
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe effect of resonance-assisted hydrogen bond on the second-order nonlinear optical properties of pyridine hydrazone photoswitches: a quantum chemistry investigation†
Douniazed
Hannachi
 *ab,
Noureddine
Khelfaoui
a,
Meriem
Zaidi
ac,
Diha
Yahiaoui
a,
Salima
Lakehal
d,
Christophe
Morell
e and
Henry
Chermette
*e
*ab,
Noureddine
Khelfaoui
a,
Meriem
Zaidi
ac,
Diha
Yahiaoui
a,
Salima
Lakehal
d,
Christophe
Morell
e and
Henry
Chermette
*e
aLaboratoire d’Électrochimie, d’Ingénierie Moléculaire et de Catalyse Redox (LEIMCR), Département d’Enseignement de Base en Technologie, Faculté de Technologie, Université Ferhat Abbas, Sétif-1, Algeria. E-mail: douniazed_hannachi@univ-setif.dz
bDépartement de Chimie, Faculté des Sciences, Université Ferhat Abbas, Setif-1, Algeria
cLaboratoire de Chimie, Ingénierie Moléculaire et Nanostructures (LCIMN), Université Ferhat Abbas Sétif 1, Sétif 19000, Algeria
dInstitut des Science de la Terre et de l’Univers, Université de Batna-2, Algeria
eUniversité de Lyon, Université Claude Bernard Lyon 1, Institut des Sciences Analytiques, UMR CNRS 5280, 69622 Villeurbanne Cedex, France. E-mail: henry.chermette@univ-lyon1.fr
First published on 5th September 2023
Abstract
The effect of hydrogen bonds on the NLO properties was not considered as essential, in particular in pyridine hydrazone systems. Yet, we show in the present study that a control of these photoswitches depends on the strength of hydrogen bonds. In this study, we investigate a selection of 18 E/Z pyridine hydrazone photoswitch molecules to explore the impact of resonance-assisted hydrogen bond (RAHB) on the NLO properties in the E/Z isomers. Using quantum calculations at the ωB97XD/6-311+g(d) level of theory, we determine various electronic parameters, reactivity descriptors, bond length alternation (BLA) values, nuclear independent chemical shift (NICS) aromaticity indices, QTAIM topology, energy of hydrogen bond (EHB), RAHB, and linear and nonlinear optical properties for these molecules. The agreement between the quantum calculations and experimental spectra is illustrated through TD-DFT calculations, showing small deviations. Contrary to conventional expectations, our findings demonstrated that the delocalization strength of the electrons and NLO properties of the Z isomers are significantly enhanced by the presence of a resonance-assisted hydrogen bond. The Z-isomer exhibited a lower excited state energy, weaker energy gap, smaller BLA value, larger dipole moment variations for the first excited state, higher ΦE→Z, and electron delocalization at the quasi-cycle closed (RAHB) compared to the E-isomer. Furthermore, we find that the hyperpolarizability value of the title photoswitches increases as the wavelength of the incident light decreases, i.e., β(695) > β(1064) > β(1340) > β(∞), and the dispersion has less effect at λ = 1064 and 1340 nm. Additionally, we observe a strong relation between the photoisomerization quantum yield (ΦE⇄Z) and static hyperpolarizability (β) of the first and second isomer, where ΦE⇄Z is proportional to β of the second isomer and inversely proportional to b of the first isomer. This inverse trend between static hyperpolarizability and photoisomerization quantum yields is attributed to the electron-withdrawing character of substituents on the Ar ring. Our research provides valuable insights into optimizing the 2nd-order NLO properties of pyridine hydrazone photoswitch molecules. By understanding the influence of hydrogen bonding on the delocalization strength of the electrons (RAHB) and the shape-dependent NLO performance, we gain the ability to design and synthesize novel photoswitch molecules with enhanced NLO characteristics.
Introduction
Molecular photoswitches are defined as chemical compounds that can reversibly be transformed from one isomer into another one with light irradiation. The two isomers differ from each other in various chemical and physical properties, such as geometrical structure, absorption spectra, oxidation/reduction potentials, magnetic properties, dielectric constant, refractive index, and others.1,2 This kind of molecules provides an invaluable tool for a large variety of applications, such as in information storage and processing,3 photo-pharmacology, photo-actuators, remote-controllable reactions, and controllable drug transport and release.4,5Nonlinear optics (NLO) is a branch of optics that deals with phenomena arising from light-induced changes in the optical properties of compounds (e.g., phase, frequency, amplitude, polarization, and path). NLO compounds are materials that exhibit nonlinear optical responses, such as second-harmonic generation (SHG), optical Kerr effect, and third-harmonic generation (THG). These compounds have a wide range of applications in different fields, such as optical communication, optical computing, optical memory, and all-optical signal processing.6–11 Photochromic compounds, such as stilbenes, azobenzenes,12–14 diarylethenes,15 spiropyrans,16 and fulgides,4,17 have excellent nonlinear optical responses (high hyperpolarizability). The combination between these two properties (photochromic and nonlinear optical) empowers generations of switchable second-order NLO materials. These classes of materials are called optical switches and have very high applications in optoelectronic and photonic technologies, including molecular-scale memory devices with multiple storage and nondestructive reading capacity.18 It is important to note that the photoswitching of second-order NLO properties makes sense only when the photoisomer compounds are thermally stable.19 From the literature, the switching of NLO properties has been performed by specific procedures including protonation/deprotonation, oxidation/reduction, and photoisomerization.20–24 In our work, we used photoisomerization to switch the NLO properties.
Recently, Mravec and collaborators25–27 designed and synthesized an extended set of 13 pyridine/quinoline hydrazones photoswitches. This new class of hydrazone-based P-type photoswitches is switchable between two isomers Z and E (see Fig. 1) and shows excellent thermal stability of both isomers. The operational wavelengths of the pyridine hydrazone structural motif shifted toward the visible region without the accompanying loss of their high thermal stability, and hydrazone 7 retains good thermal stability.25 Furthermore, the quantum-chemical calculations at the ωB97XD/def2-TZVPP level revealed a three-step inversion-rotation reaction mechanism of the thermal E-to-Z isomer. For benzoylpyridine hydrazones 1–8, 10, and 16, E-to-Z photoisomerization is more efficient compared to the Z-to-E process. From the work of Mravec et al.,25 we can note that these compounds could be used for short-lived and long-lived information storage,28 and these isomers could be utilized as photoswitch molecules.
 | ||
| Fig. 1 Chemical structures of the pyridine hydrazones photoswitches (from ref. 25 for i = 1–8, 10, and 16). | ||
In our work, we studied a series of 18 pyridine hydrazone photoswitches (Zi and Ei isomers, i = 1–18, see Fig. 1), which is divided in two subgroups, namely, one group of ten Zi and Ei isomers (where i = 1–8, 10, and 16) synthesized by Mravec et al.25 and another one we designed, consisting of eight compounds (i = 9, 11–15, and 17–18 of Ei and Zi isomers), to investigate the substitutional effect on the hydrazine and ketone parts on the structural, electronic, reactivity, optical properties, and nonlinear responses. On ring 2, we chose two electron-withdrawing (EW) namely NO2 and CN in order to study the effect of the EW character on electronic and optical properties. The main objective of this work is to show how the hydrogen bond can enhance the NLO properties of the pyridine hydrazones photoswitches and try to find a relation between photoisomerization direction (E ⇄ Z) and hyperpolarizability.
The present paper is organized as follows. In Section 1, all computational details in the corresponding section and definitions are given; in Section 2, the quantum theory of atom-in-molecules (QTAIM) is analyzed; in Section 3, the bond length alternation (BLA) is presented; in Section 4, the NICS aromaticity indices are evaluated; in Section 5, the resonance-assisted hydrogen bond are given; in Section 6, the global reactivity descriptors are studied; in Section 7, the absorption spectra are discussed; and in Section 8, the static and dynamic NLO responses of E and Z isomers are calculated; the paper ends with some concluding remarks.
Computational details
The geometries of the E and Z isomers were fully optimized using the ωB97XD density functional29,30 with 6-311+g(d) basis set.31,32 The ωB97XD is a range-separated hybrid exchange–correlation functional that includes damped atom-atom dispersion corrections.29,33 Quantum chemistry calculations were performed with Gaussian 09 program with TIGHT SCF convergence and ultra-fine integration grid.34–36 No symmetry constraints were applied and the local minima were confirmed on the potential energy surface by harmonic frequency calculations of the ground state for E and Z isomers at the same level. All the calculations were performed in the gas phase.Chemical reactivity descriptors such as chemical hardness (η), electronic chemical potential (μ), and electronegativity (χ) can be evaluated from the frontier orbital energies HOMO and LUMO (εH and εL, respectively) using the following equation.37
 | (1) |
| η = εL − εH | (2) |
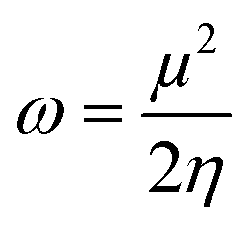 | (3) |
Time-dependent density functional theory (TD-DFT) calculations were carried out at the same level of theory to evaluate the absorption wavelengths (λ) and corresponding oscillator strengths (fosc) of electronic transitions. For TD-DFT calculations, sixty excited states were calculated.
Following the procedure proposed by LeBahers et al.,39 the excited states of interest were examined using charge-transfer indices (CT) including charge-transfer distance (dCT), transferred charge (qCT), and the variation in dipole moment between the ground and the excited states (Δμ0→n = qCT × dCT). In our work, the CT indices have been examined with MULTIWFN program.40
The electron density difference maps (EDDM) corresponding to the crucial excited states can be exactly evaluated as follows.
| Δρ(r) = ρex(r) − ρGS(r) | (4) |
On the other hand, isotropic polarizability (α), polarizability density (ρ), and first hyperpolarizability (β0) were calculated using analytical derivatives of the system energy (coupled-perturbed Kohn–Sham method)41 at the ωB97XD/6-311+g(d) level of theory. These parameters are defined as follows.8,42
 | (5) |
 | (6) |
 | (7) |
For the second harmonic generation
| βi = βiii(−2ω; ω, ω) + βijj(−2ω; ω, ω) + βikk(−2ω; ω, ω) | (8) |
| βi = βiii(−ω; ω, 0) + βijj(−ω; ω, 0) + βikk(−ω; ω, 0) | (9) |
 | (10) |
 | (11) |
 | (12) |
 | (13) |
Results and discussion
Topological study
The intramolecular H-bond is one of the most prominent features that could influence the stability of compounds.46 In this paper, we use the quantum theory of atom-in-molecules (QTAIM) to examine this bond.47,48 In this theory, the critical points (in ring (RCP) or bond (BCP)) are the positions where the gradient is null and which can be classified according to the electron density (ρ) and its Laplacian (∇2(ρ)), total electron energy density (H), kinetic electron energy density (G), and potential electron energy density (V).47,49 The QTAIM calculations were carried out for Z-isomers using the Amsterdam Density Functional (ADF18) program developed by Baerends et al.,50,51 and the results of the calculation are summarized in Table S1 (ESI†). Fig. 2 depicts the molecular graphs corresponding to the Z6 isomer, where the red circle indicates the ring critical point (RCPs) and the green circle indicates the bond critical point (BCPs). | ||
| Fig. 2 Molecular topology of the Z6-isomer. | ||
In all of the Z-isomers, ring critical point (RCP: N1–N2–C3–C4–N6⋯H) and hydrogen bonds between the ketone and hydrazine moiety are observed (see Table S1, ESI† and Fig. 2). It is interesting to note that the formation of this RCP resulted from the cyclic nature of electron current and also confirms the existence of hydrogen bonding (N1–H⋯N6).
QTAIM analysis shows that the critical point at the hydrogen bond N1–H⋯N6 (BCP) has electronic density ranges from 0.012 to 0.015 a.u., while the Laplacian of the electron densities is in the range of 0.96–0.100 a.u., G = 0.019, V = −0.014, and H = 0.005 (with the exception of the isomers substituted with Ar7 and Ar8, where the H value amounts from −0.0005 to −0.0003 a.u.), whereas for the ring critical points (RCP: N1–N2–C3–C4–N6⋯H), the values electronic density is in the range of 0.031–0.035 and its Laplacian is in the range of 0.103 to 0.109; the values of G, V, and H are 0.028, −0.027 to −0.030, and 0.05 a.u., respectively. From this value, we note that there is no effect of the substitutions (Ar moiety and EW groups (CN, NO2) at the ring 2) on the H-bond and RCP (see Table S1, ESI†). On the other hand, H-bond exhibits positive values of ∇2(ρBCP) and negative values of HBCP, which are typical of intermediate hydrogen bonds. Furthermore, in the RCP, we observe an increase in the kinetic energy (G) over the potential energy |V|, which means that the electrons are moving faster in ring N1–N2–C3–C4–N6⋯H or, in other words, the electrons are less localized. In the case of BCP H-bond (H⋯N6), we note that G ≤ |V|, indicating that the electrons are localized in this region of Z-isomers.52
On the other hand, BLA is a structural parameter, defined as the average difference between the lengths of a single bond and the adjacent multiple bonds in a π-delocalized isomer. For the title isomers, we calculated the BLAs (BLA1 and BLA2, see atom numbering in Fig. 1) and summarized them in Table 1.
| Ei | BLA1 | BLA2 | Zi | BLA1 | BLA2 | Ei | BLA1 | BLA2 | Zi | BLA1 | BLA2 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| BLA1 = 1/2(d1–2 + d3–4 − 2d2–3), BLA2 = 1/2(d1–2 + d3–5 − 2d2–3). | |||||||||||
| E1 | 0.120 | 0.126 | Z1 | 0.104 | 0.107 | E10 | 0.120 | 0.126 | Z10 | 0.104 | 0.108 |
| E2 | 0.119 | 0.125 | Z2 | 0.099 | 0.103 | E11 | 0.119 | 0.125 | Z11 | 0.100 | 0.104 |
| E3 | 0.140 | 0.143 | Z3 | 0.125 | 0.125 | E12 | 0.141 | 0.144 | Z12 | 0.113 | 0.114 |
| E4 | 0.125 | 0.130 | Z4 | 0.111 | 0.114 | E13 | 0.125 | 0.130 | Z13 | 0.111 | 0.114 |
| E5 | 0.126 | 0.132 | Z5 | 0.113 | 0.116 | E14 | 0.127 | 0.132 | Z14 | 0.114 | 0.117 |
| E6 | 0.128 | 0.132 | Z6 | 0.115 | 0.118 | E15 | 0.128 | 0.133 | Z15 | 0.116 | 0.118 |
| E7 | 0.124 | 0.128 | Z7 | 0.110 | 0.113 | E16 | 0.123 | 0.128 | Z16 | 0.111 | 0.113 |
| E8 | 0.121 | 0.126 | Z8 | 0.108 | 0.110 | E17 | 0.122 | 0.126 | Z17 | 0.110 | 0.112 |
| E9 | 0.125 | 0.130 | Z9 | 0.111 | 0.113 | E18 | 0.125 | 0.131 | Z18 | 0.112 | 0.114 |
The DFT calculation shows that in the whole set of isomers (Z and E), the BLA1 is smaller than the BLA2, which may be ascribed to the donation effect of the pyridine ring (ring 1, see Fig. 1). When comparing E1–E9 isomers to E10–E18, it is found that the BLA1 and BLA2 values of Ei-isomers substituted by NO2 group are similar to that of the Ei-isomers substituted by the CN group. For example, BLA1 = 0.128 and BLA2 = 0.132 Å for E6 and E16, respectively. We observed the same results for the Z-isomers, with the exception of Z3 and Z12 compounds, where the values are different (BLA1(Z3) = 0.125 ≠ BLA1(Z12) = 0.113 and BLA1(Z3) = 0.125 ≠ BLA2(Z12) = 0.114. From these results, we can conclude that the substitution of a NO2 by a CN group in the R position does not introduce significant changes in the bond length alternation BLA1 and BLA2 along the conjugated linker, which connects the Ar and ketone parts. On the other hand, we observe that the BLA1–2 values of the Z1–Z18 isomers are smaller than that of the corresponding E1–E18 isomers. This suggests that the π-conjugation of the Z-isomers is stronger than that of the corresponding E-isomers, which is certainly related to the existence of the hydrogen bond N1–H⋯N6 (stronger electrostatic effects in the Z-isomers). In addition, the smallest BLA1–2 values are obtained when Ar is the C6H4-p-OCH3 (Ar2) group (isomers Z2, Z11, and E2, E11), whereas the largest values are obtained for Ar3 = C6F6 (E3, E12, Z3, and Z12 isomers). Thus, the effect of the nature of the Ar group is stronger for the BLA1 and BLA2 values than the nature of the R substituent (NO2 and CN). Generally, the BLA1–2 values of Z1–Z9, Z10–18, E1–E9, and E10–18 isomers increase following the order: Ar2 < Ar1 < Ar8 < Ar7 < Ar9 < Ar4 <Ar5 < Ar6 < Ar3. From this result, we can note that the BLA1–2 values increase with the increase in the EW character of the substituents on the Ar-ring.
The calculated hydrogen bond angle and dihedral angle between the hydrazine part and rings 1 and 2 are listed in Table S2 (ESI†). Regardless of the nature of the R substituent, we observed that E to Z isomerization results in a decrease in the dihedral angle φ1 by approximately 27°, while φ2 deviates by 74°. On the other hand, for the Z isomers, it is found that the dihedral angle (φ1) measured between ring 1 and the hydrazine part is approximately 19°, which is smaller compared to the dihedral angle between ring 2 and the hydrazine part (φ2 = 44°). This decrease in angle (φ1) enhances the π-electron conjugation between ring 1 and the hydrazine part more than that between ring 2 and the hydrazine part. Additionally, the presence of hydrogen bonds leads to reduced dihedral angles between the Ar-linker-ring 2 in the Z-isomer. This observation is reinforced by the slightly lower bond length alternation (BLA2) values in the Z-isomer compared to the E-isomer.
On the other hand, for the Z isomers, the N1–H⋯N6 hydrogen bond angle (φ3) has been observed to be 130°. However, an exception is noted in Z7, Z8, Z16, and Z17, where the angle φ3 is measured at 127° (Table S2, ESI†). Notably, this angle aligns well with the IUPAC recommendations, which suggest that hydrogen bond angles should preferably be above 110°.53
Aromaticity
The local aromaticity, nonaromaticity as well as anti-aromaticity in the title compounds is assessed by NICS calculations, which was introduced by von Rague Schleyer et al.54,55 The NICS was calculated at the ring center or cages and described as the negative value of the isotropic shielding constant.56 Noted that the strongly negative NICS values (i.e., magnetically shielded) denote the presence of induced diatropic ring currents and “aromaticity”, whereas the positive values (i.e., deshielded) at the chosen point indicate paratropic ring current and “anti-aromaticity”.57 In this work, we used the NICSZZ(1) index, which is calculated to be 1 Å above the center of the ring under consideration. The results of NICSZZ(1) calculation for all the isomers considered are listed in Table 2.| NICSZZ | NICSZZ | NICSZZ | NICSZZ | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| E1 | 1 | −8.089 | Z1 | 1 | −9.396 | E10 | 1 | −8.120 | Z10 | 1 | −9.385 |
| 2 | −12.941 | 2 | −10.109 | 2 | −13.197 | 2 | −10.709 | ||||
| 3 | −9.693 | 3 | −9.387 | 3 | −9.520 | 3 | −9.405 | ||||
| QCC | 27.197 | QCC | 27.227 | ||||||||
| E2 | 1 | −8.160 | Z2 | 1 | −9.370 | E11 | 1 | −7.941 | Z11 | 1 | −9.275 |
| 2 | −13.098 | 2 | −9.958 | 2 | −12.942 | 2 | −10.621 | ||||
| 3 | −10.122 | 3 | −8.630 | 3 | −10.031 | 3 | −8.600 | ||||
| QCC | 26.980 | QCC | 27.051 | ||||||||
| E3 | 1 | −8.796 | Z3 | 1 | −10.370 | E12 | 1 | −8.802 | Z12 | 1 | −10.318 |
| 2 | −13.019 | 2 | −10.217 | 2 | −13.119 | 2 | −10.527 | ||||
| 3 | −15.667 | 3 | −15.404 | 3 | −15.846 | 3 | −15.612 | ||||
| QCC | 27.127 | QCC | 27.027 | ||||||||
| E4 | 1 | −8.406 | Z4 | 1 | −9.924 | E13 | 1 | −8.287 | Z13 | 1 | −9.827 |
| 2 | −13.065 | 2 | −10.738 | 2 | −13.843 | 2 | −11.812 | ||||
| 3 | −7.719 | 3 | −7.657 | 3 | −7.644 | 3 | −7.566 | ||||
| QCC | 27.357 | QCC | 27.406 | ||||||||
| E5 | 1 | −8.826 | Z5 | 1 | −10.058 | E14 | 1 | −8.482 | Z14 | 1 | −9.995 |
| 2 | −13.338 | 2 | −10.960 | 2 | −13.260 | 2 | −11.221 | ||||
| 3 | −7.479 | 3 | −7.216 | 3 | −7.364 | 3 | −7.265 | ||||
| QCC | 27.323 | QCC | 27.358 | ||||||||
| E6 | 1 | −8.798 | Z6 | 1 | −10.147 | E15 | 1 | −8.491 | Z15 | 1 | −9.987 |
| 2 | −13.193 | 2 | −10.989 | 2 | −13.217 | 2 | −11.256 | ||||
| 3 | −6.483 | 3 | −6.090 | 3 | −6.348 | 3 | −6.267 | ||||
| QCC | 27.459 | QCC | 27.630 | ||||||||
| E7 | 1 | −8.362 | Z7 | 1 | −9.875 | E16 | 1 | −8.324 | Z16 | 1 | −9.856 |
| 2 | −12.849 | 2 | −10.303 | 2 | −13.101 | 2 | −10.786 | ||||
| 3 | −6.512 | 3 | −6.562 | 3 | −6.335 | 3 | −6.678 | ||||
| QCC | 26.969 | QCC | 27.113 | ||||||||
| E8 | 1 | −7.902 | Z8 | 1 | −9.346 | E17 | 1 | −8.237 | Z17 | 1 | −9.230 |
| 2 | −12.916 | 2 | −9.793 | 2 | −12.893 | 2 | −10.319 | ||||
| 3 | −8.522 | 3 | −8.596 | 3 | −8.409 | 3 | −8.293 | ||||
| 1′ | −8.961 | QCC | 28.264 | 1′ | −8.984 | QCC | 28.214 | ||||
| 1′ | −9.009 | 1′ | −8.838 | ||||||||
| E9 | 1 | −8.864 | Z9 | 1 | −9.902 | E18 | 1 | −8.388 | Z18 | 1 | −9.879 |
| 2 | −13.341 | 2 | −10.205 | 2 | −13.466 | 2 | −10.747 | ||||
| 3 | −0.342 | 3 | −0.216 | 3 | −0.288 | 3 | −0.0602 | ||||
| 1′ | −13.128 | QCC | 27.615 | 1′ | −13.171 | QCC | 27.736 | ||||
| 1′ | −14.047 | 1′ | −12.639 | ||||||||
For both the isomers, the quantum calculation shows that the NICSZZ(1) index is more negative in rings 2 than in the ring 1. The aromaticity in ring 2 substituted with NO2 group is notably smaller than that substituted with the CN group (with the exception of isomers E2, E5, and E8). At the same time, the localization of NO2 group at ring 2 results in increases in the anti-aromaticity of quasi-cycle closed, with exception for the 3 and 8 compounds. However, it leads to lower stability of these isomers (Z1 to Z9) in comparison with the isomers Z10 to Z18 (CN group at the ring 2). On the other hand, the NICSZZ(1) results obtained for Ar-ring (ring 3) indicate that this ring is aromatic and less stable than the ring 2, with the exception of the compounds 3 and 12, where Ar3 exhibits a high aromaticity diatropicity and stability. The order of the NICSZZ(1) index of Ar-ring is Ar3 > Ar2 > Ar1 > Ar8 > Ar4 > Ar5 > Ar7 > Ar6 > Ar9.
The NICSZZ(1) indexes for Z isomers reveal a reduction in the aromatic behavior in rings 2 and Ar, whereas ring 1 shows an increase in the aromatic character in comparison with corresponding E isomers (Table 2). These variations in aromaticity between Z and E isomers can be attributed to the RAHB effect and pronounced anti-aromatic behavior in quasi-cycle closed in Z isomers. In general, we can conclude that the Z-isomer displays a smaller BLA value as well as larger aromatic character (ring 1) than that of the corresponding E-isomer.
Mravec et al. found that in the benzoylpyridine hydrazones 1–8, 10, and 16, the E → Z photoisomerization is more efficient compared to the Z → E process.25 We can attribute these results to the Z-isomer shape, which features an anti-aromatic ring (quasi-cycle closed) surrounded by three aromatic rings (1, 2, and Ar ring).
Resonance-assisted hydrogen bond
Based on the analyzes of the data presented in Table 3, the E-isomers exhibit a chemical shift of the bridging hydrogen at the range of δN1H,E = 5.6–8.8 ppm, and the N1–H stretching frequency in the range of σN1H,E = 3553–3581 cm−1. On the other hand, the Z isomers display significantly higher δN1H (δN1H,Z = 11.7–13.5 ppm) and lower σN1H values (σN1H,Z = 3465–3508 cm−1) compared to the corresponding E-isomer. The observed variations in Z isomers are a result of the presence of intramolecular N1–H⋯N6 hydrogen bonding, leading to the formation of a quasi-ring structure N1–N2–C3–C4–N6⋯H (the distance N1–N6 and H⋯N6 is ∼2.691 and 1.922 Å, respectively). According to the literature and our calculations, this quasi-ring structure is almost planar58–65 (see Table S2, ESI†) and distinguished by short N1–N6 distances and longer H–N1 bonds (rHN1,Z ≈ 1.018 Å and rHN1,E ≈ 1.012 Å). Furthermore, analysis using QTAIM indicates a delocalization of electrons within the quasi-cycle closed (RCP). Additionally, we observed lower δN1H,Z frequencies and downfield shift of the δN1H,Z. These findings strongly support the resonance-assisted hydrogen bond model (RAHB) proposed by Gilli et al.,66–68 and we can conclude that this quasi-cycle closed (QCC) created an RAHB phenomenon.| δ N1H | σ N1H | r N1–H | δ N1H | σ N1H | r N1–H | r H⋯N6 | r N1–N6 | E HB(V) | ||
|---|---|---|---|---|---|---|---|---|---|---|
| E1 | 6.32 | 3576.90 | 1.012 | Z1 | 12.26 | 3500.52 | 1.016 | 1.921 | 2.695 | 4.59 |
| E2 | 6.04 | 3578.16 | 1.012 | Z2 | 12.21 | 3489.31 | 1.017 | 1.919 | 2.691 | 4.59 |
| E3 | 5.62 | 3565.79 | 1.013 | Z3 | 11.70 | 3465.57 | 1.018 | 1.913 | 2.682 | 4.59 |
| E4 | 6.25 | 3579.79 | 1.012 | Z4 | 12.25 | 3496.47 | 1.017 | 1.916 | 2.693 | 4.59 |
| E5 | 6.11 | 3576.89 | 1.012 | Z5 | 12.38 | 3500.48 | 1.017 | 1.913 | 2.690 | 4.76 |
| E6 | 6.28 | 3578.79 | 1.012 | Z6 | 12.43 | 3474.69 | 1.017 | 1.907 | 2.688 | 4.76 |
| E7 | 7.00 | 3571.91 | 1.013 | Z7 | 12.32 | 3506.80 | 1.017 | 1.956 | 2.700 | 4.06 |
| E8 | 8.76 | 3555.43 | 1.014 | Z8 | 13.58 | 3481.68 | 1.018 | 1.946 | 2.691 | 4.24 |
| E9 | 5.98 | 3581.15 | 1.012 | Z9 | 12.14 | 3486.62 | 1.016 | 1.915 | 2.692 | 4.59 |
| E10 | 6.13 | 3577.43 | 1.012 | Z10 | 12.36 | 3492.43 | 1.016 | 1.914 | 2.689 | 4.76 |
| E11 | 6.22 | 3576.14 | 1.012 | Z11 | 12.18 | 3489.44 | 1.016 | 1.917 | 2.692 | 4.59 |
| E12 | 5.60 | 3562.77 | 1.014 | Z12 | 11.49 | 3478.79 | 1.017 | 1.923 | 2.687 | 4.59 |
| E13 | 6.28 | 3579.79 | 1.012 | Z13 | 12.19 | 3496.47 | 1.017 | 1.916 | 2.693 | 4.76 |
| E14 | 6.13 | 3576.36 | 1.012 | Z14 | 12.28 | 3491.46 | 1.017 | 1.913 | 2.691 | 4.76 |
| E15 | 6.23 | 3576.04 | 1.012 | Z15 | 12.37 | 3496.83 | 1.017 | 1.909 | 2.688 | 4.76 |
| E16 | 6.87 | 3576.35 | 1.013 | Z16 | 12.40 | 3507.92 | 1.017 | 1.949 | 2.695 | 4.24 |
| E17 | 8.78 | 3553.10 | 1.014 | Z17 | 13.51 | 3482.99 | 1.018 | 1.949 | 2.693 | 4.24 |
| E18 | 5.95 | 3580.07 | 1.012 | Z18 | 12.20 | 3491.47 | 1.016 | 1.917 | 2.691 | 4.59 |
We note that in the closed quasi-cycle of the Z isomer, the enhancement of conjugation results from the effective charge transfer occurring between the π-donor amine nitrogen to the pyridine nitrogen through the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bond. This charge transfer is notably more efficient compared to a similar transfer in the open quasi-cycle of the E isomer, where it is attributed to the formation of a RAHB system.
N double bond. This charge transfer is notably more efficient compared to a similar transfer in the open quasi-cycle of the E isomer, where it is attributed to the formation of a RAHB system.
Numerous methods have emerged over time to estimate the energy of hydrogen bonds. Among these, the widely employed approach for estimating EHB involves utilizing the relation established by Espinosa et al. According to their work, the potential electron energy density (V) at the BCP is directly proportional to the energy of the hydrogen bond (EHB), with an angular coefficient of 0.5.69 Despite its popularity, Espinosa's equation tends to yield a significantly overestimated energy of HB.70,71 More recently, Afonin et al. introduced a novel linear relationship between the calculated (V) and the energy (E) of hydrogen bonds.72 They achieved this utilizing empirical 1H NMR data and represented it as “eqn (14)”.
| EHB(V) = 0.277 × |V| − 0.45 | (14) |
Reactivity
The values of global reactivity descriptors chemical potential (μ), hardness (η), and electrophilicity index (ω) calculated for each Ei and Zi isomers (i = 1 to 18) are collected and presented in Table 4 and Fig. 3.| μ | η | ω | μ | η | ω | ||
|---|---|---|---|---|---|---|---|
| E1 | −4.277 | 6.617 | 1.382 | Z1 | −4.163 | 6.574 | 1.318 |
| E2 | −4.230 | 6.536 | 1.369 | Z2 | −3.969 | 6.304 | 1.249 |
| E3 | −4.736 | 7.270 | 1.542 | Z3 | −4.596 | 7.057 | 1.497 |
| E4 | −4.550 | 6.863 | 1.509 | Z4 | −4.442 | 6.792 | 1.452 |
| E5 | −4.602 | 6.868 | 1.542 | Z5 | −4.511 | 6.768 | 1.503 |
| E6 | −4.706 | 6.968 | 1.589 | Z6 | −4.630 | 6.822 | 1.571 |
| E7 | −4.380 | 6.923 | 1.385 | Z7 | −4.314 | 6.764 | 1.376 |
| E8 | −4.116 | 6.549 | 1.293 | Z8 | −4.075 | 6.414 | 1.294 |
| E9 | −4.460 | 6.709 | 1.482 | Z9 | −4.400 | 6.548 | 1.478 |
| E10 | −3.881 | 7.335 | 1.027 | Z10 | −3.903 | 6.991 | 1.089 |
| E11 | −3.850 | 7.214 | 1.027 | Z11 | −3.692 | 6.758 | 1.009 |
| E12 | −4.410 | 7.888 | 1.233 | Z12 | −4.384 | 7.382 | 1.302 |
| E13 | −4.201 | 7.495 | 1.177 | Z13 | −4.233 | 7.111 | 1.269 |
| E14 | −4.292 | 7.421 | 1.241 | Z14 | −4.330 | 7.027 | 1.334 |
| E15 | −4.534 | 7.248 | 1.418 | Z15 | −4.509 | 6.966 | 1.459 |
| E16 | −4.031 | 7.544 | 1.077 | Z16 | −4.046 | 7.186 | 1.139 |
| E17 | −3.856 | 7.004 | 1.061 | Z17 | −3.854 | 6.757 | 1.099 |
| E18 | −4.340 | 6.892 | 1.366 | Z18 | −4.292 | 6.674 | 1.380 |
 | ||
| Fig. 3 Global reactivity indices of Ei and Zi (i = 1 to 18) isomers. | ||
From these results, we can see that the E-isomers show the largest values of the chemical hardness, which signifies a greater stability and lower reactivity, whereas the smallest value of hardness are observed for the corresponding Z-isomers; this observation is in line with the experimental results of Mravec et al.25. The overall increasing order of hardness in the studied compounds is as follows: E2 < E8 < E1 < E9 < E4 < E5 < E18 < E7 < E6 < E17 < E11 < E15 < E3 < E10 < E14 < E13 < E16 < E12. The same order is observed for corresponding Z-isomers (see Fig. 3).
On the other hand, the potential chemistry of Z-isomers is larger than that of the corresponding E-isomer. These results indicate that the trend of the electrons to leave the equilibrium systems increases from Z to E isomers. In other words, the rise of the μ values indicates that the isomers with RAHB system have the greatest tendency to donate electrons.
The chemical potential values of Ei isomers increase in the following order.
E3 < E6 < E5 < E4 < E15 < E9 < E12 < E7 < E18 < E14 < E1 < E2 < E13 < E8 < E16 < E10 < E17 < E11
According to the global electrophilicity scale,56,73 the Ei and Zi isomers where i = 1 to 9 (with electron-withdrawing NO2) and i = 12, 14–15, 18, and Z13 can be classified as strong electrophiles (1.23 to 1.58 eV). Besides, the Zi and Ei isomers of i = 10, 11, 16–17, and E13 (CN substituent) display a moderate electrophilicity (1.00 to 1.17 eV).
Linear optical properties
The calculated excited state transition energies (ΔE), wavelength (λ), oscillator strengths (f), charge transfer (qCT, |e|), charge transfer distance (dCT, Å), dipole moment variation (Δμ, D), and major molecular orbital transitions of isomers are summarized in Table 5 and Table S3 (ESI†). Furthermore, the shapes of the MOs of specific excitation and EDDM for all isomers are provided in the ESI† (Fig. S1 and S2).| ΔE0→1 | Δλ0→1 | f | d CT | q CT | Δμ | |
|---|---|---|---|---|---|---|
| a Experimental results from ref. 25. | ||||||
| E1 | 3.798(3.67)a | 326 | 0.316 | 5.690 | 0.755 | 20.640 |
| E2 | 3.780 | 327 | 0.315 | 5.884 | 0.767 | 21.671 |
| E3 | 4.002 | 309 | 0.002 | 2.245 | 0.753 | 8.114 |
| E4 | 3.931 | 315 | 0.463 | 4.771 | 0.666 | 15.252 |
| E5 | 3.945 | 314 | 0.697 | 4.366 | 0.612 | 12.832 |
| E6 | 3.844 | 322 | 0.923 | 2.335 | 0.674 | 7.557 |
| E7 | 3.971(3.85)a | 312 | 0.314 | 3.549 | 0.670 | 11.126 |
| E8 | 3.556 | 348 | 0.56 | 3.659 | 0.663 | 10.590 |
| E9 | 3.537 | 350 | 0.441 | 2.002 | 0.566 | 5.440 |
| Z1 | 3.60(3.05)a | 343 | 0.673 | 5.092 | 0.710 | 17.368 |
| Z2 | 3.477 | 356 | 0.727 | 5.477 | 0.676 | 17.771 |
| Z3 | 3.878 | 319 | 0.662 | 5.239 | 0.669 | 16.830 |
| Z4 | 3.693 | 335 | 0.752 | 5.130 | 0.691 | 17.020 |
| Z5 | 3.655 | 339 | 0.879 | 5.600 | 0.688 | 18.496 |
| Z6 | 3.601 | 344 | 0.960 | 4.205 | 0.665 | 13.435 |
| Z7 | 3.751(3.20)a | 330 | 0.696 | 5.250 | 0.695 | 17.516 |
| Z8 | 3.384 | 366 | 0.706 | 5.096 | 0.660 | 16.146 |
| Z9 | 3.364 | 368 | 0.638 | 4.519 | 0.562 | 12.204 |
| E10 | 3.962 | 312 | 0.615 | 4.256 | 0.672 | 13.736 |
| E11 | 3.883 | 319 | 0.609 | 4.562 | 0.690 | 15.111 |
| E12 | 4.370 | 283 | 0.635 | 3.651 | 0.601 | 10.543 |
| E13 | 4.044 | 306 | 0.754 | 3.666 | 0.611 | 10.755 |
| E14 | 3.998 | 310 | 0.934 | 3.368 | 0.566 | 9.165 |
| E15 | 3.879 | 319 | 1.013 | 2.666 | 0.599 | 7.676 |
| E16 | 4.092 | 302 | 0.690 | 3.749 | 0.589 | 10.600 |
| E17 | 3.580 | 346 | 0.604 | 1.803 | 0.542 | 4.694 |
| E18 | 3.540 | 350 | 0.430 | 3.120 | 0.509 | 7.629 |
| Z10 | 3.642 | 340 | 0.659 | 3.551 | 0.608 | 10.370 |
| Z11 | 3.519 | 352 | 0.704 | 4.509 | 0.649 | 14.044 |
| Z12 | 3.925 | 315 | 0.627 | 3.078 | 0.561 | 8.291 |
| Z13 | 3.718 | 333 | 0.743 | 3.242 | 0.587 | 9.146 |
| Z14 | 3.6716 | 337 | 0.867 | 3.374 | 0.578 | 9.365 |
| Z15 | 3.617 | 342 | 0.958 | 0.390 | 0.558 | 1.046 |
| Z16 | 3.7940 | 326 | 0.6899 | 3.449 | 0.574 | 9.507 |
| Z17 | 3.409 | 363 | 0.680 | 2.480 | 0.552 | 6.575 |
| Z18 | 3.383 | 366 | 0.6262 | 0.740 | 0.502 | 1.783 |
Theoretically, the electron transition energy is a decisive property of optical absorption because it is closely related to the position of the maximum absorption peak (λMax). From the simulated absorption spectra (Fig. S3, ESI†), we find that the maximum absorption wavelength (λMax) of the title compounds correspond to the first excited state S0 → S1 in the range from 290 to 366 nm and presented a large oscillator strength value (with the exception of E3, which exhibits a very weak oscillator strength (f = 0.002)).
The analyses of TD-DFT results display that the absorption spectra of the E-isomers are shifted to shorter wavelengths than those of Z-isomers (see Fig. S3, ESI†). This is in agreement with the existing experimental results and theoretical study at the ωB97XD/def2-SVPP level proposed by Mravec et al.25 Furthermore, the experimental S0 → S1 transition energy of 1 and 7 isomers show red-shift energy absorption compared to TD-ωB97XD calculation by about 0.12 eV (for E1 and E7) and 0.55 eV (Z1 and Z7) (see Table 5).
For the E isomers (i = 1–9), the wavelength of the first absorption transition is very close to that of the E-isomers (10 to 18, respectively). The same observation is found for Z isomers (i from 1 to 18), indicating that the introduction of NO2 or CN group at the R position has little effect on the absorption wavelength of E and Z isomers. Taking E6 and E15 as example, the λ0→1 is 322 and 315 nm, respectively, and for Z6 and Z15, the λ0→1 = 343 nm for both (see Fig. 4). On the other hand, the TD-DFT calculation on the title isomers reveals that the low-energy transitions S0 → S1 are dominated by an electronic excitation from the HOMO to the LUMO, and the shape of these two MOs depicts a significant charge transfer (CT) from the hydrazine part and ring 1 to ring 2 for the compounds 1, E2, 3, 4, Z5–Z10, E11, 12, 13, E14, Z15, 16, Z17, and Z18, and the CT character from the hydrazine part to ring1 for the isomers Z2, Z11, and Z14 (see Table S3 and Fig. S1, S2, ESI†) and intramolecular charge transfer (ICT) in the hydrazine part and ring1 for E5, E6, E8, E9, E15, E17, and E18 isomers. For the isomers 1–9 (substituted with NO2 at the R position), the HOMO → LUMO+1 transition mainly results from the Ar group to ring1, with the exception of the isomers E5, E6, E8, E9, and Z6, which exhibit an ICT character in the ring1 and Ar groups. On the other hand, for the isomers with the CN unit at the R position, the HOMO → LUMO+1 transition can also be assigned as n, π → π* but from the Ar group to the ring2, with the exception for Z18 and E18, which exhibit an ICT character (see Table S3 and Fig. S1, ESI†).
 | ||
| Fig. 4 Molecular orbitals for S1, S2, and S4 absorption transitions. | ||
In the case of Zi and Ei isomers (i = 1 to 9), it is clear that the transition HOMO → LUMO results in a more significant dipole moment (Δμ) compared to the transition HOMO → LUMO+1. Conversely, for the remaining compounds (10 to 18) substituted with a CN group at the position R, the reverse pattern is observed (see Table S3, ESI†). Taking Z2 and Z11 as an example (see Fig. 5), for Z2, the order of Δμ values follows the order Δμ02(H → L: 20.19) > Δμ01(H → L+1: 17.77), and the reverse order is found for Z11 Δμ02(H → L+1: 21.72) > Δμ01(H → L: 14.04). In general, for the same transition HOMO → LUMO, LUMO+1, the Δμ values is larger in the 1–9 isomers than that in the corresponding 10–18 isomer, indicating that the presence of the NO2 group can enhance the Δμ and qCT values more than the CN group. For example, Z2 (17.77 D) is due to a transferred excitation charge qCT = 0.676|e|, and an associated CT distance (dCT = 5.477 Å) from HOMO to LUMO+1 (S0 → S1) is larger than that of the excitation S0 → S1 (from HOMO to LUMO) of Z11 (qCT = 0.649|e|, dCT = 4.509 Å and Δμ0→1 = 14.04 D). Generally, for the title photoswitch compounds, we note that the Z-isomer undergoes a strong CT excitation with a large CT distance (dCT) and dipole moment variation (Δμ) compared to the E-isomers counterpart (compounds 1, 2, and 15 are exceptions to this, see Table S3, ESI†). For instance, the Δμ0→1, dCT and qCT of Z4 are calculated to be 17.02 D, 5.13 Å, and 0.691|e| larger than the corresponding values of E4 (Fig. 5). Conversely, the Δμ, dCT, and qCT of Z1 and Z2 show small values with respect to that of E1 and E2 (Fig. 5 and Table S3, ESI†). It appears that the shape of the Z-isomer significantly increases the Δμ, dCT, and qCT values, which can be attributed to the electron delocalization effects in the ring N1–N2–C3–C4–N6⋯H (RAHB system).
 | ||
| Fig. 5 Electron density difference maps of Z1, E1, Z2, Z4, E4, and Z11 compounds from the ground state to the crucial excited state S0 → S1 and S0 → S2. | ||
Based on the quantum calculation, it can be observed that the strength of the hydrogen bond does not exert a significant effect on the optical properties of photoswitches (Table 5). Instead, the optical properties are influenced by conjugation, which is determined by the structure of the quasi-cycle (open or closed). It is crucial to emphasize that the opening or closing of the RAHB can significantly impact various molecular properties, including stability, acidity, basicity, reactivity, and optical properties.
Nonlinear optical properties
Dipole moment (μ, eV), isotropic polarizability (α), polarizability density (ρ a.u Å−3), nonlinear optical responses (NLO), e.g., first hyperpolarizability (β0, a.u), second harmonic generation (βSHG, a.u), electrooptic Pockels effect (βEOPE, a.u), Hyper–Rayleigh scattering (βHRS, a.u), and depolarization ratio DR (static as well as dynamic) for all isomers are provided in Tables 6 and 7 and Tables S3, S4 and S5 (see ESI†).| λ = ∞ | λ = 695 | λ = 1064 | λ = 1340 | |||||
|---|---|---|---|---|---|---|---|---|
| α | ρ | α | ρ | α | ρ | α | ρ | |
| E1 | 265 | 39 | 283 | 41 | 272 | 40 | 269 | 39 |
| E2 | 284 | 42 | 302 | 44 | 291 | 43 | 289 | 42 |
| E3 | 259 | 38 | 273 | 40 | 264 | 39 | 262 | 38 |
| E4 | 280 | 41 | 297 | 44 | 287 | 42 | 284 | 42 |
| E5 | 289 | 42 | 310 | 45 | 297 | 44 | 294 | 43 |
| E6 | 291 | 43 | 313 | 46 | 300 | 44 | 296 | 43 |
| E7 | 259 | 38 | 276 | 40 | 266 | 39 | 263 | 39 |
| E8 | 313 | 46 | 336 | 49 | 322 | 47 | 319 | 47 |
| E9 | 306 | 45 | 329 | 48 | 315 | 46 | 311 | 46 |
| Z1 | 276 | 40 | 299 | 44 | 285 | 42 | 281 | 41 |
| Z2 | 301 | 44 | 328 | 48 | 311 | 46 | 307 | 45 |
| Z3 | 270 | 40 | 288 | 42 | 277 | 41 | 274 | 40 |
| Z4 | 290 | 42 | 312 | 46 | 298 | 44 | 295 | 43 |
| Z5 | 300 | 44 | 325 | 48 | 309 | 45 | 306 | 45 |
| Z6 | 301 | 44 | 329 | 48 | 311 | 46 | 307 | 45 |
| Z7 | 268 | 39 | 289 | 42 | 276 | 41 | 273 | 40 |
| Z8 | 325 | 48 | 355 | 52 | 336 | 49 | 332 | 49 |
| Z9 | 317 | 47 | 348 | 51 | 329 | 48 | 325 | 48 |
| E10 | 265 | 39 | 282 | 41 | 272 | 40 | 270 | 40 |
| E11 | 285 | 42 | 303 | 45 | 292 | 43 | 290 | 42 |
| E12 | 259 | 38 | 270 | 40 | 264 | 39 | 262 | 38 |
| E13 | 280 | 41 | 297 | 44 | 286 | 42 | 284 | 42 |
| E14 | 290 | 43 | 310 | 45 | 298 | 44 | 295 | 43 |
| E15 | 292 | 43 | 313 | 46 | 300 | 44 | 297 | 44 |
| E16 | 260 | 38 | 276 | 40 | 266 | 39 | 264 | 39 |
| E17 | 313 | 46 | 337 | 49 | 322 | 47 | 319 | 47 |
| E18 | 306 | 45 | 329 | 48 | 315 | 46 | 312 | 46 |
| Z10 | 275 | 40 | 297 | 44 | 284 | 42 | 281 | 41 |
| Z11 | 299 | 44 | 325 | 48 | 309 | 45 | 306 | 45 |
| Z12 | 269 | 39 | 286 | 42 | 276 | 40 | 273 | 40 |
| Z13 | 288 | 42 | 310 | 45 | 297 | 44 | 294 | 43 |
| Z14 | 300 | 44 | 325 | 48 | 310 | 45 | 306 | 45 |
| Z15 | 301 | 44 | 328 | 48 | 312 | 46 | 308 | 45 |
| Z16 | 268 | 39 | 288 | 42 | 276 | 40 | 273 | 40 |
| Z17 | 324 | 48 | 353 | 52 | 335 | 49 | 331 | 49 |
| Z18 | 317 | 47 | 346 | 51 | 328 | 48 | 324 | 48 |
| μ | β 0 | β λ=∞HRS | DRλ=∞ | μ | β 0 | β λ=∞HRS | DRλ=∞ | ||
|---|---|---|---|---|---|---|---|---|---|
| E1 | 5.098 | 1608 | 756 | 3.562 | Z1 | 7.176 | 3906 | 1618 | 4.991 |
| E2 | 5.340 | 2246 | 963 | 4.48 | Z2 | 8.476 | 6498 | 2536 | 6.275 |
| E3 | 4.980 | 1024 | 474 | 3.672 | Z3 | 7.736 | 2965 | 1217 | 5.143 |
| E4 | 5.035 | 1052 | 585 | 2.693 | Z4 | 8.572 | 2980 | 1264 | 4.623 |
| E5 | 6.282 | 312 | 546 | 1.584 | Z5 | 9.924 | 2202 | 1055 | 3.428 |
| E6 | 6.604 | 1928 | 1101 | 2.607 | Z6 | 1.019 | 1530 | 1152 | 2.756 |
| E7 | 7.080 | 995 | 553 | 2.695 | Z7 | 5.343 | 2898 | 1261 | 4.294 |
| E8 | 6.047 | 1446 | 730 | 3.114 | Z8 | 7.510 | 3970 | 1674 | 4.711 |
| E9 | 3.500 | 639 | 689 | 1.735 | Z9 | 8.981 | 2570 | 1230 | 3.438 |
| E10 | 4.905 | 1510 | 702 | 3.648 | Z10 | 6.936 | 2632 | 1067 | 5.374 |
| E11 | 5.261 | 2156 | 912 | 4.660 | Z11 | 8.188 | 5017 | 1933 | 6.664 |
| E12 | 4.870 | 845 | 387 | 3.765 | Z12 | 7.649 | 1901 | 760 | 5.655 |
| E13 | 4.992 | 987 | 529 | 2.829 | Z13 | 8.516 | 1810 | 757 | 4.844 |
| E14 | 6.293 | 683 | 548 | 1.959 | Z14 | 9.853 | 1107 | 615 | 2.696 |
| E15 | 6.626 | 2293 | 1164 | 3.085 | Z15 | 1.020 | 1488 | 996 | 2.219 |
| E16 | 6.895 | 936 | 497 | 2.871 | Z16 | 5.114 | 1732 | 757 | 4.246 |
| E17 | 5.888 | 1339 | 666 | 3.196 | Z17 | 7.258 | 2390 | 1000 | 4.825 |
| E18 | 3.398 | 1126 | 757 | 2.211 | Z18 | 8.867 | 1160 | 646 | 2.689 |
The static and dynamic results for average linear polarizability 〈α〉 and dipole moment (μ) are presented in Tables 6 and 7, respectively. Molecular polarizability is the ability of its electronic system to be distorted by an external field; according to our results in Table 6, the 〈α〉 value of the Z-isomer is slightly larger than the E-isomer counterpart. On the other hand, the most polarizable isomers are 8 and 17. These results show that the presence of the Ar8 group can enhance the polarizability value. The calculation shows that the polarizability density values (ρ) are similar and not sensitive to the type of Ar and R groups. Furthermore, we observed that the effect of the incident wavelength value (λ = 1340, 1064, and 695 nm) on 〈α〉 and 〈ρ〉 is negligible.
As is well known, the hyperpolarizability is sensitive to the several parameters such as the nature of the substituent (donor/acceptor), geometry of compounds, transition dipole moment, energy gap, electronic transition, and incident wavelength (photon energy). From Table S3, ESI† and Fig. 6, one sees that the βλ=∞HRS and β0 values of E-isomers substituted by either CN group or by NO2 are very close, indicating that the replacement of NO2 by CN group (at R position) has no effect on the static hyperpolarizability. Taking E8 and E17 as examples, βλ=∞HRS(E8) = 1.09 β∞HRS(E17). But in the case of Z-isomers, the introduction of the NO2 acceptor substituent at the R position can increase the static hyperpolarizability (β∞HRS and β0) about twice more than that of the CN group, with the exception of Z6 and Z15 compounds, for which their values are close (β0(Z6) = 1.028β0 (Z15)), an effect which can be attributed to the small dipole moment (μ = 1.01 D). Furthermore, we can observe that the introduction of Ar2 groups can increase the β∞HRS and β0 compared with the Ar3 and Ar5 groups (Fig. 6 and Table 7). For example, the β0 value of E2, Z2, E11, and Z11 is about 7, 4, 3, and 4 times larger than that of E5, Z5, E14, and Z14, respectively. The β∞HRS of Z2 (2536 a.u.) and Z11 (1933) are about 7 and 5 times greater than the values of E12, respectively (Fig. 6).
 | ||
| Fig. 6 Variation of HRS hyperpolarizability in static regime of Z and E isomers. | ||
Also, in this work, we studied the correlation between the energy gap (hardness) of Zi and Ei isomers and their hyperpolarizability in the static regime. The results show that the β0[β∞HRS] values increase with the decrease in the isomer energy gap. Taking Z2 as an example, the energy gap of Z2 is the smallest (6.304 eV) and its hyperpolarizability value is the largest (β0 = 6498 a.u.), which is in good agreement with the literature.74–77
The magnitude of the hyperpolarizabilities of hydrazone photoswitches can be qualitatively rationalized using the two-state approximation, assuming that the S1 electronic excited state is the sole contributor to the sum-over-state expansion of the second-order nonlinear optical (NLO) response.78,79

Based on our findings from Table 5 and Table S3 (ESI†), it is evident that the Z isomer exhibits a lower excited energy (ΔE0→1) and higher oscillator strength (f0→1) compared to its E-isomer counterpart. Our results clearly show that a decrease in the S0 → S1 transition energy and an increase in the value of f0→1 are directly associated with an increase in the hyperpolarizabilities of hydrazone photoswitches. Taking E1 and Z1 as example, E1 has a transition energy value of 3.798 eV, while Z1 has a lower value of 3.606 eV, and the f0→1 value of Z1 is higher than that of E1. The DFT calculation shows that Z1 exhibits a larger hyperpolarizability value compared to the E1 isomer. Similarly, we observed the same trend for the other isomers. Each isomer exhibits a lower transition energy (ΔE0→1) and huge f0→1 value, generally displaying larger hyperpolarizability compared to its counterpart.
On the other hand, the larger hyperpolarizability values in the Z-isomers, compared to those in the E-isomers, can be due to the delocalization of electrons in the anti-aromatic QCC (N1–N2–C3–C4–N6⋯H) in the Z-isomers, which does not exist in the E-isomers (Fig. 1, Table 2 and Table S1, ESI†). It means that destabilizing the anti-aromatic behavior of quasi-cycle closed in the Z-isomers can reduce and grow the aromatic character in ring 2 and 1, respectively, and this leads to enhanced hyperpolarizability values.
From our calculation, we observe that when the compound is in a Z form, the BLA decreases, which indicates a better electron delocalization and, consequently, the hyperpolarization increases. Fig. 7 presents a nice linear relationship between BLA1–2 and static hyperpolarizability β∞HRS [β0] of Z-isomers. It is important to note that for the title isomers, the variations of β∞HRS values are similar to those of β0 values. Generally, the TD-DFT results indicated that the Z-isomers have low excitation energies of the first excited state and large transition dipole moments (Δμ), which are the necessary conditions to obtain high NLO response.
 | ||
| Fig. 7 Correlation between BLA and static hyperpolarizability; the red points have been omitted from the correlation line. | ||
Mravec research group showed that increasing the EW character of the substituents on the Ar-ring leads to a little increase in the efficiency of the Z → E photoisomerization, whereas a large increase in the quantum yield can be observed in the case of the back E → Z photoisomerization (in the order 3 > 5 > 4 > 7 > 1 > 6 > 2; see Fig. 8).25 In general, our quantum calculation show inverse trend between the photoisomerization quantum yields (Φ) and hyperpolarizability. Note, for instance, compound 2 (see Table 7 and Fig. 6), which has the largest hyperpolarizability and the smallest photoisomerization QY (the EW substituents on the –C6H4 is OCH3; Ar2 = –C6H4-ρ-OCH3). Furthermore, the trend photoisomerization QY is the same as that for the BLA values (see Table 1). On the other hand, we can attribute the small and large photoisomerization QY values (ΦZ→E < ΦE→Z) to the hyperpolarizability of the first and second isomer of the mechanism. In other words, the Z → E photoisomerization mechanism25 begins with the isomer having the largest hyperpolarizability (first isomer Z) and finishes with the E-isomer (second isomer) that has a small hyperpolarizability; this mechanism displays a weak ΦZ→E value. In the same context, E → Z photoisomerization starts with the E-isomers (first isomer) having the smaller hyperpolarizability than the second isomer Z and present a larger ΦE→Z value than ΦZ→E (see Tables 7, 8 and Fig. 8). Taking compound 3 as an example, the photoisomerization quantum yields of ΦE3→Z3 are larger than that of ΦZ3→E3 (Φ = 12.3 and 0.8, respectively),25 and the hyperpolarizability value of Z3 is larger than that of E3 (β[Z3] ≈ 3β[E3]) (see Table 8).
 | ||
| Fig. 8 Evolution of photoisomerization quantum yields (Φ) with the increasing EW character of the hydrazine ring (the values from the ref. 25). | ||
| Zi/Ei | η ∞HRS | η 695HRS | η 1064HRS | η 1307HRS | η 0 | η 695SHG | η 1064SHG | η 1307SHG |
|---|---|---|---|---|---|---|---|---|
| 1 | 2.140 | 14.097 | 2.385 | 2.267 | 2.429 | 13.464 | 2.541 | 2.465 |
| 2 | 2.633 | 6.485 | 3.131 | 2.883 | 2.893 | 5.854 | 3.312 | 5.713 |
| 3 | 2.567 | 5.578 | 2.980 | 2.780 | 2.895 | 5.597 | 3.224 | 3.051 |
| 4 | 2.160 | 6.191 | 2.4656 | 2.328 | 2.832 | 6.107 | 2.809 | 2.766 |
| 5 | 1.932 | 10.947 | 2.7012 | 2.357 | 7.057 | 11.286 | 5.252 | 5.943 |
| 6 | 1.046 | 6.083 | 1.034 | 1.037 | 0.793 | 4.287 | 0.733 | 0.754 |
| 7 | 2.280 | 4.939 | 2.555 | 2.434 | 2.912 | 4.863 | 2.854 | 2.837 |
| 8 | 2.293 | 0.221 | 2.792 | 2.556 | 2.745 | 0.184 | 3.059 | 2.893 |
| 9 | 1.785 | 0.212 | 2.191 | 2.012 | 4.021 | 0.076 | 4.732 | 4.496 |
| 10 | 1.519 | 7.919 | 1.739 | 1.636 | 1.743 | 7.936 | 1.915 | 1.829 |
| 11 | 2.119 | 11.437 | 2.442 | 2.280 | 2.326 | 11.102 | 2.593 | 2.450 |
| 12 | 1.963 | 4.078 | 2.237 | 2.108 | 2.249 | 4.275 | 2.492 | 2.366 |
| 13 | 1.431 | 4.866 | 1.705 | 1.577 | 1.833 | 4.982 | 2.001 | 1.908 |
| 14 | 1.122 | 8.439 | 1.667 | 1.391 | 1.620 | 9.172 | 2.681 | 2.304 |
| 15 | 0.855 | 2.092 | 0.7881 | 0.817 | 0.648 | 0.753 | 0.550 | 0.592 |
| 16 | 1.523 | 3.438 | 1.727 | 1.634 | 1.850 | 3.553 | 1.968 | 1.906 |
| 17 | 1.501 | 0.169 | 1.767 | 1.631 | 1.784 | 0.151 | 2.001 | 1.880 |
| 18 | 0.853 | 0.078 | 0.941 | 0.901 | 1.030 | 0.059 | 1.276 | 1.192 |
From our study, we can conclude that there is a proportional relationship between photoisomerization quantum yield and hyperpolarizability of the second isomer, and there is an inversely proportional relationship between the Φ and β of the first isomer. These relationships will be the object of further investigations.
According to the TD-DFT results (λMax ≡ λ0→1), we calculated the hyperpolarizability (βλHRS, βλSHG, and βλEOPE) of the studied isomers at a near resonant wavelength of 695 nm (≈2 × λMax) and a nonresonant wavelength of 1064 nm and 1340 nm. Our results from the dynamic regime present an excellent linear relationship between Hyper–Rayleigh scattering (βλHRS) and second harmonic generation (βλSHG) (see Fig. 9).
 | ||
| Fig. 9 Correlation between β∞HRS ↔ β0 and βλHRS ↔ βλSHG. | ||
As can be seen in Fig. 10, the hyperpolarizability value increases with decreasing wavelength of the incident light, i.e., β(695) > β(1064) > β(1340) > β(∞). Obviously, the βλSHG and βλHRS values dramatically increase at the 695 nm wavelength more than at λ = 1340 nm and 1064 nm, which can be attributed to the larger resonance or dispersion at 290–366 nm according to the TD-DFT results (Table S3, ESI†). As examples, Z1 exhibits the largest response at λ = 695 nm β695HRS[β695SHG] = 142![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500[337600] a.u.), while this isomer possesses small values at λ = ∞, 1340, and 1064 nm. By way of explanation, the hyperpolarizability βλHRS[βλSHG] of Z1 at 695 nm is approximately 39 and 55 times higher than at 1340 and 1064 nm, respectively. On the other hand, the magnitude of β(−ω; ω, 0) is slightly enhanced with increasing frequency of the incident light compared with βλHRS and βλSHG (Table S4, ESI†). Taking Z2 and E5 as examples, the β695EOPE value of Z2 (12100 a.u.) is evaluated to be the largest, and E5 possess a small value of β695EOPE (771 a.u.), which is almost two times larger than that of the βλEOPE at 1340 and 1064 nm.
500[337600] a.u.), while this isomer possesses small values at λ = ∞, 1340, and 1064 nm. By way of explanation, the hyperpolarizability βλHRS[βλSHG] of Z1 at 695 nm is approximately 39 and 55 times higher than at 1340 and 1064 nm, respectively. On the other hand, the magnitude of β(−ω; ω, 0) is slightly enhanced with increasing frequency of the incident light compared with βλHRS and βλSHG (Table S4, ESI†). Taking Z2 and E5 as examples, the β695EOPE value of Z2 (12100 a.u.) is evaluated to be the largest, and E5 possess a small value of β695EOPE (771 a.u.), which is almost two times larger than that of the βλEOPE at 1340 and 1064 nm.
 | ||
| Fig. 10 Calculated first hyperpolarizabilities of Z and E isomers, in the zero frequency limit and frequency-dependent fields. | ||
In most cases in static and dynamic regime (λ = ∞, 1064, and 1307 nm), the hyperpolarizability β0, βλSHG, βλHRS of Z-isomers are in the range of 2–7 times as large as those of the corresponding E-isomer. For example, the β0 value of Z5 is 2200 a.u., which is 7 times larger than that of the corresponding E5. With the exception of compounds 6, 15, and 18, where the values of Z are smaller than that of E, for instance, βλSHG(Z15)1/2βλSHG(E15) (see Table 8). On the other hand, the β695SHG [β695HRS] of Z-isomers is 2–14 times larger than that of the corresponding E-isomers. With the exception of compounds 8, 9, 17, and 18, where E8, E9, E17, and E18 have the largest β695SHG [β695HRS] value, which is about 5, 13, 7, and 17 [5, 5, 6, and 13] times as large as those of the Z-isomers Z8, Z9, Z17, and Z18, respectively (Table 8).
In addition, dynamic perturbations were introduced to explore the effect of frequency dispersion. To provide a comparison, we utilized two fundamental optical wavelengths λ = 1340 and 1064 nm (used in NLO measurements) along with 695 nm (derived from TD-DFT results). This allowed us to assess the contribution of dispersion correction to the NLO response in these isomers. To quantify this correction, we used the frequency dispersion factor between static and dynamic at a definite wavelength, which is depicted by the ratio βλHRS/β∞HRS and is listed in Table S6 (ESI†). We can observe that the dispersion of optical nonlinearity at λ = 695 nm of E8, E17, Z1, E18, Z11, E9, Z14, Z10, Z5, Z6, and Z2, respectively, have maximum frequency dispersion factor (from 115 to 32), which can be attributed to the TD-DFT results, where  . In contrast to the isomers E3 and E12 (λMax = 309 and 283 nm, respectively), the smallest dispersion factor (about 4.36) and the other isomers show a moderate values (from 6 to 29) at λ = 695 nm. As can be seen, the frequency dispersion factor at incident laser (λ = 1340 and 1064 nm) of the title compounds is in the range of 1–5.
. In contrast to the isomers E3 and E12 (λMax = 309 and 283 nm, respectively), the smallest dispersion factor (about 4.36) and the other isomers show a moderate values (from 6 to 29) at λ = 695 nm. As can be seen, the frequency dispersion factor at incident laser (λ = 1340 and 1064 nm) of the title compounds is in the range of 1–5.
Our findings indicate that the frequency dispersion factor is greater at higher frequencies compared to lower frequencies; the weak frequency of incident light should be chosen to count the 2nd NLO coefficients in the experiment.80 Regarding possible applications, in terms of writing and reading stored information on photochromic materials, the nonresonant character of NLO enables reading outside the absorption band; in this case, erasure during reading can be avoided.19 Clearly, the larger hyperpolarizability does emerge when the incident wavelength of dispersion is equal or closer to twice the wavelength in the first transition (λ0→1).
To further explore the origin of the first hyperpolarizability of the title compounds (E and Z isomers), the polarization scan of HRS intensity I2ωΨV has also been calculated, and the relationship between the I2ωΨV and polarization angle Ψ is plotted (see Fig. S5, ESI† and plotted for the compounds 1 and 5 in Fig. 11); the βJ=1 and βJ=3 are listed in Table S5 (ESI†).
 | ||
| Fig. 11 Relationship between I2ωΨV and polarization angle Ψ of Z and E isomers (green static, red λ = 1340 nm, blue λ = 1064 nm). | ||
From Fig. 12, we can conclude that the DR values are sensitive to the nature of the Ar and R substituents, the geometry of isomers (Z and E shapes), as well as the wavelength of incident light. We observed that at λ = ∞, 1064, and 1307 nm, the DR value decreases in the following order: DR (λ = 1064) > DR (λ = 1340) > DR (λ = ∞) also; from this figure, it seems that the Z-isomer exhibits larger DR than the corresponding E-isomer. In contrast, at 695 nm, the depolarization ratio of E-isomer is larger than that of the Z-isomer counterpart, which can be attributed to its resonance or dispersion at λMax.
 | ||
| Fig. 12 Evolution of the depolarization ratio (DR) in static and dynamic regime (red: Z-isomer, blue: E-isomer). | ||
On the other hand, at λ = ∞, 1064, and 1307 nm, it can be found that E4, E13, E6, Z6, E15, Z15, E7, E16, E8, E17, E9, E18, and Z18 molecules are considered as octupolar molecules with large octupolar contributions (βJ=3 > βJ=1), while E1, E10, Z10, E2, Z2, E11, Z11, E3, Z3, E12, Z12, Z4, Z13, Z5, Z7, Z16, Z8, Z17, Z9 molecules are considered as dipolar molecules and dipolar contributions, with βJ=1 larger than the βJ=3 (see Table S5 and Fig. S5, ESI†). For Z1 and E5, as illustrated in Fig. 11, the DR amounts to 4.991 and 1.584, respectively, a value close to 5 and 1.5; these values characterize ideal dipolar and typical octupolar systems, respectively.
The quantum chemical calculation shows that the Z-isomers exhibit the largest response, whereas the corresponding E-isomers exhibit smaller values at static and dynamic regimes. It is worth noting that the larger NLO responses are strongly related to the hydrogen bond, the electron-withdrawing character of the substituents on the Ar hydrazine moiety, and on the ring 2, the photoisomerization quantum yields ΦE→Z. Among the 18 E/Z isomers theoretically studied, Z2 is the most promising for future applications in the NLO field.
Conclusions
Systematic DFT and TD-DFT calculations have been carried out on extended series of substitutional derivatives of pyridine photoswitches. The structural, reactivity parameters, linear, and nonlinear optical properties of all compounds differing by a CN and NO2 substitution on the ketone fragment at the position R and Ar ring in the hydrazine fragment have been analyzed in detail.The quantum calculation indicates that the Z-isomers are proposed to be a promising candidate for the 2nd order nonlinear optical applications due to its lower excited energy, smaller BLA values, weaker energy gap, larger dipole moment variations in the first excited state, smaller electron localization in the anti-aromatic ring (QCC), and higher photoisomerization quantum yields ΦE→Z than that of the corresponding E-isomers. To the best of our knowledge, this work evidences that the delocalization strength enhanced by the resonance-assisted hydrogen bond can improve the second order NLO responses of the hydrazone photoswitches, especially due to the delocalized electrons in the anti-aromatic ring (QCC) in Z-isomers.
Regarding the NLO properties, DFT calculation indicates that the CN and NO2 groups have the same effect on the static hyperpolarizability of E-isomers. For Z-isomers, the introduction of NO2 at R position increases the β∞HRS and β0 to about twice more than that of the CN group. Furthermore, their introduction into the Ar2 group increases the hyperpolarizability (β∞HRS and β0) compared with Ar3 and Ar5 groups.
Regarding the nonlinear optical properties of isomers, we found that the frequency-dependent hyperpolarizability (λ = 695 nm) of the title isomers are larger than the static regime, revealing an eminent hyperpolarization due to the delocalized electrons in the isomers. By increasing the incident wavelengths from λ = 695 nm to λ = 1064 and 1307 nm, the dispersion of optical nonlinearity of isomers shows a smaller value than that at λ = 695 nm. Our results indicate that the resonance effect of hyperpolarizability is amplified with the decrease in the incident wavelength (λ = 695 nm). A good correlation is obtained between β∞HRS ↔ β0 and βλHRS ↔ βλSHG as well as between BLA and static hyperpolarizability.
Author contributions
All the authors discussed the results. Conceptualization and methodology: D. H. and H. C.; investigation: D. H., N. K., M. Z., D. Y., S. L., C. M. and H. C.; writing – original draft preparation: D. H. and H. C.; writing – review & editing: D. H., N. K., M. Z., D. Y., S. L.; C. M. and H. C.; data curation: D. H., N. K., M. Z., D. Y., S. L., C. M. and H. C. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the GENCI/CINES for HPC resources/computer time (Project cpt2130), and the PSMN of the ENS-Lyon for computing resources.Notes and references
- K. Matsuda and M. Irie, J. Am. Chem. Soc., 2000, 122, 7195–7201 CrossRef CAS.
- C. G. Liu, Z.-M. Su, X.-H. Guan and S. Muhammad, Mol. Phys., 2011, 115, 23946–23954 CAS.
- M. N. Chaur, D. Collado and J. M. Lehn, Chem. – Eur. J., 2011, 17, 248–258 CrossRef CAS PubMed.
- M. Schulze, M. Utecht, A. Hebert, K. Ru, P. Saalfrank and P. Tegeder, J. Phys. Chem. Lett., 2015, 6, 505–509 CrossRef CAS PubMed.
- L. Greb and J. Lehn, J. Am. Chem. Soc. Irradiat., 2014, 136, 13114–13117 CrossRef CAS PubMed.
- M. Zaidi, D. Hannachi, D. Samsar and H. Chermette, Inorg. Chem., 2021, 60, 6616–6632 CrossRef CAS PubMed.
- D. Kamli, D. Hannachi and H. Chermette, New J. Chem., 2023, 47, 1234–1246 RSC.
- M. S. Kodikara, R. Stranger and M. G. Humphrey, Coord. Chem. Rev., 2018, 375, 389–409 CrossRef CAS.
- Y. Y. Liang, B. Li, X. Xu, F. Long Gu and C. Zhu, J. Comput. Chem., 2019, 40, 971–979 CrossRef CAS PubMed.
- N. Baggi, E. Garoni, A. Colombo, C. Dragonetti, S. Righetto, D. Roberto, J. Boixel, V. Guerchais and S. Fantacci, Polyhedron, 2018, 140, 74–77 CrossRef CAS.
- C. Andraud, F. Cyril, B. Olivier, H. Chermette and P. L. Baldeck, Adv. Polym. Sci., 2008, 214, 149–203 CrossRef CAS.
- M. Schulze, M. Utecht, T. Moldt, D. Przyrembel, C. Gahl, M. Weinelt and P. Tegeder, Phys. Chem. Chem. Phys., 2015, 17, 18079–18086 RSC.
- A. Goulet-hanssens and C. J. Barrett, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 3058–3070 CrossRef CAS.
- D. J. Van Dijken, P. Kovar, S. P. Ihrig and S. Hecht, J. Am. Chem. Soc., 2015, 137, 14982–14991 CrossRef CAS PubMed.
- K. J. Chen, A. D. Laurent and D. Jacquemin, J. Phys. Chem. C, 2014, 118, 4334–4345 CrossRef CAS.
- P. Zhao, D. Wang, H. Gao, J. Zhang, Y. Xing, Z. Yang, H. Cao and W. He, Dye. Pigment., 2018, 162, 451–458 CrossRef.
- A. J. Garza, O. I. Osman, N. A. Wazzan, S. B. Khan, G. E. Scuseria and A. M. Asiri, Comput. Theor. Chem., 2013, 1022, 82–85 CrossRef CAS.
- T. Woller, P. Geerlings, F. De Proft, B. Champagne and M. Alonso, J. Phys. Chem. C, 2019, 123, 7318–7335 CrossRef CAS.
- J. A. Delaire, E. Ishow and K. Nakatani, Photoreact. Org. Thin Film., 2002, 305–329 CAS.
- L. Yan, T. Zhang and Z. Su, J. Phys. Chem. A, 2013, 117, 10783–10789 CrossRef PubMed.
- S. Ishihara, J. P. Hill, A. Shundo, G. J. Richards, J. Labuta, K. Ohkubo, S. Fukuzumi, A. Sato, M. R. J. Elsegood, S. J. Teat and K. Ariga, J. Am. Chem. Soc., 2011, 133, 16119–16126 CrossRef CAS PubMed.
- B. He and O. S. Wenger, J. Am. Chem. Soc., 2011, 133, 17027–17036 CrossRef CAS PubMed.
- Y. Zhang, H. Wang, J. Ye, X. Li and Y. Qiu, Organomet. Chem., 2019, 888, 29–36 CrossRef CAS.
- M. Samoc, N. Gauthier, M. P. Cifuentes, F. Paul, C. Lapinte and M. G. Humphrey, Angew. Chemie, 2006, 118, 7536–7539 CrossRef.
- B. Mravec, Š. Budzák, M. Medved, L. F. Pašteka, C. Slavov, T. Saßmannshausen, J. Wachtveitl, J. Kožíšek, L. Hegedüsová, J. Filo and M. Cigáň, J. Org. Chem., 2021, 86, 11633–11646 CrossRef CAS PubMed.
- B. Mravec, J. Filo, K. Csicsai, V. Garaj, M. Kemka, A. Marini, M. Mantero, A. Bianco and M. Cigáň, Phys. Chem. Chem. Phys., 2019, 21, 24749–24757 RSC.
- B. Mravec, A. Marini, M. Tommasini, J. Filo, M. Cigáň, M. Mantero, S. Tosi, M. Canepa and A. Bianco, Chem. Phys. Chem., 2021, 22, 533–541 CrossRef CAS PubMed.
- M. N. Chaur, D. Collado and J. M. Lehn, Chem. – Eur. J., 2011, 17, 248–258 CrossRef CAS PubMed.
- J. Da Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- J. Da Chai and M. Head-Gordon, J. Chem. Phys., 2008, 128, 084106–084114 CrossRef PubMed.
- G. A. Petersson and A.-L. Mohammad, J. Chem. Phys., 1991, 9, 6081–6090 CrossRef.
- G. A. Petersson, A. Bennett, T. G. Tensfeldt, M. A. Al-Laham, W. A. Shirley and J. Mantzaris, J. Chem. Phys., 1988, 89, 2193–2218 CrossRef CAS.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. OgliaroJr, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. Fox, Gaussian 09, Revision A.02, Gaussian, Wallingford, 2009 Search PubMed.
- R. A. Kendall, T. H. Dunning and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- H. Chermette, J. Comput. Chem., 1999, 20, 129–154 CrossRef CAS.
- R. G. Parr, L. V. Szentpály and S. Liu, J. Am. Chem. Soc., 1999, 121, 1922–1924 CrossRef CAS.
- T. Le Bahers, C. Adamo and I. Ciofini, J. Chem. Theory Comput., 2011, 7, 2498–2506 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- M. Kamiya, H. Sekino, T. Tsuneda, K. Hirao and M. Kamiya, J. Chem. Phys., 2005, 122, 234111 CrossRef PubMed.
- J. E. Rice, R. D. Amos, S. M. Colwell, N. C. Handy and J. Sanz, J. Chem. Phys., 1990, 93, 8828–8839 CrossRef CAS.
- R. Bersohn, P. A. O. Yoh-Han and H. L. Frisch, J. Chem. Phys., 1966, 45, 3184–3198 CrossRef CAS.
- A. Plaquet, M. Guillaume, B. Champagne, F. Castet, L. Ducasse, J. L. Pozzo and V. Rodriguez, Phys. Chem. Chem. Phys., 2008, 10, 6223–6232 RSC.
- F. Castet, E. Bogdan, A. Plaquet, L. Ducasse, B. Champagne and V. Rodriguez, J. Chem. Phys., 2012, 136, 24506–24515 CrossRef PubMed.
- H. Louis, I. B. Onyebuenyi, J. O. Odey, A. T. Igbalagh, M. T. Mbonu, E. A. Eno, A. M. S. Pembere and O. E. Offiong, RSC Adv., 2021, 11, 28433–28446 RSC.
- P. S. V. Kumar, V. Raghavendra and V. Subramanian, J. Chem. Sci., 2016, 128, 1527–1536 CrossRef CAS.
- D. Paul, J. Deb and U. Sarkar, ChemistrySelect, 2020, 5, 6987–6999 CrossRef CAS.
- E. Espinosa, I. Alkorta, J. Elguero and E. Molins, J. Chem. Phys., 2002, 117, 5529 CrossRef CAS.
- C. F. G. E. J. Baerends, T. Ziegler, A. J. Atkins, J. Autschbach, D. Bashford, A. Bérces, F. M. Bickelhaupt, C. Bo, P. M. Boerrigter, L. Cavallo, D. P. Chong, D. V. Chulhai, L. Deng, R. M. Dickson, J. M. Dieterich, D. E. Ellis, M. van Faassen, L. Fan and T. H. Fischer, ADF2016.01, SCM, Theor. Chem. Vrije Univ. Amsterdam, Netherlands, 2016, https://https//www.scm.com.
- G. Te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. Van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- M. Palusiak and T. M. Krygowski, Chem. – Eur. J., 2007, 13, 7996–8006 CrossRef CAS PubMed.
- E. Arunan, G. R. Desiraju, R. A. Klein, J. Sadlej, S. Scheiner, I. Alkorta, D. C. Clary, R. H. Crabtree, J. J. Dannenber, P. Hobza, H. G. Kjaergaard, A. C. Legon, B. Mennucci and D. J. Nesbitt, Pure Appl. Chem., 2011, 83, 1637–1641 CrossRef CAS.
- P. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao, N. J. R. V. E. Hommes, D. Erlangen and R. V. February, Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef CAS PubMed.
- P. R. Schleyer, B. Kiran, D. V. Simion and T. S. Sorensen, J. Am. Chem. Soc., 2000, 122, 510–513 CrossRef CAS.
- D. Hannachi, N. E. H. Amrane, L. Merzoud and H. Chermette, New J. Chem., 2021, 45, 13451–13462 RSC.
- J. Li and A. Y. Rogachev, Phys. Chem. Chem. Phys., 2016, 11781–11791 RSC.
- A. Ayadi, L. Mydlova, N. Zouari, M. Makowska-janusik, B. Sahraoui and A. El-ghayoury, Opt. Mater., 2016, 1–9 Search PubMed.
- R. N. Butler and S. M. Johnston, J. Chem. Soc. Perkin Trans. 1, 1984, 2109–2116 RSC.
- M. A. Al-Sheikh and M. H. Elnagdi, Molecules, 2009, 14, 4406–4413 CrossRef PubMed.
- S. M. Landge and I. Aprahamian, J. Am. Chem. Soc., 2009, 131, 18269–18271 CrossRef CAS PubMed.
- X. Su and I. Aprahamian, Org. Lett., 2011, 13, 30–33 CrossRef CAS PubMed.
- S. M. Landge, E. Tkatchouk, D. Benítez, D. A. Lanfranchi, M. Elhabiri, W. A. Goddard and I. Aprahamian, J. Am. Chem. Soc., 2011, 133, 9812–9823 CrossRef CAS PubMed.
- X. Su, M. Lõkov, A. Kütt, I. Leito and I. Aprahamian, Chem. Commun., 2012, 48, 10490–10492 RSC.
- C. Lu, B. Htan, S. Fu, C. Ma and Q. Gan, Tetrahedron, 2019, 75, 4010–4016 CrossRef CAS.
- P. Gilli, V. Bertolasi, L. Pretto, V. Ferretti and G. Gilli, J. Am. Chem. Soc., 2004, 126, 3845–3855 CrossRef CAS PubMed.
- V. Bertolasi, P. Gilli, V. Ferretti and G. Gilli, J. Am. Chem. Soc., 1991, 113, 4917–4925 CrossRef CAS.
- G. Gilli, F. Bellucci, V. Ferretti and V. Bertolasi, J. Am. Chem. Soc., 1989, 111, 1023–1028 CrossRef CAS.
- E. Espinosa, E. Molins and C. Lecomte, Chem. Phys. Lett., 1998, 285, 170–173 CrossRef CAS.
- I. Mata, I. Alkorta, E. Espinosa and E. Molins, Chem. Phys. Lett., 2011, 507, 185–189 CrossRef CAS.
- L. J. Karas, P. R. Batista, R. V. Viesser, C. F. Tormena, R. Rittner and P. R. De Oliveira, Phys. Chem. Chem. Phys., 2017, 19, 16904–16913 RSC.
- A. V. Afonin, A. V. Vashchenko and M. V. Sigalov, Org. Biomol. Chem., 2016, 14, 11199–11211 RSC.
- L. R. Domingo, M. J. Aurell, P. Pérez and R. Contreras, Tetrahedron, 2002, 58, 4417–4423 CrossRef CAS.
- L. Wang, J. Ye, H. Wang, H. Xie and Y. Qiu, Sci. Rep., 2017, 7, 1–11 CrossRef PubMed.
- K. S. Thanthiriwatte and K. M. Nalin de Silva, THEOCHEM, 2002, 617, 169–175 CrossRef CAS.
- R. V. Solomon, P. Veerapandian, S. A. Vedha and P. Venuvanalingam, J. Phys. Chem. A, 2012, 116, 4667–4677 CrossRef CAS PubMed.
- N. Hou, R. Feng and X.-H. Fang, Int. J. Quantum Chem., 2022, 122, 1–14 CrossRef.
- J. L. Oudar and D. S. Chemla, J. Chem. Phys., 1976, 66, 2664–2668 CrossRef.
- C. Tonnelé and F. Castet, Photochem. Photobiol. Sci., 2019, 18, 2759–2765 CrossRef PubMed.
- M. Zhang, C. Wang, W. Wang, N. Ma, S. Sun and Y. Qiu, J. Phys. Chem. A, 2013, 117, 12497–12510 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nj02848h |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2023 |