
Biosynthesis of indole diterpenes: a reconstitution approach in a heterologous host
Taro
Ozaki†
 a,
Atsushi
Minami
*a and
Hideaki
Oikawa
*ab
a,
Atsushi
Minami
*a and
Hideaki
Oikawa
*ab
aDepartment of Chemistry, Faculty of Science, Hokkaido University, Sapporo 060-0810, Japan. E-mail: aminami@sci.hokudai.ac.jp
bInnovation Center of Marine Biotechnology and Pharmaceuticals, School of Biotechnology and Health Sciences, Wuyi University, Jiangmen, 529020, Guangdong, China. E-mail: hoikmm1@icloud.com
First published on 2nd November 2022
Abstract
Covering: 2013 to 2022
In this review, we provide an overview elucidating the biosynthetic pathway and heterologous production of fungal indole diterpenes (IDTs). Based on the studies of six IDT biosynthesis, we extracted nature's strategy: (1) two-stage synthesis for the core scaffold and platform intermediates, and (2) late-stage modifications for installing an additional cyclic system on the indole ring. Herein, we describe reconstitution studies applying this strategy to the synthesis of highly elaborated IDTs. We also discuss its potential for future biosynthetic engineering.
 Taro Ozaki | Taro Ozaki received his PhD in 2013 from the University of Tokyo under the direction of Prof. M. Nishiyama and Prof. T. Kuzuyama. After obtaining his PhD, he worked with Prof. H. Onaka as a project assistant professor at the University of Tokyo (2013–2016) and then joined Prof. H. Oikawa's lab as an assistant professor at Hokkaido University (2016–2021). In 2021, he was appointed as an associate professor at Tohoku University. His current research focuses on the genome mining of novel natural products and identification of unique enzymes involved in their biosynthesis. |
 Atsushi Minami | Atsushi Minami received his PhD in 2007 from Tokyo Institute of Technology under the direction of Professor Tadashi Eguchi. He worked as a postdoctoral fellow with Prof. Tohru Dairi at Toyama Prefectural University in 2007. He was appointed Assistant Professor at Hokkaido University and then Associate Professor in 2015. He is a recipient of the Chemical Society of Japan Award for Young Chemists (2016). His present research focused on the biosynthesis of microbial natural products and biochemical analysis of biosynthetic enzymes. |
 Hideaki Oikawa | He received his PhD (1984) from Hokkaido University. He worked as a postdoctoral fellow with Prof. D. E. Cane at Brown University from 1984 and with Dr late K. Isono at RIKEN from 1985. He joined the Department of Agricultural Chemistry, Hokkaido University, as an Assistant Professor in 1986 and was promoted to associate professor in 1999. He moved to the Department of Chemistry, the Faculty of Science, Hokkaido University in 2003 as a professor. In 2022, he was appointed as a professor at Wuyi University, China after his retirement. His present research focuses on the genome mining of novel fungal natural products, and reaction mechanism on C–C bond forming enzymes in the natural product biosynthesis. |
1 Introduction
Fungi are prolific producers of a number of alkaloids, such as ascomycota indole alkaloids (paxilline and communesin) and basidiomycota toxins (psilocybin and amanitin) as well as ergot alkaloids (lysergic acid). Among the alkaloids, we focus on a representative fungal metabolite, indole diterpenes (IDTs, Fig. 1 and 2).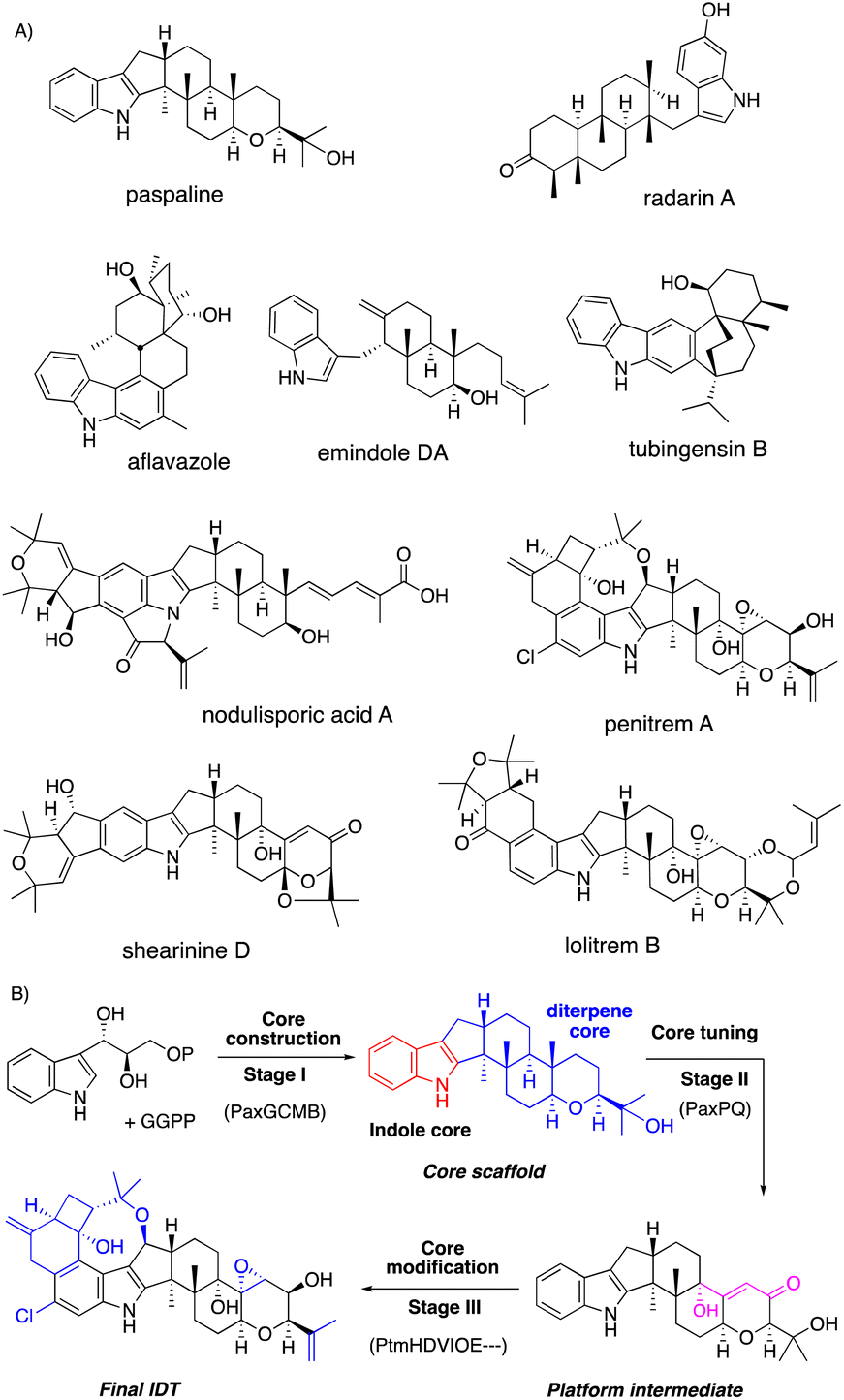 | ||
| Fig. 1 (A) Chemical structures of representative IDTs. (B) Representative three stage biosynthesis of IDTs. | ||
IDTs are well-known tremorgenic mycotoxins consisting of more than 100 family members, which are produced by various ascomycota fungi.1–5 Unique chemical structures and attractive biological activities have prompted a number of synthetic and biosynthetic studies.6 Recently, their biosynthetic gene clusters (BGCs) were identified, and most biosynthesis on highly elaborate IDTs was elucidated mainly by gene knockout and heterologous expression in versatile hosts.3–6
Based on the knowledge from biosynthetic studies of representative IDTs,3 this review systematically introduces a biosynthetic strategy. IDT biosynthesis is divided into three construction stages: (1) core construction with four minimal enzymes; (2) core tuning by the conversion to platform intermediates such as paxilline; (3) core modification generating diverse IDT end products (Fig. 1B). As described in Fig. 1B, the stepwise introduction of biosynthetic genes in parenthesis to the expression host and structure determination of intermediates from the resultant transformants enabled the characterization of the function of the introduced gene. In this way, all biosynthetic steps in stages I–III leading to the final IDT via the core scaffold and platform intermediate are elucidated.
In addition, we also introduce the reaction mechanism of the key modification enzymes involved in the late-stage biosynthesis of complex IDTs. Attractive features of the modified enzymes include promiscuous substrate specificity, which diversifies the family members. At the end of this review, we discuss its potential for future biosynthetic engineering.
2 Reconstitution of IDT BGCs
2.1 Heterologous expression systems in fungi
Early studies using heterologous expression systems in fungal hosts were mainly employed in the functional analysis of individual biosynthetic genes.7 Before 2010, successful examples of fungal expression were found in the analysis of the multifunctional enzyme genes such as iterative polyketide synthases (iPKSs), which are frequently involved in the production of fungal secondary metabolite.8,9 Although fungal expressions are usually used only in vivo analysis, yeast expression systems can be applied both in vivo and in vitro analysis.10 A representative example of yeast expression is a detailed analysis of iPKSs LovB and LovC in the lovastatin biosynthesis.9,10b More recent expression systems include powerful promoters such as amyB or alcA gene in strains of Aspergillus nidulans, and A. oryzae. However, the total biosynthesis of fungal NPs requires the introduction of multiple genes.After several natural products, such as meroterpenoid pyripyropene,11 PK-NRP related metabolite/polyketide tenellin,12 and diterpenoid aphidicolin13 are produced by the reconstitution of biosynthetic gene clusters using fungal expression host, total biosynthesis of fungal metabolites via heterologous BGC expression increases rapidly14–17 fueled by the vast amount of BGC data from various low-cost genomic data mining. While yeast expression requires the preparation of cDNA sequence for secure expression, fungal hosts splice introns of genomic DNA from foreign fungi correctly and thus cDNA sequencing is not essential. Without the optimization of the expression conditions, transformants harboring target genes produce a sufficient amount of intermediate for NMR measurements.5
As A. oryzae (AO) is naturally tolerant to various antibiotics, instead of antibiotic markers, several auxotrophic mutants of A. oryzae are introduced as hosts together with vectors having selectable markers complemented by argB (arginine), pyrG gene (uridine), sC gene (methionine), and adeA gene (adenine).5 Use of multiple vectors enabled the introduction of multiple genes, enhancing the utility of AO-system for heterologous production of NPs.15 To characterize multiple genes and large megasynthase genes, a rapid assembly method for the construction of multi-gene plasmid was also developed by homologous recombination in yeast and in vitro Gateway-recombination.18
In conventional transformation, transformed genes are randomly integrated into the AO chromosome, thus requiring a tedious screening for highly yielding transformants. To overcome this problem, we sequenced genomic DNAs from highly yielding TFs obtained in our studies and found several hot spots (HSs) where the target genes were integrated. Using a clustered, regularly interspaced, short palindromic repeat (CRISPR)-Cas9 system specifically optimized for AO,19 target genes are introduced by the knock-in method. More than 90% of the resultant TFs produced ca. 30–100 mg L−1 of intermediates.20 Marker recycling21 (removing unnecessary Cas9 plasmid) helped to suppress the off-target effect of TFs and to allow the unlimited introduction of biosynthetic genes. These HS-knock-in methods were successfully applied to the heterologous expression of biosynthetic genes for both Ascomycota and Basidiomycota fungi.22–24
2.2 Core construction: formation of a core skeleton with a minimal set of enzymes
IDTs have various types of core structures, and further modifications, such as prenylation and oxygenation (hydroxylation, epoxidation, and oxidative cyclization), yield a number of family members. Representative IDTs include the simple emindole and paspaline, as well as the more elaborated members tubingensin, shearinine, lolitrems, nodulisporic acids (NAs), and penitrems (Fig. 1).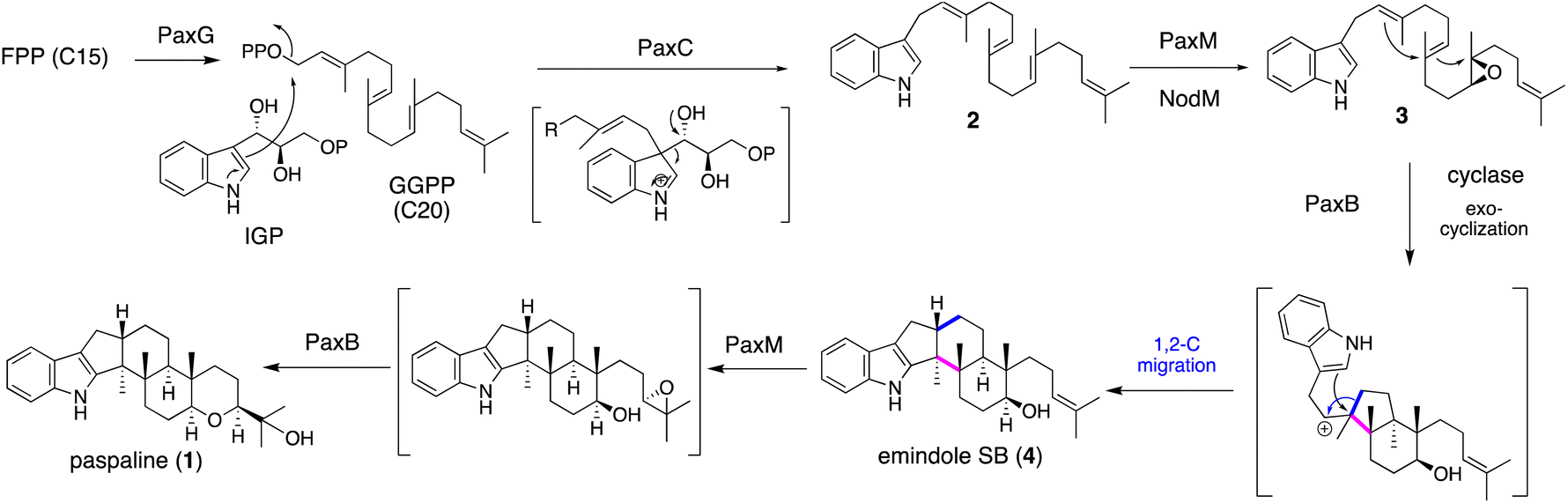 | ||
| Scheme 1 Proposed biosynthetic pathway of emindole SB (4) and paspaline (1). | ||
 | ||
| Fig. 2 Biosynthetic gene cluster of paspaline (1) and paxilline (17). | ||
To elucidate its detailed construction mechanism and supply a sufficient amount of 1, the reconstitution of paspaline gene cluster was employed in Aspergillus oryzae (AO) NSAR1. Using four plasmids with different markers, four genes paxG, paxC, paxM and paxB were introduced.29 Initially two genes paxGC were introduced and the resultant transformant AO-paxGC yielded 3-geranylgeranylindole (GGI, 2). Transformants AO-paxGCM and AO-paxGCMB introducing one or two additional genes—paxM and paxB—yielded 3 and 1, respectively, suggesting that PaxM initially oxidized the geranygeranyl side chain into bisepoxide and PaxB catalyzed epoxide opening affording 1. However, AO-paxGCM and AO-paxM with 2 generated 3, and AO-paxB converted 3 into emindole SB (4), strongly suggesting that the two rounds of cyclization proceeded in a stepwise manner (Scheme 1)29 Later, in the biosynthesis of nodulisporic acid (NA), PaxM ortholog NodM simply catalyzed monoepoxidation, leading to 4 (Section 2.4.1).30
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 for AtS2B).31 These have different molecular skeletons from those of 1. After epoxide opening in the common epoxide intermediate 3, AfB catalyzes sequential 1,2-H- and 1,2-C-migration to form 5 mainly via bicyclic carbocation 8+, while AtS2B catalyzes a 1,3-H shift of 8+ and forms tricyclic products 6 and 6a (Scheme 2).
:1 for AtS2B).31 These have different molecular skeletons from those of 1. After epoxide opening in the common epoxide intermediate 3, AfB catalyzes sequential 1,2-H- and 1,2-C-migration to form 5 mainly via bicyclic carbocation 8+, while AtS2B catalyzes a 1,3-H shift of 8+ and forms tricyclic products 6 and 6a (Scheme 2).
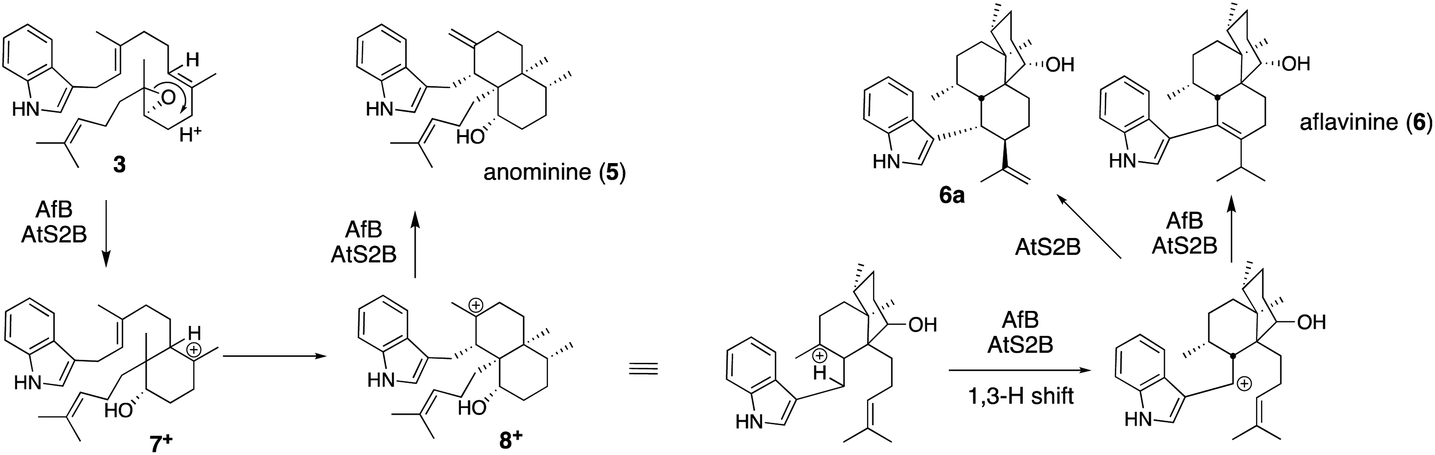 | ||
| Scheme 2 Proposed biosynthetic pathway of anominine (5) and aflavinine (6). | ||
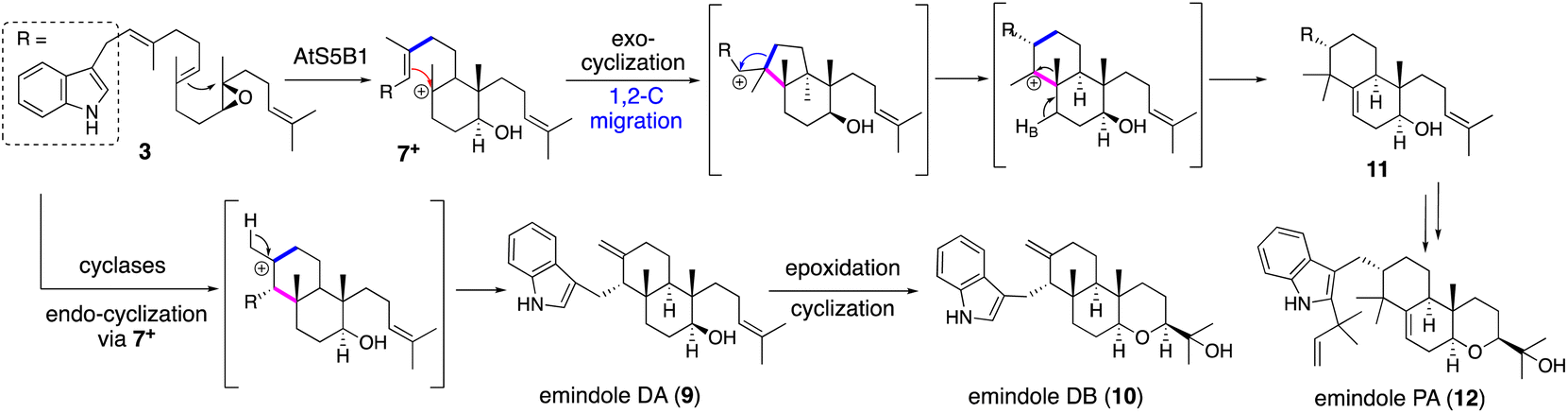 | ||
| Scheme 3 Proposed biosynthetic pathway of emindoles via two alternative cyclizations affording 9/10 and 12. | ||
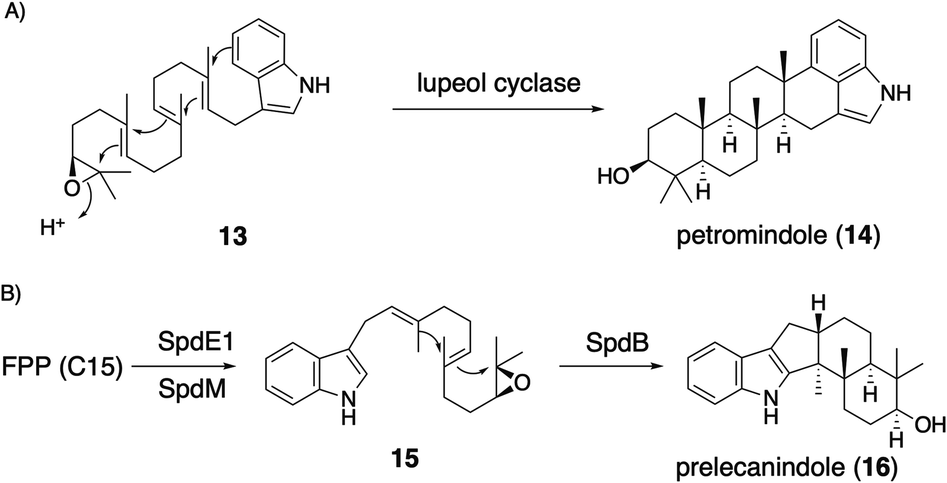 | ||
| Scheme 4 (A) Triterpene synthase-catalyzed formation of IDT core petromindole (14). (B) Proposed biosynthetic pathway of IST core 16. | ||
Applying a reconstitution strategy using a robust A. oryzae expression system to examine the early biosynthetic pathway of indole sesquiterpene (IST) sespendole36 (spd gene cluster, seven genes), two transformants, AO-spdE1 and AO-spdE1MB, were prepared. AO-spdE1 and AO-spdE1MB produced 3-farnesyl indole and prelecanindole (16) whose structure resembles 1 and thus 14 belongs to paspaline-type IDTs (Scheme 4),37 confirming that, as in the case of IDTs, a set of three genes is responsible for the core construction of IST via epoxide (15). This suggested that essentially the same set of genes as that of 1 is responsible for the IST core structure. These are examples of the diversification of indole terpenes in terms of changing the epoxide position and chain length.
2.3 Core tuning: formation of platform intermediates IDTs
The platform intermediate itself is isolated as a natural product from different sources and this intermediate is frequently used in the biosynthesis of other complex IDTs. Among the six common genes for IDT core structures (Fig. 2), four are required for the core scaffold construction. The remaining two, P450 monooxygenases, catalyze multiple oxidations, such as the oxidative elimination of the C30-angular methyl group and the introduction of the C13-tertiary hydroxy group as well as other oxidative modifications. These transformations are likely to be important for the generation of bioactive IDTs, such as shearinine/janthitrem, penitrem, and lolitrem, because the IDT core scaffold itself usually reveals poor biological activity.3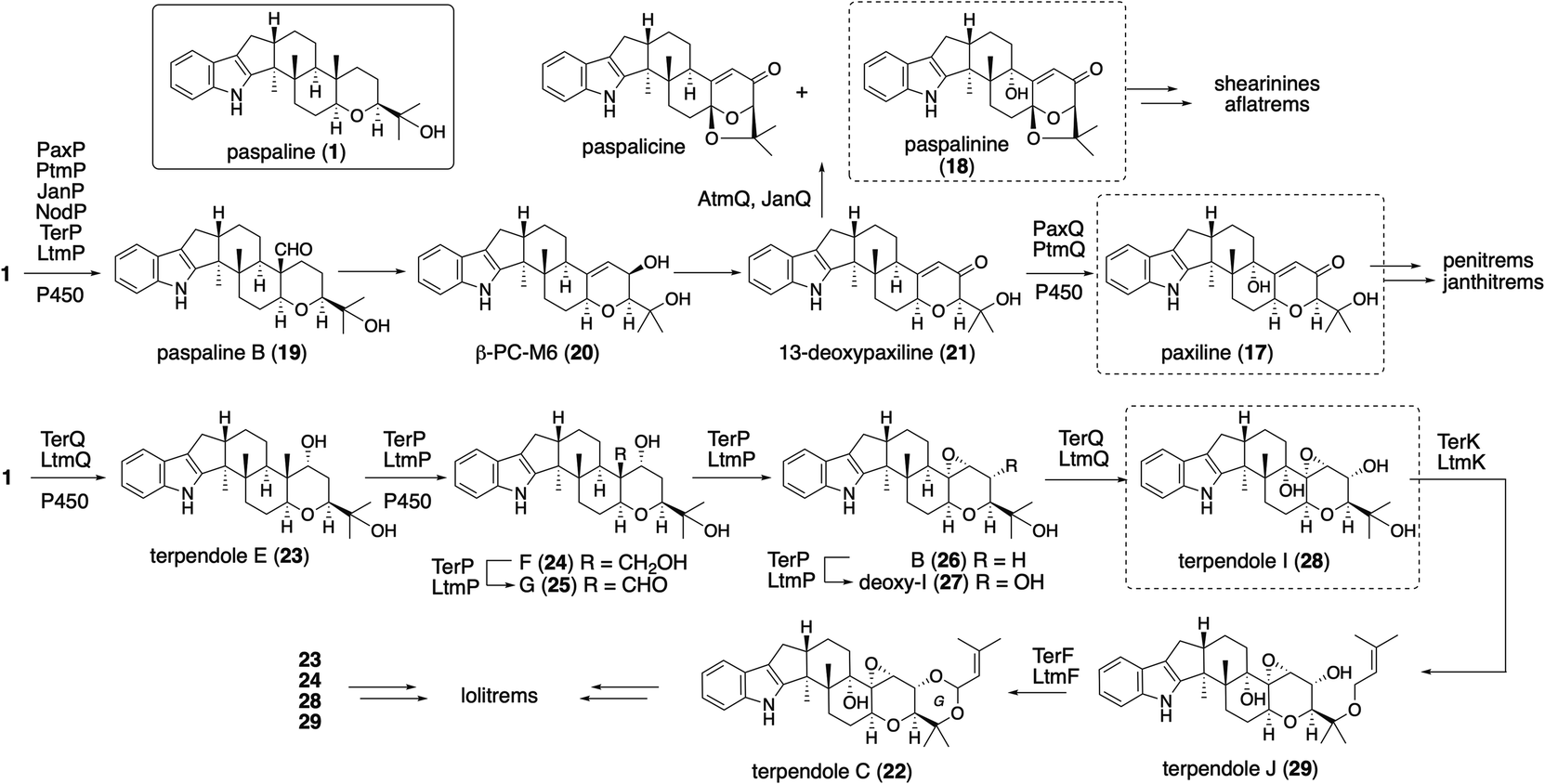 | ||
| Scheme 5 Proposed biosynthetic pathway of platform intermediates. Compounds in hashed box are platform intermediates. | ||
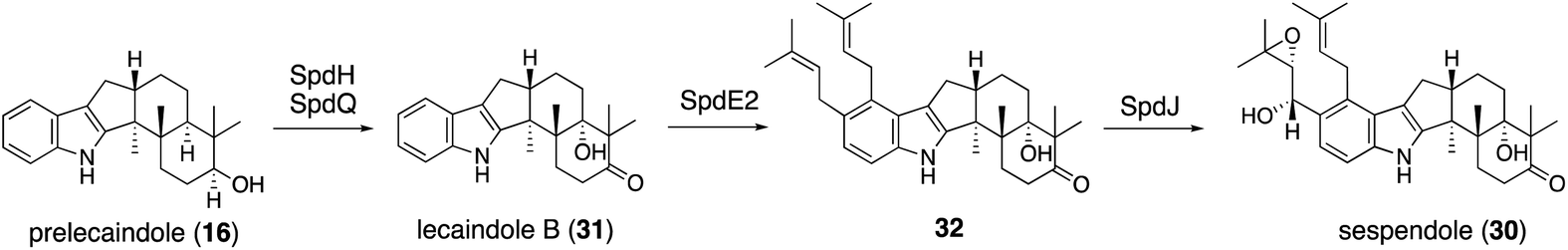 | ||
| Scheme 6 Proposed biosynthetic pathway of lecanindole B (31) and sespendole (30). | ||
Osada and co-workers found terpenedole E (23) in the extract of Chaunopycnis alba P-27 to be an inhibitor of kinesin Eg5, which is expected to be a potent anticancer drug.45 Because of the unstable production of 23, they started to study its biosynthesis. In the screening of genomic libraries from C. alba, they identified a gene cluster (ter) responsible for terpendole biosynthesis.46 The cluster consisted of an oxidative modification enzyme for paspaline core (cytochrome P450 monooxygenase, terPQ) and terpendole-specific modification enzymes (prenyltransferase and cytochrome P450, terFK).
All these genes showed significant homology to those of corresponding lolitrem biosynthetic genes (identities, 49–70%).47 Based on a series of knockout and bioconversion experiments, these results, in the case of TerP/Q, TerQ hydroxylates at C11 gave the first intermediate 23; then TerP oxidized an angular methyl to aldehyde in terpendole G (25).44 The subsequent oxidative elimination may have directly afforded the corresponding epoxide, deoxyterpendole I (27), which is further oxidized to give terpendole I (28) by TerQ (Scheme 5). Intriguingly, TerP still has the ability to oxidize 1 to 21 in the absence of TerQ. Thus, 21 isolated from C. alba is most likely a shunt metabolite. This suggests the initial C11-hydroxylation with TerQ as a characteristic point in terpendole biosynthesis.46 This unusual decarboxylative epoxide formation by TerQ is similar to P450-catalyzed decarboxylative lactone formation, which is found in fungal and bacterial gibberellin biosynthesis.48
To examine the proposed functions of TerPQ, into A. oryzae NSPlD1 CRISPR/Cas9-based genome editing (Section 2.1) was employed.20 This screening-free method readily enabled us to obtain the AO-pas, which produced 1 (>100 mg L−1 equivalent), as we reported previously.22,23 To the transformant AO-pas, terP and/or terQ were introduced. The obtained transformants, AO-pas/terQ and AO-pas/terPQ, produced 23 and 28, respectively.22 Two additional genes, prenyltransferase terF and cytochrome P450 terK, were also introduced. The obtained transformant, AO-pas/terPQFK, which produced 22, confirmed that TerF and TerK exerted the expected activities: O-prenylation and subsequent oxidative cyclic acetal formation, respectively (Scheme 5).22
2.4 Core modification: conversion to representative IDT alkaloids
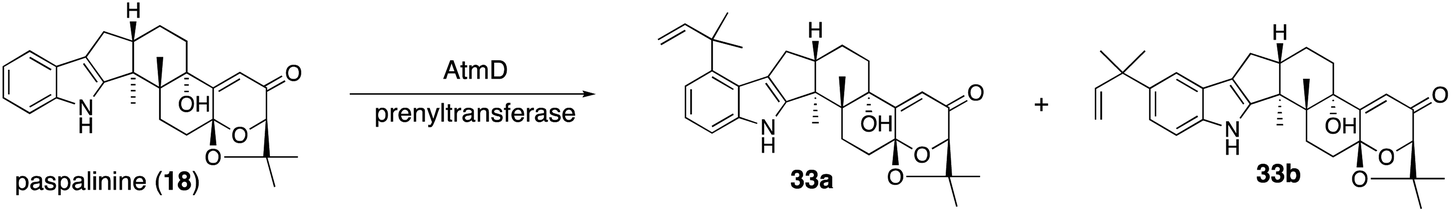 | ||
| Scheme 7 Proposed biosynthetic pathway of aflatrems (33a) and sespendole (33b). | ||
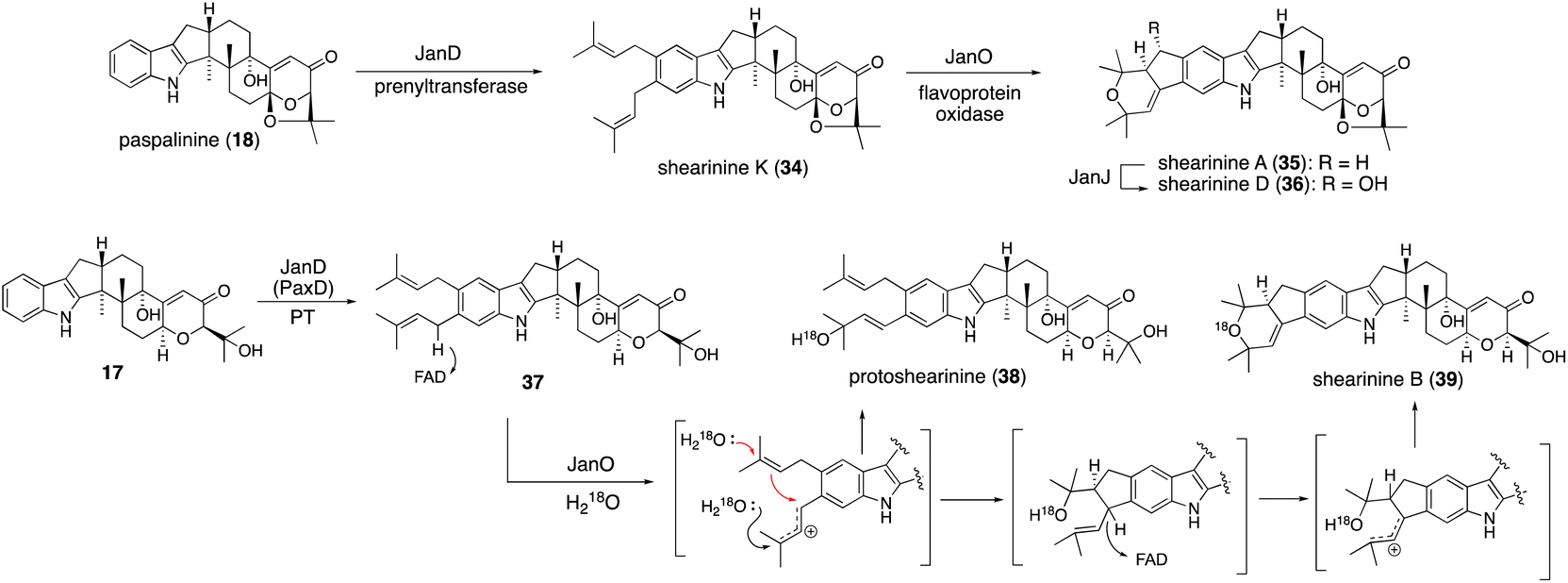
A different approach was chosen for the formal reconstitution of shearinine BGC (nine jan genes).49 In this case, genomic DNA from P. janthinellum was supplied by the collaborators and thus the producer strain was not used. Similar to the aflatrem study, three transformants AO-janQ, AO-janQDO, and AO-janQDOJ were prepared by changing a combination of the gene set. Functional analysis of these strains was conducted by the bioconversion of 13-deoxypaxilline (21), which was supplied by paxilline producer P. paxilli. The resultant transformants generated 18 (C13-hydroxylation), shearinine K (34, diprenylation)/shearinine A50,51 (35, oxidative cyclization), and shearinine D (36, hydroxylation), respectively (Scheme 8).52In vivo transformation of 21 with three transformants enabled a rapid elucidation of the late-stage biosynthetic pathway of shearinines. These data suggested that the intriguing A/B-bicyclic system was installed solely by JanO-catalyzed sequential oxidative cyclization of diprenylation product 34.52
 | ||
| Scheme 8 Proposed biosynthetic pathway of shearinines. Cyclization mechanism of JanO-catalyzed reaction was supported by several experimental evidences. | ||
To examine the intriguing bicyclic system formation, catalyzed by JanO, in vitro analysis with a recombinant JanO was employed. The incubation of 34 with JanO afforded a single product 35 (ref. 52) (Scheme 8), suggesting that the second step of this cyclization proceeded rapidly and no intermediate was accumulated. On the other hand, a JanD–JanO coupled reaction with a substrate analog 17 afforded two new products—38 and 39—with moderate yields. The structure of 39 is a sequential cyclization product corresponding to 35, while that of 38 is an allylic alcohol, which is likely a byproduct of the first step of oxidation. The incubation of 38 with JanO did not convert it into 39, confirming that 38 is a shunt product.52
When the JanO reaction with 17 was performed in a buffer containing H218O, retention of 18O in the oxidation products, 38 and 39, was observed, clearly indicating that the oxygen atoms of allylic alcohol and a cyclic ether ring originated from H218O and that a carbocation intermediate is involved in the hydride transfer of the benzylic positions. This finding is consistent with the mechanism of other vanillyl alcohol oxidase family members.53 Based on these data, an oxidative cyclization mechanism was proposed, as shown in Scheme 8. This pathway involves JanO-catalyzed two-step oxidation for the formation of the A/B rings of shearinines via a putative intermediate.52
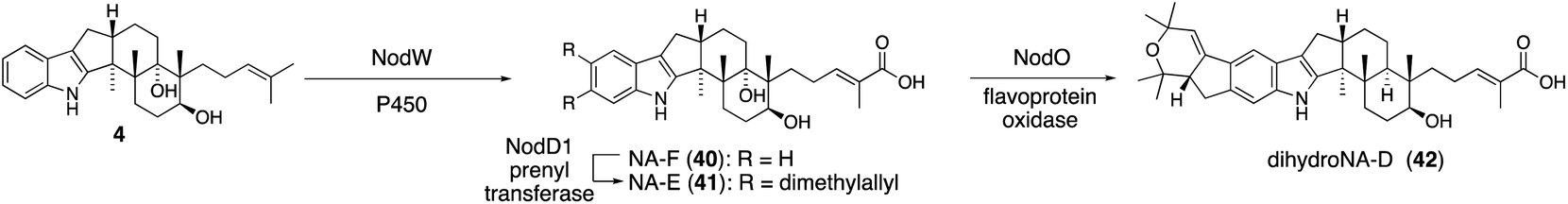
For structurally related NA54,55 biosynthesis, the corresponding NA BGC (13 nod genes) was identified, and it was found that the P. paxilli strain replacing paxCMB with nodCMB affords emindole SB (4).30 Further introduction of the P450 nodW gene gave transformant producing NA-F (40) (Scheme 9).30 In addition, functional JanD/JanO orthologs NodD1/NodO were found, strongly suggesting that the characteristic bicyclic system of dihydroNA-D (42) via41 is likely synthesized in a similar transformation.52,56
 | ||
| Scheme 9 Proposed biosynthetic pathway of NAs. NodO-catalyzed cyclization is not confirmed experimentally. | ||
 | ||
| Scheme 10 Proposed biosynthetic pathway of penitrems and PtmE-catalyzed cyclization mechanism from 46 to 47. | ||
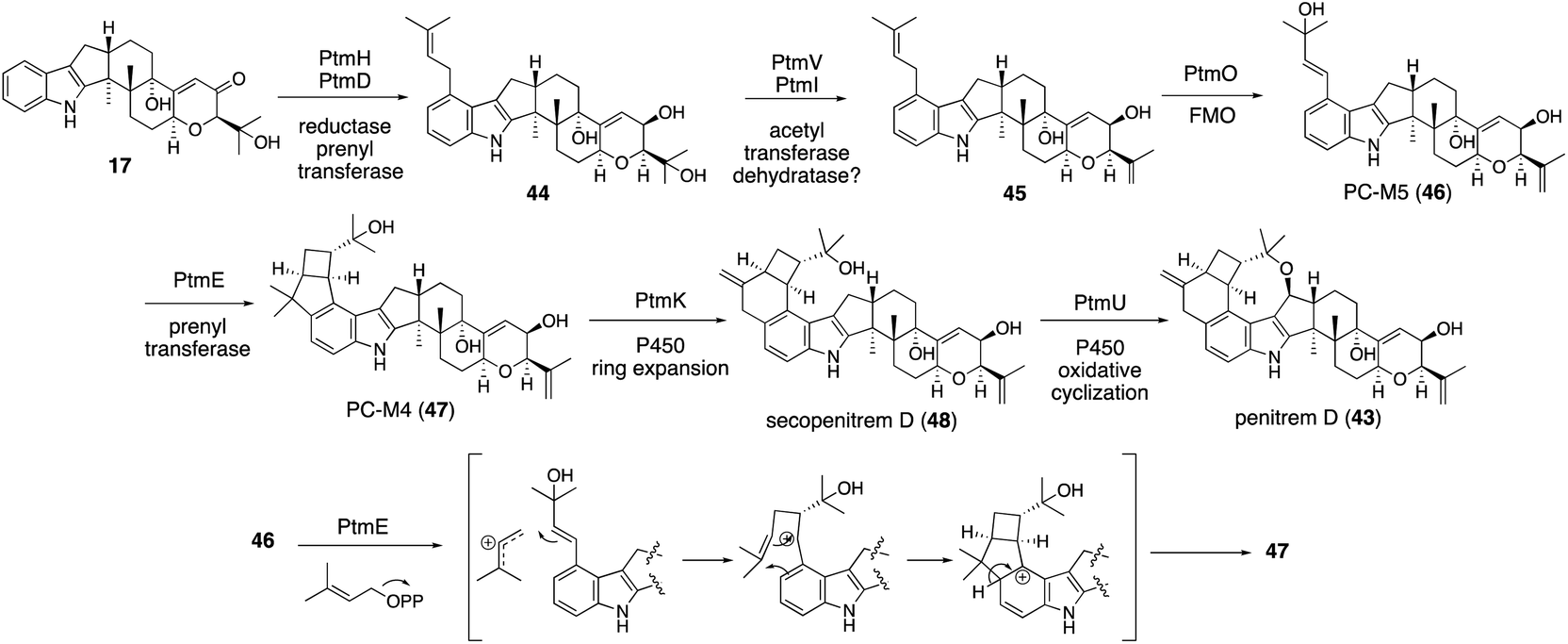
In the microbial conversion of 17 with AO-ptmHDVIE, no product other than 45 was obtained, indicating that, prior to the second prenylation with PtmE, oxidation is involved. Therefore, the biosynthetic study was then focused on the functional analysis of the oxidation enzymes to reduce the number of possible reaction pathways (Scheme 10). After various transformants were prepared using five oxidation enzyme genes in different combinations, microbial conversion of these strains with putative precursor 47 from the penitrem producer strain was performed. Structural analysis of the products from these strains revealed that 47 is an intermediate of penitrem biosynthesis, and the reaction proceeds via a five-step sequential oxidative transformation (Scheme 10): (1) ring expansion; (2) cyclic ether formation; (3) epoxidation; (4) hydroxylation; and (5) promiscuous chlorination.57 Importantly, PtmO and PtmE did not involve any transformation in the final stage. To determine their function, the AO-ptmHDVIEO prepared was incubated to afford 47via46, indicating that the two enzymes are responsible for the formation of a bicyclo[3,2,0]octane system.
Although ptm BGC also consists of ptmE/ptmO genes orthologous to janD/janO genes, the core construction mechanisms are significantly different from that of JanD/JanO. PtmD initially installs a prenyl group at C20; then, PtmO oxidized this substituent to allyl alcohol 46 in a way similar to JanO oxidation. The in vitro transformation of 45 with the recombinant flavoprotein oxidase PtmO gave a single acid-labile product, PC-M5 (46).57 Surprisingly, the incubation of 46 with the recombinant prenyltransferase PtmE in the presence of Mg2+ and DMAPP afforded a cyclization product, 47. PtmE did not catalyze any aromatic prenylation, as other prenyltransferases do. These data established that the penitrem skeletal scaffold formation proceeds from 17 to 43via the key intermediates 46 and 47. Throughout penitrem biosynthesis, the construction mechanism of the bicyclo[3,2,0]octane system is especially noteworthy. Usually, cationic cyclizations are triggered by either the protonation of olefin or the elimination of a suitable leaving group, such as diphosphate. Prenylation-initiated cationic cyclizations are a rather rare transformation in natural product biosynthesis (Scheme 10).
For lolitrem biosynthesis, similarly, AO-pas/terPQ/ltmEJ and AO-pas/terPQFK/ltmEJ are prepared from AO-pas/terPQ. These two TFs afforded lolitriol (49) and lolitrem B (57), respectively (Scheme 11, Fig. 4). Furthermore, careful examination of the products from the bioconversion of 28 with AO-ltmEJ identified epoxyalcohol 51, which was converted into 49, indicating 51 is an intermediate of 49.22
Radical cyclization using a ketyl radical derived from α,β-epoxyketone is reported for synthesizing the carbocyclic ring.62 Based on this observation, P450 catalyzing H-abstraction triggers rapid epoxide opening followed by 1,5-hydrogen transfer, proceeding to give a reactive enol radical. These radical undergoes intramolecular addition to the double bond in a 6-exo-selective manner (Scheme 11). This proposal was supported by density functional theory (DFT) calculations as well as model experiments with synthetic analogs.22
 | ||
| Scheme 11 Proposed biosynthetic pathway and mechanism of lolitrems. P450 LtmJ catalyzes highly unusual multi-step oxidative transformations. | ||
For constructing the bicyclic system of janthitrem/shearinine, penitrem, and lolitrem, a set of prenyltransferases, and oxidase are involved. However, these three types of IDT families used different mechanisms. In the janthitrem/shearinine cyclization, flavin oxidase JanO catalyzed a hydride transfer and generated a carbocation intermediate, which reacted with the adjacent prenyl group repeatedly.52 Penitrem biosynthesis uses unusual PtmE-catalyzed prenylation with another prenyl group, with the resultant carbocation triggering a sequential cyclization.57 In the third example, P450 LtmJ oxidatively installs an epoxy alcohol moiety to the one prenyl group and then the third oxidation generates a ketyl radical that is converted into the enol radical. This reacted with the adjacent prenyl double bond in an exo-selective manner, followed by oxidative cyclization to afford the bicyclic system.22 In summary, dual prenylation and oxidative cyclization represent key features in installing a unique cyclic system in indole cores. These transformations add further diversity to IDTs.
2.5 Structure diversification with promiscuous modification enzymes in late-stage biosynthesis
In natural product biosynthesis, late-stage intermediates often accumulate, and promiscuous enzymes convert them into structurally related products. These contribute to the expansion of natural product diversity. Various penitrem congeners are isolated from the producer strain,1,4,5 suggesting slow metabolite flow (PtmL-epoxidation and PtmJ-hydroxylation), which results in the accumulation of late-stage intermediates penitrems D, B, E. Promiscuous halogenase PtmN converted them into the corresponding halogenated products C, F, A. Actually, transformants AO-ptmK(LJ), AO-ptmKUN, AO-ptmKULN(O), and AO-ptmKULJN converted 47 into 48, penitrems C (52), F (54), A (56), respectively (Schemes 10 and 12).57 These results suggest that co-expressing PtmN is essential for the production of chlorinated products 52 and 54 after the formation of 43 [genes in parenthesis is not working] (Scheme 12).57 | ||
| Scheme 12 Late-stage transformation of penitrem with promiscuous halogenase PtmN. | ||
In Section 2.4.1, promiscuous JanD–JanO accepts both 17, 18, and penijanthine as a substrate. Considering NA-producing strain giving various derivatives differing right half,55 NodD1–NodO may also convert various platform intermediates (Fig. 3).52
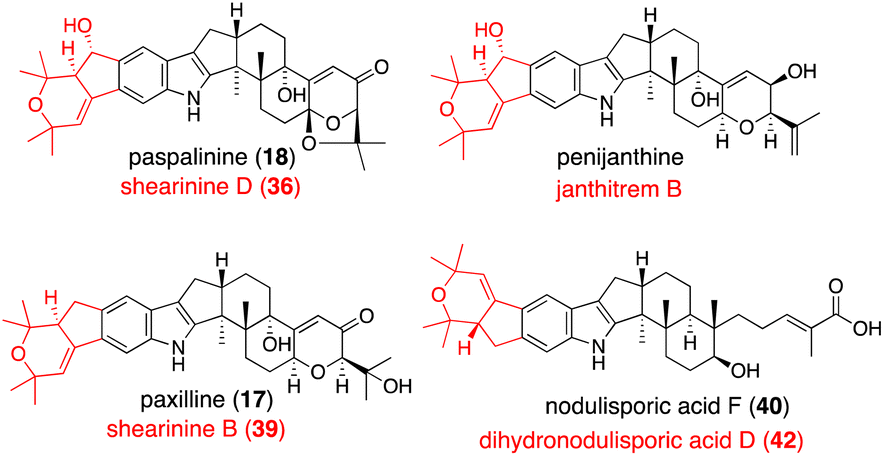 | ||
| Fig. 3 JanD–JanO and their orthologs catalyzed reaction products. Structures shown in black are the corresponding substrates. Name of their products are depicted in red. | ||
The fact that more than 10 lolitrem congeners have been isolated in nature suggests that LtmEJ can accept a diverse array of IDTs. To investigate their substrate scope, biotransformation experiments with various substrates using the transformant AO-ltmEJ were conducted. In fact, intermediate terpendoles E (23), I (28), J (29), and C (22) were converted to naturally occurring lolitremanes, lolicine A (58), lolitriol (49), lolitrems E and B (57).22 Overall, among 10 compounds tested, six IDT/IST analogs were converted into the corresponding products while four analogs lacking the C13- or C9-hydroxy group were not converted (Fig. 4). For the chemo-enzymatic synthesis of lolitrems, a wide substrate scope of LtmEJ is attractive.
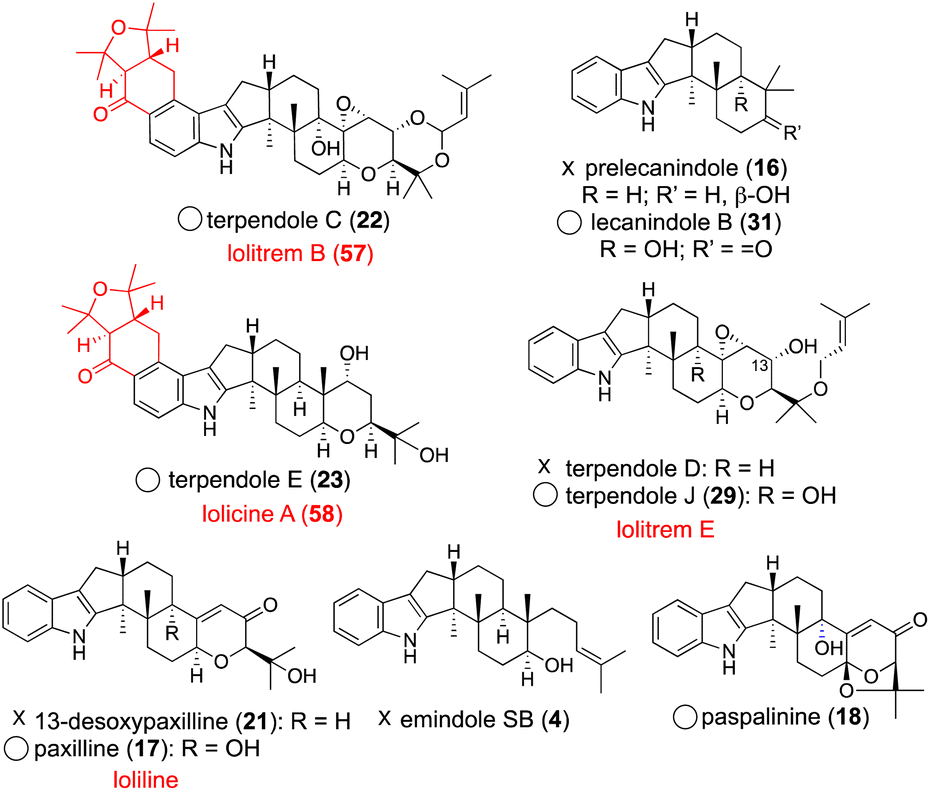 | ||
| Fig. 4 LtmE–LtmJ catalyzed reaction products. Structures shown in black are the corresponding substrates. Name of their products are depicted in red. Circles indicate accepted substrates and Xs indicate non-accepted substrates. | ||
3 Conclusion and perspective
The reconstitution approach of IDT biosynthesis successfully elucidates the IDT pathway and may trace the evolutional trajectory of IDT family members. At first, three essential enzymes install oligoprenyl side chains to the indole ring, epoxidize specific positions, and cyclize them by different modes giving diverse IDT core scaffolds (stage 1). For major paspaline type-IDTs, the resultant core is modified by two characteristic P450s to give biologically active platform intermediates (stage 2). Finally, platform intermediates are converted into highly elaborated IDTs by various modifications including characteristic diprenylation and oxidative cyclization (stage 3), which enhance the biological activity. Biosynthesis of structurally simple non-paspaline type-IDTs skips stage 2, and the core is directly modified by prenylation and other oxidative transformations. This strategy can be applied to IST biosynthesis by simply changing the substrate with the C15-side chain, as shown in the sespendole biosynthesis (Sections 2.2.4, 2.3.2, and 2.4.3).In general, a large natural product family is created by an efficient system generating diverse molecules, which have a good potential for characteristic bioactivity.63 In such a family, the history of molecular evolution may be hidden in their structures and biosynthetic pathways. In this regard, the IDT family is a good example. Because of its efficiency, the system possibly spreads over other fungi by horizontal gene transfer.64 Among the various IDTs, paspaline was chosen and intensively modified via a natural pharmacophore paxilline or related intermediates to furnish densely functionalized IDTs with potent bioactivities, such as anti-insectan, anti-virus, anti-mammal (acute toxicity against cattle through lolitrem producer-infected meadow grass).65,66 We speculate that this might be the reason why three stages of IDT biosynthesis have evolved in fungi.
As described in this review, reconstitution in a heterologous host is a very reliable strategy for elucidating the linear biosynthetic pathways of fungal IDTs. However, because of its linearity, lacking information on an intermediate of any single step does not allow us to continue the research. If a producer strain is available, gene knockout is the first choice. If the whole BGC information is available, adding all candidate genes to the host may help solve this problem.
There are still natural IDTs untapped in their biosynthesis whose structures are known such as thiersinine, thiersindole, and other IDTs, as shown in Fig. 1.67 Knowledge-based identification and pathway elucidation are possible by simply applying a heterologous expression, as shown in this review. Targeted genome mining of IDT-BGC which contains the uncharacterized gene is another application. Recently, A-domain-like genes of NRPS in a unique IDT-BGC have been characterized as an aminoacylase introducing amino acid residues to the secondary hydroxyl group.68
Recently, versatile synthesis of chiral piperidine pharmacophore by promiscuous modification enzymes from the efficient screening of their focused library has been reported.69 Regiospecific prenylation to the indole ring, halogenation, diprenylation–oxidative cyclization, and oxidative transformation to the indole core are typical modifications found in IDT biosynthesis. These can be candidates for the versatile modification enzymes of indole pharmacophore. Screening of a focused library of transformants carrying these candidate genes may identify versatile enzyme-decorating indole cores. The library may be used to convert IDT derivatives from biosynthetic study and chemical IDT synthesis6,70 into highly decorated IDTs.
In this review, we introduce nature's strategy for generating bioactive IDT congeners. This suggests that the core construction system itself has the ability to generate more diversity, but bioactivity-guided selection likely limits their structural diversity. Therefore, we could provide new unnatural IDT chemistry using the current biosynthetic system if we can find an efficient selection system and knowledge-based mutations to diversify biosynthetic enzymes.
4 Conflicts of interest
The authors declare no conflicts of interest.5 Acknowledgements
This work was financially supported by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology, Japan (JSPS KAKENHI Grant Number JP19H02891 (H. O.), JP22H02204 (A. M.), JP16H06446 (A. M.) and JP19H04635 (T. O.)) and Institute for Fermentation, Osaka (IFO), Grant Number G-2022-3-011 (A. M.).6 Notes and references
- P. S. Steyn and R. Vleggaar, Fortschr. Chem. Org. Naturst., 1985, 48, 1–80 CrossRef CAS.
- J. B. Gloer, Acc. Chem. Res., 1995, 28, 343–350 CrossRef CAS.
- S. Saikia, M. J. Nicholson, C. Young, E. J. Parker and B. Scott, Mycol. Res., 2008, 112, 184–199 CrossRef CAS.
- Comprehensive natural products III: Chemistry and Biology, ed. T. P. Begley and H.-W. Liu, Biosynthesis of indole diterpenes, Elsevier Science, 2020, vol. 2, pp. 553–576 Search PubMed.
- A. Minami, C. Liu and H. Oikawa, Heterocycles, 2016, 92, 397–421 CrossRef CAS.
- R. Tanifuji, A. Minami, H. Oguri and H. Oikawa, Nat. Prod. Rep., 2020, 37, 1098–1121 RSC.
- (a) I. Fujii, Nat. Prod. Rep., 2009, 26, 155–169 RSC; (b) Y. H. Chooi and Y. Tang, J. Org. Chem., 2012, 77, 9933–9953 CrossRef CAS PubMed.
- I. Fujii, Y. Ono, H. Tada, K. Gomi, Y. Ebizuka and U. Sankawa, Mol. Gen. Genet., 1996, 253, 1–10 CrossRef CAS PubMed.
- J. Kennedy, K. Auclair, S. G. Kendrew, C. Park, J. C. Vederas and C. R. Hutchinson, Science, 1999, 284, 1368–1372 CrossRef CAS.
- (a) C. Bond, Y. Tang and L. Li, Fungal Genet. Biol., 2016, 89, 52–61 CrossRef CAS PubMed; (b) S. M. Ma, J. W. Li, J. W. Choi, H. Zhou, K. K. Lee, V. A. Moorthie, X. Xie, J. T. Kealey, N. A. Da Silva, J. C. Vederas and Y. Tang, Science, 2009, 326, 589–592 CrossRef CAS.
- T. Itoh, K. Tokunaga, Y. Matsuda, I. Fujii, I. Abe, Y. Ebizuka and T. Kushiro, Nat. Chem., 2010, 2, 858–864 CrossRef CAS PubMed.
- M. N. Heneghan, A. A. Yakasai, L. M. Halo, Z. Song, A. M. Bailey, T. J. Simpson, R. J. Cox and C. M. Lazarus, ChemBioChem, 2010, 11, 1508–1512 CrossRef CAS.
- R. Fujii, A. Minami, T. Tsukagoshi, N. Sato, T. Sahara, S. Ohgiya, K. Gomi and H. Oikawa, Biosci., Biotechnol., Biochem., 2011, 75, 1813–1817 CrossRef CAS PubMed.
- Y. Matsuda and I. Abe, Nat. Prod. Rep., 2016, 33, 26–53 RSC.
- H. Oikawa, Biosci., Biotechnol., Biochem., 2020, 84, 433–444 CrossRef CAS PubMed.
- L. Kahlert, C. Schotte and R. J. Cox, Synthesis, 2021, 53, 2381–2394 CrossRef CAS.
- X. Zhang, J. Guo, F. Cheng and S. Li, Nat. Prod. Rep., 2021, 38, 1072–1099 RSC.
- C. M. Lazarus, K. Williams and A. M. Bailey, Nat. Prod. Rep., 2014, 31, 1339–1347 RSC.
- T. Katayama, Y. Tanaka, T. Okabe, H. Nakamura, W. Fujii, K. Kitamoto and J. Maruyama, Biotechnol. Lett., 2016, 38, 637–642 CrossRef CAS.
- C. Liu, A. Minami, T. Ozaki, J. Wu, H. Kawagishi, J. Maruyama and H. Oikawa, J. Am. Chem. Soc., 2019, 141, 15519–15523 CrossRef CAS.
- T. Katayama, H. Nakamura, Y. Zhang, A. Pascal, W. Fujii and J. I. Maruyama, Appl. Environ. Microbiol., 2019, 85, e01896-18 CrossRef PubMed.
- Y. Jiang, T. Ozaki, M. Harada, T. Miyasaka, H. Sato, K. Miyamoto, J. Kanazawa, C. Liu, J. Maruyama, M. Adachi, A. Nakazaki, T. Nishikawa, M. Uchiyama, A. Minami and H. Oikawa, Angew. Chem., Int. Ed., 2020, 59, 17996–18002 CrossRef CAS PubMed.
- Y. Jiang, T. Ozaki, C. Liu, Y. Igarashi, Y. Ye, S. Tang, T. Ye, J. I. Maruyama, A. Minami and H. Oikawa, Org. Lett., 2021, 23, 2616–2620 CrossRef PubMed.
- S. Nagamine, C. W. Liu, J. Nishishita, T. Kozaki, K. Sogahata, Y. Sato, A. Minami, T. Ozaki, C. Schmidt-Dannert, J. Maruyama and H. Oikawa, Appl. Environ. Microbiol., 2019, 85, e00409-19 CrossRef.
- J. P. Springer and J. Clardy, Tetrahedron Lett., 1980, 21, 231–234 CrossRef CAS.
- W. Acklin, F. Weibel and D. Arigoni, Chimia, 1977, 31, 63 Search PubMed.
- S. Fueki, T. Tokiwano, H. Toshima and H. Oikawa, Org. Lett., 2004, 6, 2697–2700 CrossRef CAS.
- C. Young, L. McMillan, E. Telfer and B. Scott, Mol. Microbiol., 2001, 39, 754–764 CrossRef CAS.
- K. Tagami, C. Liu, A. Minami, M. Noike, T. Isaka, S. Fueki, Y. Shichijo, H. Toshima, K. Gomi, T. Dairi and H. Oikawa, J. Am. Chem. Soc., 2013, 135, 1260–1263 CrossRef CAS PubMed.
- K. C. Van de Bittner, M. J. Nicholson, L. Y. Bustamante, S. A. Kessans, A. Ram, C. J. van Dolleweerd, B. Scott and E. J. Parker, J. Am. Chem. Soc., 2018, 140, 582–585 CrossRef CAS PubMed.
- M. C. Tang, H. C. Lin, D. Li, Y. Zou, J. Li, W. Xu, R. A. Cacho, M. E. Hillenmeyer, N. K. Garg and Y. Tang, J. Am. Chem. Soc., 2015, 137, 13724–13727 CrossRef CAS PubMed.
- T. Hosoe, T. Itabashi, N. Kobayashi, S. Udagawa and K. Kawai, Chem. Pharm. Bull., 2006, 54, 185–187 CrossRef CAS.
- K. Nozawa, S. Nakajima, K. Kawai and S. Udagawa, J. Chem. Soc., Perkin Trans. 1, 1988, 1689–1694 RSC.
- M. Ooike, K. Nozawa, S. Udagawa and K. Kawai, Chem. Pharm. Bull., 1997, 45, 1694–1696 CrossRef CAS.
- Q. Xiong, X. Zhu, W. K. Wilson, A. Ganesan and S. P. Matsuda, J. Am. Chem. Soc., 2003, 125, 9002–9003 CrossRef CAS.
- R. Uchida, Y. P. Kim, I. Namatame, H. Tomoda and S. Omura, J. Antibiot., 2006, 59, 93–97 CrossRef CAS.
- K. Kudo, C. W. Liu, T. Matsumoto, A. Minami, T. Ozaki, H. Toshima, K. Gomi and H. Oikawa, ChemBioChem, 2018, 19, 1492–1497 CrossRef CAS PubMed.
- B. Scott, C. A. Young, S. Saikia, L. K. McMillan, B. J. Monahan, A. Koulman, J. Astin, C. J. Eaton, A. Bryant, R. E. Wrenn, S. C. Finch, B. A. Tapper, E. J. Parker and G. B. Jameson, Toxins, 2013, 5, 1422–1446 CrossRef CAS.
- L. K. McMillan, R. L. Carr, C. A. Young, J. W. Astin, R. G. Lowe, E. J. Parker, G. B. Jameson, S. C. Finch, C. O. Miles, O. B. McManus, W. A. Schmalhofer, M. L. Garcia, G. J. Kaczorowski, M. Goetz, J. S. Tkacz and B. Scott, Mol. Genet. Genomics, 2003, 270, 9–23 CrossRef CAS PubMed.
- G. I. Lepesheva and M. R. Waterman, Biochim. Biophys. Acta, 2007, 1770, 467–477 CrossRef CAS PubMed.
- M. J. Nicholson, A. Koulman, B. J. Monahan, B. L. Pritchard, G. A. Payne and B. Scott, Appl. Environ. Microbiol., 2009, 75, 7469–7481 CrossRef CAS PubMed.
- K. Tagami, A. Minami, R. Fujii, C. Liu, M. Tanaka, K. Gomi, T. Dairi and H. Oikawa, ChemBioChem, 2014, 15, 2076–2080 CrossRef CAS PubMed.
- D. M. Roll, L. R. Barbieri, R. Bigelis, L. A. McDonald, D. A. Arias, L. P. Chang, M. P. Singh, S. W. Luckman, T. J. Berrodin and M. R. Yudt, J. Nat. Prod., 2009, 72, 1944–1948 CrossRef CAS PubMed.
- X. H. Huang, H. Nishida, H. Tomoda, N. Tabata, K. Shiomi, D. J. Yang, H. Takayanagi and S. Omura, J. Antibiot., 1995, 48, 5–11 CrossRef CAS.
- J. Nakazawa, J. Yajima, T. Usui, M. Ueki, A. Takatsuki, M. Imoto, Y. Y. Toyoshima and H. Osada, Chem. Biol., 2003, 10, 131–137 CrossRef CAS.
- T. Motoyama, T. Hayashi, H. Hirota, M. Ueki and H. Osada, Chem. Biol., 2012, 19, 1611–1619 CrossRef CAS.
- C. A. Young, B. A. Tapper, K. May, C. D. Moon, C. L. Schardl and B. Scott, Appl. Environ. Microbiol., 2009, 75, 2200–2211 CrossRef CAS PubMed.
- T. C. Charles, P. Hedden, M. C. Rojas and R. J. Peters, Nat. Chem. Biol., 2017, 13, 69–74 CrossRef.
- M. J. Nicholson, C. J. Eaton, C. Starkel, B. A. Tapper, M. P. Cox and B. Scott, Toxins, 2015, 7, 2701–2722 CrossRef CAS.
- G. N. Belofsky, J. B. Gloer, D. T. Wicklow and P. F. Dowd, Tetrahedron, 1995, 51, 3959–3968 CrossRef CAS.
- M. J. Xu, G. Gessner, I. Groth, C. Lange, A. Christner, T. Bruhn, Z. W. Deng, X. Li, S. H. Heinemann, S. Grabley, G. Bringmann, I. Sattler and W. H. Lin, Tetrahedron, 2007, 63, 435–444 CrossRef CAS.
- C. Liu, A. Minami, T. Dairi, K. Gomi, B. Scott and H. Oikawa, Org. Lett., 2016, 18, 5026–5029 CrossRef CAS PubMed.
- L. Sutzl, G. Foley, E. M. J. Gillam, M. Boden and D. Haltrich, Biotechnol. Biofuels, 2019, 12, 118 CrossRef.
- J. G. Ondeyka, G. L. Helms, O. D. Hensens, M. A. Goetz, D. L. Zink, A. Tsipouras, W. L. Shoop, L. Slayton, A. W. Dombrowski, J. D. Polishook, D. A. Ostlind, N. N. Tsou, R. G. Ball and S. B. Singh, J. Am. Chem. Soc., 1997, 119, 8809–8816 CrossRef CAS.
- S. B. Singh, J. G. Ondeyka, H. Jayasuriya, D. L. Zink, S. N. Ha, A. Dahl-Roshak, J. Greene, J. A. Kim, M. M. Smith, W. Shoop and J. S. Tkacz, J. Nat. Prod., 2004, 67, 1496–1506 CrossRef CAS PubMed.
- K. C. Van de Bittner, R. C. Cameron, L. Y. Bustamante, R. Bundela, S. A. Kessans, J. Vorster, M. J. Nicholson and E. J. Parker, MedChemComm, 2019, 10, 1160–1164 RSC.
- C. Liu, K. Tagami, A. Minami, T. Matsumoto, J. C. Frisvad, H. Suzuki, J. Ishikawa, K. Gomi and H. Oikawa, Angew. Chem., Int. Ed., 2015, 54, 5748–5752 CrossRef CAS PubMed.
- T. Hosoe, K. Nozawa, S. Udagawa, S. Nakajima and K. Kawai, Chem. Pharm. Bull., 1990, 38, 3473–3475 CrossRef CAS.
- T. Yamaguchi, K. Nozawa, T. Hosoe, S. Nakajima and K. I. Kawai, Phytochemistry, 1993, 32, 1177–1181 CrossRef CAS.
- J. Penn and P. G. Mantle, Phytochemistry, 1994, 35, 921–926 CrossRef CAS.
- R. T. Gallagher, A. D. Hawkes, P. S. Steyn and R. Vleggaar, J. Chem. Soc., Chem. Commun., 1984, 614–616 RSC.
- V. H. Rawal, V. Krishnamurthy and A. Fabre, Tetrahedron Lett., 1993, 34, 2899–2902 CrossRef CAS.
- A. Minami, T. Ugai, T. Ozaki and H. Oikawa, Sci. Rep., 2020, 10, 13556 CrossRef CAS.
- N. P. Keller, Nat. Rev. Microbiol., 2019, 17, 167–180 CrossRef CAS.
- C. L. Schardl, C. A. Young, J. Pan, S. Florea, J. E. Takach, D. G. Panaccione, M. L. Farman, J. S. Webb, J. Jaromczyk, N. D. Charlton, P. Nagabhyru, L. Chen, C. Shi and A. Leuchtmann, Toxins, 2013, 5, 1064–1088 CrossRef CAS PubMed.
- C. L. Schardl, C. A. Young, U. Hesse, S. G. Amyotte, K. Andreeva, P. J. Calie, D. J. Fleetwood, D. C. Haws, N. Moore, B. Oeser, D. G. Panaccione, K. K. Schweri, C. R. Voisey, M. L. Farman, J. W. Jaromczyk, B. A. Roe, D. M. O'Sullivan, B. Scott, P. Tudzynski, Z. An, E. G. Arnaoudova, C. T. Bullock, N. D. Charlton, L. Chen, M. Cox, R. D. Dinkins, S. Florea, A. E. Glenn, A. Gordon, U. Guldener, D. R. Harris, W. Hollin, J. Jaromczyk, R. D. Johnson, A. K. Khan, E. Leistner, A. Leuchtmann, C. Li, J. Liu, J. Liu, M. Liu, W. Mace, C. Machado, P. Nagabhyru, J. Pan, J. Schmid, K. Sugawara, U. Steiner, J. E. Takach, E. Tanaka, J. S. Webb, E. V. Wilson, J. L. Wiseman, R. Yoshida and Z. Zeng, PLoS Genet., 2013, 9, e1003323 CrossRef CAS PubMed.
- R. Geris and T. J. Simpson, Nat. Prod. Rep., 2009, 26, 1063–1094 RSC.
- R. M. McLellan, R. C. Cameron, M. J. Nicholson and E. J. Parker, Org. Lett., 2022, 24, 2332–2337 CrossRef CAS PubMed.
- J. R. Marshall, P. Yao, S. L. Montgomery, J. D. Finnigan, T. W. Thorpe, R. B. Palmer, J. Mangas-Sanchez, R. A. M. Duncan, R. S. Heath, K. M. Graham, D. J. Cook, S. J. Charnock and N. J. Turner, Nat. Chem., 2021, 13, 140–148 CrossRef CAS.
- D. J. Schatz, E. J. Kuenstner, D. T. George and S. V. Pronin, Nat. Prod. Rep., 2022, 39, 946–968 RSC.
Footnote |
| † Present address: Graduate School of Pharmaceutical Sciences, Tohoku University, Sendai, Japan. |
| This journal is © The Royal Society of Chemistry 2023 |