
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA complex of cobalamin with an organic peroxide†
Maria
Lehene
a,
Cezara
Zăgrean-Tuza
a,
Niculina
Hădade
 a,
Andreea
Aghion
a,
Raluca
Şeptelean
a,
Stefania D.
Iancu
b,
Adrian M.V.
Brânzanic
c and
Radu
Silaghi-Dumitrescu
*a
a,
Andreea
Aghion
a,
Raluca
Şeptelean
a,
Stefania D.
Iancu
b,
Adrian M.V.
Brânzanic
c and
Radu
Silaghi-Dumitrescu
*a
aDepartment of Chemistry, Babeş-Bolyai University, Str. Arany Janos Nr. 11, Cluj-Napoca RO-400028, Romania. E-mail: radu.silaghi@ubbcluj.ro
bFaculty of Physics, Babeş-Bolyai University, Str. Kogalniceanu 1, Cluj-Napoca RO-400084, Romania
cRaluca Ripan Institute for Research in Chemistry, Babeş-Bolyai University, Fantanele 30, 400294, Cluj-Napoca, Romania
First published on 6th September 2023
Abstract
Aquacobalamin reacts with m-chloroperoxybenzoic acid (MCPBA) to form a Co(III)-peroxo complex, as supported by UV-vis, NMR and resonance Raman spectra complemented by mass spectra and density functional calculations. At high MCPBA concentrations, degradation of cobalamin is observed as previously described.
Introduction
Peroxide complexes of biological transition metal centres are in some cases reactive intermediates in complex reaction mechanisms (e.g., in peroxidases, cytochromes P450, non-heme iron/manganese/copper monooxygenases, iron and copper bleomycin, vanadium-containing peroxidases and others).1–4 Stable peroxo complexes of biological transition metal centres are also known, such as with Co(III) cobalamin (Cbl(III)) or Co(II) bleomycin.5,6Until recently, the reactivity of cobalamin with oxidizing agents has been confined to processes where, especially with strong oxidizing agents, the corrin ring is covalently modified by oxygenation or halogenation, or where Co(I) or Co(II) are oxidized to Co(III) in an outer-sphere manner.5,7–13 Outer-sphere oxidants such as hexachloroiridate and peroxosulfate have no effect on Cbl(III) (data not shown), while hydrogen peroxide does form a stable and reversible complex, assigned as Co(III)-hydroperoxo based mostly on UV-vis and NMR spectra complemented by density functional (DFT) calculations. The UV-vis spectra (and ensuing properties, such as resonance Raman spectra) of cobalamin are notoriously insensitive to changes in the exogenous axial ligand; for instance, the maxima of aquacobalamin differ in position from those of hydroxocobalamin by less than 10 nm – and the differences between hydroxo- and hydroperoxocobalamin are even smaller. By contrast, the aromatic region of the 1H-NMR spectra does offer distinctively different signatures for each of these cobalamin complexes.5 More recently, another example of peroxide-cobalamin interaction has been described – with m-chloroperoxybenzoic acid (MCPBA).14 In this case Cbl was reported to act as a catalyst for MCPBA decomposition and subsequent oxidation of organic substrates. While such pseudoperoxidasic cycles have been reported previously for Cbl with hydrogen peroxide or tert-butyl hydroperoxide (tBuOOH) instead of MCPBA, in all of these previous instances activation of the peroxo bond was achieved via reaction with Cbl(II) rather than Cbl(III);8,9 one may expect a similar mechanism in the MCPBA case. A second, more recent example of oxygen–oxygen bond cleavage by aquacobalamin has been reported for peroxomonosulfate.15 In a related system – heptamethyl cobyrinate – a Co(III) adduct with MCPBA was detected at low temperature using UV-vis, IR, ESR, and ESI-MS.14 Somewhat related Co-peroxo complexes of geminal diols are as stable as to have been described by X-ray crystallography.16
Reported here are data showing that while a representative organic peroxide (tBuOOH) brings no significant changes to the NMR and UV-vis spectra of Cbl(III), MCPBA does yield a detectable complex, identifiable as such in UV-vis and NMR spectra corroborated with DFT calculations.
Materials and methods
Hydroxocobalamin hydrochloride (HOCbl, ≥98%) was obtained from Sigma–Aldrich (Munich, Germany) and used as received and hydrogen peroxide 30% was obtained from ChemPUR. MCPBA was from Sigma–Aldrich (Munich, Germany) and was employed in the form of a 2 M stock solution in acetone. tBuOOH was from Sigma–Aldrich (Munich, Germany), 70% solution in H2O. The buffer solution was 50 mM phosphate, pH 7, unless otherwise specified.UV-vis, NMR, resonance Raman stopped-flow and DFT data were obtained using a methodology previously described for the reaction of hydrogen peroxide with aquaCbl, as detailed below.5
UV-vis spectra were performed on a Cary 50 UV-vis spectrophotometer (Varian, Inc., Foster City, CA, USA). Stopped-flow spectra were collected on a Biologic SFM-300 system equipped with three syringes and sequential mixing, with a high-speed diode array detector. The stopped-flow data were processed with the SPECFIT32 software package (BioLogic system, Claix, France) using a single process for fitting. Time course graphs are listed in the Figures at 435 nm, specific for the peroxide adduct.
The NMR spectra were recorded immediately after preparing the samples, at 22 °C unless otherwise stated, diluting the sample (at concentrations indicated in text and Figure legends) with D2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volume ratio), on a 400 MHz Bruker instrument. The NMR titration of aquaCbl with MCPBA was performed at 20 °C on a 500 MHz Bruker Avance spectrometer. The solvent of choice was pH 7 Britton–Robinson universal buffer prepared in D2O. 1 mM aquaCbl was titrated with incremental amounts of MCPBA to final concentrations, as indicated in Fig. S9 (ESI†) caption. A water-suppression pulse sequence was used for these measurements.
:1 volume ratio), on a 400 MHz Bruker instrument. The NMR titration of aquaCbl with MCPBA was performed at 20 °C on a 500 MHz Bruker Avance spectrometer. The solvent of choice was pH 7 Britton–Robinson universal buffer prepared in D2O. 1 mM aquaCbl was titrated with incremental amounts of MCPBA to final concentrations, as indicated in Fig. S9 (ESI†) caption. A water-suppression pulse sequence was used for these measurements.
Raman spectra were measured at 22 °C immediately after preparing the samples, on a Renishaw inVia Raman spectrometer coupled with a Leica microscope. 10 μl of the sample was dropped on a microscope slide covered with aluminum foil. The 532 nm laser line with a power of 50 mW was focused on the sample by using a 5X objective. Each spectrum is represented as an average of 4 accumulations, 4 seconds each.
High-resolution mass spectra (HRMS) were recorded on an LTQ ORBITRAP XL mass spectrometer (ThermoScientific) using positive electrospray ionization. The instrument was externally calibrated. The samples were prepared at room temperature (22 °C) and then inserted into the instrument at times indicated in the Figure legends. The following conditions were used: source voltage, 3.2 kV; sheath and auxiliary gas flow, 8 and 5 arbitrary units, respectively; vaporizer temperature 50 °C, capillary temperature 275 °C, analyzer temperature 26 °C; capillary voltage, 28 V; tube lens voltage, +110 V. The number of microscans was set to three.
For DFT calculations, the Gaussian09 software package17 was employed following the methodology previously tested and described for related Cbl complexes.5 The Cbl models were truncated, with the lateral substituents on the corrin as well as the methyl groups on the benzimidazole replaced by hydrogen. Gas-phase geometries and frequency analyses were computed with the aid of the B3LYP18,19 functional at the def2-SV(P)20 double-zeta basis set level. Long-range interactions were accounted for by the use of Grimme's D3 dispersion correction.20 TD-DFT derived21 UV-Vis spectra were computed in the C-PCM solvent continuum adapted for an aqueous environment.22 For the latter property the B3PW9118,23 functional outperformed B3LYP and was subsequently employed. Data for the lowest-spin states are shown for each model. In terms of methodology choice for DFT calculations, previous studies from us,5,24–28 but more importantly from Brunold and especially from Kozlowski on related systems have each indicated advantages for a functional or another.29–42 We have, more in general, shown that even within the same class of compounds the performance of a functional can vary widely depending on the property sought, or on apparently simple changes in ligands (e.g., axial ligation in heme complexes).43–46 The methodology employed here was selected for its ability to best mimic trends in UV-vis spectra as described in our previous study on hydroperoxocobalamin, thus also allowing consistency between the two sets of data. In terms of general structural features, the cobalamin-peroxo adducts (unlike, e.g., the case of Cbl(I)) offer no particular challenge – so that the choice of functional should affect the results at the level of detail (which, nevertheless, in predicting trends in UV-vis maxima of Cbl adducts, is essential).
Results and discussion
As shown in Fig. 1, upon reaction with MCPBA the UV-vis spectrum of aquacobalamin (H2OCbl+) undergoes bathochromic shifts reminiscent of the hydroperoxocobalamin adduct.5 Isosbestic points at 461 nm, 340 and 367 nm suggest the formation of a single new product, which can in principle be assigned as a peroxoacid(MCPBA)-Cbl(III) complex analogous to the previously-reported cobalamin(III)-hydroperoxo complex. A 0.2 mM affinity for MCPBA binding to Cbl(III) may be deduced from the data in Fig. 1 (based on the absorbance changes at 351 nm; see also Fig. S2 (ESI†) for titration curves at 490 and 525 nm). At larger MCPBA concentrations (above 5–10 mM) a slight decrease in intensity in the visible region of the spectrum is observed, consistent with previous observations of a chemical reaction with degradation of MCPBA and generation of reactive redox species.14 | ||
| Fig. 1 UV-vis spectra of 50 μM aquacobalamin treated with MCPBA (upper panel) and the titration curve at pH 7 monitored at 351 nm (lower panel). | ||
No changes in the shape of the UV-vis spectrum of H2OCbl+ were observed with tBuOOH (ESI,† Fig. S1). If indeed the Cbl complexes with hydrogen peroxide and with MCPBA entail a deprotonated peroxo group, and then the pKa values may be responsible for the fact that tBuOOH does not form such a complex: the pKa of tBuOOH (12.7) is one unit higher than that of H2O2, and distinctly higher than that of MCPBA, too (7.6). At larger tBuOOH concentrations (≥10 mM), the spectrum of Cbl does decay slowly – in line with the known intrinsic instability of tBuOOH and possibly with catalysis from Cbl; however, evidence for a detectable tBuOOH-Cbl complex as an intermediate species in such reactions, if any, has so far not been identified.
The kinetics of the aquacobalamin + MCPBA reaction may be followed with stopped-flow UV-vis spectroscopy (cf.Fig. 2 and ESI,† Fig. S3 and S4) at MCPBA concentrations distinctly higher than those in Fig. 1. Between 25 and 75 mM MCPBA, the experimentally-observed changes in absorbance increase with the MCPBA concentration as expected. However, this range is too narrow to allow a meaningful estimation of a rate constant, and the trend is reversed at 100 mM MCPBA. At all concentrations, the data may be fitted with a single process, where species A (cf.Fig. 2 and Fig. S3 and S4, ESI†) is essentially identical to H2OCbl+ and species B exhibits a maximum at 560 nm, characteristic of partially degraded corrin.5,11,12,47,48 This is consistent with the behavior on Cbl in the presence of such high MCPBA concentrations more extensively described elsewhere, highlighting the occurrence of peroxide activation and ensuing oxidative activity.14
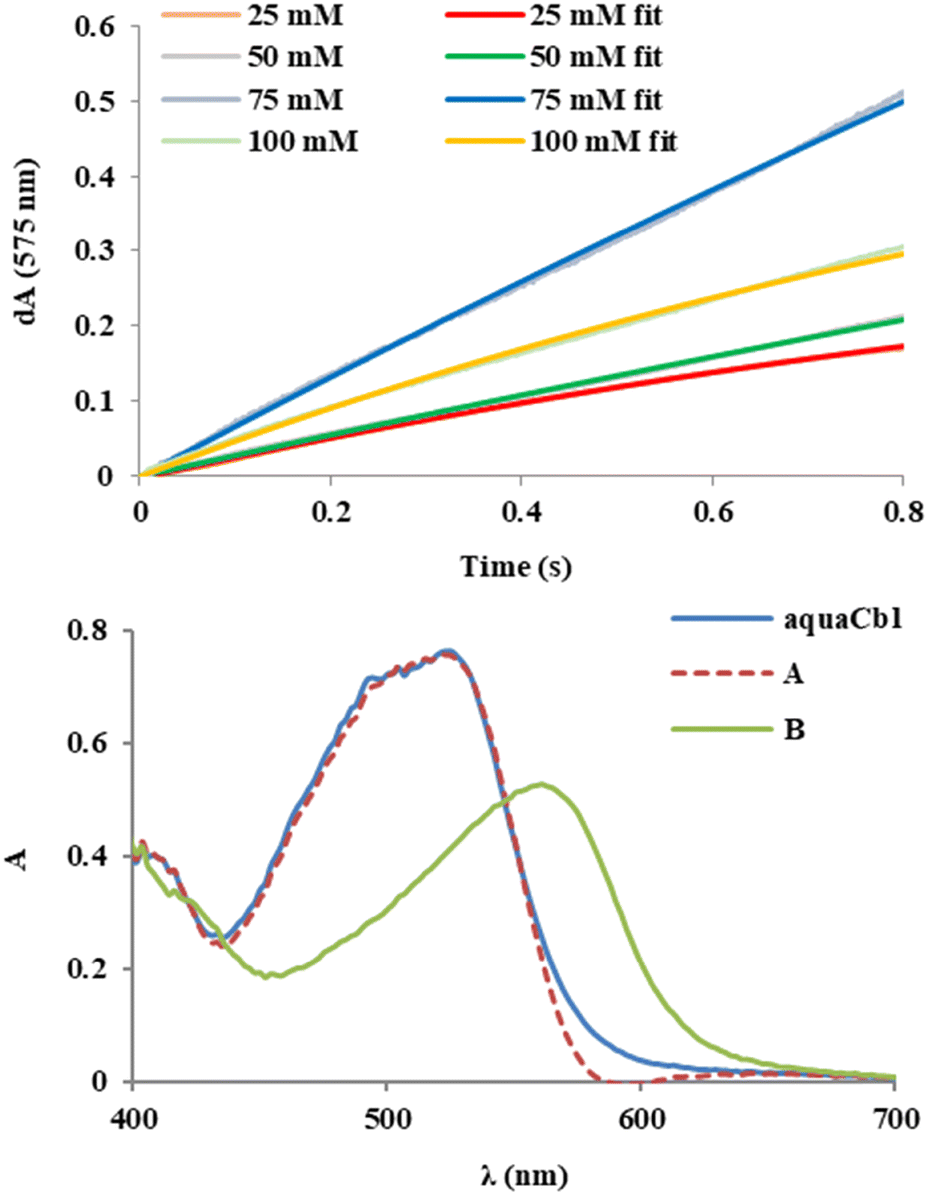 | ||
| Fig. 2 Upper panel: absorbance time courses from stopped-flow UV-vis experiments monitoring the reaction of 0.3 mM aquacobalamin with varying MCPBA concentrations at 575 nm. Lower panel: representative spectra of species A and B at pH 7, resulted from fitting of the stopped-flow data 50 mM MCPBA. See also Fig. S3 and S4 (ESI†). | ||
Fig. 3 shows NMR spectra collected for H2OCbl+ mixed with tBuOOH and with MCPBA, respectively (with further details shown in Fig. 4, and proton labelling shown in Fig. S9, ESI†). The spectrum of the Cbl-tBuOOH mixture retains almost all of the features of H2OCbl+; slight shifts are seen in the B7, R1 and C10 signals – possibly due to non-covalent interaction with tBuOOH, but decidedly not to tBuOO- coordination. A Cbl(III) complex with the neutral tBuOOH form is less likely but cannot be entirely excluded, considering the similar coordination properties of the tBuOOH hydroxyl vs. water, and the fact that density functional theory (DFT) calculations suggest much smaller spectral differences between H2O and H2O2 Cbl complexes, compared to H2O vs. a deprotonated peroxide.5 By contrast, the Cbl-MCPBA NMR spectrum is distinctly different from aqua as well as from other possible candidates (hydroxo, base-off aqua). Moreover, the MCPBA signals themselves are now all shifted compared to pure MCPBA, consistent with coordination to Cbl. The minor additional signals seen in the 7.5–6 ppm region of the Cbl-MCPBA spectrum may be due to Cbl-bound MCPBA or to degradation products in line with previous proposals.14 The concentration dependence of the Cbl-MCPBA 1H-NMR spectra (cf.Fig. 4) confirms the latter observation, with the Cbl signals mostly degraded at 100 mM MCPBA. The titration also confirms that (1) only one adduct with MCPBA is formed, and (2) saturation of Cbl occurs below 10 mM, in line with the UV-vis titration of Fig. 1.
 | ||
| Fig. 3 1H-NMR spectra of Cbl-peroxo mixtures and of reference complexes. Peaks are assigned based as previously described.5 Conditions: 5 mM Cbl(III) (aqua/hydroxo or cyano, as indicated), 5 mM ligand (MCPBA, tBuOOH), 50 mM phosphate buffer pH 7 for all samples except aqua base-off (pH 3, dimethylbenzimidazole deligated) and Cbl-OH (pH 10). | ||
 | ||
| Fig. 4 1H-NMR titration of aquaCbl with MCPBA. Conditions: concentrations indicated in the figure, pH 7 in Britton–Robinson universal buffer (40 mM each borate, acetate, phosphate). | ||
DFT geometry optimization of Cbl models with a neutral MCPBA ligand bound to Co either via the (carboxyl)OH group or via the Cl atom yield almost identical energies, with the OH slightly favoured by only ∼1 kcal mol−1, cf. Table S1 (ESI†). Since a Co-Cl(MCPBA) complex is extremely unlikely, one may further assume that the same is true for the Co–(H)OO–C(O)- isomer. This is in line with the fact that indeed organic acids are not known to be effective ligands to Cbl. On the other hand, the geometric parameters predicted for the deprotonated MCPBA as a ligand, as well as for tBuOO– and tBuOOH, are very similar to those previously reported for H2O2 and HOO– complexes of Cbl at the same level of theory (cf. Table S1 and Fig. S5, ESI†).5 Time-dependent DFT (TD-DFT) simulations of the UV-vis spectra of Co(III)-OOC(O)-MCPBA (cf. Table S2 and Fig. S6, S7, ESI†) reveal maxima at wavelengths intermediary between those of aqua and of hydroxo Cbl, which (especially taking into account the limits of the computational method for such complexes) are in line with the experimental observations.5,44 Elongation of the CoO–OH bond in a hydroperoxo peroxide-Cbl model and in a heme ferric-hydroperoxo model by 1 Å was found to entail energy costs of ∼45 kcal mol−1 in both cases, with a crossing onto the triplet/quartet state and with no major changes in partial atomic charges on the Co/Fe or on the peroxo oxygen atoms. The latter suggests, as expected, that oxygen–oxygen bond cleavage in both cases would be homolytic (hence, no change in formal oxidation state from the Fe(III)/Co(III)-hydroperoxo stage) and would lead to a hydroxyl radical and a ferryl/chromyl unit. However, as shown in Fig. 5, while the ferryl does show the classical Fe(IV)-oxo center spin density distribution (50% on each bonding partner), the chromyl unit is clearly a Co(III)-oxyl (diamagnetic Co(III) with an S = 1/2 oxyl ligand) similarly to other late transition metals such as Cu where “high-valent oxo” complexes in general tend to feature oxyl ligands, within the framework of the recently coined “oxo wall” paradigm.1,49–53
 | ||
| Fig. 5 Relative energies for S = 1 Co(III)cobalamin-hydroperoxo and S = 3/2 Fe(III)heme-hydroperoxo adducts after elongation of the O–O bonds by ∼1 Å relative to the equilibrium state. Also shown are Mulliken spin densities on the Co, Fe and oxygen atoms. Shown in parentheses are the relative energies of the low-spin states. The equilibrium geometries of the low-spin states (S = 0 and S = 1/2, respectively) are taken as references. From UB3LYP/def2-SV(P)/D3 calculations. | ||
The resonance Raman spectra of Cbl are dominated by the corrin ring and relatively insensitive to the nature of the exogenous ligand. Fig. 6 shows that the resonance Raman spectra of hydroperoxo- and MCPBA-Cbl(III) are very similar to each other, and slightly different from that of H2OCbl+, suggesting that in both cases an anionic peroxo moiety is coordinated to the corrin. The peroxo/MCPBA vs. aqua differences are observable, especially in the signals at ∼1495, 795, 725 and 630 cm−1. Possibly of more interest is the band observed for the control MCPBA sample at 873 cm−1 (cf.Fig. 6 and Fig. S8, ESI†), which is known54 to be due to the O–O stretching. This band overlaps with a shoulder in the aquaCbl spectrum. Importantly, in the MCPBA + Cbl sample the shoulder at 873 cm−1 is distinctly weaker than in the aquaCbl sample; an increase in intensity would have been expected in a sample where the signals of aquaCbl and MCPBA overlap physically without any chemical interaction. This may be taken as evidence against a scenario where aquaCbl and MCPBA are still intact. Instead, also importantly, a new weak shoulder at 866 cm−1 appears in the MCPBA + Cbl spectrum, tentatively assignable as the O–O stretching of the Cbl-MCPBA complex. For comparison, in the H2O2 + Cbl sample (also shown in Fig. 6 and Fig. S8, ESI†), a similar shift is seen compared to aquaCbl, but only to 870 cm−1.
 | ||
| Fig. 6 Resonance Raman spectra of Cbl in the presence or absence of hydrogen peroxide or MCPBA. Conditions: 1 mM H2OCbl+, 25 mM H2O2, 10 mM MCPBA, 50 mM phosphate pH 7, 22 °C. | ||
In our initial attempts to employ mass spectrometry (HRMS ESI) to detect the Co(III)-hydroperoxide complex of cobalamin, only the signal corresponding to pentacoordinated Cbl was identified, either in aqua5 or in peroxo cobalamin. However, upon recording the ESI spectra immediately after mixing Cbl with MCPBA in water (∼2 minutes from mixing to measurement), a signal could clearly be identified (cf.Fig. 7) corresponding to a Cbl-MCPBA adduct with the (m/z) found at 1500.5548 [M]+ (vs. calculated 1500.5565), alongside the expected pentacoordinated 1329.5716 [M - mCPBA]+ as well as several z = 2 signals for the Cbl-MCPBA. As shown in ESI,† Fig. S10, in a sample incubated for 2 extra minutes before injecting into the instrument, the relative abundance of the Cbl-MCPBA peak decreases and, more importantly, several new signals appear which do not fit the mass of intact Cbl complexes and are thus taken as evidence for Cbl degradation in line with above-discussed considerations. At 30 minutes after mixing, neither the intact pentacoordinated Cbl nor the Cbl-MCPBA peaks were visible. This time evolution of the mass spectra was expected: based on the NMR titration, a 10 mM concentration of MCPBA was required in the HRMS experiment in order to have the cobalt sites saturated. However, as seen in the UV-vis spectra of Fig. 1 as well as in the NMR titration of Fig. 4, at 10–20 mM there is already a decrease in intensity of the Cbl signals, suggestive of partial degradation; such degradation is expected to be accelerated by the HRMS protocol (see capillary and vaporizer temperatures in figure legends).
 | ||
| Fig. 7 Upper panel: ESI(+)-HRMS (m/z) for the Cbl-MCPBA adduct. Calculated for C69H93CoN13O17PCl 1500.5565, found: 1500.5548 [M]+, 1329.5716 [M-mCPBA]+, 675.7747 [M-mCPBA + Na]2+, 664.7855 5716 [M-mCPBA + H]2+. Lower panel: spectrum of the starting material (hydroxocobalamin). Conditions: 1 mM H2OCbl+, 10 mM MCPBA, 50 mM phosphate pH 7. | ||
Conclusions
To conclude, Cbl(III) forms a peroxoacid complex with deprotonated m-chloroperoxybenzoic acid, as supported by UV-vis, 1H-NMR and resonance Raman spectroscopic data, density functional calculations, and mass spectrometry. By contrast, tBuOOH does not appear to be able to form a peroxo complex with Cbl(III). These findings suggest that the peroxide coordination chemistry of cobalamin does extend beyond simply hydrogen peroxide,5 but that not all peroxo compounds should be expected to bind to Cbl(III).Author contributions
ML: conceptualization, investigation, validation, formal analysis, visualization, writing – original draft, review and editing; CZT: conceptualization, methodology, investigation, validation, formal analysis, visualization; NH: investigation, methodology, formal analysis, visualization; AA: investigation; RS: methodology, investigation, validation; SDI: methodology, investigation; BAMV – methodology, investigation, validation; RSD – conceptualization, investigation, validation, visualization, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding from the Romanian Ministry of Education and Research (projects PN-III-P4-ID-PCCF-2016-0142 and PN-III-P1-1.1-PD-2021-0279) and from the European Union Social Fund (Entrepreneurship for innovation through doctoral and postdoctoral research POCU/380/6/13/123886) is gratefully acknowledged. Prof S. V. Makarov (Ivanovo State University of Chemistry and Technology) and Artiom Gaina-Gardiuta (UBB) are thanked for helpful discussions.Notes and references
- R. Silaghi-Dumitrescu, Struct. Bond, 2013, 150, 97–118 CrossRef CAS PubMed.
- Y. Zhang and J. A. Gascon, J. Inorg. Biochem., 2008, 102, 1684–1690 CrossRef CAS PubMed.
- S. P. de Visser, D. Kumar, R. Neumann and S. Shaik, Angew. Chem., Int. Ed., 2004, 43, 5661–5665 CrossRef CAS PubMed.
- H. P. Hersleth, U. Ryde, P. Rydberg, C. H. Gorbitz and K. K. Andersson, J. Inorg. Biochem., 2006, 100, 460–476 CrossRef CAS PubMed.
- M. Lehene, D. Plesa, S. Ionescu-Zinca, S. D. Iancu, N. Leopold, S. V. Makarov, A. M. V. Brânzanic and R. Silaghi-Dumitrescu, Inorg. Chem., 2021, 60, 12681–12684 CrossRef CAS PubMed.
- F. Carrascoza, M. Surducan, L. A. Eriksson and R. Silaghi-Dumitrescu, Inorg. Chim. Acta, 2020, 509, 119682 CrossRef CAS.
- W. Sand, M. Dopson, G. Levicán, Á. Sandoval, A. Ferrer, J. Rivera, C. Zapata, J. Norambuena, Á. Sandoval, R. Chávez and O. Orellana, Front. Microbiol., 2016, 7, 748 Search PubMed.
- D. S. Salnikov, S. V. Makarov and O. I. Koifman, New J. Chem., 2021, 45, 535–543 RSC.
- P. W. Johns, A. Das, E. M. Kuil, W. A. Jacobs, K. J. Schimpf and D. J. Schmitz, Int. J. Food Sci. Technol., 2015, 50, 421–430 CrossRef CAS.
- G. Tsiminis, E. P. Schartner, J. L. Brooks and M. R. Hutchinson, Appl. Spectrosc. Rev., 2017, 52, 439–455 CrossRef.
- R. S. Dassanayake, M. M. Farhath, J. T. Shelley, S. Basu and N. E. Brasch, J. Inorg. Biochem., 2016, 163, 81–87 CrossRef CAS PubMed.
- I. A. Dereven’kov, S. V. Makarov, N. I. Shpagilev, D. S. Salnikov and O. I. Koifman, Biometals, 2017, 30, 57–764 CrossRef PubMed.
- I. A. Dereven’kov, D. S. Salnikov, S. V. Makarov, G. R. Boss and O. I. Koifman, Dalton Trans., 2013, 42, 15307–15316 RSC.
- J. Cheng, Y. Shiota, M. Yamasaki, K. Izukawa, Y. Tachi, K. Yoshizawa and H. Shimakoshi, Inorg. Chem., 2022, 61, 9710–9724 CrossRef CAS PubMed.
- I. A. Dereven’kov, E. S. Sakharova, V. S. Osokin and S. V. Makarov, Int. J. Mol. Sci., 2022, 23, 11907 CrossRef PubMed.
- P. Kumar, S. V. Lindeman and A. T. Fiedler, J. Am. Chem. Soc., 2019, 141, 10984–10987 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revis. E.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef.
- R. Ditchfield, Mol. Phys., 1974, 27, 789–807 CrossRef CAS.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 12947–12954 CrossRef PubMed.
- I. A. Dereven’kov, P. A. Ivlev, C. Bischin, D. S. Salnikov, R. Silaghi-Dumitrescu, S. V. Makarov and O. I. Koifman, J. Biol. Inorg. Chem., 2017, 22, 969–975 CrossRef PubMed.
- D. S. Salnikov, R. Silaghi-Dumitrescu, S. V. Makarov, R. van Eldik and G. R. Boss, Dalton Trans., 2011, 40, 9831–9834 RSC.
- M. Surducan, S. V. Makarov and R. Silaghi-Dumitrescu, Polyhedron, 2014, 78, 72–84 CrossRef CAS.
- I. A. Dereven’kov, D. S. Salnikov, S. V. Makarov, M. Surducan, R. Silaghi-Dumitrescu and G. R. Boss, J. Inorg. Biochem., 2013, 125, 32–39 CrossRef PubMed.
- D. S. Salnikov, I. A. Dereven’kov, S. V. Makarov, E. S. Ageeva, A. Lupan, M. Surducan and R. Silaghi-Dumitrescu, Rev. Roum. Chim., 2012, 57, 353–359 CAS.
- I. G. Pallares and T. C. Brunold, Inorg. Chem., 2014, 53, 7676–7691 CrossRef CAS PubMed.
- P. M. Kozlowski, V. V. Nazarenko and A. A. Jarzecki, Inorg. Chem., 2006, 45, 1424–1426 CrossRef CAS PubMed.
- A. P. Ghosh, P. Lodowski and P. M. Kozlowski, Phys. Chem. Chem. Phys., 2022, 24, 6093–6106 RSC.
- J. Kuta, S. Patchkovskii, M. Z. Zgierski and P. M. Kozlowski, J. Comput. Chem., 2006, 27, 1429–1437 CrossRef CAS PubMed.
- K. Mieda-Higa, A. Al Mamun, T. Ogura, T. Kitagawa and P. M. Kozlowski, J. Raman Spectrosc., 2020, 51, 1331–1342 CrossRef CAS.
- H. Solheim, K. Kornobis, K. Ruud and P. M. Kozlowski, J. Phys. Chem. B, 2011, 115, 737–748 CrossRef CAS PubMed.
- M. Kumar and P. M. Kozlowski, Coord. Chem. Rev., 2017, 333, 71–81 CrossRef CAS.
- A. P. Ghosh, P. Lodowski, A. Chmielowska, M. Jaworska and P. M. Kozlowski, J. Catal., 2019, 376, 32–43 CrossRef CAS.
- T. Andruniow, M. Z. Zgierski and P. M. Kozlowski, J. Am. Chem. Soc., 2001, 123, 2679–2680 CrossRef CAS PubMed.
- P. M. Kozlowski, B. D. Garabato, P. Lodowski and M. Jaworska, Dalton Trans., 2016, 45, 4457–4470 RSC.
- M. D. Liptak and T. C. Brunold, J. Am. Chem. Soc., 2006, 128, 9144–9156 CrossRef CAS PubMed.
- T. A. Stich, N. R. Buan and T. C. Brunold, J. Am. Chem. Soc., 2004, 126, 9735–9749 CrossRef CAS PubMed.
- E. D. Greenhalgh, W. Kincannon, V. Bandarian and T. C. Brunold, Biochemistry, 2022, 61, 195–205 CrossRef CAS PubMed.
- A. J. Reig, K. S. Conrad and T. C. Brunold, Inorg. Chem., 2012, 51, 2867–2879 CrossRef CAS PubMed.
- A. M. V. Brânzanic, U. Ryde and R. Silaghi-Dumitrescu, J. Inorg. Biochem., 2020, 203, 110928 CrossRef PubMed.
- A. A. A. Attia, D. Cioloboc, A. Lupan and R. Silaghi-Dumitrescu, J. Inorg. Biochem., 2016, 165, 49–53 CrossRef CAS PubMed.
- A. A. A. Attia, A. Lupan and R. Silaghi-Dumitrescu, RSC Adv., 2013, 3, 26194–26204 RSC.
- R. Silaghi-Dumitrescu and I. Silaghi-Dumitrescu, J. Inorg. Biochem., 2006, 100, 161–166 CrossRef CAS PubMed.
- H. M. Abu-Soud, D. Maitra, J. Byun, C. E. A. Souza, J. Banerjee, G. M. Saed, M. P. Diamond, P. R. Andreana and S. Pennathur, Free Radicals Biol. Med., 2012, 52, 616–625 CrossRef CAS PubMed.
- D. Maitra, I. Ali, R. M. Abdulridha, F. Shaeib, S. N. Khan, G. M. Saed, S. Pennathur and H. M. Abu-Soud, PLoS One, 2014, 9, 5–12 CrossRef PubMed.
- V. A. Larson, B. Battistella, K. Ray, N. Lehnert and W. Nam, Nat. Rev. Chem., 2020, 4, 404–419 CrossRef CAS PubMed.
- E. Andris, R. Navrátil, J. Jašík, M. Srnec, M. Rodríguez, M. Costas and J. Roithová, Angew. Chem., Int. Ed., 2019, 58, 9619–9624 CrossRef CAS PubMed.
- R. Silaghi-Dumitrescu, M. Surducan and A. Papp, Croat. Chem. Acta, 2014, 87, 75–78 CrossRef CAS.
- A. Imre, A. Mot and R. Silaghi-Dumitrescu, Cent. Eur. J. Chem., 2012, 10, 1527–1533 Search PubMed.
- R. Silaghi-Dumitrescu, Rev. Chim., 2007, 58, 461–464 CAS.
- Z. Tang, J. Xiao, F. Li, Z. Ma, L. Wang, F. Niu and X. Sun, ACS Omega, 2020, 5, 10451–10458 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: UV-vis spectra of Cbl + tBuOOH mixtures, titration curves for aquaCbl reacting with MCPBA at pH 7, monitored at 490 nm and 525 nm, representative spectra of species A and B at pH 7, room temperature, resulting from the fitting of the stopped-flow data upon the reaction of aquaCbl, absorbance time course from stopped-flow UV-vis experiments at varying MCPBA concentrations monitored at 525 nm, DFT-optimized geometries for Cbl-MCPBA (A) and Cbl-tBuOOH (B) complexes, DFT-derived data of Cbl-MCPBA and Cbl-tBuOOH models, TD-DFT-derived wavelengths (nm) and oscillator strengths (OS) for Cbl-MCPBA- and Cbl-tBuOO- models, main orbitals responsible for the main contributors to the two main bands in the TD-DFT spectra in Cbl complexes with MCPBA and tBuOOH, details of resonance Raman spectra of Cbl in the presence or absence of hydrogen peroxide or MCPBA, mass spectra of Cbl with MCPBA at 4 and at 30 minutes, and Cbl structure with atom labelling employed for 1H-NMR assignments. See DOI: https://doi.org/10.1039/d3nj03307d |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2023 |