
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Benz[e]indenyl and benz[f]indenyl molybdenum compounds: evidence of the η3-coordination mode†
Jiří
Štěpán
a,
Jaromír
Vinklárek
 a,
Ivana
Císařová
b,
Libor
Dostál
a and
Jan
Honzíček
*c
a,
Ivana
Císařová
b,
Libor
Dostál
a and
Jan
Honzíček
*c
aDepartment of General and Inorganic Chemistry, Faculty of Chemical Technology, University of Pardubice, Studentská 573, 532 10 Pardubice, Czech Republic
bDepartment of Inorganic Chemistry, Faculty of Science, Charles University in Prague, Hlavova 2030/8, 128 43 Prague 2, Czech Republic
cInstitute of Chemistry and Technology of Macromolecular Materials, Faculty of Chemical Technology, University of Pardubice, Studentská 573, 532 10 Pardubice, Czech Republic. E-mail: jan.honzicek@upce.cz
First published on 30th October 2023
Abstract
This study reports the synthesis and characterization of new molybdenum compounds bearing benz[e]indenyl and benz[f]indenyl ligands. The capability of the ligands to undergo η5-to-η3 haptotropic rearrangement was evidenced on reactions with tris(pyrazolyl)methane. Appearance of the η3-coordination mode was determined by NMR spectroscopy and X-ray structure analysis on a single crystal.
Introduction
Cyclopentadienyl compounds of transition metals have attracted considerable attention since ferrocene, (η5-Cp)2Fe (Cp = C5H5), was discovered in the early 1950s.1 Variations in the cyclopentadienyl ligand periphery often result in dramatic changes in the physical, chemical, and biological properties of the corresponding compounds.2 These changes can be attributed to the electronic and steric effects caused by the partial or full replacement of the hydrogen atoms by other groups. The extraordinary diversity of cyclopentadienyl compounds has been documented on many bonding modes described for the Cp ligand (Scheme 1).3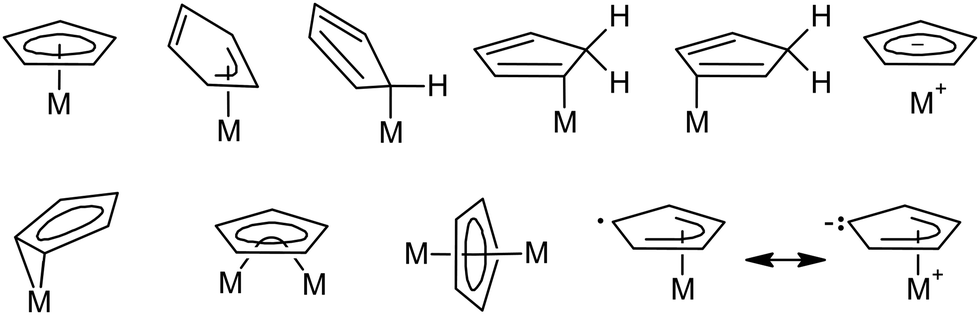 | ||
| Scheme 1 The most pronounced bonding modes of Cp. | ||
The ability of the cyclopentadienyl ligand to adjust its hapticity, i.e., the number of contiguous carbon atoms involved in bonding to the central metal, gives rise to processes known as “haptotropic shifts”. These rearrangements have been investigated on Cp and few congeners with annulated benzene rings (Cp′), mainly on indenyl (Ind = C9H7)4–12 and fluorenyl (Flu = C13H9).12–14 Electronically saturated transition metal complexes with η5-bonded extended aromatic systems exhibit a substantial increase in the reaction rates of substitution reactions, when compared to their η5-Cp counterpart that has given rise to the term “indenyl effect”.15–18 It is usually interpreted in terms of the M–Cp′ bond strength in the two relevant hapticity modes. For instance, the η5-bond is stronger for the cyclopentadienyl ligand providing ground-state stabilization for complexes with η5-Cp, when compared to their indenyl and fluorenyl analogues. In contrast, the η3- and η1-bonds are stronger for ligands with annulated benzene ring stabilizing the corresponding η3- and η1-intermediates and transition states that result in increased rates of substitution reactions19,20 and activation of coordinated ligands.21–23
Although haptotropic shifts of cyclopentadienyl, indenyl and fluorenyl ligands have been deeply investigated, revealing various distinct coordination modes, rearrangements of congeners with larger conjugated π-systems stay almost unexplored even though they are of particular importance due to the consequent implications for the design of catalytic systems.18,24 To the best of our knowledge, only η1- and η5-bonds are reported for transition metal compounds of indenyl and fluorenyl linearly or angularly annulated with benzene rings.25,26
The first occurrence of transition metal complexes bearing indenyl/fluorenyl ligands with extended conjugated π-systems appears in the literature in 1992,27 but the systematic work in this area has been started by Brintzinger et al. a few years later28 within development of metallocene-based catalysts for olefin polymerization.29,30 Later contributions have brought novel transition metal-based catalysts for polymer tailoring31,32 and ruthenium compounds suitable for catalytic racemization of secondary alcohols.33
Although the assembly of the first transition metal compounds bearing benzannulated indenyl and fluorenyl ligands started more than three decades ago, a deeper insight into their physical and chemical properties is dated much later. In 2006, Beckhaus et al. have pointed out a large helical twist of the tetrabenzo[a,c,g,i]fluorenyl ligand in titanium complexes and their increased stability when compared with fluorenyl analogues.34 A similar behavior was observed for yttrium complexes.35 Contributions of Thiel et al. on various dibenzo[c,g]fluorenyl compounds have proven a partial loss of aromaticity in the angularly annulated fluorenyl ligands that finally led to revision of the concept of aromaticity in metallocene compounds with extended π-systems.36–39
The aim of this study is to synthesize new benz[e]indenyl (e-Bind) and benz[f]indenyl (f-Bind) compounds capable of η5–η3 rearrangement. The attention will be given to 18e molybdenum(II) compounds which adjust the hapticity of the π-ligand upon coordination of an additional 2e donor.
Results and discussion
Benz[e]indene (1) and benz[f]indene (2) were prepared according literature procedures starting from benz[e]indanone28 and benz[f]indanone,40,41 respectively (for details see the ESI†). Labeling of 1 with deuterium in 1- and 3-positions was done following the standard H/D exchange protocol using the repeated lithiation/deuterolysis protocol (Scheme 2).8,42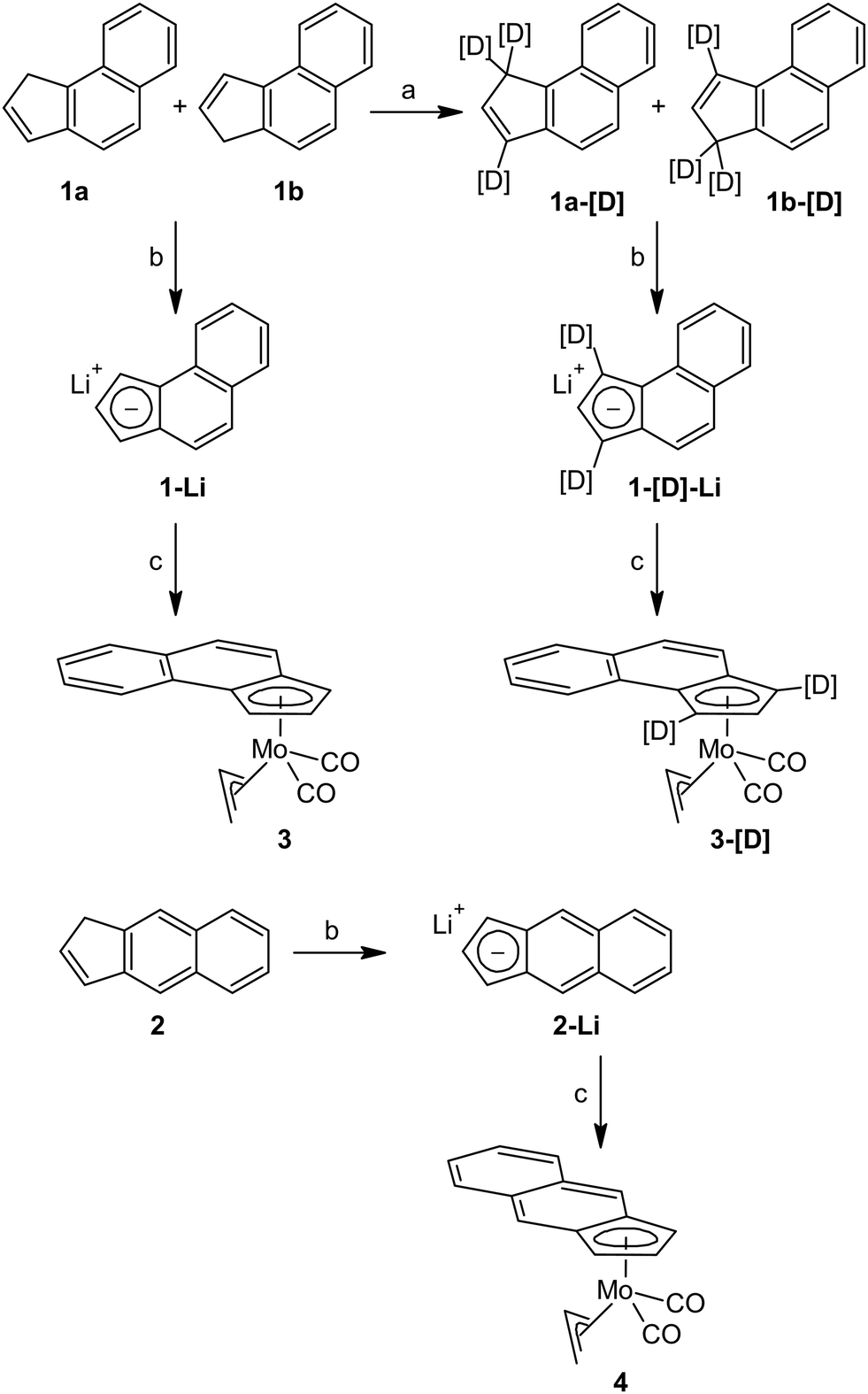 | ||
| Scheme 2 Synthesis of allyl molybdenum compounds. Reagents: (a) n-BuLi/THF, D2O, work-up (repeated three times); (b) n-BuLi/THF; and (c) [(η3-C3H5)Mo(CO)2(NCMe)2Cl]. | ||
Lithiated compounds 1-Li and 2-Li, accessible by deprotonation with n-butyllithium, were isolated and characterized by multinuclear NMR spectroscopy. The patterns of 1H and 13C APT NMR spectra verified aromatization of the five membered ring.
The values of 7Li chemical shifts, measured in thf-d8, are considerably lower than those reported for CpLi, IndLi and FluLi (Table 1).43 This is attributed to the higher electron density on the lithium atom probably caused by the higher covalency of metal–carbon bonds or the difference in the coordination sphere of the lithium atom.
Lithiated benz[e]indene (1-Li) reacts with the chloride complex [(η3-C3H5)Mo(CO)2(NCMe)2Cl] to give the benz[e]indenyl complex [(η3-C3H5)(η5-e-Bind)Mo(CO)2] (3) in medium yield; see Scheme 2. The isotopically labeled derivative [(η3-C3H5)(η5-e-1,3-[D]2-Bind)Mo(CO)2] (3-[D]) and benz[f]indenyl analogue [(η3-C3H5)(η5-f-Bind)Mo(CO)2] (4) were prepared accordingly starting from the compounds 1-[D] and 2, respectively.
Infrared spectra of the compounds 3 and 4 show CO stretching bands in the range typical for terminal carbonyl ligands. In the case of 3, the antisymmetric vibration mode is split on doublet due to the effects of the solid state.
1H NMR spectra of the compounds 3 and 4 show formation of two conformers arising from a different orientation of the η3-bonded allyl ligand. Signals of the allyl ligand were assigned to the exo-isomer (allyl ligand eclipsed with OC–Mo–CO) and the endo-isomer according to the data published for the indenyl analogue.44 At room temperature, the molar ratios (exo/endo) in 3 and 4 were found to be 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 2:1, respectively. This reveals a higher thermodynamic stability of the exo-isomer, which is in line with the data reported for [(η3-C3H5)(η5-Ind)Mo(CO)2]44 and its ring-substituted derivatives.45–47
:1 and 2:1, respectively. This reveals a higher thermodynamic stability of the exo-isomer, which is in line with the data reported for [(η3-C3H5)(η5-Ind)Mo(CO)2]44 and its ring-substituted derivatives.45–47
Solid-state structures of the compounds 3 and 4 were determined by single-crystal X-ray diffraction analysis. Both molecules have a distorted tetrahedral structure with allyl, e-Bind/f-Bind and two carbonyl ligands around molybdenum in the formal oxidation state II (Fig. 1 and 2). Geometric parameters describing the coordination sphere of molybdenum are listed in Table 2. The η3-coordinated allyl ligand is positionally disordered and was split into exo and endo conformations with occupancies 85:15 and 80:20 for 3 and 4, respectively. The e-Bind as well as f-Bind are η5-coordinated as evidenced by the small envelope fold angle [3: Ω = 2.9(3)°; 4: Ω = 5.53(18)°] and low Δ(M–C) parameter [3:0.089(3) Å; 4:0.1219(17) Å] nearing the values calculated for the indenyl analogue [(η3-C3H5)(η5-Ind)Mo(CO)2] from the X-ray data [Ω = 3.3(3)°, Δ(M–C) = 0.115(3) Å].48
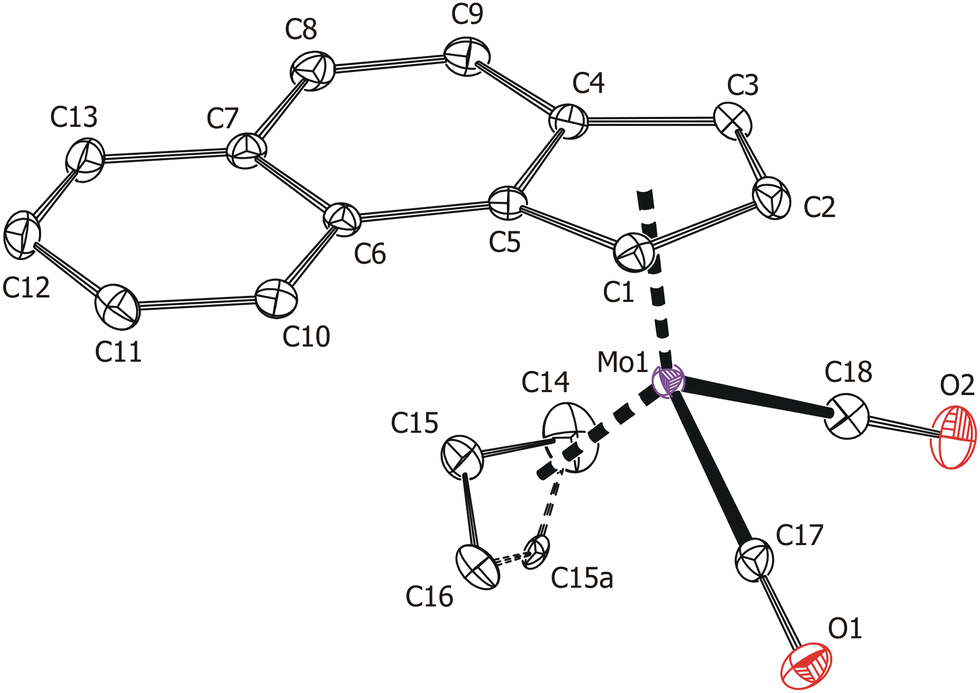 | ||
| Fig. 1 ORTEP drawing of the molecule [(η3-C3H5)(η5-e-Bind)Mo(CO)2] present in the crystal structure of 3. Thermal ellipsoids are drawn at the 30% probability level. Hydrogen atoms are omitted for clarity. | ||
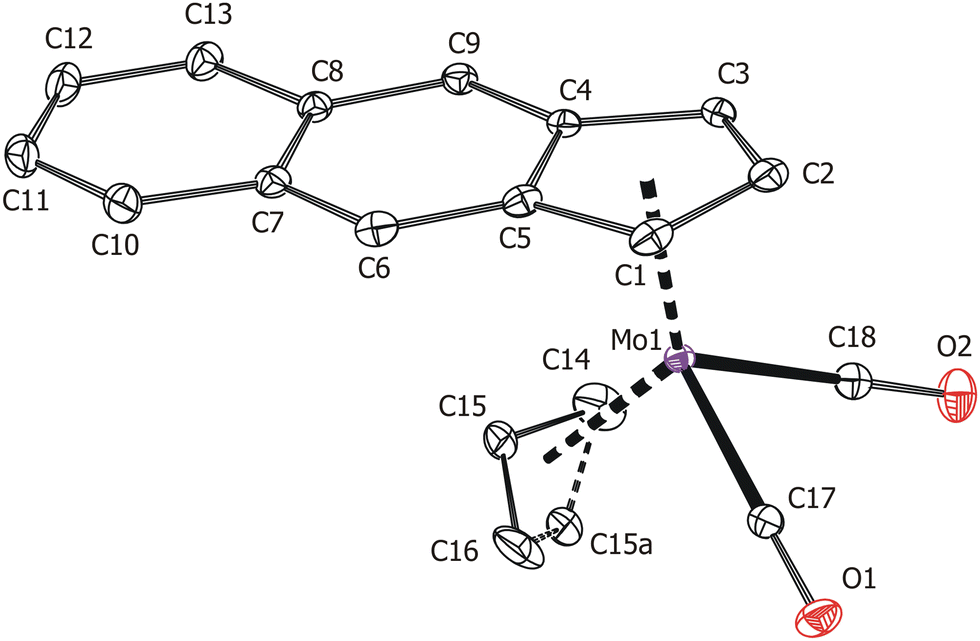 | ||
| Fig. 2 ORTEP drawing of the molecule [(η3-C3H5)(η5-f-Bind)Mo(CO)2] present in the crystal structure of 4. Thermal ellipsoids are drawn at the 30% probability level. Hydrogen atoms are omitted for clarity. | ||
| 3 | 4 | 10 | |
|---|---|---|---|
| a Cg is the center of gravity (C1–C5 for 3 and 4; C1–C3 for 10). b Envelope fold angle. c Difference in metal–carbon bonds defined as the difference between the averages of the metal–carbon distances Mo–C1, Mo–C2, and Mo–C3 and those of Mo–C4 and Mo–C5. | |||
| Mo–C(CO) | 1.939(3) | 1.942(2) | 1.958(3) |
| 1.955(3) | 1.947(2) | 1.961(3) | |
| Mo–Cga | 2.0269(11) | 2.0355(8) | 2.107(3) |
| Mo–N1 | — | — | 2.201(2) |
| Mo–N3 | — | — | 2.265(2) |
| Mo–N5 | — | — | 2.263(2) |
| C(CO)–Mo–C(CO) | 80.64(11) | 80.27(7) | 80.30(12) |
| Ωb | 2.9(3) | 5.53(18) | 21.6(3) |
| Δ(M–C)c | 0.089(3) | 0.1219(17) | 0.804(3) |
Our attempt to reach reactive species, which can undergo haptotropic rearrangement under mild conditions, led us to compounds of the formula [(η5-Cp′)Mo(CO)2(NCMe)2][BF4] (5: Cp′ = e-Bind; 6: Cp′ = f-Bind). They were prepared in situ by reaction of 3 and 4 with HBF4 in line with a protocol developed for the cyclopentadienyl analogues.49 Although synthetic difficulties preclude their isolation in the pure form, they appear as intermediates upon synthesis of cationic molybdenum compounds bearing e-Bind and f-Bind shown in Schemes 3 and 4.
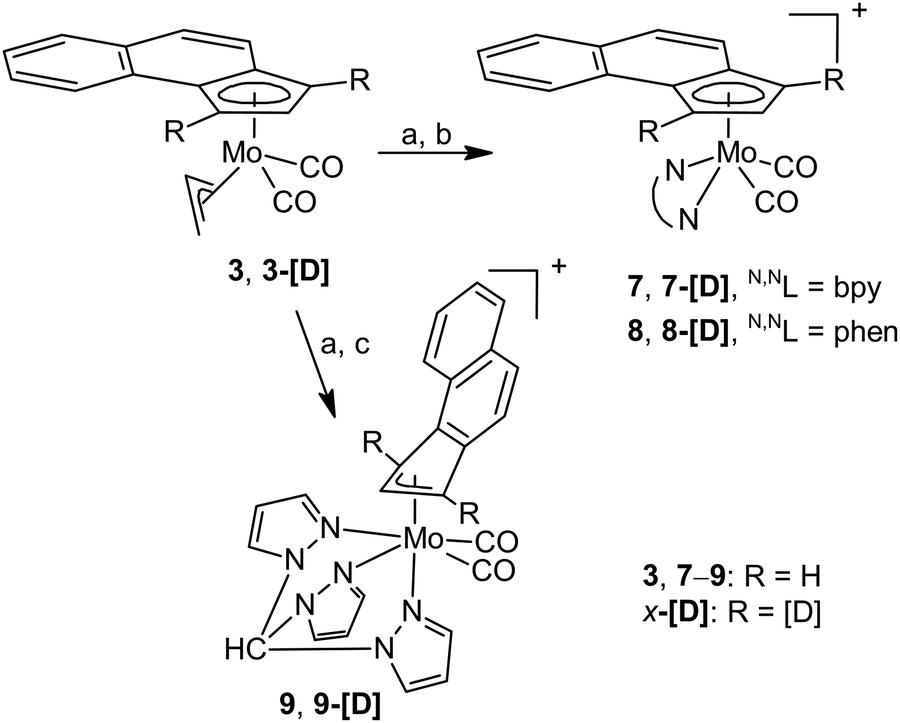 | ||
| Scheme 3 Synthesis of cationic e-Bind compounds. Reagents: (a) HBF4·Et2O/CH2Cl2, MeCN; (b) N,NL/CH2Cl2; and (c) tpm/CH2Cl2. | ||
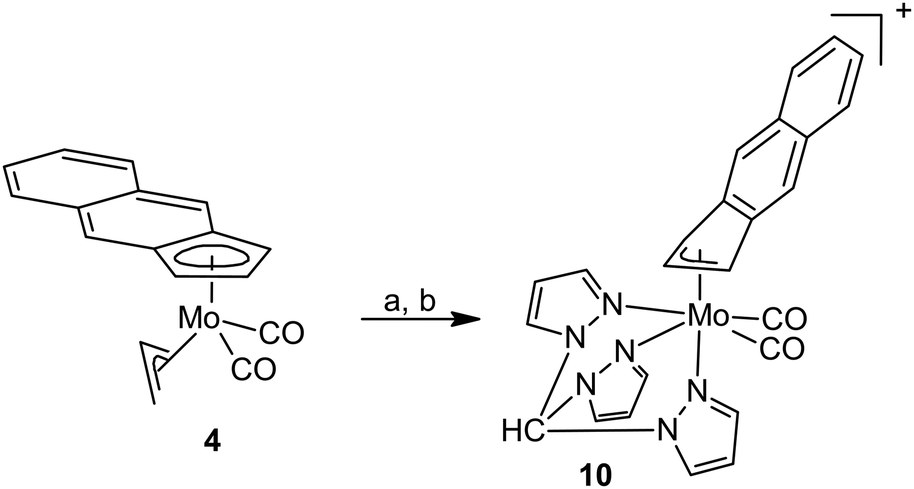 | ||
| Scheme 4 Synthesis of the compound 10. Reagents: (a) HBF4·Et2O/CH2Cl2, MeCN and (b) tmp. | ||
Stable compounds bearing η5-bonded e-Bind were prepared by reaction with bpy (7) and phen (8). 1H NMR spectra proved coordination of the bidentate ligands. They give eight signals due to lowering of the molecular symmetry. The coordination mode of the e-Bind ligand was estimated from the chemical shifts of H1/H3 of the ligand. In both compounds, the signal of H2 appears at a considerably higher (∼5.9 ppm) field than the signals of H1 (∼6.9 ppm) and H3 (7.1 ppm). Such a pattern is typical for species bearing η5-bonded ligands derived from indenyl.10,50 We note that the initial assignment, based on the assumption that 3J(H1,H2) ≈ 3J(H2,H3) > 4J(H3,H1), was verified on deuterium labeled species 7-[D] and 8-[D]. The cationic character of the compounds 7 and 8 is in line with a strong B–F stretching band observed in the infrared spectra at 1054 cm−1. Appearance of two CO stretching bands in the infrared spectra at frequencies close to cyclopentadienyl and indenyl analogues51,52 implies formation of square-pyramidal complexes which obey the 18-electron rule.
Compound 9 bearing η3-coordinated e-Bind is accessible from 3 and tris(pyrazolyl)methane (tpm) using a similar synthetic protocol as in the case of bidentate ligands. The κ3-coordination of such a tripodal tridentate ligand enables induction of η5–η3 haptotropic rearrangement of the e-Bind, which maintains 18 electrons in the valence sphere of molybdenum. Hapticity of the e-Bind ligand was estimated by 1H NMR spectroscopy. The problems with assignment of diagnostic NMR signals led us to synthesize deuterium labelled products 9-[D], which enabled distinguishing unambiguously H2 (7.90 ppm) from H1/3 (6.04 and 6.63 ppm). The appearance of H2 at a considerably lower field than H1 and H3 is typical for η3-molybdenum complexes bearing indenyl ligands.10,50 The infrared spectrum shows CO stretching modes at 1955 and 1874 cm−1.
Our attempts to synthesize cationic η5-f-Bind compounds, analogous to 7 and 8, were not successful. The suggested species were not isolated in sufficient purity probably owing to a lower stability of the intermediate 6. Nevertheless, a modified protocol enables isolation of η3-f-Bind compound 10 bearing the κ3-coordinated tpm ligand (Scheme 4).
1H and 13C APT NMR spectra of 10 show a pattern typical for Cs molecular symmetry. Full assignment of the appeared signals was done using 2D correlation techniques (1H–1H COSY, 1H–13C HSQC and 1H–13C HMBC). The η3-coordination mode of f-Bind is evident from the chemical shift of the hydrogen atom H2 and the appearance of carbon atoms C3a and C9a at a low field (145.0 ppm).
The X-ray crystallographic data proved the suggested molecular structure in the solid state (Fig. 3). The coordination sphere of the complex cation forms a distorted octahedron with one face occupied by nitrogen donor atoms of the tripodal tmp ligand. The opposite face of the distorted octahedron is occupied by η3-f-Bind and two carbonyl ligands. The coordination mode of f-Bind was elucidated from the high values of the envelope fold angle [Ω = 21.6(3)°] and Δ(M–C) parameter [0.804(3) Å] (Table 2).10 These values are close to those reported for indenyl compounds [(η3-Ind)Mo(CO)2(κ3-tpm)][BF4] [Ω = 21.9(2)°, Δ(M–C) = 0.815(2) Å] 20 and [(η3-Ind)Mo(CO)2(κ3-Tp)] [Ω = 21.4(2)°, Δ(M–C) = 0.819(3) Å] 10, implying a similarly strong indenyl effect. The Mo–N bond lengths vary in a wide range of 2.201(2)–2.265(2) Å. This is due to a strong trans effect of carbonyl ligands which weakens the bonds Mo–N3 and Mo–N5.10,20
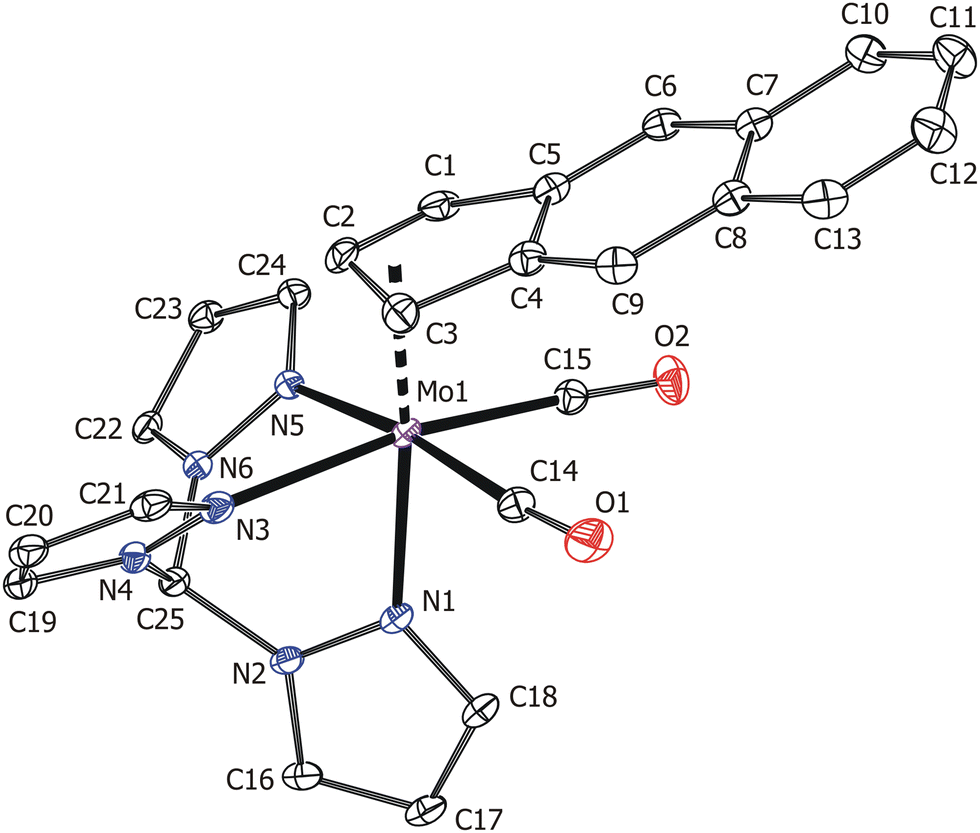 | ||
| Fig. 3 ORTEP drawing of the cation [(η3-f-Bind}Mo(CO)2(κ3-tpm)]+ present in the crystal structure of 10. Thermal ellipsoids are drawn at the 20% probability level. Hydrogen atoms are omitted for clarity. | ||
Conclusions
This study documented the appearance of stable molybdenum compounds with two distinct coordination modes (η3 and η5) for both e-Bind and f-Bind. The properties of both ligands resemble rather the indenyl ligand than cyclopentadienyl. Although compounds with η5-bonds (3, 4, 7 and 8) have their cyclopentadienyl and indenyl analogues [(η3-C3H5)(η5-Cp′)Mo(CO)2],44 [(η5-Cp′)Mo(CO)2(κ2-bpy)][BF4]51 and [(η5-Cp′)Mo(CO)2(κ2-phen)][BF4],51 the η3-species analogous to 9 and 10 was reported only for the indenyl ligand.10,20 The Cp analogue of 9 and 10 is not available due to a considerably lower tendency to undergo η5-to-η3 rearrangement.20 We note that the synthesized f-Bind compounds seem to be less stable than their e-Bind analogues, which is in line with previously reported experiments on half-sandwich titanium compounds.53Our contribution to the understanding of the coordination ability of e-Bind and f-Bind ligands could be helpful in tuning the catalytic properties of the indenyl ligand by benzannulation, as this type of modification not only increases the ligand bulkiness but also affects its electronic properties.
Experimental
Materials
Synthesis of the organometallic compounds was done under an argon atmosphere using conventional Schlenk-line techniques. The solvents were dried using standard methods.54 The reagents were purchased from commercial sources (Acros Organics) or prepared according to literature procedures: [(η3-C3H5)Mo(CO)2(NCMe)2Cl]44 and tris(pyrazolyl)methane (tpm).55 Synthetic details for benz[e]indene and benz[f]indene are given in the ESI.†Measurements
Infrared spectra were recorded on a Nicolet iS50 FTIR spectrometer in the 4000–400 cm−1 region (resolution: 1 cm−1) using a Diamond Smart Orbit ATR. The 1H, 7Li, and 13C-APT NMR spectra were recorded on Bruker Avance 400 and Bruker Avance 500 spectrometers at 300 K in thf-d8, CDCl3, acetone-d6, benzene-d6 and dmso-d6.Synthesis of benz[e]indenyllithium (1-Li)
Benz[e]indene (1; 0.33 g, 2.0 mmol) was dissolved in Et2O (12 mL), cooled at –70 °C, and treated with n-BuLi (1.6 mol L−1, 1.24 mL, 2.0 mmol) dropwise. The reaction mixture was stirred at room temperature for 2 h and volatiles were vacuum-evaporated. Yield: 0.21 g (1.2 mmol, 61%). Yellow powder. 1H NMR (500.2 MHz, thf-d8,): δ = 8.04 (d, 3J(1H,1H) = 8.0 Hz, 1H, H6/9), 7.50 (d, 3J(1H,1H) = 7.9 Hz, 1H, H6/9), 7.45 (d, 3J(1H,1H) = 8.4 Hz, 1H, H4/5), 7.08, 6.92 (2 × dd, 3J(1H,1H) = 6.3 Hz, 3J(1H,1H) = 8.0 Hz, 2H, H7,8), 6.77 (d, 3J(1H,1H) = 8.5 Hz, 1H, H4/5), 6.60 (s, 1H, H1/3), 6.46 (t, 3J(1H,1H) = 3.3 Hz, 1H, H2), 6.10 (s, 1H, H1/3). 13C APT NMR (125.8 MHz, thf-d8,): δ = 131.5, 129.6, 125.4, 123.4 (4C, C1a,3a,5a,9a), 128.0 (1C, C6/9), 123.5 (1C, C4/5), 122.8 (1C, C7/8), 122.4 (1C, C6/9), 119.3 (1C, C7/8), 113.8 (1C, C4/5), 112.9 (1C, C2), 96.7 (1C, C1/3), 94.1 (1C, C1/3). 7Li NMR (194.4 MHz, thf-d8): δ = –4.88.Synthesis of benz[f]indenyllithium (2-Li)
The reaction was performed as described for compound 1-Li but with benz[f]indene. Yield: 0.24 g (1.4 mmol, 70%). Light orange powder. 1H NMR (500.2 MHz, thf-d8,): δ = 7.69 (s, 2H, H4,9), 7.53 (dd, 3J(1H,1H) = 6.4 Hz, 4J(1H,1H) = 3.3 Hz, 2H, H5,8), 7.05 (t, 1H, 3J(1H,1H) = 3.4 Hz, H2), 6.70 (dd, 3J(1H,1H) = 6.6 Hz, 4J(1H,1H) = 3.2 Hz, 2H, H6,7), 6.06 (d, 2H, 3J(1H,1H) = 3.4 Hz, H1,3). 13C APT NMR (125.8 MHz, thf-d8,): δ = 134.3, 125.7 (2 × 2C, C1a,3a,4a,8a), 128.2 (2C, C5,8), 125.6 (1C, C2), 117.4 (2C, C6,7), 113.2 (2C, C4,9), 90.4 (2C, C1,3). 7Li NMR (194.4 MHz, thf-d8): δ = –2.35.Synthesis of [(η3-C3H5)(η5-e-Bind)Mo(CO)2] (3)
A solution of benz[e]indene (1; 1.66 g, 10.0 mmol) in THF (30 mL) was cooled to 0 °C, treated dropwise with a solution of n-BuLi in hexane (1.6 M, 6.25 mL, 10.0 mmol), and stirred for 2 h at room temperature. The mixture was cooled to –80 °C and treated dropwise with a solution of [(η3-C3H5)Mo(CO)2(NCMe)2Cl] (3.11 g, 10.0 mmol) in THF (30 mL). The mixture was stirred at room temperature overnight. The volatiles were evaporated under reduced pressure, and the crude product was extracted with hexane (5 × 70 mL) at 60 °C. The solvent was evaporated under reduced pressure. The product was recrystallized from hexane/Et2O at –80 °C and vacuum-dried. Yield: 2.82 g (7.87 mmol, 78.7%). Yellow powder. Anal. Calcd for C18H14MoO2: C, 60.35; H, 3.94. Found: C, 60.58; H, 3.77. Mp: 145–150 °C (dec). 1H NMR (400.1 MHz, CDCl3, 3:1 mixture of isomers 3a and 3b): δ = 7.78–7.68 (m, 2H of 3a and 2H of 3b, H6–9, C13H9), 7.55–7.43 (m, 2H of 3a and 2H of 3b, H6–9, C13H9), 7.34 [d, 3J(1H,1H) = 9.0 Hz, 1H of 3a, H4,5, C13H9], 7.27 [d, 3J(1H,1H) = 9.0 Hz, 1H of 3b, H4,5, C13H9], 7.02 [d, 3J(1H,1H) = 9.0 Hz, 1H of 3b, H4,5, C13H9], 6.98 [d, 3J(1H,1H) = 9.0 Hz, 1H of 3a, H4,5, C13H9], 6.36 [dd, 3J(1H,1H) = 2.8 Hz, 4J(1H,1H) = 1.5 Hz, 1H of 3a, H1,3, C13H9], 6.32 (br. s, 1H of 3b, H1,3, C13H9), 5.96 [dd, 3J(1H,1H) = 2.8 Hz, 4J(1H,1H) = 1.5 Hz, 1H of 3a, H1,3, C13H9], 5.88 (br. s, 1H of 3b, H1,3, C13H9), 5.68 [t, 3J(1H,1H) = 2.8 Hz, 1H of 3a, H2, C13H9], 5.65 (br. s, 1H of 3b, H2, C13H9), 3.27 (br. s, 2H of 3b, Hsyn, C3H5), 3.07 (br. s, 1H of 3b, Hmeso, C3H5), 2.17 [d, 3J(1H,1H) = 7.5 Hz, 1H of 3a, Hsyn, C3H5], 1.88 [d, 3J(1H,1H) = 7.3 Hz, 1H of 3a, Hsyn, C3H5], 0.84 [d, 3J(1H,1H) = 11.0 Hz, 1H of 3a, Hanti, C3H5], 0.81 [d, 3J(1H,1H) = 11.0 Hz, 1H of 3a, Hanti, C3H5], 0.29 [tt, 3J(1H,1H) = 11.0 Hz, 3J(1H,1H) = 7.4 Hz, 1H of 3a, Hmeso, C3H5], –0.85 [d, 3J(1H,1H) = 9.8 Hz, 1H of 3b, Hanti, C3H5], –0.88 [d, 3J(1H,1H) = 9.8 Hz, 1H of 3b, Hanti, C3H5]. IR(ATR-C; cm−1): 1952 s [νa(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O)]; 1927 s [νa(CO)]; 1831 vs. [νs(CO)]. Single crystals of 3 suitable for X-ray diffraction analysis were prepared by sublimation in a vacuum sealed ampoule (110 °C; 1 Pa). When the synthesis was carried out from 1-[D], 3-[D] was obtained, for which the signals at 6.36, 6.32, 5.96 and 5.88 ppm in the 1H NMR spectrum decreased in intensity and those at 5.68 and 5.65 ppm were seen as a broadened singlet.
O)]; 1927 s [νa(CO)]; 1831 vs. [νs(CO)]. Single crystals of 3 suitable for X-ray diffraction analysis were prepared by sublimation in a vacuum sealed ampoule (110 °C; 1 Pa). When the synthesis was carried out from 1-[D], 3-[D] was obtained, for which the signals at 6.36, 6.32, 5.96 and 5.88 ppm in the 1H NMR spectrum decreased in intensity and those at 5.68 and 5.65 ppm were seen as a broadened singlet.
Synthesis of [(η3-C3H5)(η5-f-Bind)Mo(CO)2] (4)
The reaction was performed as described for compound 3 but with benz[f]indene (2; 1.66 g; 10 mmol). Yield: 2.63 g (7.34 mmol, 73.4%). Yellow powder. Anal. Calcd for C18H14MoO2: C, 60.35; H, 3.94. Found: C, 60.06; H, 3.72. Mp: 140–145 °C (dec). 1H NMR (400.1 MHz, benzene-d6, 2:1 mixture of isomers 4a and 4b): δ = 7.33–7.26 (m, 2H of 4a and 2H of 4b, H5–8, C13H9), 6.99 (s, 2H of 4a and 2H of 4b, H4,9, C13H9), 6.96–6.93 (m, 2H of 4a, H5–8, C13H9), 6.90–6.88 (m, 2H of 4b, H5–8, C13H9), 5.75 (d, 3J(1H,1H) = 1.9 Hz, 2H of 4b, H1,3, C13H9), 5.67 [d, 3J(1H,1H) = 2.2 Hz, 2H of 4a, H1,3, C13H9], 5.33 [t, 3J(1H,1H) = 2.2 Hz, 1H of 4a, H2, C13H9], 5.20 [t, 3J(1H,1H) = 1.9 Hz, H2, 1H of 4b, C13H9], 3.34 [d, 3J(1H,1H) = 6.5 Hz, Hsyn, 2H of 4b, C3H5], 2.91 [tt, 3J(1H,1H) = 11.1 Hz, 3J(1H,1H) = 6.5 Hz, Hmeso, 1H of 4b, C3H5], 2.09 [d, 3J(1H,1H) = 7.4 Hz, Hsyn, 2H of 4a, C3H5], 0.77 [d, 3J(1H,1H) = 11.2 Hz, Hanti, 2H of 4a, C3H5], –0.18 [tt, 3J(1H,1H) = 11.2 Hz, 3J(1H,1H) = 7.4 Hz, Hmeso, 1H of 4a, C3H5], –0.85 [d, 3J(1H,1H) = 11.1 Hz, Hanti, 2H of 4b, C3H5]. IR(ATR-C; cm−1): 1916 vs. [νa(CO)]; 1870 vs. [νs(CO)]. Single crystals of 4 suitable for X-ray diffraction analysis were prepared by sublimation in a vacuum sealed ampoule (120 °C; 1 Pa).
Synthesis of [(η5-e-Bind)Mo(CO)2(κ2-bpy)][BF4] (7)
A solution of 3 (460 mg, 1.28 mmol) in CH2Cl2 (30 mL) was treated with MeCN (2 mL), cooled to 0 °C and treated with HBF4·Et2O (176 μL, 1.28 mmol). The reaction mixture was stirred for 2 h at room temperature. Solvents were vacuum-evaporated and the crude intermediate was washed with Et2O (3 × 20 mL) and recrystallized from an acetone/Et2O mixture at –80 °C. The purified intermediate was dissolved in CH2Cl2 (20 mL), treated with 2,2′-bipyridine (200 mg, 1.28 mmol) and stirred at room temperature for 18 h. Solvents were vacuum-evaporated and the crude product was washed with Et2O (3 × 20 mL), recrystallized from a CH2Cl2/Et2O mixture and vacuum-dried. Yield: 650 mg (1.16 mmol, 90.7%). Dark red powder. Anal. Calcd for C25H17BF4MoN2O2: C, 53.60; H, 3.06; N, 5.00. Found: C, 53.32; H, 3.11; N, 5.25. Mp: 160–165 °C (dec). 1H NMR (400.1 MHz, acetone-d6): δ = 9.51 [ddd, 3J(1H,1H) = 5.8 Hz, 4J(1H,1H) = 1.4 Hz, 5J(1H,1H) = 0.7 Hz, 1H, H2,9, bpy], 9.20 [ddd, 3J(1H,1H) = 5.7 Hz, 4J(1H,1H) = 1.5 Hz, 5J(1H,1H) = 0.7 Hz, 1H, H2,9, bpy], 8.50 [d, 3J(1H,1H) = 8.3 Hz, 1H, H5,6, bpy], 8.24 [td, 3J(1H,1H) = 7.7 Hz, 4J(1H,1H) = 1.5 Hz, 1H, H4,7, bpy], 8.19 [d, 3J(1H,1H) = 8.2 Hz, 1H, H5,6, bpy], 8.06–8.03 (m, 1H, H6,9, C13H9), 7.91 [td, 3J(1H,1H) = 7.7 Hz, 4J(1H,1H) = 1.4 Hz, 1H, H4,7, bpy], 7.76 [ddd, 3J(1H,1H) = 7.5 Hz, 3J(1H,1H) = 5.8 Hz, 4J(1H,1H) = 1.4 Hz, 1H, H3,8, bpy], 7.49 [ddd, 3J(1H,1H) = 7.5 Hz, 3J(1H,1H) = 5.8 Hz, 4J(1H,1H) = 1.3 Hz, 1H, H3,8, bpy], 7.48–7.42 (m, 2H, H7,8, C13H9), 7.15 [d, 3J(1H,1H) = 9.0 Hz, 1H, H5, C13H9], 7.08 [ddd, 3J(1H,1H) = 2.8 Hz, 4J(1H,1H) = 2.5 Hz, 4J(1H,1H) = 0.5 Hz, 1H, H3, C13H9], 6.97 [dd, 3J(1H,1H) = 9.0 Hz, 4J(1H,1H) = 0.5 Hz, 1H, H4, C13H9], 6.82 [dd, 3J(1H,1H) = 2.8 Hz, 4J(1H,1H) = 1.5 Hz, 1H, H1, C13H9], 5.88 [t, 3J(1H,1H) = 2.8 Hz, 1H, H2, C13H9]. IR(ATR-C; cm−1): 1966 vs. [νa(CO)]; 1887 vs. [νs(CO)]; 1054 s [ν(B–F)]. When the synthesis was carried out from 3-[D], 7-[D] was obtained, for which the signals at 7.08 and 6.82 ppm in the 1H NMR spectrum decreased in intensity and that at 5.88 ppm was seen as a broadened singlet.
Synthesis of [(η5-e-Bind)Mo(CO)2(κ2-phen)][BF4] (8)
The reaction was performed as described for compound 7 but with 1,10-phenanthroline (230 mg; 1.28 mmol). Yield: 660 mg (1.13 mmol, 88.3%). Dark red powder. Anal. Calcd for C27H17BF4MoN2O2: C, 55.51; H, 2.93; N, 4.80. Found: C, 55.78; H, 3.04; N, 5.01. Mp: 160–165 °C (dec). 1H NMR (400.1 MHz, acetone-d6): δ = 9.88 [dd, 3J(1H,1H) = 5.4 Hz, 4J(1H,1H) = 1.2 Hz, 1H, H2,9, phen], 9.59 [dd, 3J(1H,1H) = 5.4 Hz, 4J(1H,1H) = 1.3 Hz, 1H, H2,9, phen], 8.85 [dd, 3J(1H,1H) = 8.1 Hz, 4J(1H,1H) = 1.2 Hz, 1H, H4,7, phen], 8.52 [dd, 3J(1H,1H) = 8.2 Hz, 4J(1H,1H) = 1.2 Hz, 1H, H4,7, phen], 8.15 [dd, 3J(1H,1H = 8.1 Hz, 3J(1H,1H = 5.4 Hz, 1H, H3,8, phen], 8.13 [d, 3J(1H,1H = 8.8 Hz, 1H, H5,6, phen], 8.00 [d, 3J(1H,1H = 8.9 Hz, 1H, H5,6, phen], 7.99 [d, 3J(1H,1H) = 8.1 Hz, 1H, H6,9, C13H9], 7.86 [dd, 3J(1H,1H) = 8.2 Hz, 3J(1H,1H) = 5.4 Hz, 1H, H3,8, phen], 7.37 (ddd, 3J(1H,1H) = 7.9 Hz, 3J(1H,1H) = 7.3 Hz, 4J(1H,1H) = 1.3 Hz, 1H, H7,8, C13H9], 7.24 [ddd, 3J(1H,1H) = 7.9 Hz, 3J(1H,1H) = 7.3 Hz, 3J(1H,1H) = 1.3 Hz, 1H, H7,8, C13H9], 7.13 [ddd, 3J(1H,1H) = 2.7 Hz, 4J(1H,1H) = 1.4 Hz, 4J(1H,1H) = 0.8 Hz, 1H, H3 C13H9], 7.04 [d, 3J(1H,1H) = 8.9 Hz, 1H, H6,9, C13H9], 6.90 [dd, 3J(1H,1H) = 2.7 Hz, 4J(1H,1H) = 1.4 Hz, 1H, H1, C13H9], 6.83 [dd, 3J(1H,1H) = 9.0 Hz, 4J(1H,1H) = 0.8 Hz, 1H, H4, C13H9], 6.67 [d, 3J(1H,1H) = 9.0 Hz, 1H, H5, C13H9], 5.92 [t, 3J(1H,1H) = 2.7 Hz, 1H, H2, C13H9]. IR(ATR-C; cm−1): 1966 vs. [νa(CO)]; 1889 vs. [νs(CO)]; 1054 s [ν(B–F)]. When the synthesis was carried out from 3-[D], 8-[D] was obtained, for which the signals at 7.13 and 6.90 ppm in the 1H NMR spectrum decreased in intensity and that at 5.92 ppm was seen as a broadened singlet.
Synthesis of [(η3-e-Bind)Mo(CO)2(κ3-tpm)][BF4] (9)
The reaction was performed as described for compound 7 but with tris(pyrazolyl)methane (275 mg; 1.28 mmol). Yield: 575 mg (0.93 mmol, 72.7%). Dark red powder. Anal. Calcd for C25H19BF4MoN6O2: C, 48.57; H, 3.10; N, 13.59. Found: C, 48.22; H, 3.14; N, 13.73. Mp: 155–160 °C (dec). 1H NMR (400.1 MHz, acetone-d6): δ = 9.35 [d, 3J(1H,1H) = 2.2 Hz, 1H, H3, pz], 9.29 (s, 1H, pz3CH), 8.67 [d, 3J(1H,1H) = 2.2 Hz, 1H, H3, pz], 8.48 [d, 3J(1H,1H) = 2.2 Hz, 1H, H3, pz], 8.43 [d, 3J(1H,1H) = 2.5 Hz, 1H, H5, pz], 8.41 [d, 3J(1H,1H) = 2.5 Hz, 1H, H5, pz], 8.40 [d, 3J(1H,1H) = 2.5 Hz, 1H, H5, pz], 8.02 [d, 3J(1H,1H) = 8.2 Hz, 1H, H6,9, C13H9], 7.90 [t, 3J(1H,1H) = 3.1 Hz, 1H, H2, C13H9], 7.54 [d, 3J(1H,1H) = 8.3 Hz, 1H, H6,9, C13H9], 7.23 [ddd, 3J(1H,1H) = 8.4 Hz, 3J(1H,1H) = 6.7 Hz, 4J(1H,1H) = 1.2 Hz, 1H, H7,8, C13H9], 7.14 [ddd, 3J(1H,1H) = 8.2 Hz, 3J(1H,1H) = 6.9 Hz, 4J(1H,1H) = 1.2 Hz, 1H, H7,8, C13H9], 7.13 [d, 3J(1H,1H) = 8.0 Hz, 1H, H4,5, C13H9], 7.03 [d, 3J(1H,1H) = 8.0 Hz, 1H, H4,5, C13H9], 6.91 [dd, 3J(1H,1H) = 2.5 Hz, 3J(1H,1H) = 2.2 Hz, 1H, H4, pz], 6.72 [dd, 3J(1H,1H) = 2.5 Hz, 3J(1H,1H) = 2.2 Hz, 1H, H4, pz], 6.70 [dd, 3J(1H,1H) = 2.5 Hz, 3J(1H,1H) = 2.2 Hz, 1H, H4, pz], 6.63 [dd, 3J(1H,1H) = 3.1 Hz, 4J(1H,1H) = 3.1 Hz, 1H, H1,3, C13H9], 6.04 [dd, 3J(1H,1H) = 3.1 Hz, 4J(1H,1H) = 3.1 Hz, 1H, H1,3, C13H9]. IR(ATR-C; cm−1): 1955 vs. [νa(CO)]; 1874 vs. [νs(CO)]; 1047 s [ν(B–F)]. When the synthesis was carried out from 3-[D], 9-[D] was obtained, for which the signals at 6.63 and 6.04 ppm in the 1H NMR spectrum decreased in intensity and that at 7.90 ppm was seen as a broadened singlet.
Synthesis of [(η3-f-Bind)Mo(CO)2(κ3-tpm)][BF4] (10)
A solution of 4 (52.1 mg, 145 μmol) in CH2Cl2 (5 mL) was treated with MeCN (0.1 mL), cooled at 0 °C and treated with HBF4·Et2O (20 μL, 145 μmol). The reaction mixture was treated with tris(1-pyrazolyl)methane (31.1 mg, 145 μmol), stirred for 30 min at room temperature, then overlayered with Et2O (50 mL) and kept for 18 h to give crystals of 10 suitable for X-ray analysis. The sample for analyses was washed with Et2O (20 mL) and CH2Cl2 (20 mL) and vacuum-dried. Yield: 55 mg (89 μmol, 61.4%). Orange-red crystals. Anal. calcd for C25H19BF4MoN6O2: C, 48.57; H, 3.10; N, 13.59. Found: C, 48.26; H, 3.26; N, 13.77. Mp: 155–160 °C (dec). 1H NMR (500.2 MHz, dmso-d6): δ = 9.51 (s, 1H, pz3CH), 9.19 [d, 1H, 3J(1H,1H) = 2.2 Hz, H3, pz], 8.54 [d, 2H, 3J(1H,1H) = 2.2 Hz, H3, pz], 8.42 [d, 1H, 3J(1H,1H) = 2.5 Hz, H5, pz], 8.37 [d, 2H, 3J(1H,1H) = 2.5 Hz, H5, pz], 7.51 (m, 2H, H5,8, C13H9), 7.20 (m, 2H, H6,7, C13H9), 7.06 (s, 2H, H4,9, C13H9), 6.84 [dd, 3J(1H,1H) = 2.5 Hz, 3J(1H,1H) = 2.2 Hz, 1H, H4, pz], 6.71 [dd, 3J(1H,1H) = 2.5 Hz, 3J(1H,1H) = 2.2 Hz, 2H, H4, pz], 6.52 [t, 1H, 3J(1H,1H) = 3.6 Hz, H2, C13H9], 5.97 [d, 2H, 3J(1H,1H) = 3.6 Hz, H1,3, C13H9]. 13C APT NMR (125.8 MHz, dmso-d6): δ = 225.6 (2C, CO), 152.0 (1C, C3, pz), 146.5 (2C, C3, pz), 145.0 (2C, C3a,9a, C13H9), 135.5 (2C, C5, pz), 134.8 (1C, C5, pz), 132.0 (2C, C4a,8a, C13H9), 127.4 (2C, C5,8, C13H9), 125.1 (2C, C6,7, C13H9), 114.9 (2C, C4,9, C13H9), 109.5 (1C, C4, pz), 108.2 (2C, C4, pz), 95.0 (1C, C2, C13H9), 74.7 (2C, C1,3, C13H9), 74.4 (1C, pz3CH). IR(ATR-C; cm−1): 1951 vs. [νa(CO)]; 1867 vs. [νs(CO)]; 1024 s [ν(B–F)].
X-ray crystallography
Crystallographic data for the single crystals of the compounds 3, 4 and 10 were collected on a Bruker D8 VENTURE Kappa Duo PHOTON100 using an IμS micro-focus sealed tube with MoKα radiation (λ = 0.71073 Å) at a temperature of 150 K. The structures were solved by direct methods (SHELXT39a)56 and refined by full matrix least squares based on F2 (SHELXL201439b).57 The hydrogen atoms on carbon were fixed into idealized positions (riding model) or found on the difference Fourier map and all refined under rigid body assumption with assigned temperature factors Hiso(H) = 1.2 Ueq(pivot atom). The allyl ligands in 3 and 4 crystals are disordered over two positions.X-ray crystallographic data have been deposited with the Cambridge Crystallographic Data Centre (CCDC) under deposition numbers 2295997–2295999 for 3, 4 and 10, respectively, and can be obtained free of charge from the centre via its website (https://www.ccdc.cam.ac.uk/structures/).
Author contributions
The authors contributed equally.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to acknowledge the financial support to the Faculty of Chemical Technology, University of Pardubice (Project No. VA390018).References
- T. J. Kealy and P. L. Pauson, Nature, 1951, 168, 1039–1040 CrossRef CAS.
- P. Štěpnička, Ferrocenes: Ligands, Materials and Biomolecules, John Wiley & Sons, Chichester, 2008 Search PubMed.
- J. M. O’Connor and C. P. Casey, Chem. Rev., 1987, 87, 307–318 CrossRef.
- L. Orian, M. Swart and F. M. Bickelhaupt, Chem. Phys. Chem., 2014, 15, 219–228 CrossRef CAS PubMed.
- B. M. Trost and Michael C. Ryan, Angew. Chem., Int. Ed., 2017, 56, 2862–2879 CrossRef CAS PubMed.
- L. F. Veiros and M. J. Calhorda, Dalton Trans., 2011, 40, 11138–11146 RSC.
- S. M. Bruno, M. Pillinger, A. A. Valente and I. S. Gonçalves, J. Organomet. Chem., 2022, 970–971, 122372 CrossRef CAS.
- O. Mrózek, J. Vinklárek, Z. Růžičková and J. Honzíček, Eur. J. Inorg. Chem., 2016, 5250–5264 CrossRef.
- O. Mrózek, L. Šebestová, J. Vinklárek, M. Řezáčová, A. Eisner, Z. Růžičková and J. Honzíček, Eur. J. Inorg. Chem., 2016, 519–529 CrossRef.
- J. Honzíček, J. Vinklárek, M. Erben, J. Lodinský, L. Dostál and Z. Padělková, Organometallics, 2013, 32, 3502–3511 CrossRef.
- R. W. Baker, Organometallics, 2018, 37, 433–440 CrossRef CAS.
- K. J. Evans, P. A. Morton, C. Luz, C. Miller, O. Raine, J. M. Lynam and S. M. Mansell, Chem. – Eur. J., 2021, 27, 17824–17833 CrossRef CAS PubMed.
- E. Kirillov, S. Kahlal, T. Roisnel, T. Georgelin, J. Y. Saillard and J. F. Carpentier, Organometallics, 2008, 27, 387–393 CrossRef CAS.
- L. Hirneise, D. A. Buschmann, C. Maichle-Mössmer and R. Anwander, Organometallics, 2022, 41, 962–976 CrossRef CAS.
- M. J. Calhorda, C. C. Romão and L. F. Veiros, Chem. – Eur. J., 2002, 8, 868–875 CrossRef CAS PubMed.
- L. Orian, L. P. Wolters and F. M. Bickelhaupt, Chem. – Eur. J., 2013, 19, 13337–13347 CrossRef CAS PubMed.
- K. T. Nguyen, E. E. Lane, C. D. McMillen, J. A. Pienkos and P. S. Wagenknecht, Organometallics, 2020, 39, 670–678 CrossRef CAS.
- L. Atkin and D. L. Priebbenow, Angew. Chem., Int. Ed., 2023, 62, e202302175 CrossRef CAS PubMed.
- S. Brydges, N. Reginato, L. P. Cuffe, C. M. Seward and M. J. McGlinchey, C. R. Chim, 2005, 8, 1497–1505 CrossRef CAS.
- J. Honzíček, I. Honzíčková, J. Vinklárek and Z. Růžičková, J. Organomet. Chem., 2014, 772–773, 299–306 CrossRef.
- J. Honzíček, F. A. A. Paz and C. C. Romão, Eur. J. Inorg. Chem., 2007, 2827–2838 CrossRef.
- C. A. Gamelas, N. A. G. Bandeira, C. C. L. Pereira, M. J. Calhorda, E. Herdtweck, M. Machuqueiro, C. C. Romão and L. F. Veiros, Dalton Trans., 2011, 10513–10525 RSC.
- J. Honzíček, C. C. Romão, M. J. Calhorda, A. Mukhopadhyay, J. Vinklárek and Z. Padělková, Organometallics, 2011, 30, 717–725 CrossRef.
- V. V. Izmer, A. Y. Lebedev, D. S. Kononovich, I. S. Borisov, P. S. Kulyabin, G. P. Goryunov, D. V. Uborsky, J. A. M. Canich and A. Z. Voskoboynikov, Organometallics, 2019, 38, 4645–4657 CrossRef CAS.
- F. Pammer and W. R. Thiel, Coord. Chem. Rev., 2014, 270–271, 14–30 CrossRef CAS.
- M. J. McGlinchey, Molecules, 2022, 27, 3882 CrossRef CAS PubMed.
- K. Albrecht, O. Reiser, M. Weber and A. de Meijere, Synlett, 1992, 521–524 CrossRef CAS.
- U. Stehling, J. Diebold, R. Kirsten, W. Roll, H. H. Brintzinger, S. Jungling, R. Mulhaupt and F. Langhauser, Organometallics, 1994, 13, 964–970 CrossRef CAS.
- D. E. Babushkin and H. H. Brintzinger, Chem. – Eur. J., 2007, 13, 5294–5299 CrossRef CAS PubMed.
- N. Schneider, M. E. Huttenloch, U. Stehling, R. Kirsten, F. Schaper and H. H. Brintzinger, Organometallics, 1997, 16, 3413–3420 CrossRef CAS.
- D. J. Arriola, M. Bokota, R. E. J. Campbell, J. Klosin, R. E. LaPointe, O. D. Redwine, R. B. Shankar, F. J. Timmers and K. A. Abboud, J. Am. Chem. Soc., 2007, 129, 7065–7076 CrossRef CAS PubMed.
- X. Zhao, X. Luo, B. Li, H. Song, S. Xu and B. Wang, Eur. Polym. J., 2008, 44, 3264–3270 CrossRef CAS.
- D. Mavrynsky, R. Sillanpää and R. Leino, Organometallics, 2009, 28, 598–605 CrossRef CAS.
- K. Schröder, D. Haase, W. Saak, A. Lützen, R. Beckhaus, S. Wichmann and J. Schellenberg, Organometallics, 2006, 25, 3824–3836 CrossRef.
- J. Sun, D. J. Berg and B. Twamley, Can. J. Chem., 2017, 95, 363–370 CrossRef CAS.
- F. Pammer, Y. Sun, C. May, G. Wolmershäuser, H. Kelm, H. J. Krüger and W. R. Thiel, Angew. Chem., Int. Ed., 2007, 46, 1270–1273 CrossRef CAS PubMed.
- F. Pammer, Y. Sun and W. R. Thiel, Organometallics, 2008, 27, 1015–1018 CrossRef CAS.
- F. Pammer, Y. Sun, D. Weismann, H. Sitzmann and W. R. Thiel, Chem. – Eur. J., 2010, 16, 1265–1270 CrossRef CAS PubMed.
- F. Pammer, Y. Sun, M. Sieger, J. Fiedler, B. Sarkar and W. R. Thiel, Organometallics, 2010, 29, 6165–6168 CrossRef CAS.
- C. L. Becker and M. L. McLaughlin, Synlett, 1991, 642 CrossRef CAS.
- M. L. Morris, C. L. Becker, F. R. Fronczek, W. H. Daly and M. L. McLaughlin, J. Org. Chem., 1994, 59, 6484–6486 CrossRef.
- W. E. Noland, L. L. Landucci and V. Kameswaran, J. Org. Chem., 1980, 45, 3456–3461 CrossRef CAS.
- R. H. Cox and H. W. Terry Jr., J. Magn. Reson., 1974, 14, 317–322 CAS.
- J. W. Faller, C. C. Chen, M. J. Mattina and A. Jakubowski, J. Organomet. Chem., 1973, 52, 361–386 CrossRef CAS.
- I. Honzíčková, J. Vinklárek, C. C. Romão, Z. Růžičková and J. Honzíček, New J. Chem., 2016, 40, 245–256 RSC.
- O. Mrózek, L. Melounková, L. Dostál, I. Císařová, A. Eisner, R. Havelek, E. Peterová, J. Honzíček and J. Vinklárek, Dalton Trans., 2019, 48, 11361–11373 RSC.
- O. Mrózek, L. Dostál, I. Císařová, J. Honzíček and J. Vinklárek, Dalton Trans., 2019, 48, 12210–12218 RSC.
- I. S. Gonçalves, L. F. Veiros, C. A. Gamelas, C. Cabrita, M. J. Calhorda, C. F. G. C. Geraldes, J. Green, E. Packham, M. G. B. Drew, V. Félix, A. G. Santos and C. C. Romão, J. Organomet. Chem., 2015, 792, 154–166 CrossRef.
- J. Honzíček, P. Kratochvíl, J. Vinklárek, A. Eisner and Z. Padělková, Organometallics, 2012, 31, 2193–2202 CrossRef.
- M. J. Calhorda, C. A. Gamelas, I. S. Gonçalves, E. Herdtweck, C. C. Romão and L. F. Veiros, Organometallics, 1998, 17, 2597–2611 CrossRef CAS.
- J. Honzíček, J. Vinklárek, Z. Padělková, L. Šebestová, K. Foltánová and M. Řezáčová, J. Organomet. Chem., 2012, 716, 258–268 CrossRef.
- C. C. L. Pereira, P. J. Costa, M. J. Calhorda, C. Freire, S. S. Rodrigues, E. Herdtweck and C. C. Romão, Organometallics, 2006, 25, 5223–5234 CrossRef CAS.
- P. Foster, J. C. W. Chien and M. D. Rausch, Organometallics, 1996, 15, 2404–2409 CrossRef CAS.
- W. L. F. Armarego and D. D. Perrin, Purification of Laboratory Chemicals, Butterworth-Heinemann, Oxford, 1996 Search PubMed.
- S. Juliá, J. M. del Mazo, L. Avila and J. Elguero, Org. Prep. Proc. Int., 1984, 16, 299–307 CrossRef.
- G. M. Sheldrick, Acta Crystallogr. Sect. A, 2015, A71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr. Sect. C, 2015, C71, 3–8 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2295997–2295999. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3nj04406h |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2023 |