
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
2-Pyridine cyclic triimidazole as a chelating and bridging ligand in mono- and hexa-nuclear Re(I) complexes with emissive properties in solution and in the solid state†
Daniele
Malpicci
 a,
Daniele
Maver
a,
Daniela
Maggioni
ab,
Pierluigi
Mercandelli
a,
Lucia
Carlucci
ab,
Elena
Cariati
ab,
Patrizia
Mussini
a and
Monica
Panigati
*ab
a,
Daniele
Maver
a,
Daniela
Maggioni
ab,
Pierluigi
Mercandelli
a,
Lucia
Carlucci
ab,
Elena
Cariati
ab,
Patrizia
Mussini
a and
Monica
Panigati
*ab
aDipartimento di Chimica, Università degli Studi di Milano, Via Golgi 19, 20133 Milano, Italy. E-mail: monica.panigati@unimi.it
bConsorzio INSTM, Via G. Giusti 9, 50121 Firenze, Italy
First published on 27th October 2023
Abstract
Two luminescent Re(I) complexes, namely mononuclear fac-[ReCl(CO)3(TT-Py)] (1) and hexanuclear [{Re(μ-Cl)(CO)3(μ-TT-Py)}{Re2(μ-Cl)2(CO)6}]2 (2), were isolated in high yields by a one-pot reaction between [ReCl(CO)5] and different amounts of nitrogen-rich 2-pyridine cyclic triimidazole (TT-Py), which acts as a chelating ligand in both derivatives. The metallo-supramolecular cyclic structure of 2, which can be described as a metallacycle in which two mononuclear units of 1 act as bridging ligands towards two “Re2(μ-Cl)2(CO)6” fragments, was confirmed by single-crystal X-ray diffraction analysis. Both complexes were fully characterized in solution by 1D and 2D multinuclear NMR spectroscopy. 1 and 2 exhibit photoluminescence at room temperature in solution, with a broad unstructured emission in the 530–570 nm range. The nature of the excited states involved in the electronic absorption spectrum of 1 was ascertained by a combined density functional theory and time-dependent density functional theory (TD-DFT) study. Both compounds exhibit strong aggregation induced emission enhancement (AIEE), which is more pronounced for 2 leading to a solid state photoluminescence quantum yield of 2.5% that is one of the highest values ever reported for Re(I) polynuclear metallacycles so far. The versatility of TT-Py to adopt variegated coordination modes, previously observed for Cu(I) and Cu(II) derivatives, is confirmed supporting further use of this ligand to obtain new and peculiar emissive mono-, polynuclear and extended coordination systems.
1. Introduction
Pairing transition metals with different organic chromophores has proven to be a successful strategy to obtain materials with diversified and intriguing photophysical and optoelectronic properties, dominated by the presence of low-lying triplet (π,π*) ligand-centered (3LC) and/or metal-to-ligand charge transfer (3MLCT) transitions. A variety of hybrid metal–organic and organometallic compounds have been employed in organic light emitting devices, imaging, sensing, solar energy conversion and photocatalysis.1In this regard, the search for new exo-donor multidentate ligands appears particularly interesting in view of isolating different luminescent polynuclear metal complexes. Interestingly, some of us have recently reported on the versatility of triimidazol[1,2-a:1′,2′-c:1′′,2′′-e][1,3,5]triazine, TT, able to coordinate as a mono-, bi- or tri-dentate ligand, affording luminescent metal complexes and coordination polymers.2 The functionalization of TT with a pyridinic fragment results in an enhancement of the TT photoluminescence properties and its coordination ability. In particular, 3-(pyridin-2-yl)triimidazo[1,2-a:1′,2′-c:1′′,2′′-e][1,3,5]triazine, TT-Py (Chart 1), revealed excitation dependent multicomponent emissive behaviour3 and varied (mono-dentate, chelating-bidentate and μ-bridging chelating) coordination modes towards Cu(II)2e and Cu(I).4 In fact, TT-Py, due to the presence of two nitrogen atoms, namely that on the pyridine ring (N6′ in Chart 1) and the other on the imidazolic unit (N5) at an appropriate distance, is able to act as a chelating ligand. Moreover, additional nitrogen atoms of the triimidazole moiety are available to obtain molecular or extended coordination structures.
 | ||
| Chart 1 Molecular structure of TT-Py. | ||
Besides, tricarbonyl rhenium(I) complexes containing chelating diimine ligands, of the general formula fac-[Re(CO)3(N^N)X]n+ (N^N = 1,10-phenanthroline or 2,2′-bipyridine, X = anionic or neutral monodentate ligand, with n = 0 or 1, respectively), have been extensively investigated due to their interesting excited state behaviour.5 More than four decades after the first study reported by Wrighton and Morse on these Re(I) complexes,6 there is still a lot of interest in their photochemical and photophysical properties related to 3MLCT excited states, which can be modulated by the ligand and the medium.7 This tunability, together with chemical stability and synthetic flexibility, makes these Re(I) compounds interesting for numerous applications (i.e., luminescent probes for sensing and biological labelling,8 electroluminescent devices,9 photocatalysts or photosensitizers in solar energy conversion processes such as H2 evolution10 and photocatalytic and electrocatalytic CO2 reduction11 and dye sensitized solar cells12).
Photochemistry of these carbonyl complexes has also played a key role in the development of molecules capable of controlled CO release for biological applications such as treatment of cardiovascular diseases and bacterial infections.13
Moreover, fac-Re(CO)3 represents an interesting and versatile metallic fragment in one-step and selective syntheses of metallacycles characterized by high stability and solubility.14 Most of these metallacycles display interesting photophysical and redox properties leading to their applications in host–guest chemistry, photo-catalysis, biological activity and functional molecular devices.15
Based on these results, we decided to explore the coordination properties of TT-Py with Re(I).
Accordingly, here we report on the synthesis, and structural and photophysical characterization of two luminescent complexes obtained by reacting TT-Py and [ReCl(CO)5] in different stoichiometric ratios. The relationship between the structure and emission properties is also analyzed using density functional theory and time dependent density functional theory (TD-DFT) calculations.
2. Results and discussion
2.1. Synthesis of rhenium complexes
Previous results indicated the versatility of TT-Py, able to behave towards Cu(II) and Cu(I) both as a chelating ligand2e and as a bridging ligand through the seven-membered chelating ring and one of the three nitrogen atoms of triimidazole.4 We therefore investigated the same versatility towards Re(I) by using different metal![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ligand stoichiometric ratios.
:ligand stoichiometric ratios.
The mononuclear [ReCl(CO)3(TT-Py)] (1) and hexanuclear [{Re(μ-Cl)(CO)3(μ-TT-Py)}{Re2(μ-Cl)2(CO)6}]2 (2) derivatives, as shown in Chart 2, were isolated in high yields by treating TT-Py with different amounts of [ReCl(CO)5] in toluene under reflux.
 | ||
| Chart 2 Molecular structures of 1 and 2. | ||
In particular, 1 was isolated in >60% yield when TT-Py was reacted with one equivalent of [ReCl(CO)5] (see eqn (1) and Scheme 1).
| [ReCl(CO)5] + TT-Py → [Re(Cl)(CO)3(TT-Py)] + 2CO | (1) |
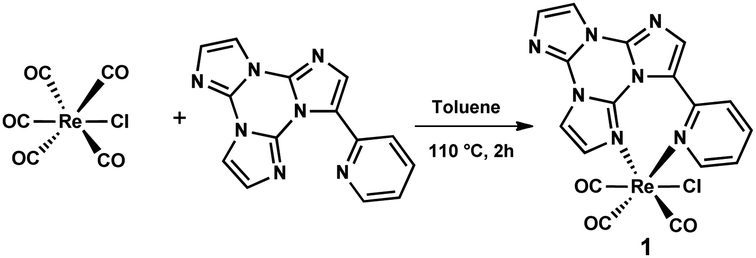 | ||
| Scheme 1 Synthesis of complex 1. | ||
FTIR monitoring of the reaction after a few minutes showed the carbonyl bands of the starting complex together with another three bands in the carbonyl region (ν(CO) at 2027s, 1925s, and 1890s cm−1) (Fig. S1, ESI†), attributable to a reaction intermediate. However, at the end of the reaction, namely after 2 hours, the FTIR spectrum showed three new bands at 2021s, 1909s, and 1892s cm−1 (Fig. S1, ESI†), belonging to complex 1. Both sets of carbonyl frequencies indicate the formation of mononuclear fac-rhenium tricarbonyl complexes,16 where L is a nitrogen ligand and X is a halide. Therefore, also considering that at the beginning of the reaction TT-Py is in large excess (as a result of the slow dissolution of [ReCl(CO)5]), the initial set of carbonyl stretching could belong to an intermediate of the general formula fac-Re(CO)3(L)2(X) containing two monodentate terminally coordinated TT-Py ligands. This intermediate very rapidly loses one of the two TT-Py ligands giving complex 1 where, due to the optimized coordination of the chelating TT-Py ligand, an increase of the electron density on the metal is expected. In agreement, the shift at lower wavenumbers of complex 1 clearly suggests the presence of a more electron rich Re atom. Complex 1 is poorly soluble in toluene and, therefore, it gradually precipitated in three hours from the reaction mixture as a white powder.
Besides, by the reaction of TT-Py with two equivalents of [ReCl(CO)5], in toluene solution under reflux, an equimolar mixture of 1 and hexanuclear species 2 was obtained in one hour. Conversely, 2 was quantitatively obtained by refluxing in toluene one equivalent of TT-Py with three equivalents of [ReCl(CO)5], according to eqn (2) (see Schemes 2 and 3). 2 is highly soluble in toluene and, only by adding n-hexane, it is precipitated as a white-cream solid (isolated yield > 70%).
| 6 [ReCl(CO)5] + 2 TT-Py → [{Re(μ-Cl)(CO)3(μ-TT-Py)}{Re2(μ-Cl)2(CO)6}]2 + 12 CO | (2) |
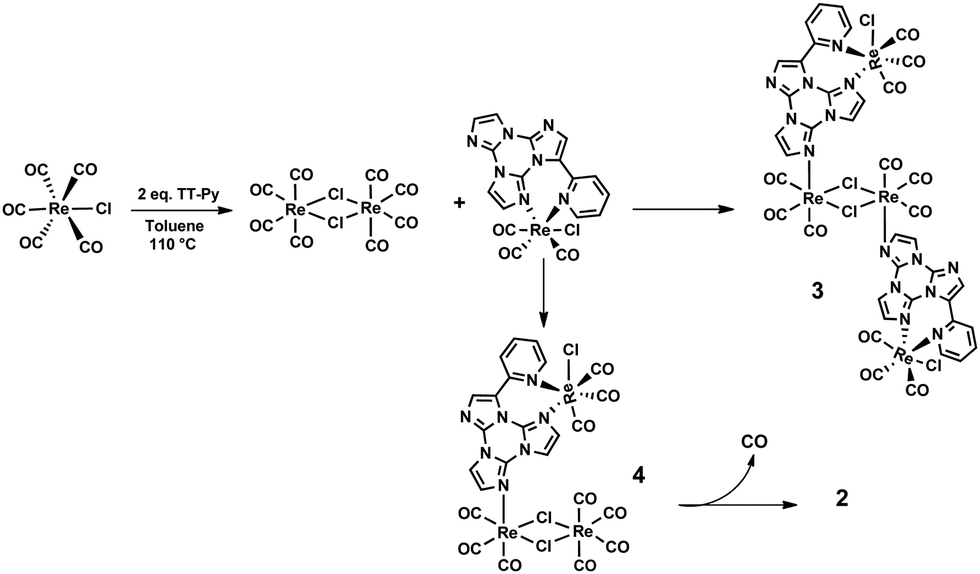 | ||
| Scheme 2 Intermediate of the reaction with 2 equivalents of [ReCl(CO)5]. | ||
 | ||
| Scheme 3 Dimerization reaction of 4. | ||
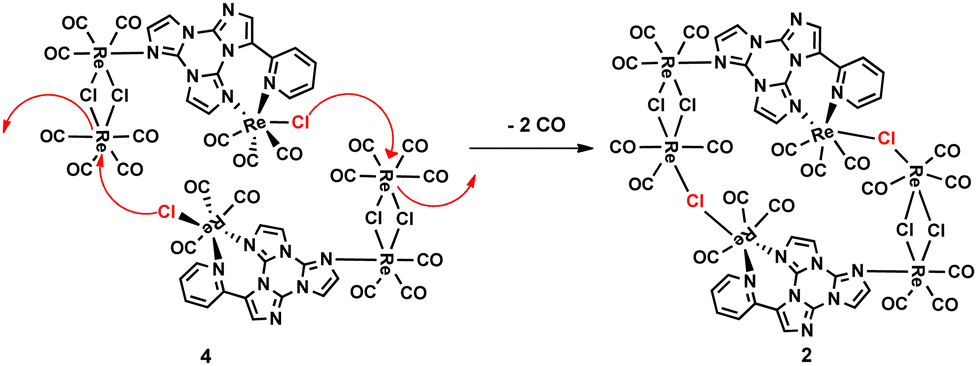
It is interesting to note that a trace amount of 2 was also detected by FTIR in the mother liquor of the reaction of TT-Py with one equivalent of [ReCl(CO)5] after the complete precipitation of 1. The formation of complex 2 confirmed the possibility of TT-Py to act as a bridging ligand, and the possible mechanism of formation could be described as reported in Scheme 2. In particular, at the beginning of the reaction with two equivalents of the metal precursor, the concomitant formation of complex 1 and the dinuclear complex [Re2(μ-Cl)2(CO)8] is observed (FTIR evidence). This dinuclear species, which can be easily obtained by refluxing [ReCl(CO)5] in an inert solvent,17 undergoes a decarbonilation reaction faster than [ReCl(CO)5], affording the unsaturated [Re2(μ-Cl)2(CO)6] fragment (see Scheme 2), which further reacts with complex 1, affording two possible intermediates (3 or 4) which cannot be isolated nor spectroscopically revealed. The possible formation of complex 3, containing two mononuclear complexes terminally coordinated to the [Re2(μ-Cl)2(CO)6] fragment (see Scheme 2), whose ratio Re/TT-Py is 2/1, is unfavourable due to the progressive precipitation of 1 in toluene. In contrast, the other possible, more feasible, intermediate 4, containing only one terminally coordinated mononuclear species and in which the ratio of Re/TT-Py is 3/1, is not stable and can then dimerize, to give complex 2, by the fast decarbonilation of the “Re(CO)4” unit and the following bridging of the chloride ligand (see Scheme 3).
From the molecular point of view, 2 can also be considered as a dimeric structure in which two mononuclear 1 units act as bridging ligands towards two “Re2(μ-Cl)2(CO)6” fragments. The coordination features of TT-Py in 2 resemble those observed in a related Cu(I) coordination polymer4 where the ligand acts as a bridging-chelating fragment through the pyridine and imidazole nitrogen atoms.
The nature of both 1 and 2 was established on the basis of spectroscopic data (FTIR and NMR), and was confirmed by single-crystal X-ray diffraction analysis. For complex 2, a ν(CO) pattern more complex than that of 1 was observed, in agreement with the different symmetries of the many “Re(CO)3” units present in 2 (see the Experimental section and Fig. S2, ESI†).
Similar to many mononuclear rhenium carbonyl complexes, 1 is stable in the solid state, at room temperature and under daylight for different months, if stored under nitrogen. The same stability was also observed in solution, both in non-donor solvents, such as toluene or CH2Cl2, and in coordinating solvents such as acetonitrile or DMSO (see Fig. S3–S5 in the ESI†). In contrast, complex 2 is less stable in the solid state, even under nitrogen, as shown by the slight change in its FTIR spectrum after one month (See Fig. S6, ESI†). Moreover, complex 2 is completely unstable in a donor solvent such as acetonitrile and in weakly coordinating solvents such as THF and MeOH. 1H-NMR and FTIR spectra (see Fig. S7–S9, ESI†) clearly indicated that 2 undergoes rapid fragmentation to complex 1 and to [ReCl(CO)3(L)2], with L = CH3CN, THF, and MeOH, according to eqn (3) (with t1/2 values of few minutes in CH3CN).
| [{Re(μ-Cl)(CO)3(μ-TT-Py)}{Re2(μ-Cl)2(CO)6}]2 + 8L → 2[Re(Cl)(CO)3(TT-Py)] + 4[ReCl(CO)3(L)2] | (3) |
2.2. Multinuclear NMR characterization
1H NMR spectra of 1 and TT-Py in DMSO-d6 are compared in Fig. 1. Only a partial attribution of the protonic signals could be obtained by the analysis of the 1H COSY experiment (Fig. S10, ESI†), since the four CH of imidazole rings B and C (Chart 1) are indistinguishable. After complexation, most of the signals displayed a downfield shift (Δδ ∼ 0.2 ppm) with the expected largest value for the Hortho of the pyridine ring (H5′, 8.66 ppm, Δδ = 0.73 ppm). The notable shift displayed by CH at the α position to the pyridine nitrogen atom was mainly due to the charge redistribution after coordination of the pyridine to the metal center (see Tables S1 and S2, ESI†).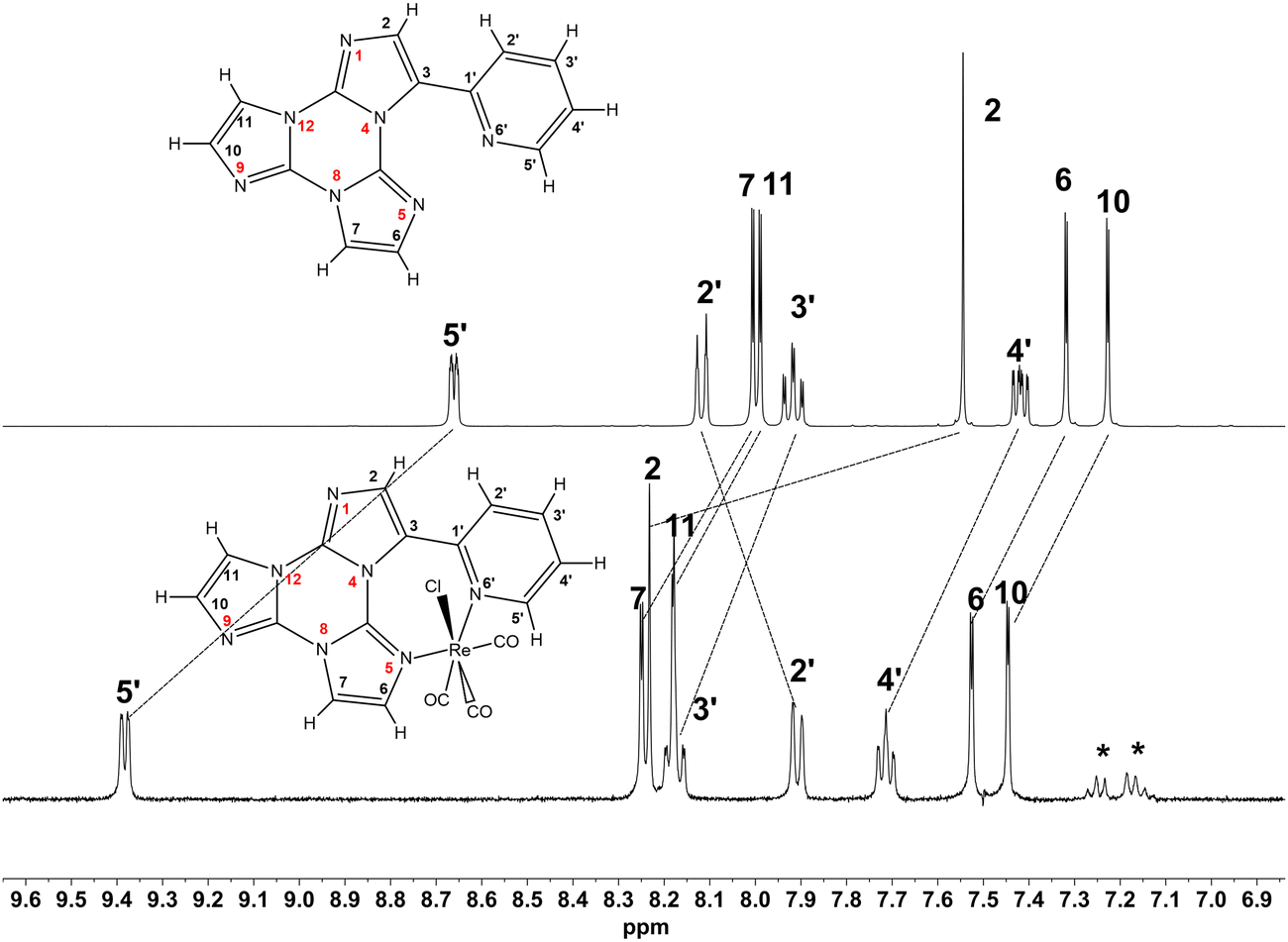 | ||
| Fig. 1 1H-NMR spectra of TT-Py (upper trace) and 1 (lower trace) (298 K, DMSO-d6, 7.05 T). Asterisks mark some toluene residue from the synthesis. | ||
Despite their distance from coordinating nitrogen atoms N6’ and N5, H2′ and H2 were highly perturbed upon complexation, showing an upfield (Δδ = −0.21 ppm) and a downfield shift (Δδ = 0.68 ppm), respectively. These coordination shifts could be ascribed to a major change in their magnetic environment upon re-orientation of the pyridine ring from a free to bonded form (see Paragraph 2.3). In fact, different from its geometry in 1, in TT-Py, H2′ points towards the imidazole nitrogen N5 as evidenced from its crystal structure previously reported by some of us.3
To achieve a complete attribution of the protonic signals of 1, we exploited 15N NMR spectroscopy and carried out 2D 1H-15N heteronuclear long-range correlation (HMBC) experiments (Fig. S11 and S12, ESI†). As expected, the coordinating imidazole nitrogen N5 showed a substantial change of its chemical shift before and after complexation (−157.3 vs. −211.6 ppm, respectively). The attribution of the N5 chemical shift allowed the unequivocal assignment of H6, and consequently all the other protons of rings B and C. In addition, all the 13C signals were attributed through heteronuclear scalar 2D NMR experiments (Fig. S13 and S14, ESI†). The assignment of all the experimental resonances of 1 was validated by DFT calculations. The whole experimental and calculated 1H, 13C and 15N NMR chemical shifts are summarized in Table S1 (ESI†).
The 1H NMR spectrum of 2 was acquired on a sample dissolved in toluene-d8 owing to its insolubility in more polar solvents such as DMSO-d6. This hampered a direct comparison with the signals of 1 in the same solvent. The 1H spectrum showed the same splitting pattern observed in 1. The complete attribution of all the protonic signals of 2 was accomplished by multinuclear 2D NMR experiments (see Fig. S15–S18, ESI†). In particular, due to an accidental overlap with the aromatic signals of toluene, it was possible to identify the two imidazole protonic signals only by exploiting 2D spectroscopy, and in particular 2D 1H–13C HSQC (see Fig. S16, ESI†). Nevertheless, for complex 1, 15N spectroscopy was crucial for the attribution of the 1H NMR signals, since the typical shift of the N9 signal after complexation (from −151.5 to −209.2 ppm) allowed the discrimination of the proton signals of rings B and C, being H10 and H11 immediately recognized and attributed (2D 1H–15N HMBC, Fig. S17, ESI†). To complete the attribution of the quaternary carbon signals, a 1H–13C HMBC experiment was acquired as well (Fig. S18, ESI†).
The complete attribution of the protonic signals highlighted some notable differences in the 1H resonance positions with respect to that of the free ligand in the same solvent (see Fig. 2 and S19, ESI†).
 | ||
| Fig. 2 1H-NMR spectra of TT-Py (upper trace) and 2 (lower trace) (298 K, toluene-d8, 7.05 T). Asterisks mark some signals of a small amount of 1. | ||
Some downfield shifts were easily ascribed to coordination (H5′, H10 and H11). On the other hand, H2, H2′ and H3′ displayed a considerable upfield shift (Δδ = −0.702, −2.27, and −0.835 ppm, respectively), which were not observed in 1. A possible origin of these important upfield shifts, assuming a negligible change in the atomic charge distribution upon coordination, could derive from the interaction of toluene with these protons.
The polynuclear nature of 2, initially suggested by a diffusion NMR experiment (1H DOSY spectroscopy, Fig. S20, ESI†) showing a smaller diffusion coefficient for 2 than for 1, was fully confirmed by single-crystal X-ray diffraction analysis (vide infra).
2.3. Structural characterization
The structures of 1 and 2 were established by single-crystal X-ray diffraction analysis. Fig. 3 and 4 show pictures of the two species, while Table S3 (ESI†) reports some relevant geometrical parameters. In mononuclear complex 1, the rhenium atom attains an octahedral geometry and bears three terminal carbonyl ligands in a facial arrangement, one terminal chloro ligand and two nitrogen atoms of the chelating TT-Py ligand, namely a nitrogen atom of the pyridyl substituent (N7, labelled as N6′ in the molecular scheme of NMR characterization) and a nitrogen atom of the triimidazotriazine moiety (N3, labelled as N5 in the molecular scheme of NMR characterization). The coordination geometry of the rhenium atom resembles that observed in similar fac-[ReCl(CO)3(N^N)] complexes. The conformation adopted by the TT-Py ligand is significantly different from that shown by the free ligand in the solid state.18 In particular, to allow the ligand to chelate, the pyridyl substituent must rotate around the TT-Py bond (as evidenced by the N7–C13–C5–N2 dihedral angle of 46.8°, to be compared to values in the range of 144.4–156.1° found in the different polymorphs of the free ligand) and tilt to accommodate the rhenium atom (as evidenced by the N7–C13–C5 bond angle of 123.45°, ten degrees larger than the values in the range of 112.46–113.71° found for the free ligand). A similar conformation was adopted by TT-Py in previously reported species, in which it similarly acts as a chelating ligand, namely [Cu2I2(TT-Py)]n4 and [Cu(μ-NO3)(NO3)(TT-Py)]n.2e Hexanuclear species 2 (as found in 2·4(n-hexane) crystal) contains two [ReCl(CO)3(TT-Py)] moieties whose geometry is quite similar to that observed in mononuclear complex 1 (see Table S3, ESI†). The two [ReCl(CO)3(TT-Py)] moieties bridge two additional [Re2(μ-Cl)2(CO)6] fragments through the chloro ligand and a nitrogen atom of the triimidazotriazine moiety (corresponding to N5 in 1). The geometry of these trans-disubstituted fragments is similar to that observed in similar species, such as [Re2(μ-Cl)2(CO)6L2] (L = MeCN19 and THF20). Hexanuclear species 2 shows a non-crystallographically imposed C2 symmetry. | ||
| Fig. 3 View of the rhenium complex [ReCl(CO)3(TT-Py)] (1) as found in its crystal structure, with a partial labelling scheme. Ellipsoids are drawn at a 50% probability level. | ||
 | ||
| Fig. 4 View of the rhenium complex [{Re(μ-Cl)(CO)3(μ-TT-Py)}{Re2(μ-Cl)2(CO)6}]2 (2) as found in the crystal structure of its n-hexane solvate, with a partial labelling scheme. Ellipsoids are drawn at a 50% probability level. | ||
From the crystal structures obtained so far using TT-Py as the ligand towards Cu(II), Re(I) and Cu(I), it is evident that the versatility of this simple molecule means it is able to adopt different coordination modes to give mononuclear, polynuclear and extended 1D structures. In particular, in paddle-wheel dinuclear [Cu2(CH3COO)4(TT-Py)2], TT-Py behaved as monodentate through one imidazole nitrogen;2e in [Cu(μ-NO3)(NO3) (TT-Py)], it acted as a chelating ligand,4 and in the coordination polymer [Cu2I2(TT-Py)]n, it was found to bridge through the seven-membered chelating ring and one of the three nitrogen atoms of triimidazole. Therefore, additional interesting structural motifs are expected by investigating the coordination ability of this ligand with other metals.
2.4. Electrochemical characterization
Complexes 1 and 2 were investigated by cyclic voltammetry (CV) and selected data are shown in Fig. 5. The study was performed in CH3CN solution, which in the case of complex 2 enabled in situ monitoring of its fast solvolysis into smaller electroactive molecules. | ||
| Fig. 5 (a) Cyclic voltammetry of 1 and (b) that of 2 in CH3CN, [NBu4]PF6 0.1 M, using a GC electrode of 3 mm in diameter. Scan rate: 0.2 V s−1. Potentials are reported vs. the standard Fc+|Fc redox couple. | ||
As previously reported21, the TT-Py free ligand shows a peculiar first reduction two-peak system at about −2.8 V and −3.1 V vs. Fc+|Fc, which was assigned to a complex electrode process including a series of electron transfer (ET) and chemical steps, starting with radical anion formation, mainly pyridine-centered and kinetically facile. The CV pattern of complex 1 (Fig. 5a) exhibits a quite similar first reduction two-peak system, which points to its first reduction being ligand centered (see also the DFT computational part). Such a two-peak system exhibits a large shift to less negative potentials respect to the free ligand case (−2.14 V and −2.50 V vs. Fc+|Fc, for C1 and C2, respectively), in agreement with the electron withdrawing effect of the coordinated rhenium center, which makes the ligand much electron poorer.
The first monoelectronic and chemically and electrochemically quasi reversible oxidation peak of complex 1, at 1.07 V vs. Fc+|Fc, (peak A1 in Fig. 5a) is related to oxidation of the rhenium center, also consistently with the nature of the HOMO of complex 1 (see below). At the same time, ligand oxidation, observed at ∼1.1 V in the case of the free ligand,21 because of coordination, must be shifted to more positive potentials, thus possibly corresponding to the second and/or third oxidation peak observed at ∼1.6 V/∼1.9 V (peaks A2 and A3 in Fig. 5a).22
Different from complex 1, which is stable in the working medium,23 complex 2 undergoes fast solvolysis, affording complex 1 and [Re(CO)3Cl(CH3CN)2]. In fact, the CV patterns show the progressive decrease of the oxidation and reduction peaks of 2, together with the increase of peaks attributed to the decomposition fragments (Fig. 5b).
In particular, in the cathodic range, complex 2 shows a similar two-peak system, as observed for complex 1, with the first reduction peak at a less negative potential (−2.01 V, C1′ in Fig. 5b). This feature can be related to the coordination of the nitrogen of the imidazolic ring with the rhenium atom, making the ligand in complex 2 electron poorer than that in complex 1. Solvolysis starts immediately (the C1 peak is already present after one minute; see also NMR evidence, Fig. S9, ESI†) and is complete after about 20 minutes. Along this period, peak C1′ gradually disappears and the first reduction pattern becomes that of complex 1 (C1 and C2 peaks). Moreover, the decomposition process is accompanied by the gradual appearance of another reduction peak at −2.96 V (see C3 in Fig. 5b), which is consistent with the Re(I)–Re(0) reduction in [Re(CO)3Cl(CH3CN)2].11f It is interesting to note that this reduction is lacking in the CV pattern of complex 1, due to the higher electron donor capability of TT-Py, making the rhenium center electron richer and shifting the reduction potential out of the solvent window.
In the anodic part, complex 2 shows two different rhenium oxidation peaks (while A2′ and A3′ are likely related to the oxidation of the TT-Py ligand, see above). The first one (A1 in Fig. 5b), lying at the same potential of complex 1, corresponds to the oxidation of the two rhenium centers coordinated to the TT-Py ligand, whose coordination environment is the same as in complex 1. In addition, the peak position supports the lack of electronic communication between the two units forming complex 2. The second oxidation peak, observed at a more positive potential (at ∼1.4 V, A1′ peak in Fig. 5b), should correspond to the oxidation of the four electron poorer rhenium centers belonging to the “Re(μ-Cl)2Re” units. During the solvolysis process, A1′ gradually disappears while A1 remarkably increases.
This feature can be justified considering that besides the formation of complex 1, upon solvolysis, each “Re(μ-Cl)2Re” moiety affords two [Re(CO)3Cl(CH3CN)2], which should undergo oxidation at the same potential of complex 1, being coordinated to three CO, one Cl and two N ligands (although belonging to two acetonitrile molecules instead of a TT-Py ligand).24
2.5. Photophysical characterization
The absorption spectra of the two complexes in diluted CH2Cl2 solutions (ca. 10−5 M) are depicted in Fig. 6a and can be compared with that of the free ligand in the same solvent. TT-Py displays two bands at 235 and 290 nm;3 similarly, 1 shows two broad bands at 247 and 301 nm (with a shoulder at 314 nm) which, according to the electrochemical characterization and DFT calculations, correspond to the admixture of different monoelectronic excitations. In particular, the lower energy band, centred at 301 nm, is due to contributions from metal-to-ligand and halogen-to-ligand charge transfer (1MLCT + 1XLCT), with a component of intra-ligand charge transfer (ILCT) from triimidazotriazine to the pyridyl substituent (see Table 1 and the computational paragraph). Complex 1 displays another broad band, at 247 nm, which is attributed, according to DFT computations, to main ligand-centered monoelectronic transitions. However, upon increasing the solvent polarity, from CH2Cl2 to MeCN, a clear solvatochromic effect, expected for charge transfer transitions, was not observed (Fig. S21, ESI†). This feature could be associated with the different nature of the charge transfer electronic transitions contributing in different ways to each of these absorption bands. | ||
| Fig. 6 (a) UV-Vis absorption spectra of 1 (solid lines) and 2 (dotted lines) in CH2Cl2 at 298 K. (b) Emission spectra in the aerated toluene solution at 298 K. | ||
| Complex | λ abs [nm] | λ em [nm] | λ em [nm] | Φa [%] | Φb [%] | τ [ns] | τ [μs] | k r [s−1] | k nr [s−1] |
|---|---|---|---|---|---|---|---|---|---|
| a In aerated toluene solution (λex = 365 nm). b In the solid state (λex = 365 nm). | |||||||||
| 1 | 247, 301, 314 | 583 | 532 | 0.4 | 12.0 | 483 | 85, 143 | 8.28 × 103 (1.05 × 103)b | 2.06 × 106 (6.15 × 103)b |
| 2 | 234, 264, 320, 346 | 597 | 563 | 0.01 | 2.5 | 500 | 15, 35, 82 | 2.00 × 102 (5.68 × 102)b | 1.99 × 106 (2.21 × 104)b |
In agreement with the more complex molecular structure of 2, a larger number of absorption components are observed. Indeed, the spectrum of 2 displays absorptions at 234, 264 and 320 nm, with a shoulder at 346 nm. Unfortunately, a clear attribution of the components was also hampered by the instability of this complex in polar donor solvents, such as MeCN, and by its very low solubility in the most common non-donor polar solvents. However, the electrochemical characterization clearly indicated that the involvement of the nitrogen N9 of the imidazolic ring, and the chlorine atoms, in coordination with the “Re(μ-Cl)2Re” moiety makes easier the reduction on the aromatic ligand, affording a reduction of the HOMO–LUMO gap and a red shift of the corresponding absorption bands. Therefore, we could tentatively ascribe the low energy bands of complex 2 to the same transitions observed for complex 1.
The photophysical behaviour of both compounds was measured in solution and in the solid state. Upon excitation at 365 nm in toluene (ca. 10−5 M) at 298 K, 1 and 2 display a broad unstructured emission in the visible region (583 and 597 nm, respectively, see Fig. 6b and Table 1). The emission was independent of the excitation wavelengths (see excitation spectra in Fig. S22, ESI†). Taking into account the computation of the singlet–triplet excitation through TD-DFT calculations (see Computational details and Table S5 in the ESI†) and considering the character of the MOs involved (see Fig. S24, ESI†), the T1 state responsible for the emission confidently possesses a mixture of metal-to-ligand charge transfer (MLCT) and intraligand (IL) characters as already reported for [Cu2I2(TT-Py)]n4 and for some Re(I) complexes containing diimine pyridinic ligands.5,7 The reduction of the photoluminescence quantum yield (Φ) on going from 1 to 2 (0.4% and 0.01%, respectively), followed by the lengthening of the excited state lifetime (483 and 500 ns, respectively), affords for 2 a kr value of one order of magnitude lower than that of 1 (see Table 1), while the knr values are identical. This behaviour, which is opposite to what predicted by the energy gap law (where a decrease of the emission energy results in an increase of the non-radiative rate constant knr),25 may suggest that the electronic transitions, responsible for the observed emission in both complexes, are not the same. This difference is in agreement with the more complex molecular and electronic structure of 2, which, as in the case of the absorption spectrum, hampered an unequivocal attribution of the nature of the excited state responsible for the observed emission.
In the solid state, both derivatives maintain the structureless profile with a concomitant important hypsochromic shift of the emission maxima (532 vs. 583 and 563 vs. 597 nm, for 1 and 2, respectively, see Fig. S23, ESI†). This rigidochromic effect, usually observed for emission from polar excited states, less stabilized by the lack of environment mobility,6 suggests a charge transfer character of the emissive state for both complexes. Moreover, the long lifetimes observed for both, in the order of tens of microseconds (see Table 1), agree with the phosphorescent character of their radiative deactivation.
Together with the shift, both complexes also exhibit an increase in the photoluminescence quantum yield on moving to the solid state. In particular, for 1, Φ increases from 0.4 to 12% (see Table 1), while, for 2, this behaviour is much more pronounced with Φ equal to 2.5%, a value almost 250 times higher than in toluene solution. The emission enhancement of 2 is accompanied by a strong decrease of the radiationless rate constant (knr) which drops from 1.99 × 106 to 2.21 × 104 s−1, going from solution to the solid state, whilst the radiative rate constant (kr) maintains the same order of magnitude. For complex 1, the reduction of knr is of three order of magnitude on moving from solution to the solid state (2.06 × 106vs. 6.15 × 103 s−1). These data agree with the hypothesis that the restriction of the vibrational motions is most likely responsible for the enhancement of the emission observed in the solid state, for both complexes, indicating the presence of the aggregation induced enhancement emission (AIEE) effect.26 Indeed, none the alternative mechanisms proposed to explain the aggregation induced emission appear to be at work here, as the structural data rule out any close intermolecular interactions. Moreover, looking at the structure of 2, and considering its very low stability also in poorly coordinated solvents (see above), it is reasonable to suppose that a possible reversible bridge opening and fast interchange between bridging and terminal coordination of the chloride ligands occurs, affording a fast and reversible opening of the circular structure. These features could explain the stronger AIEE effect observed for 2 whose emission, differently from 1, is almost completely quenched in solution and stronger in the solid state due to the lower mobility imposed by the more rigid environment.
It is important to note that, while AIEE behaviour has already been reported for several Re(I) mononuclear complexes (with Φ up to 59%),27 and dinuclear complexes (with Φ up to 56%),7f far less examples of polynuclear compounds, containing more than two rhenium atoms and based on Re(I) molecular rectangles (with Φ of around 0.65%) have appeared in the literature.28 Among these latter, Φ reported for 2 represents a record.
2.6. Computational study
The electronic structure of complex 1 and its absorption spectrum were computed employing density functional theory and time-dependent density functional theory. Fig. S24 (ESI†) shows some relevant molecular orbital plots while Tables S4 and S5 (ESI†) report some calculated electronic transition properties.According to the calculations, in 1, the rhenium d orbitals mainly contribute to the three highest occupied molecular orbitals (HOMOs), lying close together in energy. The three HOMOs show, in addition, significant antibonding interactions with the π orbitals of the halogen atom and bonding contributions from the π* orbitals of the carbonyl ligands. Lying at lower energy, HOMO−3 and HOMO−4 are ligand π orbitals, mainly located on the pyridyl substituent (HOMO−3) and the triimidazotriazine rings (HOMO−4). The three lowest unoccupied molecular orbitals (LUMOs) are π* orbitals delocalized all over the organic ligand. In particular, the LUMO and LUMO+1 show an important contribution from the pyridyl substituent while LUMO+2 is delocalized over the triimidazotriazine rings only.
The band at lower energy in the absorption spectrum of 1 can be assigned to the superposition of two electronic transitions, S0 → S4 and S0 → S5 (computed at 305 and 299 nm, and experimentally observed at 301 and 246 nm, respectively, in CH2Cl2, see above), corresponding to the admixture of HOMO → LUMO+1 and HOMO−3 → LUMO monoelectronic excitations. According to the nature of the orbitals involved, and taking into account the difference between the computed atomic charges for the ground and the excited states, both transitions imply a significant metal-to-ligand and halogen-to-ligand charge transfer. The lower-energy transition, however, shows also an important intraligand character (in particular, a net charge transfer from the triimidazotriazine rings to the pyridyl substituent). Two intense electronic transitions, S0 → S19 and S0 → S21 (computed at 251 and 244 nm, respectively), are responsible for the peak experimentally observed at ca. 250 nm, see above. They correspond to some complex admixtures of many monoelectronic excitations, mainly HOMO−3 → LUMO+2 and HOMO−4 → LUMO+1. Both electronic transitions can be described as ligand-centered. However, the first one mainly implies a redistribution of the electron density within the TT rings while the second one shows also a sizeable charge transfer from the triimidazotriazine moiety to the pyridyl substituent.
3. Conclusions
The versatility of the nitrogen-rich derivative of cyclic triimidazole, TT-Py, bearing a 2-pyridine substituent on the triimidazole scaffold, was exploited together with the attractive properties of the robust and stable fac-Re(CO)3 core for the synthesis of mononuclear complex 1 and polynuclear 2. This latter represents one of the few examples of Re(I) hexanuclear derivatives reported in the literature so far.291 and 2 could be selectively obtained by varying the TT-Py/[ReCl(CO)5] stoichiometric ratio and were fully characterized in solution by FTIR and multinuclear NMR spectroscopies. This last one was particularly useful for confirming that the nature of the two complexes in solution was the same as the one observed in the solid state by single-crystal X-ray diffraction.
As previously reported for Cu(I) and Cu(II) derivatives with the same ligand, in both 1 and 2, the rotation of the pyridine fragment around the TT-Py bond allowed the formation of a seven-member ring with the “Re(CO)3Cl” unit. Moreover, in 2, as already reported for Cu(I), TT-Py also acted as a bridging ligand through one nitrogen atom of the triimidazole core affording, through the very simple one-pot synthetic approach, the hexanuclear cyclic structure. In this case, thanks to the bridging coordination ability of chloro ancillary ligand, no 1D coordination chain was obtained.
Both complexes displayed interesting photophysical properties in solution and in the solid state where 2 showed a record value of Φ among polynuclear rhenium(I) complexes. These results represent a springboard for the use of other pyridinic substituted TT derivatives as ligands to be exploited for the obtainment of both molecular and, desirably, extended Re(I) tricarbonyl luminescent materials.
4. Experimental
4.1. General procedures and instruments
All the manipulations were carried out under an inert atmosphere (N2) using dried glassware with the Schlenk technique. The solvents were distilled by standard methods before use. FTIR spectra were acquired using a PerkinElmer Frontier spectrometer in solution. Elemental analyses were performed on a PerkinElmer CHN2400 instrument. ESI-MS spectra were recorded on a LCQ Fleet ion trap mass spectrometer (Thermo Fisher).The deuterated solvents (CIL) for NMR spectroscopy were used as received. NMR spectra were acquired using a Bruker Avance DRX-400 spectrometer equipped with a Bruker 5 mm BBI Z-gradient probe head with a maximum gradient strength of 53.5 G cm−1, and operating at 400.13, 100.62, and 40.55 MHz for 1H, 13C and 15N, respectively. The 90° pulses were calibrated resulting in the following pulse lengths: 8.5 μs (1H), 13 μs (13C), and 28 μs (15N). NMR samples were prepared by dissolving a few milligrams in 600 μL of either DMSO-d6 or toluene-d8 to obtain clear solutions in a disposable NMR tube. 1H NMR spectra were recorded by accumulating 32 transients with a recovery delay of 10 s on a sweep width of 15 ppm. 15N NMR spectra were referenced to external CH3NO2. The experiments were all collected at 300 K using standard 1D and 2D sequences of the Bruker library. 1H diffusion spectroscopy (1H DOSY) was carried out by using the sequence ledbpgp2s incorporating bipolar gradient pulses, incrementing the gradient strength (G) in 16 steps from 5% to 95% by following a sine-shaped function. A diffusion time of 100 ms and a gradient pulse duration of 4 ms were used. TT-Py was synthetized as reported in the literature.3
4.2. Synthesis of the complexes
4.3. Crystal structure analysis
Single-crystal X-ray diffraction data were collected using a Bruker ApexII CCD diffractometer using graphite-monochromated Mo Kα radiation (λ = 0.71073 Å). Low-temperature data collection was performed using a Cryostream 600 system (Oxford Cryosystems). For all the species, a full sphere of reciprocal space was collected, granting data completeness up to at least (sinθ)/λ = 0.625 Å−1. CCDC 2214632 and 2214633 contain the supplementary crystallographic data for the paper.† These data can be obtained free of charge from the Cambridge Crystallographic Data Centre.
4.4. Electrochemical characterization
Electrochemical investigation into complexes 1 and 2 was carried out by cyclic voltammetry at 0.2 V s−1 (and also at different scan rates in the case of stable complex 1), in CH3CN at a 2.5 × 10−4 M concentration with 0.1 M tetrabutylammonium hexafluorophoshate TBAPF6 (Aldrich) as the supporting electrolyte, in a glass minicell (working volume 3 cm3), at 298 K. The working electrode was a GC disk (AMEL, diameter 3 mm), a Pt wire acts as the counter electrode and an aqueous saturated calomel electrode SCE acts as the operating reference electrode, inserted in a glass compartment ending with a porous frit and filled with the working medium, to prevent KCl and water leakage from the reference electrode into the working solution. Nitrogen was bubbled through a presaturator filled with the working solvent and then into the working cell in order to remove oxygen from the solution; during measurements, it was kept above the working solution. The reversible CV pattern of the intersolvental reference redox couple ferricinium|ferrocene was recorded under the same operating conditions, determining its E°′Fc+|Fc formal potential as the forward and backward peak averages, to which the potentials were afterwards referred, thus eliminating the intersolvental junction potential. The measurements were carried out using an Autolab potentiostat managed using a PC with GPES software. The ohmic drop was corrected by the positive feedback method.4.5. Photophysical characterization
Photophysical measurements were carried out in air-equilibrated toluene solutions at room temperature or in the solid state. Electronic absorption spectra of solutions were recorded using an Agilent Model 8543 spectrophotometer at room temperature, using quartz cells with a 1.0 cm path length. Steady-state emission spectra were recorded using an Edinburgh FLS980 spectrometer equipped with a 450 W ozone-free xenon arc lamp, double grating excitation and emission monochromators (2 × 300 mm focal length) and a Hamamatsu R928P photomultiplier tube. Time-resolved measurements were performed using the time-correlated single-photon counting (TCSPC) option on the FLS980, using a microsecond flash Xe-lamp as the excitation source (60 W, 0.1/100 Hz). The emission was collected using a multichannel plate MCP-PMT Hamamatsu H10720-01 single-photon-counting detector. The photons collected at the detector were correlated using a time-to-amplitude converter (TAC) to the excitation pulse. The data analysis was performed using the commercially available F980 software (Edinburgh Instruments). The goodness of the data fitting was assessed by minimizing the reduced chi-squared function (χ2). Photoluminescence quantum yields (Φ) were collected for an optically diluted solution (<10−5 M) using wavelength scanning with a Hamamatsu C11347-11 Quantaurus-QY absolute PL quantum yield spectrometer, equipped with a xenon light source (150 W), a monochromator, and a Spectralon integrating sphere, and employing the commercially available U6039-05 PLQY measurement software (Hamamatsu Photonics Ltd, Shizuoka, Japan). The photoluminescence quantum yields were measured by exciting the samples between 340 and 400 nm.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work is supported by the Università Degli Studi di Milano (projects PSR2021_DIP_005_PI_CDPIN and PSR2021_DIP_005_PI_DCARL). The use of instrumentation purchased through the Regione Lombardia-Fondazione Cariplo joint SmartMatLab Project is gratefully acknowledged.References
- For OLED devices see for instance: (a) H.-T. Mao, G.-F. Li, G.-G. Shan, X.-L. Wang and Z.-M. Su, Coord. Chem. Rev., 2020, 413, 213283 CrossRef CAS; (b) X. Wu, M. Zhu, D. W. Bruce, W. Zhu, T. Fleetham, G. Li and J. Li, Adv. Mater., 2017, 29, 1601861 CrossRef PubMed ; For imaging and sensing see for instance: ; (c) C.-C. L. Lee and K.-W. K. Lo, J. Am. Chem. Soc., 2022, 144, 14420 CrossRef PubMed; (d) C.-C. L. Lee and K.-W. K. Lo, Chem. Asian J., 2022, 17, e202200840 CrossRef PubMed ; For solar energy conversion see for instance: ; (e) A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed ; For photocatalysis see for instance: ; (f) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- (a) E. Cariati, A. Forni, E. Lucenti, D. Marinotto, A. Previtali, S. Righetto, C. Botta, V. Bold, V. C. Kravtsov and M. S. Fonari, Chem. – Asian J., 2019, 14, 853–858 CrossRef CAS PubMed; (b) M. S. Fonari, V. C. Kravtsov, V. Bold, E. Lucenti, E. Cariati, D. Marinotto and A. Forni, Cryst. Growth Des., 2021, 21, 4184–4200 CrossRef CAS; (c) E. Lucenti, E. Cariati, A. Previtali, D. Marinotto, A. Forni, V. Bold, V. C. Kravtsov, M. S. Fonari, S. Galli and L. Carlucci, Cryst. Growth Des., 2019, 19, 1567–1575 CrossRef CAS; (d) D. Malpicci, E. Lucenti, A. Forni, D. Marinotto, A. Previtali, L. Carlucci, P. Mercandelli, C. Botta, S. Righetto and E. Cariati, Inorg. Chem. Front., 2021, 8, 1312–1323 RSC; (e) E. Melnic, V. C. Kravtsov, E. Lucenti, E. Cariati, A. Forni, N. Siminel and M. S. Fonari, New J. Chem., 2021, 45, 9040–9052 RSC.
- E. Lucenti, A. Forni, A. Previtali, D. Marinotto, D. Malpicci, S. Righetto, C. Giannini, T. Virgili, P. Kabacinski and L. Ganzer, Chem. Sci., 2020, 11, 7599–7608 RSC.
- D. Malpicci, D. Blasi, D. Marinotto, A. Forni, E. Cariati, E. Lucenti and L. Carlucci, Crystals, 2023, 13, 149 CrossRef CAS.
- (a) A. Vogler and H. Kunkely, Coord. Chem. Rev., 2000, 200–202, 991–1008 CrossRef CAS; (b) A. Kumar, S.-S. Sun and A. J. Lees, Top. Organomet. Chem., 2010, 29, 1–35 CAS; (c) A. Vlček, Top. Organomet. Chem, 2010, 29, 73–114 CrossRef.
- M. Wrighton and D. L. Morse, J. Am. Chem. Soc., 1974, 96, 998–1003 CrossRef CAS.
- (a) C.-C. Ko, A.-W.-Y. Cheung, L.-T.-L. Lo, J.-W.-K. Siu, C.-O. Ng and S.-M. Yiu, Coord. Chem. Rev., 2012, 256, 1546–1555 CrossRef CAS; (b) H. Takeda, K. Koike, T. Morimoto, H. Inumaru, O. Ishitani, R. V. Eldik and G. Stochel, Adv. Inorg. Chem., 2011, 137–186 CrossRef CAS; (c) C. Daniel, Coord. Chem. Rev., 2015, 282–283, 19–32 CrossRef CAS; (d) A. Vlcek and S. Zalis, Coord. Chem. Rev., 2007, 251, 258–287 CrossRef CAS; (e) R. A. Kirgan, B. P. Sullivan and D. P. Rillema, Top. Curr. Chem., 2007, 281, 45–100 CrossRef CAS; (f) M. Panigati, M. Mauro, D. Donghi, P. Mercandelli, P. Mussini, L. De Cola and G. D’Alfonso, Coord. Chem. Rev., 2012, 256, 1621–1643 CrossRef CAS.
- (a) K. K.-W. Lo, K. Y. Zhang and S. P.-Y. Li, Eur. J. Inorg. Chem., 2011, 3551–3568 CrossRef CAS; (b) K. K.-W. Lo, Acc. Chem. Res., 2015, 48, 2985–2995 CrossRef CAS PubMed; (c) V. Fernandez-Moreira, F. L. Thorp-Greenwood and M. P. Coogan, Chem. Commun., 2010, 46, 186 RSC; (d) K. K.-W. Lo, Top. Organomet. Chem., 2010, 29, 115 CrossRef CAS; (e) E. A. Hillard and G. Jaouen, Organometallics, 2011, 30, 20 CrossRef CAS; (f) S. Cauteruccio, E. Licandro, M. Panigati, G. D’Alfonso and S. Maiorana, Coord. Chem. Rev., 2019, 386, 119–137 CrossRef CAS.
- (a) G.-W. Zhao, J.-H. Zhao, Y.-X. Hu, D.-Y. Zhang and X. Li, Synth. Met., 2016, 212, 131–141 CrossRef CAS; (b) For OLED devices see for instance: X. Li, D. Zhang, H. Chi, G. Xiao, Y. Dong, S. Wu, Z. Su, Z. Zhang, P. Lei, Z. Hu and W. Li, Appl. Phys. Chem., 2010, 97, 263303 Search PubMed; (c) For polymer light emitting devices see for instance: L. Qian, D. Bera and P. H. Holloway, Appl. Phys. Lett., 2007, 90, 103511 CrossRef; (d) M. Mauro, C.-H. Yang, C.-Y. Shin, M. Panigati, C.-H. Chang, G. D’Alfonso and L. De Cola, Adv. Mater., 2012, 24, 2054–2058 CrossRef CAS PubMed.
- (a) M. Schulz, M. Karnahl, M. Schwalbe and J. G. Vos, Coord. Chem. Rev., 2012, 256, 1682–1705 CrossRef CAS; (b) A. Zarkadoulas, E. Koutsouri, C. Kefalidi and C. A. Mitsopoulou, Coord. Chem. Rev., 2015, 304–305, 55–72 CrossRef CAS.
- (a) C. Sun, S. Prosperini, P. Quagliotto, G. Viscardi, S. S. Yoon, R. Gobetto and C. Nervi, Dalton Trans., 2016, 45, 14678–14688 RSC; (b) S. Sung, D. Kumar, M. Gil-sepulcre and M. Nippe, J. Am. Chem. Soc., 2017, 139, 13993–13996 CrossRef CAS PubMed; (c) D. A. Popov, J. M. Luna, N. M. Orchanian, R. Haiges and C. A. D. C. S. C. Marinescu, Dalton Trans., 2018, 47, 17450–17460 RSC; (d) R. Kamata, H. Kumagai, Y. Yamazaki, G. Sahara and O. Ishitani, ACS Appl. Mater. Interfaces, 2019, 11, 5632–5641 CrossRef CAS PubMed; (e) S. Yang, W. Hu, X. Zhang, P. He, B. Pattengale, C. Liu, M. Cendejas, I. Hermans, X. Zhang and J. Zhang, et al. , J. Am. Chem. Soc., 2018, 140, 14614–14618 CrossRef CAS PubMed; (f) E. Quartapelle Procopio, A. Boni, L. Veronese, M. Marcaccio, P. Mercandelli, G. Valenti, M. Panigati and F. Paolucci, ChemElectroChem, 2021, 8, 2065–2069 CrossRef CAS.
- (a) A. S. Polo, M. K. Itokazu and N. Y. Murakami Iha, Coord. Chem. Rev., 2004, 248, 1343–1361 CrossRef CAS; (b) L. Veronese, E. Quartapelle Procopio, T. Moehl, M. Panigati, K. Nonomura and A. Hagfeldt, Phys. Chem. Chem. Phys., 2019, 21, 7534 RSC.
- L. C.-C. Lee, K.-K. Leung and K. K.-W. Lo, Dalton Trans., 2017, 46, 16357 RSC.
- (a) R. Q. Snurr, J. T. Hupp and S. T. Nguyen, AIChE J., 2004, 50, 1090–1095 CrossRef CAS; (b) S. T. Nguyen, D. L. Gin, J. T. Hupp and X. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 11849–11850 CrossRef CAS PubMed; (c) P. H. Dinolfo and J. T. Hupp, Chem. Mater., 2001, 13, 3113–3125 CrossRef CAS.
- D. Gupta and M. Sathiyendiran, Chemistry Select, 2018, 3, 7439–7458 CAS.
- R. Carballo, E. Garcia-Martinez, G. Pereiras-Gabian and E. M. Vazquez-Lopez, Z. Naturforsch., 2003, 58b, 1021 CrossRef.
- E. W. Abel, G. B. Hargreaves and G. Wilkinson, J. Chem. Soc., 1958, 3149 RSC.
- Two polymorphs and a pseudopolymorph of the free ligand have been characterized, see ref 3.
- A. C. C. Wong, G. Wilkinson, B. Hussain, M. Motevalli and M. B. Hursthouse, Polyhedron, 1988, 7, 1363–1370 CrossRef CAS.
- J. Mukiza, T. I. A. Gerber, E. C. Hosten and R. Betz, Z. Kristallogr. - New Cryst. Struct., 2014, 229, 355–356 CAS.
- M. Magni, E. Lucenti, A. Previtali, P. R. Mussini and E. Cariati, Electrochim. Acta, 2019, 317, 272–280 CrossRef CAS.
- It is worthwhile noting that the electrochemical characterization of free ligand was performed in dimethyl formamide, unlike the present study in acetonitrile; however, in both cases potentials were normalized vs the formal potential of the intersolvental reference standard Fc+|Fc measured in the same conditions, which eliminates offsets related to differences in the intersolvental junction potentials; moreover, the effects of solvents DMF and CH3CN in electrode processes are usually similar.
- Complex 1 is stable in the working CH3CN + 0.1 M TBAPF6 medium and its electrode processes result in negligible surface conditioning, a favourable feature enabling to record many subsequent CV patterns with no need of frequent electrode polishing.
- It is worthwhile underlining that, although CV patterns have been normalized by concentration besides electrode surface and square root of potential scan rate, we would refrain from a quantitative comparison of complex 2, complex 1 and ligand peak current densities for a deeper insight in the electrode process mechanisms. In fact, besides some concentration uncertainty due to the very small weighed quantities and/or to stability/solubility issues, diffusion coefficients should remarkably decrease from ligand to complex 1 and complex 2, resulting in significantly lower current densities at equal concentration; and in the case of complex 2 also issues concerning simultaneous availability of all redox active sites should be taken into consideration.
- J. V. Caspar and T. J. Meyer, J. Phys. Chem., 1983, 87, 952–957 CrossRef CAS.
- (a) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC; (b) J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429–5479 CrossRef CAS PubMed.
- (a) A. Poirot, C. Vanucci-Bacqué, B. Delavaux-Nicot, N. Leygue, N. Saffon-Merceron, F. Alary, F. Bedos-Belval, E. Benoist and S. Fery-Forgues, Dalton Trans., 2021, 50, 13686 RSC; (b) J. Wang, A. Poirot, B. Delavaux-Nicot, M. Wolff, S. Mallet-Ladeira, J. P. Calupitan, C. Allain, E. Benoista and S. Fery-Forgues, Dalton Trans., 2019, 48, 15906 RSC.
- B. Manimaran, P. Thanasekaran, T. Rajendran, R.-J. Lin, I.-J. Chang, G.-H. Lee, S.-M. Peng, S. Rajagopal and K.-L. Lu, Inorg. Chem., 2002, 41, 5323–5325 CrossRef CAS PubMed.
- (a) P. Thanasekaran, C.-C. Lee and K.-L. LU, Acc. Chem. Res., 2012, 45, 1403–1418 CrossRef CAS PubMed; (b) R. Nagarajaprakash, R. Govindarajan and B. Manimaran, Dalton Trans., 2015, 44, 11732 RSC.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2214632 and 2214633. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3nj02823b |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2023 |