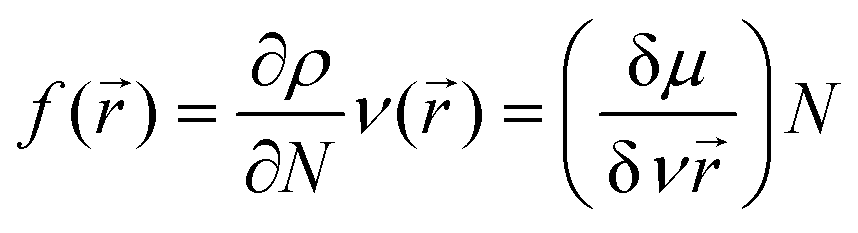DOI:
10.1039/D3NJ02910G
(Paper)
New J. Chem., 2023,
47, 17853-17870
Synthesis, characterization, biological evaluation, DFT and molecular docking studies of (Z)-2-((2-bromo-4-chlorophenyl)imino)methyl)-4-chlorophenol and its Co(II), Ni(II), Cu(II), and Zn(II) complexes†
Received
23rd June 2023
, Accepted 12th September 2023
First published on 19th September 2023
Abstract
An equimolar reaction of 5-chlorosalicylaldehyde and 2-bromo-4-chloroaniline yielded the Schiff base (Z)-2-((2-bromo-4-chlorophenyl)imino)methyl)-4-chlorophenol) (HL), which was used for complexation to Co2+, Ni2+, Cu2+, and Zn2+ metal salts. Elemental and thermogravimetric analyses, conductivity measurements, powder X-ray diffraction, nuclear magnetic resonance (1H and 13C), infrared, ultraviolet-visible, energy dispersive X-ray-scanning electron and mass spectroscopies confirm the Schiff base structure and show mono-nuclear homoleptic complexes of the type ML2 for all metal salts used. The crystal structures of Ni2+ and Cu2+ complexes revealed a perfect square planar geometry around the metal ions, with the ligand acting as bidentate through oxygen and nitrogen atoms of the phenolic and azomethine groups, respectively. The antimicrobial potential of the compounds was evaluated on some selected pathogenic bacteria consisting of Gram-positive (Staphylococcus aureus and Bacillus subtilis) and Gram-negative (Klebsiella pneumonioe and Pseudomonas aeruginosa) strains using an in vitro assay. Antioxidant activity was evaluated using the DPPH assay. The complexes showed enhanced activity over the free Schiff base ligand in all the assays. Toxicity studies on WISH-ATCC-CCL-25, human epithelial amnion (normal liver cell lines), and MRC-5-ATCC-CCL-171 (normal human lung fibroblast cell lines) revealed that at lower concentrations, the complexes did not affect the cell lines. A computational study was deployed to investigate the electronic properties of the ligands and the complexes relating to their stability, reactivity, and biological potential. The computational data corroborated sufficiently with the experimental findings. Molecular docking studies demonstrated the compounds' mechanism of action and identified potential binding sites consistent with the in vitro assays. Hirshfeld surface analysis was also performed on selected compounds to reveal qualitative and quantitative intermolecular interactions within the topology crystal network of crystal structures.
1. Introduction
Research and development of metallodrugs in medical inorganic chemistry spans back a century with pioneering work such as that of Paul Ehrlich on Salvarsan®, an arsenic-based antibacterial agent that complemented mercury based drugs.1,2 While the compound has many side effects and was difficult to handle (due to its air instability), it remained a primary method of treatment against syphilis for three decades until the emergence of Penicillin® in the 1940s. Another pioneering example is cis-platin, with some major developmental strides surfacing over the last few decades on clinical trials of these chemotherapeutic drugs.1–3 The development for metallo-based drugs is slow, primarily due to their more severe side effects, as opposed to the organic/biological alternatives that dominate the market. Thus, there is a research drive to look beyond the initial success of cis-platin and develop metallo-based drugs with fewer side effects. While current medications have proven successful in the fight against a number of diseases, there is concern over a sharp rise in bacterial antibiotic resistance,4–7 which could render medications that were once effective useless. The problem of bacterial resistance to antimicrobial agents is a multifaceted public health threat that must be addressed through coordinated efforts.8,9 Several factors have been linked to this threat, the most notable of which are natural selection and mutations, which cause bacteria to develop resistance pathways that can alter drug binding sites within a molecular target.10–12 To combat this phenomenon, several compounds are being researched for their potential use as lead compounds to combat bacterial resistance,13 amongst these inorganic and organometallic complexes of first row transition metals have recently gained additional attention because of their therapeutic properties.14 According to research, both the metal and the ligand play a pivotal role in the overall biological properties of the complexes, i.e. each metal has its own mode of action against a specific biological system, while a coordinated ligand aids in complex stabilization and improvement of mechanism of action against the target.15 One of the strategies in drug design and development process, is the incorporation of electron withdrawing groups such as a nitro group or halogen atoms within a molecule due to their ability to improve membrane penetration and increase the drug's lifespan through slowing the carbolic pathway, which is responsible for the decline of potency against organisms.16 In addition, the presence of electron-rich sites within a protein that contains oxygen, nitrogen, and sulphur atoms with aromatic moieties allows halogen atoms to form halogen bonds for favourable interactions with the receptor.17 As a particular example, halogen substituted Schiff bases and their metal complexes are known to improve lipid membrane penetration and increase the lipophilic nature of molecules, thereby improving the biological activities of their corresponding metal complexes. In several instances, halogen substituted Schiff base ligands and their metal complexes were investigated for a variety of biological activities ranging from anticancer to antibacterial using various human cell lines and bacteria, as well as the DPPH radical scavenging assay.16–19 The results revealed a wide range of activity, which attributed to the ligands' electronic properties. In a similar trend, we recently reported three halogen-substituted Schiff bases and evaluated their potentials on Gram-positive and Gram-negative bacteria, as well as antioxidant activity using the DPPH radical assay,20 demonstrating remarkable activity on both assays.
Taking the aforementioned factors into consideration, as well as our pursuit of lead compounds that can aid in combating the devastating effect of bacterial antibiotic resistance, we report here the synthesis and characterisation of a novel Br/Cl-substituted Schiff base ligand and its complexes with Co2+, Ni2+, Cu2+, and Zn2+ salts. All compounds were evaluated for antibacterial and antioxidant activities, and their level of toxicity was assessed on healthy cell lines.
2. Experimental
2.1. Chemicals and instrumentation
The chemicals and reagents used in this experiment were obtained from Merck Life Sciences (Pty) Ltd and used directly as received. These include 5-chlorosalicylaldehyde, 2-bromo-4-chloroanilline, and metal salts (CoCl2·6H2O, Ni(OAc)2·4H2O, Cu(OAc)2, and Zn(NO3)2·6H2O. Standard spectroscopic techniques were used for the characterization and structural elucidation of the synthesized compounds.
2.2. Synthesis of the ligand and its complexes
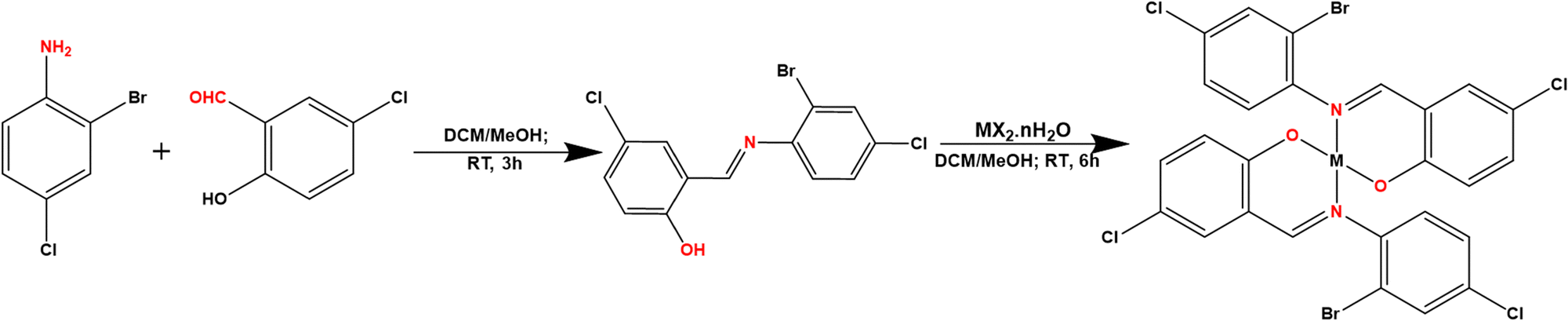
2.2.1. Synthesis of 2-(((2-bromo-4-chlorophenyl)imino)methyl)-5-chlorophenol (HL).
The ligand was synthesized using the previously described procedures.21 In summary, a 20 mL methanol solution of 5-chlorosalicyldehyde (1.00 g, 6.39 mmol, 1 eq.) was reacted with a 20 mL methanolic solution of 2-bromo-4-chloroaniline (1.32 g, 6.39 mmol, 1 eq.), and three drops of formic acid were added. The solution was stirred at room temperature for 3 hours, and the precipitate form was filtered, washed with ether, dried, and re-crystallized in dichloromethane (Scheme 1).20,22 Yield: 1.06 g (51.3%); m.p. 78 °C; 1H NMR (500 MHz, DMSO-d6): δ = 7.03 (d, 1H, J = 9.0 Hz, Ar), 7.49 (t, 1H, J = 6.0 Hz, Ar), 7.61–7.59 (m, 2H, J = 7.5 Hz, Ar), 7.76 (d, 1H, J = 5.0 Hz, Ar), 7.90 (d, 1H, J = 3.0 Hz, Ar), 8.94 (s, 1H, HC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 12.72 (s, 1H, OH); 13C{H}NMR (125 MHz, DMSO-d6): δ = 118.7, 119.9, 120.3, 121.0, 122.6, 129.9, 131.1, 131.8, 132.0, 133.3, 145.1, 158.8: (Ar–C); 163.3 (CN); IR: 3018, 1618, 1363,1092,760 cm−1; UV (CH3CN, 10−3 M): 259, and 361 nm; CHN Anal. calc. for C13H8BrCl2NO: C, 45.26; H, 2.34; N, 4.06; expt.: C, 46.45; H, 2.62; N, 4.14; m/z [M + H]+: calc. for C13H8BrCl2NO = 343.9245; expt. = 343.9250.
N), 12.72 (s, 1H, OH); 13C{H}NMR (125 MHz, DMSO-d6): δ = 118.7, 119.9, 120.3, 121.0, 122.6, 129.9, 131.1, 131.8, 132.0, 133.3, 145.1, 158.8: (Ar–C); 163.3 (CN); IR: 3018, 1618, 1363,1092,760 cm−1; UV (CH3CN, 10−3 M): 259, and 361 nm; CHN Anal. calc. for C13H8BrCl2NO: C, 45.26; H, 2.34; N, 4.06; expt.: C, 46.45; H, 2.62; N, 4.14; m/z [M + H]+: calc. for C13H8BrCl2NO = 343.9245; expt. = 343.9250.
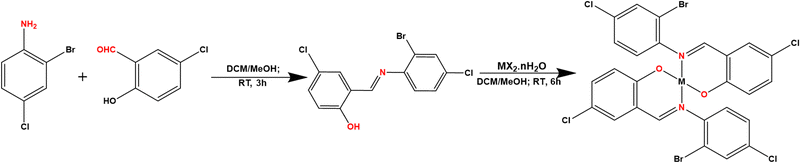 |
| | Scheme 1 Synthetic pathway for the preparation of the ligand and the complexes: MX·nH2O (M = Co, Ni, Cu or Zn; X = OAc, Cl or NO3; n = 0, 4 or 6). | |
2.2.2. Synthesis of Co(II), Ni(II), Cu(II), and Zn(II) complexes.
The solution of HL (0.284 g, 0.825 mmol, 2 eq.) in 20 mL of dichloromethane was mixed with one equivalent each of the metal salts: CoCl2·6H2O (0.098 g, 0.413 mmol), Ni(OAc)2·4H2O (0.103 g, 0.413 mmol), Cu(OAc)2 (0.075 g, 0.413 mmol), and Zn(NO3)2·6H2O (0.123 g, 0.413 mmol) in 20 mL of methanol, respectively. The four solutions were stirred overnight at room temperature. For those that precipitate out, the precipitate was filtered, washed with methanol (10 × 2 mL) and ether (10 × 2 mL) and in the absence of precipitate, the solution was concentrated (using rotary evaporator) to obtain the solid product. The crude solid products were re-crystallized by slow vapor diffusion of hexane into dichloromethane with complexes of Cu and Ni providing crystals suitable for single crystal X-ray diffraction analyses. Complexes of Zn2+ and Co2+ yielded micro-crystalline material. The reaction procedure is illustrated in Scheme 1.
Bis (Z)-2-((4-bromo-4-chlorobenzylidene)amino)-4-chlorophenolato cobalt(II) (CoL2).
Yield: 0.190 g, (61.1%); m.p. 282–284 °C; IR: 1600, 1355, 1044, 789, 554, 423 cm−1; UV: (CH3CN, 10−3 M): 238, 278, 410 nm; CHN anal. calc. for C26H18Br2Cl4CoN2O2: C, 41.81; H, 1.89; N, 3.75; expt.: C, 41.69; H, 1.79; N, 3.71; m/z [M]+: calc. for C26H18Br2Cl4CoN2O2 = 742.7497; expt. = 742.6346.
Bis (Z)-2-((4-bromo-4-chlorobenzylidene)amino)-4-chlorophenolato nickel(II) (NiL2).
Yield: 0.247 g (58.8%); m.p. 263–266 °C; 1H NMR (500 MHz, DMSO-d6): δ = 6.12 (d, 1H, J = 9.0 Hz, Ar), 6.76–6.78 (m, 1H, J = 9.0 Hz, Ar), 7.00 (d, 1H, J = 8.5 Hz, Ar), 7.37 (t, 1H, J = 8.5 Hz, Ar), 7.48 (d, 1H, J = 6.0 Hz, Ar), 7.66 (d, 1H, J = 5.0 Hz, Ar), 8.55 (s, 1H, HCN); 13C{H}NMR (125 MHz, DMSO-d6): δ = 116.1, 118.9, 120.8, 122.6, 125.7, 128.1, 128.5, 131.3, 132.7, 134.5; 160.5, (Ar); 188.9 (![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) N); IR: 1635, 1342, 1203, 784, 524, 455 cm−1; UV: (CH3N, 10−3 M): 256, 327, 402 nm; CHN anal. calc. for C26H14BrCl4N2NiO2: C, 41.82; H, 1.89; N, 3.75; expt.: C, 39.94; H, 2.04; N, 3.03; m/z [M]+, Calc. for C26H14Br2Cl4N2NiO2 = 741.7530; expt. = 741.6118.
Bis (Z)-2-((4-bromo-4-chlorobenzylidene)amino)-4-chlorophenolato copper(II) (CuL2).
Yield: 0.3438 g (68.8%); m.p. 226–268 °C; IR: 1612, 1352, 1074, 823, 554, 428 cm−1; UV: (CH3N, 10−3 M): 287, 384, 487 nm; CHN Anal. calc. for C26H14Br2Cl4CuN2O2: C, 41.55; H, 1.88; N, 3.73; expt.: C, 40. 92; H, 2.15; N, 3.73; m/z [M + H]+: calc. for C26H14Br2Cl4N2NiO2 = 747.7550; expt. = 747.5822.
Bis (Z)-2-((4-bromo-4-chlorobenzylidene)amino)-4-chlorophenolato zinc(II) (ZnL2).
Yield: 0.172 g (51.6%); m.p. 258–261 °C; 1H NMR (500 MHz, DMSO-d6): δ = 6.57 (d, 1H, Ar), 7.11 (s, 1H, Ar), 7.22 (d, 1H, J = 6.0 Hz, Ar), 7.45 (d, 1H, J = 8.0 Hz, Ar), 7.64 (s, 1H, Ar), 7.76 (s, 1H, Ar), 8.73 (s, 1H, HCN); 13C{H}NMR (125 MHz, DMSO-d6): δ = 116.0, 119.1, 121.0, 125.6, 128.1, 130.0, 131.7, 133.1, 134.6; 145.0, (Ar); 188.9 (N); IR:1652, 1320, 1097, 754, 537, 468 cm−1; UV: (CH3N, 10−3 M): 280, 382 nm; CHN anal. calc. for C26H14BrCl4N2O2Zn: C, 41.45; H, 1.87; N, 3.75; expt.: C, 41.17; H, 1.78; N, 3.64; m/z [M]+, calc. for C26H14BrCl4N2O2Zn = 747.7468; expt. = 747.5897.
N); IR: 1635, 1342, 1203, 784, 524, 455 cm−1; UV: (CH3N, 10−3 M): 256, 327, 402 nm; CHN anal. calc. for C26H14BrCl4N2NiO2: C, 41.82; H, 1.89; N, 3.75; expt.: C, 39.94; H, 2.04; N, 3.03; m/z [M]+, Calc. for C26H14Br2Cl4N2NiO2 = 741.7530; expt. = 741.6118.
Bis (Z)-2-((4-bromo-4-chlorobenzylidene)amino)-4-chlorophenolato copper(II) (CuL2).
Yield: 0.3438 g (68.8%); m.p. 226–268 °C; IR: 1612, 1352, 1074, 823, 554, 428 cm−1; UV: (CH3N, 10−3 M): 287, 384, 487 nm; CHN Anal. calc. for C26H14Br2Cl4CuN2O2: C, 41.55; H, 1.88; N, 3.73; expt.: C, 40. 92; H, 2.15; N, 3.73; m/z [M + H]+: calc. for C26H14Br2Cl4N2NiO2 = 747.7550; expt. = 747.5822.
Bis (Z)-2-((4-bromo-4-chlorobenzylidene)amino)-4-chlorophenolato zinc(II) (ZnL2).
Yield: 0.172 g (51.6%); m.p. 258–261 °C; 1H NMR (500 MHz, DMSO-d6): δ = 6.57 (d, 1H, Ar), 7.11 (s, 1H, Ar), 7.22 (d, 1H, J = 6.0 Hz, Ar), 7.45 (d, 1H, J = 8.0 Hz, Ar), 7.64 (s, 1H, Ar), 7.76 (s, 1H, Ar), 8.73 (s, 1H, HCN); 13C{H}NMR (125 MHz, DMSO-d6): δ = 116.0, 119.1, 121.0, 125.6, 128.1, 130.0, 131.7, 133.1, 134.6; 145.0, (Ar); 188.9 (N); IR:1652, 1320, 1097, 754, 537, 468 cm−1; UV: (CH3N, 10−3 M): 280, 382 nm; CHN anal. calc. for C26H14BrCl4N2O2Zn: C, 41.45; H, 1.87; N, 3.75; expt.: C, 41.17; H, 1.78; N, 3.64; m/z [M]+, calc. for C26H14BrCl4N2O2Zn = 747.7468; expt. = 747.5897.
2.3. Single-crystal X-ray diffraction analysis
Crystallographic data of HL, NiL2, and CuL2, were collected on a APEXII with Mo Kα (λ = 0.71073) radiation at 293 K. To process raw data, Bruker SAINT was used for the integration of the collected frames.23 Thereafter, absorption effects were reduce using SADABS,24 and the structures were solved using SHELXT.25 SHELXL,26 was used to refine them. The crystal structure graphics were created using Mercury software.27 Non-hydrogen atoms were refined on F2 isotropically and then anisotropically by least squares method. All hydrogen atoms were placed geometrically and refine through a ridding approximation.28
2.4. Hirshfeld surface analysis
Crystal explorer 17.5 software was utilized to obtain Hirshfeld surface (HS) as well as two-dimensional fingerprint plots and interactions energies at the B3LYP/BGDZVP level of theory.29 The HS and two-dimensional fingerprint plots were generated using high resolution and d norm functions using a color scale of −0.0701 to 1.798 a.u., a reciprocal touch is displayed, translated in the range of 0.6–2.6 for the former. To obtain the interaction energy topology network and its quantitative values, the Tonto method was used to calculate the wavefunctions to obtain the interaction energy topology network and its quantitative values.
2.5. Biological evaluation
2.5.1.
In vitro antibacterial studies.
The free ligand, its complexes, the standard drug (ciprofloxacin®), and dimethyl sulfoxide (carrier solvent) were tested for antibacterial activity against Gram-positive (Staphylococcus aureus, ATCC-25923, and Bacillus subtilis, ATCC-23857) and Gram-negative (Escherichia coli, ATCC-25922, and Klebsiella pneumoniae, ATCC-13883) pathogenic bacteria.30,31 The bacteria isolates were inoculated uniformly on a plate containing agar and incubated at 37 °C for 24 hours. Thereafter, a paper disc encapsulated with various concentrations of the standard and synthesized compounds was introduced. Effect of the compounds on the organism was estimated via the inhibition zone and expressed in millimetres. The experiment was replicated three times, and the result are presented as mean ± standard deviations.
2.5.2. Determination of minimum inhibitory concentration (MIC).
The MIC of the synthesized compounds and the control were assessed using modified broth dilution method.10,32,33 In brief, each of the test compounds and the control were dissolved in DMSO, and diluted two-fold to obtain a stock solution concentration range of 512 to 0.25 μg mL−1. From the stock solution, 100 μL was introduced into a microplate (96-well) containing broth solution (90 μL), followed by addition of 10 μL of bacteria inoculum (1 × 106 CFU mL−1) which resulted to concentration range of 250 to 0.125 μg mL−1. The plates were covered and incubated for 24 hours at 37 °C. Minimum inhibitory concentration was estimated visually as the lowest concentration at which no bacterial growth observed. Ciprofloxacin, and mixture of broth and DMSO were used as positive and negative controls, respectively.
2.5.3. Antioxidant studies.
Radical scavenging ability of the synthesized compounds was evaluated using a DPPH protocol.34,35 The ligand, its complexes, and the control (ascorbic acid) were dissolve in dimethyl sulfoxide to a various concentration ranging from 20 to 100 μg mL−1. Thereafter, 1 mL of each concentration was mixed with 3 mL solution (0.1 mM) of the DPPH radical in methanol. The mixture was incubated in a dark room for 30 minutes, and the absorbance was determined using spectrophotometer at wavelength of 517 nm. The measurement was carryout in triplicate, and percentage scavenging activity (SA) was estimated.
2.5.4. Toxicity studies.
Toxicity effect of the complexes was evaluated on normal human cell lines comprises of (WISH-ATCC-CCL-25, human epithelial amnion (liver cell lines), and MRC-5-ATCC-CCL-171, human lung fibroblast cell lines), using MTT method.36,37 Samples concentrations of 50 and 100 μM were used against these cell lines. 200 μL representing ((1 × 104 cells per well) was seeded into 96-well plates and incubated at 37 °C for 24 hours in 5% CO2 atmosphere. After the incubation, medium was replaced, and the cells were treated with various concentrations of the complexes for 24 hours. 8 g mL−1 of methyl methanesulphonate (MMS) and 10% DMSO were used as positive and negative controls, respectively, with doxorubicin serving as an internal control. The plates were incubated for 72 h at 37 °C in 5% CO2 after being treated with controls or complexes. The medium was replaced after 3 days with 2 μL of 3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) (5 mg mL−1), and the cells were incubated for 3 h. The proportion of metabolically active cells was compared to untreated controls using a mitochondrial conversion of MTT to formazan crystals dissolved in dimethyl sulfoxide (DMSO). Spectrometer was used measure the absorbance at 570 nm. The cell viability was expressed as a mean ± standard deviations in comparison to the control.
2.6. Computational studies
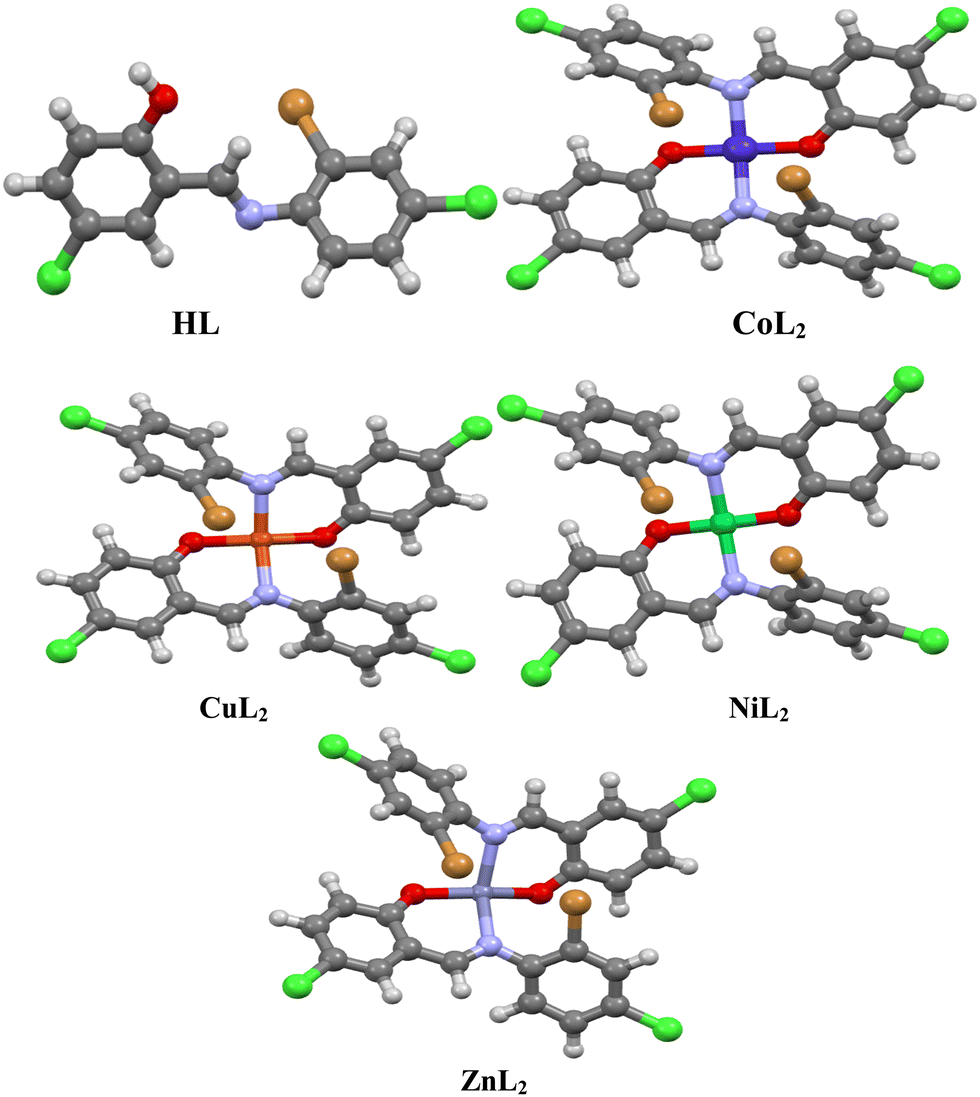
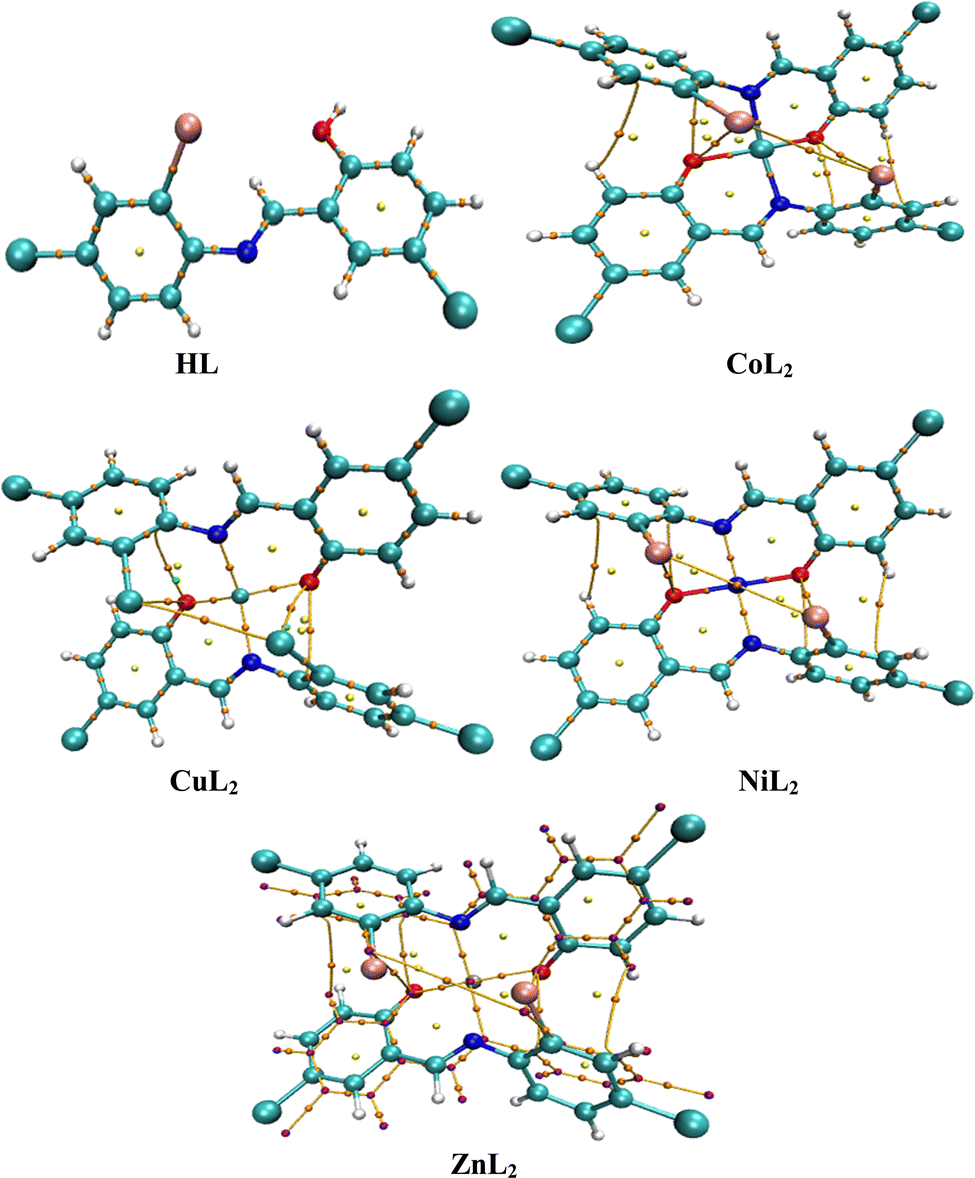
DFT calculations on the compounds were performed using Gaussian 09 software, which was constructed and optimized at the M06-2x/6-311G(d,p) level of theory.38 The polarizable continuum model (PCM) describes solvation effects in the solvent phase (MeOH). Furthermore, frequency calculations were performed to ensure the obtained structures converged to a minimum (Fig. 1). By converting the optimized structures to CCDC Mercury files, we obtain M-N bond distances of 2.004 Å (CoL2), 2.036 Å (CuL2), 1.975 Å (NiL2), and 2.136 Å (ZnL2), while M–O bond distances of 1.893 Å (CoL2), 1.918 Å (CuL2), 1.867 Å (NiL2), and 1.977 Å (ZnL2) were obtained. From the optimized structures, fchk files were created and used as input for multiwfn39 to perform quantum theory of atoms in molecules (QTAIM) analysis. In addition to this, VMD40 and gnuplot41 software was used to analyze results of the QTAIM analysis. Moreover, multiwfn software was used for MEP, CDFT, and ELF analyses. Furthermore, quantitative information about NCI was also extracted using multiwfn software so that qualitative images from VMD and gnuplot software could be produced.
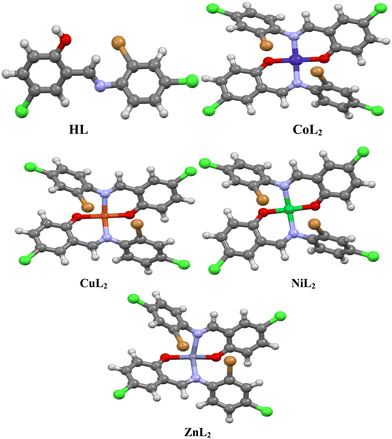 |
| | Fig. 1 Optimized structures of the ligand, and its complexes visualized using Mercury. | |
2.7. Molecular docking studies
2.7.1. Preparation of the complexes for docking studies.
The optimized geometries of CoL2, NiL2, CuL2, and ZnL2 were used for the docking study using Auto-dock tool software.42
2.7.2. Preparation of tyrosyl-tRNA synthetase (PDB ID: 1jij) and type IIA topoisomerase (PDB: 2xct).
The crystal structures of tyrosyl-tRNA synthetase (PDB ID: 1jij),43 and type IIA topoisomerase (PDB: 2xct)44 were obtained from the online source at (https://www.rcsb.org). They were examined for the presence of foreign material before saving in PDB format using Pymol software.45 The docking programs AutoDock Tools (ADT) version 1.5.6 and AutoDock version 4.2.6 were used for molecular docking.46 During docking calculations, Kollmann charges were applied to proteins and DNA. RESP charges for complexes were calculated using the RED Server.47,48 The VMD program,47 and UCSF Chimera49 were used to visualize and analyze the binding mode as well as interactions in the binding pocket of the obtained poses.
3. Results and discussions
3.1. Chemistry
The halogen-substituted ON donor ligand (HL) was synthesized via treatment of 5-chlorosalicylaldehyde with 2-bromo-4-chloroaniline in a methanolic medium using formic acid as a dehydrating agent at ambient temperature. The reaction of HL with metal salts of Co2+, Ni2+, Cu2+, and Zn2+ in a mixture of dichloromethane and methanol at ambient temperature produces mononuclear homoleptic complexes of the form ML2 (Scheme 1). The ligand and its complexes were found to be stable in the presence of air and moisture and are soluble in polar solvents. Conductivity study of the complexes in 10−3 M solution of dimethyl sulfoxide show values in the range of 3.48–5.64 Ω−1 cm2 mol−1, revealing non-electrolyte property.50,51 The complexes show elevated melting points their ligand; this could be due to the formation of new compounds with different physical and chemical properties, which aid in the formation of the complexes.
3.2. Characterization of the ligand and its complexes
The structure of the ligand and its complexes were elucidated and confirmed using various spectroscopic techniques which include 1H and 13C NMR, IR, UV-Vis, and SCXRD. (Discussion and detail spectra in the ESI†). The IR spectra of the compounds (Fig. S8, S14, S23, S29, and S37, ESI†) showed the characteristic band at (1652–1600) cm−1 which indicated the presence of a –CN group in the compounds. The 1H and 13C NMR spectra of the ligand and its diamagnetic Ni(II) and Zn(II) complexes shows peaks that accounted for all protons and carbons in the compounds (Fig. S6, S7, S21, S22, S36, and S37, ESI†). The electronic spectra of the ligand and the complexes are presented in (Fig. S9, A15, S24, S30, and S30, ESI†), respectively. To further elucidate their structure and examine their chemical composition, powder X-ray diffraction (PXRD), scanning electron microscopy (SEM), and energy dispersive X-ray (EDX) analyses were performed. The diffractogram and spectra are presented in (Fig. S11, S12, S17–S19, S26, S27, S32, S33, and S39–S41, ESI†) for HL, CoL2, NIL2, CuL2, and ZnL2, respectively.
3.2.1. Microanalysis and mass spectra.
Table 1 shows the analytical and some selected physicochemical properties of the compounds. To validate the stoichiometric composition of the ligand and its complexes, the elemental CHN composition was quantified using microanalysis, and the results corroborate well with the proposed structures.
Table 1 Physicochemical data of the ligand and its complexes
| Compounds |
Empirical formula |
m.p. (°C) |
Yield (%) |
Conductivity (Ω−1 cm−2 mol−1) |
Anal. found (calc.) |
| C |
H |
N |
|
HL
|
C13H8BrCl2NO |
78–80 |
51.3 |
— |
46.45 (45.26) |
2.62 (2.34) |
4.14 (4.06) |
|
CoL2
|
C26H14Br2Cl4CoN2O2 |
282–284 |
61.0 |
3.48 |
41.61 (41.81) |
1.79 (1.87) |
3.71 (3.72) |
|
NiL2
|
C26H14Br2Cl4N2NiO2 |
263–266 |
58.8 |
4.53 |
39.94 (41.82) |
2.04 (1.89) |
3.03 (3.75) |
|
CuL2
|
C26H14Br2Cl4CuN2O2 |
226–268 |
68.8 |
5.46 |
40.92 (41.55) |
2.15 (1.88) |
3.73 (3.73) |
|
ZnL2
|
C26H14Br2Cl4N2O2Zn |
258–261 |
52.6 |
5.64 |
41.17 (41.45) |
1.78 (1.87) |
3.64 (3.72) |
Furthermore, mass spectroscopy was used to confirm the compounds' proposed structures. The mass spectra of the ligand and its complexes (see ESI†) revealed molecular ion peaks that corresponded to the proposed structures' molecular weights. The HL spectrum (Fig. S13, ESI†) shows a peak at m/z = 343.9250, which represent [M + H]+. Other peaks at m/z = 345.9243, 347.9194, and 348.9249 correspond to the molecular weight of the that match two bromine isotopes (79Br and 81Br).
In addition, the complexes' mass spectra (Fig. S20, S28, S34, and S42, ESI†) show molecular ion peaks that correspond to their proposed molecular weights and other complex fragments. Table 2 summarizes the m/z peak values of the ligand and complexes supporting the proposed 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 metal to ligand ratio. In addition, the result of the NMR study of the ligand and its Ni(II) and Zn(II) shows that nitrogen and oxygen atoms of the ligand coordinated to metal ions as bidentate, affirming 1:2 metal to ligand ratio (Table 3).
:2 metal to ligand ratio. In addition, the result of the NMR study of the ligand and its Ni(II) and Zn(II) shows that nitrogen and oxygen atoms of the ligand coordinated to metal ions as bidentate, affirming 1:2 metal to ligand ratio (Table 3).
Table 2 Mass spectrometric data of the ligand and its complexes
| Compounds |
Calculated mass |
Found mass |
Peak assignment |
|
HL
|
343.9245 |
343.9250 |
[M + H]+ |
|
CoL2
|
742.7508 |
742.6346 |
[M]+ |
|
NiL2
|
741.7530 |
741.6118 |
[M]+ |
|
CuL2
|
747.7550 |
747.5822 |
[M + H]+ |
|
ZnL2
|
747.7468 |
747.5897 |
[M]+ |
Table 3
1H NMR spectral data (δ, ppm) of the ligand and its NiL2 and ZnL2 complexes
| Compounds |
OH |
HCN |
Aromatic protons |
|
HL
|
12.72 |
8.94 |
7.03–7.90 |
|
NiL2
|
— |
8.55 |
6.12–7.66 |
|
ZnL2
|
— |
8.73 |
6.57–8.73 |
3.2.2 Description of crystal structures.
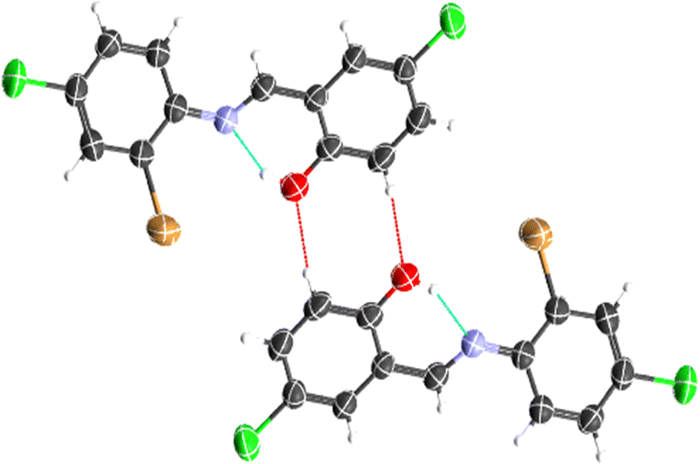
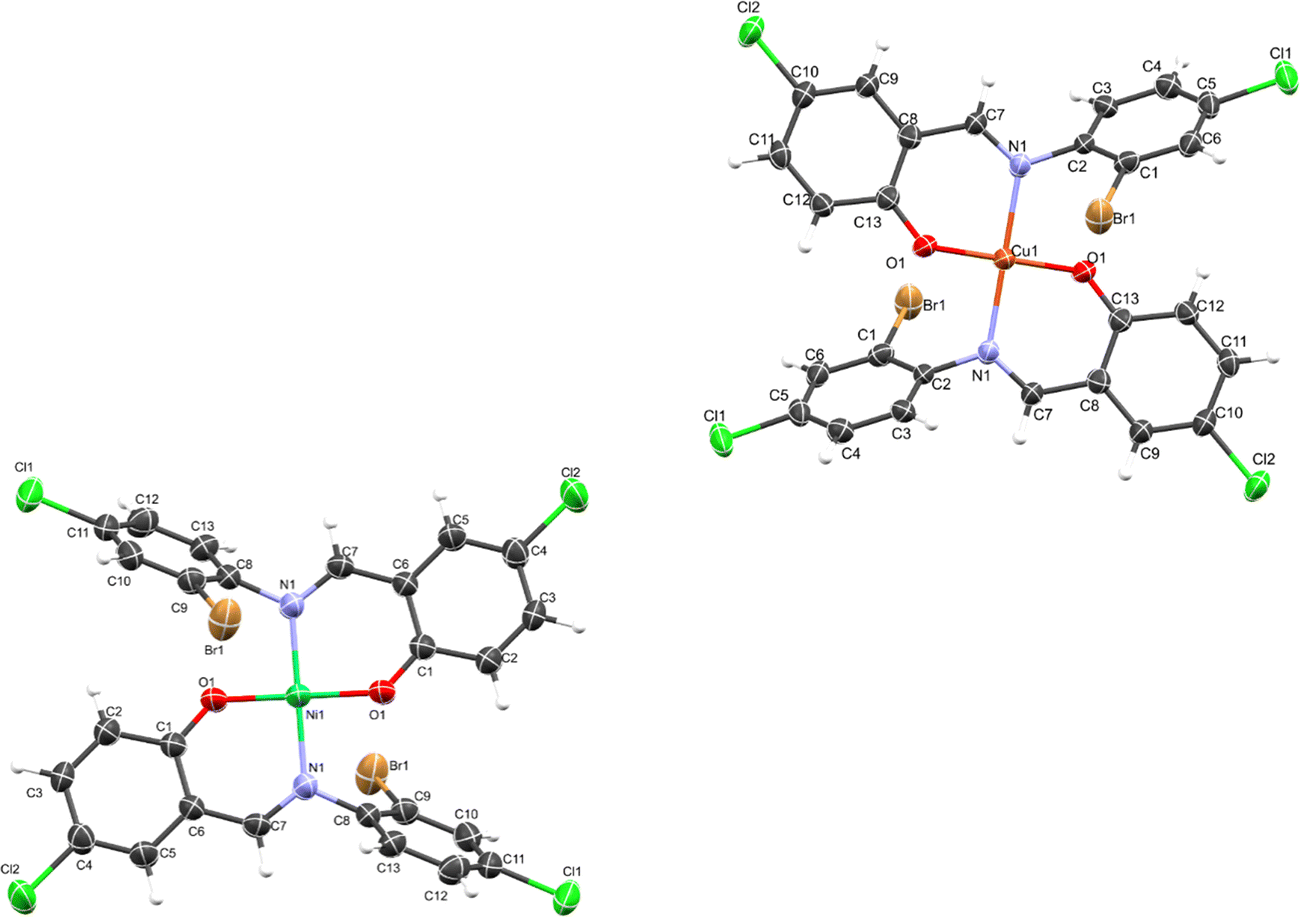
The crystal structure of HL crystallizes in a monoclinic space group P21/c with a single monomeric unit in the asymmetric unit stabilized by an intramolecular O–H⋯N hydrogen bond as shown in Fig. 2. The dihedral angle between the two phenyl rings is 8.2(11)° which is larger than (E)-1-(2-nitrophenyl)-N-(o-tolyl)methanamine.52 The bond parameters for this compound are comparable to similar structures in literature.20,53,54 Further analysis shows that there exists a non-classical C2B–H2B⋯O1A hydrogen bond in the crystal packing of the compound (Fig. 2). The crystal structures of NiL2 and CuL2 also pack in the monoclinic, and monomeric asymmetric unit with space group P21/c (Fig. 3). The geometry around the central metal atom is near an ideal square planar with a slight distortion on bond angles ranging from 87.45(15) to 180.0° and 89.61(14) to 180°, respectively. These values are comparable to those published for similar compounds, including various intramolecular bond characteristics.55 The geometric orientation of the anilinyl rings was also found to be almost orthogonal to the aryl ring.
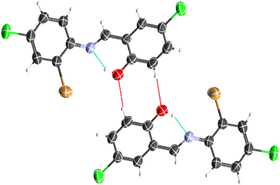 |
| | Fig. 2 Representation of intramolecular O1–H1⋯N1 and intermolecular C2B–H2B⋯O1A hydrogen bonds in the ligand. | |
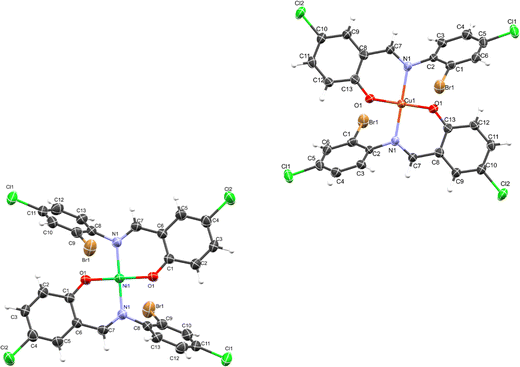 |
| | Fig. 3 ORTEP diagram of NiL2 and CuL2 with thermal ellipsoids drawn at a 50% probability. | |
The Cu–N and Cu–O bond distance is 1.968(3) and 1.880(4) Å respectively, while the Ni–O and Ni–N bond distances is 1.812(3) and 1.873(3) Å, respectively. These distances agree with similar structures previously reported.56–58 Non-classical hydrogen bond exists in the crystal system of the lattice structure (Fig. 2). The detailed crystallographic data and refinement parameters of the compounds are presented in Table 4.
Table 4 Crystal data and structure refinement for HL, NiL2 and CuL2
| Identification code |
HL2
|
NiL2
|
CuL2
|
| Empirical formula |
C13H8BrCl2NO |
C26H14Br2Cl4N2NiO2 |
C26H14Br2Cl4CuN2O2 |
| Formula weight |
345.01 |
746.724 |
751.576 |
| Temperature/K |
273.15 |
172.98 |
173.01 |
| Crystal system |
monoclinic |
Monoclinic |
Monoclinic |
| Space group |
P21/c |
P21/c |
P21/c |
|
a/Å |
14.366(3) |
9.974(2) |
9.966(4) |
|
b/Å |
3.9097(8) |
10.356(2) |
10.774(4) |
|
c/Å |
23.118(4) |
12.438(3) |
11.925(5) |
|
α/° |
90 |
90 |
90 |
|
β/° |
90.068(7) |
105.626(6) |
103.479(8) |
|
γ/° |
90 |
90 |
90 |
| Volume/Å3 |
1298.5(4) |
1237.3(5) |
1245.1(9) |
|
Z
|
4 |
2 |
2 |
|
ρ
calc g cm−3 |
1.765 |
2.004 |
2.005 |
|
μ/mm−1 |
3.562 |
4.476 |
4.546 |
|
F(000) |
680.0 |
733.4 |
735.4 |
| Crystal size/mm3 |
0.259 × 0.172 × 0.062 |
0.19 × 0.15 × 0.09 |
0.55 × 0.2 × 0.18 |
| Radiation |
MoKα (λ = 0.71073) |
Mo Kα (λ = 0.71073) |
Mo Kα (λ = 0.71073) |
| 2Θ range for data collection/° |
3.524 to 55.796 |
4.24 to 57.7 |
5.16 to 57.08 |
| Index ranges |
−18 ≤ h ≤ 18 |
−13 ≤ h ≤ 13 |
−13 ≤ h ≤ 13 |
| −5 ≤ k ≤ 5 |
−14 ≤ k ≤ 13 |
−14 ≤ k ≤ 14 |
| −30 ≤ l ≤ 30 |
−16 ≤ l ≤ 16 |
−15 ≤ l ≤ 12 |
| Reflections collected |
19688 |
49546 |
15573 |
| Independent reflections |
3035 [Rint = 0.0700, RSigma = 0.0472] |
3214 [Rint = 0.1652, RSigma = 0.0732] |
3144 [Rint = 0.1047, RSigma = 0.0931] |
| Data/restraints/parameters |
3035/0/164 |
3214/0/169 |
3144/0/169 |
| Goodness-of-fit on F2 |
1.069 |
1.066 |
0.997 |
| Final R indexes [I ≥ 2σ(I)] |
R
1 = 0.0631, wR2 = 0.1280 |
R
1 = 0.0530, wR2 = 0.1088 |
R
1 = 0.0481, wR2 = 0.0926 |
| Final R indexes [all data] |
R
1 = 0.1007, wR2 = 0.1431 |
R
1 = 0.1250, wR2 = 0.1380 |
R
1 = 0.1183, wR2 = 0.1147 |
| Largest diff. peak/hole/e Å−3 |
0.69/−0.56 |
1.40/−0.60 |
1.16/−1.36 |
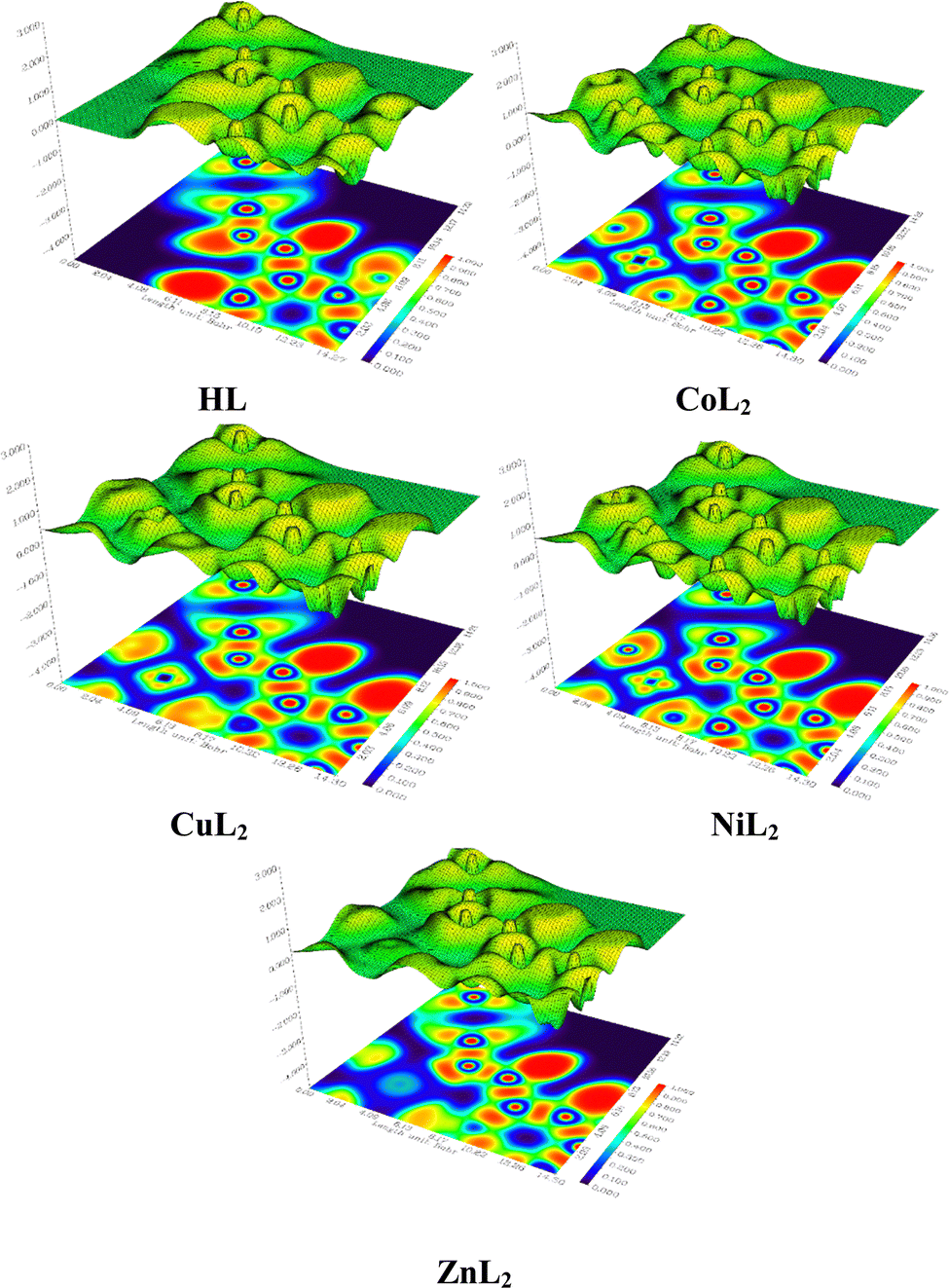
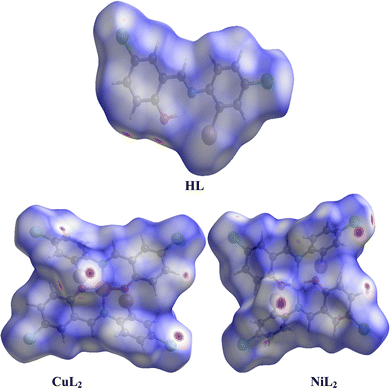
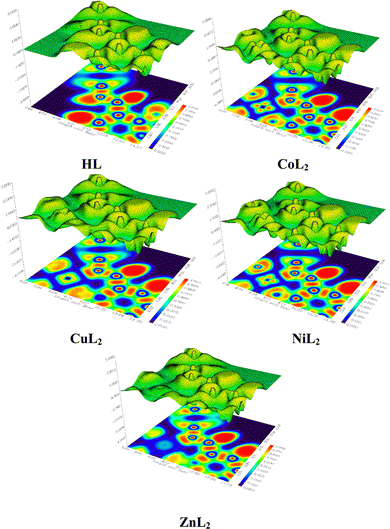
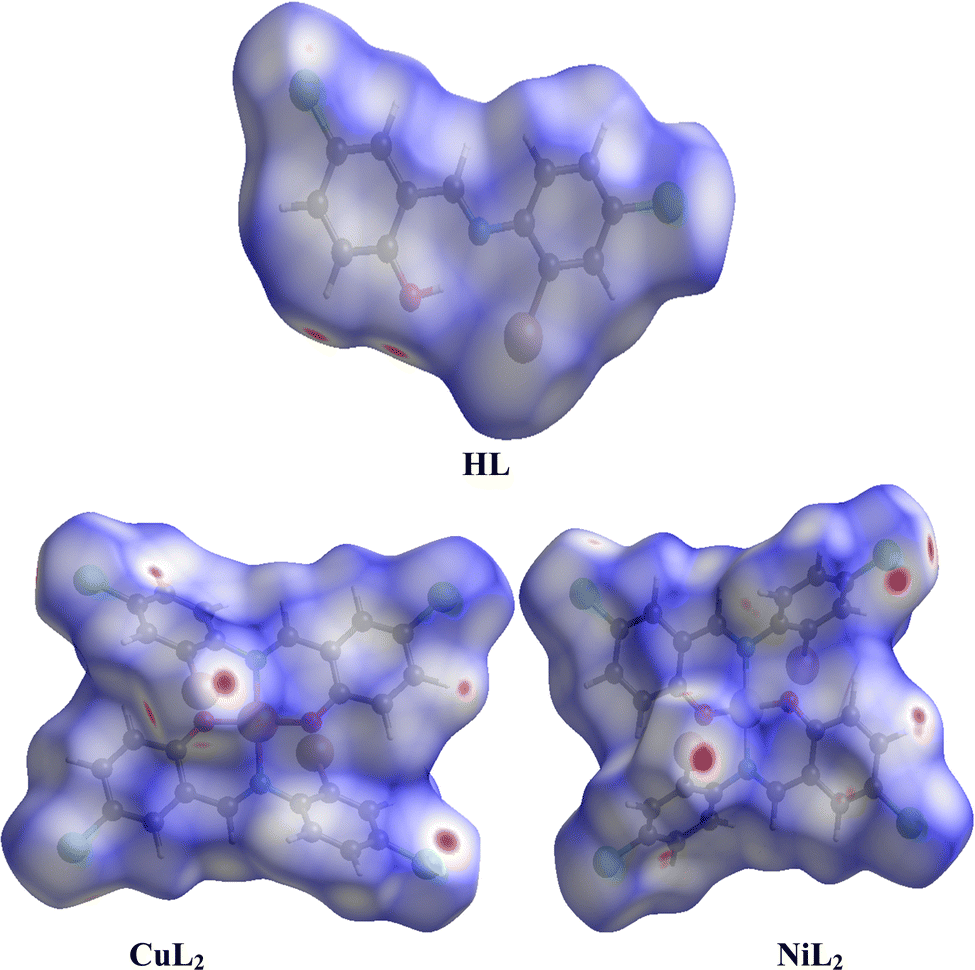
3.2.3 Hirshfeld surface analysis and two-dimensional fingerprint plots.
The Hirshfeld surface analysis (HSA) is a powerful tool that can visualize and interpret intermolecular contacts in molecular crystals.59 The Hirshfeld surface (HS) covers the outer contour of the space occupied by a molecule in a crystalline environment and reveals a visual map that represents these interactions by displaying different colored isosurfaces. These isosurfaces show the separation distances between the surface and the nearest exterior (de) or interior (di) nucleus and can show the surface characteristics of various types of interactions. The normalized contact distance (dnorm) can be calculated using eqn (1) as follows:| | | dnorm = di − rvdWi/rvdWi+ de − rvdWe/rvdWe | (1) |
where rvdWiand rvdWe are the van der Waals radii of the atoms. Red patches on the dnorm map of the HS indicate short interatomic interactions, whereas white areas show contacts that are longer than the sum of van der Waals radii and blue areas show no interatomic contacts because of huge dnorm between surrounding atoms.60–62 In this case, the intermolecular interaction between the crystal structures of the ligand and its Ni(II) and Cu(II) complexes was assessed using Hirshfeld surface analysis. The compounds' 3D Hirshfeld surface was mapped onto the dnorm shape index, as seen in Fig. 4. The ligand exhibits two deep red spots on the dnorm surface, but the Ni(II) and Cu(II) complexes exhibit several deep red spots on the Hirshfeld surface. These suggest the presence of strong and medium contacts and close-contact interactions caused by intermolecular hydrogen bonding.
 |
| | Fig. 4 Hirshfeld surface of the ligand and its Cu(II) and Ni(II) complexes. | |
In addition to HSA, two-dimensional fingerprint plots can be generated after obtaining the HS map, providing quantitative analysis of atomic interactions between molecules in molecular crystals (Fig. S43, ESI†). A significant feature of these plots is the number of atomic contributions to these interactions that exceed 1% in both complexes, of which the H⋯H interaction dominates. A stronger H⋯H interaction is observed in the nickel complex than the copper complex, while a weaker H⋯H interaction is observed in the free ligand. Strong C⋯H, Cl⋯H, and H⋯C interactions that exceed 10% atomic contributions are also observed in both complexes, particularly C⋯H interactions. Other than Cl⋯H, these interactions were observed to be significantly weaker in the free ligand, where both C⋯H and H⋯C made less than 10% atomic contributions. However, the C⋯C interactions made substantially larger atomic contributions in the free ligand, where they exceeded 10%, while below 3% atomic contributions were observed in the complexes. These results may provide a clue to the interaction between these compounds and receptor sites in biological systems.62
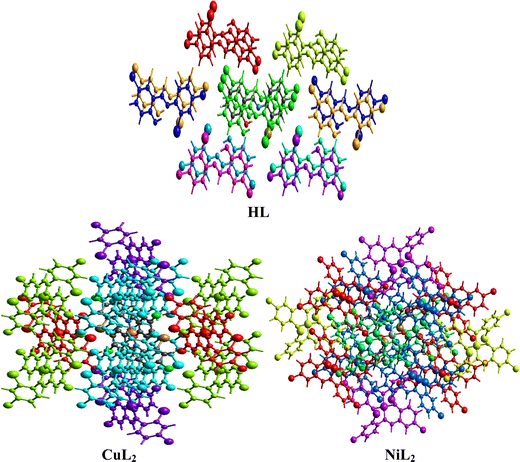
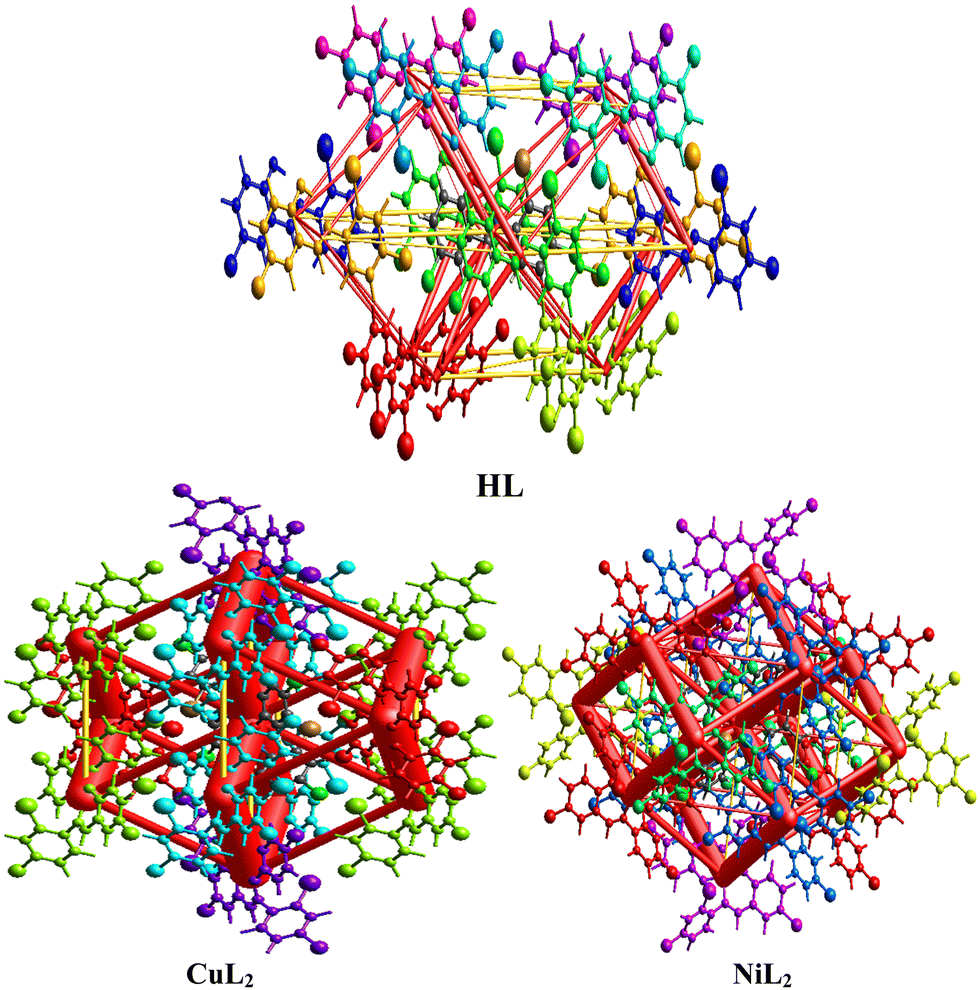
Interaction energy topology network.
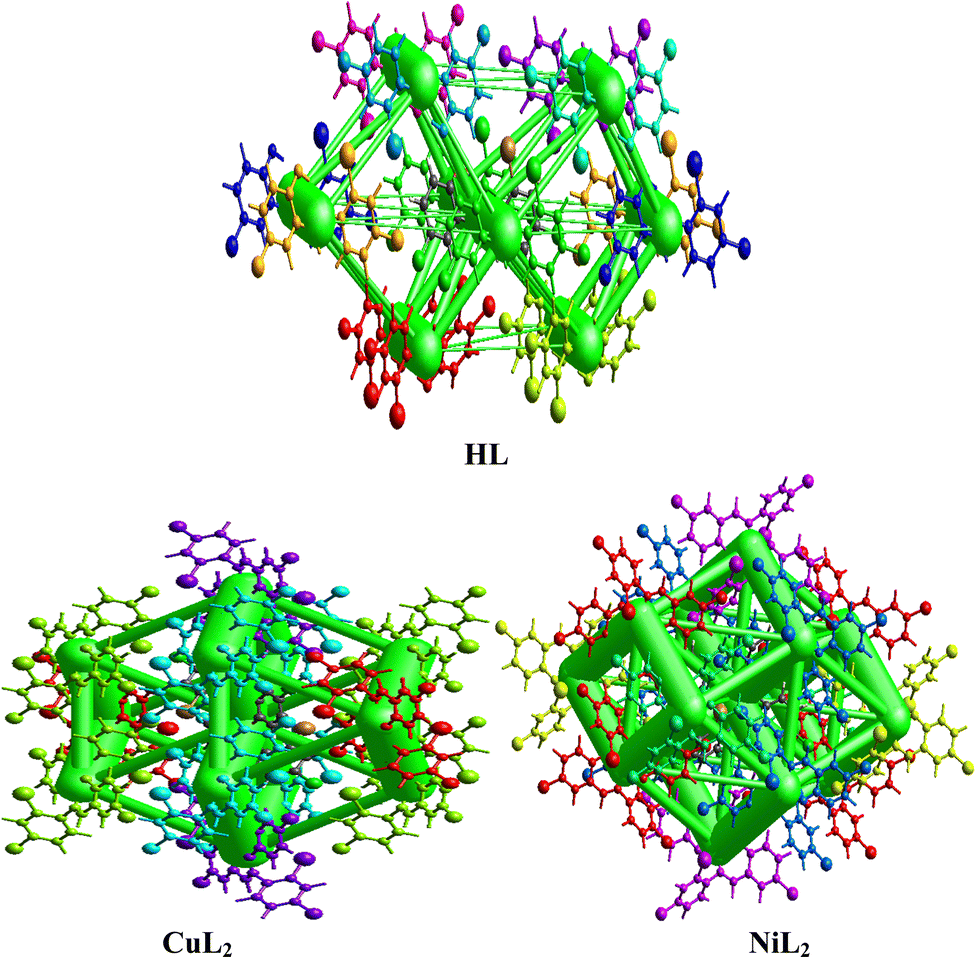
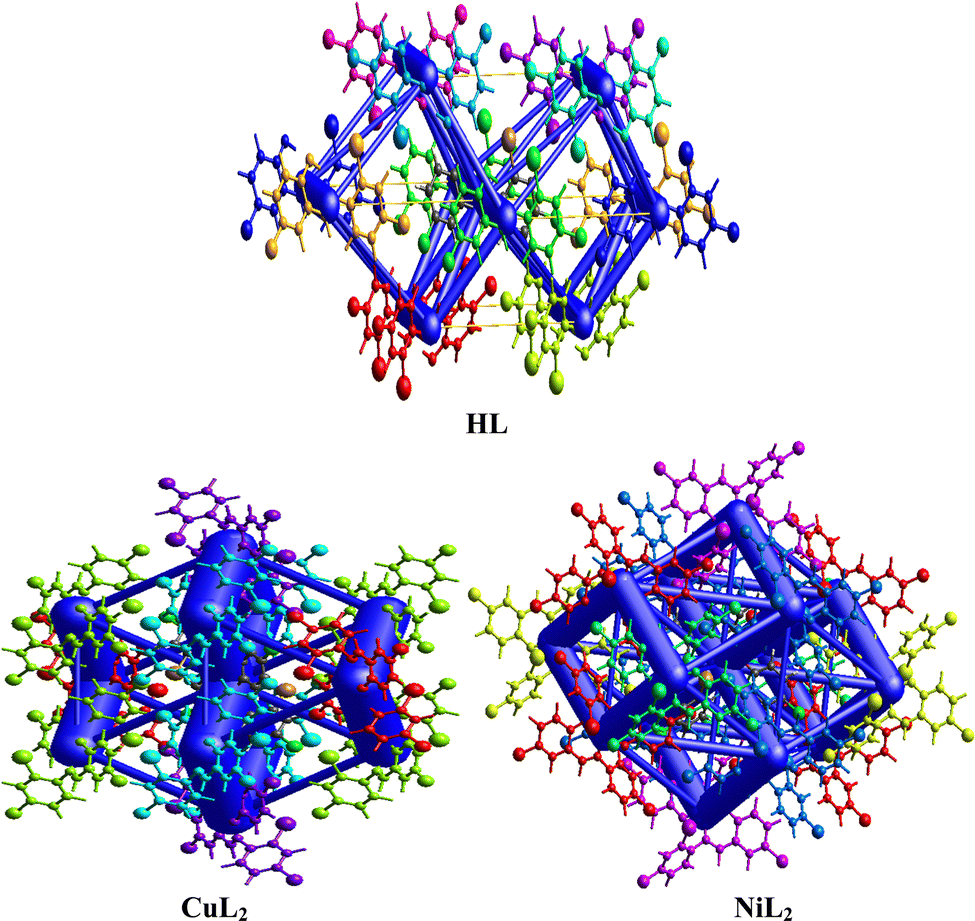
The interaction energy topology network was generated using the Tonto method by calculating the wavefunction and producing the images for the free ligand and complexes in Fig. 5, wherein similar colors correspond to neighboring compounds that interact with each other in the topology network. The symmetry operations (Sym op) for these colors are observed in Tables S2–S4 (ESI†), in which R is the separation distance between molecular centroids (mean atomic position). The vector characteristics of each interacting molecule influence its interaction with its neighboring molecule within the molecular crystal, causing a perturbation that leads to an energy breakdown that can be expressed by the total energy (Etot) in eqn (2):| | | Etot = Eele + Epol + Edis + Erep | (2) |
where Eele, Epol, Edis, and Erep represent electrostatic, polarization, dispersive, and repulsive energies, respectively. By joining each interatomic interaction in the molecular crystal, an interaction energy framework can be produced for coulombic, dispersive, and total energies (Fig. 6–8). Although a relationship between R and Etot is observed for CuL2 (short R values induce higher Etot values), this relationship does not seem to hold for the ligand and NiL2 as a few outliers are observed (Tables S2–S4, ESI†).
 |
| | Fig. 5 Interaction topology network for the ligand and its Cu(II) and Ni(II) complexes. | |
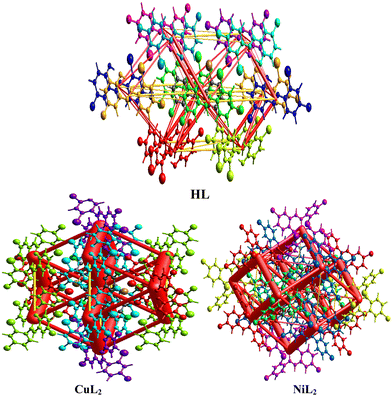 |
| | Fig. 6 Coulombic interaction energy frameworks between the atoms within the interaction energy topology network of the ligand and its Cu(II) and Ni(II) complexes. | |
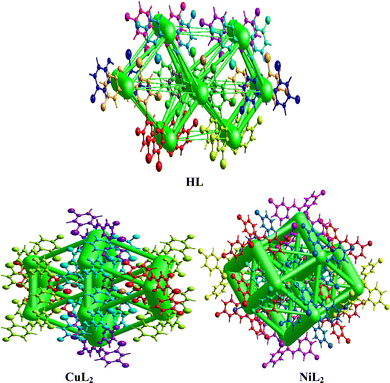 |
| | Fig. 7 Dispersion interaction energy framework between the atoms within the interaction topology network for the ligand and its Cu(II) and Ni(II) complexes. | |
 |
| | Fig. 8 Total interaction energy framework between the atoms within the interaction topology network within the ligand and its Cu(II) and Ni(II) complexes. | |
3.3. Biological evaluations
3.3.1. Antimicrobial activity.
The antibacterial efficacy of the free ligand and its complexes, compared with ciprofloxacin, (Cipro), was screened on some selected bacteria consisting of Gram-positive S. aureus (Sa), B. subtilis (Bs), Gram-negative E. coli (Ec), and K. pneumonioe (Kb) using the in-vitro disc diffusion method63 at concentrations of 50 and 100 μM, respectively. The representative culture plates are presented in Fig. S44 (ESI†).
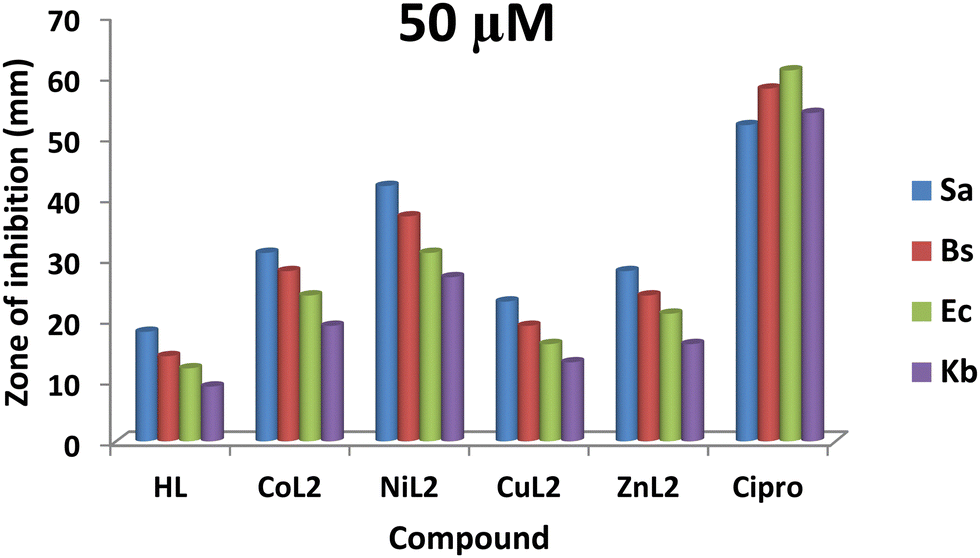
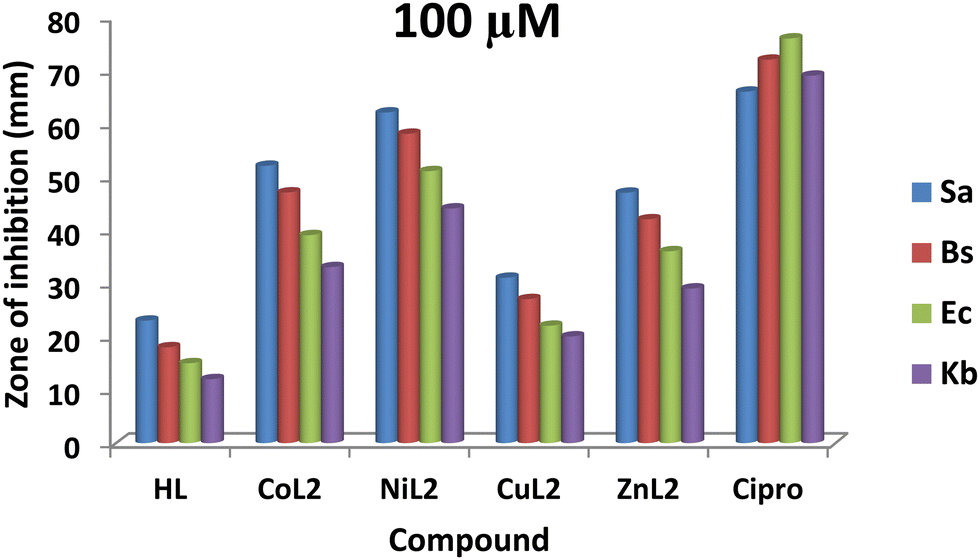
The zones of inhibition obtained from this study are shown in Fig. 9 and 10. From the results, all the complexes demonstrated higher activity on the tested bacteria compared to the free ligand. The activity was found to be concentration-dependent, with the complexes still having a greater inhibitory effect than the free ligand. However, the standard drug outperformed the complexes at both concentrations. The compounds' activity was higher on Gram-positive pathogens than on Gram-negative pathogens, with S. aureus being the most susceptible to all at both concentrations. Similarly, NiL2 showed enhanced activity on all the tested bacteria compared to the other complexes, with zones of inhibition of 42 ± 1.1, 370 ± 0.6, 31 ± 1.5, and 27 ± 2.1 mm at a concentration of 50 μM on S. aureus, B. subtilis, E. coli, and K. pneumonioe, respectively (Fig. 9). At 100 μM, the zones of inhibition of 62 ± 0.9, 57 ± 1.3, 51 ± 2.0, and 44 ± 1.7 mm were observed for S. aureus, B. subtilis, E. coli, and K. pneumonioe, respectively (Fig. 10). CuL2 has the least activity among the complexes, with an inhibition zone of 28 ± 0.9, 24 ± 1.6, 21 ± 2.2, and 16 ± 1.8 mm at a concentration of 50 μM on S. aureus, B. subtilis, E. coli, and K. pneumonioe, respectively (Fig. 9). Furthermore, at a concentration of 100 μM, S. aureus, B. subtilis, E. coli, and K. pneumonioe were inhibited to 31 ± 2.2, 27 ± 3.1, 22 ± 0.7, and 20 ± 1.4 mm, respectively (Fig. 10). The ligand, on the other hand, showed zones of inhibition of 18 ± 0.8, 14 ± 0.8, 12 ± 1.1, and 9 ± 1.6 mm, respectively, at 50 μM, Fig. 9. Also, at 100 μM, zones of inhibition of 23 ± 0.4, 18 ± 1.9, 15 ± 1.3, and 12 ± 2.2 mm were observed on S. aureus, B. subtilis, E. coli, and K. pneumonioe, respectively (Fig. 10). The higher activity of compounds on Gram-positive pathogens could mean that the compounds are single-spectrum antimicrobial agents rather than broad-spectrum agents, and the trend of activity among the compounds is in the order NiL2 > CoL2 > ZnL2 > CuL2 > HL. This implies that the ligand's antimicrobial efficacy increases with metal ion coordination, and that each metal has its own influence on the ligand's activity. Chelation theory could be used to explain this phenomenon. Metal ion coordination to a ligand result in a significant decrease in polarity, resulting in donor groups overlapping. As a result, electron delocalization within the ligand frequently increased, increasing the lipophilicity of the complexes. The increased lipophilicity of the complexes promotes bacterial lipid membrane penetration, giving advantages to inhibition.11,64–67
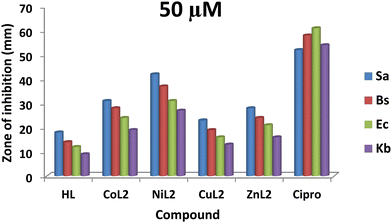 |
| | Fig. 9 The result of in vitro antibacterial activity (mm) of the ligand and its complexes on the bacteria at concentration of 50 μM. | |
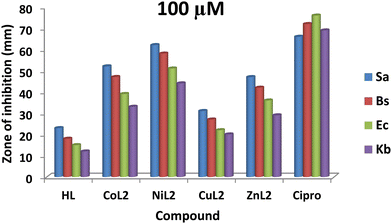 |
| | Fig. 10 The result of in vitro antibacterial activity (mm) of the ligand and its complexes on the bacteria at concentration of 100 μM. | |
3.3.2. Minimum inhibitory concentration (MIC).
Following the observation of an increase in antibacterial activity of complexes relative to the free ligand in the in vitro assay, the ligand, complexes, and control were subjected to MIC studies. The MIC was determined using the broth dilution method68 twofold, ranging from 512 to 0.250 μg mL−1 (96 well plates shown in Fig. S45, ESI†), and the results are shown in Table 5. From these results, the MIC values of the free ligand and its CuL2 complex were not detected at a maximum concentration of 256 μg mL−1 on all the tested pathogens. However, CoL2 showed MICs of 256 μgmL−1 for all the tested pathogens. while NiL2 had MICs of 8, 32, 128, and 128 μg mL−1 on S. aureus, B. subtilis, E. coli, and K. pneumonioe, respectively. Similarly, ZnL2 showed MICs of 256 μg mL−1 on S. aureus and B. subtilis, but MICs on E. coli and K. pneumoniae were not detected at the maximum dilution. The control drug, on the other hand, displayed MICs of 0.5 μg mL−1 on S. aureus and B. subtilis and MICs of 2 μg mL−1 on E. coli and K. pneumonioe, respectively. From these studies, NiL2 remains the most active among the complexes.
Table 5 The MICs (μg mL−1) of the ligand and it complexesa
| Compound |
Sa |
Bs |
Ec |
Kp |
|
Test sample.
DMSO considered due to its usage as a vehicle carrier for the test compounds.
Positive control.ND = not detected at 256 μg mL−1; – = no activity. |
|
HL
|
ND |
ND |
ND |
ND |
|
CoL2
|
256 |
256 |
256 |
256 |
|
NiL2
|
8 |
32 |
128 |
128 |
|
CuL2
|
ND |
ND |
ND |
ND |
|
ZnL2
|
256 |
256 |
ND |
ND |
| DMSOb |
— |
— |
— |
— |
| Ciproc |
0.5 |
0.5 |
2 |
2 |
3.3.3. Antioxidant study.
The radical scavenging activity of the free ligand, its complexes, and control (ascorbic acid) was evaluated using DPPH assay. The percentage of DPPH scavenging activity, IC50 values of the test compounds and the control are presented in Table 6. From the results, it is observed that the radical scavenging ability of the compounds is concentration dependent. However, the complexes demonstrated enhanced activity compared to the free ligand, which is due to the influence of the metal ion. A similar observation was reported by other researchers.69–71 Like the antimicrobial results, the positive control shows higher radical scavenging activities when compared to the ligand and its complexes. The values of the half-inhibitory concentration (IC50) of the tested compounds and the positive control as shown in Table 6 unveil the strength of the compounds. As a radical eliminator, the higher the IC50 value, the lower the radical scavenging activity of a compound. The IC50 values of the ligand, complexes and ascorbic acid are 109.2, 66.5, 57.2, 84.3, and 52.1 μg mL−1, respectively. Based on the IC50 values it can be concluded that only NiL2 show higher DPPH radical scavenging activity than the positive control (Table 6).
Table 6 DPPH assay radical scavenging activity and IC50 of the ligand and its complexes
| Conc. (μgmL−1) |
Radical scavenging activity (%) |
|
HL
|
CoL2
|
NiL2
|
CuL2
|
ZnL2
|
AA |
| 20 |
8.2 |
21.4 |
25.3 |
11.4 |
15.6 |
28.2 |
| 40 |
15.7 |
28.7 |
34.5 |
18.9 |
23.4 |
42.1 |
| 60 |
24.5 |
46.3 |
49.3 |
36.2 |
39.8 |
58.3 |
| 80 |
37.2 |
67.2 |
75.6 |
47.0 |
54.6 |
81.5 |
| 100 |
48.5 |
73.4 |
83.5 |
61.3 |
66.1 |
88.5 |
| IC50 |
109.2
|
66.5
|
57.2
|
84.3
|
75.3
|
52.1
|
3.3.4. Toxicity study.
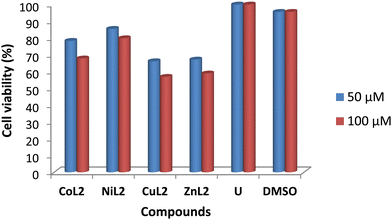
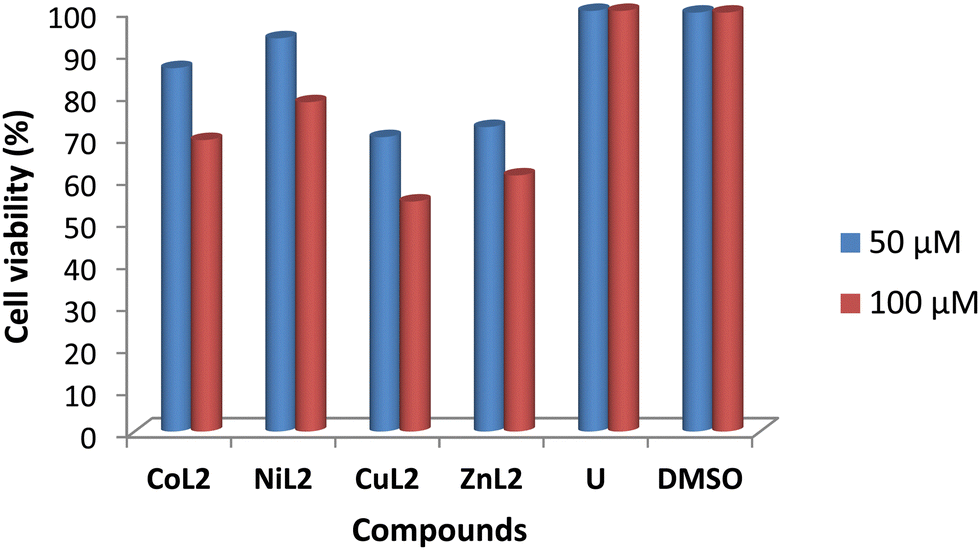
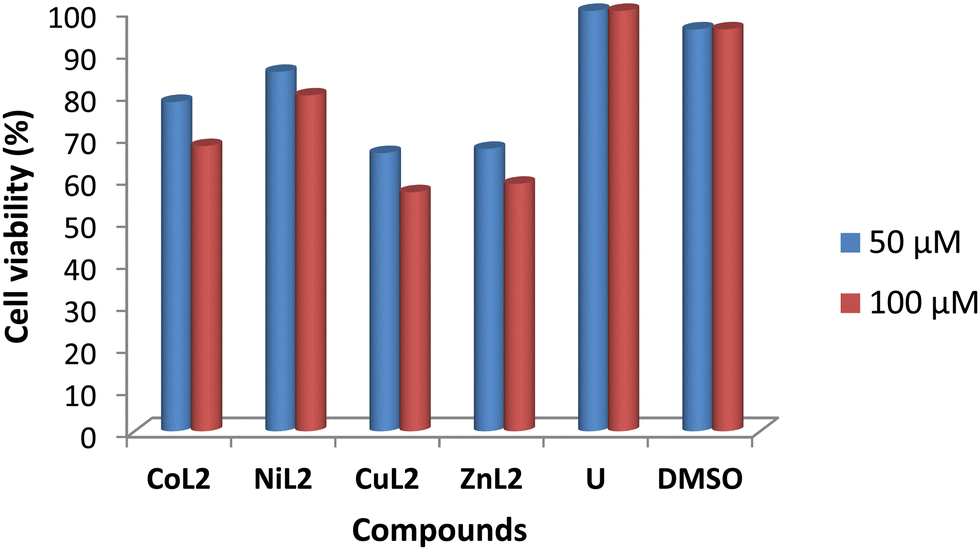
The toxicity profile of the complexes was assessed using concentrations of 50 and 100 μM on two human cell lines, WISH-ATCC-CCL-25, human epithelial amnion (normal liver cell lines and MRC-5-ATCC-CCL-171, normal human lung fibroblast cell lines. The compounds' activity is measured in terms of cell viability after 48 hours of exposure. The results are presented in Fig. 11 and 12. The toxicity of the complexes on cell lines revealed that at 50 μM, cell viability of WISH cell lines was 86.4 and 93.5% after treatment with CoL2 and NiL2, respectively. Treatment of the same cell line with CuL2 and ZnL2 at 50 μM, on the other hand, resulted in viability of 70 and 72.4%, respectively (Fig. 11). CoL2, NiL2, CuL2, and ZnL2 had viabilities of 69.3, 78.3, 54.7, and 60.9% at 100 μM, respectively on WISH cell lines (Fig. 11). The toxicity profile of the complexes on the MRC-5 cell lines is depicted in Fig. 12. The results show that at a concentration of 50 μM, CoL2, NiL2, CuL2, and ZnL2 reduced the cell viability to 78.3, 85.5, 66.2, and 67.2%, respectively. Similarly, at 100 μM, after treatment of MRC-5 cell lines with CoL2 and NiL2, cell viability of 67.8 and 79.9% were observed, respectively, and 56.9 and 58.7% for CuL2 and ZnL2, respectively. In general, the results show that CoL2 and NiL2 complexes are less toxic to the two cell lines than CuL2 and ZnL2 complexes, with CuL2 being the most toxic to both cell lines. The lower toxicity of CoL2 and NiL2 on the cell lines provides a platform for further investigations of the compounds as potential antibacterial agents.
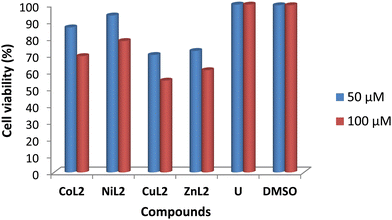 |
| | Fig. 11 Viability of WISH cell lines (% in relation to untreated cells) after treatment with 50 and 100 μM compound's solution. DMSO included due to its use as a solvent carrier for the test compounds. | |
 |
| | Fig. 12 Viability of MRC-5 cell line (% in relation to untreated cells) after treatment with 50 and 100 μM compound's solution. | |
3.4. Computational study
3.4.1. Non-covalent interactions (NCI).
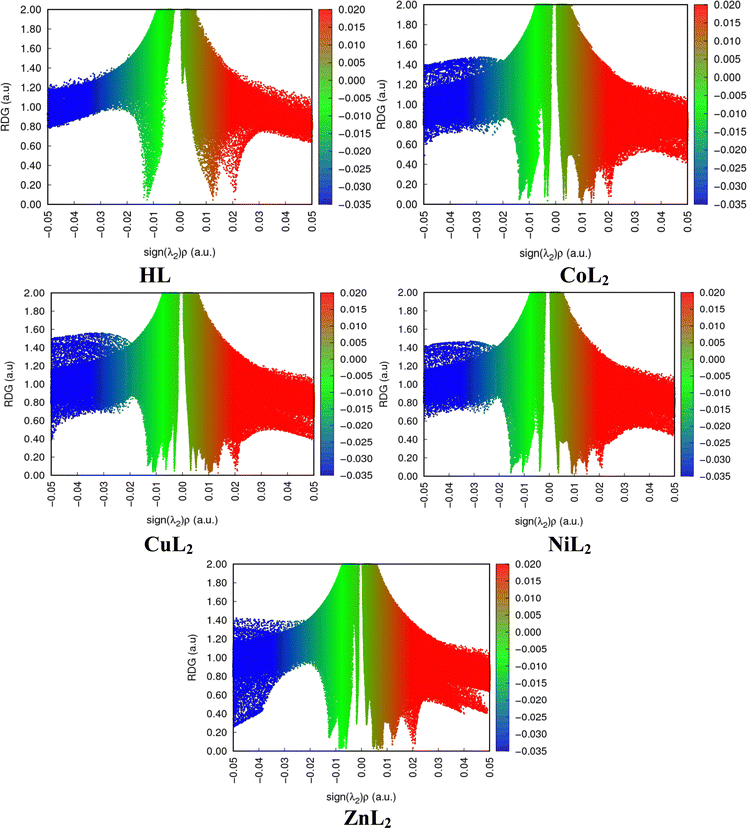
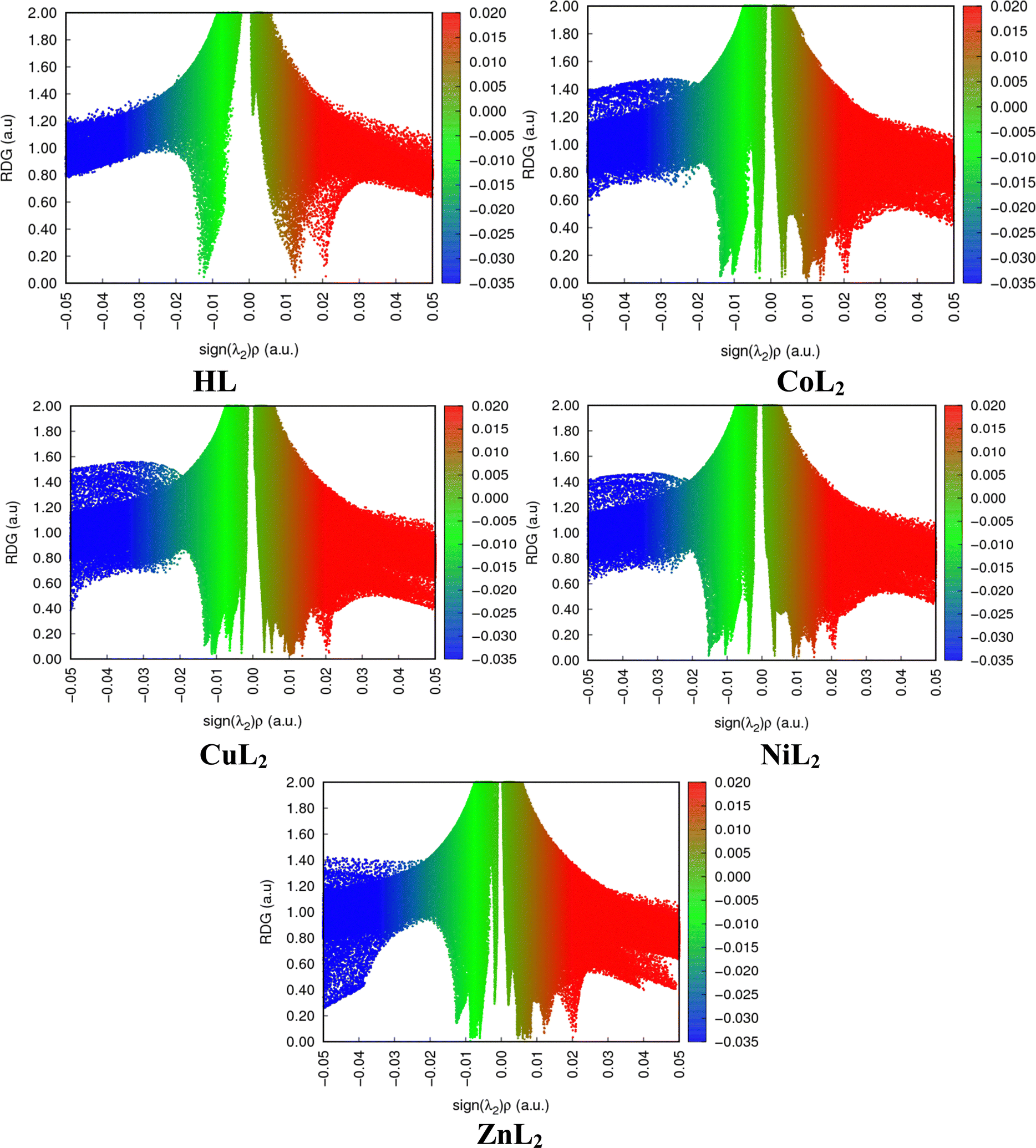
All biological systems depend on molecular recognition, protein stability, specificity, and efficiency of enzymatic activities, which are all stabilized by non-covalent interactions (NCI). They instantly cleave upon formation and depend on factors such as properties of interacting groups or atoms, the distances between them, and the media in which they occur.72 They play significant roles in crystal engineering and constructing multidimensional structures.73 Using the NCI index as a tool, these interactions can be analyzed and can reveal both qualitative and quantitative information about electron densities through an index s which locates the region in space where the reduce density gradient (RDG) is close to zero and forms well-defined troughs (Fig. 13). These RDG zones are known as isosurfaces and are characterized by well-defined density values (ρ) that result from minute gradients along each of them. They signify weak interactions when both s and are characterized as weak. When both s and ρ are defined as weak, then they denote weak interactions. As different colors reveal different interactions, a red color indicates steric repulsion between electrons, green colors van der Waals attractive forces and hydrogen bonding is blue. The intensity of the colors is a measure of the strength of the interaction, with more intense colors indicating stronger interactions. The strength of the interaction is measured by the index ρ, which assigns a trough to the strongest interaction where the highest trough and all forces are denoted by the eigen value λ2.74 A larger quantity of well-defined troughs is observed in the complexes than in the free ligand. In all compounds though, it appears as if repulsive forces dominate.
 |
| | Fig. 13 Two-dimensional reduced density gradient scatter maps for compounds. | |
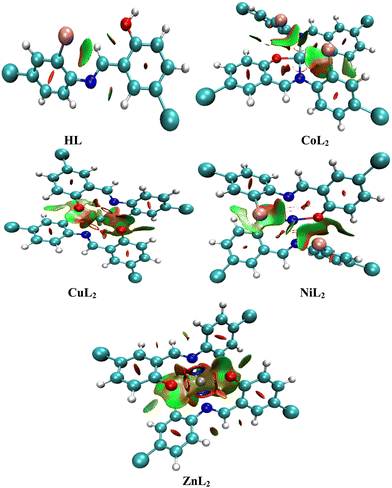
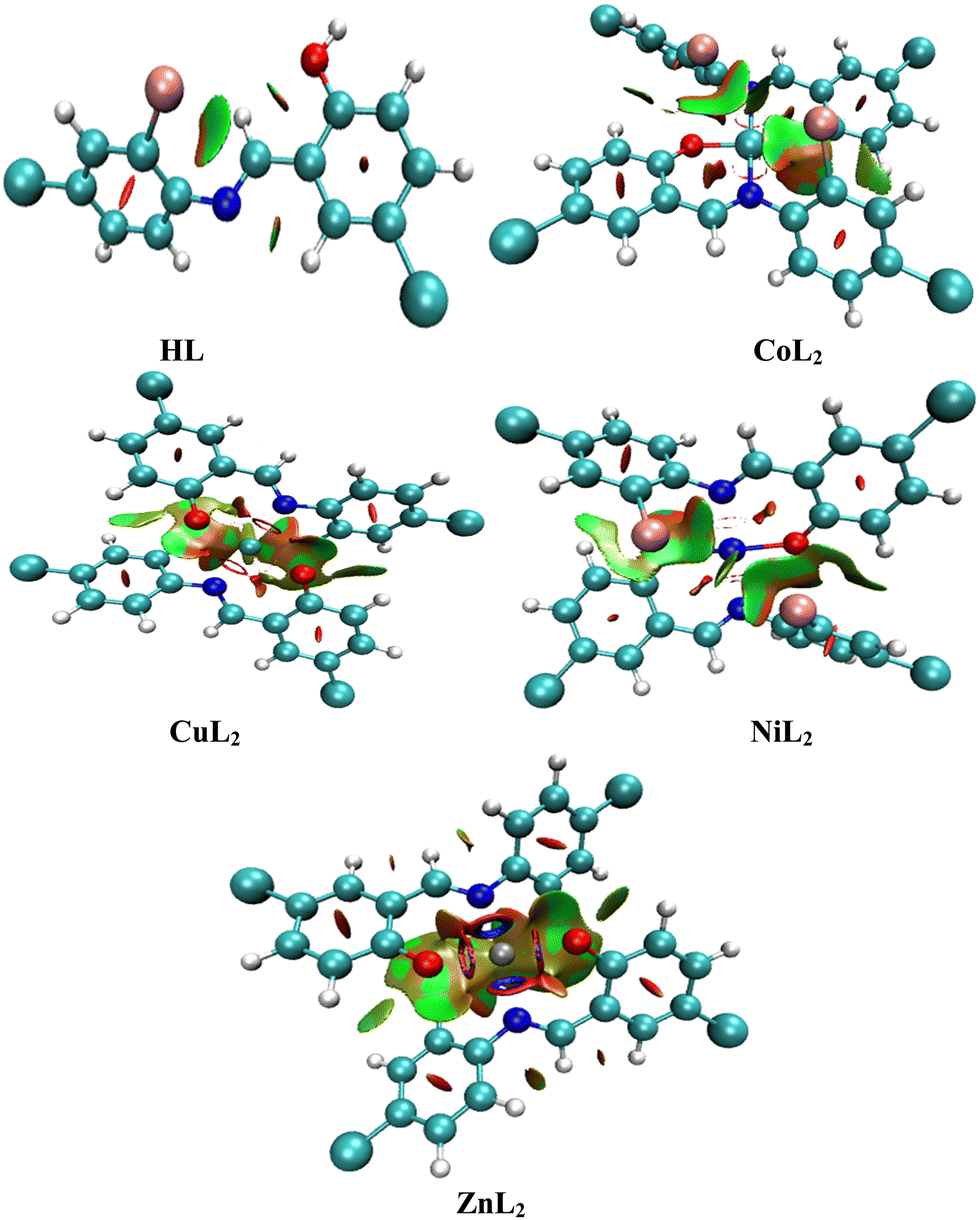
The locations of these interactions on each compound in three dimensions (Fig. 14) are established by observing red cigar-shape isosurfaces in the center of each aromatic ring for ring closure, while the multi colored (red/green) extended sheets reveals van der Waals repulsive and attractive forces.75 In addition to this, the folded sheets represent intra-ligand and metal–ligand interactions which is stabilized through a multicentric building up of electron density around the chelate ring where the red color represents steric stain and is counter balanced by a green part that denotes steric crowding.74,76 This indicates that the degree of homogenous electron distribution in compounds causes the difference in biological properties observed between the ligand and the complexes.77 A multicentric building up of electron density predominates in the Zn complex. This observation agreed with the reported literature work. Although, the multicentric building up of electron density was completely absent in the nickel complex, while it was present in the zinc complex in this study.78
 |
| | Fig. 14 Three-dimensional isosurfaces that stabilize the compounds through non-covalent interactions. | |
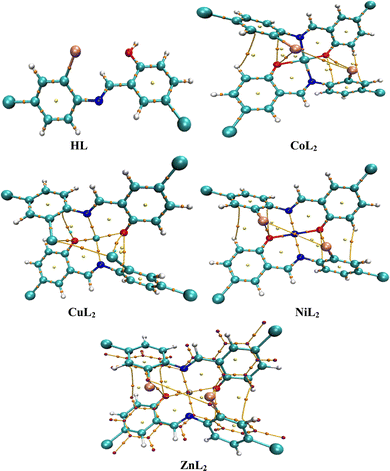
No atomic-interaction-lines (AIL) were detected for the ligand, but several for the complexes exist (Fig. 15). As these lines depicts interaction routes, contact routes between them are often measured via bond critical points (BCP).39 C⋯H, C⋯O, Br⋯O and Br⋯Br predominates in both complexes. Other than the latter interaction, similar interactions were observed for palladium complexes and explains the sharing of electron density between the center of each metal and the ligand.79
 |
| | Fig. 15 Atomic interaction lines and bond critical points between atoms in each compound. | |
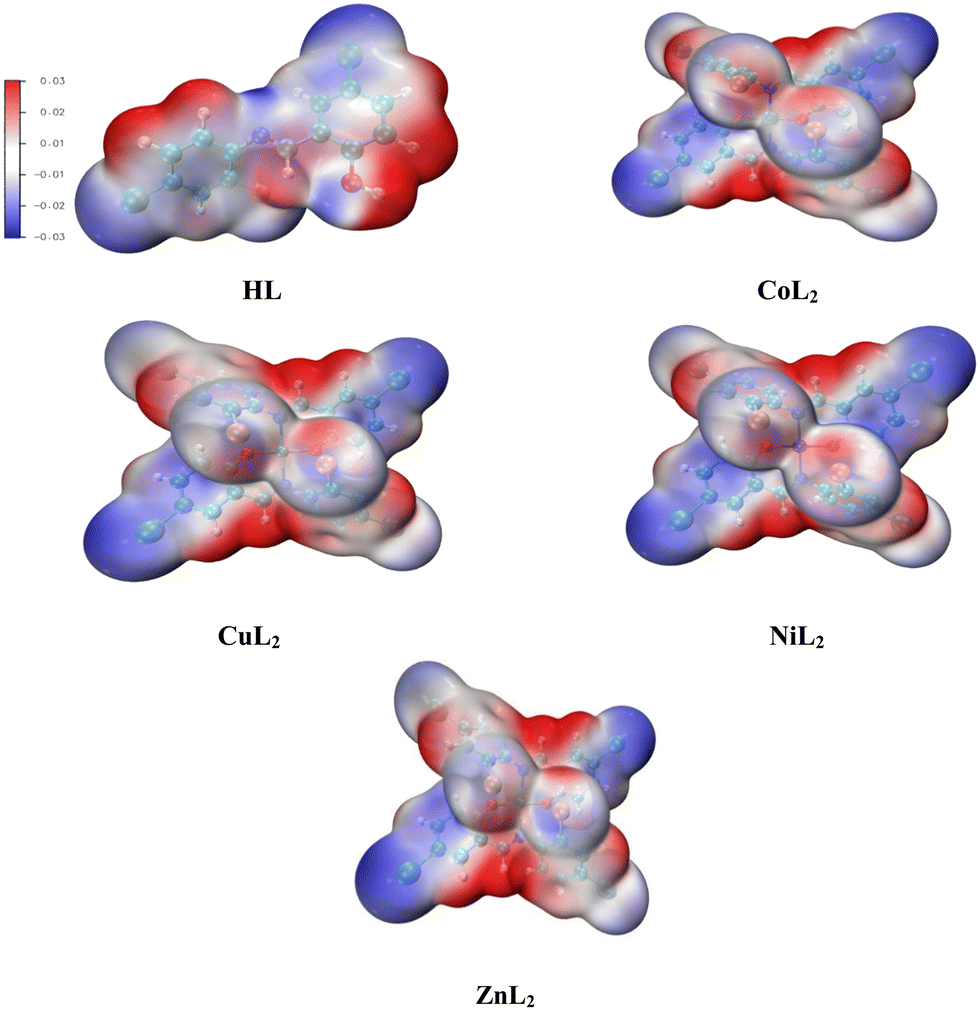
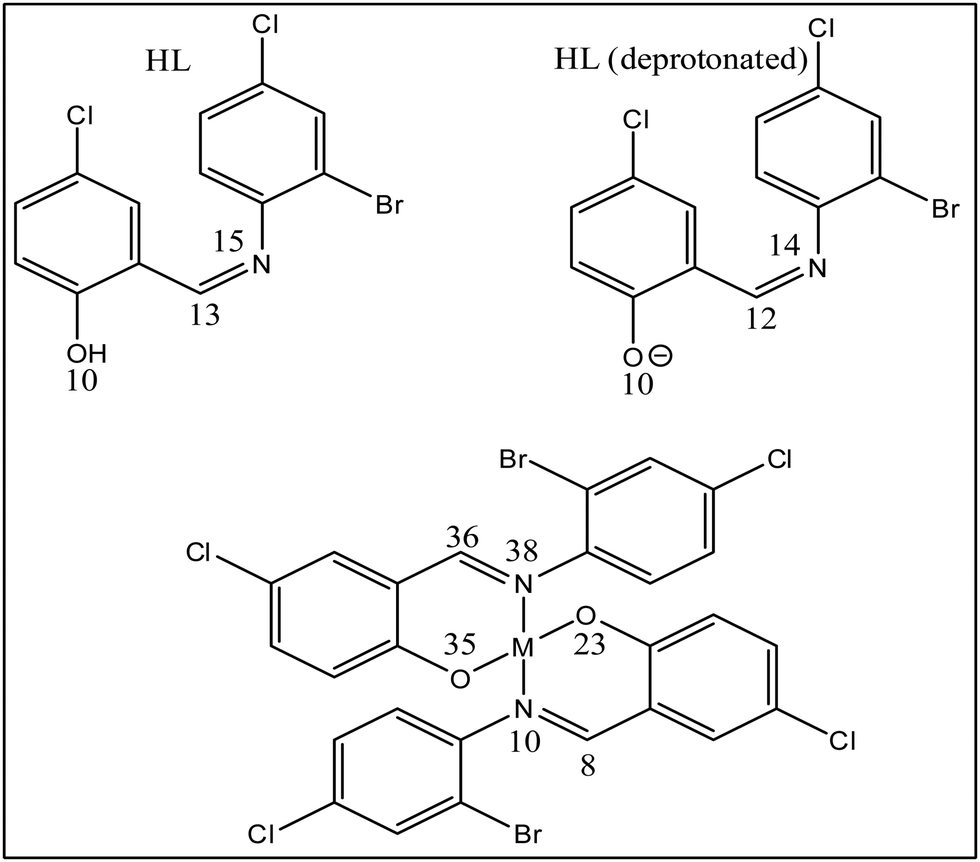
3.4.2. Molecular Electrostatic Potential (MEP).
Molecular Electrostatic Potential (MEP) is a chemical descriptor that studies the reactive sites on compounds by measuring variations in electron densities across these sites. In doing so, it displays an electron density map with blue, green, yellow, orange, and red colors (Fig. 16). The reduction of electron concentrations on these sites in the order blue > green > yellow > orange > red indicates that blue denotes a nucleophilic attack, while red reveals an electrophilic attack.80,81 When comparing the neutral ligand to the deprotonated ligand, the inductive effect on the latter causes the para substituted deprotonated oxygen to remove electrons, resulting in an electron flow to the imine moiety. We observe a larger electron flow towards C12 and N14 (Table 7). The electron densities in the complexes drop considerably on the C8 and C36, while it increases significantly on the lone pairs on the N10 and N38 of the imine moiety. This occurs due to a reduction in electron densities on O10 due to bond formation. As a significantly lower concentration of electrons on O23 and O35 on the copper complex is observed, a larger flow of electrons occurs towards N10 and N38 of this complex (Fig. 17 shows the position of these atoms in the compounds). When comparing the cobalt and copper complexes to the nickel and zinc complexes, higher concentrations of electrons are observed in the latter on the donor atoms.
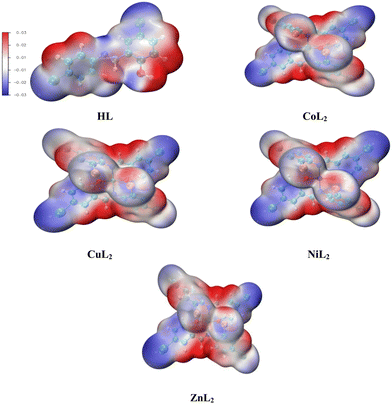 |
| | Fig. 16 Electron density map for the molecular electrostatic potential of the compounds. | |
Table 7 Molecular electrostatic potential values (−1 × 10−3) on the imine moieties
| Atom |
L2 |
L2 (deprotonated) |
CoL2
|
CuL2
|
NiL2
|
ZnL2
|
| N10/14/15/38 |
201 |
−91.5 |
−244 |
−264 |
261 |
255 |
| C8/12/13/36 |
282 |
−254 |
−122 |
−127 |
−185 |
113 |
| O10/23/35 |
552 |
471 |
−210 |
−127 |
−11.2 |
−45.5 |
 |
| | Fig. 17 Atomic numbering on the imine moieties of the compounds; M = Co(II), Ni(II), Cu(II), and Zn(II), respectively. | |
3.4.3. Electron localization function (ELF).
A reference electron is used to determine the probability of finding an electron pair. This reference electron must lie in the same vicinity and have the same spin as its associated Fermi hole. This allows the revelation of the description of the spatial localization of these electron pairs, where a smaller probability induces a higher localization of the reference electron. Moreover, the extraction of information regarding the spatial electron localization from its kinetic energy density through Becke and Edgecombe's dimensionless scalar electron localization function (ELF) can be obtained using eqn (3):| | 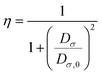 | (3) |
where 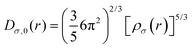 .
.
Using eqn (3), a description of the kinetic energy density of a homogeneous electron gas with the spin density locally equal to ρσ(r) where the ELF are restricted to the range 0 ≤ η ≤ 1 can be revealed. The electron is completely localized if η = 1, and if 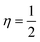 , a homogeneous electron gas-like pair probability (electron delocalization) occurs. Borders between electron pairs result from a value close to zero.82,83 The topological interpretation of molecular space which aids in identifying bonds, lone pairs and core regions can clarify the molecular and atomic shell structures through ELF. Red colors reveal highly localized electrons, while dark colors indicate regions where electrons are not highly localized and green colors indicate delocalized electrons (Fig. 18). When investigating the C–N–C plane, we observe that electrons become more localized in the region 0–6 Bohr in both x and y-directions upon complexation. The free ligand shows no localized electrons in this region though. When comparing the cobalt and copper complexes, electrons are more localized in the former in this vicinity. Electrons are more localized at 2.04 Bohr in the x-direction in the nickel complex than in the other complexes at this position. For the zinc complex, electrons are not highly localized in the 0–6 Bohr range in the x-direction. We also observe that electrons become highly localized in the region 12.26–14.30 Bohr in the x-direction.
, a homogeneous electron gas-like pair probability (electron delocalization) occurs. Borders between electron pairs result from a value close to zero.82,83 The topological interpretation of molecular space which aids in identifying bonds, lone pairs and core regions can clarify the molecular and atomic shell structures through ELF. Red colors reveal highly localized electrons, while dark colors indicate regions where electrons are not highly localized and green colors indicate delocalized electrons (Fig. 18). When investigating the C–N–C plane, we observe that electrons become more localized in the region 0–6 Bohr in both x and y-directions upon complexation. The free ligand shows no localized electrons in this region though. When comparing the cobalt and copper complexes, electrons are more localized in the former in this vicinity. Electrons are more localized at 2.04 Bohr in the x-direction in the nickel complex than in the other complexes at this position. For the zinc complex, electrons are not highly localized in the 0–6 Bohr range in the x-direction. We also observe that electrons become highly localized in the region 12.26–14.30 Bohr in the x-direction.
 |
| | Fig. 18 Two and three-dimensional electron localization maps for the ligand and complexes. | |
3.4.4. Conceptual density functional theory (CDFT).
CDFT is a useful alternative chemical descriptor to MEP. In addition to this, CDFT also detect sites where radical attacks occurs as well as measuring chemical potential, ionization potential (IP), electron affinity (EA), electrophilicity index (ω), nucleophilicity index (N), chemical hardness (η), global softness (δ), electronegativity (χ), and energy differences (Table S5, ESI†). As reactive oxygen species (ROS) are inactivated by mopping up radical attacks, identifying these sites can be used as input for structure–activity relationships in molecular docking analyses.84,85 The index δ can be used to measure the ability of an atom or a group of atoms to receive electrons,86 while the resistance of an atom to charge transfer (CT) is measured by the index η.84 The parameter ω predicts their stabilization energies, which can be useful in estimating the biological activities of compounds.87–89 The index N measures the organic molecules electrophilic and nucleophilic behavior.90 When an electron through the electron accepting power of an accepting molecule “jump” from the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO), it releases energy which can be measured by EA.85 The IP knocks out an electron from its HOMO through its electron donating power.91 A compound becomes more stable at larger HOMO–LUMO energy gaps.92 A study by Bulat et al., debunked previous studies that suggested that an electron that occupies the HOMO level will automatically be excited to the LUMO level as this study revealed that an electron's position in atomic space and the contour of the molecule also play a role in excitations.93 Nevertheless, it is still widely accepted that HOMO–LUMO energy gaps play a significant role in electron excitations.94 Comparing the ligand to the complexes, we observe greater stability, larger IP, lower EA, larger η, lower δ and ω indices in the former. In the cobalt and copper complexes no IP, EA, η, δ and ω indices were obtained. In the free ligand, electrons are highly localized throughout the compound and upon excitation becomes less localized across the chloro and hydroxy groups, and in the complexes, a similar picture is revealed (Fig. S46, ESI†).
CDFT calculations were performed to obtain a descriptor called the Fukui Function, which relates the reactivity/selectivity of a specific site of local quantities on atomic sites on a molecule. It is defined as follows.
| | 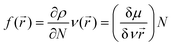 | (4) |
The condensed Fukui Functions (
f−,
f+ and
f0) and charge density difference (CDD) are depicted in
Table 9. The Fukui Function can be calculated using the following equations.
| | f+(![[r with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0072_20d1.gif) ) = qr(N + 1) − qr(N) ) = qr(N + 1) − qr(N) | (5) |
for a nucleophilic attack
| | | f−() = qr(N) − qr(N − 1) | (6) |
for an electrophilic attack
| | | f0() = qr(N + 1) − qr(N − 1) | (7) |
for a radical attack.
The charge density difference (CCD) is the difference between f+() and f−(). Since the quantitative values for the reactivities agree with the quantitative values for MEP analyses, it was worthwhile to observe the radical attacks (Table 8). Other than N15 in the free ligand, low radical attacks are observed in all the compounds.
Table 8 Conceptual density functional theory parameters for the compounds
| Atom |
f
−
|
f
+
|
f
0
|
CDD |
|
HL
|
| N15 |
0.0622 |
0.0836 |
0.0729 |
0.0214 |
| C13 |
0.0302 |
0.0947 |
0.0625 |
0.0645 |
|
CoL2
|
| N10 |
−1.26 |
1.26 |
0.004 |
2.53 |
| C8 |
−1.38 |
1.38 |
0.0005 |
2.76 |
|
CuL2
|
| N10 |
−1.59 |
1.59 |
−0.0011 |
3.18 |
| C8 |
−1.75 |
1.76 |
0.0053 |
3.50 |
|
NiL2
|
| N10 |
0.0143 |
0.0249 |
0.0196 |
0.0106 |
| C8 |
0.0175 |
0.0442 |
0.0308 |
0.0267 |
|
ZnL2
|
| N10 |
0.0147 |
0.0289 |
0.0218 |
0.0143 |
| C8 |
0.0101 |
0.0565 |
0.0333 |
0.0464 |
Table 9 Binding affinity (kcal mol−1) for docked complexes
| Compound |
Proteins |
| 1JIJ |
2CXT |
|
CoL2
|
4.21 |
−5.22 |
|
NiL2
|
0.57 |
−7.25 |
|
CuL2
|
14.63 |
−3.72 |
|
ZnL2
|
6.59 |
−3.11 |
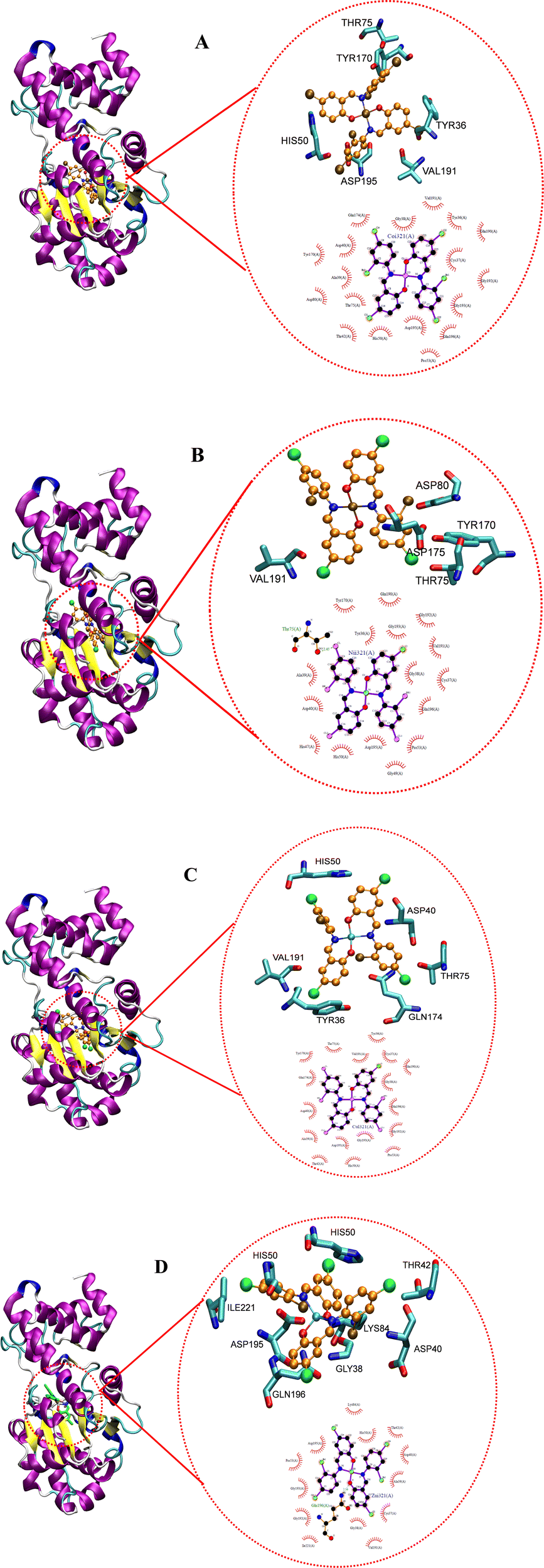
3.5. Molecular docking study
Docking studies reveal the interactions that occur between a biomolecule and a receptor's active site, as well as the quantitative binding energy that results from these interactions.95 The complexes were docked against tyrosyl t-RNA synthase (PDB: 1jij) and type IIA topoisomerase (PDB: 2xct) proteins to further extrapolate the mechanism of action of the complexes against the pathogens used for the antibacterial study and to correlate the in vitro and MIC studies. Table 9 and Fig. 19 and 20 show the obtained results and docking interaction patterns. The complexes' interactions with these proteins revealed a varying degree of binding mode within the active sites. After interaction with tyrosyl t-RNA synthase, NiL2 shows the lowest binding energy (0.57 kcal mol−1), while CuL2 has the highest binding energy (14.63 kcal mol−1) (Table 9). CoL2 and ZnL2 have binding energies of 4.21 and 6.59 kcal mol−1, respectively (Table 9). From this result, NiL2 strongly interacted with the active site of the receptor, and this occurred through His50 and halogen atoms of the complexes forming hydrogen bonds with the amino acid residues Tyr36 and Thr75 (Fig. 19). The strong interactions in NiL2 support the antibacterial activity finding. Similarly, the highest binding energy of CuL2 showed weak interaction within the active site of the protein, and this could be responsible for the weak antibacterial activity demonstrated by the compound. CoL2 and ZnL2 binding energies were ranked second and third, respectively. This is consistent with the antibacterial activity trends demonstrated by the compounds. In general, all complexes bind to the receptors through a halogen atom, forming a hydrogen bond with the amino acid residue.
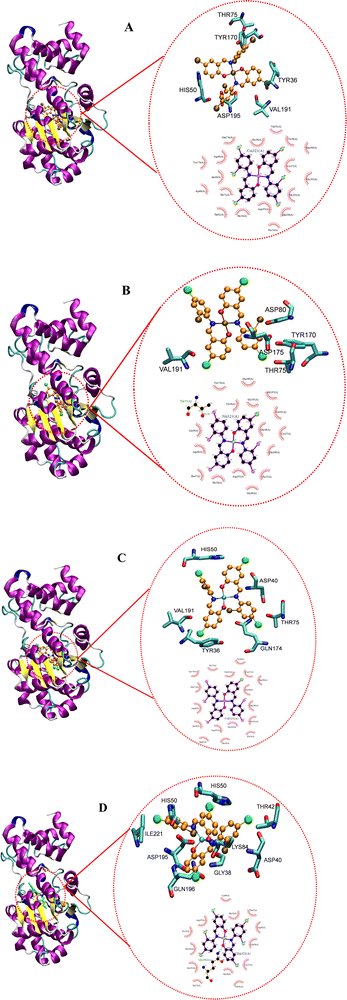 |
| | Fig. 19 Docked conformation of (A) CoL2, (B) NiL2, (C) CuL2 and (D) ZnL2 in tyrosyl t-RNA synthases. | |
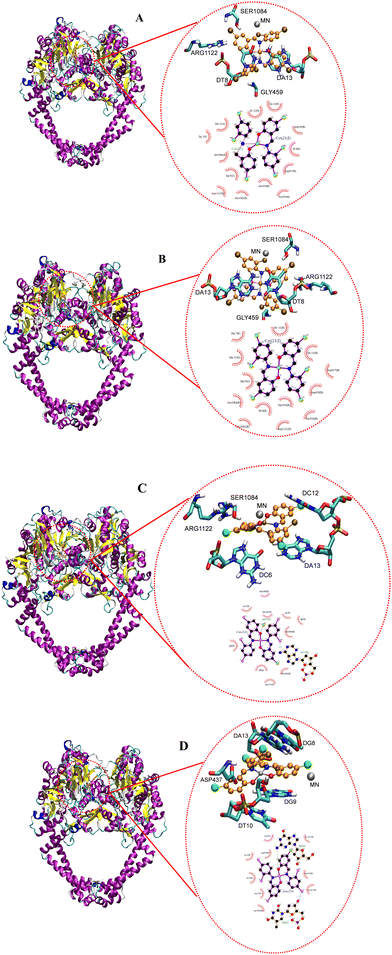 |
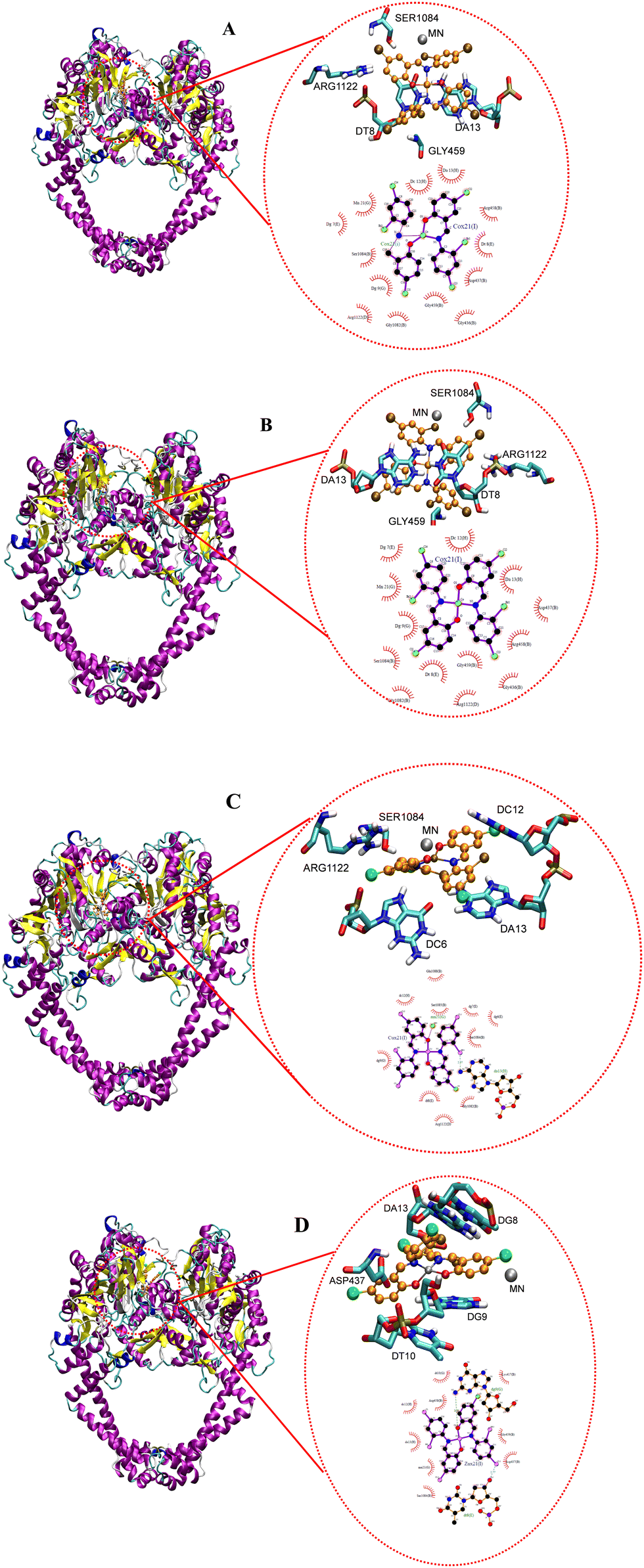
| | Fig. 20 Docked conformation of (A) CoL2, (B) NiL2, (C) CuL2 and (D) ZnL2 in type IIA topoisomerase. | |
Fig. 20 depicts the complexes' interaction with type IIA topoisomerase. This interaction also revealed that NiL2 has the lowest binding energy (−7.25 kcal mol−1) and forms a halogen-amino acid hydrogen bond via Arg1122 and DC12. This suggests a strong interaction with the receptor and is consistent with the experimental result. CoL2, CuL2, and ZnL2 binding energies were found to be −5.22, −3.11, and −3.72 kcal mol−1, respectively (Table 9). This corresponds to the level of interaction in the following order: CoL2 > ZnL2 > CuL2. This pattern is consistent with the experimental in vitro and MIC results. Overall, the docking study demonstrates that the complexes bind to the receptor in a manner consistent with the experimental results and that they correlate well.
4. Conclusions
In this study, we have synthesized an ON donor halogen substituted Schiff base ligand and its Co(II), Ni(II), Cu(II), and Zn(II) complexes. The formation of the ligand and its complexes were established via various spectroscopic and analytical techniques. Based on these characterization techniques, the ligand acted as bidentate and coordinated to the metal ions through nitrogen and oxygen atoms of the imine and phenolate groups, respectively, giving rise to square planar geometry around the metal ion in all the complexes. The geometry of the ligand and its Ni(II) and Cu(II) complexes were obtained using single crystal X-ray diffraction (SCXRD) analysis, while structures of Co(II) and Cu(II) complexes were elucidated using additional characterization techniques such as SEM, EDX, and powder X-ray diffraction (PXRD) analysis. The ligand and it complexes were screened for their antibacterial potential against selected pathogenic bacteria consisting of Gram-positive (Staphylococcus aureus and Bacillus subtilis) and Gram-negative (Klebsiella pneumonioe and Pseudomonas aeruginosa) strains using an in vitro assay. Antioxidant activity was evaluated using the DPPH assay. In all the assays, the complexes showed enhanced activity over the un-complexed Schiff base. Toxicity studies on normal human cell lines WISH-and MRC-5 cell lines revealed that at lower concentrations, the complexes had no effect on the cell lines. Although only crystal structures of the free ligand, NiL2 and CuL2 were obtained, performing Hirshfeld surface analysis on them showed H⋯H percentage atomic contributions for intermolecular interactions that were consistent with biological activities. In addition to this, the computational investigation provides information regarding the electronic properties of the ligand and its complexes such as stability, reactivity, and biological potentials using molecular electrostatic potential maps, conceptual density functional theory calculations and non-covalent-interactions analysis. An electron localization function (ELF) analysis along the C–N–C plane revealed a correlation between the ELF distance and biological activities, where shorter ELF distances corresponded to enhanced biological activities. These computational data reinforced the experimental findings. Molecular docking studies demonstrated the compounds' mechanism of action and identified potential binding sites. The obtained results are consistent with the in vitro assays.
Authorship contributions
I. Waziri: conceptualization, supervision, writing original draft, data curation/analysis, review and editing, HM. Masena: experiment, data curation and analysis, TL. Yusuf: crystal refinement, review, and editing, LC. Coetzee: computational study, review and editing, AS. Adeyenka: computational study, review and editing, AJ. Muller: conceptualization, supervision, formal analysis, review, editing, and funding.
Conflicts of interest
The authors affirm that they do not have any competing interests of any type that would affect the research presented here.
Acknowledgements
We thank the South African National Research Foundation (NRF) (Grant No. 111706) for running expenses. HM Massena is grateful for funding from the University of Johannesburg. The authors would like to acknowledge the Center for High Performance Computing (CHPC), South Africa, for providing computational resources for this research project. We also thank Dr B. Vatsha of the Department of Chemical Sciences, University of Johannesburg, for the single crystal data collection. Dr I. Waziri and Dr T. L. Yusuf are grateful to the URC for the award of PDRF.
References
- K. D. Mjos and C. Orvig, Chem. Rev., 2014, 114, 4540–4563 CrossRef CAS.
- P. Ehrlich and A. Bertheim, Ber. Dtsch. Chem. Ges., 1912, 45, 756–766 CrossRef CAS.
- Z. Faghih, A. Neshat, A. Wojtczak, Z. Faghih, Z. Mohammadi and S. Varestan, Inorg. Chim. Acta, 2018, 471, 404–412 CrossRef CAS.
- M. E. Cooley, L. Davis and J. Abrahm, Cancer Nurs., 1994, 17, 283–293 CrossRef CAS PubMed.
- S. Ghosh, Bioorg. Chem., 2019, 88, 102925 CrossRef CAS PubMed.
- L. Qi, Q. Luo, Y. Zhang, F. Jia, Y. Zhao and F. Wang, Chem. Res. Toxicol., 2019, 32, 1469–1486 Search PubMed.
- A. Evans and K. A. Kavanagh, J. Med. Microbiol., 2021, 70 CrossRef CAS PubMed.
- U. Hofer, Nat. Rev. Microbiol., 2019, 17, 3 CrossRef CAS PubMed.
- W. H. Organization, Weekly Epidemiological Record = Relevé épidémiologique hebdomadaire, 2000, 75, 336 Search PubMed.
- I. Waziri, M. A. Isa, M. Sonopo, D. B. G. Williams and A. Muller, Bioorg. Med. Chem. Lett., 2021, 52, 128381 CrossRef CAS PubMed.
- W. C. Reygaert, AIMS Microbiol., 2018, 4, 482 CAS.
- J. M. Munita and C. A. Arias, Microbiol. Spectrum, 2016, 4, 4.2.15 CrossRef.
- S. Alghamdi, S. U. Rehman, N. T. Shesha, H. Faidah, M. Khurram and S. U. Rehman, Molecules, 2020, 25, 5685 CrossRef CAS.
- R. K. Sodhi and S. Paul, Cancer Therapy Oncol. Int. J., 2019, 14, 25–32 Search PubMed.
- K. L. Haas and K. J. Franz, Chem. Rev., 2009, 109, 4921–4960 CrossRef CAS PubMed.
- N. Kordestani, H. A. Rudbari, A. R. Fernandes, L. R. Raposo, A. Luz, P. V. Baptista, G. Bruno, R. Scopelliti, Z. Fateminia and N. Micale, Dalton Trans., 2021, 50, 3990–4007 RSC.
- H. Kargar, F. Aghaei-Meybodi, M. R. Elahifard, M. N. Tahir, M. Ashfaq and K. S. Munawar, J. Coord. Chem., 2021, 74, 1534–1549 CrossRef CAS.
- A. S. Basaleh, H. B. Howsaui, A. A. Sharfalddin and M. A. Hussien, Results Chem., 2022, 4, 100445 CrossRef CAS.
- L. H. Abdel-Rahman, M. S. S. Adam, N. Al-Zaqri, M. R. Shehata, H. E.-S. Ahmed and S. K. Mohamed, Arabian J. Chem., 2022, 15, 103737 CrossRef CAS.
- I. Waziri, T. L. Yusuf, E. Akintemi, M. T. Kelani and A. Muller, J. Mol. Struct., 2023, 1273, 134382 CrossRef CAS.
- I. Waziri, T. L. Yusuf, H. A. Zarma, S. O. Oselusi, L.-C. C. Coetzee and A. S. Adeyinka, Inorg. Chim. Acta, 2023, 552, 121505 CrossRef CAS.
- T. A. Alorini, A. N. Al-Hakimi, S. E.-S. Saeed, E. H. L. Alhamzi and A. E. Albadri, Arabian J. Chem., 2022, 15, 103559 CrossRef CAS.
- A. Bruker and B. Saint, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 467 CrossRef.
- T. Schulz, K. Meindl, D. Leusser, D. Stern, J. Graf, C. Michaelsen, M. Ruf, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2009, 42, 885–891 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens and M. Towler, J. Appl. Crystallogr., 2020, 53, 226–235 CrossRef CAS PubMed.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
- C. B. Pinto, B. L. Rodrigues and L. H. Dos Santos, J. Appl. Crystallogr., 2021, 54, 1600–1605 CrossRef CAS.
- A. Bauer, Am. J. Clin. Pathol., 1966, 45, 149–158 CrossRef.
- A. L. N. Pagning, M. Lateef, L. A. Tapondjou, J.-R. Kuiate, D. Ngnokam and M. S. Ali, Phytochem. Lett., 2016, 16, 121–128 CrossRef.
- S. Janowska, D. Khylyuk, S. Andrzejczuk and M. Wujec, Molecules, 2022, 27, 3161 CrossRef CAS PubMed.
- J. N. Eloff, Planta Med., 1998, 64, 711–713 CrossRef CAS PubMed.
- A. Xing, D. Zeng and Z. Chen, J. Mol. Struct., 2022, 1253, 132209 CrossRef CAS.
- A. Kilic, R. Söylemez and V. Okumuş, J. Organomet. Chem., 2022, 960, 122228 CrossRef CAS.
- S.-Y. Chen, X.-H. Jiang, R.-X. Liu, Y. Huang, W.-Y. Shen, Y.-H. Jiang, K.-B. Huang and Y.-C. Liu, Inorg. Chim. Acta, 2021, 516, 120171 CrossRef CAS.
- M. Tyszka-Czochara, A. Adach, T. Grabowski, P. Konieczny, P. Pasko, J. Ortyl, T. Świergosz and M. Majka, Int. J. Mol. Sci., 2021, 22, 1802 CrossRef CAS.
-
M. Frisch, F. Clemente, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino and G. Zhe, Gaussion 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009, pp. 20–44 Search PubMed.
- T. Lu and F. Chen, J. Theor. Comput. Chem., 2012, 11, 163–183 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- J. A. Nylander, J. C. Wilgenbusch, D. L. Warren and D. L. Swofford, Bioinformatics, 2008, 24, 581–583 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CAS.
- X. Qiu, C. A. Janson, W. W. Smith, S. M. Green, P. McDevitt, K. Johanson, P. Carter, M. Hibbs, C. Lewis and A. Chalker, Protein Sci., 2001, 10, 2008–2016 CrossRef CAS PubMed.
- B. D. Bax, P. F. Chan, D. S. Eggleston, A. Fosberry, D. R. Gentry, F. Gorrec, I. Giordano, M. M. Hann, A. Hennessy and M. Hibbs, Nature, 2010, 466, 935–940 CrossRef.
-
L. Schrödinger and W. DeLano, The PyMOL molecular graphics system, version 2, 2020.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- G. Morris, R. Huey, W. Lindstrom, M. Sanner and R. Belew, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
- E. Vanquelef, S. Simon, G. Marquant, E. Garcia, G. Klimerak, J. C. Delepine, P. Cieplak and F.-Y. Dupradeau, Nucleic Acids Res., 2011, 39, W511–W517 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- W. J. Geary, Coord. Chem. Rev., 1971, 7, 81–122 CrossRef CAS.
- K. V. Katti, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2005, 35, 1–2 CrossRef CAS.
- S. A. Olagboye, T. L. Yusuf, S. D. Oladipo and S. J. Zamisa, Z. Kristallogr. - New Cryst. Struct., 2020, 235, 833–836 CrossRef CAS.
- S. D. Oladipo, T. L. Yusuf, S. J. Zamisa, M. Shapi and T. J. Ajayi, J. Mol. Struct., 2021, 1241, 130620 CrossRef CAS.
- S. D. Oladipo, T. L. Yusuf, S. J. Zamisa, G. F. Tolufashe, K. A. Olofinsan, Z. Tywabi-Ngeva and N. Mabuba, Eur. J. Chem., 2021, 12, 204–215 CrossRef CAS.
- M. Bashir, I. Yousuf, M. Ahmad, F. Arjmand and S. Tabassum, Polyhedron, 2023, 229, 116189 CrossRef CAS.
- T. L. Yusuf, D. C. Akintayo, S. D. Oladipo, A. A. Adeleke, K. Olofinsan, B. Vatsha and N. Mabuba, New J. Chem., 2022, 46, 12968–12980 RSC.
- T. L. Yusuf, S. D. Oladipo, S. Zamisa, H. M. Kumalo, I. A. Lawal, M. M. Lawal and N. Mabuba, ACS Omega, 2021, 6, 13704–13718 CrossRef CAS PubMed.
- S. A. Olagboye, T. L. Yusuf, S. D. Oladipo and S. J. Zamisa, Z. Kristallogr. - New Cryst. Struct., 2020, 235, 689–692 CrossRef CAS.
- S. D. Kanmazalp, O. E. Doĝan, V. Taşdemir, N. Dege, E. Aĝar and I. O. Fritsky, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2019, 75, 362 CrossRef PubMed.
- M. A. Raza, D. Necmi, O. E. Doğan, A. Tuğgan and S. H. Sumrra, J. Mol. Struct., 2021, 1226, 129330 CrossRef CAS.
- C. Mchiri, L.-C. C. Coetzee, F. Chandoul, A. Jedidi, A. S. Adeyinka, N. Magwa, T. Roisnel, S. Ben Moussa and H. Nasri, Molecules, 2022, 27, 3833 CrossRef CAS PubMed.
- I. Waziri, M. T. Kelani, M. O. Oyedeji-Amusa, A. K. Oyebamiji, L.-C. C. Coetzee, A. S. Adeyinka and A. J. Muller, J. Mol. Struct., 2023, 1276, 134756 CrossRef CAS.
- J. Hudzicki, Am. Soc. Microbiol., 2009, 15, 55–63 Search PubMed.
- T. D. Tavares, J. C. Antunes, J. Padrão, A. I. Ribeiro, A. Zille, M. T. P. Amorim, F. Ferreira and H. P. Felgueiras, Antibiotics, 2020, 9, 314 CrossRef CAS PubMed.
- Z. H. Chohan and M. Hanif, Appl. Organomet. Chem., 2011, 25, 753–760 CrossRef CAS.
- V. Uivarosi, Molecules, 2013, 18, 11153–11197 CrossRef CAS PubMed.
- M. Claudel, J. V. Schwarte and K. M. Fromm, Chemistry, 2020, 2, 849–899 CrossRef CAS.
- I. Wiegand, K. Hilpert and R. E. Hancock, Nat. Protoc., 2008, 3, 163–175 CrossRef CAS PubMed.
- S. B. Bukhari, S. Memon, M. M. Tahir and M. Bhanger, J. Mol. Struct., 2008, 892, 39–46 CrossRef.
- E. Rodríguez-Arce and M. Saldías, Biomed. Pharmacother., 2021, 143, 112236 CrossRef.
- N. Turan, K. Buldurun, Y. Alan, A. Savci, N. Çolak and A. Mantarcı, Res. Chem. Intermed., 2019, 45, 3525–3540 CAS.
- S. Konishi, Y. Kashiwagi, G. Watanabe, M. Osaki, T. Katashima, O. Urakawa, T. Inoue, H. Yamaguchi, A. Harada and Y. Takashima, Polym. Chem., 2020, 11, 6811–6820 RSC.
- M. Ashfaq, M. N. Tahir, K. S. Munawar, R. Behjatmanesh-Ardakani and H. Kargar, J. Mol. Struct., 2022, 1261, 132952 CrossRef CAS.
- I. Waziri, T. L. Yusuf, H. A. Zarma, S. O. Oselusi, L.-C. C. Coetzee and A. S. Adeyinka, Inorg. Chim. Acta, 2023, 121505 CrossRef CAS.
- B. Aslam, W. Wang, M. I. Arshad, M. Khurshid, S. Muzammil, M. H. Rasool, M. A. Nisar, R. F. Alvi, M. A. Aslam and M. U. Qamar, Infect. Drug Resist., 2018, 11, 1645 CrossRef CAS.
- R. Chaudret, Phys. Chem. Chem. Phys., 2014, 16, 9876–9891 RSC.
- J. Chakraborty, Heliyon, 2022, 8, e11408 CrossRef CAS PubMed.
- I. Waziri, G. A. Mala, I. M. Wakil, L.-C. C. Coetzee and H. Garba, Arid Zone J. Basic Appl. Res., 2023, 2(1), 30–58 CrossRef.
- R. F. Bader and H. Essén, J. Chem. Phys., 1984, 80, 1943–1960 CrossRef CAS.
- I. Kostova, N. Trendafilova and G. Momekov, J. Inorg. Biochem., 2005, 99, 477–487 CrossRef CAS PubMed.
- R. H. H. Salih, A. H. Hasan, N. H. Hussen, F. E. Hawaiz, T. B. Hadda, J. Jamalis, F. A. Almalki, A. S. Adeyinka, L.-C. C. Coetzee and A. K. Oyebamiji, J. Mol. Struct., 2023, 1282, 135191 CrossRef CAS.
- V. Tsirelson and A. Stash, Chem. Phys. Lett., 2002, 351, 142–148 CrossRef CAS.
- F. Feixas, E. Matito, J. Poater and M. Solà, Chem. Soc. Rev., 2015, 44, 6434–6451 RSC.
- R. K. Roy, S. Krishnamurti, P. Geerlings and S. Pal, J. Phys. Chem. A, 1998, 102, 3746–3755 CrossRef CAS.
- P. K. Chattaraj and S. Giri, Annu. Rep. Sect. “C” (Phys. Chem.), 2009, 105, 13–39 RSC.
- P. K. Chattaraj, U. Sarkar and D. Roy, J. Chem. Educ., 2007, 84, 354 CrossRef CAS.
- R. G. Parr and R. G. Pearson, J. Am. Chem. Soc., 1983, 105, 7512–7516 CrossRef CAS.
- R. G. Parr, Lv Szentpály and S. Liu, J. Am. Chem. Soc., 1999, 121, 1922–1924 CrossRef CAS.
- C.-G. Zhan, J. A. Nichols and D. A. Dixon, J. Phys. Chem. A, 2003, 107, 4184–4195 CrossRef CAS.
- A. Innasiraj, B. Anandhi, Y. Gnanadeepam, N. Das, F. Paularokiadoss, A. V. Ilavarasi, C. D. Sheela, D. R. Ampasala and T. C. Jeyakumar, J. Mol. Struct., 2022, 1265, 133450 CrossRef CAS.
- L. R. Domingo and P. Pérez, Org. Biomol. Chem., 2011, 9, 7168–7175 RSC.
- D. F. Lewis, Inflammopharmacology, 2003, 11, 43–73 CrossRef CAS PubMed.
- L.-C. C. Coetzee, A. S. Adeyinka and N. Magwa, Energies, 2022, 15, 4913 CrossRef CAS.
- H. AlRabiah, S. Muthu, F. Al-Omary, A.-M. Al-Tamimi, M. Raja, R. R. Muhamed and A. A.-R. El-Emam, Maced. J. Chem. Chem. Eng., 2017, 36, 59–80 CAS.
- T. Pantsar and A. Poso, Molecules, 2018, 23, 1899 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary crystallographic data. CCDC 2257235 (HL), 2257236 (NiL2), and 2257237 (CuL2) contains the supplementary crystallographic data for these compounds. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3nj02910g |
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Hlonepho M.
Masena
*a,
Hlonepho M.
Masena
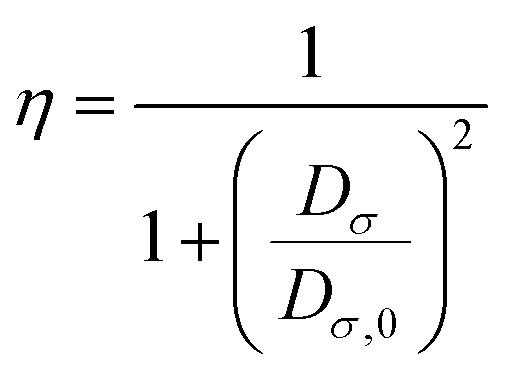
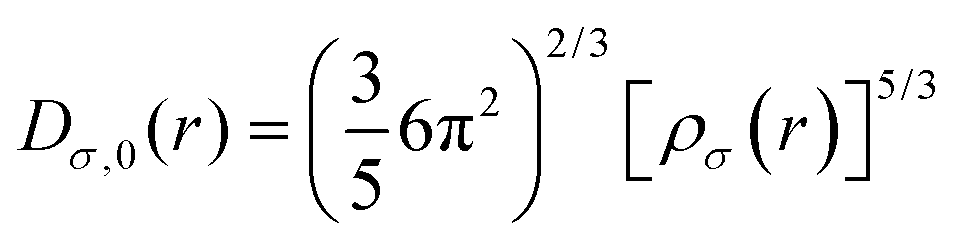 .
.
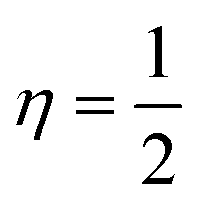 , a homogeneous electron gas-like pair probability (electron delocalization) occurs. Borders between electron pairs result from a value close to zero.82,83 The topological interpretation of molecular space which aids in identifying bonds, lone pairs and core regions can clarify the molecular and atomic shell structures through ELF. Red colors reveal highly localized electrons, while dark colors indicate regions where electrons are not highly localized and green colors indicate delocalized electrons (Fig. 18). When investigating the C–N–C plane, we observe that electrons become more localized in the region 0–6 Bohr in both x and y-directions upon complexation. The free ligand shows no localized electrons in this region though. When comparing the cobalt and copper complexes, electrons are more localized in the former in this vicinity. Electrons are more localized at 2.04 Bohr in the x-direction in the nickel complex than in the other complexes at this position. For the zinc complex, electrons are not highly localized in the 0–6 Bohr range in the x-direction. We also observe that electrons become highly localized in the region 12.26–14.30 Bohr in the x-direction.
, a homogeneous electron gas-like pair probability (electron delocalization) occurs. Borders between electron pairs result from a value close to zero.82,83 The topological interpretation of molecular space which aids in identifying bonds, lone pairs and core regions can clarify the molecular and atomic shell structures through ELF. Red colors reveal highly localized electrons, while dark colors indicate regions where electrons are not highly localized and green colors indicate delocalized electrons (Fig. 18). When investigating the C–N–C plane, we observe that electrons become more localized in the region 0–6 Bohr in both x and y-directions upon complexation. The free ligand shows no localized electrons in this region though. When comparing the cobalt and copper complexes, electrons are more localized in the former in this vicinity. Electrons are more localized at 2.04 Bohr in the x-direction in the nickel complex than in the other complexes at this position. For the zinc complex, electrons are not highly localized in the 0–6 Bohr range in the x-direction. We also observe that electrons become highly localized in the region 12.26–14.30 Bohr in the x-direction.