
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructure elucidation of 4-carboxy-3-deoxyanthocyanidins formed from thermal degradation of hydroxyphenyl-pyranoanthocyanins†
Danielle M.
Voss
 a,
Fenfen
Tang
a,
Kenneth M.
Riedl
abc,
Gonzalo
Miyagusuku-Cruzado
a,
Siyu
Yao
a,
Luis
Rodriguez-Saona
a,
Emmanuel
Hatzakis
a and
M. Monica
Giusti
*a
a,
Fenfen
Tang
a,
Kenneth M.
Riedl
abc,
Gonzalo
Miyagusuku-Cruzado
a,
Siyu
Yao
a,
Luis
Rodriguez-Saona
a,
Emmanuel
Hatzakis
a and
M. Monica
Giusti
*a
aThe Ohio State University, Department of Food Science and Technology, 2015 Fyffe Rd., Columbus, OH 43210, USA. E-mail: giusti.6@osu.edu; Fax: +1 614-292-0218; Tel: +1 614-247-8016
bThe Ohio State University Comprehensive Cancer Center, 2015 Fyffe Rd., Columbus, OH 43210, USA
cBeaconPoint Labs, 150 N Research Campus Dr, Kannapolis, NC 28081, USA
First published on 18th October 2023
Abstract
Hydroxyphenyl-pyranoanthocyanins are vibrantly colored, anthocyanin-derived polyphenols found in some wines and aged juices. Upon heating (90 °C), solutions of 10-catechyl-pyranoanthocyanins (a type of hydroxyphenyl-pyranoanthocyanins) formed a unique, color producing degradation compound. This compound was produced in the greatest quantities at pH 1 and pH 3 where its formation contributed to retention of color intensity despite extensive heating conditions (up to 34 hours). We elucidated the structure of this colored compound and investigated if other hydroxyphenyl-pyranoanthocyanins produced similar colored thermal degradation products. Analysis of the isolated 10-catechyl-pyranoanthocyanin degradation compound by uHPLC-PDA, high resolution MS/MS (QTOF), FTIR, 1D and 2D NMR (1H, COSY, HMBC, HSQC), spectrophotometry, and color analysis revealed that it was a 4-carboxy-3-deoxyanthocyanidin with a catechol substitution, named 4-carboxy-3-deoxycyanidin. It produced an exact mass of 315.0502 consistent with a chemical formula of C16H11O7+. NMR allowed for structural assignments, and key FTIR discriminating bands supported the presence of aromatic rings and the carboxylic acid moiety. The color of 4-carboxy-3-deoxycyanidin transitioned from yellow (λvis-max = 479 nm) at pH 1 to magenta (λvis-max = 545 nm) at pH 9.2, analogous with a flavylium system. Similarly, unique, colored degradation compounds formed from three other hydroxyphenyl-pyranoanthocyanins (10-p-hydroxyphenyl-, 10-guaiacyl-, and 10-syringyl-pyranoanthocyanins) each with a m/z 398 units lower than their parent pyranoanthocyanin. These findings revealed the formation of a unique class of pyranoanthocyanin derived flavylium compounds: 4-carboxy-3-deoxyanthocyanidins.
1. Introduction
Pyranoanthocyanins (PACNs) are a class of polyphenolic pigments found in aged red wine where they contribute to the characteristic tawny color.1–3 They have also been found in products such as aged juices4,5 and strawberries6 albeit in minor amounts. The chemical structure of a PACN is comprised of an anthocyanin backbone with a second pyran ring attached at the C4 and C5–OH group (Fig. 1). This ring can form through a multistep cycloaddition reaction between an anthocyanin and a reactive cofactor such as an enolizable compound or one with a vinyl group.2,7,8 Of the cofactors used for PACN formation, hydroxycinnamic acids and their decarboxylated 4-vinylphenol counterparts form PACNs most efficiently under accelerated conditions, producing appreciable yields of hydroxyphenyl-PACNs within several days.9,10 As efficient production methods continue to be developed, interest is growing in using hydroxyphenyl-PACNs as colorants in food and consumer packaged goods due to their vibrant color which is stable to heat,11,12 pH changes,13 and bleaching14 and their antioxidant capacity.13,15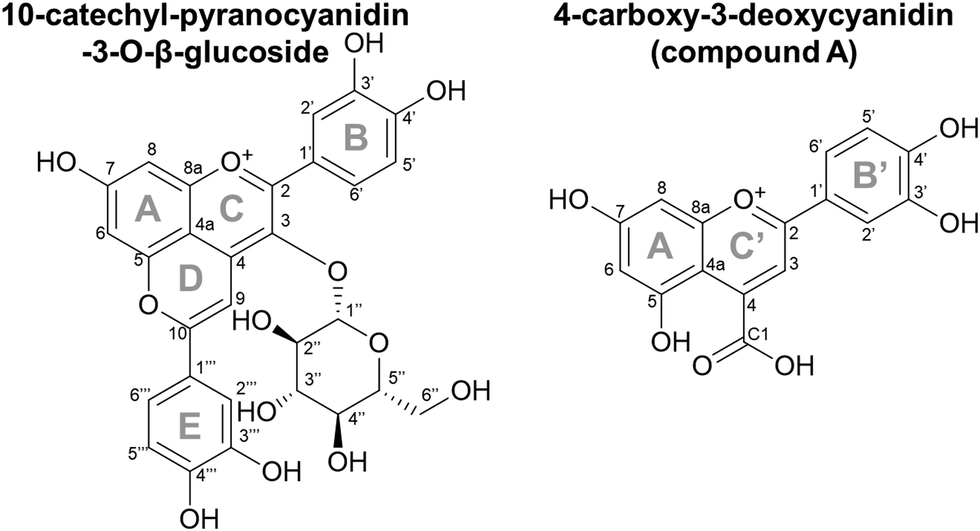 | ||
| Fig. 1 Chemical structures for 10-catechyl-pyranocyanidin-3-O-β-glucoside and 4-carboxy-3-deoxycyanidin (compound A). Ring B′ and C′ on compound A were derived from the E and D rings, respectively, of 10-catechyl-pyranocyanidin-3-O-β-glucoside. | ||
Recently, we reported unusual behavior of 10-catechyl-PACNs, a type of hydroxyphenyl-PACN, during thermal treatment. With heat, a unique degradation compound that absorbed light in the visible range (λvis-max = 478 nm) formed.11,12 Its formation likely contributed to the remarkable color stability to heat for 10-catechyl-PACNs, behavior contrasted with the rapid color loss with heat for the precursor anthocyanins and the formation of only colorless degradation compounds from 10-carboxy- and 10-methyl-PACNs.11,12 This unique colored degradation compound formed from 10-catechyl-PACNs irrespective of the core anthocyanin type (i.e. cyanidin vs. malvidin) and was thought to contain parts of the A, D, and E ring of the parent PACN,11,12 leading us to hypothesize that other hydroxyphenyl-PACNs could produce similar colored compounds.
Therefore, the objective of this experiment was to elucidate the chemical structure and evaluate the formation, color, and stability characteristics of the colored 10-catechyl-PACN degradation compound, referred to as compound A hereafter. In addition, we determined if the same type of compound is formed from three other hydroxyphenyl-PACNs. As hydroxyphenyl-PACNs have been found in products such as juice and wine4,5,16 and with consideration for their use as colorants for consumer goods, it is necessary to further identify the degradation compound(s) as they could potentially form in PACN-containing products.
2. Experimental section
2.1. Materials
Elderberry (Sambucus nigra) anthocyanin color powder from DDW The Color House (Louisville, KY, USA) was used as the source of anthocyanins (primarily comprised of cyanidin-3-glucoside and cyanidin-3-sambubioside).17 Caffeic acid, ferulic acid, and sinapic acid were obtained from TCI Chemicals (Portland, OR, USA). p-Coumaric acid was obtained from MP Biomedicals (Solon, OH, USA). Luteolinidin chloride standard was obtained from Extrasynthese (Genay, France). Deuterated MeOH was obtained from Acros Organics (Fair Lawn, NJ, USA).2.2. Hydroxyphenyl-pyranoanthocyanin formation, isolation, and thermal degradation compound detection
Hydroxyphenyl-PACNs were formed following the procedure in Miyagusuku-Cruzado et al. with slight modifications.9 Briefly, elderberry anthocyanins (∼500 μM final concentration) were mixed with p-coumaric acid, caffeic acid, ferulic acid, or sinapic acid at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30 M ratio in water adjusted to a pH of 3.1 with HCl with 0.1% potassium sorbate and 0.1% sodium benzoate preservatives. The solutions were incubated (45 °C, ∼30 days) to form 10-p-hydroxyphenyl-, 10-catechyl-, 10-guaiacyl-, and 10-syringyl-PACNs, respectively. Incubation ended when there were little to no anthocyanins remaining. Chemical yields for PACN formation were calculated based on the PDA peak area for the PACNs at the end of the incubation period divided by the initial peak are for the ACNs as described in Miyagusuku-Cruzado et al.9 The formed hydroxyphenyl-PACNs were semi-purified with solid phase extraction on C18 cartridges18 and isolated using semi-preparatory HPLC. A Shimadzu Prominence semi-preparatory HPLC comprised of two LC-6AD pumps, CBM-20A controller, SIL-20A autosampler, and SPD-M20A PDA detector was used (Columbia, MD, USA). Separation conditions included a 12 mL min−1 flow rate under gradient conditions of A: 4.5% formic acid in H2O and B: acetonitrile and a Synergi Max-RP 80 Å (250 mm × 21.2 mm i.d., 4 μm) or Luna PFP 100 Å (250 mm × 21.2 mm i.d., 5 μm) column (both from Phenomenex, Torrence, CA, USA). From the collected and diluted eluate, HPLC solvents were removed with solid phase extraction.
:30 M ratio in water adjusted to a pH of 3.1 with HCl with 0.1% potassium sorbate and 0.1% sodium benzoate preservatives. The solutions were incubated (45 °C, ∼30 days) to form 10-p-hydroxyphenyl-, 10-catechyl-, 10-guaiacyl-, and 10-syringyl-PACNs, respectively. Incubation ended when there were little to no anthocyanins remaining. Chemical yields for PACN formation were calculated based on the PDA peak area for the PACNs at the end of the incubation period divided by the initial peak are for the ACNs as described in Miyagusuku-Cruzado et al.9 The formed hydroxyphenyl-PACNs were semi-purified with solid phase extraction on C18 cartridges18 and isolated using semi-preparatory HPLC. A Shimadzu Prominence semi-preparatory HPLC comprised of two LC-6AD pumps, CBM-20A controller, SIL-20A autosampler, and SPD-M20A PDA detector was used (Columbia, MD, USA). Separation conditions included a 12 mL min−1 flow rate under gradient conditions of A: 4.5% formic acid in H2O and B: acetonitrile and a Synergi Max-RP 80 Å (250 mm × 21.2 mm i.d., 4 μm) or Luna PFP 100 Å (250 mm × 21.2 mm i.d., 5 μm) column (both from Phenomenex, Torrence, CA, USA). From the collected and diluted eluate, HPLC solvents were removed with solid phase extraction.
For degradation compound analysis, isolates of either 10-p-hydroxyphenyl-pyranocyanidin-3-sambubioside, 10-catechyl-pyranocyanidin-3-sambubioside, 10-guaiacyl-pyranocyanidin-3-sambubioside, or 10-syringyl-pyranocyanidin-3-sambubioside were mixed in water with a small concentration of MeOH to aid in solubility, and the pH was adjusted to 3.0 ± 0.8 and absorbance of ∼1 at λvis-max. The samples were heated in a 90 °C water bath for 15 hours as performed in Voss et al. with each sample heated in duplicate.11 Pigment identities and their degradation compound were identified using a Nexera-I LC2040 ultra high performance liquid chromatography system with photodiode array detection run tandem to a LCMS 8040 triple quadrupole mass spectrometer with electrospray ionization (uHPLC-PDA-ESI-MS/MS; Shimadzu, Columbia, MD, USA). Chromatographic separation was achieved with a Restek Pinnacle DB IBD column (50 mm × 2.1 mm i.d., 1.9 μm; Bellefonte, PA, USA), 50 °C column oven, and a binary gradient of solvent A: 4.5% formic acid in H2O and solvent B: acetonitrile at a 0.25 mL min−1 flow at the following percentages: 0–20% B from 0 to 12 min, 20–35% B from 12 to 15 min, and 35–45% B from 15 to 16 minutes followed by column equilibration. MS/MS was conducted in positive ion mode with 2 L min−1 nebulizing gas, 11 L min−1 drying gas, 230 °C desolvation line, and 200 °C heat block. Key MS/MS programs included selective ion monitoring, product ion scans (35 eV collision energy), and a neutral loss scan for m/z of 294 (35 eV collision energy) for the sambubioside moiety. Data was processed using LabSolutions (v.5.80; Shimadzu, Columbia, MD, USA).
2.3. Compound A formation across pH values
Isolated 10-catechyl-pyranocyanidin-3-sambubioside (Section 2.2) was heated for up to 34 hours at four pH values to monitor the kinetic rate of PACN degradation and compound A formation. A stock solution of 10-catechyl-pyranocyanidin-3-sambubioside was prepared with 50% MeOH and 50% H2O and aliquoted at a dilution factor 15.04 into buffers: pH 1 (0.025 M KCl adjusted with HCl), pH 3.0 (citric acid–Na2HPO4), pH 5.0 (citric acid–Na2HPO4), and pH 7.0 (citric acid–Na2HPO4) all within ± 0.1 pH unit. The PACN-buffer solutions were distributed into brown microcentrifuge tubes, flushed with N2, and placed in a water bath set to 90 °C. At each heating time point, a microcentrifuge tube was removed from heat, chilled in ice water for 2 minutes, and then let sit at room temperature for 2 minutes prior to measurements. Heating was stopped after degradation (or formation) rates plateaued or after 34 hours (pH 3).After removing from heat, the samples were spiked with HCl to decrease the pH below 1 prior to HPLC-PDA analysis. Acidification favored the flavylium cation for both PACN and compound A thus removing the effect of pH on quantification. HPLC-PDA analysis was performed with a Shimadzu Prominence HPLC with two LC-20AD pumps, CBM-20A controller, SPD-M20A PDA detector, SIL-20AC autosampler, and CTO-20A column oven (Columbia, MD, USA). Separation was achieved with a Kinetex EVO C18 column (150 mm × 4.6 mm i.d., 5 μm; Phenomenex, Torrance, CA, USA) and a binary gradient of solvent A: 4.5% formic acid H2O and solvent B: 4.5% formic acid acetonitrile at a 1.0 mL min−1 flow rate. The following gradient was used: 0–37% B from 0 to 10 min followed by 9 minutes of column equilibration. The peak areas of the PACNs (starting 10-catechyl-pyranocyanidin-3-sambubioside and formed 10-catechyl-pyranocyanidin-3-glucoside) and compound A were integrated using the 450–550 nm max plot. Each sample was injected in duplicate, and the mean values were used for calculations and analyses. Linear regression analysis of PACN degradation and compound A formation was performed using GraphPad Prism (v.10.0.2). A first order-kinetic fit was used to model PACN degradation. Either a first (pH 1) or zero (pH 3) order model was used for compound A formation with the best model selected based on a sum of squares F-test. Yields of compound A were calculated using the following formula:
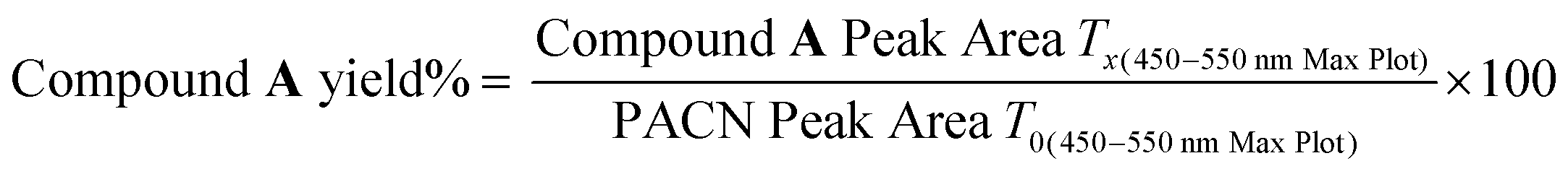
 (chroma), hab (hue angle) color coordinates under D65 illuminant and 10° observer angle using ColorBySpectra software.19 Thermal heating was performed with 3 experimental replicates but pH 3, 34 hour and pH 5, 2 hour measurements had only 2 replicates.
(chroma), hab (hue angle) color coordinates under D65 illuminant and 10° observer angle using ColorBySpectra software.19 Thermal heating was performed with 3 experimental replicates but pH 3, 34 hour and pH 5, 2 hour measurements had only 2 replicates.
2.4. Formation and isolation of compound A for structure elucidation
Large amounts of compound A were needed for structure elucidation. For this purpose, formed 10-catechyl-PACNs (from Section 2.2) were semi-purified to remove remaining ACNs, cofactors, and preservatives (potassium sorbate and sodium benzoate) present from the PACN formation process using cation exchange chromatography20 and solid phase extraction on C18 cartridges.18 This semi-purified solution of 10-catechyl-PACNs was adjusted to a pH of 3.0 ± 0.05 with HCl and heated for up to 22 hours in a water bath set to 90 °C. The heated PACNs were filtered to remove the formed precipitate and semi-purified again with solid phase extraction, and formed compound A was isolated with semi preparatory HPLC as previously described. The semi-preparatory HPLC eluate containing compound A was processed and dried. For high resolution MS, nuclear magnetic resonance (NMR), and spectrophotometric and color analyses, the HPLC eluate containing compound A was processed using solid phase extraction and then dried with a Vacufuge Plus vacufuge (Eppendorf, Enfield, CT, USA). For Fourier transform infrared (FTIR) analysis, the eluate was dried directly using N2 evaporation.2.5. Structure elucidation and characterization of compound A
A multipronged approach was employed to determine the chemical structure and the color characteristics of compound A including uHPLC-PDA, high resolution mass spectrometry, 1D and 2D NMR, FTIR, spectrophotometry, and color measurement. Prior to each analysis, the identity and purity of compound A was confirmed with the uHPLC-PDA-ESI-MS/MS system, column, column oven temperature, flow rate, and mobile phase solvents described in Section 2.2. Peak purity was determined based on the equation below: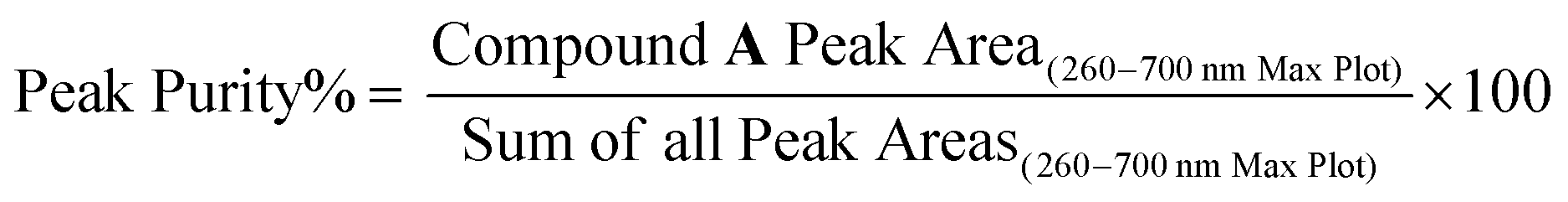 000 resolution and 3 Hz. For MS/MS acquisition, both 6 and 3 Hz were used with 10, 20, and 40 eV collision energy. Profile mass data was acquired and centroided using MassHunter Acq software (ver.B.09) and analyzed with MassHunter Qual (ver.B.10; Agilent Technologies, Santa Clara, CA, USA). Calibration of the mass axis was performed using ESI-L solution with HP321 for positive ion mode (Agilent Technologies, Santa Clara, CA, USA), and within-run calibration used m/z 1033 from HP921 and m/z 112 from trifluoroacetic acid. SIRIUS software (v.4) was used for determination of the chemical formula from the exact mass.21
000 resolution and 3 Hz. For MS/MS acquisition, both 6 and 3 Hz were used with 10, 20, and 40 eV collision energy. Profile mass data was acquired and centroided using MassHunter Acq software (ver.B.09) and analyzed with MassHunter Qual (ver.B.10; Agilent Technologies, Santa Clara, CA, USA). Calibration of the mass axis was performed using ESI-L solution with HP321 for positive ion mode (Agilent Technologies, Santa Clara, CA, USA), and within-run calibration used m/z 1033 from HP921 and m/z 112 from trifluoroacetic acid. SIRIUS software (v.4) was used for determination of the chemical formula from the exact mass.21
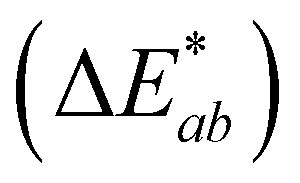 was calculated from the L*, a*, and b* coordinates, and a
was calculated from the L*, a*, and b* coordinates, and a  was the cutoff for an obvious color change.23 For data analysis, the results were evaluated as means ± standard deviation of the 9 data points (3 experimental replicates × 3 technical replicates) to represent the spread of the data. A 1-way nested ANOVA with Tukey's multiple comparison test was used to evaluate the effect of pH on each color coordinate at time 0 with a p value <0.05 considered statistically significant. Absorbance stability over time was evaluated by comparing the percent change in absorbance intensity at the initial λvis-max for each technical replicate. GraphPad Prism (v.9.5.1) was used for statistical analysis and graphing (Boston, MA, USA).
was the cutoff for an obvious color change.23 For data analysis, the results were evaluated as means ± standard deviation of the 9 data points (3 experimental replicates × 3 technical replicates) to represent the spread of the data. A 1-way nested ANOVA with Tukey's multiple comparison test was used to evaluate the effect of pH on each color coordinate at time 0 with a p value <0.05 considered statistically significant. Absorbance stability over time was evaluated by comparing the percent change in absorbance intensity at the initial λvis-max for each technical replicate. GraphPad Prism (v.9.5.1) was used for statistical analysis and graphing (Boston, MA, USA).
3. Results and discussion
3.1. Formation of compound A across pH
The thermal stability of 10-catechyl-pyranocyanidin-3-sambubioside and the formation rates of compound A were dependent on the pH of the solution. From pH 1 to 7, PACNs degraded following a first-order kinetic model. Compound A formed at these pH values; however, formation was only modeled at pH 1 (first order) and pH 3 (zero order) due to both increasing and decreasing concentrations through heating at pH 5 and 7 (Fig. 2). Assessment of pH 9 was not included because preliminary work did not detect the formation of compound A at that pH. At pH 1 and 3, heating 10-catechyl-pyranocyanidin-3-sambubioside resulted in partial acid hydrolysis to produce 10-catechyl-pyranocyanidin-3-glucoside (revealed by uHPLC-PDA-ESI-MS/MS analysis) (Fig. S1, ESI†). Both peak areas were included in the PACN degradation models. At pH 1, total PACN loss coincided with the formation of compound A as both compounds reached their kinetic plateau at ∼12 hours (Fig. 2). In the subsequent 15 hours of heating there were only slight changes in compound A levels with largest yields obtained at the final time point of 27 hours (22.3 ± 1.1%). At pH 3, 10-catechyl-pyranocyanidin-3-sambubioside showed the greatest thermal stability, with some PACNs remaining with 34 hours of 90 °C heat (Fig. 2). Concentration of compound A continued to increase at the final time point of 34 hours with a yield of 17.9 ± 0.7%. As no concentration plateau was reached within this period, formation was modeled with a zero-order kinetic model. At pH 5, some 10-catechyl-pyranocyanidin-3-sambubioside remained by 27 hours of heating. The levels of compound A did not inversely mirror that of PACN degradation, however (Fig. 2). The greatest yields of compound A (7.7 ± 0.2%) were measured at 8 hours with yields decreasing over the subsequent 19 hours of heating. At pH 7, 10-catechyl-pyranocyanidin-3-sambubioside showed the lowest thermal stability, decreasing to barely detectable levels by 12 hours of heating. Only a small amount of compound A formed (highest yield of 1.0 ± 0.2% measured at 1 hour), but it was no longer detectable at 12 hours. Protocatechuic acid was identified to form with heating at each pH value (1, 3, 5, 7) by comparison of retention time and PDA spectra with an analytical standard. | ||
| Fig. 2 Changes in PDA peak area (from 450–550 nm max plot) for pyranoanthocyanins (PACN) including starting 10-catechyl-pyranocyanidin-3-sambubioside and formed 10-catechyl-pyranocyanidin-3-glucoside (pH 1 & 3) and compound A with 90 °C heat. Data points show mean ± standard deviation. Table shows results of kinetic model for PACN degradation and compound A formation. | ||
The stability of solution color during heating was influenced both by the levels of PACN and compound A. Before heating, all four solutions of 10-catechyl-pyranocyaindin-3-sambubioside were orange in color (hab = 44–46°) with color intensity and λvis-max decreasing slightly with increasing pH. With 90 °C heat, all solutions lost orange color intensity marked by increasing L* and decreasing 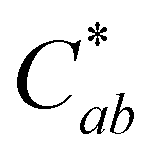 and absorption at λvis-max (Fig. 3). The pH 5 and 7 solutions were very pale at the final measured time point (27 and 12 hours, respectively) with
and absorption at λvis-max (Fig. 3). The pH 5 and 7 solutions were very pale at the final measured time point (27 and 12 hours, respectively) with 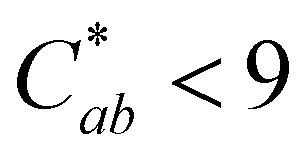 (Fig. 3). At this point, these solutions had low levels of color producing compounds (PACNs and compound A; Fig. S1, ESI† and Fig. 2). However, for the solutions at pH 1 and pH 3, the formation of appreciable quantities of compound A mitigated the overall color loss. A clear hypsochromic shift in λvis-max (501 to ∼481 nm) and increase in hab by ∼23° occurred (Fig. 3), coinciding with the formation trends of compound A in Fig. 2. For pH 1 solution, no detectable levels of PACNs remained after 12 hours, yet
(Fig. 3). At this point, these solutions had low levels of color producing compounds (PACNs and compound A; Fig. S1, ESI† and Fig. 2). However, for the solutions at pH 1 and pH 3, the formation of appreciable quantities of compound A mitigated the overall color loss. A clear hypsochromic shift in λvis-max (501 to ∼481 nm) and increase in hab by ∼23° occurred (Fig. 3), coinciding with the formation trends of compound A in Fig. 2. For pH 1 solution, no detectable levels of PACNs remained after 12 hours, yet 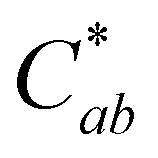 remained constant near 21 and the solution produced a yellow color over the subsequent 15 hours of heating, clearly demonstrating the important role of compound A on color retention in heated PACNs.
remained constant near 21 and the solution produced a yellow color over the subsequent 15 hours of heating, clearly demonstrating the important role of compound A on color retention in heated PACNs.
 | ||
Fig. 3 Absorbance and color change of 10-catechyl-pyranocyanidin-3-sambubioside solutions in four pH buffers with 90 °C heating. Spectra show mean absorbance values taken with a 5 nm step, and L*, 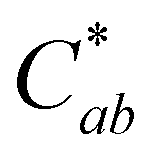 , and hab data points show mean ± standard deviation. , and hab data points show mean ± standard deviation. | ||
3.2. Identification of 10-catechyl-pyranocyanidin-3-O-β-glucoside with nuclear magnetic resonance
The identity and structure of 10-catechyl-pyranocyanidin-3-O-β-glucoside—a precursor to compound A—was confirmed, validating our PACN formation procedure, identification from uHPLC-PDA-MS, and NMR process. From LC-PDA-MS analysis, the λvis-max (505 nm) and m/z (581) matched the identified 10-catechyl-pyranocyanidin-3-glucoside in Voss et al. which reported a λvis-max of 505 nm and an exact mass of 581.1283.11 The 1H and 13C NMR assignments of 10-catechyl-pyranocyanidin-3-glucoside were conducted using various 1D and 2D NMR experiments (Table 1). For the sugar moiety, there was a doublet at 4.78 ppm that was attributed to the anomeric proton H1′′ of the sugar moiety. The 3J coupling value of 7.7 Hz and the chemical shift indicated the presence of a β sugar. This signal had a cross peak in the COSY spectrum with a doublet of doublets at 3.68 ppm, which was assigned to hydrogen H2′′ of the sugar ring.| 10-Catechyl-pyranocyanidin-3-O-β-glucoside | 4-Carboxy-3-deoxycyanidin (compound A) | ||||
|---|---|---|---|---|---|
| Atom number | δ 1H ppm, J (Hz) | δ 13C ppm | Atom number | δ 1H ppm, J (Hz) | δ 13C ppm |
| 2 | N/A | 162.4 | |||
| 3 | N/A | 134.4 | |||
| 4 | N/A | Not identified | 4 | N/A | Not identified |
| 4a | N/A | 108.0 | 4a | N/A | 108.0 |
| 5 | N/A | 153.3 | 8a | N/A | 160.0 |
| 6 | 7.06, d (1.3) | 100.6 | 8 | 6.95, d (2.1) | 96.4 |
| 7 | N/A | 168.4 | 7 | N/A | 171.7 |
| 8 | 7.10, d (1.3) | 101.2 | 6 | 6.63, d (2.1) | 103.9 |
| 8a | N/A | 154.2 | 5 | N/A | Not identified |
| 9 | 7.89, s | 97.6 | 3 | 8.00, s | 108.5 |
| 10 | N/A | 168.6 | 2 | N/A | 172.7 |
| 1′ | N/A | 121.6 | |||
| 2′ | 7.82, d (2.2) | 117.5 | |||
| 3′ | N/A | 145.9 | |||
| 4′ | N/A | 152.2 | |||
| 5′ | 6.94, d (8.5) | 116.0 | |||
| 6′ | 7.88, dd (8.5, 2.2) | 125.0 | |||
| 1′′ | 4.78, d (7.7) | 105.4 | |||
| 2′′ | 3.68, dd (7.7) | 74.8 | |||
| 3′′ | 3.40, m | 77.1 | |||
| 4′′ | 3.28, m | 71.2 | |||
| 5′′ | 3.15, m | 78.0 | |||
| 6′′ | 3.65 dd (11.4), 3.38 dd (11.4) | 62.2 | |||
| 1′′′ | N/A | 122.2 | 1′ | N/A | 121.3 |
| 2′′′ | 7.62, d (2.2) | 114.7 | 2′ | 7.76, d (2.1) | 115.6 |
| 3′′′ | N/A | 147.1 | 3′ | N/A | 148.2 |
| 4′′′ | N/A | 153.5 | 4′ | N/A | 156.7 |
| 5′′′ | 6.99, d (8.4) | 116.7 | 5′ | 7.04, d (8.6) | 117.3 |
| 6′′′ | 7.70 dd (8.4, 2.2) | 122.3 | 6′ | 7.92, dd (8.6, 2.1) | 125.4 |
| C1 (Carboxyl) | N/A | 168.5 | |||
Similarly, by combining the COSY and HMBC spectra, the signals of the remaining sugar hydrogens were assigned. The carbon chemical shifts of the sugar were assigned using the HSQC-DEPT and HMBC spectra, and the results are shown in Table 1. The 1H NMR spectrum of the PACN was characterized by the presence of nine protons in the aromatic area (Fig. S2, ESI†). The singlet proton at 7.89 ppm and the anomeric proton shared an HMBC correlation peak with a quaternary carbon (C3) at 134.4. Thus, this singlet was assigned to H9 in the D ring. In addition, as shown by the COSY analysis in Fig. S3 (ESI†), the protons at 7.88 and 6.94 ppm formed an AB system, while the protons 7.70 and 6.99 ppm formed a second AB system. Additional splitting indicated the presence of protons in three AX systems. The carbon signals were assigned by the HSQC-DEPT (Fig. S4, ESI†) and HMBC (Fig. S5, ESI†). For example, the quaternary carbon C3 of the C ring appeared at 134.4 ppm while the quaternary carbon C10 of the D ring appeared at 168.6 as assigned based on its HMBC correlations with H2′′′, H6′′′, and H9. These 1H and 13C NMR shifts are similar to those reported for similar molecules such as 10-catechyl-pyranomalvidin-3-glucoside and 10-catechyl-pyranoluteolinidin with exception of the known structural differences in the B ring substitutions and C3 position, respectively.13,24,25
3.3. Structure elucidation of compound A
The efficient formation method of 10-catechyl-PACNs from caffeic acid cofactor developed previously9 enabled the production of large quantities of 10-catechyl-PACNs and subsequently large quantities of compound A required for structure elucidation experiments. The isolated compound A formed for these experiments had the same LCMS characteristics (λvis-max = 478 ± 1 nm, m/z = 315) as the compound previously reported supporting the reproducibility of formation and the retention of the compound through isolation.11,12 Using a multifaceted approach including uHPLC-PDA, high resolution MS/MS, 1D and 2D NMR, FTIR, spectrophotometry, and color analyses, the chemical structure of the 10-catechyl-PACN degradation compound (compound A) was determined to correspond to 4-carboxy-2-(3,4-dihydroxyphenyl)-5,7-dihydroxychromenylium (Fig. 1). The compound is a 3-deoxyanthocyanidin with a carboxylic acid substitution at carbon 4. As the two ortho hydroxyl substitutions in the B′ ring are in the same position as observed for stable conformers of cyanidin,26 compound A was named 4-carboxy-3-deoxycyanidin. Detailed analyses and conclusions from each characterization step are reported below.The NMR spectra showed several shifts consistent with acyl chains. The main impurity signals are a triplet (0.896 ppm) that is typical for a methyl group of acyl chains and a broad signal at 1.29 ppm belonging to the methylene hydrogens of the chains. These signals have correlations in COSY further confirming that they belong to the same chains. These are believed to be contaminants from the alkyl C18 chains in the separatory cartridges used to concentrate compound A after semi-preparatory HPLC isolation. This was supported with FTIR analysis of the cartridge methanolic eluent, showing two small bands at ∼2922 and 2852 cm−1 consistent with CH stretching for CH3 and CH2 groups (data not shown).27–29 As they do not absorb UV or visible light, they did not appear on the 260–700 nm max plot from uHPLC-PDA analysis used to determine purity.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching of aromatic rings.27,30 The band at 1402 cm−1 may relate to symmetrical stretching of the COO− group as previous work has correlated these vibrations to bands ∼1380–1390 cm−1.31–33 While compound A was dried with acidic MeOH, it is possible for the carboxylic acid moiety to be present both in its protonated (COOH) and deprotonated (COO−) form as compound A is believed to have a low pKa (discussed in Section 3.3.4). Both the 1324 and 1275 cm−1 bands may relate to the C(phenolic)–O(H) vibrations.30–32,34 The band at 1151 cm−1 may relate to COC stretching on the C ring of compound A.30,34–36 The slight differences in our reported wavenumbers compared to previous FTIR analysis of phenolic compounds may be due to the unique structure of compound A. These observations of the FTIR spectrum suggested that the differentiating structural attributes between compound A and luteolinidin are related to substitutions on the aromatic ring, specifically the C′ ring, and supported the presence of a carboxylic acid moiety.
C stretching of aromatic rings.27,30 The band at 1402 cm−1 may relate to symmetrical stretching of the COO− group as previous work has correlated these vibrations to bands ∼1380–1390 cm−1.31–33 While compound A was dried with acidic MeOH, it is possible for the carboxylic acid moiety to be present both in its protonated (COOH) and deprotonated (COO−) form as compound A is believed to have a low pKa (discussed in Section 3.3.4). Both the 1324 and 1275 cm−1 bands may relate to the C(phenolic)–O(H) vibrations.30–32,34 The band at 1151 cm−1 may relate to COC stretching on the C ring of compound A.30,34–36 The slight differences in our reported wavenumbers compared to previous FTIR analysis of phenolic compounds may be due to the unique structure of compound A. These observations of the FTIR spectrum suggested that the differentiating structural attributes between compound A and luteolinidin are related to substitutions on the aromatic ring, specifically the C′ ring, and supported the presence of a carboxylic acid moiety.
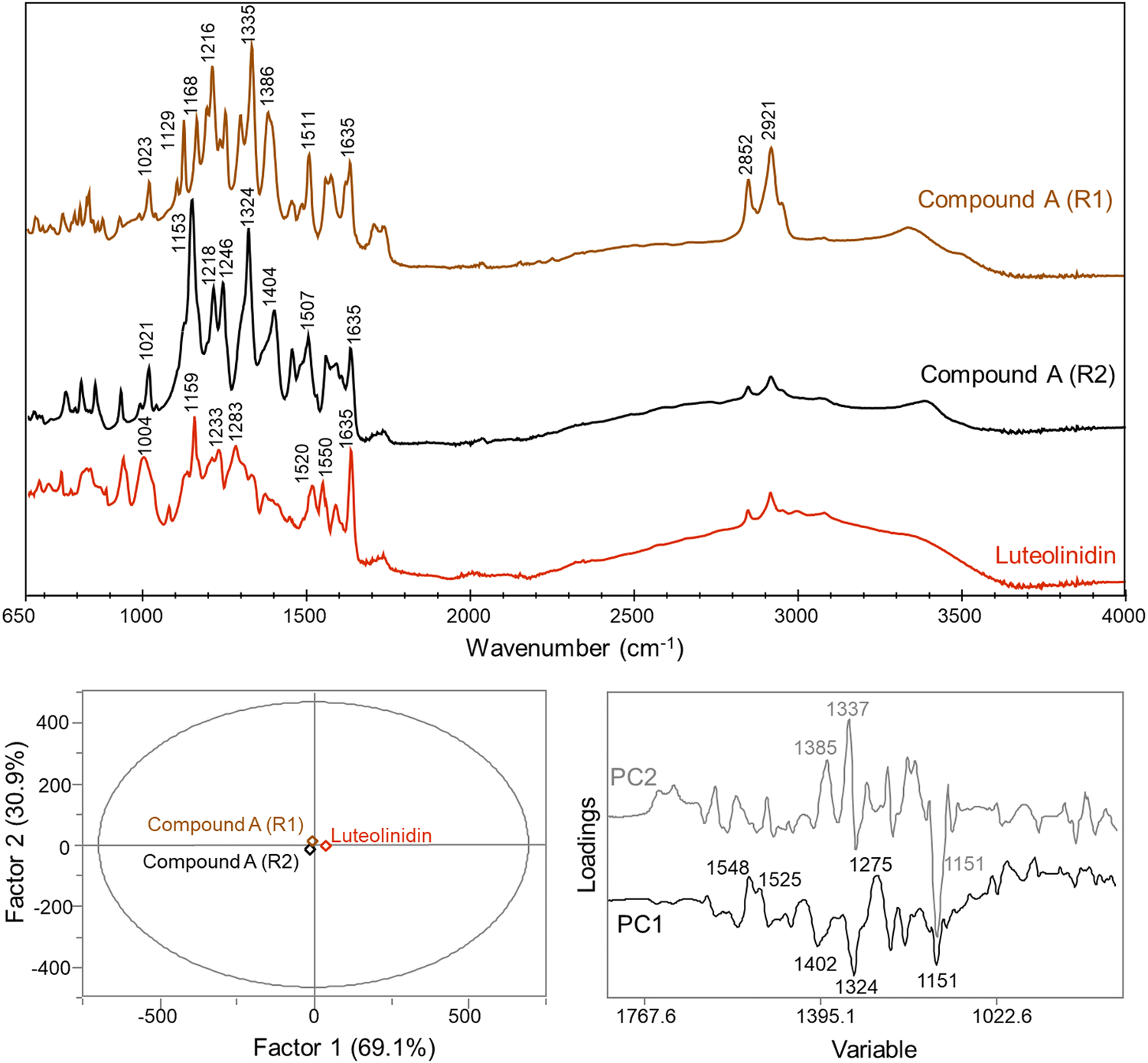 | ||
| Fig. 4 FTIR spectra for 4-carboxy-3-deoxycyanidin (compound A) repetition 1 (R1) and 2 (R2) and luteolinidin standard and the calculated PCA plot and differentiating loadings between the three samples. | ||
PCA factor 2 accounted for differences between the two replicates of compound A with key differentiation bands at 1385, 1337, and 1151 cm−1 (Fig. 4). Differences between the two replicates may be due to changes in non-covalent interactions that could be altered and disrupted during the re-solution and drying process. The bands at 1385 and 1337 cm−1 may be related either to the COO− group or C(phenolic)–O(H) group, as previously discussed, with both functional groups highly prone to intermolecular interactions. Previous work with phenolic compounds has shown that shifts within this range may be due to complexation with metal ions.31,36,37 Trace metals may have been present in the sample and changes in the complexation of compound A with these ions may have occurred with the subsequent re-dissolution and drying cycle. These differences between the two measurements for compound A support the structural assignment as 4-carboxy-3-deoxycyanidin.
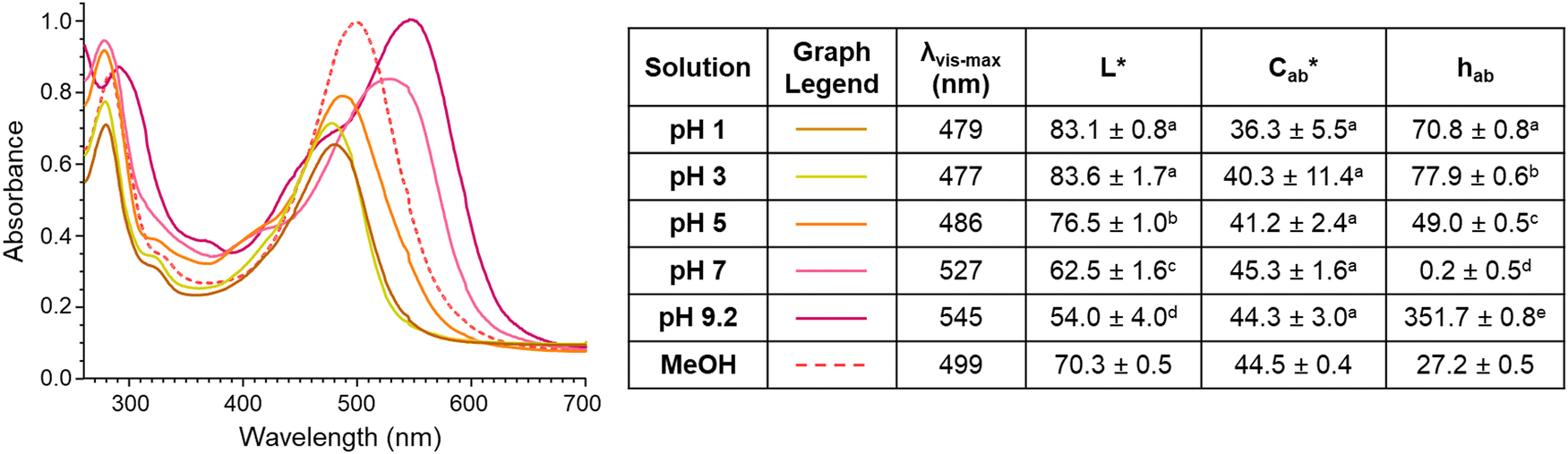 | ||
Fig. 5 Spectral and CIELAB L*, 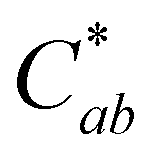 , hab values of 4-carboxy-3-deoxycyanidin (compound A) in pH buffers 1–9.2 and MeOH. Spectra shows mean absorption values and color characteristics are expressed as mean ± standard deviation. Different letters show statistical difference (p < 0.05) within a column (MeOH not included). , hab values of 4-carboxy-3-deoxycyanidin (compound A) in pH buffers 1–9.2 and MeOH. Spectra shows mean absorption values and color characteristics are expressed as mean ± standard deviation. Different letters show statistical difference (p < 0.05) within a column (MeOH not included). | ||
The different colors produced by compound A across pH values is consistent with the pH-dependent structure chemistry of flavylium systems As the pH of the environment increases, the flavylium cation can be deprotonated to form a quinoidal base, and the flavylium cation can by hydrated followed by tautomerization to form the hemiketal and chalcone, with the extent of occurrence for each of these reactions dependent on the type and number of substitutions.38,39
Previous work by other authors with the synthetic 4-carboxy-7-hydroxy-4′-methoxy flavylium chloride (CHMF) compound showed that at low pH values, the carboxylic carbon could be deprotonated (pKa 0.73) to form a zwitterion, with this change causing a hypsochromic shift in λvis-max.40 As pH increased for CHMF zwitterion, the anionic quinoidal base formed (∼pKa 4.8), accompanied by a bathochromic shift in λvis-max.40 These color changes match those observed for compound A from pH 1–7 and suggest similar structural changes occurred with pH adjustments. The color of compound A continued to shift to a more magenta hue with increasing pH, noted by decreasing hab and increasing λvis-max. These color changes may be due to continued deprotonation of hydroxyl substitutions to form the dianionic quinoidal base structure and re-equilibration of the multistate chemical structures.39,41
There was surprisingly no color fading in the mildly acidic pH range (pH 3–7) for compound A. Instead, as pH increased, the color became darker, supported by significant decreases in L* (p < 0.05) (except between pH 1 and 3 (p = 0.99)) and increases in absorbance intensity at the λvis-max. There was no significant change in 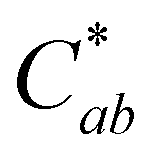 with increasing pH (p = 0.43), indicating that the color vibrancy remained the same among pH values. For flavylium compounds, lack of a substitution at C3 position and/or presence of a substitution at C4 are structural attributes both shown to improve color retention across pH values.41–43 These substitutions are not expected to eliminate color loss as related compounds do show some hydration-initiated color fading in the mildly acidic pH range.41,43,44 However, this reaction may be masked in our findings as the initial spectral measurement for compound A was recorded after 30 minutes of equilibration time, and the color was visually observed to fade during this period for pH 1 and 3. Nevertheless, as pH increased, deprotonation was likely the dominant reaction occurring rather than hydration. This supports conclusions by previous authors working with related 4-substituted flavyliums.41,43,44 The current spectral observations and the analogous behavior to related compounds provided additional support for identification of compound A as 4-carboxy-3-deoxycyanidin.
with increasing pH (p = 0.43), indicating that the color vibrancy remained the same among pH values. For flavylium compounds, lack of a substitution at C3 position and/or presence of a substitution at C4 are structural attributes both shown to improve color retention across pH values.41–43 These substitutions are not expected to eliminate color loss as related compounds do show some hydration-initiated color fading in the mildly acidic pH range.41,43,44 However, this reaction may be masked in our findings as the initial spectral measurement for compound A was recorded after 30 minutes of equilibration time, and the color was visually observed to fade during this period for pH 1 and 3. Nevertheless, as pH increased, deprotonation was likely the dominant reaction occurring rather than hydration. This supports conclusions by previous authors working with related 4-substituted flavyliums.41,43,44 The current spectral observations and the analogous behavior to related compounds provided additional support for identification of compound A as 4-carboxy-3-deoxycyanidin.
3.4. Isolated compound A color stability at 25 °C
While color intensity for isolated compound A was retained across pH values, the color noticeably faded in 24 hours by at least 44% in all pH solutions tested except for pH 7 (Fig. 6). At pH 7, the color was remarkably stable at room temperature, with a loss of absorption at λvis-max of just ∼7%. This exceptional pH 7 stability is consistent with the findings in Sweeny and Iacobucci for a synthetic 4-carboxy-3-deoxyanthocyanidin, which differed in structure from compound A only by substitutions in the B′ ring.45 The color of compound A was also stable in MeOH, in which the absorbance dropped by ∼2% and there was no observable color change over 24 hours (Fig. 6).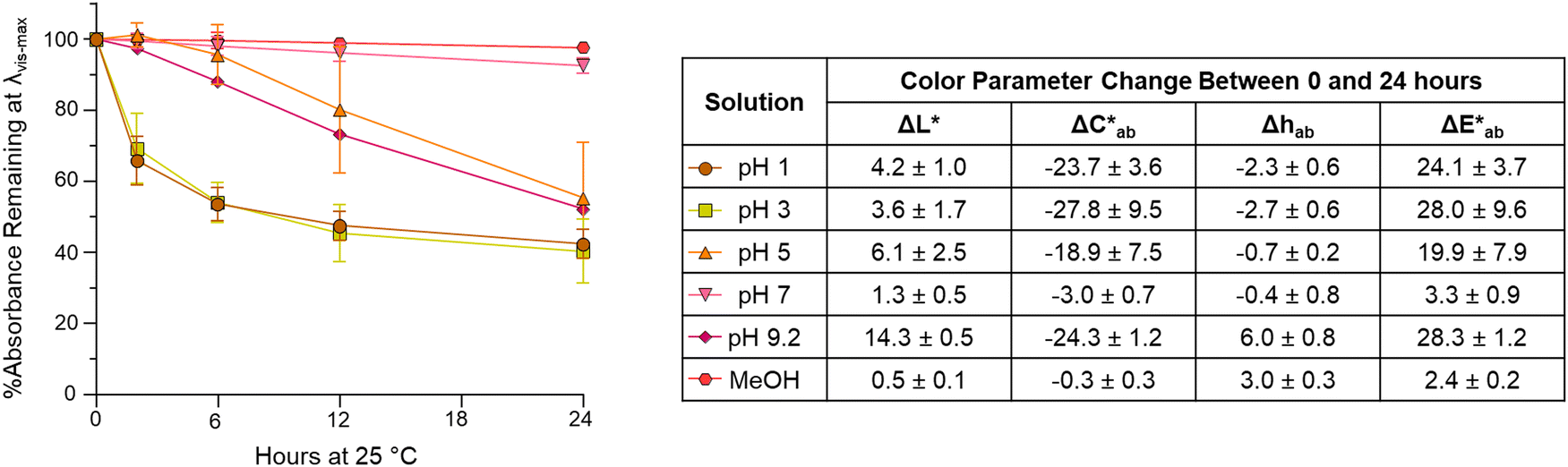 | ||
| Fig. 6 Color stability of 4-carboxy-3-deoxycyanidin (compound A) in pH from 1–9.2 buffers and MeOH over 24 hours. The graph shows the percent absorption change at λvis-max, and the table displays the change in CIELAB color coordinates with results presented as means with standard deviation. | ||
Stability at pH 1 and 3 followed similar trends with absorbance at λvis-max decreasing rapidly (∼30–34%) in the first 2 hours. In contrast, at pH 5, most absorbance decrease happened after 6 hours. Only two experimental replicates were included for stability analysis at pH 5 as one experimental replicate showed different behavior; this replicate showed no decrease in absorbance at λvis-max and a 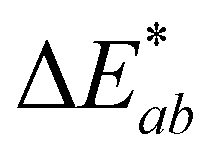 of only 0.52. Additional research is needed to investigate the stability observed for this one experimental replicate. At all 3 acidic pH values, absorbance decreased at all wavelengths during the 24 hours (Fig. S10, ESI†), and the observable color change was driven by color fading (increasing L* and decreasing
of only 0.52. Additional research is needed to investigate the stability observed for this one experimental replicate. At all 3 acidic pH values, absorbance decreased at all wavelengths during the 24 hours (Fig. S10, ESI†), and the observable color change was driven by color fading (increasing L* and decreasing 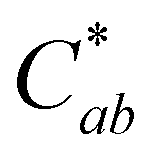 ) (Fig. 6). Different trends were observed at pH 9.2. Here, the spectral shape changed over time as absorbance increased in the ∼260–280 nm and 320–400 nm ranges while decreasing at the λvis-max (Fig. S10, ESI†). In addition to color fading (increasing L* and decreasing
) (Fig. 6). Different trends were observed at pH 9.2. Here, the spectral shape changed over time as absorbance increased in the ∼260–280 nm and 320–400 nm ranges while decreasing at the λvis-max (Fig. S10, ESI†). In addition to color fading (increasing L* and decreasing  ), the hue increased by ∼6 units over 24 hours (Fig. 6). These changes in spectral shape and hue angle may indicate that the chemical structure equilibriums are transforming over 24 hours in the high alkaline pH range.
), the hue increased by ∼6 units over 24 hours (Fig. 6). These changes in spectral shape and hue angle may indicate that the chemical structure equilibriums are transforming over 24 hours in the high alkaline pH range.
3.5. Identification of degradation compounds from hydroxyphenyl-pyranoanthocyanins and insight into the degradation mechanism
In addition to 10-catechyl-PACNs, 10-p-hydroxyphenyl-, 10-guaiacyl-, and 10-syringyl-PACNs derived from cyanidin-3-sambubioside were formed with chemical yields from elderberry ACN extracts at ∼33.2%, 45.2%, and 39.1%, respectively. The pigments were isolated, and the reported PDA spectra, m/z, and MS/MS values of the four hydroxyphenyl-PACNs, differing only in E ring substitutions, matched the expected values and data reported for similar PACNs (Fig. 7).46 During heating, each hydroxyphenyl-PACN formed a unique, colored degradation compound (peak 2d, 3d, 4d, 5d in Fig. 7) with the compound from 10-catechyl-pyranocyanidin-3-sambubioside (peak 3d) matching compound A. The differences in characteristics (λvis-max, m/z, retention time) among the four degradation compounds followed similar patterns observed for the parent PACNs. Namely, 4-carboxy-3-deoxypelargonidin (peak 2d) had the lowest λvis-max, 4-carboxy-3-deoxymalvidin (peak 5d) had the highest λvis-max, and all spectra included minor peaks near 280 nm and 324–340 nm (Fig. 7). The shape of the spectra of 4-carboxy-3-deoxypelargonidin (peak 2d) showed a slight shoulder near 415 nm, and while the wavelengths are hypsochromically shifted, the spectral shape is similar to the appearance of pelargonidin anthocyanins which often have a characteristic shoulder near 440 nm.47 Each degradation compound had a m/z 398 units lower than their parent PACN which would support the loss of the sambubioside sugar moiety and B ring (Fig. 8). Additionally, the m/z difference among the compounds was the same as for the parent PACNs, indicating that the degradation compounds differed in structure by the E ring substitutions. Protocatechuic acid (peak 1) formed in all 4 of the heated PACNs (Fig. 7), tentatively identified based on the spectral characteristics and retention time. Protocatechuic acid is reported to form from the B ring of the heated pigment and has previously been found in solutions of heated cyanidin anthocyanins and cyanidin-derived PACNs.12,48,49 Interestingly, our structure for peak 5d, 4-carboxy-3-deoxymalvidin, matches the theoretical compound proposed as a stable flavylium compound in Amić et al.;50 these authors proposed a λvis-max of 497.8 nm for the compound, which is slightly higher than our experimental value of 491 nm.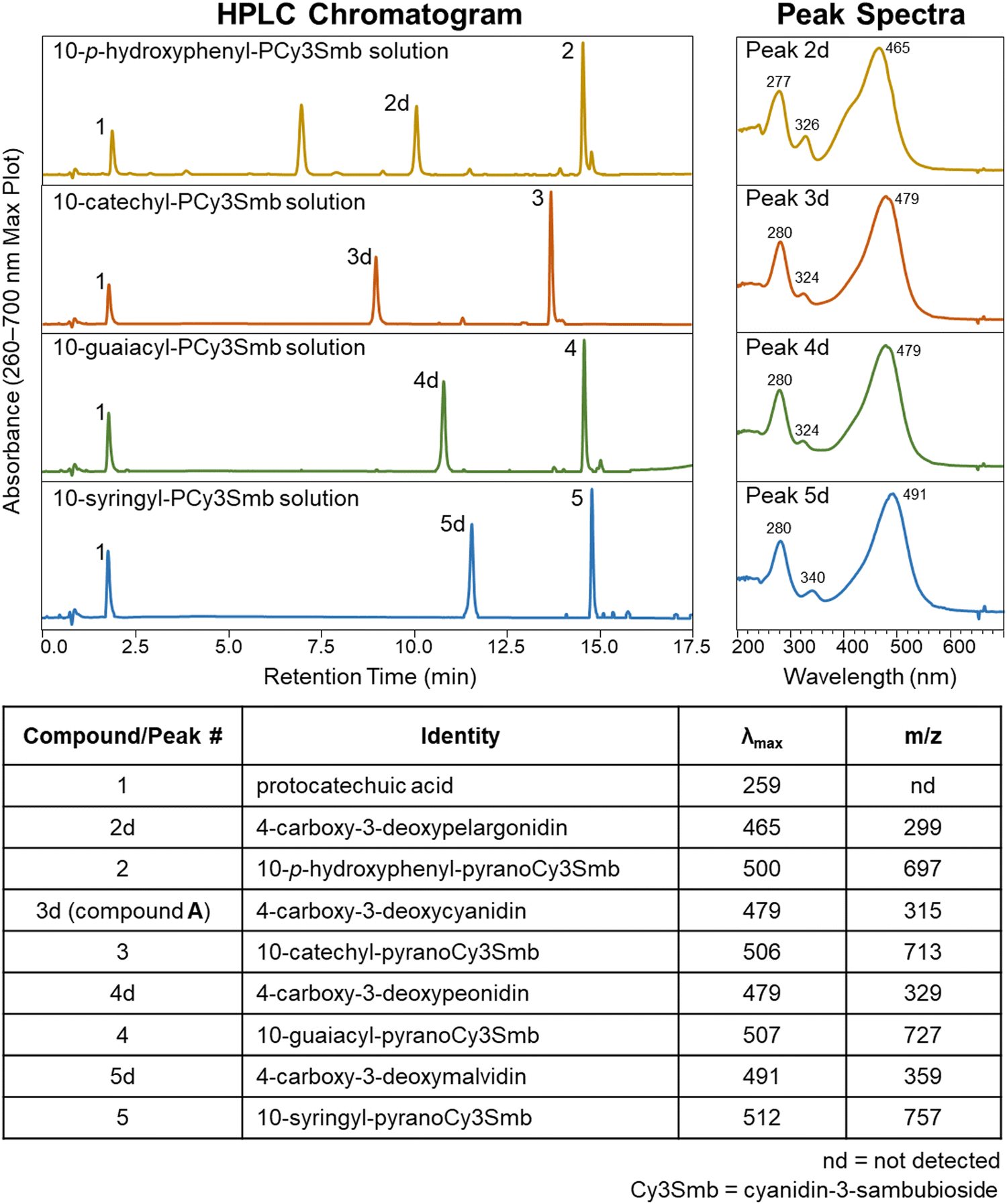 | ||
| Fig. 7 uHPLC-PDA-MS characteristics of hydroxyphenyl-pyranoanthocyanins (peaks 2–5) and their thermal degradation compounds (peaks 1 and 2d–5d) following 15 hours of 90 °C heating at pH 3.0. 10-p-Hydroxyphenyl-pyranoCy3Smb solution contained an impurity (∼6.8 min) present both before and after heating. Chromatographic conditions are provided in Section 2.2. | ||
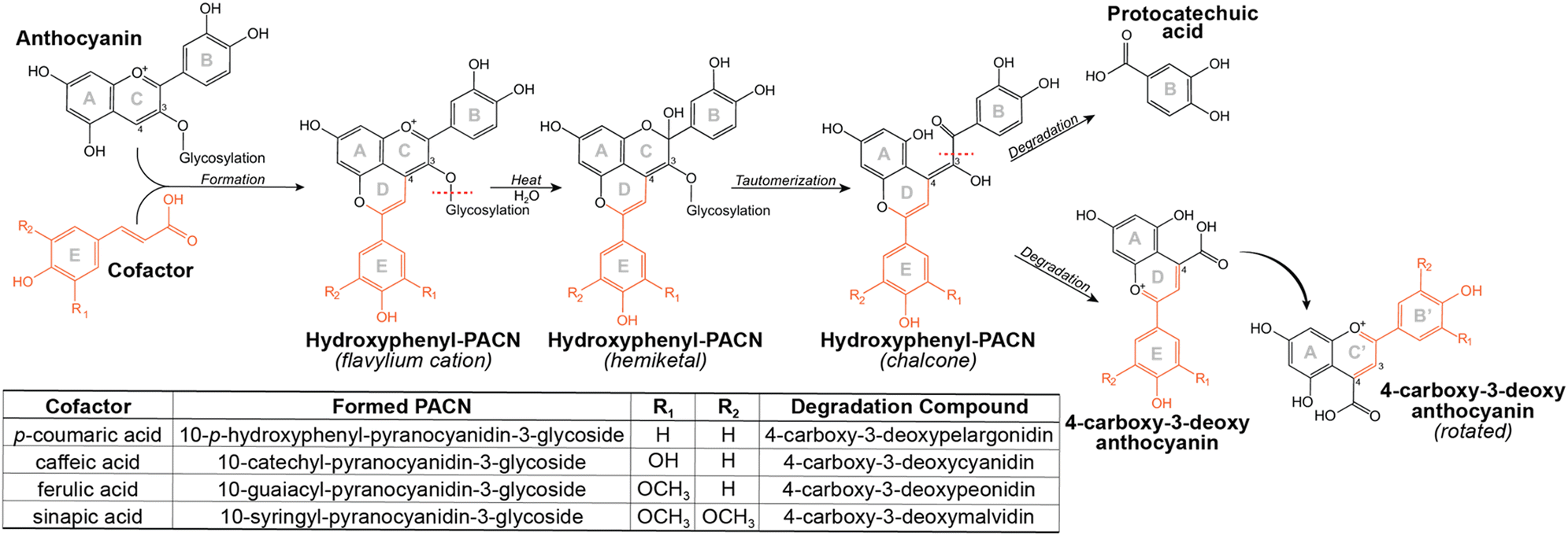 | ||
| Fig. 8 Proposed schematic for hydroxyphenyl-PACN formation and thermal degradation into the 4-carboxy-3-deoxyanthocyanidin degradation compound. Rings B′ and C′ correspond to ring E and D of the parent compound, respectively. | ||
Elucidation of the structure of compound A/peak 3d provided supporting evidence for the proposed thermal degradation pathway for PACNs (Fig. 8). This pathway is postulated to involve hydration at C2 of the flavylium cation PACN to form a hemiketal, tautomerization to the chalcone, and subsequent breakage to form the B ring containing protocatechuic acid and 4-carboxy-3-deoxyanthocyanidin, based on the present findings, ACN thermal degradation pathway,49,51 and hypotheses in Voss et al. and Voss et al.11,12 Additionally, uHPLC-PDA-MS analysis of heated 10-catechyl-pyranomalvidin-3-glucoside/3-rutionside PACNs detected MS signals in both positive (m/z = 481.1) and negative (m/z = 479.1) mode consistent with a hydrated PACN aglycone (+18 m/z units from the aglycone) which corresponded to a small PDA peak absorbing in the 300–400 nm region (Fig. S11, ESI†). These signals were found only in the heated PACN sample with LCMS analysis using non-acidified mobile phases and supported the possibility of a chalcone or hemiketal forming during heating.
The similarities among the degradation compounds from the four hydroxyphenyl-PACNs and identification of protocatechuic acid in all four heated samples suggested that the PACNs degraded in the same manner and the structure of the colored degradation compounds are the same apart from the substitutions on the B′ ring (Fig. 8).
4. Conclusion
The colored degradation compound obtained from heat stress of 10-catechyl-PACNs was discovered to be a 4-carboxy-3-deoxycyanidin, as evidenced by uHPLC-PDA, high resolution-MS/MS, NMR, FTIR, spectrophotometry, and color results. Similar compounds were observed to form from heating other hydroxyphenyl-PACNs (10-p-hydroxyphenyl, 10-guaiacyl, 10-syringyl). These observations show the discovery of a unique class of 4-substituted flavylium compounds, the 4-carboxy-3-deoxyanthocyanidins, formed from nature-derived ingredients.Data availability
The data supporting this experiment has been included in the manuscript and supplements. Additional data may be shared upon reasonable request.Conflicts of interest
The authors declare no competing interests.Acknowledgements
The authors would like to thank Molly J. Davis for editing this manuscript. This research was supported in part by the USDA National Institute of Food and Agriculture, Hatch Project OHO01423, Accession number 1014136 and by state and federal funds appropriated to The Ohio State University, College of Food, Agricultural, and Environmental Sciences, Ohio Experiment Station through the CFAES IGP Graduate Program (Proposal 2022-079).References
- P.-J. Cameira-dos-Santos, J. M. Brillouet, V. Cheynier and M. Moutounet, J. Sci. Food Agric., 1996, 70, 204–208 CrossRef.
- H. Fulcrand, P.-J. Cameira-dos-Santo, P. Sarni-Manchado, V. Cheynier and J. Favre-Bonvin, J. Chem. Soc., Perkin Trans. 1, 1996, 735–739 RSC.
- X.-K. Zhang, Y.-B. Lan, Y. Huang, X. Zhao and C.-Q. Duan, Food Chem., 2021, 339, 127795 CrossRef PubMed.
- S. Hillebrand, M. Schwarz and P. Winterhalter, J. Agric. Food Chem., 2004, 52, 7331–7338 CrossRef PubMed.
- M. Schwarz, V. Wray and P. Winterhalter, J. Agric. Food Chem., 2004, 52, 5095–5101 CrossRef PubMed.
- Ø. M. Andersen, T. Fossen, K. Torskangerpoll, A. Fossen and U. Hauge, Phytochemistry, 2004, 65, 405–410 CrossRef PubMed.
- M. Schwarz, T. C. Wabnitz and P. Winterhalter, J. Agric. Food Chem., 2003, 51, 3682–3687 CrossRef PubMed.
- V. De Freitas and N. Mateus, Anal. Bioanal. Chem., 2011, 401, 1467–1477 CrossRef PubMed.
- G. Miyagusuku-Cruzado, D. M. Voss and M. M. Giusti, Int. J. Mol. Sci., 2021, 22, 6708 CrossRef PubMed.
- G. Miyagusuku-Cruzado, D. M. Voss, T. N. Ortiz-Santiago, Y. Chen and M. M. Giusti, Food Chem., 2023, 427, 136705 CrossRef PubMed.
- D. M. Voss, G. Miyagusuku-Cruzado and M. M. Giusti, npj Sci. Food, 2022, 6, 16 CrossRef PubMed.
- D. M. Voss, G. Miyagusuku-Cruzado and M. M. Giusti, Food Chem., 2023, 403, 134305 CrossRef PubMed.
- J. Sun, X. Li, H. Luo, L. Ding, X. Jiang, X. Li, R. Jiao and W. Bai, J. Agric. Food Chem., 2020, 68, 2783–2794 CrossRef PubMed.
- A. E. Håkansson, K. Pardon, Y. Hayasaka, M. de Sa and M. Herderich, Tetrahedron Lett., 2003, 44, 4887–4891 CrossRef.
- J. Azevedo, J. Oliveira, L. Cruz, N. Teixeira, N. F. Brás, V. De Freitas and N. Mateus, J. Agric. Food Chem., 2014, 62, 7002–7009 CrossRef PubMed.
- M. Á. Pozo-Bayón, M. Monagas, M. C. Polo and C. Gómez-Cordovés, J. Agric. Food Chem., 2004, 52, 1300–1306 CrossRef PubMed.
- X. Wu, L. Gu, R. L. Prior and S. McKay, J. Agric. Food Chem., 2004, 52, 7846–7856 CrossRef PubMed.
- L. E. Rodríguez-Saona and R. E. Wrolstad, in Current Protocols in Food Analytical Chemistry, ed. R. E. Wrolstad, T. E. Acree, H. An, E. A. Decker, M. H. Penner, D. S. Reid, S. J. Schwartz, C. F. Shoemaker and P. Sporns, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2001, p. F1.1.1 Search PubMed.
- J. E. Farr and M. M. Giusti, ColorBySpectra-Academic License, Tech License Express, Ohio State Innovation Foundation, Columbus, OH, 2017 Search PubMed.
- G. Miyagusuku-Cruzado, D. M. Voss and M. M. Giusti, Polyphenols Communications, 2021, vol. 1, pp. 113–115. https://www.groupepolyphenols.com/wp-content/uploads/2021/10/Polyphenols-Communications-e-vol-1-no-1.pdf Search PubMed.
- K. Dührkop, M. Fleischauer, M. Ludwig, A. A. Aksenov, A. V. Melnik, M. Meusel, P. C. Dorrestein, J. Rousu and S. Böcker, Nat. Methods, 2019, 16, 299–302 CrossRef PubMed.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef.
- J. M. Obón, M. R. Castellar, M. Alacid and J. A. Fernández-López, J. Food Eng., 2009, 90, 471–479 CrossRef.
- A. Vallverdú-Queralt, E. Meudec, N. Ferreira-Lima, N. Sommerer, O. Dangles, V. Cheynier and C. Le Guernevé, Food Chem., 2016, 199, 902–910 CrossRef PubMed.
- L. Cruz, J. L. C. Sousa, A. Marinho, N. Mateus and V. de Freitas, Tetrahedron Lett., 2017, 58, 159–162 CrossRef.
- M. Rusishvili, L. Grisanti, S. Laporte, M. Micciarelli, M. Rosa, R. J. Robbins, T. Collins, A. Magistrato and S. Baroni, Phys. Chem. Chem. Phys., 2019, 21, 8757–8766 RSC.
- C. Shiroma-Kian, D. Tay, I. Manrique, M. M. Giusti and L. E. Rodriguez-Saona, J. Agric. Food Chem., 2008, 56, 9835–9842 CrossRef PubMed.
- M. K. Trivedi, A. B. Dahryn Trivedi and H. S. Khemraj Bairwa, Nat. Prod. Chem. Res., 2015, 3, 1000186 Search PubMed.
- M. Lucarini, A. Durazzo, J. Kiefer, A. Santini, G. Lombardi-Boccia, E. B. Souto, A. Romani, A. Lampe, S. F. Nicoli, P. Gabrielli, N. Bevilacqua, M. Campo, M. Morassut and F. Cecchini, Foods, 2020, 9, 10 CrossRef PubMed.
- O. Abbas, G. Compère, Y. Larondelle, D. Pompeu, H. Rogez and V. Baeten, Vib. Spectrosc., 2017, 92, 111–118 CrossRef.
- X. H. Guan, C. Shang and G. H. Chen, Chemosphere, 2006, 65, 2074–2081 CrossRef PubMed.
- M. V. Biber and W. Stumm, Environ. Sci. Technol., 1994, 28, 763–768 CrossRef PubMed.
- M. I. Tejedor-Tejedor, E. C. Yost and M. A. Anderson, Langmuir, 1992, 8, 525–533 CrossRef.
- G. Tondi and A. Petutschnigg, Ind. Crops Prod., 2015, 65, 422–428 CrossRef.
- S. Agatonovic-Kustrin, D. W. Morton and A. P. M. Yusof, Mod. Chem. Appl., 2013, 1, 1000110 Search PubMed.
- H. Dong, X. Yang, J. He, S. Cai, K. Xiao and L. Zhu, RSC Adv., 2017, 7, 53385–53395 RSC.
- M. C. Alvarez-Ros, S. Sánchez-Cortés and J. V. García-Ramos, Spectrochim. Acta, Part A, 2000, 56, 2471–2477 CrossRef PubMed.
- R. Brouillard and J.-E. E. Dubois, J. Am. Chem. Soc., 1977, 99, 1359–1364 CrossRef.
- L. Cruz, N. Basílio, N. Mateus, V. De Freitas and F. Pina, Chem. Rev., 2022, 122, 1416–1481 CrossRef PubMed.
- L. Paulo, A. A. Freitas, P. F. Da Silva, K. Shimizu, F. H. Quina and A. L. Maçanita, J. Phys. Chem. A, 2006, 110, 2089–2096 CrossRef PubMed.
- J. Baranac, D. Amic and V. Vukadinovic, Food Chem., 1990, 38, 932–936 CrossRef.
- J. M. Awika, L. W. Rooney and R. D. Waniska, J. Agric. Food Chem., 2004, 52, 4388–4394 CrossRef PubMed.
- D. Davidović-Amić, D. Amić and N. Trinajstić, Croat. Chem. Acta, 1994, 67, 163–170 Search PubMed.
- G. Mazza and R. Brouillard, J. Agric. Food Chem., 1987, 35, 422–426 CrossRef.
- J. G. Sweeny and G. A. Iacobucci, J. Agric. Food Chem., 1983, 31, 531–533 CrossRef.
- D. Blanco-Vega, F. J. López-Bellido, J. M. Alía-Robledo and I. Hermosín-Gutiérrez, J. Agric. Food Chem., 2011, 59, 9523–9531 CrossRef PubMed.
- M. M. Giusti and R. E. Wrolstad, J. Food Sci., 1996, 61, 322–326 CrossRef.
- E. Sadilova, R. Carle and F. C. Stintzing, Mol. Nutr. Food Res., 2007, 51, 1461–1471 CrossRef PubMed.
- L. Cabrita, V. Petrov and F. Pina, RSC Adv., 2014, 4, 18939–18944 RSC.
- D. Amić, D. Davidović-Amić, D. Bešlo, B. Lučić and N. Trinajstić, J. Chem. Inf. Comput. Sci., 1999, 39, 967–973 CrossRef.
- R. Brouillard and B. Delaporte, J. Am. Chem. Soc., 1977, 99, 8461–8468 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nj02233a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2023 |