
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of Pt(II) phosphinocarboxylate complexes with auxiliary arylcarbene ligands and factors that control their stereochemistry†
Filip
Horký
a,
Johannes
Soellner
b,
Jiří
Schulz
a,
Ivana
Císařová
a,
Thomas
Strassner
 *b and
Petr
Štěpnička
*a
*b and
Petr
Štěpnička
*a
aDepartment of Inorganic Chemistry, Faculty of Science, Charles University, Hlavova 2030, 128 40 Prague, Czech Republic. E-mail: stepnic@natur.cuni.cz
bPhysikalische Organische Chemie, Technische Universität Dresden, Bergstrasse 66, 01069 Dresden, Germany. E-mail: thomas.strassner@chemie.tu-dresden.de
First published on 20th September 2023
Abstract
Orthoplatinated complexes [Pt(C^C*)(acac)] (1R), in which C^C* is orthoplatinated 3-R-1-phenyl-1H-benzo[d]imidazol-2-ylidene and R = Me and Ph, reacted with 1′-(diphenylphosphino)ferrocene-1-carboxylic acid (Hdpf) under protonation of the acetylacetonate ligand (acac) to produce the corresponding phosphinocarboxylate bischelate complexes [Pt(C^C*)(dpf-κ2O,P)] (2R) as single isomers with trans-P,C(carbene) geometry. The compounds were fully characterized by elemental analysis, spectroscopic methods, single-crystal X-ray diffraction analysis, and cyclic voltammetry. In addition, DFT calculations were used to determine differences in energy and the bonding situation between 2R and the hypothetical geometric isomers 3R with a trans-P,C(phenyl) arrangement. The experimental and theoretical results are consistent with the antisymbiosis effect observed in complexes of soft metal ions, namely with weakening of Pt–C bonds by strongly trans-influencing ligands.
Introduction
Reactions between Pd and Pt-acetylacetonate (acac) complexes and Brønsted acids typically proceed under protonation of the acac ligand, which leads to its decoordination and liberation of two coordination sites at the metal.1 In this way, the readily accessible acetylacetonate complexes can serve as convenient precursors of diverse transition metal compounds.2 Examples include products obtained through reactions between palladium(II) acetylacetonate and imidazolium salts to produce carbene complexes3 as well as analogous reactions with oxalic amidinates,4 2,5-diamino-1,4-benzoquinonediimine,5 1-(isoquinolin-1-yl)-2-naphtholes,6 and porphyrins bearing enaminoketone moieties at the periphery,7 or reactions between the corresponding platinum(II) complex and secondary phosphine oxides8 or benzoquinonediimines.9Reactions of acetylacetonate complexes with protic difunctional proligands (X^YH) that produce X,Y-chelate complexes are particularly attractive. For instance, the Pd-acetylacetonate complex [(LNC)Pd(acac)], where LNC stands for orthometallated 2-(dimethylamino-κN)phenyl-κC1 ligand, reacted with amino acids to generate N,O-chelate complexes of the type [(LNC)Pd(NH2CH(R)CO2-κ2N,O)].10 In our research, we used a similar approach to prepare P,O-chelate complexes from phosphinoferrocene carboxylic and phosphonic acids.11,12 In these reactions, we consistently observed the formation of a single isomer in which the phosphine moiety and phenyl group from the LNC ligand (as the donors with the largest trans influence13) mutually occupy cis positions, in line with the antisymbiosis (or transphobia) concept.14 Now we considered analogous complexes obtained from cyclometallated Pt(II)-carbene complexes [(C^C*)Pt(acac)] (1 in Scheme 1) because their orthoplatinated C^C* ligands less significantly differentiate the two remaining coordination sites by trans influence,15 which can result in dichotomy in reactivity. Compounds 1 were studied as tuneable photoluminescent materials16 but have only rarely been used as synthetic precursors thus far.
 | ||
| Scheme 1 General formula of compounds 1 and reactions investigated in this work. | ||
In this contribution we build upon our recent research focused on the reactions of type 1 complexes with α-donor substituted acetic acids,17 now aiming on the reactions with 1′-(diphenylphosphino)ferrocene-1-carboxylic acid (Hdpf).18 In particular, we report the results of our reactivity studies using two Pt(II) precursors, detailed structural characterization of the resulting complexes, and DFT calculations focused on the differences in energy and bonding situation between the product isomers.
Results and discussion
Syntheses and structural characterization
Reactions of complexes 1R with Hdpf produced the respective phosphinocarboxylate complexes 2R with trans-P,C(carbene) geometry (Scheme 2), which were isolated in approximately 70% yields. In these isomers, the phosphine moiety and the aryl group of the C^C* ligand (C), which exerts a higher trans influence than the carbene moiety (C*),15 occupy mutually cis positions. Isomers 3R with the trans-P,C(phenyl) arrangement have not been detected.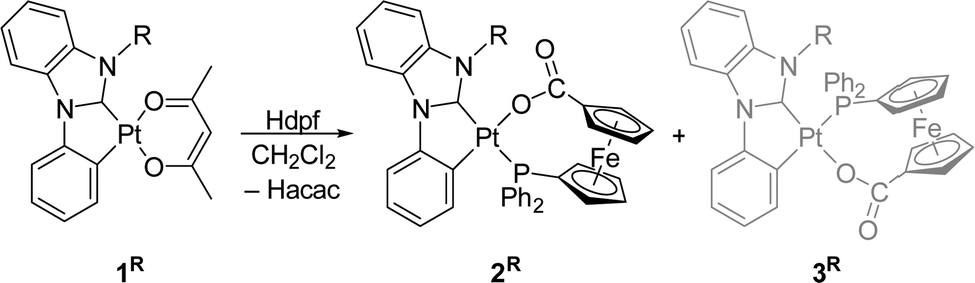 | ||
| Scheme 2 Synthesis of complexes 2R (R = Me, Ph). Isomeric compounds 3R were not detected. | ||
The compounds were characterized by multinuclear NMR and IR spectroscopy, mass spectrometry, and elemental analysis. In addition, the solid-state structures of 2Me·CH2Cl2 and 2Ph·1/2AcOEt were determined by single-crystal X-ray diffraction analysis. In their 1H and 13C NMR spectra, the complexes showed the expected signals, including the diagnostic resonances due to carboxylate and carbene carbons at δC 175.82 and 178.48 ppm for 2Me and at δC 174.27 and 178.24 ppm for 2Ph (the carbene resonances were observed as doublets due to coupling with the proximal phosphine moiety, 2JPC = 145–146 Hz). The IR spectra displayed intense bands at 1578 cm−1 attributable to the νas mode of the carboxylate ligands. The 31P NMR resonances were observed at 19.4 (2Me) and 18.9 (2Ph) ppm as singlets with 195Pt satellites (1JPtP = 2964 and 2990 Hz).19 The compounds showed no significant luminescence in PMMA matrix at room temperature (quantum yields < 5%), which is indeed in line with our previous observations20 and can be rationalised by quenching by the ferrocene moiety.21
The structures of the complexes are depicted in Fig. 1, and Table 1 provides selected geometric parameters. The compounds present square-planar coordination around the Pt(II) centres. The Pt-donor distances are similar to those determined for precursors 1R (ref. 22) and the phosphinocarboxylate complex [(LPh)Pt(Ph2PCH2CO2-κ2O,P)], where LPh stands for the C^C* ligand arising from 1Ph;17 the differences in the parameters obtained for 2Me and 2Ph are only small and can be explained by steric factors. The benzimidazol-2-ylidene fragments and the platinated benzene rings are almost coplanar (the interplanar angles are 9.06(7)° and 2.63(9)° in 2Me and 2Ph, respectively), while the phenyl substituent in 2Ph is twisted to minimize steric congestion (interplanar angle: 68.65(8)°). The ferrocene moieties exhibit negligible tilting and adopt conformations near to eclipsed (see τ angles in Table 1). The carboxylate groups retain partly localized character (C41–O1 < C41–O2) and are twisted from the planes of their parent cyclopentadienyl rings (by 25.9(2)° and 22.5(2)° in 2Me and 2Ph, respectively) to ensure that O2 approaches the Pt centre.
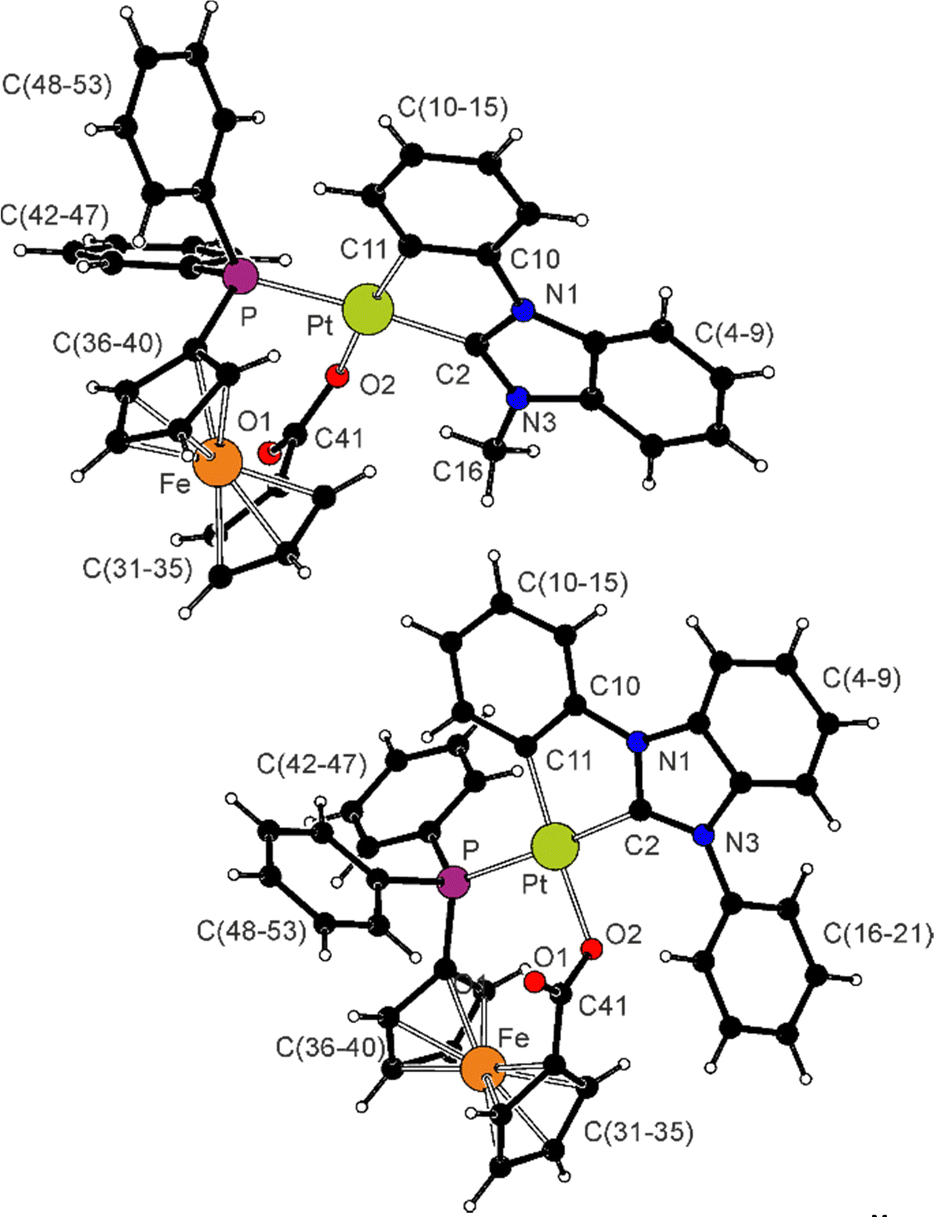 | ||
| Fig. 1 Views of the complex molecules in the crystal structures of 2Me·CH2Cl2 (top) and 2Ph·1/2AcOEt (bottom). Displacement ellipsoid plots are available in the ESI.† | ||
| Parametera | 2Me·CH2Cl2 | 2Ph·1/2AcOEt |
|---|---|---|
| a Tilt is the dihedral angle of the least-square cyclopentadienyl planes C(31–35) and (C36–40), τ denotes the torsion angle C31–Cg1–Cg2–C36, where Cg2 and Cg2 stand for the centroids of the rings C(31–35) and (C36–40), respectively. b The range of the Fe–C(31–40) bonds. | ||
| Pt–P | 2.3305(5) | 2.3192(7) |
| Pt–O2 | 2.117(1) | 2.093(1) |
| Pt–C2 | 2.016(2) | 2.004(2) |
| Pt–C11 | 2.011(2) | 2.016(2) |
| P–Pt–O2 | 86.27(3) | 87.08(4) |
| P–Pt–C11 | 100.16(5) | 97.11(6) |
| C2–Pt–C11 | 79.83(7) | 79.70(8) |
| C2–Pt–O2 | 93.98(6) | 96.10(7) |
| Fe–C (range)b | 2.026(2)–2.060(2) | 2.022(2)–2.069(2) |
| Tilt | 1.0(1) | 6.4(1) |
| τ | −12.3(1) | −4.0(1) |
| P–C36 | 1.796(2) | 1.824(2) |
| P–C42/P–C48 | 1.827(2)/1.836(2) | 1.823(2)/1.825(2) |
| C41–O1/C41–O2 | 1.235(2)/1.290(2) | 1.234(2)/1.291(2) |
| O1–C41–O2 | 122.2(2) | 126.1(2) |
The redox properties of complexes 2R were studied by cyclic voltammetry at a glassy carbon disc electrode using dichloromethane solutions containing Bu4N[PF6] as the supporting electrolyte. The compounds displayed similar but rather complicated behaviour (Fig. 2). Initially, they underwent diffusion-controlled oxidation [indicated by the anodic peak potential (ipa) increasing linearly with ν1/2, ipa ∝ ν1/2, where ν is scan rate], which was essentially reversible when scanned separately (i.e., when the switching potential was set after the first oxidation) and at relatively faster scan rates (ν ≥ 100 mV s−1). At slower scan rates, however, the reversibility markedly decreased (Fig. 3), suggesting that the redox change is followed by chemical reaction(s) that convert the oxidized molecule into other species. The oxidation occurred at E°′ 0.26 V for 2Me and at 0.21 V for 2Ph (E°′ determined at ν = 100 mV s−1, when the redox change appears essentially reversible; values vs. ferrocene/ferrocenium standard), which is more positive than for Hdpf (0.31 V in MeCN).18a Considering the nature of HOMO (Fig. 4), the oxidation was attributed to ferrocene/ferrocenium redox transition (vide infra).
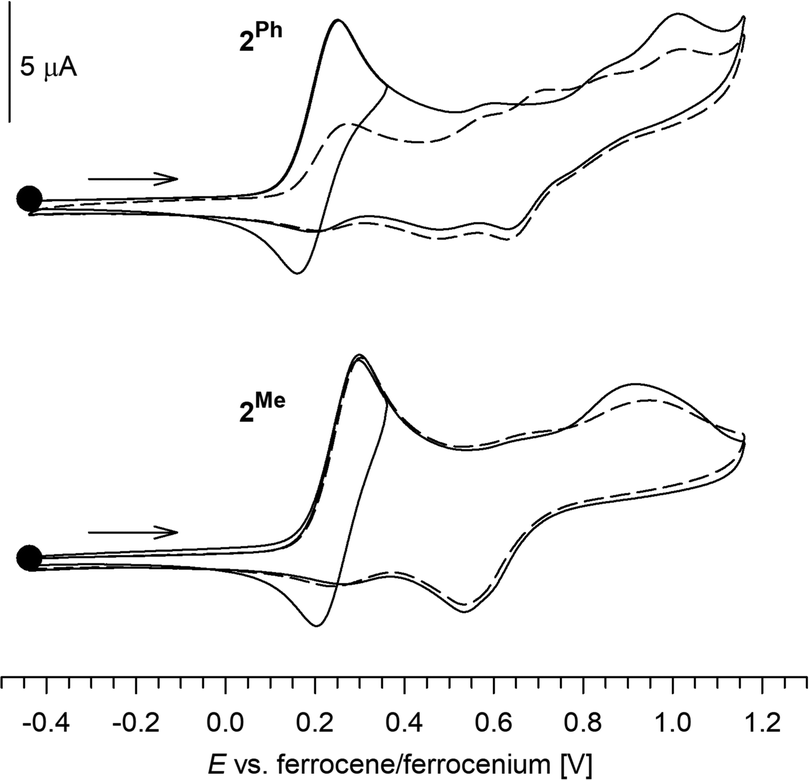 | ||
| Fig. 2 Cyclic voltammograms of 2Me and 2Ph as recorded at a glassy carbon electrode in dichloromethane at a 100 mV s−1 scan rate. The second scan is shown by a dashed line. | ||
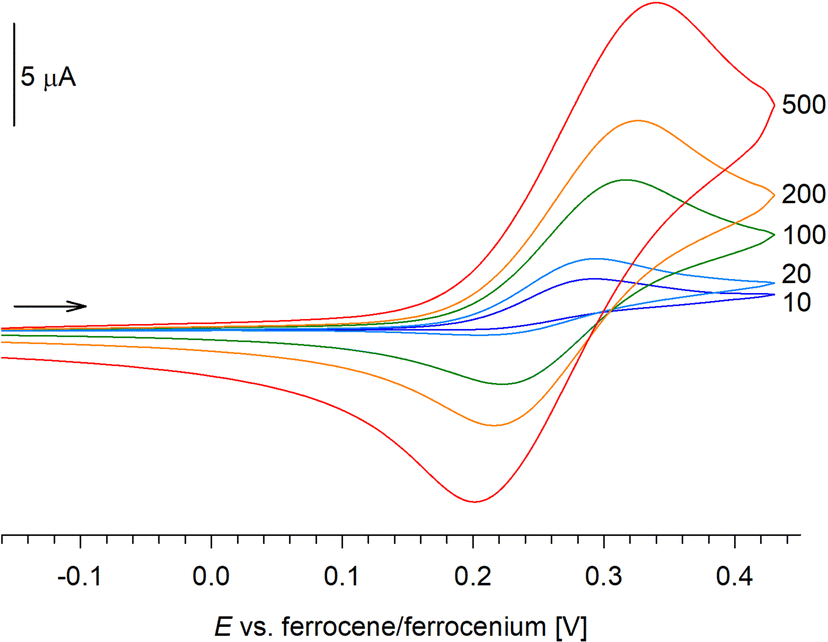 | ||
| Fig. 3 Cyclic voltammograms of 2Me recorded at varying scan rate (values in mV s−1). A similar Figure for 2Ph is available in ESI.† | ||
 | ||
| Fig. 4 (top) Frontier orbitals of 2Me (contour maps with isosurfaces at ±0.04 a.u.) at the B3LYP(d3bj)/6-311+G(d,p):LanL2TZ(Pt) level of theory, and (bottom) the electron difference map ρ(2Me)–ρ(2Me+) mapped at the equilibrium geometry of 2Me (isosurface at ±0.02 a.u.). A similar diagram for 2Ph is available in the ESI.† | ||
After the scan range was expanded towards more positive potentials, additional weaker redox waves were observed (Fig. 2). These redox transitions were associated with ill-defined, irreversible reduction steps and differed for the two compounds. In the cathodic region, no defined redox waves were detected. However, after scanning towards positive potentials (oxidation), weak irreversible waves were observed in the cathodic region, attributable to decomposition products.
DFT calculations
The electronic structures of 2R and 3R were studied by computational methods (DFT) at the B3LYP(d3bj)/6-311+G(d,p):LanL2TZ(Pt) level of theory. The frontier orbitals of 2Me and 2Ph exhibit almost identical spatial contours (Fig. 4). Their composition was analysed by the Natural Atomic Orbitals (NAO) approach, revealing that the HOMO of both complexes is localized almost exclusively on the ferrocene ligand and consists mainly of the iron 3d orbitals (≈85%) with smaller contributions from the carbon 2p orbitals constituting the π-system of the cyclopentadienyl rings (≈11%). This result already suggested that the initial electrochemical oxidation of 2R is a ferrocene-centred process. This was further corroborated by following the change in the electron density associated with electron removal, ρ(2R)–ρ(2R+), mapped at the equilibrium geometry of 2R,23 which was located exclusively at the iron atom (Fig. 4).Conversely, the LUMO in both compounds is highly delocalized, comprising mostly the π-system of the benzimidazole moiety with contributions from the vacant p-orbital of the carbene atom (≈25%), the platinum ion (≈15%, 6p and 5d) and the phosphorus atom (≈3%, 3p).
Consistent with the experimental observation, the computations suggested that 2R are the thermodynamically favoured isomers (Table 2). The trend is maintained even when the dispersion effects are included in the calculations, albeit with a different impact on the two compound pairs. The inclusion of solvation phenomena significantly decreased the energy difference between the isomers to approximately 5 kcal mol−1. The slightly higher energy difference that was estimated for 2Ph/3Ph can be rationalized by steric factors, namely, by possible steric congestion in 3Ph, in which the PPh2 group and the phenyl substituent are directed towards each other.24
| Method | 2Me/3Me | 2Ph/3Ph |
|---|---|---|
| a Determined as ΔG = G(2R) − G(3R) at 298 K. For details, see Experimental. | ||
| B3LYP/6-31G(d) | −7.51 | −6.69 |
| B3LYP/6-311+G(d,p) | −7.76 | −7.08 |
| B3LYP/6-311+G(d,p)+dispersion | −8.45 | −4.05 |
| B3LYP/6-311+G(d,p)+PCM | −5.22 | −5.75 |
The Pt-donor distances calculated for 2R reasonably corresponded with the experimental values (Table 3). More importantly, a comparison of the data calculated for 2R and the hypothetical isomers 3R revealed longer Pt–P and shorter Pt–O bonds for the latter isomers, in line with the stronger trans influence of the Pt-bound phenyl group. In contrast, the Pt–C bonds did not differ significantly in the two isomers.
| Complex | Pt–P | Pt–O | Pt–C(carbene) | Pt–C(Ph) |
|---|---|---|---|---|
| a Experimental values are reproduced from Table 1. Calculated data at the B3LYP/6-311+G(d,p) level of theory. | ||||
| 2Me (exp.) | 2.3305(5) | 2.117(1) | 2.016(2) | 2.011(2) |
| 2Me (DFT) | 2.437 | 2.189 | 2.005 | 2.025 |
| 3Me (DFT) | 2.519 | 2.100 | 1.978 | 2.027 |
| 2Ph (exp.) | 2.3192(7) | 2.093(1) | 2.004(2) | 2.016(2) |
| 2Ph (DFT) | 2.439 | 2.172 | 1.991 | 2.029 |
| 3Ph (DFT) | 2.541 | 2.100 | 1.974 | 2.024 |
Furthermore, the lower Mulliken charges at platinum in 2R (cf.2Me/3Me 0.579/0.665, and 2Ph/3Ph 0.428/0.507) suggested a stronger P → Pt donation in these isomers. The charges at the Pt-bound carboxylate oxygens were −0.338/−0.128 for 2Me/3Me and −0.125/−0.045 for 2Ph/3Ph. Compared to the predominantly covalent Pt–P bonds, the Pt–O bonds are more ionic and thus less sensitive to length variation.
The bonding situation in isomers 2 and 3 was further analysed using the intrinsic bond orbital (IBO) approach,25 which provides a representation of the Kohn–Sham wavefunction in terms of the more intuitive localized orbitals. A comparison of the IBOs representing the donor–acceptor interactions in 2Me and 3Me (Fig. 5) clearly shows the mutual trans-influence of the donor atoms in both isomers. The stronger σ-donor ligands tend to polarize the electron density of the M–L bond towards the trans-positioned ligand. Thus, the coordination of the phosphine group in 3Me (as expressed by the assigned partial charges: P 1.73/Pt 0.18) is weakened compared to that of 2Me (P 1.67/Pt 0.26) due to the strong trans influence of the X-type26 phenyl ligand. In contrast, the carbene-platinum bond appears insensitive to the influence of the phosphine group in the trans position (2Me: C 1.53/Pt 0.41 vs.3Me: C 1.51/Pt 0.43) and the Pt → P back-bonding is negligible, although slightly enhanced in 2Me (apparently at the expense of the weakened back-donation to the carbene ligand). The preferred mutual arrangement of the coordinated chelating ligands is thus trans-P,C(carbene), which reflects the relative σ-donor strength of the coordinated donor atoms and places the most ionic (Pt–O) and the most covalent coordination bonds (Pt–CPh) opposite to each other.
 | ||
| Fig. 5 Selected intrinsic bond orbitals (IBOs) of 2Me and 3Me. Values in parentheses indicate the fraction of bonding electrons assigned to the individual atoms. The carbon atoms are labelled as in the crystal structure. Similar diagrams for 2Ph and 3Ph are available in the ESI.† | ||
Conclusions
Compounds [Pt(C^C*)(acac)] (1), where C^C*stands for a chelating benzimidazole-2-ylidene ligand with an orthoplatinated phenyl substituent, react cleanly with 1′-(diphenylphosphino)-ferrocene-1-carboxylic acid (Hdpf) under proton transfer to the acetylacetonate and coordination of the formed dpf− anion as an O,P-chelating ligand. Of the two possible geometric isomers, which differ by the mutual orientation of the two chelating ligands, only the isomer with a trans-P,C(carbene) arrangement was detected and isolated due to a destabilizing effect exhibited by strongly trans-influencing donor moieties from the chelating ligands (viz., the phenyl and phosphine). The analysis of the bonding situation by DFT calculations and the IBO approach revealed a weakening of the covalent Pt–C interactions by the strongly donating trans-ligand, in line with the transphobia concept.Experimental
Materials and methods
All syntheses were performed under an argon atmosphere using standard Schlenk techniques. Complexes 1Me and 1Ph (ref. 22) and Hdpf18a were prepared according to procedures described in literature. Anhydrous dichloromethane was obtained from a Pure Solv MD5 solvent purification system (Innovative Technology, USA). Solvents used for workup and chromatography (Lach-Ner, Czech Republic) were utilized without further purification.NMR spectra were recorded with a Varian INOVA 400 spectrometer operating at 399.95, 100.58, and 161.90 MHz for 1H, 13C, and 31P, respectively. The chemical shifts (δ in ppm) are expressed relative to SiMe4 as an internal standard (1H and 13C) and to 85% aqueous H3PO4 as an external reference (31P). In addition to the standard notation of signal multiplicity,27 vt and vq are used to denote virtual triplets and quartets arising from AA′BB′ and AA′BB′X (A, B = 1H; X = 31P) spin systems constituted by the ferrocene cyclopentadienyl rings, respectively. ESI mass spectra were recorded with an AmaZon SL (Bruker) ion trap spectrometer using samples dissolved in HPLC-grade acetonitrile. IR spectra were recorded in diffuse reflectance mode (DRIFTS) using a Nicolet FTIR 205 spectrometer. The emission spectra were recorded with a Hamamatsu Quantaurus spectrometer, model C9920-02, using samples dispersed in PMMA matrix. Elemental analyses were performed with a PerkinElmer 2400 Series II CHNS/O analyser. The presence of residual solvent was confirmed by NMR analysis.
Electrochemical measurements were performed at ambient temperature using an μAUTOLAB III instrument (Eco Chemie) and a three-electrode cell equipped with a glassy carbon disc (2 mm diameter) working electrode, a platinum sheet auxiliary electrode, and a Ag/AgCl (3 M KCl) reference electrode. The samples were dissolved in anhydrous dichloromethane to generate a solution containing 1 mM of the analysed compounds and 0.1 M Bu4N[PF6] as the supporting electrolyte. The solutions were deaerated with argon before measurements and then maintained under an argon blanket. Decamethylferrocene (Alfa-Aesar) was added as an internal standard during the final scans, and the redox potentials were subsequently converted to the ferrocene/ferrocenium scale by subtracting 0.548 V.28
Syntheses
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). A single orange band was collected and evaporated, leaving solvated complex 2Me as an orange solid. Yield of 2Me·1/2CH2Cl2:41 mg (72%). The single crystal of 2Me·CH2Cl2 used for structure determination was grown from dichloromethane/isohexane.
:1). A single orange band was collected and evaporated, leaving solvated complex 2Me as an orange solid. Yield of 2Me·1/2CH2Cl2:41 mg (72%). The single crystal of 2Me·CH2Cl2 used for structure determination was grown from dichloromethane/isohexane.
1H NMR (399.95 MHz, CDCl3): δ 4.22 (vt, J′ = 1.9 Hz, 2H, fc), 4.37 (s, 3H, Me), 4.37–4.39 (m, 2H, fc), 4.51–4.52 (m, 2H, fc), 5.33 (vt, J′ = 1.9 Hz, 2H, fc), 6.35 (td, J = 7.5, 1.3 Hz, 1H, C6H4), 6.56 (ddd with 195Pt satellites, JPtC ≈ 60 Hz, J = 7.8, 2.3, 1.3 Hz, 1H, C6H4), 6.99 (td, J = 7.5, 1.3 Hz, 1H, C6H4), 7.32–7.59 (m, 10H, PPh2), 7.80–7.86 (m, 4H, C6H4), 8.04–8.09 (m, 1H, C6H4). 13C{1H} NMR (100.58 MHz, CDCl3): δ 32.55 (s, Me), 70.97 (s, CH of fc), 71.75 (d, 1JPC = 57 Hz, C–P of fc), 72.20 (d, JPC = 7 Hz, CH of fc), 72.62 (s, CH of fc), 75.44 (d, JPC = 11 Hz, CH of fc), 80.54 (s, C-COO of fc), 111.49 (s, CH of C6H4), 111.88 (s, CH of C6H4), 112.31 (s with 195Pt satellites, JPtC ≈ 30 Hz, CH of C6H4), 118.95 (d, 3JPC = 6 Hz, C–N of C6H4), 123.62 (s, CH of C6H4), 123.93 (br s with 195Pt satellites, JPtC ≈ 60 Hz, 2× CH of C6H4), 125.01 (s, CH of C6H4), 128.33 (d, JPC = 11 Hz, CH of PPh2), 130.45 (s, CH of PPh2), 132.20 (d, 1JPC = 52 Hz, C–P of PPh2), 134.34 (d, JPC = 12 Hz, CH of PPh2), 135.92 (d, 4JPC = 6 Hz, C–N of C6H4), 139.35 (d, 4JPC = 8 Hz, CH of C6H4), 148.78 (d, 2JPC = 2 Hz, C–Pt of C6H4), 175.82 (s, COO), 178.48 (d, 2JPC = 145 Hz, NCN). The signal due to the second C–N of C6H4 was not detected. 31P{1H} NMR (161.90 MHz, CDCl3): δ 19.4 (s with 195Pt satellites, 1JPtP = 2964 Hz, PPh2). IR (DRIFTS): ν = 3409 br m, 3053 br m, 2953 br m, 1598 s, 1478 s, 1436 s, 1380 s, 1362 m, 1324 s, 1247 w, 1178 m, 1098 m, 1052 m, 1030 m, 999 w, 922 w, 804 w, 791 w, 749 s, 696 s, 673 w, 631 w, 540 m, 522 s, 507 s, 476 s cm−1. HRMS (ESI+) calc. for C37H30FeN2O2PPt ([M + H]+): 816.1037, found: 816.1032. Anal. calc. for C37H29FeN2O2PPt·1/2CH2Cl2 (833.4): C 53.67, H 3.56, N 3.37%. Found: C 53.66, H 3.38, N 3.40%.
:65 mg (67%). The crystal of 2Ph·AcOEt used for X-ray diffraction analysis was grown from dichloromethane/ethyl acetate/hexane.
1H NMR (399.95 MHz, CDCl3): δ 3.91 (vq, J′ = 2.0 Hz, 2H, fc), 4.13 (vt, J′ = 2.0 Hz, 2H, fc), 4.31–4.42 (m, 2H, fc), 5.05 (br vt, J′ = 1.8 Hz, 2H, fc), 6.37 (td, J = 7.6, 1.3 Hz, 1H, C6H4), 6.65 (ddd with 195Pt satellites, JPtC ≈ 60 Hz, J = 7.7, 2.2, 1.3 Hz, 1H, C6H4), 7.01 (td, J = 7.7, 1.3 Hz, 1H, C6H4), 7.12 (ddd, J = 8.2, 1.2, 0.2 Hz, 1H, C6H4), 7.31–7.41 (m, 7H, 1H of C6H4 and 6H of PPh2), 7.48–7.61 (m, 6H, 5H of NPh and 1H C6H4), 7.65 (dd, J = 7.9, 1.3 Hz, 1H, C6H4), 7.80–7.85 (m, 4H, PPh2), 8.13 (dt, J = 8.3, 0.9 Hz, 1H, C6H4).13C{1H} NMR (100.58 MHz, CDCl3): δ 70.67 (s, CH of fc), 71.35 (d, JPC = 7 Hz, CH of fc), 72.73 (s, CH of fc), 75.36 (br d, 1JPC ≈ 55 Hz, C–P of fc), 75.74 (d, JPC = 10 Hz, CH of fc), 78.97 (s, C–COO of fc), 111.83 (s, CH of C6H4), 112.59 (s with 195Pt satellites, JPtC ≈ 30 Hz, CH of C6H4), 112.74 (s, CH of C6H4), 120.96 (d, 3JPC = 9 Hz, C–N of C6H4), 123.39 (s, CH of C6H4), 124.13 (s, CH of C6H4), 124.23 (d with 195Pt satellites, JPtC ≈ 60 Hz, JPC = 2 Hz, CH of C6H4), 125.19 (s, CH of C6H4), 127.94 (s, CH of NPh), 128.23 (d, JPC = 11 Hz, CH of PPh2), 129.37 (s, CH of NPh), 129.71 (s, CH of NPh), 130.03 (d, 4JPC = 3 Hz, C–N of C6H4), 130.43 (d, JPC = 2 Hz, CH of PPh2), 131.60 (d, 1JPC = 51 Hz, C–P of PPh2), 134.95 (d, JPC = 12 Hz, CH of PPh2), 135.36 (s, C–N of NPh), 137.27 (d, 4JPC = 5 Hz, C–N of C6H4), 139.89 (d, JPC = 8 Hz, CH of C6H4), 148.69 (d, 2JPC = 2 Hz, C–Pt of C6H4), 174.27 (s, COO), 178.24 (d, 2JPC = 146 Hz, NCN). 31P{1H} NMR (161.90 MHz, CDCl3): δ 18.9 (s with 195Pt satellites, 1JPtP = 2990 Hz, PPh2). IR (DRIFTS): ν = 3412 br m, 3054 br m, 1598 s, 1502 m, 1473 m, 1458 s, 1436 m, 1412 m, 1355 m, 1323 s, 1252 w, 1177 m, 1097 m, 1029 m, 921 w, 821 w, 803 w, 772 w, 748 s, 696 s, 645 w, 619 w, 599 w, 538 w, 521 m, 506 s cm−1. HRMS (ESI+) calc. for C42H32FeN2O2PPt (M+): 878.1193, found: 878.1198. Anal. calc. for C42H32FeN2O2PPt·1/2CH2Cl2 (895.4): C 56.59, H 3.65, N 3.13%. Found: C 56.52, H 3.53, N 2.99%.
X-Ray crystallography
The diffraction data (±h ±k ±l, θmax ≈ 27.5°) were collected on a Bruker D8 VENTURE Kappa Duo diffractometer equipped with a PHOTON III detector and a Cryostream Cooler (Oxford Cryosystems) using Mo Kα radiation (λ = 0.71073 Å). The structures were solved by direct methods (SHELXT-201429) and refined by a full-matrix least-squares routine on F2 (SHELXL-201730). All nonhydrogen atoms were refined with anisotropic displacement parameters. The hydrogen atoms were included in their theoretical positions with their Uiso(H) set to a multiple of Ueq(C) of their bonding carbon atom (1.2 times for CH and CH2 groups and 1.5 times for methyl groups). The solvent molecules in the structure of the solvate 2Ph·1/2AcOEt were severely disordered in space near the crystallographic inversion centres, and their contribution to the overall scattering was therefore removed using PLATON SQUEEZE.31 All geometric parameters and structural diagrams were obtained using the PLATON program.32Selected crystallographic data and structure refinement parameters are available in the ESI.† The numerical values were rounded to one decimal place with respect to their estimated standard deviations (ESDs).
DFT computations
The Gaussian 16, Rev. A.0333 program package was used to perform all quantum chemical calculations and the hybrid functional B3LYP34 was employed as an established and reliable method to calculate transition metal compounds35 together with the double-ξ (dz) 6-31G(d)36 and triple-ξ (tz) 6-311+G(d,p) basis sets.35,37 Platinum was described by the LANL2TZ ECP and basis set.38 Dispersion forces were simulated by using the D3 dispersion correction with Becke-Johnson damping (D3BJ).39 Solvent calculations used the PCM SCRF model and the solvent dichloromethane.40All structures were optimized without any restrictions, employing the default grid (UltraFine). All local minima were verified as true minima by the absence of negative eigenvalues in the vibrational frequency analysis, providing thermochemical data at 298.15 K. If not stated otherwise, all discussed values are the ΔG298 values. GaussView41 and Molden42 were used for visualization. The coordinates of the optimized structures are available in ESI.†
Orbital composition analysis based on the Natural Atomic Orbitals (NAO)43 (at the B3LYP(d3bj)/6-311+G(d,p):LanL2TZ(Pt) level of theory) was performed using the Multiwfn software package (version 3.8).44 Molecular orbitals were visualized using the Avogadro program.45 Intrinsic bond orbital (IBO) analysis (at the B3LYP(d3)/def2-TZVP:sdd(Pt) level of theory)46–48 and visualization of the obtained orbitals were performed using IboView software.49
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support for this work was provided through the Institutional Strategy of TU Dresden “The Synergetic University” and the Charles University Research Centre program (project UNCE/SCI/014). Computational resources were provided by the e-INFRA CZ project (ID: 90254), supported by the Ministry of Education, Youth and Sports of the Czech Republic. We also thank the ZIH Dresden for allocating computational time to their high-performance computing system.References
- For early studies, see: (a) R. G. Pearson and D. A. Johnson, J. Am. Chem. Soc., 1964, 86, 3983 CrossRef CAS; (b) K. Saito and K. Masuda, Bull. Chem. Soc. Jpn., 1698, 41, 384 CrossRef; (c) A. J. C. Nixon and D. R. Eaton, Can. J. Chem., 1978, 56, 1928 CrossRef CAS.
- J. Vicente and M. T. Chicote, Coord. Chem. Rev., 1999, 193–195, 1143 CrossRef CAS.
- (a) N. Marion, E. C. Ecarnot, O. Navarro, D. Amoroso, A. Bell and S. P. Nolan, J. Org. Chem., 2006, 71, 3816 CrossRef CAS PubMed; (b) N. Marion, P. de Frémont, I. M. Puijk, E. C. Ecarnot, D. Amoroso, A. Bell and S. P. Nolan, Adv. Synth. Catal., 2007, 349, 2380 CrossRef CAS.
- (a) D. Walther, T. Döhler, N. Theyssen and H. Görls, Eur. J. Inorg. Chem., 2001, 2049 CrossRef CAS; (b) S. Rau, K. Lamm, H. Görls, J. Schöffel and D. Walther, J. Organomet. Chem., 2004, 689, 3582 CrossRef CAS.
- J.-P. Taquet, O. Siri, P. Braunstein and R. Welter, Inorg. Chem., 2006, 45, 4668 CrossRef CAS PubMed.
- R. H. Howard, C. Alonso-Moreno, L. M. Broomfield, D. L. Hughes, J. A. Wright and M. Bochmann, Dalton Trans., 2009, 8667 RSC.
- (a) S. Richeter, C. Jeandon, R. Ruppert and H. J. Callot, Chem. Commun., 2002, 266 RSC; (b) S. Richeter, C. Jeandon, J.-P. Gisselbrecht, R. Ruppert and H. J. Callot, J. Am. Chem. Soc., 2002, 124, 6168 CrossRef CAS PubMed.
- N. Allefeld, J. Bader, B. Neumann, H.-G. Stammler, N. Ignat’ev and B. Hoge, Inorg. Chem., 2015, 54, 7945 CrossRef CAS PubMed.
- L. Lavaud, Z. Chen, M. Elhabiri, D. Jacquemin, G. Canard and O. Siri, Dalton Trans., 2017, 46, 12794 RSC.
- (a) R. Navarro, J. García, E. P. Urriolabeitia, C. Cativiela and M. D. Diaz de Villegas, J. Organomet. Chem., 1995, 490, 35 CrossRef CAS; (b) O. Cantín, C. Cativiela, M. D. Díaz-de-Villegas, R. Navarro and E. P. Urriolabeitia, Tetrahedron: Asymmetry, 1996, 7, 2695 CrossRef.
- (a) M. Zábranský, I. Císařová and P. Štěpnička, Eur. J. Inorg. Chem., 2017, 2557 CrossRef; (b) P. Štěpnička, M. Zábranský and I. Císařová, J. Organomet. Chem., 2017, 846, 193 CrossRef; (c) F. Horký, I. Císařová, J. Schulz and P. Štěpnička, J. Organomet. Chem., 2019, 891, 44 CrossRef.
- For similar reactions with Rh(I)-acetylacetonates, see: (a) A. Jegorov, B. Kratochvíl, V. Langer and J. Podlahová, Inorg. Chem., 1984, 23, 4288 CrossRef CAS; (b) P. Štěpnička and I. Císařová, J. Chem. Soc., Dalton Trans., 1998, 2807 RSC.
- (a) T. G. Appleton, H. C. Clark and L. E. Manzer, Coord. Chem. Rev., 1973, 10, 335 CrossRef CAS; (b) F. R. Hartley, Chem. Soc. Rev., 1973, 2, 163 RSC.
- (a) R. G. Pearson, Inorg. Chem., 1973, 12, 712 CrossRef CAS; (b) J. Vicente, A. Arcas, D. Bautista and P. G. Jones, Organometallics, 1997, 16, 2127 CrossRef CAS.
- S. Fuertes, A. J. Chueca and V. Sicilia, Inorg. Chem., 2015, 54, 9885 CrossRef CAS PubMed.
- T. Strassner, Acc. Chem. Res., 2016, 49, 2680 CrossRef CAS PubMed.
- M. Zábranský, J. Soellner, F. Horký, I. Císařová, P. Štěpnička and T. Strassner, Eur. J. Inorg. Chem., 2019, 2284 CrossRef.
- (a) J. Podlaha, P. Štěpnička, J. Ludvík and I. Císařová, Organometallics, 1996, 15, 543 CrossRef CAS; (b) P. Štěpnička, Eur. J. Inorg. Chem., 2005, 3787 CrossRef.
- These values are larger than 1JPtP determined for a 2Me-type complex featuring (diphenylphosphino)acetate ligand (1JPtP = 2736 Hz; ref. 17). In addition, the larger 1JPtP constant for 2Ph corresponds with the shorter Pt–P bond in this compound: P. G. Waddell, A. M. Z. Slawin and J. D. Woollins, Dalton Trans., 2010, 39, 8260 Search PubMed.
- O. Bárta, P. Pinter, I. Císařová, T. Strassner and P. Štěpnička, Eur. J. Inorg. Chem., 2020, 575 CrossRef.
- S. Fery-Forgues and B. Delavaux-Nicot, J. Photochem. Photobiol. A: Chem., 2000, 132, 137 CrossRef CAS.
- A. Tronnier, A. Pöthig, S. Metz, G. Wagenblast, I. Münster and T. Strassner, Inorg. Chem., 2014, 53, 6346 CrossRef CAS PubMed.
- For a description and applications of this approach, see: (a) J. Schulz, F. Uhlík, J. M. Speck, I. Císařová, H. Lang and P. Štěpnička, Organometallics, 2014, 33, 5020 CrossRef CAS; (b) K. Škoch, I. Císařová, F. Uhlík and P. Štěpnička, Dalton Trans., 2018, 47, 16082 RSC; (c) K. Škoch, J. Schulz, I. Císařová and P. Štěpnička, Organometallics, 2019, 38, 3060 CrossRef; (d) P. Vosáhlo, J. Schulz, I. Císařová and P. Štěpnička, Dalton Trans., 2021, 50, 6232 RSC.
- The energy difference between the HOMO and LUMO calculated from the FMO eigenvalues obtained after geometry optimisation are 4.06 eV for 2Me, 3.94 eV for 3Me, 4.03 eV for 2Ph, and 2.88 eV for 3Ph.
- (a) G. Knizia, J. Chem. Theory Comput., 2013, 9, 4834 CrossRef CAS PubMed; (b) G. Knizia and J. E. M. N. Klein, Angew. Chem., Int. Ed., 2015, 54, 5518 CrossRef CAS PubMed.
- M. L. H. Green, J. Organomet. Chem., 1995, 500, 127 CrossRef CAS.
- R. M. Silverstein, F. X. Webster and D. J. Kiemle, Spectrometric Identification of Organic Compounds, Wiley, New York, 7th edn, 2005, ch. 3, p. 127 Search PubMed.
- F. Barrière and W. E. Geiger, J. Am. Chem. Soc., 2006, 128, 3980 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 Search PubMed.
- A. L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9 CrossRef CAS PubMed.
- (a) A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS; (b) A. L. Spek, Acta Crystallogr. D: Biol. Crystallogr., 2009, 65, 148 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Rev. A.03, Gaussian Inc., Wallingford, CT, 2016 Search PubMed.
- (a) A. D. Becke, Phys. Rev. A, 1988, 38, 3098 CrossRef CAS PubMed; (b) A. D. Becke, J. Chem. Phys., 1993, 98, 1372 CrossRef CAS; (c) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS; (d) B. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200 CrossRef CAS; (e) P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623 CrossRef CAS; (f) S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157 CrossRef CAS PubMed.
- (a) J.-P. Blaudeau, M. P. McGrath, L. A. Curtiss and L. Radom, J. Chem. Phys., 1997, 107, 5016 CrossRef CAS; (b) R. Ditchfield, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 724 CrossRef CAS; (c) M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. DeFrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654 CrossRef CAS; (d) M. S. Gordon, Chem. Phys. Lett., 1980, 76, 163 CrossRef CAS; (e) P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213 CrossRef CAS; (f) G. A. Petersson, A. Bennett, T. G. Tensfeldt, M. A. Al-Laham, W. A. Shirley and J. Mantzaris, J. Chem. Phys., 1988, 89, 2193 CrossRef CAS; (g) V. A. Rassolov, J. A. Pople, M. A. Ratner and T. L. Windus, J. Chem. Phys., 1998, 109, 1223 CrossRef CAS; (h) M. J. Frisch, J. A. Pople and J. S. Binkley, J. Chem. Phys., 1984, 80, 3265 CrossRef CAS.
- (a) T. Clark, J. Chandrasekhar, G. W. Spitznagel and P. V. R. Schleyer, J. Comput. Chem., 1983, 4, 294 CrossRef CAS; (b) R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650 CrossRef CAS; (c) A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639 CrossRef CAS; (d) L. A. Curtiss, M. P. McGrath, J.-P. Blaudeau, N. E. Davis, R. C. Binning and L. Radom, J. Chem. Phys., 1995, 103, 6104 CrossRef CAS.
- (a) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270 CrossRef CAS; (b) W. R. Wadt and J. P. Hay, J. Chem. Phys., 1985, 82, 284 CrossRef CAS; (c) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299 CrossRef CAS; (d) L. E. Roy, P. J. Hay and R. L. Martin, J. Chem. Theory Comput., 2008, 4, 1029 CrossRef CAS PubMed.
- (a) S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456 CrossRef CAS PubMed; (b) S. Grimme, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 211 CAS.
- (a) V. Barone and M. J. Cossi, J. Phys. Chem. A, 1998, 102, 1995 CrossRef CAS; (b) M. Cossi, N. Rega, G. Scalmani and V. J. Barone, J. Comput. Chem., 2003, 28, 669 CrossRef PubMed; (c) J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999 CrossRef CAS PubMed.
- R. D. Dennington, T. A. Keith and J. M. Millam, Gaussview, version 5.0.8, Semichem Inc., Shawnee Mission KS, 2009 Search PubMed.
- G. Schaftenaar and J. H. Noordik, J. Comp.-Aid. Mol. Design, 2000, 14, 123 CrossRef CAS PubMed.
- T. Lu and F. Chen, Acta Chim. Sin., 2011, 69, 2393 CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580 CrossRef CAS PubMed.
- (a) Avogadro: An Open-Source Molecular Builder and Visualization Tool, version 1.2.0, https://avogadro.openmolecules.net/ Search PubMed; (b) M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, J. Cheminf., 2012, 4, 17 CrossRef CAS PubMed.
- (a) F. Weigend, F. Furche and R. Ahlrichs, J. Chem. Phys., 2003, 119, 12753 CrossRef CAS; (b) F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- (a) A. D. Becke and E. R. A. Johnson, J. Chem. Phys., 2005, 123, 154101 CrossRef PubMed; (b) S. Grimme, J. Antony, S. Ehrlich and H. A. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed; (c) S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456 CrossRef CAS PubMed.
- D. Andrae, U. Haeussermann, M. Dolg, H. Stoll and H. Preuss, Theor. Chim. Acta, 1990, 77, 123 CrossRef CAS.
- G. Knizia, https://www.iboview.org.
Footnote |
| † Electronic supplementary information (ESI) available: Summary of crystallographic parameters, additional structure diagrams, electrochemical data and results from IBO analysis, copies of the NMR spectra, and cartesian coordinates for the DFT-optimized structures of 2R and 3R. CCDC 2284526 and 2284527. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3nj03729k |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2023 |