site. The lid region is variable in different TE domains composed of primarily one to four helices.
 | ||
| Fig. 5 Structures of NRPS thioesterase domains. (A) The structure of the EntF complex between the PCP and the thioesterase (PDB, 3TEJ) shows the PCP binding to the N-terminal face of the catalytic domain, with the pantetheine reaching into the active site where it interacts with the Asp–His–Ser catalytic triad. The lid loop region (pink) folds over the active site, forming part of the channel through which the pantetheine passes. (B) Active site of the thioesterase domain of NocB captured an acyl enzyme intermediate analog (PDB 6OJD) illustrates the binding of the peptide in a pocket formed by the lid loop region. Dashed lines indicate the catalytic triad, as well as an arginine from the lid region, interacting with the phosphonate moiety. | ||
Attempts to capture substrate in the active site have been disappointing for many thioesterase domains. Two approaches to overcome this difficulty were designed to capture a covalent adduct with the catalytic serine. In one approach, a phosphonate warhead was designed at C-terminus of tripeptide substrate to covalently link phosphonate to the catalytic serine.152,157 In another complementary approach, the catalytic serine was replaced by diamino propionate (DAP) using genetic engineering to allow the substrate to be covalently linked to the nucleophilic amine of the DAP residue.151 The ligand-bound structures revealed that oxyanion hole is provided by the amide backbone of the neighboring residues on the loop after β3 strand. Notably, several NRPS thioesterase domains that have unusual functions, like the thioesterase domains of ObiF26,128 and SulM,76 which perform β-lactone and β-lactam ring formation respectively, have cysteine at the position of serine in catalytic triad.
The lid region in NRPS thioesterase domains has been proposed to play a role in solvent exclusion from the active site150 or substrate positioning and specificity.152,155 In several domains, the lid region was found to be flexible by molecular dynamics simulations or various conformations in NMR. SrfTE crystal structures revealed open and closed conformations of lid region;108 however, similar conformations were not observed in other thioesterase domains. The flexibility of lid region was also proposed to play a role in interactions with the carrier domain and phosphopantetheine arm, and in substrate channel formation. The cavity between lid region and the core forms channel for phosphopantetheine arm and substrate loading (from β2 strand side).
The active site of some thioesterase domains catalyze unusual reactions. The thioesterase domain of the nocardicin NRPS (NocTE) performs an epimerization reaction on hydroxy-phenyl glycine (HPG) residue prior to hydrolytic cleavage of the nocardicin product.152,158 The structure has been solved bound to a phosphonate analog that illustrates the binding position of three amino acids of the nocardicin molecule. In the NocTE active site (Fig. 5B), the histidine from the catalytic triad is proposed to deprotonate the L-HPG and reprotonate from opposite side for epimerization. This is achieved through movement of the phenyl ring of the HPG residue while the remainder of the peptide chain remains relatively static. On the other hand, the thioesterase domain of skyllamycin biosynthesis performs both epimerization and macrocyclization.153 Apart from similar role of deprotonation by the histidine in Skyxy-TE, structural and mutational analysis revealed role of two additional residues that played a role in the epimerization and cyclization. Finally, the thioesterase domain from valinomycin NRPS is involved in oligomerization of intermediates by a reverse transfer pathway,151 as proposed for other oligomerizing TE domains.159 The lid region of Vlm TE showed conformation rearrangements, especially the first α-helix, to direct the dodecadepsipeptide back to the active site to favor the cyclization of product.
2.5. Epimerization and cyclization domains
NRPS epimerization domains convert L-amino acids to the D-configuration. The epimerization domain shares a structural architecture with condensation domains, including the conserved histidine motif, which is critical for catalytic activity in both domains, as well as the overall floor loop and bridge region.160Two types of epimerization domains have been identified, canonical and non-cannonical. Canonical epimerization domains share sequence homology with condensation domains, especially the important HHxxxD motif. Known epimerization structures are the C-term epimerization domain of TycA (PDB 2XHG)160 and the C-term epimerization domain of TycB (PDB 6TA8).161 Non-canonical domains resemble N-methylation domains in which they can be embedded in Asub domain, as for the pyochelin NRPS PchE.162 These domains contain an N-terminal helical bundle and a C-terminal Rossmann fold.
The two terminal domains of GrsA (PDB 5ISX)163 provide a structure of an epimerization domain in complex with its PCP domain partner. In this structure, the PCP domain is oriented into the V-shaped cavity of the epimerization domain. An extended 20 amino acid linker provided the necessary flexibility to allow the PCP and epimerization domains to adopt a catalytically relevant conformation. Here the complex illustrates the binding interface between the donor PCP and the downstream epimerization domain and also identifies amino acids residues of the PCP that play a role in the protein interface.
Multiple structures of NRPS cyclization domains have been determined, including domains from the hybrid PKS/NRPS epothilone20 and bacillamide164 clusters, and two cyclization domains from the yersiniabactin pathway.165,166 The structures illustrate the conventional condensation domain fold containing the N- and C-terminal lobes.141 These structures offer insights into the catalytic mechanism for the two-step condensation and cyclodehydration reactions. Residues that were implicated by biochemical studies are not positioned near the reacting atoms, suggesting that proper substrate orientation may promote catalysis of the condensation reaction, as suggested in early studies for condensation domains.145,164 For the cyclodehydration step, an aspartic acid not belonging to the conserved DXXXD motif appears to orient and deprotonate the cysteine side chain to promote cyclization.164,165 Recent structures of larger, multidomain NRPS proteins containing cyclization domains, described below, provide views of the active site and interfaces with carrier domains.
2.6. Reductase domains
The first NRPS reductase domain structure was solved from an unknown NRPS cluster in M. tuberculosis.67 The structure (PDB 4DQV) revealed a Rossmann fold at the N-terminus, similar to that of the short-chain dehydrogenase/reductase (SDR) superfamily of proteins,167 and a unique C-terminal subdomain (Fig. 6). The reductase domain has a central β-sheet with seven parallel β-strands that is surrounded by five α-helices on each side. The C-terminal subdomain is composed of six α-helices and two small β-strands on top and at the interface between two subdomains. Structural comparisons with SDR superfamily showed both N-terminal and C-terminal domain have insertions in NRPS reductase domains. The N-terminal subdomain contains an insertion of helix-turn-helix motif between β3 and β4 strands. Compared to the SDR family, the C-terminal subdomain is ∼40 residues larger resulting from two insertions that form a loop and an α-helix. NRPS reductase domain structures solved from other bacteria and archea similarly showed these features of Rossmann fold with unique C-terminal subdomain and insertions.168–170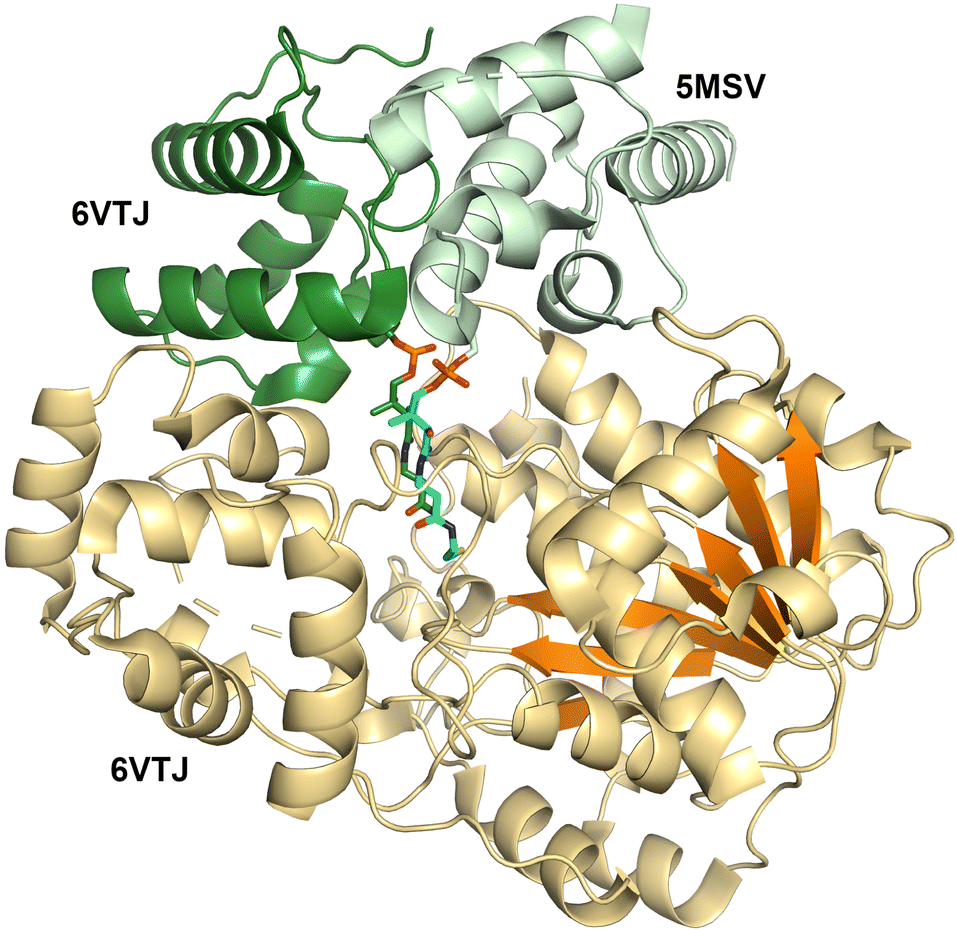 | ||
| Fig. 6 Structure of the NRPS reductase domain. The reductase domain (orange) from an archaeal NRPS (PDB 6VTJ) shows two domains, with a Rossmann fold on the right capped by a domain composed of α-helices. The active site is located at the interface between the two domains. The binding site of the PCP domain is shown for the archaeal protein (PDB 6VTJ, dark green) and the corresponding position in the superimposed structure of a carboxylic acid reductase (PDB 5MSV, light green). | ||
The canonical Thr/Ser–Tyr–Lys catalytic triad of the SDR superfamily and the NADPH-binding motif TGxxGxxG located in N-terminal Rossmann fold have been observed in all NRPS reductase domains.170 The threonine from the catalytic triad is present at the end of β5-strand, while tyrosine and lysine are on an α-helix before the β6-strand.
Structures of the holo-PCP-R didomains (Fig. 6) indicate a substrate binding pocket between two subdomains (PDB 5MSV and 6VTJ).91,168 Additionally, docking with substrate and molecular dynamics followed by mutagenesis studies support a role for the C-terminal subdomain in substrate binding.67,170 SAXS studies showed C-terminal subdomain closing towards the N-terminal subdomain upon substrate or NADPH binding in Mtb-R domain.171 Furthermore, two loops from the N-terminal Rossmann fold referred to as gating and catalytic loops showed different conformations in NADPH-free and bound states indicating concerted loop movements linked to C-terminal subdomain movement.172 Through interactions with C-terminal subdomain and linker regions, these loops were proposed to control NADPH binding and offloading of product.
Surprisingly, two PCP-reductase didomain structures revealed different interfaces for PCP interaction with reductase domain. The CAR structure (PDB 5MSV) showed that PCP interacts mainly with N-terminal subdomain, while the archaeal PCP-R didomain structure (PDB 6VTJ) showed the interaction is with a helix-turn-helix motif on the C-terminal. Although, the archaeal PCP-R didomain showed a higher buried surface area of ∼1250 Å2 compared to ∼950 Å2 for CAR–PCP-R interface, the orientation of PCP conserved Ser residue and phosphopantetheine were similar and presumed to be catalytically competent.
2.7. Methyltransferase domains
The cyclic depsipeptide thiocoraline is composed of two N-terminal 3-hydroxyquinaldic acid-capped tetrapeptides that are joined through thioester linkages between the N-terminal cysteine residue and the carboxylate at the C-terminus of the other peptide, as well as a disulfide between the internal Cys residues in the third position.173 The tetrapeptide is built through the activity of two dimodular NRPS enzymes, TioR and TioS that each incorporate two residues. TioS contains two modules that incorporate cysteine residues, each containing a methyltransferase domain inserted into the adenylation Asub domain. The methyltransferase domain of the second module catalyzes both N- and S-methylation of the loaded cysteine residue. The interrupted adenylation domain of the second module of TioS with the inserted methyltransferase domain has been structurally characterized (PDB 5WMM), illustrating a two-domain, dumbbell-shaped architecture (Fig. 7).174 Here the Acore and the methyltransferase domains are separated by the Asub domain. The 380-residue methyltransferase domain contains an N-terminal Rossmann fold, similar to other SAM-dependent methyltransferase enzymes. The domain contains an extended C-terminal region of ∼110 residues harboring four β-strands and several surrounding helices that expand the Rossmann domain. Notably, the active site of the methyltransferase domain is opened toward the catalytic face of the adenylation domain providing facile access of the loaded substrate.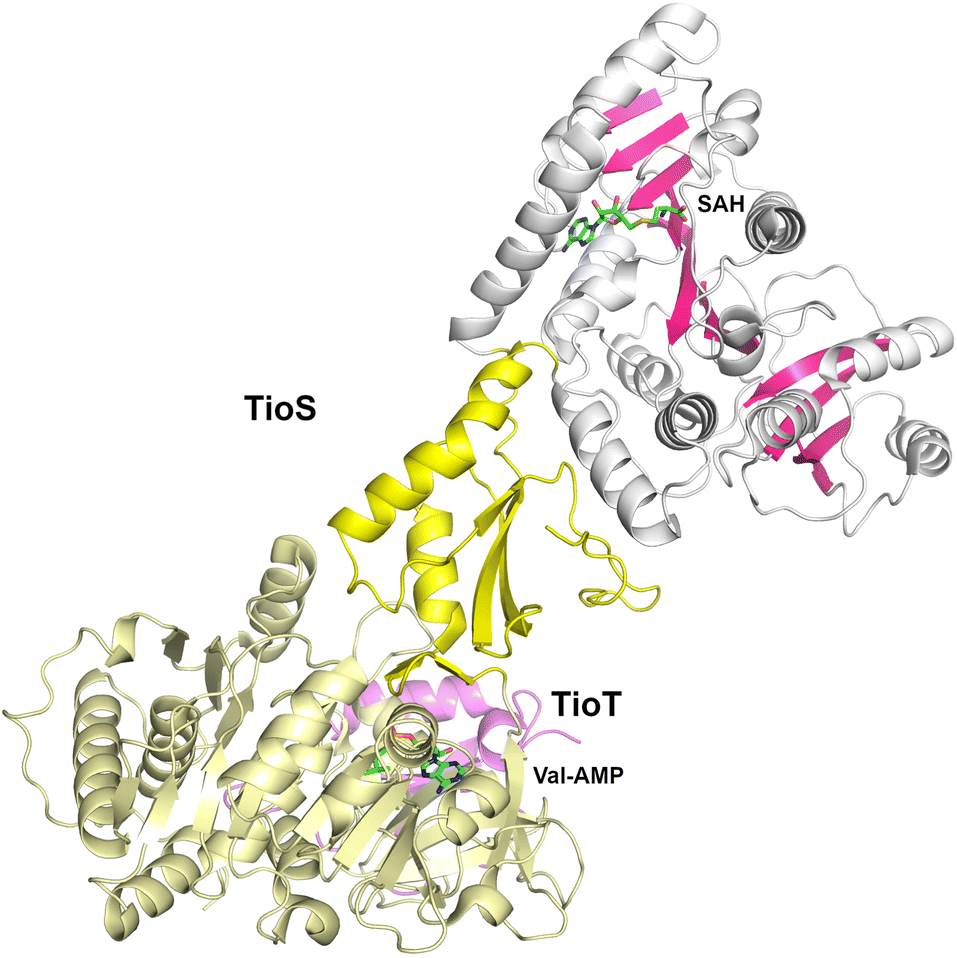 | ||
| Fig. 7 The TioS adenylation domain with an interrupted methyltransferase domain. The structure (PDB 5WMM) of TioS complexed with the TioT MLP (violet) shows that, bound to a valinyl-AMP intermediate, the adenylation domain adopts the thioester-forming conformation. The methyltransferase domain (dark pink strands) bound to S-adenosylhomocysteine only makes interactions with the Asub domain. | ||
3. Structural studies of nonribosomal peptide synthetase modules
In Section 2, we described the catalytic domains, as well as the complexes formed between catalytic domains with carrier proteins. We focus here on structures containing two or more catalytic domains that provide insight into the multidomain architecture of the NRPS assembly lines.3.1. Structures of NRPS termination modules
The SrfA-C structure (Fig. 8A) provided views of the other domains as well.175 The apo-PCP domain was located near the condensation domain, identifying a putative position for the binding the acceptor (downstream) PCP binding site. The thioesterase domain was positioned with the active site directed towards the condensation and adenylation domain active sites, although not in a position that could accommodate binding of the PCP without reorganization of the domains. Modeling the phosphopantetheine onto the PCP showed that it could reach the active site of the condensation domain. The authors noted, however, that significant conformational rearrangements would be necessary to deliver the pantetheine to the adenylation and thioesterase domains for substrate loading or release.
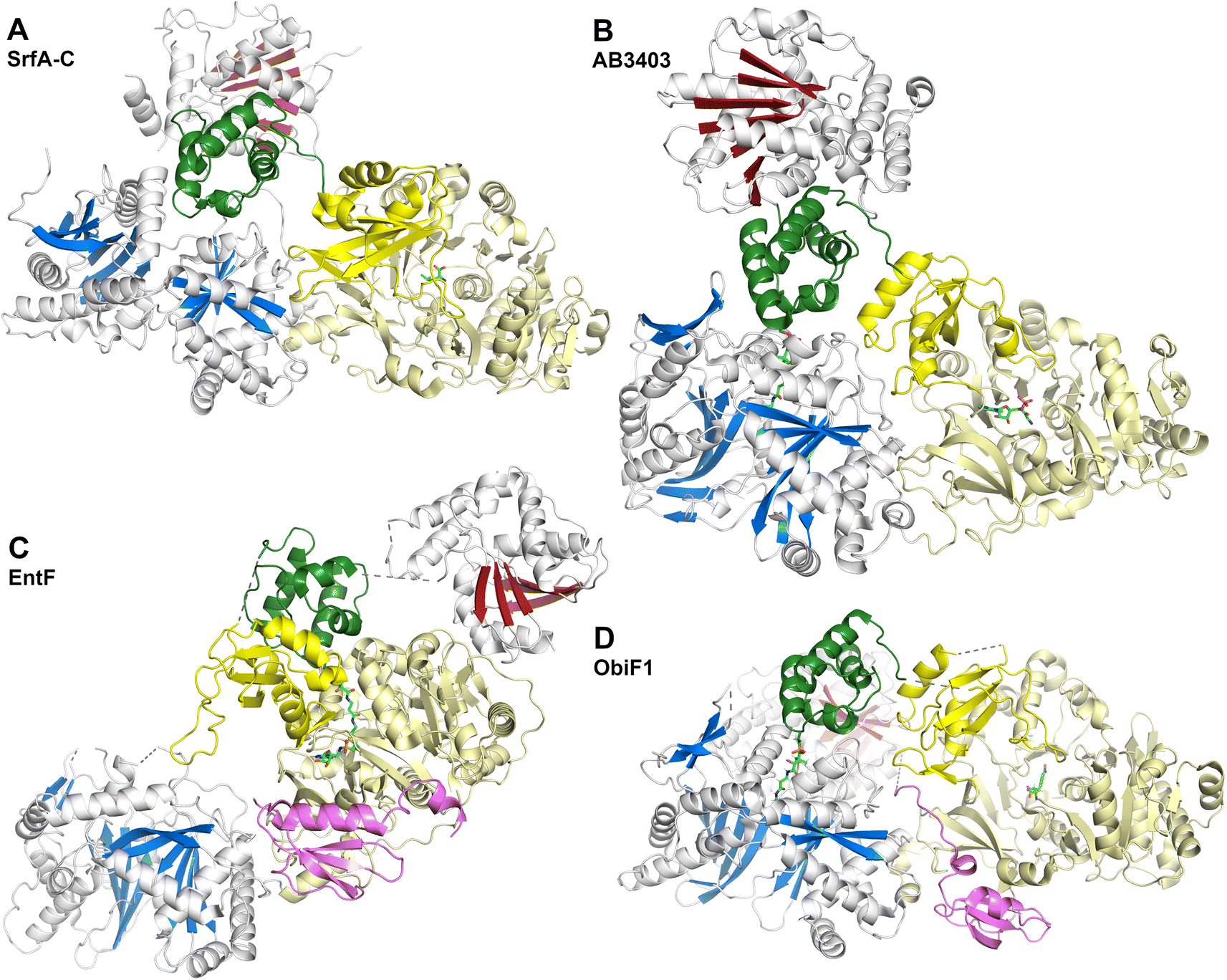 | ||
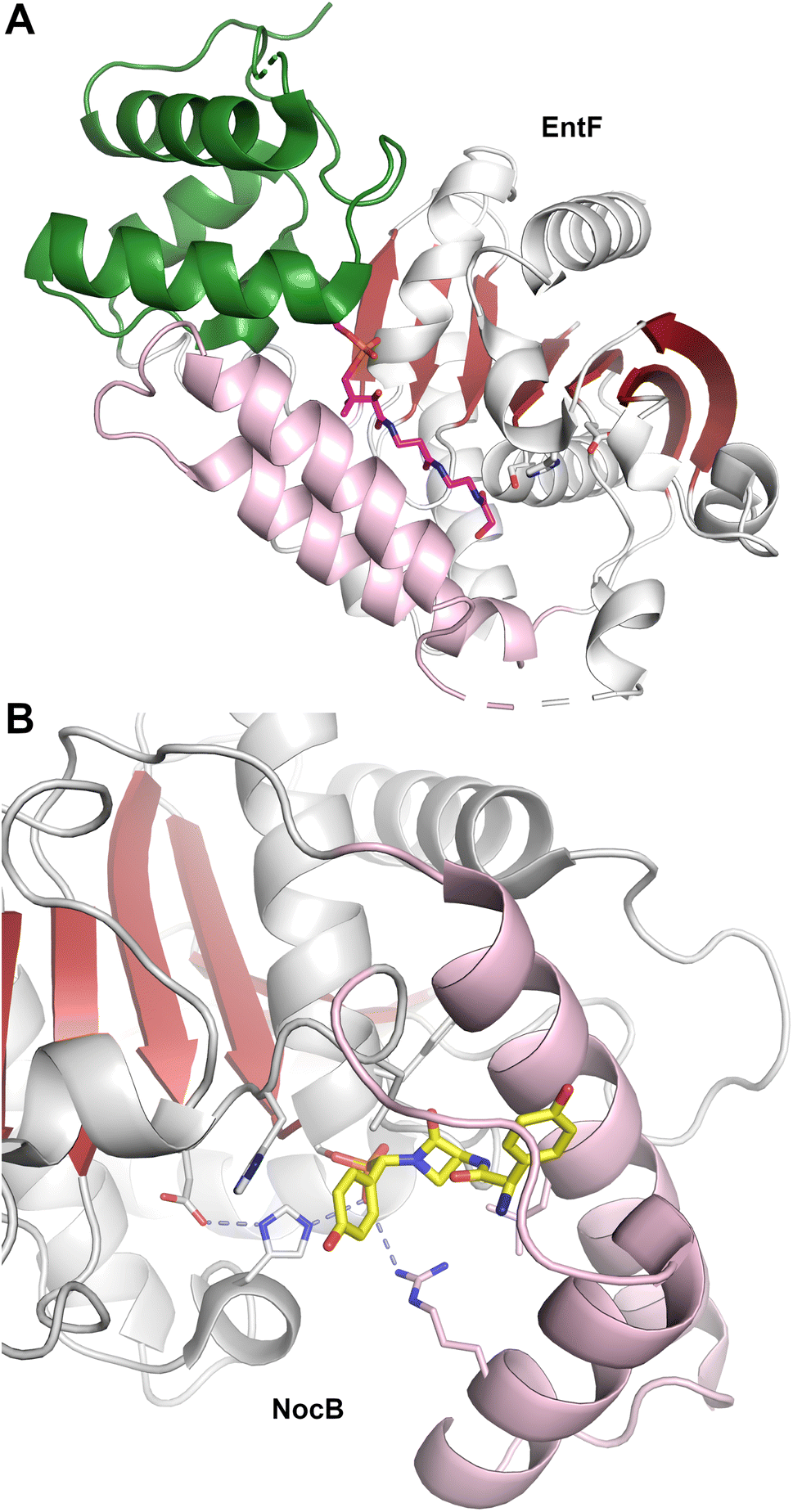
| Fig. 8 NRPS termination modules. Four structures of NRPS proteins with the C-A-T-TE architecture including (A) SrfA-C (PDB 2VSQ), (B) AB3403 (PDB 4ZXI), (C) EntF (PDB 5JA1), and (D) BdObiF1 (PDB 6N8E). All structures illustrate the condensation (blue), adenylation (yellow), PCP (green) and thioesterase (red) domains. EntF and BdObiF also contain MLP proteins. The E. coli MLP YbdZ is bound to EntF, while the BdObiF1 contains a terminal MLP that is C-terminal to the thioesterase domain. Pantetheine and additional ligands are shown with green atoms. Note that in this orientation, the thioesterase domain of ObiF is behind the condensation domain. | ||
These two proteins, AB3403 and EntF were both solved with the PCP in the holo state, illustrating the pantetheine in the active site of the condensation (AB3403) and adenylation (EntF) domains. The AB3403 (Fig. 8B) structure was similar to SrfA-C, positioning the PCP in a position near the acceptor site of the condensation domain. More compelling, EntF was crystallized in the thioester-forming state (Fig. 8C). Combined the structures provided views of the loading and peptide bond forming states within the module.
The didomain platform consisting of the condensation and Acore domains were similar to those of SrfA-C. This supported the prediction175 that, other than minor rotations between the two domains, they likely formed a organized foundation about which the dynamic domains could move. The pantetheine of holo-AB3403 reached into the active site of the condensation domain, positioning the thiol of the pantetheine near the conserved histidine of the HHxxxDG motif. In one AB3403 structure, the adenylation domain contained AMP and glycine, the latter as a representative substrate present in the active site. The adenylation domain aligned very well with the structure of gramicidin S synthetase106 in the adenylate-forming conformation. The AB3403 structure thus demonstrated that the adenylation and condensation domains can simultaneously adopt their catalytic conformations for adenylate and peptide bond formation, respectively.
The thioesterase domain of AB3403 was positioned so that it cradled the back face (opposite the pantetheinylation site) of the PCP. In this position, the thioesterase domain made no interactions with the adenylation or condensation domains.
The structure of EntF143,176 illustrated the position of the PCP bound to the adenylation domain. The structure was obtained with the serine adenosine vinylsulfonamide mechanism-based inhibitor.109,123 In EntF, the Asub domain was positioned in the thioester-forming conformation. The orientation of the PCP domain of EntF, particularly the α2 helix was similar to the PCP structures observed previously. The position of the thioesterase domain was highly dynamic, in one crystal form being sufficiently disordered in the crystal lattice to prevent inclusion in the model. Single particle reconstructions by negative stain cryo-electron microscopy supported this, as the core domains, attributed to the condensation and adenylation domains were relatively well conserved, while the presumed thioesterase domain adopted multiple positions.143 In a second crystal form (PDB 5JA1 and 5JA2),176 the thioesterase adopted a strikingly different conformation, interacting with the face of the adenylation domain that placed it opposite the condensation domain. This almost linear arrangement placed the catalytic nucleophile of the thioesterase domain ∼90 Å from the homologous position in AB3403.
EntF also served as a system to gain insights into NRPS MLP domains. EntF was solved in the absence143 and presence176 of two MLPs, its natural partner YbdZ from E. coli and PA2412, an MLP from the pyoverdine biosynthetic cluster of P. aeruginosa. The structures confirmed the prior interface seen in SlgN1.139 Importantly, no significant structural changes were noticed in the EntF adenylation domain in the presence or absence of the MLP, suggesting that the interaction did not have a structural impact that could be detected crystallographically.
Obafluorin is produced from two building blocks, a 2,3-dihydroxybenzoic acid and the nhPhe unit. Upon formation of the amide between the two, the thioesterase domain catalyzes formation of the β-lactone ring from the hydroxyl of the homophenylalanine derivative (Fig. 1).26,27 The Burkholdaria diffusa obafluorin system encompasses ObiF2 for a free-standing aryl adenylating enzyme, the carrier protein ObiD, and ObiF1, which contains the C-A-PCP-TE-MLP architecture.
What is perhaps most interesting about the ObiF structure from Burkholderia was the presence of the MLP domain. Most MLPs exist as free-standing proteins, although some, including the SlgN1 protein that was structurally characterized, exist as N-terminal fusions with the partner adenylation domain.139 The unusual ObiF1 protein from B. diffusa contains the MLP appended to the end of the module. Here, a linker joining the thioesterase domain to the MLP passes over a cleft formed between the condensation and adenylation domains, allowing the MLP to form the conserved interface with the adenylation domain, similar to those seen in prior MLP-adenylation structures.139 This was tested biochemically with truncated and mutated enzymes to confirm that this interaction supported function of the ObiF NRPS. The observation that integrated MLPs may interact with non-neighboring adenylation domains increases the potential binding partners for MLPs seen previously and suggests that predictions about MLP dependent interactions need to be experimentally tested and validated.
3.2. The didomain structures of teixobactin synthetase support the core condensation–adenylation domain platform
Complimenting the prior structures of termination modules, a series of structures from the teixobactin biosynthetic proteins provide additional views of the platform formed by the condensation and Acore domains.177 Teixobactin is a cyclic peptide antibiotic composed of 11 residues, including the non-proteinogenic enduracididine residue and a macrocycle between the C-terminal carboxylate and a threonine residue at the eighth position.178 The 11 modules are spread over two proteins, Txo1 and Txo2. The structures of the C-A didomain of the first module of Txo2 and the C-Acore didomain of the third module of Txo1 were solved.177 In the structure from Txo2, the Asub domain of the adenylation domain adopted the adenylate forming conformation. In both structures of the didomain fragments from Txo1 and Txo2, the interface between the adenylation and condensation domains were conserved with those seen previously in the termination modules. Thus, in diverse proteins from multiple sources, the conservation of this interface supported the original prediction175 of a core module structure, built from this platform that would be accessed by the mobile carrier domain.3.3. Structures of DhbF suggests limited interactions between NRPS modules
While these termination modules illustrate the conformational dynamics within a single NRPS module, studies to explore the conformational flexibility between modules can provide greater insights into the multimodular nature of NRPSs and the structural features that guide the passage of the peptide down the assembly line. Structures of DhbF, a dimodular protein that incorporates the final two amino acids of the peptide siderophore bacillibactin, provided the first insight into the interaction between an NRPS module and the downstream condensation domain. DhbF contains two modules for glycine and threonine as well as a C-terminal thioesterase domain to adopt a C-A-PCP-C-A-PCP-TE architecture. A tridomain crystal structure was solved with a protein construct containing the adenylation and PCP of the first module and the condensation domain of the second module.179 This structure (PDB 5U89), solved with an MLP bound to the adenylation domain thus spanned the modular boundaries. Additionally, negative stain EM envelopes of the complete didomain protein were also provided. The crystal structure of the A-PCP-C protein was solved with a glycyl vinylsulfonamide inhibitor, showing the PCP pantetheine bound to the adenylation domain. While the trapped pantetheine adopted the trajectory observed with prior thioester-forming conformations within the adenylation domain, the Asub did not form the thioester-forming conformation, instead showing an open position not previously observed. The PCP similarly did not adopt the same orientation seen in the prior A-PCP complexes. Most importantly, the downstream condensation domain interacts only with the PCP, making no contacts to the adenylation domain from the previous module. The dynamic nature of the cross-module protein was supported by single particle reconstructions of the full length DhbF dimodular protein (lacking the C-terminal thioesterase domain), which illustrated multiple overall configurations that lacked a consistent interface between the first and second modules.3.4. Dynamic structures of the dimodular NRPS involved in linear gramicidin synthesis
To date, the clearest views of the structures of a dimodular NRPS derive from the study of LrgA from the linear gramicidin synthetase pathway (we note parenthetically that linear gramicidin is distinct from the macrocyclic gramicidin S discussed above, Section 2.2.1). The linear gramicidin NRPS system spans four proteins, with the initial two modules encoded on LrgA, a dimodule protein with an initiation module consisting of a formyltransferase domain (F), adenylation domain, and thiolation domain, and an elongation domain harboring a condensation, adenylation, PCP and an inactive epimerization domain (E*), resulting in a complete F-A-PCP-C-A-PCP-E* architecture.74 In the reaction catalyzed by LgrA, valine and glycine are the native substrates and the intermediate Val–Gly dipeptide product is then passed to the N-terminal condensation domain of LgrB, and onward until the linear gramicidin is released by the terminal reductase domain of LgrD.An initial study89 reported the structure of the first module of LgrA. Five structures were solved and showed that the formylation and adenylation domains formed a didomain structure reminiscent of the condensation–adenylation platform in prior structures. The interface between the formyltransferase and adenylation domains, however, was smaller than observed in the more extensive C-A didomain platforms and the orientation of the formyltransferase domain was rotated slightly compared to the conventional condensation domain position. The structures supported a dynamic organization, showing structures of the adenylation domain adopting the adenylate-forming conformation (PDB 5ES5), and an F-A-PCP tridomain in both the thiolation (PDB 5ES8) and formylation (PDB 5ES9) conformations. The thiolation state employed a valine aminopantetheine analog, depicting the active site of the adenylation domain. These structures captured the large movement required by the PCP and Asub domain to transition from one catalytic state to the next. The delivery of the PCP to the upstream formylation domain required a more open state of the Asub domain than seen in the adenylation state, involving a rotation of ∼180°.
In a second study employing the complete dimodule LgrA protein (lacking the inactive C-terminal epimerization-like domain) four new structures were determined, providing views of the PCP as it migrated through different catalytic states.144 An initial structure shows the adenylation domain of a four module F-A-PCP-C protein in the adenylate-forming conformation (PDB 6MFW). Unlike the termination modules, the PCP is not positioned in the upstream domain, the formylation domain in this case, but instead is positioned bound to the donor site of the downstream condensation domain. A second structure (Fig. 9A) depicts the first adenylation domain of the F-A-PCP-C-A protein in the thiolation conformation (PDB 6MG0). With two independent molecules in the asymmetric unit, the two domains of the downstream module adopt widely different conformations, suggesting limited interactions between the consecutive NRPS modules.
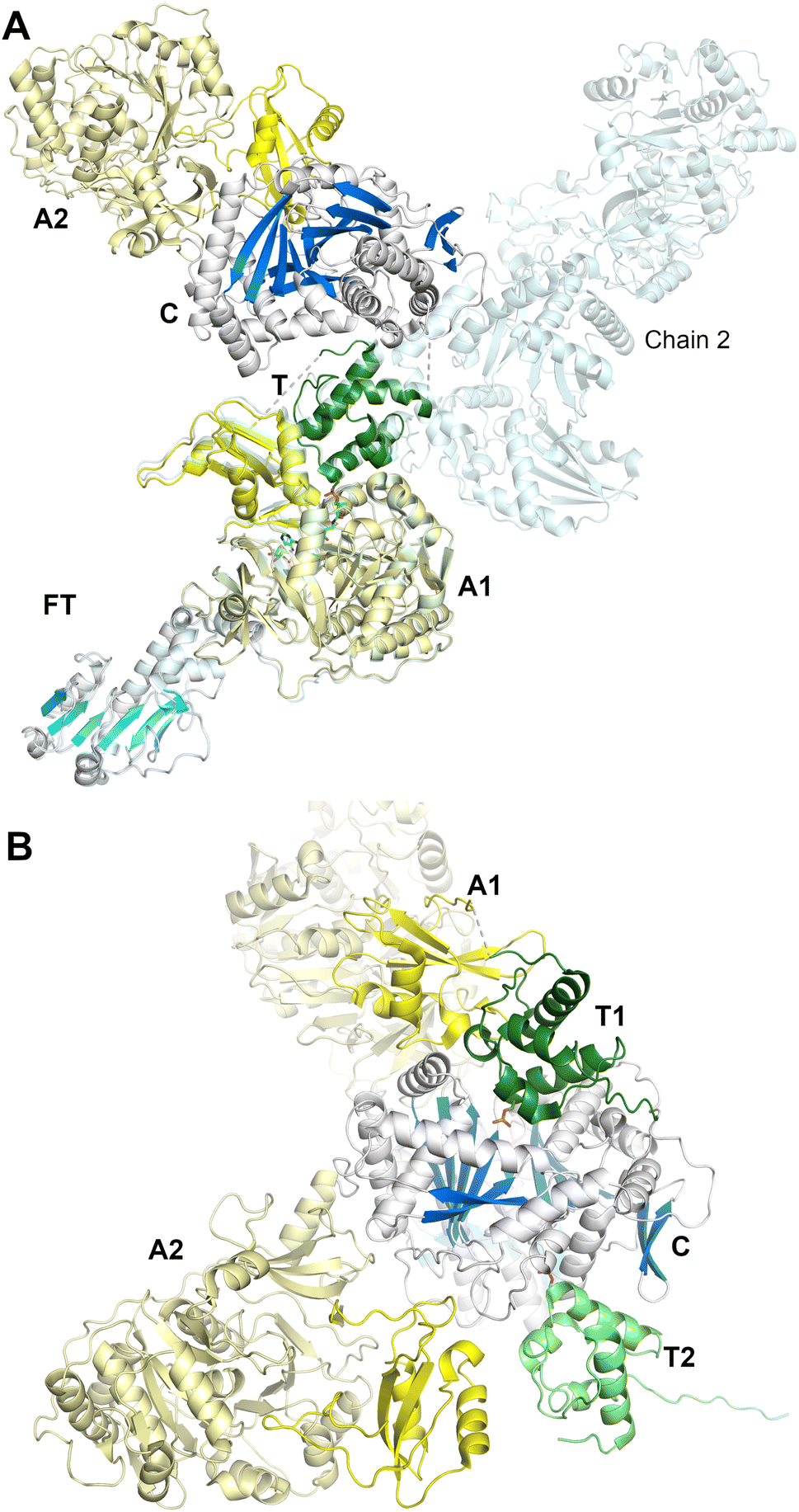 | ||
| Fig. 9 Structures of the LgrA dimodule. (A) The structure of the first five domains, F-A-T-C-A, of LgrA (PDB 6MG0). The N-terminal 193 residues form the formyltransferase domain (cyan). The holo-PCP is bound to the first adenylation domain in the thioester-forming conformation. The condensation and adenylation domains from the second module form limited interactions the first module. The crystallographic asymmetric unit contained two chains; chain B is superimposed on chain A via the F-A didomain, highlighting the highly dynamic nature of the dimodular architecture. (B) A second structure of LgrA that additionally contained the C-terminal PCP domain (PDB 6MFZ). Here both PCP domains interact with the condensation domain of module 2 in a conformation suitable for peptide bond formation. The phosphate groups of the pantetheine cofactors orient the two PCP domains. Note that the formyltransferase domain is positioned behind the condensation domain in this view. | ||
Finally, a six-domain structure depicts the entire F-A-PCP-C-A-PCP dimodular enzyme in a peptide bond-forming conformation with both carrier protein domains interacting functionally with the condensation domain (PDB 6MFZ). Here (Fig. 9B), the first adenylation domain adopts the adenylate-forming conformation, allowing the first PCP domain to adopt the donor site of the downstream condensation domain. The second module C-A-PCP aligns reasonably well with the structures of SrfA-C or AB3403, with the adenylation domain in the adenylate-forming conformation and the PCP projecting into the acceptor site of the condensation domain. Combined, these remarkable structures illustrate the static nature of the C-Acore and F-Acore, while the PCP and Asub domain are dynamic allowing the dimodular protein to adopt the necessary conformational states to deliver the PCP domains to the neighboring catalytic domains of each catalytic state.144
3.5. Structures of FmoA3 and BmdBC with cyclization domains
The FmoA3 protein involved in biosynthesis of chloroindole capped peptides JBIR-34 and JBIR-35 was the first NRPS module structure determined that contained a cyclization domain.180 Three crystal structures of the module with a Cy-A-PCP architecture were obtained using the PCP domain pantetheine serine mutant lacking additional ligands (PDB 6LTA), and complexed with AMPPNP (PDB 6LTB) or α-methyl-L-seryl-AMP (PDB 6LTC). In the structures bound to AMPPNP (Fig. 10A), the apo-PCP is positioned near the acceptor position of the cyclization domain, although not in the previously observed catalytic states and the Asub domain was disordered and not part of the final model. The crystal structures form head-to-tail dimers, supported by the cryo-EM structure (EMD-30440) and size exclusion chromatography. These structures thus demonstrated that the C-A platform could also be involved in intermodular interactions with another protein molecule. This interface is formed by residues from the C-lobe of condensation domain, as well as the C-A linker, highlighting how the NRPS module platform can serve for interactions with other mobile (sub)domains, and potentially for dimer stabilization.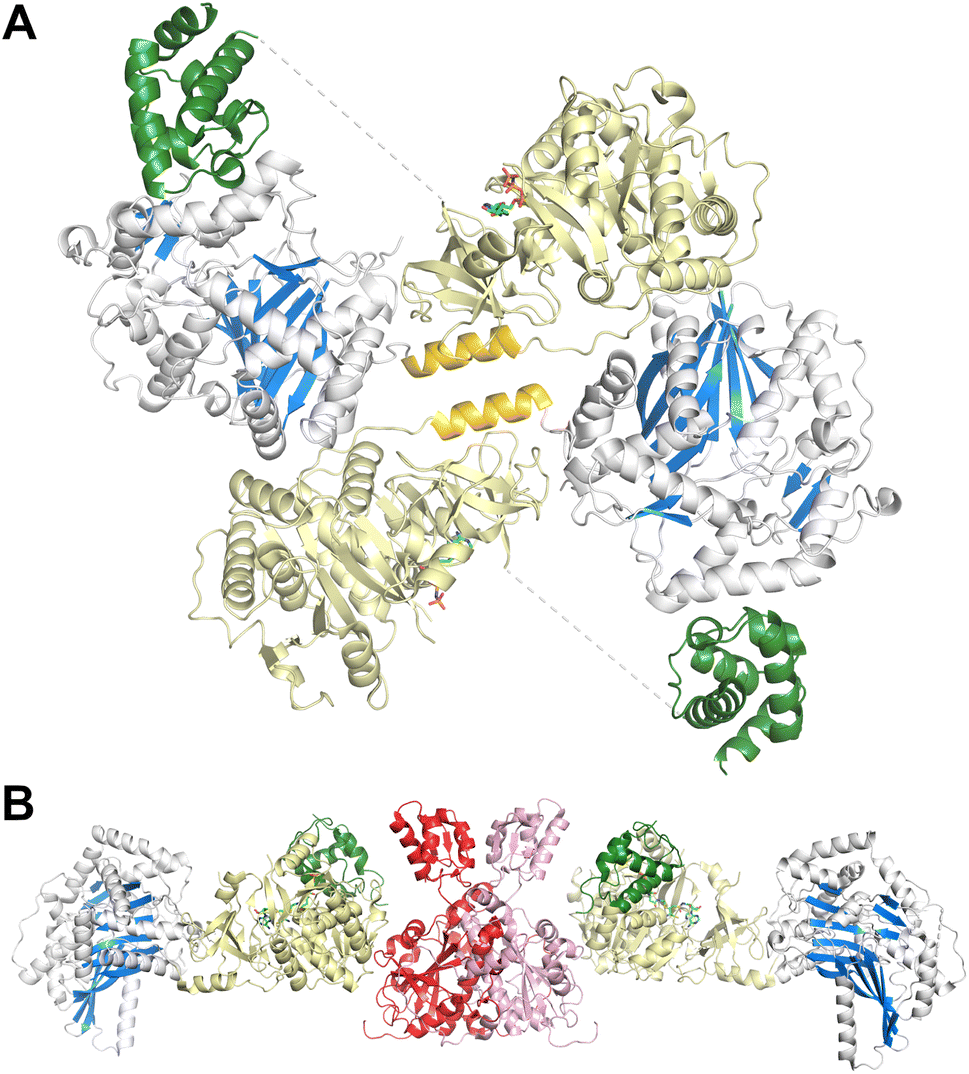 | ||
| Fig. 10 Dimeric NRPS structures. (A) The three domain NRPS FmoA3 crystallized as a dimeric structure (PDB 6LTB), which was confirmed by size exclusion chromatography and cryo electron microscopy. The dimer interface involved the condensation and Acore domains, as well as an α-helix (gold) in the linker spanning the two domains. The Asub domain was disordered and the apo-PCP was positioned near the condensation domain, although in neither the acceptor nor donor site. (B) The complex of the BmdC oxidase and the BmdB tridomain protein forms an extended dimer (PDB 7LY7). The BmdC oxidase forms a dimer; each chain interacts with the Acore domain of the BmdB NRPS. The holo-PCP interacts with the inhibitor in the adenylation domain. The Asub domain is disordered. | ||
A subsequent structure of the BmdB module 2 (BmdBM2) with a similar Cy-A-PCP architecture showed a different mode of NRPS module dimerization that required a free-standing, dimeric oxidase domain, BmdC.181 Two structures of the complex of module 2 of BmdB with BmdC from the bacillamide NRPS system were determined, including a cysteine-vinylsulfonamide adenylate inhibited X-ray crystallography structure also bound to the flavin cofactor in the oxidase domain (PDB 7LY7) and a cryo-EM (PDB 7LY4) structure bound only to the flavin. While the Asub domain was disordered, the PCP adopts a position similar to the thioester-forming state seen with EntF. The BmdC oxidase structure showed a conventional dimer formation seen in other flavin reductase domain proteins.182 The BmdB–BmdC complex structure showed a BmdC dimer at the center and two chains of BmdB module 2 interacting with each BmdC subunit, forming an elongated dimeric structure. BmdC interacts with BmdB through the Acore subdomain (Fig. 10B). As some homologous NRPSs contain an oxidase domain embedded within the Asub subdomain, this was recapitulated functionally with the insertion of the oxidase domain of BmdC into BmdB, resulting in a functional protein that was competent for production of the oxidized bacillamide product.181
3.6. Structures of the pyochelin NRPS protein, PchE
Pyochelin is a peptide siderophore produced by Pseudomonas aeruginosa that is composed of a salicyl cap followed by thiazoline and thiazole rings each derived from cyclization of cysteine residues.78,85 Like the enterobactin siderophore pathway, pyochelin biosynthesis begins with a free-standing adenylation domain, PchD, that loads the salicylate onto an N-terminal carrier domain of PchE.78,183 The PchE architecture is PCP-Cy-A-(E)-A-PCP domains, with an epimerization domain with homology to methyltransferase domains inserted into the Asub domain that converts the stereochemistry of the thiazoline ring.184Cryo-EM of PchE classified particles into three states in the catalytic cycle.162 In the first state (PDB 7EN1 and 7EN2) the downstream PCP is bound to the acceptor position of the cyclization domain with the adenylation domain adopting the adenylate-forming conformation. These core domains of this conformation therefore approximate the models seen in AB3403, SrfA-C, and ObiF1. In a second conformation (PDB 7EN2), the upstream PCP binds at the donor site of the cyclization domain providing the conformation of the cyclization domain bound with both partner PCPs (Fig. 11). In another model, the downstream PCP shows the loaded pantetheine in a post-condensation state, illustrating a bound salicylthiazoline thioester.
 | ||
| Fig. 11 The structure of PchE in the peptide bond forming conformation. PchE, an NRPS from pyochelin biosynthesis, contains a PCP-Cy-A-(Ep)-PCP organization, with an epimerization domain inserted into the Asub domain. The structure (PDB 7EN2) lacks the pantetheine cofactors but highlights the structure of both PCP domains bound to the cyclization domain. | ||
In the other major conformation (PDB 7EMY), the core PchE domains adopt a conformation similar to that seen in the thioester-forming conformation of EntF. In this model, the upstream PCP is loaded with the salicyl group providing a view of the loaded pantetheine binding in the cyclization active site. The downstream PCP domain, however, was not observed in the structure. The structure of the cyclization domain shows the conventional fold seen in the condensation domain family of proteins. The structure interfaces that bind the donor or acceptor PCP domains are relatively static, showing only minor orientation changes of several residues at the interface.
In all of the conformations, the epimerization domain, which is inserted into the Asub domain, appears to follow the Asub trajectory, adopting different positions in the overall modular structure. The interface between the Asub and epimerization domain remains constant, suggesting they rotate as a rigid body.
Importantly, the structures also provided views of ligands bound to the pantetheine in the structures giving insight into the active site residues that provide catalytic and binding activity. The structures thus identify a binding pocket for the salicyl moiety of the donor PCP and the salicylthiazoline group of the acceptor in the post-condensation state. Residues that are identified interacting with the ligands were mutated to alanines, confirming predicted roles. The overall structure of the active site was described as a “Y-shaped substrate-binding tunnel”, with both panteteine moieties approaching from opposite sides of the active site and the base of the pocket providing room for the peptide ligands.
4. Structures of modular nonribosomal peptide synthetases provide insight into a highly dynamic structural cycle
These structures provide a foundation for understanding the conformational changes necessary for the NRPS structural cycle, the coordinated series of events that load and deliver the biosynthetic intermediates to the neighboring catalytic domains. The views provided by multiple structural techniques and multiple NRPSs from different sources illustrate conserved structural features within one module and dynamic interactions across modular boundaries.4.1. The core platform of multidomain NRPS proteins
The condensation–adenylation domains are a consistent feature, providing, as first postulated with SrfA-C,175 a core platform around which the other domains can migrate to adopt the necessary catalytic states. The C-A interface is conserved in the homologous Cy-A proteins and, interestingly, the non-homologous formyltransferase domains also adopt a similar position relative to the adenylation domain.In addition to the NRPS termination modules, the condensation–adenylation didomain structures from internal module of Txo1 (ref. 177) and the complete dimodule structures from LgrA showed that second module with C-A-PCP architecture144 and also have similar C-A platforms. The platform thus appears stable within internal elongation modules of multi-module proteins as well. Moreover, the platform was also found fairly rigid at different conformations of the dimodule in substrate donation, thiolation or condensation states of LgrA,144 as well as the Cy-A-PCP enzymes, FmoA3 and BmdBC, and the ArPCP-Cy-A-(E)-PCP architecture in PchE showed similar C-A platform at different states of catalytic cycle.162,180,181 Combined, these studies demonstrate the conserved condensation-Acore conformation, allowing other mobile domains to rearrange for the catalytic cycle of an NRPS module.
4.2. Preferred states of NRPS C-A-PCP modules
Including the PCP domain to consider the structure of the C-A-PCP tridomain conformation, the NRPS module appears to have two conformations that are commonly observed and may represent favored, stable, states (Fig. 12). The first places the Asub domain in the adenylate-forming conformation and places the PCP in the acceptor site of the condensation domain. Indeed, this adenylate-forming conformation is most frequently observed in adenylation domains in isolation supporting the relative stability of this conformation.38 This preferred NRPS conformation may thus be driven by the adenylation domain. This state (State 1) was first observed with the termination modules SrfA-C, AB3403, and ObiF1. More recently, this state is seen in additional multidomain proteins that contain additional domains, including the PchE,162 the second module of the LgrA dimodular protein,144 and perhaps by the structure of FmoA3,180 although here the PCP adopts only a similar conformation.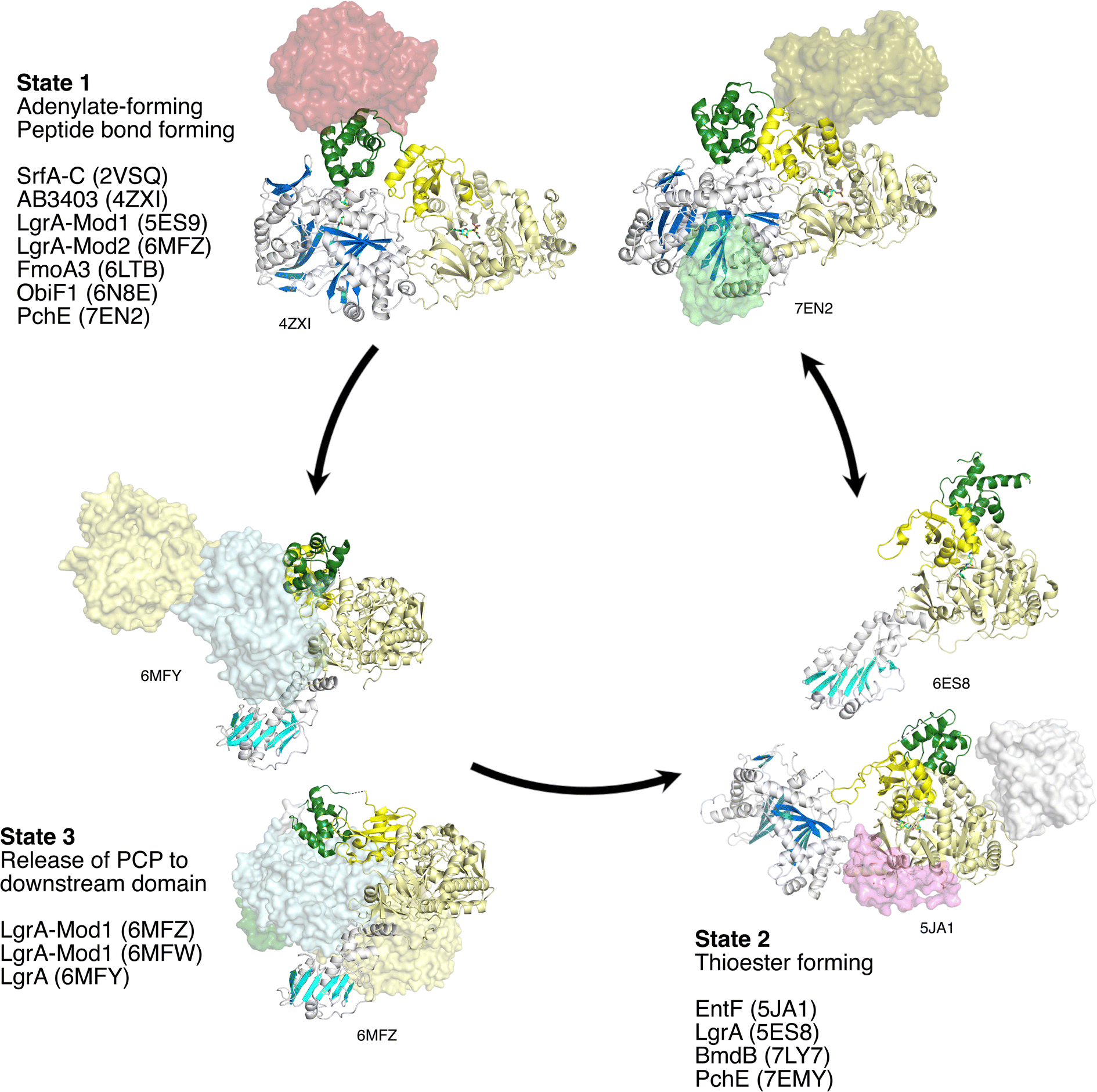 | ||
| Fig. 12 Structural cycle of the NRPS module. To transit through the catalytic cycle, an NRPS module must allow the PCP to visit the adenylation domain for substrate loading, the condensation domain for peptide extension, and then release the PCP to the downstream domain. Multiple proteins have been structurally characterized in State 1, in which the adenylation domain adopts the adenylate forming conformation, while the PCP is positioned in the acceptor site of the condensation domain. Similarly, many multidomain modular proteins have been crystallized in State 2, in which the Asub domain has rotated into the thioester-forming conformation, enabling the PCP to deliver the pantetheine cofactor to the adenylation domain active site for loading. Return of the now loaded PCP to the condensation domain allows peptide extension. Finally, the PCP is released in State 3 to the downstream module, or thioesterase domain. | ||
The second common conformation for an NRPS module is State 2. Here, domain alternation of the Asub positions the adenylation domain into the reactive thioester-forming conformation, allowing the Acore and Asub to form the binding interface to bind the PCP and direct the pantetheine into the active site for substrate loading. First observed with EntF, this state has been observed multiple times, including the first F-A-PCP module of LgrA,89 where the condensation domain is replaced with a formyltransferase domain, and the Cy-A-PCP BmdB,181 although the Asub domain was disordered in the structure.
The Asub rotation that is responsible for the adenylation domain to adopt the two catalytic conformations of the adenylation domain38 is the primary conformational change of the central core of the module. As the PCP is positioned downstream of the Asub domain, the movement of this subdomain repositions the PCP facilitating its migration between the condensation and adenylation domains. The PCP and the Asub domain do not move as a rigid body, but rather the movement of the Asub domain creates a binding interface for the carrier, directing the pantetheine into the active site to attack the adenylate intermediate. In contrast, the embedded epimerization domain in PchE does move with Asub as a rigid body as observed in multiple conformations.162 While only a single example, this does suggest that the core catalytic conformations are compatible with interrupted adenylation domains.
It is reasonable to conclude that State 1 and State 2 are favored positions of an NRPS module. Importantly, in State 1, both the condensation and adenylation domains adopt catalytically-competent states, with the loaded pantetheine directed into the condensation domain for peptide bond formation and the adenylation domain in the adenylate-forming conformation.
The two catalytic states of the adenylation domain, controlled by the domain alternation strategy of the Asub domain, transition the PCP domain between the core adenylation and condensation domains. In State 1, the amino acid reacts with ATP in the adenylation domain forming the adenylate intermediate. Release of pyrophosphate accompanies rotation of the Asub into the adenylation domain to adopt State 2 for loading of the activated amino acid onto the pantetheine. The loaded carrier then returns to State 1, placing the PCP close to the acceptor site of the upstream condensation domain. Here it can insert the aminoacyl pantetheine into the condensation domain. Upon delivery of the peptide or amino acid on the upstream PCP domain, peptide bond formation can occur, transferring the incoming amino acid or peptide to the amine of the downstream amino acid, and extending the peptide by one unit. These two features, the ability to catalyze simultaneously two reactions in State 1 and the Asub domain rotation between the two preferred conformational states that naturally deliver the downstream PCP to the adenylation and condensation domains, increases the efficiency of the NRPS structural cycle.10,143
This feature of the NRPS module is further supported by a series of elegant studies that have used biophysical approaches to explore the dynamics and equilibrium of the core domains. In these studies,185–187 small molecule fluorescent probes and fluorescent protein domains were employed to monitor via fluorescence energy transfer experiments the relative orientations of NRPS domains in solution. Studies with a adenylation PCP didomain support the formation of the two catalytic conformations supporting the domain alternation hypothesis,38 noting that addition of excess PPi drives the Asub towards the adenylate-forming conformation.185 Additionally, these studies invoke a new, intermediate conformation that is used for threading the pantetheine group into the adenylation domain for the thioester-forming reaction.187 Interestingly, the authors note that one structure of DhbF (PDB 5U89)179 adopts a plausible partially closed structure. Finally, this FRET-based approach has recently been employed to investigate a full condensation-adenylation-PCP module, illustrating a preference of the loaded PCP to adopt a conformation in the condensation domain, awaiting delivery of the upstream peptide.186 Inclusion of a downstream condensation domain in a C-A-PCP-C, or indeed the inclusion of a full downstream module, while technically challenging, will undoubtedly offer more insight into the equilibrium of the loaded PCP between three potential catalytic domains.
4.3. Interactions between NRPS modules appear less well-structured
While States 1 and 2 allow amino acid activation and loading, as well as peptide bond formation, a third state involves interactions between the PCP and the downstream module. The features that govern the delivery of a PCP domain to the downstream module are less well-defined. We first consider delivery of the PCP to the thioesterase domain. While this domain is often considered to be a component of the final module, the specialized termination module,188 for structural considerations, the thioesterase domain could be considered an extra downstream module, albeit one that contains only single domain. Indeed, some early reviews189 insightfully considered the thioesterase domain as its own module.NRPS termination is thus the simplest case to consider for release of the extended peptide and delivery of the PCP to the next module. As observed in the structures of different termination modules, the thioesterase domain is not tightly bound to the core platform of the termination module, suggesting that there is not a consistent conformational change that drives the PCP delivery to the thioesterase for product release. The mobility and lack of the thioesterase domain in different structures of termination modules as well as the need to release fully the PCP from the condensation domain, suggests that in the termination modules, the PCP completely disengages from the platform formed by the condensation and Acore domains, freeing the PCP and thioesterase to interact productively.
Perhaps more interesting than the interaction with the downstream thioesterase domain is the delivery of the PCP to a downstream module. While there are quite a few structures of PCP-C didomains that illustrate the position of the donor PCP binding to the N-lobe of the condensation domain, these structures do not really offer insights into the intermodular interactions. For that, it is necessary to examine the structures harboring domains that span the modular structural boundaries, specifically containing, at minimum, a A-PCP-C architecture. Here the structures that provide views of such a construct are those of DhbF and LgrA.144,179 With both systems, the downstream module, represented by the condensation domain in DhbF or the downstream C-A-PCP of the larger LgrA proteins (Fig. 12), does not interact with the adenylation domain of the upstream module at all. This suggests that, while tethered through the PCP and the associated linkers, the modules do not interact functionally in any organized manner and adopt multiple positions.144,179 State 3 is therefore not likely to be a uniform “state” at all, and the conformations of multiple modules will highlight dynamic conformations. In fact, single particle EM of EntF143 or DhbF179 also support this, with multiple conformational states observed in both systems.
5. Conclusions and future directions
Combined, the structural studies of NRPSs point to an orderly structural cycle within a module, contrasting with more dynamic conformations of multimodular proteins. As we look toward the future, we briefly discuss recent advances in the broader field of structural biology and how they will impact the study of NRPSs. We then conclude with a discussion of some remaining questions in the field of NRPS structural biology, and the role that structural biology of NRPS proteins has played and will continue to play in the discovery of novel natural products.5.1. Impact of cryo-EM on NRPS structural biology
Structural biology is in the middle of a technical revolution in which cryo electron microscopy (cryo-EM) now rivals X-ray crystallography in its ability to reveal atomic level details about protein structures.190,191 This dramatic improvement in resolution has come about because of improvements in technology, including detectors and processing algorithms, and improvements in availability, further enabled by the creation of resources that provide opportunities to the structural biology community for training and access.192–195As described earlier, several NRPSs have been probed with cryo-EM and negative stain EM, including the low resolution class averages of EntF143 and DhbF,179 and more recently the atomic resolution structures of FmoA3 (ref. 180) and PchE.162 Where the former supported the dynamic conformational states that exist across modules, the structures of FmoA3 and PchE herald a potential new era for the investigation of large multidomain NRPSs that are recalcitrant to crystallization.
The dynamic nature of multidomain and multimodule NRPS proteins remains a challenge to structural characterization. As technical improvements in the field of cryo-EM continue, the tool may provide the ability to classify and characterize individual conformational states toward an ultimate goal of visualizing the trajectories between different catalytic conformations adopted by NRPSs as the carrier protein delivers its cargo to the active sites of neighboring domains.
In this regard, the structure of PchE162 was examined with cryoDRGN196 to model the heterogeneity that exists in a population of particles observed by cryo-EM. This approach uses a neural network to model both individual states and continuous trajectories in an unbiased manner. Analysis of the dimeric PchE enzyme with cryoDRGN provided views of six discrete steps in the structural catalytic cycle as the two carrier proteins from the N- and C-termini of the PCP-Cy-A-(E)-PCP protein migrated through their respective catalytic conformations for loading, peptide bond formation, and epimerization.162
5.2. Impact of artificial intelligence methods in NRPS structural studies
In addition to cryo-EM, a second significant advance in the field of structural biology recently is the use of artificial intelligence tools on the prediction of protein structures from sequence. The foundation built by ∼200000 experimental protein structures and the decades long effort of the Community Assessment of Structure Prediction (CASP) community197 resulted in the use machine learning and artificial intelligence for the high reliability prediction of structures. The achievements by the AlphaFold198 and RoseTTAFold199 teams allow for the prediction of models with high accuracy for many targets.
NRPS enzymes provide exciting opportunities as well as challenges to employ these tools for additional discovery. At its most basic, the creation of predictions of domain structures may facilitate the design of protein constructs of individual domains for functional and structural studies, providing guidance in the design of domain boundaries and truncation sites. More exciting, the predictions of structures of domains that catalyze unconventional chemical steps (for that particular NRPS domain) should provide plausible hypotheses for testing in validation of the features responsible for these distinct activities.
Multidomain catalytic machines like the NRPSs and PKSs may also provide an opportunity to further develop these tools for the proper prediction of the organization of multidomain proteins. With a wealth of data on the structures of individual domains and increasing awareness of the organization of distinct catalytic states, these modular enzymes are attractive targets for optimizing approaches to predict larger organizational principles that enable predictions of multidomain structures. On the other hand, unlike stable multidomain proteins that may adopt a single, lowest energy state, the dynamic systems of natural product biosynthesis require multiple transient interactions between protein domains, so tools that seek a single, energetically favorable conformation may be challenged to identify multiple conformations.
5.3. The impact of NRPS structural biology on natural product discovery
Efforts to discover and characterize new NRPSs, or exploit existing NRPS pathways, may enable the isolation of novel bioactive compounds. In the first approach, the diverse biosynthetic gene clusters identified from sequencing individual species and metagenomic samples that have yet to be characterized can be used to computationally or experimentally identify novel active compounds.13,14,16,200 In the second approach, well-characterized NRPS clusters can be engineered to create novel molecules in a directed or random strategy.40,201–203 Both of these approaches benefit from prior structural studies on NRPS proteins and warrant continued studies to understand more fully the relationship between protein sequence, structure, and function, including both substrate specificity and catalytic activity.Finding novel natural product biosynthetic gene clusters is relatively straightforward, given the large size and sequence conservation of gene encoding NRPS enzymes as well as tools such as AntiSMASH16,204 that allow mining DNA sequences for the presence of different classes of natural product biosynthetic clusters. AntiSMASH, and its related derivatives, provides not only information about the existence of the cluster and the genes contained therein, it also provides homology searches and can, in instances where a well-characterized pathway exists, identify potential products. While clearly identifying NRPS clusters within a genome, truly novel pathways with limited homology to known biosynthetic gene clusters pose a larger challenge. A clear understanding of the unique features of a predicted sequence and the implication for protein structure and function would facilitate predictions about the product that results from a predicted biosynthetic cluster.
Notably, while the accurate determination of a product from a novel gene cluster is most desired, it has been shown that it is not an absolute requirement to find active molecules. Predictions from biosynthetic gene clusters harbored in the human microbiome were used to inform chemical synthesis of potential products.205 A collection of 157 synthetic-bioinformatic natural products (syn-BNP) were chemically synthesized, guided by reasonable predictions derived from the NRPS architecture and predicted substrate specificity. This approach identified nine molecules with antibacterial activity that had not previously been identified. The molecules were chemically diverse; combined, they contain macrocycles of 18–39 atoms, and 14 of 20 proteinogenic amino acids as well as several nonstandard residues. More recently, this approach identified a topoisomerase inhibitor containing a p-aminobenzoic acid building block.206 Related approaches have also shown promising results to predict accurately the final chemical model using the gene sequence, including neighboring accessory proteins, and the insights from domain structures.15 As the sequence, structure, function relationship gets clearer, the predictions will presumably also improve and the rate of finding accurate products with novel activities may also increase.
In addition to supporting natural product discovery through genome mining and predictions, structures of NRPSs have also provided insights used to engineer novel enzymes and pathways. In particular, structural studies have guided the design of boundaries between, and sometimes within, domains to enable altering specificity while maintaining proper domain interfaces. Recent studies have emphasized the important role played by the condensation domain in enabling proper function of an engineered module. The introduction of exchange units (XU) that encompass the A-PCP-C tridomain have allowed for the generation of novel catalysts.207 However, this initial approach was somewhat limited by the specificity of the condensation domain, which must match the residue introduced by the subsequent condensation domain. This limitation has in some instances been addressed by considering the subdomain architecture of the condensation domain, identifying different junctions to allow small exchange units (XUC) that consist of the CAsub-A-PCP-CDsub region, where CAsub and CDsub represent the acceptor and donor subdomains of the condensation domain, respectively.208 Heterologous expression of the engineered clusters enabled high production titres of the novel peptide products in several distinct proof-of-concept experiments. In a related approach, maintenance of the compatibility of the CDsub region of the condensation domain with the downstream adenylation domain allowed efficient substitution of an adenylation domain in both pyoverdine and tyrocidine NRPS systems.209 Combined, these studies and others56,210 are providing insights into the structural features that guide progress through the NRPS pathway and the complementary roles that individual domains play to provide specificity for the incorporation of novel building blocks into a peptide intermediate. A complete understanding of these features will identify changes that can be made to active site pockets to enable the production of novel peptide products.
5.4. Structural characterization of unusual NRPS proteins
Finally, while the conventional NRPS systems have proven to be rich targets for structural study, novel biosynthetic gene clusters continue to provide unusual systems that remain to be understood. Investigation of the structures of these novel targets serves to inform the discovery of novel compounds through both bioinformatic mining and and engineering efforts.In contrast, in lysobactin biosynthesis, the LybB NRPS also harbors C-terminal tandem thioesterase domains. The first domain is sufficient to catalyze cyclization of the peptide while the second thioesterase preferentially catalyzes hydrolysis of the linear peptide.212 Additionally, the second thioesterase domain catalyzed more efficient release of misprimed acetyl-pantetheine groups, suggesting that this domain functioned more like a Type II proof-reading thioesterase domain.
In some cases, when tandem domains are present, one of the catalytic domains is nonfunctional, exhibiting truncations or smaller substitutions to critical catalytic residues. The PyrG protein that encodes the fourth module for the biosynthesis of pyridomycin, a hybrid NRPS-PKS antibiotic, for example contains tandem adenylation domains.213 The first adenylation domain lacks the critical residues in the phosphate-binding A3 motif and the conserved catalytic lysine. The individual domains were expressed alone and in tandem, confirming the role of the second adenylation domain in activating the isoleucine substrate.
Other examples of tandem NRPS domains include auriculamide biosynthesis, where the first module of AurA contains tandem adenylation domains, where the first domain appears dispensible,214 the tandem cyclization domains of the anguibactin63 and vibriobactin46 NRPSs, and the iterative fungal NRPS responsible for beauvericin production contains tandem PCP domains in the terminal module.215
The thalassospiramide peptides, a family of hybrid NRPS-PKS depsipeptides, contain multiple modules that lack adenylation domains.220 In one proposed system, the carrier domain of the fifth module is loaded by the adenylation domain of module 2. Additionally, this unusual pathway employs module skipping and a “pass-back” strategy in which the peptide at module 4 is attacked by the loaded substrate at module 2, resulting in products containing multiple copies of the modified peptide produced iteratively by the NRPS and PKS modules 2–4. Combined, this pathway is highly promiscuous, resulting in the production of a panel of over two dozen lipopeptides in both the native and heterologous producing organisms.
An unusual condensation domain was recently examined in the NRPS system that is responsible for the production of the antimetabolite methoxyvinylglycine.227 Here the condensation domain catalyzes the α,β-dehydration of a PCP-bound β-hydroxy-γ-methoxyglutamate derivative, leading to decarboxylation and the formation of L-2-amino-4-methoxy-trans-3-butenoic acid. The structure of this condensation domain informed mutagenesis experiments to explore the roles of canonical amino acids and other residues present in the active site.
In the production of the ansamycin antibiotics, the three domain A-PCP-TE protein AstC activates a D-alanine residue that is loaded onto the carrier domain.228 Rather than catalyzing hydrolysis or cyclization, the TE domain then catalyzes the transfer of this residue to the hydroxyl of a macrolactam polyketide precursor that has been produced by an associated PKS protein.
Instances of unusual architecture and activity are seen in the icosalide biosynthetic pathway of several Burkholderia species.229 In addition to the lipoinitiating starter condensation domain that installs a fatty acid, the icosalide system contains tandem condensation domains at the third module, in which a second starter condensation domain of the third module N-acylates the loaded serine residue at the downstream carrier domain. The installed β-hydroxy fatty acid then serves as the nucleophile for the first condensation domain of this module to capture the lipopeptide from module 2.
Ultimately, the structural foundation that enables the unusual architecture and activities of the modular NRPSs in many cases remains to be explored. Structural studies supported by biochemical analysis can illustrate how variations in catalytic residues facilitate the novel chemistry that is carried out. In addition to variation in specific catalytic activity, structural studies can also inform our understanding of the interactions of NRPS domains with non-neighboring carrier domains when needed. While it might be expected that interactions are driven through complementary protein interfaces, questions remain as to what allows for the correct balance of promiscuity and selectivity. For example, the ability of an adenylation domain to load multiple carrier proteins as seen in capreomycin and viomycin biosynthesis,230 requires the ability to recognize the appropriate carrier partners while retaining sufficient discrimination to prevent undesirable loading of alternate carrier domains.
5.5. Conclusions
The fascinating NRPS enzymes and the modular catalytic strategy that results in the production of novel peptide natural products has intrigued the chemical biology and enzymology communities for decades. However, genome mining suggests that we have only scratched the surface of the full breadth of NRPS products that exist in nature. Continued discovery of these clusters will undoubtedly raise additional questions that can be answered by the careful structural and functional exploration of proteins that do not fall into conventional rules governed by previously characterized systems. This remains an exciting time in the field of natural product biosynthesis and the combination of new computational and experimental tools heralds a new era of natural product discovery.6. Author contributions
All authors were involved in writing sections of the individual draft. KDP and AMG were responsible for editing and revisions to the final version.7. Conflicts of interest
The authors have no conflicts of interest to declare.8. Acknowledgement
Work in our lab is funded by a grant from the National Institutes of General Medicine (GM136235) from the National Institutes of Health.9. References
- C. T. Walsh, Nat. Chem. Biol., 2015, 11, 620–624 CrossRef CAS PubMed.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2020, 83, 770–803 CrossRef CAS PubMed.
- M. Montalban-Lopez, T. A. Scott, S. Ramesh, I. R. Rahman, A. J. van Heel, J. H. Viel, V. Bandarian, E. Dittmann, O. Genilloud, Y. Goto, M. J. Grande Burgos, C. Hill, S. Kim, J. Koehnke, J. A. Latham, A. J. Link, B. Martinez, S. K. Nair, Y. Nicolet, S. Rebuffat, H. G. Sahl, D. Sareen, E. W. Schmidt, L. Schmitt, K. Severinov, R. D. Sussmuth, A. W. Truman, H. Wang, J. K. Weng, G. P. van Wezel, Q. Zhang, J. Zhong, J. Piel, D. A. Mitchell, O. P. Kuipers and W. A. van der Donk, Nat. Prod. Rep., 2021, 38, 130–239 RSC.
- M. Dell, K. L. Dunbar and C. Hertweck, Nat. Prod. Rep., 2022, 39, 453–459 RSC.
- J. C. Corpuz, J. O. Sanlley and M. D. Burkart, Synth. Syst. Biotechnol., 2022, 7, 677–688 CrossRef PubMed.
- S. Bonhomme, A. Dessen and P. Macheboeuf, Open Biol., 2021, 11, 200386 CrossRef CAS PubMed.
- J. M. Reimer, A. S. Haque, M. J. Tarry and T. M. Schmeing, Curr. Opin. Struct. Biol., 2018, 49, 104–113 CrossRef CAS PubMed.
- T. Izore and M. J. Cryle, Nat. Prod. Rep., 2018, 35, 1120–1139 RSC.
- B. R. Miller and A. M. Gulick, Methods Mol. Biol., 2016, 1401, 3–29 CrossRef CAS PubMed.
- A. M. Gulick, Curr. Opin. Chem. Biol., 2016, 35, 89–96 CrossRef CAS PubMed.
- R. D. Süssmuth and A. Mainz, Angew. Chem., Int. Ed. Engl., 2017, 56, 3770–3821 CrossRef PubMed.
- C. T. Walsh, H. Chen, T. A. Keating, B. K. Hubbard, H. C. Losey, L. Luo, C. G. Marshall, D. A. Miller and H. M. Patel, Curr. Opin. Chem. Biol., 2001, 5, 525–534 CrossRef CAS PubMed.
- M. H. Medema, Nat. Prod. Rep., 2021, 38, 301–306 RSC.
- M. H. Medema, T. de Rond and B. S. Moore, Nat. Rev. Genet., 2021, 22, 553–571 CrossRef CAS PubMed.
- M. A. Skinnider, N. J. Merwin, C. W. Johnston and N. A. Magarvey, Nucleic Acids Res., 2017, 45, W49–W54 CrossRef CAS PubMed.
- K. Blin, S. Shaw, A. M. Kloosterman, Z. Charlop-Powers, G. P. van Wezel, M. H. Medema and T. Weber, Nucleic Acids Res., 2021, 49, W29–W35 CrossRef CAS PubMed.
- X. Hao, S. Li, J. Ni, G. Wang, F. Li, Q. Li, S. Chen, J. Shu and M. Gan, J. Nat. Prod., 2021, 84, 2990–3000 CrossRef CAS PubMed.
- H. G. Smith, M. J. Beech, J. R. Lewandowski, G. L. Challis and M. Jenner, J. Ind. Microbiol. Biotechnol., 2021, 48, kuab018 CrossRef CAS PubMed.
- E. Dehling, J. Ruschenbaum, J. Diecker, W. Dorner and H. D. Mootz, Chem. Sci., 2020, 11, 8945–8954 RSC.
- D. P. Dowling, Y. Kung, A. K. Croft, K. Taghizadeh, W. L. Kelly, C. T. Walsh and C. L. Drennan, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 12432–12437 CrossRef CAS PubMed.
- H. Liu, L. Gao, J. Han, Z. Ma, Z. Lu, C. Dai, C. Zhang and X. Bie, Front. Microbiol., 2016, 7, 1801 Search PubMed.
- Z. Lv, W. Ma, P. Zhang, Z. Lu, L. Zhou, F. Meng, Z. Wang and X. Bie, Synth. Syst. Biotechnol., 2022, 7, 989–1001 CrossRef CAS PubMed.
- R. F. Little and C. Hertweck, Nat. Prod. Rep., 2022, 39, 163–205 RSC.
- D. A. Herbst, C. A. Townsend and T. Maier, Nat. Prod. Rep., 2018, 35, 1046–1069 RSC.
- P. Paiva, F. E. Medina, M. Viegas, P. Ferreira, R. P. P. Neves, J. P. M. Sousa, M. J. Ramos and P. A. Fernandes, Chem. Rev., 2021, 121, 9502–9553 CrossRef CAS PubMed.
- J. E. Schaffer, M. R. Reck, N. K. Prasad and T. A. Wencewicz, Nat. Chem. Biol., 2017, 13, 737–744 CrossRef CAS PubMed.
- T. A. Scott, D. Heine, Z. Qin and B. Wilkinson, Nat. Commun., 2017, 8, 15935 CrossRef CAS PubMed.
- J. Crosby and M. P. Crump, Nat. Prod. Rep., 2012, 29, 1111–1137 RSC.
- A. C. Mercer and M. D. Burkart, Nat. Prod. Rep., 2007, 24, 750–773 RSC.
- K. Reuter, M. R. Mofid, M. A. Marahiel and R. Ficner, EMBO J., 1999, 18, 6823–6831 CrossRef CAS PubMed.
- R. H. Lambalot, A. M. Gehring, R. S. Flugel, P. Zuber, M. LaCelle, M. A. Marahiel, R. Reid, C. Khosla and C. T. Walsh, Chem. Biol., 1996, 3, 923–936 CrossRef CAS PubMed.
- J. Beld, E. C. Sonnenschein, C. R. Vickery, J. P. Noel and M. D. Burkart, Nat. Prod. Rep., 2014, 31, 61–108 RSC.
- F. Rusnak, W. S. Faraci and C. T. Walsh, Biochemistry, 1989, 28, 6827–6835 CrossRef CAS PubMed.
- C. Rausch, T. Weber, O. Kohlbacher, W. Wohlleben and D. H. Huson, Nucleic Acids Res., 2005, 33, 5799–5808 CrossRef CAS PubMed.
- T. Stachelhaus, H. D. Mootz and M. A. Marahiel, Chem. Biol., 1999, 6, 493–505 CrossRef CAS PubMed.
- G. S. Patil, P. Kinatukara, S. Mondal, S. Shambhavi, K. D. Patel, S. Pramanik, N. Dubey, S. Narasimhan, M. K. Madduri, B. Pal, R. S. Gokhale and R. Sankaranarayanan, Elife, 2021, 10, e70067 CrossRef CAS PubMed.
- K. Turgay, M. Krause and M. A. Marahiel, Mol. Microbiol., 1992, 6, 529–546 CrossRef CAS PubMed.
- A. M. Gulick, ACS Chem. Biol., 2009, 4, 811–827 CrossRef CAS PubMed.
- M. A. Marahiel, T. Stachelhaus and H. D. Mootz, Chem. Rev., 1997, 97, 2651–2674 CrossRef CAS PubMed.
- A. Stanišić and H. Kries, ChemBioChem, 2019, 20, 1347–1356 CrossRef PubMed.
- T. Stachelhaus, H. D. Mootz, V. Bergendahl and M. A. Marahiel, J. Biol. Chem., 1998, 273, 22773–22781 CrossRef CAS PubMed.
- S. Dekimpe and J. Masschelein, Nat. Prod. Rep., 2021, 38, 1910–1937 RSC.
- S. A. Samel, G. Schoenafinger, T. A. Knappe, M. A. Marahiel and L. O. Essen, Structure, 2007, 15, 781–792 CrossRef CAS PubMed.
- E. D. Roche and C. T. Walsh, Biochemistry, 2003, 42, 1334–1344 CrossRef CAS PubMed.
- T. A. Keating, C. G. Marshall, C. T. Walsh and A. E. Keating, Nat. Struct. Biol., 2002, 9, 522–526 CAS.
- C. G. Marshall, N. J. Hillson and C. T. Walsh, Biochemistry, 2002, 41, 244–250 CrossRef CAS PubMed.
- K. Bloudoff, D. Rodionov and T. M. Schmeing, J. Mol. Biol., 2013, 425, 3137–3150 CrossRef CAS PubMed.
- C. Rausch, I. Hoof, T. Weber, W. Wohlleben and D. H. Huson, BMC Evol. Biol., 2007, 7, 78 CrossRef PubMed.
- M. J. Wheadon and C. A. Townsend, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2026017118 CrossRef CAS PubMed.
- L. Zhong, X. Diao, N. Zhang, F. Li, H. Zhou, H. Chen, X. Bai, X. Ren, Y. Zhang, D. Wu and X. Bian, Nat. Commun., 2021, 12, 296 CrossRef CAS PubMed.
- K. Bloudoff and T. M. Schmeing, Biochim. Biophys. Acta, 2017, 1865, 1587–1604 CrossRef CAS PubMed.
- M. E. Horsman, T. P. Hari and C. N. Boddy, Nat. Prod. Rep., 2016, 33, 183–202 RSC.
- B. T. Caswell, C. C. de Carvalho, H. Nguyen, M. Roy, T. Nguyen and D. C. Cantu, Protein Sci., 2022, 31, 652–676 CrossRef CAS PubMed.
- C. A. Shaw-Reid, N. L. Kelleher, H. C. Losey, A. M. Gehring, C. Berg and C. T. Walsh, Chem. Biol., 1999, 6, 385–400 CrossRef CAS PubMed.
- M. Kotowska and K. Pawlik, Appl. Microbiol. Biotechnol., 2014, 98, 7735–7746 CrossRef CAS PubMed.
- M. Kaniusaite, J. Tailhades, E. A. Marschall, R. J. A. Goode, R. B. Schittenhelm and M. J. Cryle, Chem. Sci., 2019, 10, 9466–9482 RSC.
- A. M. Gehring, I. Mori, R. D. Perry and C. T. Walsh, Biochemistry, 1998, 37, 11637–11650 CrossRef CAS PubMed.
- T. Duerfahrt, K. Eppelmann, R. Muller and M. A. Marahiel, Chem. Biol., 2004, 11, 261–271 CrossRef CAS PubMed.
- T. Stachelhaus and C. T. Walsh, Biochemistry, 2000, 39, 5775–5787 CrossRef CAS PubMed.
- X. Gao, S. W. Haynes, B. D. Ames, P. Wang, L. P. Vien, C. T. Walsh and Y. Tang, Nat. Chem. Biol., 2012, 8, 823–830 CrossRef CAS PubMed.
- S. W. Haynes, B. D. Ames, X. Gao, Y. Tang and C. T. Walsh, Biochemistry, 2011, 50, 5668–5679 CrossRef CAS PubMed.
- W. L. Kelly, N. J. Hillson and C. T. Walsh, Biochemistry, 2005, 44, 13385–13393 CrossRef CAS PubMed.
- M. Di Lorenzo, M. Stork, H. Naka, M. E. Tolmasky and J. H. Crosa, BioMetals, 2008, 21, 635–648 CrossRef CAS PubMed.
- J. Zhang, N. Liu, R. A. Cacho, Z. Gong, Z. Liu, W. Qin, C. Tang, Y. Tang and J. Zhou, Nat. Chem. Biol., 2016, 12, 1001–1003 CrossRef CAS PubMed.
- K. Haslinger, M. Peschke, C. Brieke, E. Maximowitsch and M. J. Cryle, Nature, 2015, 521, 105–109 CrossRef CAS PubMed.
- L. Du and L. Lou, Nat. Prod. Rep., 2010, 27, 255–278 RSC.
- A. Chhabra, A. S. Haque, R. K. Pal, A. Goyal, R. Rai, S. Joshi, S. Panjikar, S. Pasha, R. Sankaranarayanan and R. S. Gokhale, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 5681–5686 CrossRef CAS PubMed.
- J. A. Read and C. T. Walsh, J. Am. Chem. Soc., 2007, 129, 15762–15763 CrossRef CAS PubMed.
- D. J. Wilson, C. Shi, A. M. Teitelbaum, A. M. Gulick and C. C. Aldrich, Biochemistry, 2013, 52, 926–937 CrossRef CAS PubMed.
- M. A. Wyatt, W. Wang, C. M. Roux, F. C. Beasley, D. E. Heinrichs, P. M. Dunman and N. A. Magarvey, Science, 2010, 329, 294–296 CrossRef CAS PubMed.
- Y. Li, K. J. Weissman and R. Muller, J. Am. Chem. Soc., 2008, 130, 7554–7555 CrossRef CAS PubMed.
- F. Kopp, C. Mahlert, J. Grunewald and M. A. Marahiel, J. Am. Chem. Soc., 2006, 128, 16478–16479 CrossRef CAS PubMed.
- J. E. Becker, R. E. Moore and B. S. Moore, Gene, 2004, 325, 35–42 CrossRef CAS PubMed.
- N. Kessler, H. Schuhmann, S. Morneweg, U. Linne and M. A. Marahiel, J. Biol. Chem., 2004, 279, 7413–7419 CrossRef CAS PubMed.
- M. Z. Ansari, J. Sharma, R. S. Gokhale and D. Mohanty, BMC Bioinf., 2008, 9, 454 CrossRef PubMed.
- R. Li, R. A. Oliver and C. A. Townsend, Cell Chem. Biol., 2017, 24, 24–34 CrossRef CAS PubMed.
- K. J. Labby, S. G. Watsula and S. Garneau-Tsodikova, Nat. Prod. Rep., 2015, 32, 641–653 RSC.
- L. E. Quadri, T. A. Keating, H. M. Patel and C. T. Walsh, Biochemistry, 1999, 38, 14941–14954 CrossRef CAS PubMed.
- T. A. Ronnebaum, J. S. McFarlane, T. E. Prisinzano, S. J. Booker and A. L. Lamb, Biochemistry, 2019, 58, 665–678 CrossRef CAS PubMed.
- D. A. Miller, L. Luo, N. Hillson, T. A. Keating and C. T. Walsh, Chem. Biol., 2002, 9, 333–344 CrossRef CAS PubMed.
- G. Weber, K. Schorgendorfer, E. Schneider-Scherzer and E. Leitner, Curr. Genet., 1994, 26, 120–125 CrossRef CAS PubMed.
- S. Sivanathan and J. Scherkenbeck, Molecules, 2014, 19, 12368–12420 CrossRef PubMed.
- B. Yuan, Z. Wu, W. Ji, D. Liu, X. Guo, D. Yang, A. Fan, H. Jia, M. Ma and W. Lin, J. Biol. Chem., 2021, 100822, DOI:10.1016/j.jbc.2021.100822.
- H. M. Patel and C. T. Walsh, Biochemistry, 2001, 40, 9023–9031 CrossRef CAS PubMed.
- T. A. Ronnebaum and A. L. Lamb, Curr. Opin. Struct. Biol., 2018, 53, 1–11 CrossRef CAS PubMed.
- C. Reimmann, H. M. Patel, L. Serino, M. Barone, C. T. Walsh and D. Haas, J. Bacteriol., 2001, 183, 813–820 CrossRef CAS PubMed.
- K. M. Meneely, T. A. Ronnebaum, A. P. Riley, T. E. Prisinzano and A. L. Lamb, Biochemistry, 2016, 55, 5423–5433 CrossRef CAS PubMed.
- G. Schoenafinger, N. Schracke, U. Linne and M. A. Marahiel, J. Am. Chem. Soc., 2006, 128, 7406–7407 CrossRef CAS PubMed.
- J. M. Reimer, M. N. Aloise, P. M. Harrison and T. M. Schmeing, Nature, 2016, 529, 239–242 CrossRef CAS PubMed.
- G. G. Gross, Eur. J. Biochem., 1972, 31, 585–592 CrossRef CAS PubMed.
- D. Gahloth, M. S. Dunstan, D. Quaglia, E. Klumbys, M. P. Lockhart-Cairns, A. M. Hill, S. R. Derrington, N. S. Scrutton, N. J. Turner and D. Leys, Nat. Chem. Biol., 2017, 13, 975–981 CrossRef CAS PubMed.
- R. R. Forseth, S. Amaike, D. Schwenk, K. J. Affeldt, D. Hoffmeister, F. C. Schroeder and N. P. Keller, Angew. Chem., Int. Ed. Engl., 2013, 52, 1590–1594 CrossRef CAS PubMed.
- M. Winkler, Curr. Opin. Chem. Biol., 2018, 43, 23–29 CrossRef CAS PubMed.
- Y. Hai, A. M. Huang and Y. Tang, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 10348–10353 CrossRef CAS PubMed.
- B. N. Lee, S. Kroken, D. Y. Chou, B. Robbertse, O. C. Yoder and B. G. Turgeon, Eukaryotic Cell, 2005, 4, 545–555 CrossRef CAS PubMed.
- D. Yan, Q. Chen, J. Gao, J. Bai, B. Liu, Y. Zhang, L. Zhang, C. Zhang, Y. Zou and Y. Hu, Org. Lett., 2019, 21, 1475–1479 CrossRef CAS PubMed.
- D. E. Ehmann, A. M. Gehring and C. T. Walsh, Biochemistry, 1999, 38, 6171–6177 CrossRef CAS PubMed.
- S. Guo and J. K. Bhattacharjee, Mol. Genet. Genomics, 2003, 269, 271–279 CrossRef CAS PubMed.
- S. Hartwig, C. Dovengerds, C. Herrmann and B. T. Hovemann, FEBS J., 2014, 281, 5147–5158 CrossRef CAS PubMed.
- A. Richardt, T. Kemme, S. Wagner, D. Schwarzer, M. A. Marahiel and B. T. Hovemann, J. Biol. Chem., 2003, 278, 41160–41166 CrossRef CAS PubMed.
- T. Izore, J. Tailhades, M. H. Hansen, J. A. Kaczmarski, C. J. Jackson and M. J. Cryle, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 2913–2918 CrossRef PubMed.
- L. Di Vincenzo, I. Grgurina and S. Pascarella, FEBS J., 2005, 272, 929–941 CrossRef CAS PubMed.
- K. Yamanaka, C. Maruyama, H. Takagi and Y. Hamano, Nat. Chem. Biol., 2008, 4, 766–772 CrossRef CAS PubMed.
- Y. Hamano, T. Arai, M. Ashiuchi and K. Kino, Nat. Prod. Rep., 2013, 30, 1087–1097 RSC.
- M. J. Jaremko, T. D. Davis, J. C. Corpuz and M. D. Burkart, Nat. Prod. Rep., 2020, 37, 355–379 RSC.
- E. Conti, T. Stachelhaus, M. A. Marahiel and P. Brick, EMBO J., 1997, 16, 4174–4183 CrossRef CAS PubMed.
- J. J. May, N. Kessler, M. A. Marahiel and M. T. Stubbs, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 12120–12125 CrossRef CAS PubMed.
- S. D. Bruner, T. Weber, R. M. Kohli, D. Schwarzer, M. A. Marahiel, C. T. Walsh and M. T. Stubbs, Struct., 2002, 10, 301–310 CrossRef CAS PubMed.
- A. M. Gulick and C. C. Aldrich, Nat. Prod. Rep., 2018, 35, 1156–1184 RSC.
- B. R. Branchini, M. H. Murtiashaw, R. A. Magyar and S. M. Anderson, Biochemistry, 2000, 39, 5433–5440 CrossRef CAS PubMed.
- A. R. Horswill and J. C. Escalante-Semerena, Biochemistry, 2002, 41, 2379–2387 CrossRef CAS PubMed.
- A. S. Reger, J. M. Carney and A. M. Gulick, Biochemistry, 2007, 46, 6536–6546 CrossRef CAS PubMed.
- R. Wu, J. Cao, X. Lu, A. S. Reger, A. M. Gulick and D. Dunaway-Mariano, Biochemistry, 2008, 47, 8026–8039 CrossRef CAS PubMed.
- A. Goyal, P. Verma, M. Anandhakrishnan, R. S. Gokhale and R. Sankaranarayanan, J. Mol. Biol., 2012, 416, 221–238 CrossRef CAS PubMed.
- C. A. Mitchell, C. Shi, C. C. Aldrich and A. M. Gulick, Biochemistry, 2012, 51, 3252–3263 CrossRef CAS PubMed.
- L. W. Hamoen, H. Eshuis, J. Jongbloed, G. Venema and D. van Sinderen, Mol. Microbiol., 1995, 15, 55–63 CrossRef CAS PubMed.
- A. M. Gulick, V. J. Starai, A. R. Horswill, K. M. Homick and J. C. Escalante-Semerena, Biochemistry, 2003, 42, 2866–2873 CrossRef CAS PubMed.
- A. S. Reger, R. Wu, D. Dunaway-Mariano and A. M. Gulick, Biochemistry, 2008, 47, 8016–8025 CrossRef CAS PubMed.
- G. L. Challis, J. Ravel and C. A. Townsend, Chem. Biol., 2000, 7, 211–224 CrossRef CAS PubMed.
- H. Kries, J. Pept. Sci., 2016, 22, 564–570 CrossRef CAS PubMed.
- J. A. Sundlov and A. M. Gulick, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2013, 69, 1482–1492 CrossRef CAS PubMed.
- J. A. Sundlov, C. Shi, D. J. Wilson, C. C. Aldrich and A. M. Gulick, Chem. Biol., 2012, 19, 188–198 CrossRef CAS PubMed.
- C. Qiao, D. J. Wilson, E. M. Bennett and C. C. Aldrich, J. Am. Chem. Soc., 2007, 129, 6350–6351 CrossRef CAS PubMed.
- E. M. Alexander, D. F. Kreitler, V. Guidolin, A. K. Hurben, E. Drake, P. W. Villalta, S. Balbo, A. M. Gulick and C. C. Aldrich, ACS Infect. Dis., 2020, 6, 1976–1997 CrossRef CAS PubMed.
- J. C. Corpuz, L. M. Podust, T. D. Davis, M. J. Jaremko and M. D. Burkart, RSC Chem. Biol., 2020, 1, 8–12 RSC.
- A. Miyanaga, S. Kurihara, T. Chisuga, F. Kudo and T. Eguchi, ACS Chem. Biol., 2020, 15, 1808–1812 CrossRef CAS PubMed.
- A. Miyanaga, F. Kudo and T. Eguchi, Curr. Opin. Chem. Biol., 2022, 71, 102212 CrossRef CAS PubMed.
- D. F. Kreitler, E. M. Gemmell, J. E. Schaffer, T. A. Wencewicz and A. M. Gulick, Nat. Commun., 2019, 10, 3432 CrossRef PubMed.
- D. J. Edwards and W. H. Gerwick, J. Am. Chem. Soc., 2004, 126, 11432–11433 CrossRef CAS PubMed.
- A. H. Soeriyadi, S. E. Ongley, J. C. Kehr, R. Pickford, E. Dittmann and B. A. Neilan, Chembiochem, 2022, 23, e202100574 CrossRef CAS PubMed.
- L. E. Quadri, J. Sello, T. A. Keating, P. H. Weinreb and C. T. Walsh, Chem. Biol., 1998, 5, 631–645 CrossRef CAS PubMed.
- A. S. Eustaquio, S. M. Li and L. Heide, Microbiol., 2005, 151, 1949–1961 CrossRef CAS PubMed.
- E. J. Drake, J. Cao, J. Qu, M. B. Shah, R. M. Straubinger and A. M. Gulick, J. Biol. Chem., 2007, 282, 20425–20434 CrossRef CAS PubMed.
- S. Lautru, D. Oves-Costales, J. L. Pernodet and G. L. Challis, Microbiology, 2007, 153, 1405–1412 CrossRef CAS PubMed.
- M. Wolpert, B. Gust, B. Kammerer and L. Heide, Microbiology, 2007, 153, 1413–1423 CrossRef CAS PubMed.
- G. W. Buchko, C. Y. Kim, T. C. Terwilliger and P. J. Myler, Tuberc., 2010, 90, 245–251 CrossRef CAS PubMed.
- E. A. Felnagle, J. J. Barkei, H. Park, A. M. Podevels, M. D. McMahon, D. W. Drott and M. G. Thomas, Biochemistry, 2010, 49, 8815–8817 CrossRef CAS PubMed.
- W. Zhang, J. R. Heemstra Jr, C. T. Walsh and H. J. Imker, Biochemistry, 2010, 49, 9946–9947 CrossRef CAS PubMed.
- D. A. Herbst, B. Boll, G. Zocher, T. Stehle and L. Heide, J. Biol. Chem., 2013, 288, 1991–2003 CrossRef CAS PubMed.
- D. A. Alonzo, C. Chiche-Lapierre, M. J. Tarry, J. Wang and T. M. Schmeing, Nat. Chem. Biol., 2020, 16, 493–496 CrossRef CAS PubMed.
- A. D. Gnann, K. Marincin, D. P. Frueh and D. P. Dowling, Curr. Opin. Chem. Biol., 2022, 72, 102228 CrossRef PubMed.
- R. Veevers and S. Hayward, Biophys. Physicobiol., 2019, 16, 328–336 CrossRef CAS PubMed.
- E. J. Drake, B. R. Miller, C. Shi, J. T. Tarrasch, J. A. Sundlov, C. L. Allen, G. Skiniotis, C. C. Aldrich and A. M. Gulick, Nature, 2016, 529, 235–238 CrossRef CAS PubMed.
- J. M. Reimer, M. Eivaskhani, I. Harb, A. Guarne, M. Weigt and T. M. Schmeing, Science, 2019, 366, eaaw4388 CrossRef CAS PubMed.
- K. Bloudoff, D. A. Alonzo and T. M. Schmeing, Cell Chem. Biol., 2016, 23, 331–339 CrossRef CAS PubMed.
- M. J. Chu Yuan Kee, S. R. Bharath, S. Wee, M. W. Bowler, J. Gunaratne, S. Pan, L. Zhang and H. Song, Sci. Rep., 2022, 12, 5353 CrossRef CAS PubMed.
- T. Izore, Y. T. Candace Ho, J. A. Kaczmarski, A. Gavriilidou, K. H. Chow, D. L. Steer, R. J. A. Goode, R. B. Schittenhelm, J. Tailhades, M. Tosin, G. L. Challis, E. H. Krenske, N. Ziemert, C. J. Jackson and M. J. Cryle, Nat. Commun., 2021, 12, 2511 CrossRef CAS PubMed.
- D. C. Cantu, Y. Chen and P. J. Reilly, Protein Sci., 2010, 19, 1281–1295 CrossRef CAS PubMed.
- A. Koglin, F. Lohr, F. Bernhard, V. V. Rogov, D. P. Frueh, E. R. Strieter, M. R. Mofid, P. Guntert, G. Wagner, C. T. Walsh, M. A. Marahiel and V. Dotsch, Nature, 2008, 454, 907–911 CrossRef CAS PubMed.
- S. A. Samel, B. Wagner, M. A. Marahiel and L. O. Essen, J. Mol. Biol., 2006, 359, 876–889 CrossRef CAS PubMed.
- N. Huguenin-Dezot, D. A. Alonzo, G. W. Heberlig, M. Mahesh, D. P. Nguyen, M. H. Dornan, C. N. Boddy, T. M. Schmeing and J. W. Chin, Nature, 2019, 565, 112–117 CrossRef CAS PubMed.
- K. D. Patel, F. B. d'Andrea, N. M. Gaudelli, A. R. Buller, C. A. Townsend and A. M. Gulick, Nat. Commun., 2019, 10, 3868 CrossRef PubMed.
- J. H. Yu, J. Song, C. B. Chi, T. Liu, T. T. Geng, Z. H. Cai, W. D. Dong, C. Shi, X. Y. Ma, Z. Y. Zhang, X. J. Ma, B. Y. Xing, H. W. Jin, L. R. Zhang, S. W. Dong, D. H. Yang and M. Ma, ACS Catal., 2021, 11, 11733–11741 CrossRef CAS.
- D. P. Frueh, H. Arthanari, A. Koglin, D. A. Vosburg, A. E. Bennett, C. T. Walsh and G. Wagner, Nature, 2008, 454, 903–906 CrossRef CAS PubMed.
- Y. Liu, T. Zheng and S. D. Bruner, Chem. Biol., 2011, 18, 1482–1488 CrossRef CAS PubMed.
- H. B. Claxton, D. L. Akey, M. K. Silver, S. J. Admiraal and J. L. Smith, J. Biol. Chem., 2009, 284, 5021–5029 CrossRef CAS PubMed.
- F. B. d’Andrea and C. A. Townsend, Cell Chem. Biol., 2019, 26, 878–884.e8 CrossRef PubMed.
- N. M. Gaudelli and C. A. Townsend, Nat. Chem. Biol., 2014, 10, 251–258 CrossRef CAS PubMed.
- K. M. Hoyer, C. Mahlert and M. A. Marahiel, Chem. Biol., 2007, 14, 13–22 CrossRef CAS PubMed.
- S. A. Samel, P. Czodrowski and L. O. Essen, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2014, 70, 1442–1452 CrossRef CAS PubMed.
- C. D. Fage, S. Kosol, M. Jenner, C. Öster, A. Gallo, M. Kaniusaite, R. Steinbach, M. Staniforth, V. G. Stavros, M. A. Marahiel, M. J. Cryle and J. R. Lewandowski, ACS Catal., 2021, 11, 10802–10813 CrossRef CAS.
- J. Wang, D. Li, L. Chen, W. Cao, L. Kong, W. Zhang, T. Croll, Z. Deng, J. Liang and Z. Wang, Nat. Commun., 2022, 13, 592 CrossRef CAS PubMed.
- W. H. Chen, K. Li, N. S. Guntaka and S. D. Bruner, ACS Chem. Biol., 2016, 11, 2293–2303 CrossRef CAS PubMed.
- K. Bloudoff, C. D. Fage, M. A. Marahiel and T. M. Schmeing, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 95–100 CrossRef CAS PubMed.
- A. D. Gnann, Y. Xia, J. Soule, C. Barthelemy, J. S. Mawani, S. N. Musoke, B. M. Castellano, E. J. Brignole, D. P. Frueh and D. P. Dowling, J. Biol. Chem., 2022, 102454, DOI:10.1016/j.jbc.2022.102454.
- S. H. Mishra, A. K. Kancherla, K. A. Marincin, G. Bouvignies, S. Nerli, N. Sgourakis, D. P. Dowling and D. P. Frueh, Sci. Adv., 2022, 8, eabn6549 CrossRef CAS PubMed.
- K. L. Kavanagh, H. Jornvall, B. Persson and U. Oppermann, Cell. Mol. Life Sci., 2008, 65, 3895–3906 CrossRef CAS PubMed.
- S. Deshpande, E. Altermann, V. Sarojini, J. S. Lott and T. V. Lee, J. Biol. Chem., 2021, 296, 100432 CrossRef CAS PubMed.
- M. A. Wyatt, M. C. Mok, M. Junop and N. A. Magarvey, Chembiochem, 2012, 13, 2408–2415 CrossRef CAS PubMed.
- J. F. Barajas, R. M. Phelan, A. J. Schaub, J. T. Kliewer, P. J. Kelly, D. R. Jackson, R. Luo, J. D. Keasling and S. C. Tsai, Chem. Biol., 2015, 22, 1018–1029 CrossRef CAS PubMed.
- A. S. Haque, K. D. Patel, M. V. Deshmukh, A. Chhabra, R. S. Gokhale and R. Sankaranarayanan, J. Struct. Biol., 2014, 187, 207–214 CrossRef CAS PubMed.
- P. Kinatukara, K. D. Patel, A. S. Haque, R. Singh, R. S. Gokhale and R. Sankaranarayananan, J. Struct. Biol., 2016, 194, 368–374 CrossRef CAS PubMed.
- F. Lombo, A. Velasco, A. Castro, F. de la Calle, A. F. Brana, J. M. Sanchez-Puelles, C. Mendez and J. A. Salas, Chembiochem, 2006, 7, 366–376 CrossRef CAS PubMed.
- S. Mori, A. H. Pang, T. A. Lundy, A. Garzan, O. V. Tsodikov and S. Garneau-Tsodikova, Nat. Chem. Biol., 2018, 14, 428–430 CrossRef CAS PubMed.
- A. Tanovic, S. A. Samel, L. O. Essen and M. A. Marahiel, Science, 2008, 321, 659–663 CrossRef CAS PubMed.
- B. R. Miller, E. J. Drake, C. Shi, C. C. Aldrich and A. M. Gulick, J. Biol. Chem., 2016, 291, 22559–22571 CrossRef CAS PubMed.
- K. Tan, M. Zhou, R. P. Jedrzejczak, R. Wu, R. A. Higuera, D. Borek, G. Babnigg and A. Joachimiak, Curr. Res. Struct. Biol., 2020, 2, 14–24 CrossRef PubMed.
- L. L. Ling, T. Schneider, A. J. Peoples, A. L. Spoering, I. Engels, B. P. Conlon, A. Mueller, T. F. Schaberle, D. E. Hughes, S. Epstein, M. Jones, L. Lazarides, V. A. Steadman, D. R. Cohen, C. R. Felix, K. A. Fetterman, W. P. Millett, A. G. Nitti, A. M. Zullo, C. Chen and K. Lewis, Nature, 2015, 517, 455–459 CrossRef CAS PubMed.
- M. J. Tarry, A. S. Haque, K. H. Bui and T. M. Schmeing, Structure, 2017, 25, 783–793 CrossRef CAS PubMed.
- Y. Katsuyama, K. Sone, A. Harada, S. Kawai, N. Urano, N. Adachi, T. Moriya, M. Kawasaki, K. Shin-Ya, T. Senda and Y. Ohnishi, Angew. Chem., Int. Ed. Engl., 2021, 60, 14554–14562 CrossRef CAS PubMed.
- C. M. Fortinez, K. Bloudoff, C. Harrigan, I. Sharon, M. Strauss and T. M. Schmeing, Nat. Commun., 2022, 13, 548 CrossRef CAS PubMed.
- J. Hu, W. Chuenchor and S. E. Rokita, J. Biol. Chem., 2015, 290, 590–600 CrossRef CAS PubMed.
- C. L. Shelton, K. M. Meneely, T. A. Ronnebaum, A. S. Chilton, A. P. Riley, T. E. Prisinzano and A. L. Lamb, J. Biol. Inorg. Chem., 2022, 27, 541–551 CrossRef CAS PubMed.
- H. M. Patel, J. Tao and C. T. Walsh, Biochemistry, 2003, 42, 10514–10527 CrossRef CAS PubMed.
- J. Alfermann, X. Sun, F. Mayerthaler, T. E. Morrell, E. Dehling, G. Volkmann, T. Komatsuzaki, H. Yang and H. D. Mootz, Nat. Chem. Biol., 2017, 13, 1009–1015 CrossRef CAS PubMed.
- J. Rüschenbaum, W. Steinchen, F. Mayerthaler, A. L. Feldberg and H. D. Mootz, Angew. Chem., Int. Ed. Engl., 2022, 61, e202212994 CrossRef PubMed.
- F. Mayerthaler, A. L. Feldberg, J. Alfermann, X. Sun, W. Steinchen, H. Yang and H. D. Mootz, RSC Chem. Biol., 2021, 2, 843–854 RSC.
- M. Strieker, A. Tanovic and M. A. Marahiel, Curr. Opin. Struct. Biol., 2010, 20, 234–240 CrossRef CAS PubMed.
- D. E. Cane, C. T. Walsh and C. Khosla, Science, 1998, 282, 63–68 CrossRef CAS PubMed.
- S. P. Muench, S. V. Antonyuk and S. S. Hasnain, IUCrJ, 2019, 6, 167–177 CrossRef CAS PubMed.
- Y. Cheng, Science, 2018, 361, 876–880 CrossRef CAS PubMed.
- C. M. Zimanyi, M. Kopylov, C. S. Potter, B. Carragher and E. T. Eng, Trends Biochem. Sci., 2022, 47, 106–116 CrossRef CAS PubMed.
- E. Y. D. Chua, J. H. Mendez, M. Rapp, S. L. Ilca, Y. Z. Tan, K. Maruthi, H. Kuang, C. M. Zimanyi, A. Cheng, E. T. Eng, A. J. Noble, C. S. Potter and B. Carragher, Annu. Rev. Biochem., 2022, 91, 1–32 CrossRef PubMed.
- U. Baxa, T. J. Edwards, M. Hutchison, A. D. Wier, J. Finney, H. Wang and S. Subramaniam, Microsc. Today, 2020, 28, 12–17 CrossRef PubMed.
- D. K. Clare, C. A. Siebert, C. Hecksel, C. Hagen, V. Mordhorst, M. Grange, A. W. Ashton, M. A. Walsh, K. Grunewald, H. R. Saibil, D. I. Stuart and P. Zhang, Acta Crystallogr., Sect. D: Struct. Biol., 2017, 73, 488–495 CrossRef CAS PubMed.
- E. D. Zhong, T. Bepler, B. Berger and J. H. Davis, Nat. Methods, 2021, 18, 176–185 CrossRef CAS PubMed.
- J. Pereira, A. J. Simpkin, M. D. Hartmann, D. J. Rigden, R. M. Keegan and A. N. Lupas, Proteins, 2021, 89, 1687–1699 CrossRef CAS PubMed.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Zidek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Nature, 2021, 596, 583–589 CrossRef CAS PubMed.
- M. Baek, F. DiMaio, I. Anishchenko, J. Dauparas, S. Ovchinnikov, G. R. Lee, J. Wang, Q. Cong, L. N. Kinch, R. D. Schaeffer, C. Millan, H. Park, C. Adams, C. R. Glassman, A. DeGiovanni, J. H. Pereira, A. V. Rodrigues, A. A. van Dijk, A. C. Ebrecht, D. J. Opperman, T. Sagmeister, C. Buhlheller, T. Pavkov-Keller, M. K. Rathinaswamy, U. Dalwadi, C. K. Yip, J. E. Burke, K. C. Garcia, N. V. Grishin, P. D. Adams, R. J. Read and D. Baker, Science, 2021, 373, 871–876 CrossRef CAS PubMed.
- M. H. Medema, mSystems, 2018, 3, e00182-17 CrossRef PubMed.
- M. Winn, J. K. Fyans, Y. Zhuo and J. Micklefield, Nat. Prod. Rep., 2016, 33, 317–347 RSC.
- A. S. Brown, M. J. Calcott, J. G. Owen and D. F. Ackerley, Nat. Prod. Rep., 2018, 35, 1210–1228 RSC.
- K. A. Bozhüyük, J. Micklefield and B. Wilkinson, Curr. Opin. Microbiol., 2019, 51, 88–96 CrossRef PubMed.
- K. Blin, M. H. Medema, R. Kottmann, S. Y. Lee and T. Weber, Nucleic Acids Res., 2017, 45, D555–D559 CrossRef CAS PubMed.
- J. Chu, B. Koirala, N. Forelli, X. Vila-Farres, M. A. Ternei, T. Ali, D. A. Colosimo and S. F. Brady, J. Am. Chem. Soc., 2020, 142, 14158–14168 CrossRef CAS PubMed.
- Z. Wang, N. Forelli, Y. Hernandez, M. Ternei and S. F. Brady, Nat. Commun., 2022, 13, 842 CrossRef CAS PubMed.
- K. A. J. Bozhüyük, F. Fleischhacker, A. Linck, F. Wesche, A. Tietze, C. P. Niesert and H. B. Bode, Nat. Chem., 2018, 10, 275–281 CrossRef PubMed.
- K. A. J. Bozhüyük, A. Linck, A. Tietze, J. Kranz, F. Wesche, S. Nowak, F. Fleischhacker, Y. N. Shi, P. Grun and H. B. Bode, Nat. Chem., 2019, 11, 653–661 CrossRef PubMed.
- M. J. Calcott, J. G. Owen and D. F. Ackerley, Nat. Commun., 2020, 11, 4554 CrossRef CAS PubMed.
- A. Stanisic, A. Husken, P. Stephan, D. L. Niquille, J. Reinstein and H. Kries, ACS Catal., 2021, 11, 8692–8700 CrossRef CAS.
- D. Mandalapu, X. Ji, J. Chen, C. Guo, W. Q. Liu, W. Ding, J. Zhou and Q. Zhang, J. Org. Chem., 2018, 83, 7271–7275 CrossRef CAS PubMed.
- J. Hou, L. Robbel and M. A. Marahiel, Chem. Biol., 2011, 18, 655–664 CrossRef CAS PubMed.
- T. Huang, L. Li, N. L. Brock, Z. Deng and S. Lin, Chembiochem, 2016, 17, 1421–1425 CrossRef CAS PubMed.
- D. Braga, D. Hoffmeister and M. Nett, Beilstein J. Org. Chem., 2016, 12, 2766–2770 CrossRef CAS PubMed.
- D. Yu, F. Xu, S. Zhang and J. Zhan, Nat. Commun., 2017, 8, 15349 CrossRef CAS PubMed.
- M. S. Kim, M. Bae, Y. E. Jung, J. M. Kim, S. Hwang, M. C. Song, Y. H. Ban, E. S. Bae, S. Hong, S. K. Lee, S. S. Cha, D. C. Oh and Y. J. Yoon, Angew. Chem., Int. Ed. Engl., 2021, 60, 19766–19773 CrossRef CAS PubMed.
- M. Bernhardt, S. Berman, D. Zechel and A. Bechthold, Chembiochem, 2020, 21, 2659–2666 CrossRef CAS PubMed.
- S. Planckaert, B. Deflandre, A. M. de Vries, M. Ameye, J. C. Martins, K. Audenaert, S. Rigali and B. Devreese, Microbiol. Spectrum, 2021, 9, e0057121 CrossRef PubMed.
- S. Wang, W. D. G. Brittain, Q. Zhang, Z. Lu, M. H. Tong, K. Wu, K. Kyeremeh, M. Jenner, Y. Yu, S. L. Cobb and H. Deng, Nat. Commun., 2022, 13, 62 CrossRef CAS PubMed.
- J. J. Zhang, X. Tang, T. Huan, A. C. Ross and B. S. Moore, Nat. Chem. Biol., 2020, 16, 42–49 CrossRef CAS PubMed.
- J. M. Davidsen and C. A. Townsend, Chem. Biol., 2012, 19, 297–306 CrossRef CAS PubMed.
- N. M. Gaudelli, D. H. Long and C. A. Townsend, Nature, 2015, 520, 383–387 CrossRef CAS PubMed.
- D. H. Long and C. A. Townsend, Biochemistry, 2018, 57, 3353–3358 CrossRef CAS PubMed.
- S. Wang, Q. Fang, Z. Lu, Y. Gao, L. Trembleau, R. Ebel, J. H. Andersen, C. Philips, S. Law and H. Deng, Angew. Chem., Int. Ed. Engl., 2021, 60, 3229–3237 CrossRef CAS PubMed.
- R. A. Oliver, R. Li and C. A. Townsend, Nat. Chem. Biol., 2018, 14, 5–7 CrossRef CAS PubMed.
- C. A. Townsend, Curr. Opin. Chem. Biol., 2016, 35, 97–108 CrossRef CAS PubMed.
- J. B. Patteson, C. M. Fortinez, A. T. Putz, J. Rodriguez-Rivas, L. H. Bryant 3rd, K. Adhikari, M. Weigt, T. M. Schmeing and B. Li, J. Am. Chem. Soc., 2022, 144, 14057–14070 CrossRef CAS PubMed.
- G. Shi, N. Shi, Y. Li, W. Chen, J. Deng, C. Liu, J. Zhu, H. Wang and Y. Shen, ACS Chem. Biol., 2016, 11, 876–881 CrossRef CAS PubMed.
- B. Dose, S. P. Niehs, K. Scherlach, L. V. Florez, M. Kaltenpoth and C. Hertweck, ACS Chem. Biol., 2018, 13, 2414–2420 CrossRef CAS PubMed.
- E. A. Felnagle, A. M. Podevels, J. J. Barkei and M. G. Thomas, Chembiochem, 2011, 12, 1859–1867 CrossRef CAS PubMed.
- L. Du, Y. He and Y. Luo, Biochemistry, 2008, 47, 11473–11480 CrossRef CAS PubMed.
- L. Du and Y. Luo, F1000Research, 2014, 3, 106 Search PubMed.
- H. Yonus, P. Neumann, S. Zimmermann, J. J. May, M. A. Marahiel and M. T. Stubbs, J. Biol. Chem., 2008, 283, 32484–32491 CrossRef CAS PubMed.
- E. J. Drake, B. P. Duckworth, J. Neres, C. C. Aldrich and A. M. Gulick, Biochemistry, 2010, 49, 9292–9305 CrossRef CAS PubMed.
- J. Neres, C. A. Engelhart, E. J. Drake, D. J. Wilson, P. Fu, H. I. Boshoff, C. E. Barry 3rd, A. M. Gulick and C. C. Aldrich, J. Med. Chem., 2013, 56, 2385–2405 CrossRef CAS PubMed.
- T. V. Lee, L. J. Johnson, R. D. Johnson, A. Koulman, G. A. Lane, J. S. Lott and V. L. Arcus, J. Biol. Chem., 2010, 285, 2415–2427 CrossRef CAS PubMed.
- A. Miyanaga, J. Cieslak, Y. Shinohara, F. Kudo and T. Eguchi, J. Biol. Chem., 2014, 289, 31448–31457 CrossRef CAS PubMed.
- H. Kaljunen, S. H. Schiefelbein, D. Stummer, S. Kozak, R. Meijers, G. Christiansen and A. Rentmeister, Angew. Chem., Int. Ed. Engl., 2015, 54, 8833–8836 CrossRef CAS PubMed.
- J. C. Henderson, C. D. Fage, J. R. Cannon, J. S. Brodbelt, A. T. Keatinge-Clay and M. S. Trent, ACS Chem. Biol., 2014, 9, 2382–2392 CrossRef CAS PubMed.
- A. Scaglione, M. R. Fullone, L. C. Montemiglio, G. Parisi, C. Zamparelli, B. Vallone, C. Savino and I. Grgurina, FEBS J., 2017, 284, 2981–2999 CrossRef CAS PubMed.
- O. Vergnolle, H. Xu, J. M. Tufariello, L. Favrot, A. A. Malek, W. R. Jacobs, Jr. and J. S. Blanchard, J. Biol. Chem., 2016, 291, 22315–22326 CrossRef CAS PubMed.
- A. Tripathi, S. R. Park, A. P. Sikkema, H. J. Cho, J. Wu, B. Lee, C. Xi, J. L. Smith and D. H. Sherman, Chembiochem, 2018, 19, 1595–1600 CrossRef CAS PubMed.
- F. Ishikawa, A. Miyanaga, H. Kitayama, S. Nakamura, I. Nakanishi, F. Kudo, T. Eguchi and G. Tanabe, Angew. Chem., Int. Ed. Engl., 2019, 58, 6906–6910 CrossRef CAS PubMed.
- T. P. Fedorchuk, A. N. Khusnutdinova, E. Evdokimova, R. Flick, R. Di Leo, P. Stogios, A. Savchenko and A. F. Yakunin, J. Am. Chem. Soc., 2020, 142, 1038–1048 CrossRef CAS PubMed.
- I. G. Lee, C. Song, S. Yang, H. Jeon, J. Park, H. J. Yoon, H. Im, S. M. Kang, H. J. Eun and B. J. Lee, Acta Crystallogr., Sect. D: Struct. Biol., 2022, 78, 424–434 CrossRef CAS PubMed.
- I. H. Chen, T. Cheng, Y. L. Wang, S. J. Huang, Y. H. Hsiao, Y. T. Lai, S. I. Toh, J. Chu, J. D. Rudolf and C. Y. Chang, Chembiochem, 2022, e202200563, DOI:10.1002/cbic.202200563.
- E. J. Drake, D. A. Nicolai and A. M. Gulick, Chem. Biol., 2006, 13, 409–419 CrossRef CAS PubMed.
- X. F. Tan, Y. N. Dai, K. Zhou, Y. L. Jiang, Y. M. Ren, Y. Chen and C. Z. Zhou, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2015, 71, 873–881 CrossRef CAS PubMed.
- J. M. Reimer, M. N. Aloise, H. R. Powell and T. M. Schmeing, Acta Crystallogr., Sect. D: Struct. Biol., 2016, 72, 1130–1136 CrossRef CAS PubMed.
- A. Greule, T. Izore, D. Iftime, J. Tailhades, M. Schoppet, Y. Zhao, M. Peschke, I. Ahmed, A. Kulik, M. Adamek, R. J. A. Goode, R. B. Schittenhelm, J. A. Kaczmarski, C. J. Jackson, N. Ziemert, E. H. Krenske, J. J. De Voss, E. Stegmann and M. J. Cryle, Nat. Commun., 2019, 10, 2613 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2023 |