
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly diastereoselective Heck–Matsuda reaction with pyrazolyl diazonium salts†
F.
Bellina
ab,
F.
Berti
a,
S. M.
Bertozzi
c,
T.
Bandiera
 a and
F.
Bertozzi
*a
a and
F.
Bertozzi
*a
aD3-PharmaChemistry, Fondazione Istituto Italiano di Tecnologia, Via Morego 30, Genoa 16163, Italy. E-mail: fabio.bertozzi@iit.it
bDepartment of Chemistry and Industrial Chemistry, University of Genova, Genoa 16146, Italy
cAnalytical Chemistry Facility, Fondazione Istituto Italiano di Tecnologia, Via Morego 30, Genoa 16163, Italy
First published on 18th May 2023
Abstract
The Heck–Matsuda (HM) reaction is a powerful synthetic approach cut out for C–C bonds formation under mild conditions. We demonstrated that pyrazolyl diazonium salts are suitable reagents in this protocol, allowing us to deliver highly substituted cyclopentenols and cyclopentenamines with an excellent degree of diastereoselectivity and a control of enantioselectivity.
Functionalized 5-membered carbocycles represent a valuable and versatile chemical motif since they are widespread in natural and pharmaceutical compounds.1 Substituted cyclopentene scaffolds with controlled, defined stereochemistry could be envisaged as synthetically rewarding building blocks, despite their challenging preparation.2 In our group, we recognized substituted 4-(pyrazol-4-yl)cyclopent-2-en-1-ols and 4-(pyrazol-4-yl)cyclopent-2-en-1-amines as advanced, chemically tractable building blocks to achieve high complexity molecules with well-defined stereochemistry (Fig. 1).
 | ||
| Fig. 1 From pyrazolyl diazonium salts to highly substituted cyclopentenyl derivatives. | ||
These scaffolds offer reactive moieties such as the allylic hydroxyl/amino functionalities, which can undergo conventional chemical transformations through essential reactions.3 Ultimately, the possibility of inserting a diversely substituted pyrazole onto the cyclopentenyl scaffold makes the resulting compounds of broad interest, as the pyrazole is a valuable heterocycle frequently found in biologically active compounds and a key structural unit crucial in the modern medicinal chemistry portfolio.4,5 Although the C-4 position of these heterocycles is the most nucleophilic, allowing direct cross-coupling modifications via C–H activation,6 the insertion of a functionalized cycloaliphatic portion onto this carbon atom has not yet been fully disclosed.
Taking into account the potential, broad applications of substituted 4-pyrazolyl-1-cyclopenten-amines/-ols, we embarked on this challenging synthetic goal by exploiting a classical palladium-catalysed approach. Due to its versatility and compatibility with several functional groups, we envisaged that the Heck reaction could be a suitable tool to address our synthetic objective.7 Although the development of the Heck reaction dates back to 1968,8 this approach and its variations still represent a powerful tool to functionalize olefins in synthesizing both natural and pharmaceutical compounds.7 In addition to bromides and iodides, several pseudohalides can react as coupling reagents offering alternatives to installing the new C–C bond. Matsuda and co-workers9 demonstrated that aryl diazonium salts could react under mild palladium-catalysed conditions offering many valuable advantages. This synthetic variation, named Heck–Matsuda (HM) reaction, works without structurally complex ligands, under relatively mild conditions, and with high levels of regio- and stereo- control in the final products.10 Although phenyl-based diazonium salts are considered strategic reagents to achieve complex compounds, the preparation of the corresponding heteroaryl analogs and their application11 in palladium-catalysed coupling reactions have received limited attention.12
Herein, we describe the synthesis of a series of 4-diazonium pyrazolyl tetrafluoroborate salts 1 and their application in a highly stereoselective HM reaction. Based on the previously reported data using phenyl-diazonium salts,13 cyclopent-3-en-1-ol (4) was selected as a benchmark olefin, which allowed affording substituted 4-(pyrazol-4-yl)cyclopent-2-en-1-ol derivatives 2 with a high degree of cis-stereocontrol (Scheme 1).
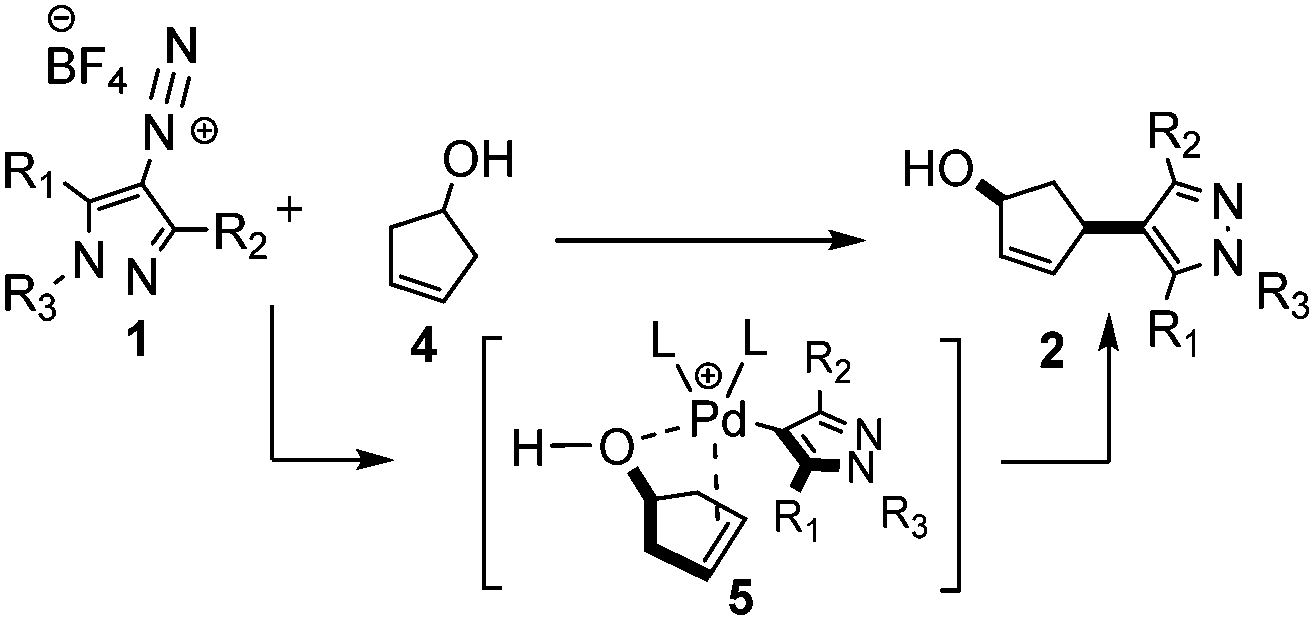 | ||
| Scheme 1 Outcome and proposed mechanism of the HM reaction with pyrazolyl diazonium tetrafluoroborate salts 1. | ||
To achieve this aim, we prepared our model substrate 1a by a three-step synthesis starting from commercial 3,5-dimethyl-4-nitro pyrazole (6a) (Scheme 2). The desired 1-phenyl-4-amino-3,5-dimethyl pyrazole (7a) was produced in excellent overall yield (90%) by sequentially applying a standard Chan–Lam protocol to insert the phenyl moiety in position N-1 of the pyrazole, followed by nitro group reduction using ammonium formate in the presence of Pd/C. Following classical conditions, the amino group of 7a was converted into the stable diazonium salt 1a in good yield (80%).
 | ||
| Scheme 2 Synthesis of diazonium salt 1a. | ||
With diazonium salt 1a in hand, to explore the feasibility and versatility of this synthetic approach, we began our investigation by carefully screening a few conditions and experimental parameters. We first monitored the scope of this protocol applying conditions previously reported in the work of Matsuda.9 We explored a few changes, such as the temperature, the solvent, and the palladium species. However, these initial attempts provided only traces of the desired product 2a, as a consequence of a fast diazonium salt decomposition promoted by sodium acetate (NaOAc) as a mild base (Table 1, entries 1 and 2). To avoid this undesired side-reaction we then focused on developing a base-free protocol. This strategy led to the expected coupling product 2a (Table 1, entry 3) when the reaction was conducted in EtOH at 60 °C and in the presence of 10% of Pd(dba)2. The HM product 2a turned out to be a mixture of the cis- and trans-diastereoisomers, with the cis-isomer as the major component (dr = 98.8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.2). The relative configuration of the major cis-isomer was assigned by analysing the 1H-NMR spectra of similar stereoisomers reported in the literature13 and of the corresponding trans-stereoisomer obtained after Mitsunobu reaction (see the ESI†). These findings are in line with the prediction of a model where electrostatic non-covalent interactions occur in the coordination complex 5 (Scheme 1), as proposed in previous studies.13
:1.2). The relative configuration of the major cis-isomer was assigned by analysing the 1H-NMR spectra of similar stereoisomers reported in the literature13 and of the corresponding trans-stereoisomer obtained after Mitsunobu reaction (see the ESI†). These findings are in line with the prediction of a model where electrostatic non-covalent interactions occur in the coordination complex 5 (Scheme 1), as proposed in previous studies.13
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Solvent | Catalyst (%mol) | T (°C) | Time (h) | Yielda | drb |
| a Isolated yield. b dr: diastereomeric ratio, calculated by UPLC-MS analysis for cis-isomer. c 4 (2 eq.) and NaOAc (3 eq.). d 4 (1.5 eq.). e CaCO3 (1.1 eq.) was added. f 2,6-di-tert-butyl-4-methylpyridine (1.1 eq.) was added. | ||||||
| 1 | DCMc | Pd(dba)2 (5%) | r.t. | 1 | <5% | — |
| 2 | MeCNc | Pd(dba)2 (5%) | r.t. | 1 | <5% | — |
| 3 | EtOH | Pd(dba)2 (10%) | 60 | 0.5 | 50% | 98.8:1.2 |
| 4 | EtOH | Pd(dba)2 (10%) | 40 | 3 | 53% | 99.4:0.6 |
| 5 | EtOH | Pd(dba)2 (10%) | r.t. | 24 | 25% | 99.6:0.4 |
| 6 | EtOH | Pd(dba)2 (5%) | 40 | 7 | 32% | — |
| 7 | EtOHd | Pd(dba)2 (10%) | 40 | 6 | 31% | — |
| 8 | EtOH | Pd2(dba)3 (10%) | 40 | 4 | 39% | 97.9:2.1 |
| 9 | EtOH | Pd(OAc)2 (10%) | 40 | 8 | 30% | 99.9:0.1 |
| 10 | MeOH | Pd(dba)2 (10%) | 40 | 2 | 38% | 97.0:3.0 |
| 11 | iPrOH | Pd(dba)2 (10%) | 40 | 28 | 17% | 98.1:1.9 |
| 12 | MeCN | Pd(dba)2 (10%) | 40 | 24 | — | — |
| 13 | EtOHe | Pd(dba)2 (10%) | 40 | 3.5 | 50% | 99.3:0.7 |
| 14 | EtOHf | Pd(dba)2 (10%) | 40 | 3.5 | 54% | 99.2:0.8 |

While lowering the temperature from 60 °C to 40 °C increased the reaction time (from 30 minutes to 3 h), marginally affecting the yield and the diastereoselectivity (entry 4), carrying out the reaction at room temperature resulted only in partial conversion after one day (entry 5). The reduction in the amount of either the catalyst (5% mol) or the olefin (1.5 eq.) drastically decreased the overall yield (32% and 31%, respectively; entries 6 and 7). Replacing the initially identified catalyst with Pd2(dba)3 or Pd(OAc)2 led to lower yields and longer times in both cases, although with slightly higher diastereomeric ratio for the diacetate salt (entries 8 and 9).
The effect of the solvent on both the overall yield and stereochemical outcome was also investigated. While MeOH led to a faster reaction (ca. 2 h) but with a significant formation (ca. 25%) of a side-product (entry 10), the use of either iPrOH or MeCN resulted in poor or no reactivity (Table 1, entries 11 and 12). Finally, the effect of different types of bases was evaluated. CaCO3 and 2,6-di-tert-butyl-4-methylpyridine (DTBMP) were selected to explore whether the heteroaryl diazonium salt 1a could efficiently react in their presence. However, they turned out to be ineffective, as the overall outcome was similar to the base-free conditions (entries 13 and 14). Although still preliminary, the use of Pd(dba)2 (10% mol) in EtOH at 40 °C (Table 1, entry 4) was chosen as the benchmark reaction conditions, being a good compromise between overall yield, dr, and versatility. Having set the parameters suited to exploit pyrazolyl diazonium salts in the HM approach, we focused on studying the scope of this methodology.
Firstly, we synthesized a few N-substituted 3,5-dimethylpyrazolyl diazonium salts 1b–j (see the ESI†) in a similar manner as reported for 1a (Scheme 2). Interestingly, we observed that all these compounds showed good chemical stability, allowing simple isolation and storage without losing their reactivity. The thermal stability of some pyrazolyl diazonium tetrafluoroborates was also assessed by TGA analyses, showing for all of them a high degree of stability (see the ESI†). Using the optimized conditions, we challenged salts 1b–j in our synthetic protocol (Scheme 3). Diazonium salts 1b–d bearing electron-withdrawing groups on the aryl gave slightly lower yields (30–50%) compared to the unsubstituted phenyl analog 1a, probably due to their higher susceptibility to de-diazotization under these conditions. Compound 1d was also tested in a one-pot protocol (diazotization/HM reaction, see the ESI†) providing the corresponding cyclopentenol 2d in an overall acceptable yield. The presence of a methyl substituent in ortho, metha, or para position of diazonium salts 1e–g resulted in the corresponding products 2e–g in good to high yields. The 78% yield observed for the para-methyl phenyl analog 2g suggests a possible steric effect of the substituent on the phenyl ring in the reactive Pd-complex. The electron-donating p-methoxy group, as in diazonium salt 1h, led to poor yield. To expand the scope of this approach, we investigated benzylic groups as N-substituent of the pyrazole, introducing a benzyl and a p-methoxy benzyl functionality (1i,j); they gave similar results to their aryl analogs 1a,h, affording cyclopentenols 2i,j with comparable yields. Unfortunately, attempting to use either unsubstituted (R3 = H) or tosyl (R3 = p-tolylsulfonyl) substituted pyrazoles resulted in no or low reactivity, probably due to the formation of an unreactive complex of the reactive species with the palladium.14
 | ||
| Scheme 3 Effect of N-substitution of pyrazolyl-diazonium salts in the HM reaction. *One-pot protocol; **DTBMP (1.1 eq.) was added. | ||
We then prepared a series of unsubstituted, symmetrical, and unsymmetrical alkyl-substituted N-phenyl-pyrazolyl diazonium salts 1k–p to potentially unveil and verify the effects of the substituents at 3- and 5-position of the pyrazole on the yield and the stereochemical outcome (Scheme 4).
 | ||
| Scheme 4 Heck–Matsuda with substituted N-phenyl-pyrazolyl diazonium salts. | ||
In our optimized HM reaction conditions, 3,5-unsubstituted pyrazolyl diazonium tetrafluoroborates 1k,l led to complete conversion in short times (ca. 1.5–2h), delivering the desired products 2k,l in moderate to good yields and nearly complete cis-stereoselectivity. Although this protocol led to the desired cyclopentenol 2k in an acceptable 32% yield, by using diazonium salt 1k we also isolated a by-product (ca. 10% isolated yield) not detected in the previously investigated cases (see compound S16, the ESI†).15 Unsymmetrical pyrazolyl-diazonium salts 1m,n worked nicely, giving the corresponding substituted cyclopentenols 2m and 2n in good yields (67% and 43%, respectively) and high cis-stereoselectivity. Unfortunately, when testing hindered analogs, such as the 3,5-diethyl- (1o) or the 5-isobutyl-3-methyl (1p) substituted pyrazolyl diazonium salts, many decomposition products were observed with only trace amounts of the desired products detected in the crude mixture. Finally, to assess the robustness and efficiency of our protocol, we tested the reaction of cyclopent-3-en-1-ol (4) with bromo-phenyl diazonium salt 1b at a gram-scale, affording 2b with high cis-diastreoselectivity in moderate yield, similarly to the milligram-scale reaction (see the ESI†).
Having assessed the conditions to use pyrazolyl diazonium salts in the HM protocol for the highly diastereoselective synthesis of substituted cyclopentenols, we decided to broaden the scope of this approach to afford the corresponding cyclopentenamines 3 (Table 2). We initially aimed to prepare cis isomer 3b through a concise and elegant approach by applying the HM reaction conditions used to produce cyclopentene 2b. In the first attempt, HM product 9b was afforded as a mixture of diastereoisomers with the cis isomer as major component (dr = 77:23, Table 2, entry 1). We then tested other solvents compatible with a base-free reaction protocol (e.g., DMA and MeCN/H2O). Notably, while DMA led to a diastereomeric mixture with the trans isomer as major component, the use of MeCN/H2O (1:1) gave the phthalimido-protected derivative 9b with complete inversion of stereochemistry (cis:trans dr < 1:99) (Table 2, entries 2 and 3). The corresponding cyclopentenamine 3b was afforded in a straightforward and quantitative manner by deprotection of 9b with methylhydrazine in EtOH.

To unambiguously confirm the stereochemistry of trans cyclopent-2-en-1-amine 3b, we converted cis-cyclopentenol 2b into the corresponding trans isomer by a simple Mitsunobu reaction using phathalimide, followed by methylhydrazine deprotection (see the ESI†). In a similar manner, by using unsubstituted N-bromophenyl-pyrazolyl diazonium salt 1l, trans-cyclopentenamine 3l was afforded in an acceptable yield (49% over two-steps) with complete diastereocontrol (Table 2, entry 4).
As concluding experiments, we decided to preliminary challenge our Pd-catalysed heteroaryl HM protocol in the presence of a chiral ligand to investigate whether our pyrazolyl diazonium salts could react under a stereoselective control, delivering the desired products with induction of enantioselectivity. As initial experiments, we reproduced the conditions where PyBOX and PyOX, as chiral ligands, were successfully applied in a HM protocol using phenyl diazonium salts.13,16 Remarkably, whereas the dimethyl pyrazole derivative 1b reacted poorly in the presence of selected chiral ligands, cis-stereoisomer 2l was exclusively obtained using ligand L1 in an acceptable yield (33%) and a promising enantiomeric excess (64% e.e.) (Scheme 5, see also the ESI†). Although still very preliminary, these findings suggest that an enantioselective induction could be obtained with heteroaryl diazonium salts, affording the final products with control of both diastereo- and enantioselectivity.
 | ||
| Scheme 5 Enantioselective HM reaction with substituted pyrazolyl diazonium salts 1b,l. | ||
In conclusion, we demonstrated that 4-diazonium pyrazole salts are suitable reagents for the Pd-catalysed HM reaction. They have been found to stereoselectively provide either 1,4-disubstituted cis-cyclopentenols or 1,4-disubstituted trans-cyclopentenamines, as key synthetic intermediates. Although the current work may well be at a preliminary stage, we envisaged that it could represent an adequate protocol allowing the synthesis of highly substituted heterocyclic compounds, with potential use in medicinal chemistry and chemical biology. Further developments, primarily targeting increased overall yields and enantioselectivity may provide valuable insights on how to expand the scope and the stereochemical outcome of this approach. The ambition of the present work is mainly to stimulate further investigations of these marginally explored heteroaryl reactive species in other metal-catalysed coupling reactions to achieve complex molecular architectures.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank S. Venzano for the compounds’ handling, Dr L. Goldoni and M. Veronesi for NMR technical support, and Dr G. Pugliese for the TGA analyses.Notes and references
- B. Heasley, Curr. Org. Chem., 2014, 18, 641–686 CrossRef CAS.
- J.-R. Chen, X.-Q. Hu, L.-Q. Lu and W.-J. Xiao, Chem. Rev., 2015, 115, 5301–5365 CrossRef CAS PubMed.
- J. Zhang, J. Liao, Y.-F. Wei, G. Cheng and R. Luo, Mini-Rev. Org. Chem., 2018, 15, 476–487 CrossRef CAS.
- K. Karrouchi, S. Radi, Y. Ramli, J. Taoufik, Y. N. Mabkhot, F. A. Al-aizari and M. H. Ansar, Molecules, 2018, 23, 134 CrossRef PubMed.
- G. Li, Y. Cheng, C. Han, C. Song, N. Huang and Y. Du, RSC Med. Chem., 2022, 13, 1300–1321 RSC.
- R. Goikhman, T. L. Jacques and D. Sames, J. Am. Chem. Soc., 2009, 131, 3042–3048 CrossRef CAS PubMed.
- T. Ghosh, D. Biswas and S. Bhakta, Chem. – Asian J., 2022, 17, e202200725 CAS.
- R. F. Heck, J. Am. Chem. Soc., 1968, 90, 5518–5526 CrossRef CAS.
- K. Kikukawa, K. Nagira, F. Wada and T. Matsuda, Tetrahedron, 1981, 37, 31–36 CrossRef CAS.
- (a) F.-X. Felpin, L. Nassar-Hardy, F. Callonnec and E. Fouquet, Tetrahedron, 2011, 67, 2815–2831 CrossRef CAS; (b) J. G. Taylor, A. V. Moro and C. R. D. Correia, Eur. J. Org. Chem., 2011, 1403–1428 CrossRef CAS.
- (a) A. A. Fadda, R. Rabie and H. A. Etman, J. Heterocycl. Chem., 2017, 54, 1015–1023 CrossRef CAS; (b) J. Wu, Y. Gu, X. Leng and Q. Shen, Angew. Chem., Int. Ed., 2015, 54, 7648–7652 CrossRef CAS PubMed.
- K. Kiyoshi, M. Kdji, N. Kazuhiko, W. Fumio and M. Tsutomu, Chem. Lett., 1980, 551–552 Search PubMed.
- R. A. Angnes, J. M. Oliveira, C. C. Oliveira, N. C. Martins and C. R. D. Correia, Chemistry, 2014, 20, 13117–13121 CrossRef CAS PubMed.
- C. Amoah, C. Obuah, M. K. Ainooson, C. K. Adokoh and A. Muller, New J. Chem., 2021, 45, 3716–3726 RSC.
- R. Uma, C. Crévisy and R. Grée, Chem. Rev., 2003, 103, 27–52 CrossRef CAS PubMed.
- C. C. Oliveira and C. R. D. Correia, Chem. Rec., 2021, 21, 2688–2701 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures for intermediates and final compounds’ synthesis, complete characterization data and copies of 1H, 13C, COSY, HSQC and 19F NMR spectra (pdf) of compounds 1a–n, 2a–n, 3b,l and S16, chiral HPLC analyses of racemic and enantio-enriched compound 2l, TGA analysis of salts 1a,d,f,i,k-m. See DOI: https://doi.org/10.1039/d3nj01724a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2023 |