
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Curiouser and curiouser: progress in understanding the programming of iterative highly-reducing polyketide synthases
Russell J.
Cox
 *
*
Institute for Organic Chemistry and BMWZ, Leibniz University of Hannover, Schneiderberg 38, 30167, Hannover, Germany. E-mail: russell.cox@oci.uni-hannover.de
First published on 11th May 2022
Abstract
Covering: 1996–2022
Investigations over the last 2 decades have begun to reveal how fungal iterative highly-reducing polyketide synthases are programmed. Both in vitro and in vivo experiments have revealed the interplay of intrinsic and extrinsic selectivity of the component catalytic domains of these systems. Structural biology has begun to provide high resolution structures of hr-PKS that can be used as the basis for their engineering and reprogramming, but progress to-date remains rudimentary. However, significant opportunities exist for translating the current level of understanding into the ability to rationally re-engineer these highly efficient systems for the production of important biologically active compounds through biotechnology.
 Russell J. Cox | Russell Cox was born in 1967 in the New Forest in the UK where he grew up. He studied chemistry at the University of Durham, and then worked with Prof. David O'Hagan at the same institution for his Ph.D., studying the biosynthesis of fungal metabolites. Post-doctoral periods with Professor John Vederas FRS in Edmonton Alberta, and Professors David Hopwood FRS and Tom Simpson FRS at Norwich and Bristol in the UK were followed by his appointment as a lecturer in the School of Chemistry at the University of Bristol where he rose to become full Professor of Organic and Biological Chemistry. He moved to become Professor of Microbiological Chemistry at the Leibniz Universität Hannover in Germany in 2013. He has served as an editorial board member of Natural Product Reports until 2012, and has been past chair of the Directing Biosynthesis series of scientific conferences. He is currently chair of the editorial board of RSC Advances, and Head of the Institute for Organic Chemistry at the Leibniz Universität Hannover. |
1 Fungal PKS
All known functional fungal polyketide synthases (PKS) are iterative systems, of either Type I or Type III architectures. Type III systems, analogous to the classical plant chalcone synthases, are known to occur in fungi where they catalyse relatively simple reactions.1 While modular Type I PKS have been detected in some fungal genomes,2 and heterologous expression of bacterial modular PKS has been shown to be possible in fungi,3 the overwhelming majority of fungal PKS are Type I iterative systems. These are classified as non-reducing (nr-PKS), partially reducing (pr-PKS) and highly reducing (hr-PKS) systems.4 The function and engineering of nr-PKS that form primarily (poly)aromatic compounds has been extensively reviewed elsewhere.5 Here, the focus is on hr-PKS that synthesise primarily aliphatic chains and rings.Highly-reducing PKS are capable of producing a wide range of structures. These vary from the diketide sidechains of lovastatin 1a and compactin 1b,6 to the C34 chain of phaeospelide A 2 (ref. 7) and the C41 chain of T-toxin 3 (Fig. 1).8 They form the starter units for compounds such as the sorbicillins (e.g. epoxysorbicillinol 4),9 resorcylic acid lactones (e.g. dehydrozearalenol 5 and hypothemycin 6),10 dihydroxy-phenylacetyl lactones (e.g. dehydrocurvularin 7) and the backbones of dimeric compounds such as the maleidrides (e.g. byssochlamic acid 8).11 Highly-reducing PKS can vary the starter unit as observed in squalestatin S1 9 (ref. 12) and strobilurin A 10 (ref. 13) where benzoate is used rather than the much more common acetate. Methylation is achieved at β-positions by an integral C-methyltransferase (C-MeT) that uses S-adenosyl methionine (SAM) as cofactor and the programming of both frequency and distribution of methyls is controlled. Many hr-PKS act alone or in collaboration with a specific hydrolase system, but in some cases specific off-loading systems are fused at the C-terminus: for example carnitine acyl transferase (cAT).14 The most common of these is a single-module NRPS that can release its product either reductively to form the large class of fungal pyrrol-2-ones exemplified by pseurotin A 11 and the cytochalasans (e.g. cytochalasin E 12)15 or via a Dieckmann cyclisation (DKC) to form 3-acyl tetramic acids such as the cyclopiazonic acid precursor 13 or pretenellin A 14.16 An increasing number of these systems also seem to be able to control olefin geometry as in the EZE trienes observed in the strobilurin precursor 15 (ref. 13) and EZ diene compounds such as islandic acid 16.17 Remarkable ZEZ trienes such as soppiline B 17 have recently been reported.18 The released polyketides may be linear as in the tetraketide sidechain of squalestatin S1 9,19 or cyclic as in the precursor 18 of brefeldin A 19,20 and in many cases cyclisation by Diels–Alder reaction to form the characteristic decalins of lovastatin 1a or the octahydro-isoindole cores of the cytochalasans such as 12 are observed in the case of polyene polyketides.
 | ||
| Fig. 1 Diverse structures of typical compounds derived from pathways featuring fungal hr-PKS. | ||
Genome sequences of fungi show that genes encoding hr-PKS are common. For example, in a recent detailed analysis of the genomes of 13 Hypoxylaceae species, between 8 and 26 hr-PKS encoding genes were found in each organism, with an additional 1 to 6 hr-PKS-NRPS genes.21 These genes encode proteins that have a domain architecture highly homologous to vertebrate fatty acid synthases (vFAS). In order from the N- to C-termini the domain structure normally includes: keto synthase (KS); acyl transferase (AT); dehydratase (DH); C-methyl transferase (C-MeT); ΨKR (vide infra); enoylreductase (ER); ketoreductase (KR); and acyl carrier protein (ACP). This domain organisation is also found in some cyanobacterial modular PKS: for example the CurJ module of the curacin PKS from Lyngbya majuscula also features the KS-AT-DH-CMeT-ΨKR-ER-KR-ACP organisation.22 Thus the very wide structural diversity of polyketides synthesised by hr-PKS stems from a series of iterative proteins with highly conserved domain organisation.
Such systems are clearly highly programmed (and presumably programmable) and yet from a purely sequence and structure-based analysis very few clues exist as to how this programme is encoded and executed or how it could be rationally manipulated. In the cases of the fungal nr-PKS it is now well understood how starter unit selection, chain-length control and cyclisation regioselectivity are controlled and there are strong hypotheses for how methylation fidelity is programmed.5 Despite the importance of fungal hr-PKS in providing a very wide range of bioactive metabolites, information regarding programming mechanisms has been slow to accumulate. However, recent advances in the areas of structural biology, in vitro enzymology and rational in vivo engineering have started to shed light on this fascinating area.
2 Overall structures of fungal hr-PKS
For many years no structural data was available regarding any component of fungal hr-PKS. However, despite this deficiency end-to-end sequence homology with vFAS suggested the close structural analogy of these proteins. Such comparisons allowed the estimation of putative domain boundaries, for example. Informatic tools such as the Udwary–Merski algorithm,23 developed for multi-domain proteins in general, also enabled the prediction of domain boundaries in vFAS and hr-PKS. Sequence-based alignments with Type II PKS and FAS proteins again illuminate these features.The first structural breakthrough came with the publication of the vFAS structure at 3.2 Å in 2008.24,25 Although this structure lacked the ACP and chain-releasing thiolesterase (TE) domain, it provided the first hard evidence of the overall domain organisation (Fig. 2A). The overall structure is divided into two main regions. The KS forms a homodimer as the core of the bottom section of vFAS. The AT is not dimeric, but is bonded to the KS via a ca. 90 residue linker region. At the C-terminus of the AT a ca. 55 residue linker extends back across the KS to form a short flexible connection to the top region, for which the first domain is the dimeric DH. Remarkably, downstream of the DH, the vFAS possesses a non-functional C-MeT domain (ΨC-MeT), that does not possess a functional SAM-binding site. The presence of this ΨC-MeT domain very firmly links the structure to the fungal hr-PKS where the C-MeT is present and often fully active.
 | ||
| Fig. 2 Structures of vFAS and LNKS dimeric proteins. (A) Crystal structure of vFAS (from pdb 2vz9); (B) vFAS dimer calculated by AlphaFold2 showing position of ACP and TE; (C) Cryo-EM structure of LNKS (pdb 7cpy) showing position of LovC (light green). Note that in LNKS the lower KS-AT didomain is rotated ca. 180° with respect to the upper β-processing domains, but otherwise possess a highly similar quaternary structure to vFAS. In all cases, one monomer is shown as cartoons and one as solid. Structures to right show top view of β-processing domains. | ||
After the vFAS ΨC-MeT comes a structural (non catalytic) domain of the KR known as ΨKR. This is followed by the ER and then the catalytic part of the KR. It appears that the ER domain may have been inserted into a pre-existing KR. In sequence terms the ACP follows directly downstream of the KR on a short linker that presumably allows the ACP to visit the various active sites of the catalytic domains. The lack of the ACP (and its downstream thiolesterase [TE] domain) in the vFAS crystal structure was attributed to its likely conformational mobility.24 A second structure, apparently of a FAS/PKS involved in mycocerosic acid biosynthesis in Mycobacterium smegmatis shows a highly similar domain organisation,26 although because it lacks any kind of C-MeT domain it is a relatively poor model of the fungal hr-PKS.
The structure of vFAS has acted as a useful template for the construction of threaded structures of hr-PKS in the absence of true structures of these proteins. The inability of numerous groups to obtain crystals and structures of complete and active fungal hr-PKS is linked to the great difficulty in obtaining soluble active protein in the first instance, and adequately diffracting crystals of likely highly flexible proteins in the second. However, this problem has been very recently solved with the disclosure of a cryo-EM structure of the lovastatin nonaketide synthase (LNKS) obtained at 2.9 Å (Fig. 2C). This structure displays the same domain organisation as vFAS, aligning particularly well in the bottom part of the structure where KS and AT are almost identically structured in the two systems. Differences in the top part of the structure probably reflect large differences in the ER between the two synthases where the LNKS ER is inactive and replaced by a trans-acting ER, LovC, that the cryo-EM structure shows docked to the AT. Further differences focus on the C-MeT that is active in LNKS but inactive in vFAS. Never-the-less, the top parts of the structure show the same overall distribution.
A key lack of all structures obtained to date has been the ACP and its downstream domains (TE in the case of vFAS, a condensation domain in the case of LNKS). However, the very significant advance in protein structure prediction by the AlphaFold2 algorithm27 has improved this situation. It is notable that predicted monomeric structures for the human FAS do contain the ACP and TE domains. Dimeric structures can be assembled by alignment with the vFAS crystal structure and these show the ACP positioned conveniently close to all required active sites (Fig. 2B).
3 Divide and conquer: in vitro studies of catalytic domains
In iterative PKS the growing acyl chain, held by the ACP, must visit the different catalytic domains for each successive chemical transformation. Some of these visits must always happen, such as to the KS/AT for chain extension, but others are optional, for example to the ER. It therefore seems reasonable that individual catalytic domains should possess some intrinsic selectivity. For example, selectivity for the ACP-bound acyl group chain-length, methylation pattern or the extent of prior β-processing reactions might be a key programming mechanism. Similarly, KS/AT might select against particular intermediates either to prevent intermediates from being extended too early, or to halt chain construction at the programmed chain-length. All hr-PKS studied to-date appear to be highly selective, making few, if any, 'mistakes'. In order to probe such questions of intrinsic selectivity individual domains have been cloned, expressed, purified and studied in vitro using synthetic substrates and classical enzyme kinetics. These substrates are normally SNACs (Fig. 3B) or pantetheines (Fig. 8) that mimic the ACP's phosphopantetheinyl prosthetic group. A complimentary approach is to study complete PKS in vitro in which various functions are disabled, by for example, lack of cofactors, or site-specific mutation. In these experiments interactions of the catalytic domain under study could also occur with other nearby catalytic domains. In some cases structural biology or detailed modelling has supported these studies. | ||
| Fig. 3 (A) Overall reactions of LNKS + LovC; (B) individual and competition assays of the LNKS C-MeT and KR domains. | ||
3.1 KS and starter unit selection
Hr-PKS KS domains have not yet been assayed directly in vitro for their (e.g. chain-length) selectivity, but they are active in fully developed hr-PKS systems in vitro and assumed to extend chains as required. For example LNKS + the trans-ER LovC constructs the monacolin L skeleton 20in vitro in the presence of all required cofactors (Fig. 3A).Non-reducing PKS often possess starter unit acyl transferase (SAT)28 domains that select the starter unit, allowing nr-PKS to show a high degree of flexibility in starter unit selection. In contrast, no hr-PKS are known to possess specialised starter unit selection domains. This coincides with the observation that almost all hr-PKS products are initiated by acetate. However, rare examples of initiation by benzoate (e.g. squalestatin hexaketide 9 and strobilurin 10) and propionate (e.g. pseurotin A 11) are known. In the case of the pseurotin system the PsoA hr-PKS-NRPS has been expressed and purified. Propionyl CoA was shown to be the preferred starter unit, but in its absence acetyl-, isobutyryl-, hexanoyl- and octanoyl-CoAs could serve as low efficiency starter units, all giving the expected pentaketide pyrone products.29
3.2 C-Methyltransferase
While vFAS proteins possess an inactive C-MeT domain, fungal hr-PKS usually have a complete C-MeT and methylation is a key programmed feature. LNKS has been expressed successfully in yeast as an intact protein.30 An H985A mutant of LNKS was generated (LNKS DH0) to disable dehydration and downstream β-processing reactions, meaning that the activities of the C-MeT and KR could be probed using synthetic SNAC substrates (Fig. 3B), and differentiated by the presence or absence of the C-MeT (SAM) and KR (NADPH) cofactors (Fig. 3B).31 The C-MeT of LNKS DH0 shows remarkable selectivity towards unsaturated tetraketide substrates in the presence of SAM.The presence of active C-MeT and KR catalysts in this assay also allowed crucial competition assays to be devised. Thus, in the presence of both SAM and NADPH products from reduction alone (e.g. +2 mass units), from methylation alone (+14 mass units) and from both methylation and reduction (+16 mass units) can be detected and quantified by mass spectrometry. In the case of a triketide substrate 31 the KR reaction was ca. 10 times more effective than methylation, but in the case of the tetraketide 21, all observed products were methylated by LNKS DH0.
Almost all known hr-PKS C-MeT domains methylate a maximum of once after each extension cycle. However, the Tv6-931 PKS from Trichoderma virens displays a double methylation ability.14 The PKS was expressed in yeast and purified for in vitro assays. In the presence of all required substrates and cofactors Tv6-931 builds a β-keto tetraketide and at the final stage of synthesis it methylates twice to give the ACP-bound tetraketide 36 (Fig. 4). The Tv6-931 PKS is also unusual in having an integral carnitine acyl transferase (cAT)14 off-loading domain downstream of the normal terminal ACP. This domain reversibly releases the polyketide using a polyol as ester 37. Further detail was gathered by using synthetic β-keto SNAC substrates. These assays showed that triketides could be monomethylated, but not dimethylated, whereas tetraketide substrates could give both possible products.
 | ||
| Fig. 4 In vitro activity of the Tv6-931 hrPKS and its cAT off-loading domain. | ||
3.3 Ketoreductase
The KR domain of vFAS appears to be unselective, reducing β-carbonyls after every extension reaction. Ketones are almost never observed in the structures of highly reduced polyketides, except at β-positions of fully extended polyketides (see discussion below). It therefore seems likely that KR domains in hr-PKS show low intrinsic substrate selectivity. This has been demonstrated in the case of the LNKS DH0 mutant in vitro (Fig. 3B). In the absence of SAM the kinetics of the KR reaction were studied for a variety of SNAC β-keto thiolesters 27–28. The KR appears to bind SNAC substrates significantly less well than the previously assayed C-MeT, possibly indicating that protein–protein interactions between KR and the ACP are more important than between ACP and C-MeT. However, no significant substrate selectivity was observed in terms of either chain-length or the presence or absence of an α-methyl group (Fig. 3B).An unusual case of KR stereoselectivity was observed for the fungal polyketide hypothemycin 6, a derivative of dehydrozearalenol (DHZ, 5, Fig. 5A). This compound is produced by a collaborating PKS system that consists of the hr-PKS Hpm8 that builds a hexaketide starter unit 36 for the nr-PKS Hpm3 that catalyses three more extensions to 37, then 2,7 ring closure, aromatisation and release via formation of a macrolide.32 The hexaketide produced by Hpm8 features two alcohols at C-13 (produced at the tetraketide stage) and C-17 (produced at the diketide stage). The C-17 alcohol is S-configured and this is unexpected (Section 3.7).
 | ||
| Fig. 5 (A) Overall reactivity of the hypothemycin collaborating PKS; (B) reactions of the Hpm8 KR domain; (C) reprogramming the stereoselectivity of the Hpm8 KR domain. | ||
The Hpm8 PKS was expressed in yeast and purified for in vitro assays with synthetic SNAC substrates 22 and 38–40 (Fig. 5B). Results of in vitro assays showed an inversion in stereoselectivity: the diketide gives products resulting from β-re reduction; but for triketides and longer, ‘normal’ β-si reduction was observed, indicating that the shorter and longer substrates must bind in the KR active site in opposite orientations (Fig. 6C).
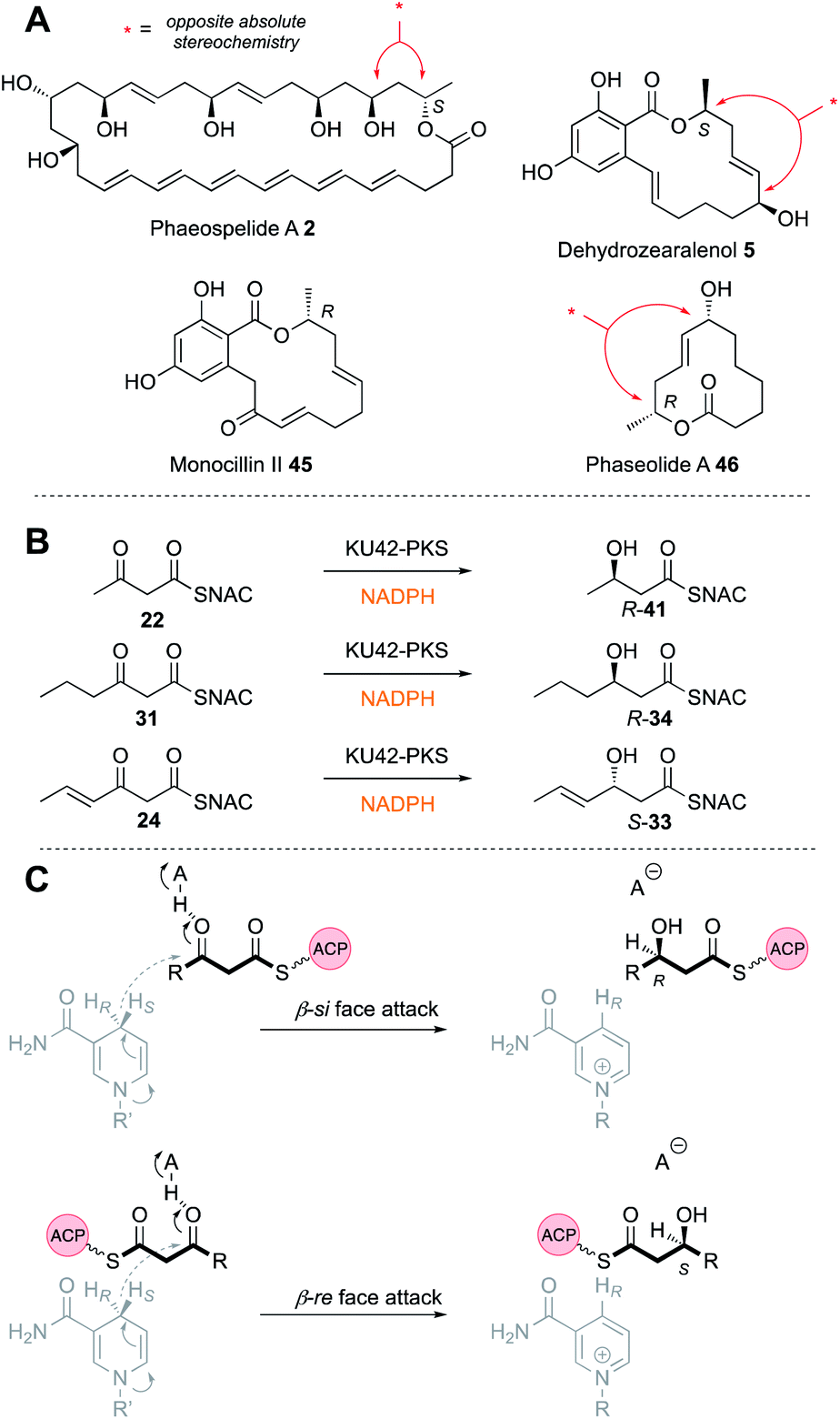 | ||
| Fig. 6 (A) Compounds featuring opposite alcohol stereochemistries; (B) in vitro activity of the KU42 KR; (C) possible mechanism of KR stereoselectivity. | ||
A different case is observed for the Rdc5 PKS involved in the biosynthesis of monocillin II 45 (Fig. 6A). Here, the diketide is reduced normally at the β-si face. Both the Rdc5 and Hpm8 KRs appear to belong to the B-type KR family33,34 and are thus expected to reduce β-si so the structural determinant for the unusual reduction of diketides by the Hpm8 KR was investigated by constructing Hpm8 chimeras with donated fragments from Rdc5.
This successfully identified a small region (helix-α4) of the KR responsible for the observed inversion of stereoselectivity for diketides (Fig. 5C). When expressed with the Hpm3 nr-PKS in yeast the chimeric system could produce diastereomeric products, although as mixtures of diastereomers indicating that downstream functions (e.g. the Hpm3 TE that catalyses macrolactonisation) are also likely to be stereoselective, limiting the effect of the Hpm8 KR engineering step.
The same reverse in selectivity by a KR has been observed in vivo for the phaeospelide A 2 hrPKS ApmlA that produces a diketide S-alcohol, but all subsequent alcohols are of opposite absolute configuration.7 An opposite case appears to be that of phaseolide A 46 where the first KR appears to give an R alcohol at the diketide stage and later gives an S alcohol for the tetraketide intermediate.35 A similar change in KR selectivity has also been observed for the KU42 hrPKS from the basidiomycete Punctularia strigosozonata (Fig. 6B). In vitro this PKS reduces its diketide substrate 22 to give the ‘normal’ 3R product R-41; but the triketide 24 is reduced with opposite stereoselectivity. Remarkably, the KR is highly sensitive to the presence of the 4-alkene at the triketide stage: the saturated homolog 31 undergoes normal reduction.
3.4 Dehydratase
Dehydratase (DH) domains show high stereoselectivity, but their substrate (e.g. chain-length) selectivities have not been investigated in depth in vitro. The DH from the squalestatin tetraketide synthase (SQTKS) has been cloned and expressed in E. coli and shown to be dimeric. In the native SQTKS the DH is only ever presented with α-methylated diketide substrates, but in vitro it can also dehydrate non-α-methylated diketides.36Stereoselectivity is tight at both the α- and β-positions: only 2R, 3R synthetic diketide substrates 47 are dehydrated in vitro to give the E-product 48. Docking of model substrates into a threaded model of the SQTKS DH, using the CurF DH22 as a template, showed that the putative active site catalytic residues align well with the α-proton and β-hydroxyl for the 2R,3R stereoisomer, but satisfactory docked poses could not be obtained for the other stereoisomers. This correlates with the observation that the other stereoisomers did not appear to inhibit the DH reaction, suggesting that they cannot enter the active site. In some cases it appears that 3S-alcohols can be eliminated to give Z-olefins. This appears to be the case in the KU42-PKS involved in the construction of EZ dienes (Fig. 7B).48
 | ||
| Fig. 7 DH stereochemistry. (A) The DH domain of SQTKS catalyses elimination from only the 2R,3R stereoisomer. Docked model of 47 in the active site of SQTKS DH; (B) 2S-syn elimination to generate Z-olefin. | ||
Observation of the structures of many highly-reduced polyketides show that β-alcohols are rare substituents. Notable exceptions are the resorcylic acid lactones (RAL) and dihydroxyphenylacetate lactones (DAL) where an alcohol is usually left intact on the starter unit. For example, during the biosynthesis of the dehydrozearalenol starter unit 36 a 3S-alcohol is left intact after the first extension and KR when the DH must be inactive (Fig. 5B). This is in agreement with the in vitro studies of the SQTKS DH that does not eliminate 3S alcohols. But the Hpm8 hr-PKS has another trick – it also does not react with tetraketide alcohols even of the ‘correct’ 3R stereochemistry (Fig. 5B), although tri-, penta and hexaketides are eliminated. Compounds such as T-toxin 3 and related compounds feature many intact β-alcohols, but the PKS in these cases have been barely investigated in terms of selectivity. Thus, while many hr-PKS DH appear to be unselective to chain-length, it is clearly possible for some systems to evolve sophisticated selectivity.
3.5 Enoylreductase
Both cis and trans type enoylreductases (ER) are known in hr-PKS systems. PKS-NRPS almost always feature trans-ER, as found in the pretenellin A 14 PKS where the function of a degraded cis-ER domain is replaced by a trans-ER.Some systems such as the fusarin PKS-NRPS are similar in that the cis-ER is non-functional, but the lack of a trans-ER means that the PKS produces a polyene.41 Apparently, stand-alone PKS such as lovastatin nonaketide synthase (LNKS) can also feature trans-ER, but such systems are now recognised as deriving from PKS-NRPS that have lost all, or most, of the NRPS components. LNKS, for example, possesses a condensation (C) domain downstream of its ACP and is clearly a PKS-NRPS that has lost the A, T and R/DKC domains more normally present.37
Both cis- and trans-acting ER domains have been investigated in vitro. The cis-ER from the squalestatin 9 tetraketide synthase (SQTKS) has been investigated in detail (Fig. 8). It exists as a dimer in solution in agreement with structural models based on vFAS (Fig. 2).38 Its activity in vitro is easily followed and quantified by measuring the consumption of NADPH.
 | ||
| Fig. 8 Substrate selectivity of the isolated SQTKS ER domain. | ||
The SQTKS ER displays a remarkably broad substrate selectivity, accepting a wide range of chain-lengths and methylation patterns (Fig. 8). It can reduce Z-alkenes and other substrates it would never be exposed to normally such as α-ethyl groups and odd-carbon chain-lengths 49–51. In fact kcat/KM measurements showed that the most highly favoured triketide substrate 52 is not even a natural intermediate on the pathway to SQTK itself, and has kcat/KM > 700 s−1 μmol−1 L compared to a value of ca. 150 s−1 μmol−1 L for native triketide substrate 51.
However, kcat/KM values fall for longer substrates and here methylation pattern and stereochemistry become important. For example the SQTKS ER can process pentaketides that lack round-1 and round-2 methyls (e.g. ca. 50 s−1 μmol−1 L for 55), but kinetic analysis showed that although the squalestatin tetraketide 53 itself can enter (and inhibit) the ER active site, it cannot reach the productive s-cis conformation for reduction.
Indeed, the rate of reduction by the ER may reflect the time it takes for short (diketide) and long (tetra- and penta-ketide) substrates to reach a productive binding conformation, whereas α-methyl triketides are processed quickly. In terms of stereochemistry, the ER uses the 4′-pro-R hydrogen of NADPH and adds it to the substrate's 3-Re face – both identical to the reactions catalysed by the vFAS ER. At the α-carbon it appears that the isolated SQTKS ER cannot control the face-selectivity of reprotonation in in vitro assays, giving α-racemic products, but in the full protein protonation clearly occurs from the α-Re face of the intermediate enol(ate), again identical to the vFAS reaction (Section 3.7).
A threaded structural model of the SQTKS ER was developed based on the vFAS ER structure. This shows a deep and narrow pocket that accommodates both the cofactor and the substrate (Fig. 9). Cofactor docking shows that NADPH interacts with conserved residues in the Rossmann fold, and exposes the 4′-pro-R hydrogen for reaction consistent with experimental results. Substrate docking shows that the enoyl moiety of the substrate must take up an s-cis conformation (Fig. 9B) and this is consistent with the observed stereochemistry of the reduction. Squalestatin tetraketide 53 itself cannot reach this conformation in the WT enzyme and cannot be reduced (Fig. 9A).
 | ||
| Fig. 9 Docked structures of 53 in the active site of SQTKS ER domain. (A) WT enzyme showing unproductive s-trans conformation; (B) mutant enzyme in which 53 can reach a productive s-cis conformation. | ||
No apparent acid is present for the reprotonation step (i.e. 92 to 93, Fig. 14), suggesting that this may be achieved by a water molecule that cannot be properly oriented in the isolated domain in vitro, but which presumably can perform the required α-Re protonation in the complete SQTKS.
The substrate binding-pocket of SQTKS ER is defined by several non-polar amino acids. Mutation of these led to the creation of enzymes with altered intrinsic substrate selectivities.39 In particular, expansion of the pocket by replacement of large non-polar residues with alanines led to ERs with enhanced selectivity for pentaketides in vitro and allows SQTK 53 itself to reach a productive s-cis conformation (Fig. 9B).
The trans-ER LovC, from the lovastatin system, has also been investigated. Recent structural work has shown that LovC docks with a specific region of the AT domain of LNKS that brings the trans-ER within range of the LNKS ACP (Fig. 2C).40 Many hr-PKS are able to function without the presence of a trans-ER, producing polyenes (e.g. fusarin C).41 In the case of the tenellin PKS-NRPS, however, the presence of the trans-ER is required for the PKS to display correct programming fidelity.16 In the absence of the trans-ER programming elements such as chain-length and methylation are impeded such that the TenS hr-PKS produces mixtures of products. In the case of LNKS itself, polyene pyrones are produced in the absence of LovC, or when the LNKS-LovC binding region is mutated.40
LovC itself has been studied in vitro (Fig. 10) and crystal-lographically.42 It is a member of the MDR family, but unusually exists in solution as a monomer, and this is consistent with the fact that it binds to the LNKS AT domain. It uses NADPH and transfers the 4′-pro-R hydrogen, although the stereoselectivity at the substrate β-position was not investigated. LovC does possess some intrinsic substrate selectivity. Like the SQTKS cis-ER, LovC can reduce non-intermediates such as the ketone 64, and preliminary kinetic analysis shows that di- 61 and tri-ketide 62 SNAC substrates are not reduced, but a non-methylated enoyl tetraketide 63 is reduced in agreement with the proposed biosynthetic pathway.
 | ||
| Fig. 10 In vitro reactions of LovC. | ||
3.6 Off-loading
In vFAS the only programmed step is catalysed by the C-terminal off-loading thiolesterase (TE) domain. In vitro studies of the TE show that it recognises C16 and C18 chains and transfers them to a conserved TE-active site serine, before hydrolysis with H2O.43 It is therefore conceivable that off-loading components of hr-PKS could control chain-length by recognising and cleaving polyketide intermediates. In the case of hr-PKS-NRPS such hypotheses have been probed in vivo (Section 4.4) rather than in vitro. Almost all other hr-PKS do not contain an integral TE like the vFAS (although rare exceptions are known, including carnitine acyl transferases cAT14). Hr-PKS most often interact with a range of trans-acting off-loading systems including hydrolases, acyl transferases and rare PLP-dependent domains.44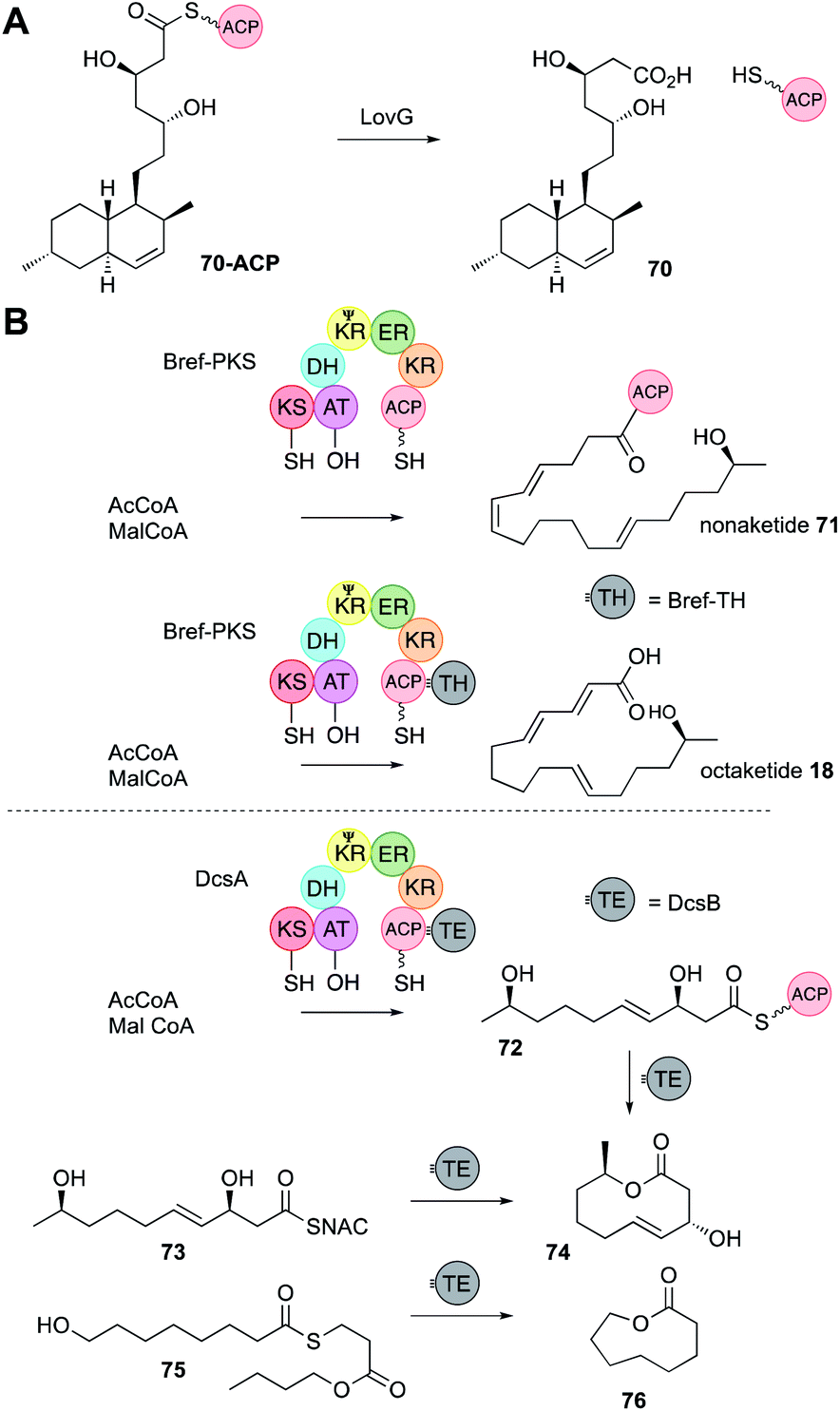 | ||
| Fig. 11 PKS off-loading reactions. (A) Function of LovG; (B) non-cyclising and cyclising release systems. | ||
Rarely, hr-PKS have integral thiolesterase (TE) domains located downstream of the ACP. In two cases recently investigated these have been shown to catalyse off-loading to amino acids.48 The TE itself was shown to have low selectivity, accepting butyryl-, β-keto-hexanoyl- and β-hydroxyoctanoyl SNACs as a substrate in vitro and passing them to both the N and S atoms of cysteine and homocysteine. It therefore seems probable that the TE accepts any polyketide residing on the ACP at the end of the hr-PKS synthesis.
 | ||
| Fig. 12 (A) Function of the acyl transferase LovD; (B) function of the collaborating hr-PKS and nr-PKS AtCURS1/AtCURS2 involved in dehydrocurvularin biosynthesis. | ||
SAT domains of nr-PKS appear to operate very similarly. These domains have been shown to recognise ACPs of their cognate hr-PKS and they most often accept fully elaborated polyketides (e.g.80) as in the case of the dehydrocurvularin PKS pair AtCURS1 and AtCURS2 (Fig. 12B). The tetraketide 80 is transferred to AtCURS2 where it is further extended and cyclised to 81. The enzymology and selectivity of SAT domains has been recently extensively reviewed elsewhere.5
 | ||
| Fig. 13 (A) Function of the aspyridone ApdS + ApdC PKS-NRPS; (B) formation of pyrones by ApdS; (C) function of the cyclopiazonic acid PKS-NRPS CpaS. | ||
The final step for ApdA and CpaS is Dieckmann off-loading (DKC or R* domain)52 to tetramic acids. In the case of pre-aspyridone 82, tyrosine is the amino acid; in the case of cyclopiazonic acid, tryptophan is used. Experiments in which no SAM was provided to the aspyridone system made unmethylated and unsaturated polyketide pyrones 85 showing that the trans-acting ER ApdC requires methylated substrates to act, and that unlike the TenS system (Section 4.4) the off-loading NRPS cannot recognise incorrect substrates and these are (presumably slowly) extended to β,δ diketones that self-release as pyrones such as 86 and 87 (e.g. Fig. 13B).
The hr-PKS and NRPS modules of ApdS were also produced separately, and in vitro these can interact and function correctly in trans to make the expected tetraketide tetramic acids 82 and 13. On its own the ApdS PKS makes and releases a pentaketide pyrone 87 – presumably ApdS slowly extends unreacted tetraketides that would normally be off-loaded by the NRPS. Remarkably the ApdS hr-PKS also functions in trans with the CpaS NRPS that provides tryptophan to build the tetraketide tetramic acid 89.51
Other examples also illustrate the selectivity of the off-loading NRPS. For example, in the case of the pseurotin 11 synthase a trans-acting C-MeT methylates the growing PKS at the tetraketide stage. In coexpression experiments in yeast,29 in the absence of the C-MeT partner, the PKS is still functional, but releases pyrones in the same way as ApdS. These assays were recapitulated in vitro, but because the β,δ diketopentaketide intermediates spontaneously cyclise to pyrones in vitro it was not possible to test this directly with the isolated PsoA NRPS module and synthetic substrates. However, the aspyridone NRPS module was shown to behave similarly – it can only accept γ-methylated substrates, although it seems insensitive to chain-length or methylation at other positions.30
3.7 Overall stereoselectivity of hr-PKS
Almost all of the stereoselective steps during the β-processing steps of hr-PKS biosynthesis have been measured. For example, the KR of SQTKS reduces β-carbonyls (e.g. 90) at their si faces to provide β-R alcohols 47.36 This is achieved using the 4′-pro-S hydrogen of NADPH.53 Sequence analysis of the SQTKS and vFAS KR domains show that they are B-type reductases consistent with this observation (conserved LDD motif).33,34,54 The SQTKS DH is selective for 2R,3R substrates and catalyses an overall syn elimination to give an E product 48.36 Finally, ER uses the 4′-pro-R hydride of NADPH to reduce the β-carbon at its re face. Final reprotonation occurs at the α-re face of the intermediate enol(ate) 92. All of these steps are identical to those previously measured for vFAS.36 Intriguingly, hr-PKS also catalyse a methyl-transfer, presumably by attack of an intermediate enol(ate) 22 on SAM. However, no studies on the stereochemical preferences of the C-MeT step have yet been published and it is unknown whether the α-methyl-β-keto product 90 would be configurationally stable or rapidly racemised prior to the KR step (Fig. 14). | ||
| Fig. 14 Overall stereochemistry of the first β-processing cycle of SQTKS. Bold arrows indicate experimentally determined route. | ||
4 Activity of full hr-PKS: in vivo studies
While in vitro studies of individual hr-PKS activities can give detailed information regarding the intrinsic selectivities of some catalytic domains, numerous problems have prevented wider exploitation of this methodology. In the past it has been difficult to predict correct domain boundaries, and it is frequently observed that isolated domains are insoluble and inactive after heterologous expression – presumably because isolated domains reveal large exposed hydrophobic surfaces.Isolated domains may be damaged in terms of their substrate or stereoselectivities as observed for the SQTKS ER because of missing cooperative protein partners. Heterologous expression and purification of active full hr-PKS also remains highly challenging.
In vitro assays require the development of synthetic substrates and often sophisticated analytical methodology that may not be widely accessible. It has also been infeasible to use acylated ACP substrates for these assays and synthetic SNAC and pantetheine substrates cannot illuminate the importance of possible protein interactions with the ACP.
The alternative is to work with hr-PKS in vivo, either in native or heterologous hosts. Such experiments allow specific questions regarding programming to be asked in the context of the complete synthase. Heterologous expression systems are particularly useful in this respect as they can be combined with rapid engineering systems and standardised fermentation, isolation and analytical platforms. The rapid formation of chimaeric hr-PKS has been key to many of these experiments, however, it often requires high levels of sequence identity (>75%) between donor and acceptors PKS to give functional hybrid synthases. Never-the-less, the results of such experiments have begun to provide useful information.
4.1 KS-AT
The KS-AT component of both vFAS and hr-PKS forms an independent structural unit separated from the β-processing domains by a short unstructured linker (Fig. 2). Structures of both systems suggest there may be no significant contact between the KS-AT and the DH. Exchange of KS-AT domains between the tenellin synthetase (TenS, acceptor) and the desmethylbassianin synthetase (DmbS, donor) hr-PKS showed that these domains play no role in PKS programming – the TenS KS-AT can produce chain-lengths from C6 to C14 in various circumstances (Fig. 15).55 In other iterative PKS, such as the Type II PKS involved in the biosynthesis of bacterial polycyclic aromatics, it is known that KSαKSβ heterodimers are involved in chain-length control,56 but in the pretenellin 14/preDMB A 95/militarinone C 96 hr-PKS this is clearly not the case where the activity of the KR quite clearly controls chain-length (vide infra).57 Results of experiments to-date suggest that the hr-PKS KS/AT are unprogrammed, as they are in the vFAS where the off-loading TE domain controls chain-length.43 Swaps of ACP showed that it too is not involved in programming.54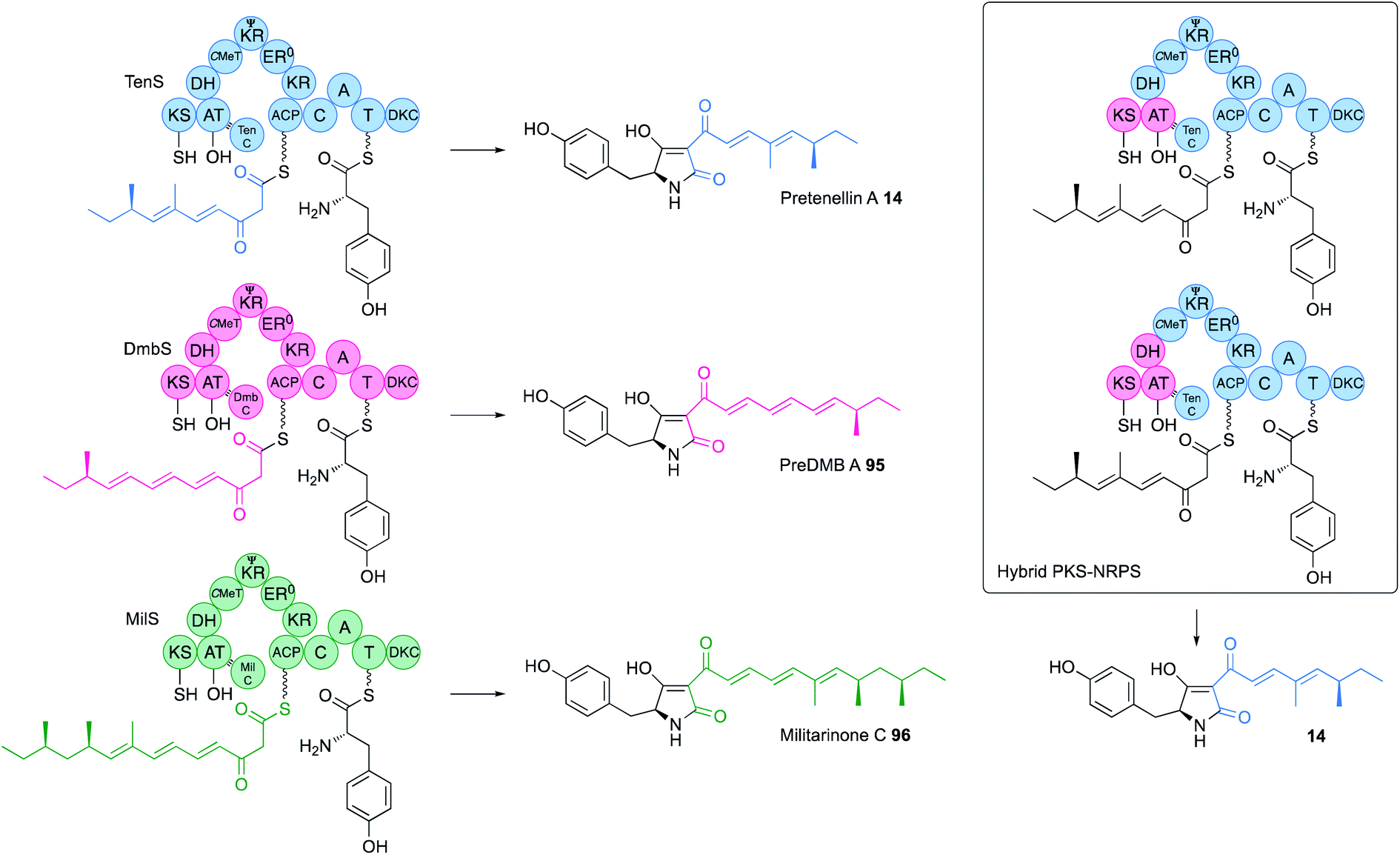 | ||
| Fig. 15 Function of the TenS, DmbS and MilS PKS-NRPS systems. | ||
Similarly, in the LNKS system the KS-AT-ACP continues to extend incorrectly formed polyketides, for example resulting from the absence of the trans-ER LovC until β,δ-diketones are formed that self-release as pyrones. In some cases, AT-domains must be involved in starter unit selection, for example in the cases of SQTKS synthase or the prestrobilurin 15 synthase where benzoate is used, but the molecular basis for this selectivity has not yet been examined.
4.2 C-MeT
Domain swaps of the C-MeT domain have been achieved in the TenS/DmbS system (Fig. 16). In the pretenellin A 14 case, methylation occurs at the diketide and triketide stages, but for preDMB A 95 it only occurs at the diketide stage. When the DmbS C-MeT was inserted into the TenS background monomethylated products 97 were formed, although around 20% of the dimethylated product 14 was also observed (Fig. 16). Detailed modelling of the TenS and DmbS C-MeT domains, that are ca. 85% identical, show that almost all of the differences occur at solvent exposed surfaces. Other minor changes are observed in the hydrophobic cores, but residues in contact with the substrate and cofactor are identical in both the TenS and DmbS C-MeT domains. It was thus concluded that the intrinsic selectivities of these domains are likely to be highly similar and that extrinsic factors, such as protein–protein interactions with adjacent ΨKR, KR, DH and ER may impact on relative rates of reaction with ACP-bound substrates, explaining the observation of mixtures of methylated products.56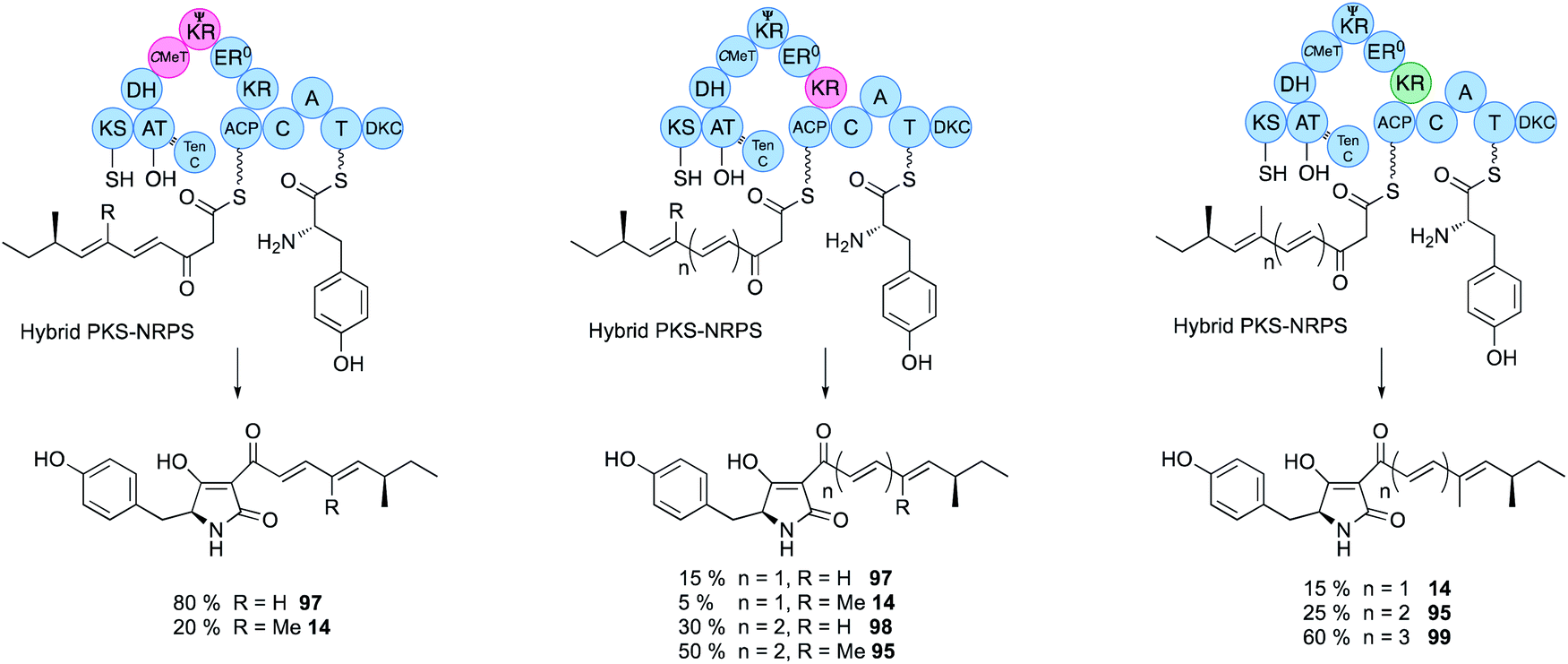 | ||
| Fig. 16 Hybrid PKS-NRPS engineering to control chain length and methylation pattern. | ||
4.3 ΨKR–KR
The ΨKR sequence forms a ca. 115-residue structural domain of the overall KR. The KR sequence itself contains the cofactor and substrate-binding motifs. In structural terms it appears that the ER domain has inserted into the KR sequence, but allowing ΨKR and KR to remain in contact (Fig. 2). Domain swaps of the complete ΨKR and its sub-fragments from DmbS into TenS make almost no difference to the TenS programme. However, swap of the entire KR, or various sub-fragments result in a change in chain length from pentaketides 14/97 to hexaketides 95/98 (Fig. 16). When the KR (and sub-fragments as short as 12-residues) from the militarinone hr-PKS (heptaketide) was inserted, then the heptaketide 99 was formed.56Extensive heterologous expression, stereoselective synthesis, and detailed analytical work has been applied to three families of hr-PKS that produce poly-hydroxylated polyketides such as phaeospelide A 2 and related compounds. This has allowed a stereochemical rule to be devised that has significant predictive power, based on the known stereochemical preferences of KR domains (Fig. 14).34
4.4 DH, ER and off-loading domains
Swap of DH domains between TenS and DmbS resulted in no change in programming.54 In these systems the DH must always be active after every extension cycle and these results are consistent with it being unselective. ER selectivity has only been investigated in vivo for the TenS and DmbS trans-ER systems.Swap of these made no difference to programming – in both cases the enacted programme was that of the core hr-PKS.58 However, in all systems investigated to date, lack of a trans-ER leads to loss of fidelity of the overall hr-PKS. In the case of TenS shorter and mis-methylated chains are constructed in the absence of TenC and passed to the off-loading NRPS, that can correctly react them with tyrosine to form tetramic acids. In the cases of LNKS and the ACE1 PKS-NRPS59 lack of the trans-ER in each case results in unreduced polyenes that fail to be reduced by the KR and eventually tricarbonyls spontaneously release as pyrones, similar to the aspyridone case (Fig. 13).
Chimaeric hr-PKS-NRPS systems have been extensively studied in vivo via heterologous expression. In the TenS/DmbS system the NRPS modules could be exchanged with no change in programme, showing that the off-loading NRPS module does not control chain-length of the polyketide.54 This mirrors the in vitro results obtained for the CpaS and ApdS hr-PKS-NRPS systems investigated in vitro (Section 3.6.3). Similar results have been observed when exchanging the NRPS modules of CcsA (involved in cytochalasin E 12 synthesis in Aspergillus clavatus) and Syn2 (from the biosynthesis of an unknown compound in Magnaporthe grisea) – a clean swap of the amino acid was observed with no change in the hr-PKS programme.60
Hydrolases are known to interact with hr-PKS to off-load linear (e.g. squalestatin hexaketide 9) or cyclic products (e.g. brefeldin A 19). Remarkable compounds such as phaeospelide A 2 are produced by a single hr-PKS and a cyclising TE.7 In this case expression of the PKS ApmlA alone resulted in no product production, probably indicating that the complete PKS product waits on the ACP, blocking further synthesis until it can be released. Co-expression with the TE releases the cyclised product and allows further turn-over. However, it is not yet known whether the action of the TE can affect the programme of the hrPKS. Also recently reported, isolated trans-acting condensation (C) domains have been shown to be involved in the release of linear polyketides, and appear not to affect the basic hr-PKS programme.34
A large family of collaborating hr-PKS + nr-PKS systems are known in fungi.61 Such systems are responsible for the biosynthesis of compounds such as the sorbicillinoids,62 some azaphilones,63 resorcylic acid lactones (RAL) and dihydroxyphenyl acetic acid lactones (DAL).64 In these systems the hr-PKS effectively synthesises a starter unit for the nr-PKS. A specialised starter unit acyl transferase (SAT) domain on the nr-PKS is matched to the ACP of the hr-PKS and must presumably dock with it in order to transfer the polyketide from the hr-PKS to the nr-PKS. Extensive domain-swap work has shown that in most cases the polyketide transfer step does not affect the programme of the hr-PKS itself. It appears that the PKS usually enacts its programme and the completed polyketide then waits on the ACP until it is removed by the SAT. A pertinent example comes from the combination65 of genes encoding hr-PKS and nr-PKS involved in the biosynthesis of dehydrocurvularin 7 (AtCURS1 & AtCURS2 respectively)66 and the asperfuranone intermediate 100 (AfoG & AfoE respectively, Fig. 17A).61 AfoG is an hr-PKS that synthesises a doubly methylated tetraketide 101. Normally, this is extended four more times to 102 by the nr-PKS AfoE that also cyclises and reductively releases to make preasperefuranone 100. Replacement of AfoE with an engineered AtCURS2 that bears the AfoE SAT domain results in a new octaketide 104 (after hydrolytic release with ethanol) built from the expected AfoG tetraketide starter 101 (Fig. 17B).
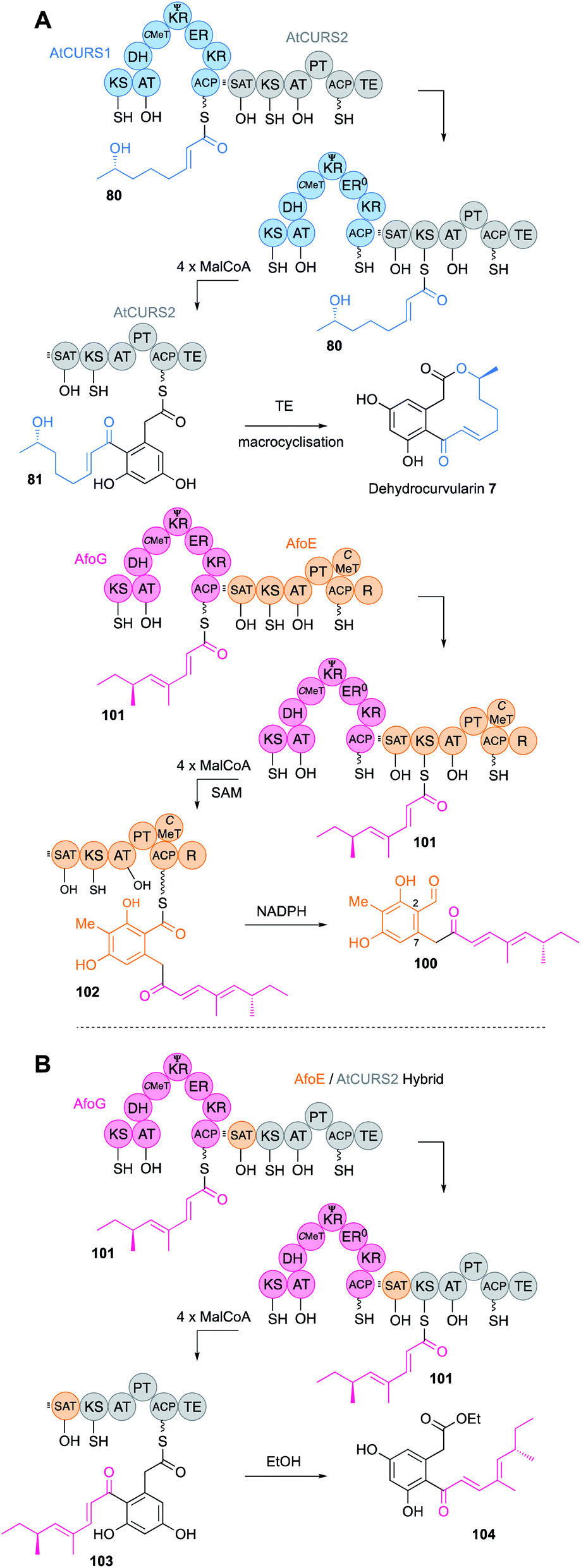 | ||
| Fig. 17 (A) Function of AtCurS and Afo PKS; (B) hybrid AtCurS and Afo PKS. | ||
However, recently an example has been discovered where the SAT does influence the chain-length of the hr-PKS.62 In this case an hr-PKS/nr-PKS pair (RrDALS1/RrDALS2) was shown to be barely active when expressed in yeast (Fig. 18). However, replacing the TE domain of RrDALS2 with that from AtCURS2 gave a fully functional system that made triketide 106 primed heptaketides 108 in good titre. But use of complete AtCURS2 resulted in production of a tetraketide 109 primed octaketide 111, as the main product. Presumably, the SAT of AtCURS2 only slowly removes starter units from RrDALS1, allowing it time to extend triketides 106 to tetraketides 109. A related off-loading system was recently described in which a Type III PKS intercepts a nonaketide intermediate of an hr-PKS during the biosynthesis of soppiline B 17 (Fig. 19). This was investigated by heterologous expression in A. oryzae. In the absence of the Type III PKS off-loading step, however, the polyketide is extended one more time and reduced at the β-position to be released as soppiline A 112.18
 | ||
| Fig. 18 Hybrid DAL and RAL PKS systems. | ||
 | ||
| Fig. 19 Soppilines. | ||
4.5 PKS that produce two different chains
Programming by hr-PKS is normally tightly controlled, usually by the PKS itself, or more rarely by the off-loading mechanism. In either case a single polyketide product is normally observed from correctly functioning systems. However, three hr-PKS systems are known that can produce more than one programmed product.In the first case a bacterial hr-PKS-NRPS enzyme (IkaA) is known to be involved in the biosynthesis of the polycyclic tetramate macrolactam (PTM) ikarugamycin via tetramic acid 115 in Streptomyces sp. Tü6239 (Fig. 20A).67 Although bacterial in origin, IkaA shows high homology to fungal hr-PKS-NRPS (e.g. IkaA is 34% identical to LNKS over 80% of its length) making it possible that these proteins share a common ancestor. IkaA has been expressed in E. coli where it produces a non-methylated polyunsaturated β-keto hexaketide 113. This appears to be connected to the ε-amine of L-ornithine. Then the PKS synthesises a different hexaketide 114 that has not been reduced and dehydrated after the final extension and this β-keto chain is linked to the α-amine in the usual way, followed by tetramic acid formation. In vitro assay of a closely related system that makes the PTM known as HSAF from the same precursor 115 showed that the fully processed hexaketide 113 is probably made first, attached to the more sterically available ε-amine of L-ornithine, before the β-keto hexaketide 114 is synthesised and attached to the α-amine. Such selectivity could arise if the C-domain of the NRPS has some substrate preference for β-keto thiolesters as observed in many of the fungal hr-PKS-NRPS systems.
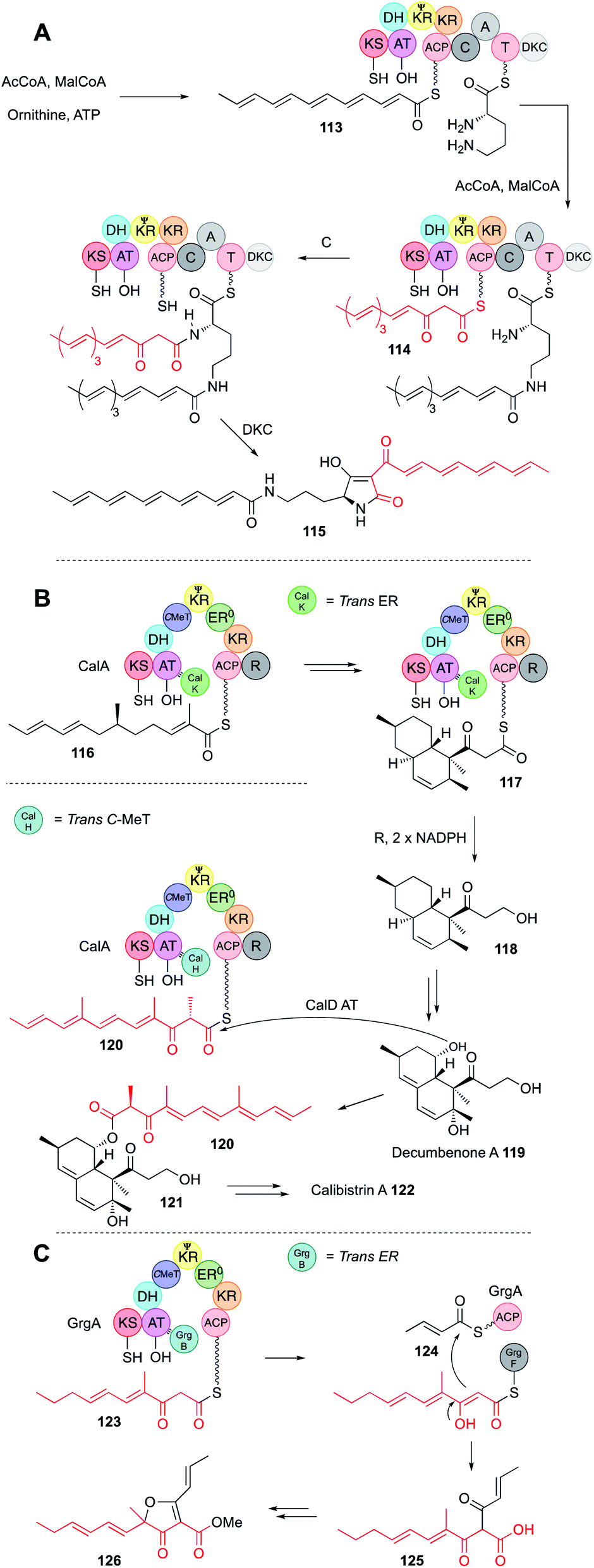 | ||
| Fig. 20 (A) Function of the IkaA hr-PKS-NRPS; (B) biosynthesis of calibistrin A 122; (C) biosynthesis of gregatin A 126. | ||
A second example of hr-PKS producing two different polyketides is observed during calbistrin A 122 biosynthesis.68 The hr-PKS (CalA) is highly unusual in that it works with a trans-ER (CalK) and a trans-C-MeT (CalH), despite having a fully functional C-MeT domain of its own. A further unusual feature is a reductive release domain (R) located downstream of the ACP. Here it appears that CalA + CalK produces a typical dimethylated hexaketide 116 that can undergo Diels–Alder cyclisation to form a decalin while attached to the ACP.
The decalin is extended one time to the β-keto species 117 and then reductively released to give 118. The decalin is then tailored to decumbenone A 119. In parallel, CalA + CalH produces a trimethylated β-keto hexaketide 120 that is transferred by the CalD acyl transferase to decumbenone 119 to form intermediate 121. Presumably, binding of CalH displaces CalK, possibly at the same AT binding site that CalK occupies, as the resultant polyketide is unreduced. Remarkably, the methylation pattern of the second polyketide 120 is altered, and it appears that CalH methylates after the final extension reaction, similar to the trans-C-MeT involved in pseurotin 11 biosynthesis (Section 3.6). Since hr-PKS are dimeric, an intriguing possibility is that CalK binds to one monomer while CalH binds to the other. However, further mechanistic and selectivity questions remain to be answered in this fascinating case.
Finally, the biosynthesis of gregatin A 126 also involves the production of two different chains by a single hr-PKS (Fig. 20C).69 Here, GrgA + the trans-ER GrgB make a β-keto monomethylated pentaketide 123. GrgA alone makes an unmethylated unsaturated diketide 124, and the α/β hydrolase GrgF links and releases them as 125 by an intriguing Claisen reaction.
5 Overall view
Programming in hr-PKS appears to be an emergent property, arising because of the interplay of both intrinsic and extrinsic selectivities of the component catalytic domains of the synthase towards the growing polyketide. Intrinsic properties arise because of the inherent selectivities of domains for particular substrates at two levels. First, domains can only enact their fundamental chemistry – e.g. DH domains cannot catalyse chemical reactions of ketones, although a ketone might reside in a DH active site. Second, catalytic domains possess substrate selectivities that can distinguish different chain-lengths and functionalisation patterns (e.g. the trans-ER of TenS) and stereoselectivities that can vary for different substrates (e.g. the KR of Hpm8). The level of selectivity can be very narrow, effectively allowing only a single substrate to react (e.g. the C-MeT domain of LNKS), or very broad, allowing even unnatural substrates to react effectively (e.g. the ER of SQTKS). In the case of some domains the selectivity can be ‘gating’. For example some DH appear not to react with 3S-alcohols, leaving the alcohol intact (e.g. in RAL systems). Other DH, however, can respond by catalysing normal 2-pro-S-syn-elimination of 3S-alcohols to give a Z-olefin (e.g. Section 3.3).48 Such features are observable, but not yet predictable.A key feature of the model is that at certain points during the extension and β-processing cycle different catalytic domains must compete for a single ACP-bound substrate. A good example of this is during the biosynthesis of the pretenellin A 14 polyketide. After the first chain extension to give the first diketide 127 three fates await the ACP-bound substrate. Off-loading to the NRPS would give 128 and ultimately a diketide tetramic acid such as 13 observed during the biosynthesis of cyclopiazonic acid (Fig. 21). C-MeT and KR also compete for 127. In the case of the hr-PKS involved in pretenellin A 14, pre-DMB A 95 and militarinone C 96 (blue, red and green respectively in Fig. 21), the methyltransferase is fastest and 127 is formed in preference to 128 or 130. Methylketone 127 can still be reduced, and DH and ER seem unselective so that diketide 129 is formed, and this is extended again to β-keto triketide 132.
 | ||
| Fig. 21 Competition for ACP-bound intermediates during hr-PKS function. Colour codes same as Fig. 15. | ||
Once again, competition is possible. The triketide 132 could be a substrate for: off-loading (i.e. chain termination); the KS (i.e. further chain-growth); C-methylation; or keto-reduction.
Here, C-MeT out-competes the other possibilities for TenS and MilS to give 133, but for the closely related DmbS system, KR beats C-MeT, giving a singly methylated triketide 134. KR and DH then eliminate to an α,β-unsaturated triketide. The triketide is then extentend to tetraketide 135 and competition between catalytic fates is again possible. For MilS the C-MeT is fastest giving 136, but for TenS and DmbS KR is fastest giving 137.
The difference in methylation selectivity is unlikely to be intrinsic to the C-MeT domains because extensive modelling suggests their active sites must be almost identical: differences in the KR affect the competition. Thus, the apparent methylation programming is an extrinsic property arising from competition between KR and C-MeT.
Direct KR/C-MeT competition has been nicely demonstrated in vitro for the LNKS (Section 3.2). In the case of TenS, the KR can control chain-length because when it fails to act, the only remaining possibility is off-loading. Once again such processes have been observed, but it is not yet possible to predict in any given case.
This also leads to the conclusion that off-loading is often (but not always) the slowest step of the overall construction cycle. It seems likely that intermediates that pause on the ACP, or wait here for extended periods can be released by the appropriate off-loading mechanism. However, several cases have been demonstrated where off-loading does impact the programme of the hr-PKS. Again, such processes are not yet predictable.
The question of chain-termination is an interesting one, i.e. when is a polyketide complete? In other iterative PKS such as the bacterial Type II PKS it is clear that KsαKSβ70 controls chain-length (rather than the number of cycles),71 but there is no evidence yet that the KS domains of hr-PKS have any role in chain-length determination. In the pretenellin A 14 system (TenS) the failure of the KR to react clearly signals the end of construction and off-loading, which seems to have little if any control in this case, must be faster than extension to a β,δ-diketone. In other cases, such as SQTKS (Section 3.5) it seems that failure of the cis-ER at the tetraketide stage triggers chain termination. In vitro results show that the final enoyl tetraketide 53 cannot be reduced, but it is unclear exactly why it cannot be further extended by the KS.
It is also interesting to consider the response of hr-PKS to ‘errors’ in construction. When domains compete for ACP-bound substrates there is a possibility for ‘incorrect’ steps to occur. This can also be simulated in vitro by lack of appropriate cofactors for example. Similarly, hr-PKS are known to lose their programming fidelity when the trans-ER is missing – loss of binding by this protein presumably changes other protein–protein interactions within the hr-PKS. In many cases, and in the case of vFAS itself, it appears that β-processing stalls when chain-building runs awry (e.g. lack of cofactors), but the KS can slowly extend β-keto substrates to β,δ-diketo thiolesters that can self-release by pyrone formation. This is an effective mechanism to unblock the hr-PKS/FAS system. However, lack of programming fidelity could also be regarded as an advantage for iterative PKS. The possibility to synthesise several related compounds gives these systems the ability to rapidly evolve,72,73 or to form hyper-efficient synthases for the synthesis of complex metabolites such as calibistrin A 122 (Fig. 20B). It may not be coincidental that hr-PKS systems are predominantly found in eukaryotes where the use of monocistronic operons allows expression levels of individual synthase components to be independently controlled.71
6 Perspective and outstanding questions
The high similarity in sequence, domain organisation, and overall structure between vFAS and fungal hr-PKS emphasises the very close relationships between these systems. Details of the stereoselectivity of the individual steps of fungal hr-PKS shows them to be identical in all respects to those known for vFAS, further emphasising this point. It is tempting to assume that hr-PKS are more highly evolved, and more programmed, versions of vFAS. The vFAS itself displays very little programming: all domains are active in all cycles and the only programmed event, the release of a particular chain-length (C16 or C18) by hydrolysis, is determined by the off-loading TE that is usually absent from the hr-PKS. Another view is that vFAS are deprogrammed versions of hr-PKS. This view is supported by the fact that vFAS possess a fossilised and inactive C-MeT domain, presumably derived from some ancestral functional precursor more like the hr-PKS.Despite significant advances in the structural biology of hr-PKS and vFAS, the molecular determinants of intrinsic, and especially extrinsic, selectivities remain very difficult to determine and near impossible to predict. Outstanding questions remaining to be resolved include, inter alia: the mechanism of starter unit selection; the stereochemistry of C-methylation and whether KR domains can racemise α-methylated positions; the role of protein–protein interactions with the ACP; how skipped patterns of functionalisation are controlled; what terminates chain growth.
Bioinformatic methodology offers some predictive ability. There is now enough knowledge of the links between particular compounds and their hr-PKS, and especially the biosynthetic gene cluster (BGC) within which the PKS is encoded, to be able to predict the general class of compound produced from an hr-PKS sequence. For example a recent detailed survey of hr-PKS-NRPS systems has revealed interesting and useful relationships in this class of fungal metabolite.74 Never-the-less, prediction of: starter unit selection; chain-length; methylation pattern; hydroxylation pattern and stereochemistry; and olefin presence and geometry for any given hr-PKS seems unachieveable at present.
Despite these difficulties, hr-PKS offer a potentially highly programmable platform for the production of bioactive secondary metabolites that is extremely efficient in comparison to bacterial modular systems. In some cases hr-PKS have been reprogrammed (at least semi-rationally) to produce new compounds, but the emerging understanding that the entire programme of hr-PKS is an emergent property of the interactions and competition of individual catalytic domains that possess highly diverse (and at present highly unpredictable) selectivities means that rational efforts to engineer these systems will remain extremely difficult. However, other approaches, such as machine learning and directed evolution, hold out much more promise for the eventual widespread use of hr-PKS components in biotechnology.
7 Conflicts of interest
There are no conflicts to declare.8 Notes and references
- J. C. Navarro-Muñoz and J. Collemare, Front. Microbiol., 2020, 10, 3018 CrossRef PubMed
.
- E. Thynne, O. L. Mead, Y.-H. Chooi, M. C. McDonald and P. S. Solomon, Genome Biol. Evol., 2019, 11, evz037 CrossRef PubMed
- J. Feng, M. Hauser, R. J. Cox and E. Skellam, J. Fungi, 2021, 7, 1085 CrossRef CAS PubMed
- L. E. H. Bingle, T. J. Simpson and C. M. Lazarus, Fungal Genet. Biol., 1999, 26, 209–223 CrossRef CAS PubMed
- R. J. Cox and E. J. Skellam, Compr. Nat. Prod. III, 2020, 1, 266–312 CAS
- J. Kennedy, K. Auclair, S. G. Kendrew, C. Park, J. C. Vederas and C. R. Hutchinson, Science, 1999, 284, 1368–1372 CrossRef CAS PubMed
- Y. Morishita, H. Zhang, T. Taniguchi, K. Mori and T. Asai, Org. Lett., 2019, 21, 4788–4792 CrossRef CAS PubMed
- G. Yang, M. S. Rose, B. G. Turgeon and O. C. Yoder, Plant Cell, 1996, 8, 2139–2150 CAS
- A. M. Harned and K. A. Volp, Nat. Prod. Rep., 2011, 28, 1790–1810 RSC
- X. Wang, C. Wang, L. Duan, L. Zhang, H. Liu, Y.-M. Xu, Q. Liu, T. Mao, W. Zhang, M. Chen, M. Lin, L. A. Gunatilaka, Y. Xu and I. Molnar, J. Am. Chem. Soc., 2019, 141(10), 4355–4364 CrossRef CAS PubMed
- A. J. Szwalbe, K. Williams, D. E. O'Flynn, A. M. Bailey, N. P. Mulholland, J. L. Vincent, C. L. Willis, R. J. Cox and T. J. Simpson, Chem. Commun., 2015, 51, 17088–17091 RSC
- B. Bonsch, V. Belt, C. Bartel, N. Duensing, M. Koziol, C. Lazarus, A. Bailey, T. Simpson and R. Cox, Chem. Commun., 2016, 52, 6777–6780 RSC
- R. Nofiani, K. de Mattos-Shipley, K. E. Lebe, L.-C. Han, Z. Iqbal, A. M. Bailey, C. L. Willis, T. J. Simpson and R. J. Cox, Nat. Commun., 2018, 9, 3940 CrossRef PubMed
- L. Hang, M. Tang, C. J. Harvey, C. G. Page, J. Li, Y. Hung, N. Liu, M. E. Hillenmeyer and Y. Tang, Angew. Chem., Int. Ed., 2017, 56, 9556–9560 CrossRef CAS PubMed
- H. Zhang, V. Hantke, P. Bruhnke, E. J. Skellam and R. J. Cox, Chem.–Eur. J., 2021, 27, 3106–3113 CrossRef CAS PubMed
- L. M. Halo, J. W. Marshall, A. A. Yakasai, Z. Song, C. P. Butts, M. P. Crump, M. Heneghan, A. M. Bailey, T. J. Simpson, C. M. Lazarus and R. J. Cox, ChemBioChem, 2008, 9, 585–594 CrossRef CAS
- T. Shimizu, S. Hiranuma, T. Watanabe and M. Kirihara, Heterocycles, 1994, 38, 243 CrossRef CAS
- A. Kaneko, Y. Morishita, K. Tsukada, T. Taniguchi and T. Asai, Org. Biomol.
Chem., 2019, 17, 5239–5243 RSC
- K. E. Lebe and R. J. Cox, Chem. Sci., 2019, 10, 1227–1231 RSC
- A. O. Zabala, Y.-H. Chooi, M. Choi, H.-C. Lin and Y. Tang, ACS Chem. Biol., 2014, 9, 1576–1586 CrossRef CAS PubMed
- E. Kuhnert, J. C. Navarro-Muñoz, K. Becker, M. Stadler, J. Collemare and R. J. Cox, Stud. Mycol., 2021, 99, 100118 CrossRef CAS PubMed
- Z. Chang, N. Sitachitta, J. V. Rossi, M. A. Roberts, P. M. Flatt, J. Jia, D. H. Sherman and W. H. Gerwick, J. Nat. Prod., 2004, 67, 1356–1367 CrossRef CAS PubMed
- D. W. Udwary, M. Merski and C. A. Townsend, J. Mol. Biol., 2002, 323, 585–598 CrossRef CAS PubMed
- T. Maier, M. Leibundgut and N. Ban, Science, 2008, 321, 1315–1322 CrossRef CAS PubMed
- D. A. Herbst, C. A. Townsend and T. Maier, Nat. Prod. Rep., 2018, 35, 1046–1069 RSC
- D. A. Herbst, R. P. Jakob, F. Zähringer and T. Maier, Nature, 2016, 531, 533 CrossRef CAS PubMed
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Nature, 2021, 596, 583–589 CrossRef CAS PubMed
- J. M. Crawford, B. C. Dancy, E. A. Hill, D. W. Udwary and C. A. Townsend, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 16728–16733 CrossRef CAS PubMed
- Y. Zou, W. Xu, Y. Tsunematsu, M. Tang, K. Watanabe and Y. Tang, Org. Lett., 2014, 16, 6390–6393 CrossRef CAS PubMed
- S. M. Ma, J. W. Li, J. W. Choi, H. Zhou, K. K. Lee, V. A. Moorthie, X. Xie, J. T. Kealey, N. A. D. Silva, J. C. Vederas and Y. Tang, Science, 2009, 326, 589–592 CrossRef CAS PubMed
- R. A. Cacho, J. Thuss, W. Xu, R. Sanichar, Z. Gao, A. Nguyen, J. C. Vederas and Y. Tang, J. Am. Chem. Soc., 2015, 137, 15688–15691 CrossRef CAS PubMed
- H. Zhou, Z. Gao, K. Qiao, J. Wang, J. C. Vederas and Y. Tang, Nat. Chem. Biol., 2012, 8, 331–333 CrossRef CAS PubMed
- J. Zheng and A. T. Keatinge-Clay, MedChemComm, 2012, 4, 34–40 RSC
- J. Takino, A. Kotani, T. Ozaki, W. Peng, J. Yu, Y. Guo, S. Mochizuki, K. Akimitsu, M. Hashimoto, T. Ye, A. Minami and H. Oikawa, Angew. Chem., Int. Ed., 2021, 60, 23403–23411 CrossRef CAS PubMed
- Y. Morishita, T. Sonohara, T. Taniguchi, K. Adachi, M. Fujita and T. Asai, Org. Biomol. Chem., 2020, 18, 2813–2816 RSC
- E. Liddle, A. Scott, L.-C. Han, D. Ivison, T. J. Simpson, C. L. Willis and R. J. Cox, Chem. Commun., 2017, 53, 1727–1730 RSC
- L. Wang, M. Yuan and J. Zheng, Synth. Syst. Biotechnol., 2018, 4, 10–15 CrossRef PubMed
- D. M. Roberts, C. Bartel, A. Scott, D. Ivison, T. J. Simpson and R. J. Cox, Chem. Sci., 2016, 8, 1116–1126 RSC
- O. Piech and R. J. Cox, RSC Adv., 2020, 10, 18469–18476 RSC
- J. Wang, J. Liang, L. Chen, W. Zhang, L. Kong, C. Peng, C. Su, Y. Tang, Z. Deng and Z. Wang, Nat. Commun., 2021, 12, 867 CrossRef CAS PubMed
- Z. Song, R. J. Cox, C. M. Lazarus and T. J. Simpson, ChemBioChem, 2004, 5, 1196–1203 CrossRef CAS PubMed
- B. D. Ames, C. Nguyen, J. Bruegger, P. Smith, W. Xu, S. Ma, E. Wong, S. Wong, X. Xie, J. W. Li, J. C. Vederas, Y. Tang and S.-C. C. Tsai, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11144–11149 CrossRef CAS PubMed
- C. Lin and S. Smith, J. Biol. Chem., 1978, 253, 1954–1962 CrossRef CAS PubMed
- R. Gerber, L. Lou and L. Du, J. Am. Chem. Soc., 2009, 131, 3148–3149 CrossRef CAS PubMed
- W. Xu, Y.-H. H. Chooi, J. W. Choi, S. Li, J. C. Vederas, N. A. D. Silva and Y. Tang, Angew. Chem., Int. Ed., 2013, 52, 6472–6475 CrossRef CAS PubMed
- A. O. Zabala, Y.-H. Chooi, M. Choi, H.-C. Lin and Y. Tang, ACS Chem. Biol., 2014, 9, 1576–1586 CrossRef CAS PubMed
- D.-W. Gao, C. S. Jamieson, G. Wang, Y. Yan, J. Zhou, K. N. Houk and Y. Tang, J. Am. Chem. Soc., 2021, 143, 80–84 CrossRef CAS PubMed
- M.-C. Tang, C. R. Fischer, J. V. Chari, D. Tan, S. Suresh, A. Chu, M. Miranda, J. Smith, Z. Zhang, N. K. Garg, R. P. S. Onge and Y. Tang, J. Am. Chem. Soc., 2019, 141, 8198–8206 CrossRef CAS PubMed
- M. J. Meehan, X. Xie, X. Zhao, W. Xu, Y. Tang and P. C. Dorrestein, Biochemistry, 2011, 50, 287–299 CrossRef CAS PubMed
- X. Xie, K. Watanabe, W. A. Wojcicki, C. C. C. Wang and Y. Tang, Chem. Biol., 2006, 13, 1161–1169 CrossRef CAS PubMed
- W. Xu, X. Cai, M. E. Jung and Y. Tang, J. Am. Chem. Soc., 2010, 132, 13604–13607 CrossRef CAS PubMed
- X. Liu and C. T. Walsh, Biochemistry, 2009, 48, 8746–8757 CrossRef CAS PubMed
-
H. Yao, In Vitro Investigation of Multi-domain Fragments of Squalestatin Tetraketide Synthase, Ph.D. thesis, University of Hannover, 2019
-
O. Piech, Computational and In Vitro Study of Isolated Domains from Fungal Polyketide Synthases, Ph.D. thesis, University of Hannover, 2020
- K. M. Fisch, W. Bakeer, A. A. Yakasai, Z. Song, J. Pedrick, Z. Wasil, A. M. Bailey, C. M. Lazarus, T. J. Simpson and R. J. Cox, J. Am. Chem. Soc., 2011, 133, 16635–16641 CrossRef CAS PubMed
- C. Bisang, P. F. Long, J. Corte's, J. Westcott, J. Crosby, A.-L. Matharu, R. J. Cox, T. J. Simpson, J. Staunton and P. F. Leadlay, Nature, 1999, 401, 502 CrossRef CAS PubMed
- X.-L. Yang, S. Friedrich, S. Yin, O. Piech, K. Williams, T. J. Simpson and R. J. Cox, Chem. Sci., 2019, 10, 8478–8489 RSC
- M. N. Heneghan, A. A. Yakasai, K. Williams, K. A. Kadir, Z. Wasil, W. Bakeer, K. M. Fisch, A. M. Bailey, T. J. Simpson, R. J. Cox and C. M. Lazarus, Chem. Sci., 2011, 2, 972–979 RSC
- Z. Song, W. Bakeer, J. W. Marshall, A. A. Yakasai, R. Khalid, J. Collemare, E. Skellam, D. Tharreau, M.-H. Lebrun, C. M. Lazarus, A. M. Bailey, T. J. Simpson and R. J. Cox, Chem. Sci., 2015, 6, 4837–4845 RSC
- M. L. Nielsen, T. Isbrandt, L. M. Petersen, U. H. Mortensen, M. R. Andersen, J. B. Hoof and T. O. Larsen, PLoS One, 2016, 11, e0161199 CrossRef PubMed
- E. Skellam, Nat. Prod. Rep., 2022, 39, 754–783 RSC
- A. al Fahad, A. Abood, K. M. Fisch, A. Osipow, J. Davison, M. Avramović, C. P. Butts, J. Piel, T. J. Simpson and R. J. Cox, Chem. Sci., 2013, 5, 523–527 RSC
- Y.-M. Chiang, E. Szewczyk, A. D. Davidson, N. Keller, B. R. Oakley and C. C. C. Wang, J. Am. Chem. Soc., 2009, 131, 2965–2970 CrossRef CAS PubMed
- C. Wang, X. Wang, L. Zhang, Q. Yue, Q. Liu, Y. Xu, A. A. L. Gunatilaka, X. Wei, Y. Xu and I. Molnár, J. Am. Chem. Soc., 2020, 142, 17093–17104 CrossRef CAS PubMed
- J. Bai, Y. Lu, Y. Xu, W. Zhang, M. Chen, M. Lin, L. A. Gunatilaka, Y. Xu and I. Molnár, Org. Lett., 2016, 18, 1262–1265 CrossRef CAS PubMed
- Y. Xu, P. Espinosa-Artiles, V. Schubert, Y. Xu, W. Zhang, M. Lin, L. A. Gunatilaka, R. Süssmuth and I. Molnár, Appl. Environ. Microbiol., 2013, 79, 2038–2047 CrossRef CAS PubMed
- J. Antosch, F. Schaefers and T. A. Gulder, Angew. Chem., Int. Ed., 2014, 53, 3011–3014 CrossRef CAS PubMed
- H. Tao, T. Mori, X. Wei, Y. Matsuda and I. Abe, Angew. Chem., Int. Ed., 2021, 60, 8851–8858 CrossRef CAS PubMed
- W.-G. Wang, H. Wang, L.-Q. Du, M. Li, L. Chen, J. Yu, G.-G. Cheng, M.-T. Zhan, Q.-F. Hu, L. Zhang, M. Yao and Y. Matsuda, J. Am. Chem. Soc., 2020, 142, 8464–8472 CrossRef CAS PubMed
- R. McDaniel, S. Ebert-Khosla, D. A. Hopwood and C. Khosla, Science, 1993, 262, 1546–1550 CrossRef CAS PubMed
- T. Nicholson, C. Winfield, J. Westcott, J. Crosby, T. Simpson and R. Cox, Chem. Commun., 2003, 686–687 RSC
- A. A. Yakasai, J. Davison, Z. Wasil, L. M. Halo, C. P. Butts, C. M. Lazarus, A. M. Bailey, T. J. Simpson and R. J. Cox, J. Am. Chem. Soc., 2011, 133, 10990–10998 CrossRef CAS PubMed
- Z. Wasil, K. A. Pahirulzaman, C. Butts, T. J. Simpson, C. M. Lazarus and R. J. Cox, Chem. Sci., 2013, 4, 3845–3856 RSC
- A. Minami, T. Ugai, T. Ozaki and H. Oikawa, Sci. Rep., 2020, 10, 13556 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2023 |