
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient reducibility of layered polysilane (SiH)n for selective recovery of platinum ions from aqueous media†
Masataka
Ohashi
 *,
Yasutomo
Goto
and
Hideyuki
Nakano
*,
Yasutomo
Goto
and
Hideyuki
Nakano
Toyota Central R&D Labs., Inc. (TCRDL), Nagakute, Aichi 480-1192, Japan. E-mail: m-ohashi@mosk.tytlabs.co.jp
First published on 6th May 2023
Abstract
To achieve sustainable economic growth, it is essential to have a resource recycling system that uses energy and resources efficiently, eliminates waste, and maximizes their value. In particular, efficient recovery and recycling technology for scarce resources is extremely important because it leads to a stable supply of resources, environmental conservation, and reduction of mining energy. In this study, we demonstrate the one-pot and selective recovery of platinum (Pt) ions from aqueous media containing various coexisting ions using a two-dimensional silicon material. Specifically, Pt ions dissolved in artificial seawater were recovered using layered polysilane (SiH)n (LpSi), which is a stacking of hydrogen-terminated monatomic silicon layers. Hydrosilyl groups (![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Si–H) in LpSi selectively reduced Pt ions to form Pt nanoparticles (Pt-NPs) on LpSi with a particle size less than 200 nm. The selective recovery of Pt ions using LpSi is effective even for low concentrations of Pt ions around 10 ppm, and Pt-NPs with sizes of less than 1 nm were recovered on LpSi. The formed Pt-NPs can be isolated from LpSi by simple treatment with an alkaline solution. Moreover, the resulting Pt-NP-loaded LpSi catalysed the hydrogen evolution reaction in water splitting as an electrocatalyst, and hydrogen evolution proceeded quantitatively for the supplied charges.
Si–H) in LpSi selectively reduced Pt ions to form Pt nanoparticles (Pt-NPs) on LpSi with a particle size less than 200 nm. The selective recovery of Pt ions using LpSi is effective even for low concentrations of Pt ions around 10 ppm, and Pt-NPs with sizes of less than 1 nm were recovered on LpSi. The formed Pt-NPs can be isolated from LpSi by simple treatment with an alkaline solution. Moreover, the resulting Pt-NP-loaded LpSi catalysed the hydrogen evolution reaction in water splitting as an electrocatalyst, and hydrogen evolution proceeded quantitatively for the supplied charges.
Introduction
Platinum group metals (PGMs: the collective name for platinum (Pt), palladium (Pd), rhodium (Rh), iridium (Ir), ruthenium (Ru), and osmium (Os)) are classified as precious metals and are rare resources with high industrial value, and are also essential resources for the realization of a low carbon society and the development of next generation technologies. Among them, Pt is in particularly high demand, and is widely used in applications such as automotive catalysis, chemical catalysis, electronic devices, and pharmaceuticals, as well as jewellery and investment goods.1 Furthermore, global demand for Pt is expected to increase with the global trend toward a hydrogen energy society, including the widespread use of fuel cell technologies and hydrogen production using renewable energy sources.2 For this reason, along with a reduction of Pt usage and the development of alternative materials, the development of selective Pt recovery technologies3 is attracting much attention.For the recovery and reuse of Pt from industrial waste containing high concentrations of Pt, industrialized separation and recovery recycling chains have been established that use precipitation separation or solvent extraction methods.1 However, the recycling rates for Pt are not sufficient, and in particular, the recovery of Pt from liquid waste containing low concentrations of Pt (<10 ppm Pt) generated in the recycling processes is rarely implemented due to its high cost and environmental impact.4 Moreover, Pt contained in automobile catalysts5 and anticancer drugs6,7 has been released into the aquatic environment in recent years, and thus Pt that exceeds its natural abundance ratio has been detected in estuaries and rivers around densely populated areas.8,9 Such Pt solutions with low concentrations also have the potential to be utilized as new sources of Pt to compensate for resource depletion or supply concerns. However, the recovery of low-concentration Pt presents many challenges in terms of both cost and environmental impact, and thus practical application requires the development of inexpensive, low-energy-consumption, and low-environmental-impact Pt recovery technologies.
Layered polysilane (LpSi) is a laminated material consisting of hydrogen-terminated silicon monoatomic layers stacked by van der Waals interactions (Fig. 1).10 We have previously reported that dense hydrosilyl (Si–H) groups in the interlayer space of LpSi reduce some metal ions such as Au3+, Cu2+, and Pd2+ in aqueous solution, which results in the formation of metal nanoparticles on LpSi.11 The reduction reaction with LpSi as a reducing agent proceeds for metal ions that have a more positive standard electrode potential (E0) than that for the Si–H groups on LpSi. Therefore, we consider that Pt ions are reduced to metallic Pt by LpSi because Pt ions have a more positive E0 (E0 > 0.7 V) than Cu2+ (E0 for Cu2+/Cu: 0.337 V), which is already confirmed to be reduced by LpSi. Moreover, LpSi is an easily prepared solid material composed of silicon (Si) and hydrogen (H), which are abundant elements in nature; therefore, it is expected to be a low-cost and environmentally friendly Pt ion recovery agent compared to conventional adsorbents and chemical reducing agents for precious metal ions.
 | ||
| Fig. 1 Structure model of LpSi (A) and (B) the monoatomic silicon layer of LpSi. | ||
Herein, we focus on the recovery of Pt ions from aqueous media by the reducibility of LpSi. Assuming the recovery of Pt ions from the natural environment, hexachloroplatinic acid (H2PtCl6) dissolved in artificial seawater containing various ion species at high concentrations was recovered by LpSi, which resulted in Pt nanoparticles (Pt-NPs) loaded on LpSi (Pt/LpSi). Furthermore, the isolation of Pt-NPs from Pt/LpSi and the hydrogen evolution reaction (HER) in water splitting using Pt/LpSi as an electrocatalyst were demonstrated.
Experimental
Preparation of layered polysilane (LpSi)
LpSi was prepared according to the method described by Yamanaka et al.,12 in which calcium disilicide (CaSi2)13 was immersed in 37% HCl and stirred continuously at −30 °C for 7 days. The resultant LpSi was recovered with a membrane filter, rinsed with a cold HCl and dilute HF solution under an Ar atmosphere, and then vacuum-dried at room temperature for 24 h in the dark.Recovery of Pt4+ from artificial seawater using LpSi
Artificial seawater (ASW) was prepared by mixing commercially purchased sea salt (MARINE ART SF-1, Osaka Yakken Co., Ltd) with distilled water. The prepared ASW is an aqueous solution with pH 7.5–8.0 and contains salts and minerals present in seawater. LpSi was added to an ASW solution containing hexachloroplatinic acid (H2PtCl6) and stirred vigorously at room temperature and normal pressure. The resulting suspension was filtered with a membrane filter. The recovered powder was washed with distilled water and dried under vacuum at room temperature overnight.Isolation of recovered Pt from Pt/LpSi
An aqueous solution of sodium hydroxide (NaOH) with dispersed Pt/LpSi was placed in a sealed Teflon vessel and heated at 65 °C for 24 h. After heating, the suspension was filtered through a porous polytetrafluoroethylene membrane to collect the Pt-NPs isolated from Pt/LpSi. The obtained Pt-NPs were dispersed in distilled water and dropped onto a germanium plate to prepare a sample for SEM observation.Electrochemical measurements
A Pt/LpSi electrode (Pt/LpSi/CP) was fabricated by rubbing Pt/LpSi onto the water-repellent carbon paper (CP) using a spatula, and then coating it with an ethanol dispersion of multi-walled carbon nanotubes (MWCNTs). For comparison with Pt/LpSi/CP, a LpSi electrode (LpSi/CP), in which LpSi was fixed on CP, and a MWCNT electrode (MWCNTs/CP), in which only MWCNTs were coated on the CP surface, were also fabricated using the same procedure. Cyclic voltammograms were obtained using an electrochemical flow cell. The fabricated sample electrode, Pt foil, and an Ag/AgCl electrode were used as the working (WE), counter (CE), and reference (RE) electrodes, respectively. WE and CE were separated by a Nafion 117 membrane, and an Ar-saturated aqueous solution of potassium sulphate (K2SO4) was used as the electrolyte.Electrochemical hydrogen evolution reaction (HER)
The HER under constant potential electrolysis was monitored by in situ measurements using the above-mentioned electrochemical flow cell in combination with gas chromatography (GC). The electrochemical flow cell was directly connected to a GC system equipped with an autosampler, and the evolved hydrogen (H2) was fed from the electrochemical flow cell to the GC by a constant Ar flow. The amount of evolved H2 was estimated by multiplying the concentration of products determined using GC, the Ar flow rate, and the measurement interval.Characterization
UV-Visible (UV-Vis) absorption spectra were recorded using an Ocean Optics FLAME-S spectrometer coupled with an Ocean Optics halogen light source (HL-2000LL). Powder X-ray diffraction (XRD) measurements were performed using a Rigaku RINT-TTR diffractometer with Cu Kα radiation generated at 50 kV and 300 mA. Out-of-plane XRD patterns for the samples were measured at a scanning rate of 2° per min. Fourier transform infrared (FTIR) spectra were recorded on a Nicolet FT-IR Avatar360 spectrometer with a diamond attenuated total reflection (ATR) unit. Data sampling was repeated 64 times with 2 cm−1 resolution. Scanning electron microscopy (SEM) and energy-dispersive X-ray spectroscopy (EDS) analysis were performed using a Hitachi SU3500 electron microscope with an acceleration voltage of 15 kV and a Horiba EMAX Evolution X-Max 150 energy-dispersive X-ray micro analyser system. The samples were attached to the SEM stage using conductive carbon double-faced tape without further treatment such as gold sputtering of the surface. High-resolution transmission electron microscopy (HRTEM), high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM), and energy-dispersive X-ray spectroscopy (EDS) analyses were performed using a JEOL JEM-2100F field-emission electron microscope with an acceleration voltage of 200 kV. The samples for observation were prepared by dropping a suspension of sample dispersed in alcohol onto a copper or nickel grid and evaporating the solvent in air. X-ray photoelectron spectroscopy (XPS) was conducted using a Ulvac-Phi Quantera SXM spectrometer with a monochromatic Al Kα X-ray source (1486.6 eV), a photoelectron takeoff angle of 45°, an analysis area with a diameter of 200 μm, and charge-up correction using the C 1s 285 eV peak. Electrochemical measurements were performed using a BioLogic SP-150 potentiostat electrochemical workstation.Results and discussion
Selective recovery of platinum ions using LpSi
By adding LpSi (Si–H: 0.96 mmol) to an ASW solution containing H2PtCl6 (Pt4+: 0.06 mmol, 1950 ppm), the colour of the suspended reaction mixture gradually changed from yellow to green. After the reaction, a green powder and a colourless filtrate were obtained by filtration of the reaction solution (Fig. 2). The UV-visible absorption spectrum of the filtrate showed no characteristic absorption bands around 400 and 460 nm attributable to H2PtCl6 (Fig. 2B). In contrast, ASW containing H2PtCl6 was stirred under the same conditions without the addition of LpSi; however, the concentration of H2PtCl6 in the ASW remained the same (Fig. S1, ESI†) and no precipitates were formed in the ASW. | ||
| Fig. 2 (A) Digital camera images of LpSi before (a) and after (b) the recovery reaction. (B) UV-Vis spectra and digital camera images (inset) of (a) H2PtCl6 (0.06 mmol) dissolved in ASW (6 mL) and (b) filtrate obtained from the reaction of LpSi with H2PtCl6 in ASW. | ||
SEM/EDS analysis revealed the green powder to consist of plate-like particles characteristic of LpSi (Fig. 3A), and carbon (C), oxygen (O), sodium (Na), silicon (Si), chlorine (Cl) and Pt were detected (Fig. 3B). The elements O, Na, Cl, Si, and Pt were mainly detected on the plate-like particles (Fig. S2, ESI†). The Na and Cl are assumed to be derived from the ASW, although their apparent atomic weight ratios against Si were much smaller than that for Pt, and no other elements originating from the ASW were detected (Table S1, ESI†). These results clearly indicate that most of the Pt ions in the ASW were selectively recovered by LpSi to form Pt-loaded LpSi (Pt/LpSi).
 | ||
| Fig. 3 SEM image (A) and EDS spectrum (B) of Pt/LpSi. FTIR spectra (C) and XRD patterns (D) for LpSi (a) and Pt/LpSi (b). The XRD pattern for standard fcc Pt was obtained from JCPDS files (JCPDS No. 65-2868). | ||
FTIR spectra and XRD patterns for Pt/LpSi and LpSi are shown in Fig. 3C and D, respectively. The FTIR spectrum of LpSi shows characteristic stretching vibration peaks that correspond to Si–H (2100 cm−1), Si–O–Si (1000–1100 cm−1), and Si–Si (500–800 cm−1); however, the Si–H stretching vibration peak is absent in the spectrum of Pt/LpSi. The XRD pattern for Pt/LpSi showed no peaks associated with LpSi but the appearance of new peaks at 2θ = 39.8, 46.3, and 67.5°, which correspond to the (111), (200), and (220) planes of face-centered cubic (fcc) Pt. The Pt 4f XPS spectrum of Pt/LpSi shows two peaks at 71.5 eV (Pt 4f7/2) and 74.5 eV (Pt 4f5/2), which are in good agreement with the previously reported Pt 4f peaks for metallic Pt (Pt0) with surface-adsorbed oxygen (Fig. S3, ESI†).14 The reduction of Pt ions to Pt0 by LpSi is considered to proceed by radical species due to the cleavage of Si–Si bonds or by the Si–H groups acting as reducing agents. However, in the FTIR spectra of Pt/LpSi, the Si–H stretching vibration disappeared while the Si–Si stretching vibration remained and the Si–O–Si stretching vibration increased. Therefore, the reduction of Pt ions is mainly driven by the Si–H groups on LpSi, and the Si–H groups are oxidised to silicate. In addition, the disappearance of the characteristic structures of LpSi caused by Pt loading suggests that Pt ions react with Si–H groups in the interlayer space of LpSi.
Characterization of recovered Pt
HAADF-STEM, HRTEM, and EDS analyses were performed to determine the distribution and shape of the recovered Pt particles on LpSi. Many spherical particles with a wide size distribution of 30–200 nm (Fig. S4, ESI†) were observed as bright spots on the plate-like particles in the HAADF-STEM image of Pt/LpSi (Fig. 4a). EDS analysis revealed that Pt is present in the spherical particles, while Si and O are present in the plate-like particles (Fig. 4b–e). These results indicate that recovered Pt ions are reduced to Pt0 and form Pt-NPs on LpSi. HRTEM observations suggested that these Pt-NPs are polycrystalline and consist of smaller Pt-NPs with sizes of approximately 5–10 nm (Fig. 4f), and lattice fringes corresponding to the interplanar spacing of the (111) plane of Pt (d = 0.224 nm) were observed in the Pt-NPs (Fig. 4g and Fig. S5, ESI†).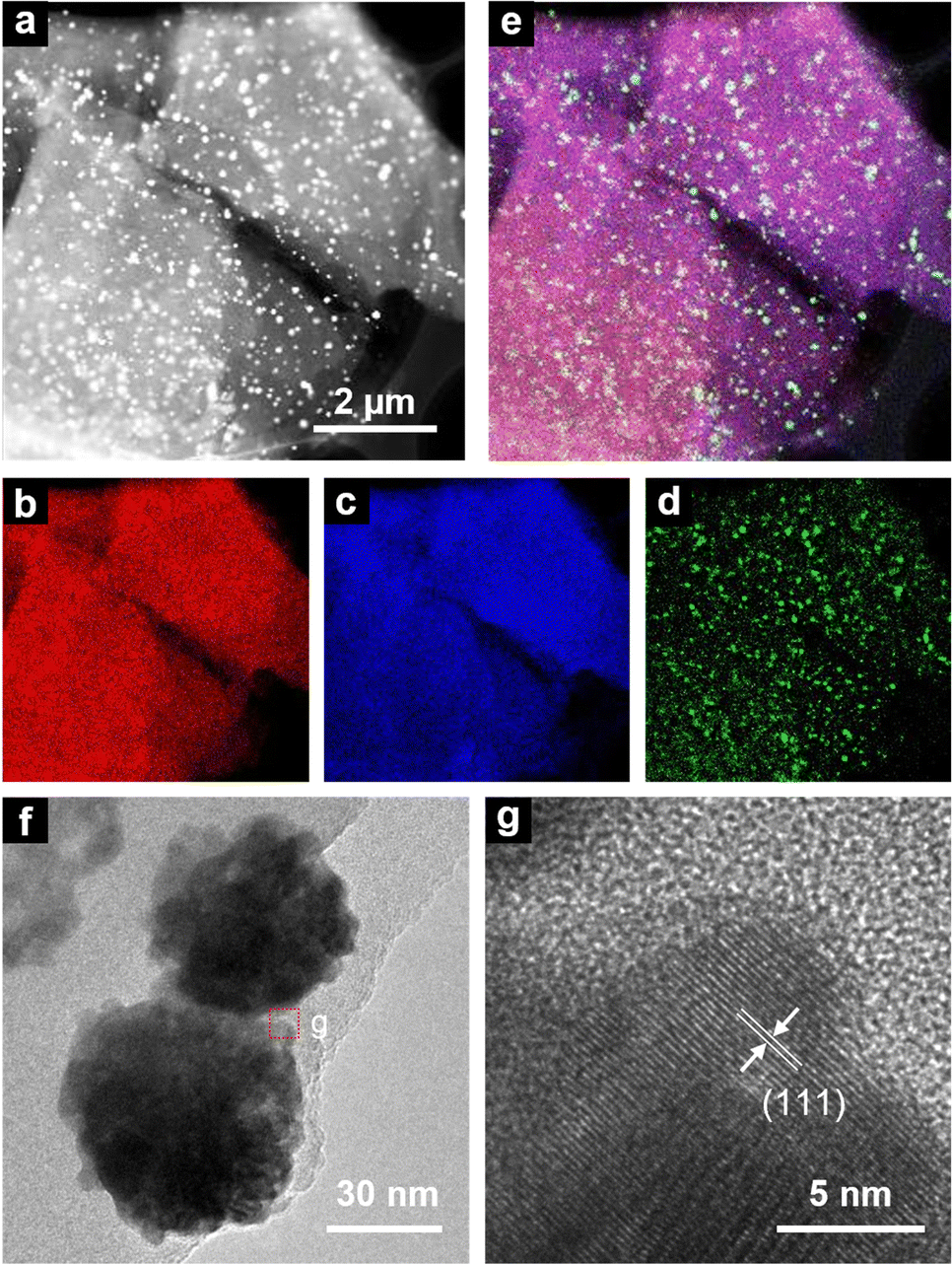 | ||
| Fig. 4 HAADF-STEM (a) and HRTEM (f, g) images of Pt/LpSi and elemental mapping images of the area shown in (a): Si K edge (b), O K edge (c), Pt M edge (d), and overlay (e). | ||
Stoichiometric relationship for the reduction of Pt ions with LpSi
To investigate the reduction efficiency of Pt ions by LpSi, various amounts of LpSi, equivalent to 0.03–0.96 mmol of Si–H groups, were added to ASW containing 0.06 mmol Pt4+. Fig. S6 (ESI†) shows the correlation between the amount of unrecovered Pt4+ remaining in the filtrate from each reaction and the amount of Si–H in the added LpSi. The amount of unrecovered Pt4+ in the filtrate decreased linearly with increasing amount of LpSi added, and LpSi equivalent to 0.7 mmol of Si–H groups was required to completely recover 0.06 mmol of Pt4+. The stoichiometric ratio for the reduction of Pt4+ to Pt0 indicates that the reduction reaction is completed with 4 equivalents of one-electron reductant, i.e., Si–H groups, against Pt4+, while the reduction of Pt4+ with LpSi requires 12 equivalents of Si–H groups, which is approximately 3 times the stoichiometric ratio. The requirement for an excess amount of Si–H groups above the stoichiometric ratio for the reduction of Pt4+ is assumed to be due to the decrease in the density of Si–H groups by the partial oxidative decomposition of LpSi in air and in the ASW. The chemical equilibrium of Pt ionic species in the ASW is also considered to be caused by the requirement for excess Si–H groups because Pt ions form a variety of complex ions at pH 7.5–8.4. Among them, PtCl42− and PtCl5OH2− are thermodynamically stable species in seawater,15 and the thermal equilibrium between them is dominated by the Pt4+ species (PtCl5OH2−) rather than the Pt2+ species (PtCl42−).16 The same recovery experiment was performed using distilled water as a solvent; however, the reduction efficiency of Pt4+ by LpSi was almost the same as in that in ASW, which suggests that the coexistent ionic species in ASW have little effect on the reduction reaction of Pt4+ by LpSi.Isolation of recovered Pt from LpSi
To reuse the recovered Pt, isolation from the recovery agent is generally required. In Pt/LpSi, recovered Pt-NPs can be isolated by dissolving LpSi using an alkali treatment; Pt/LpSi was suspended in 1 M NaOH and heated at 65 °C, and a black powder was then collected by filtration. SEM/EDS analysis revealed that the black powder comprised irregularly shaped particles formed by the aggregation of Pt-NPs with a particle size less than 200 nm, in which little Si was present (Fig. 5 and Fig. S7, ESI†). The isolated Pt-NPs aggregated easily due to their high surface energy, which allowed them to be collected by a simple filtration process. | ||
| Fig. 5 SEM/EDS analysis of isolated Pt-NPs. SEM images (a), elemental mapping images of Si (b), O (c) and Pt (d) and EDS spectra of the SEM observation field (e). *Ge signals derived from the SEM plate used to load the observation sample. | ||
Recovery of low-concentration Pt ions in ASW
Conventional industrialized Pt recovery processes are rarely used to recover Pt from solutions with Pt concentrations below 10 ppm due to profitability issues. LpSi is a relatively inexpensive material that can selectively recover Pt ions, even from a solution containing many contaminants; therefore, it is expected to be applied to the recovery of low-concentration Pt ions. The addition of LpSi (Si–H: 1.75 mmol) to ASW (20 mL) containing 9.75 ppm of Pt4+ (1.0 μmol) yielded a light-yellow powder (denoted as Low-Pt/LpSi) that was almost the same colour as LpSi (Fig. 6A). The HAADF-STEM image of Low-Pt/LpSi in Fig. 6B shows bright spots with sizes of <0.5 nm on the plate-like LpSi particles, and the EDS spectra in Fig. 6C, taken around the bright spots, reveals the presence of Pt, although no other elements from ASW were detected. Based on the size of the bright spots, most of the recovered Pt was considered to be loaded on LpSi as monoatomic Pt. The XRD pattern for Low-Pt/LpSi in Fig. S8 (ESI†) shows no Pt peaks. This is due to the small particle size and extremely low loading of Pt-NPs, which supports the HAADF-STEM observations. The disappearance of diffraction peaks associated with the (100) and (110) planes in LpSi, and the appearance of a broad silica halo at around 2θ = 22° indicate that the Si framework was oxidized and amorphized during the recovery reaction. In addition, diffraction peaks associated with (001) planes had similar intensities before and after Pt recovery, which indicates that the stacking structure of the Si layers is retained, even after Pt recovery due to the extremely low loading of Pt-NPs on LpSi. These results indicate that LpSi can selectively recover even extremely low concentrations of Pt ions in ASW. Moreover, it is suggested that the size of Pt-NPs formed on LpSi can be controlled by the concentration of Pt ions in the aqueous media. | ||
| Fig. 6 (A) Digital camera images of LpSi and Low-Pt/LpSi, (B) HAADF-STEM images of Low-Pt/LpSi and (C) EDS spectrum for the area marked with a red dashed line in (B). The inset shows a magnified view of the EDS spectrum in the energy range of 1–3 keV. *Ni signals derived from the TEM grid. | ||
Electrochemical catalysis application of Pt/LpSi
Pt-NPs have been used as catalysts for various chemical reactions. In the as-prepared Pt/LpSi, Pt-NPs are loaded on LpSi in a highly dispersed state because the surface of LpSi prevents aggregation of the Pt-NPs, so that Pt/LpSi has potential application as a catalyst for chemical reactions without isolation treatment of Pt-NPs. To evaluate the catalytic activity of Pt/LpSi, an electrochemical HER was performed in neutral media using Pt/LpSi as an electrocatalyst. An electrode was fabricated by rubbing Pt/LpSi onto carbon paper and coating it with MWCNTs. Electrodes with LpSi or MWCNTs fixed to carbon paper were also fabricated (Fig. S9, ESI†) and evaluated in the same way for comparison with the Pt/LpSi electrode.The HER proceeded by application of a constant voltage of −1.5 V vs. Ag/AgCl to three electrodes consisting of Pt/LpSi, LpSi, and MWCNTs, and the amount of H2 generated by each electrode was found to increase linearly with electrolysis time. Pt/LpSi exhibited a reduction current of 8–13 mA (total charge: 35.7 C) and generated 186.8 μmol of H2 during 1 h of potentiostatic electrolysis (Fig. 7 and Fig. S10, ESI†). The current efficiency for H2 evolution was 101%, which suggested that the charge supplied to Pt/LpSi was quantitatively utilized for the HER. On the other hand, the LpSi and MWCNT electrodes exhibited reduction currents of 0.58 mA (total charge: 2.09 C) and 2.2 mA (total charge: 7.81 C) under the same conditions, respectively. The amounts of generated H2 and the current efficiencies were 5.46 μmol and 50.4%, and 28.0 μmol and 69.2% for the LpSi and MWCNT electrodes, respectively, which were significantly lower than that for Pt/LpSi. Cyclic voltammograms for Pt/LpSi, LpSi, and MWCNTs also support the results of the HER evaluation (Fig. 7 inset). The reduction current for Pt/LpSi increased from ca. −0.5 V vs. RHE and reached −37 mA at −1.44 V vs. RHE, whereas LpSi and MWCNTs exhibited an increase in the onset voltages and smaller current values than that for Pt/LpSi. These results clearly indicate that the as-prepared Pt/LpSi catalyses the HER as an electrocatalyst. An applied voltage of 0.938 V was required for Pt/LpSi to obtain a reduction current of 10 mA, which is approximately 36 times higher than that for previously reported commercial Pt-NPs/carbon (25.9 mV@10 mA),17 and indicates that Pt/LpSi requires a higher overpotential for the HER. This is presumably due to the low conductivity of the oxidized LpSi loaded with Pt-NPs. To reduce the overpotential of Pt/LpSi, it is essential to improve the electrical conductivity of Pt/LpSi by chemical doping of the Si framework of LpSi.
 | ||
| Fig. 7 Hydrogen evolution profiles and cyclic voltammograms (inset) for Pt/LpSi (a), LpSi (b) and MWCNTs (c). The electrochemical HER was carried out under a potentiostatic condition of −1.5 V vs. Ag/AgCl (−0.94 V vs. RHE). The vertical axis of (B) expresses the cathodic current as negative. | ||
XRD patterns for the Pt/LpSi electrode before and after the HER evaluation show diffraction peaks due to CP and fcc Pt with no significant changes (Fig. S11, ESI†). Cyclic voltammograms show that the onset voltage for Pt/LpSi is almost the same before and after the HER evaluation, although the reduction current after the HER evaluation increases at more negative applied voltages than −0.5 V vs. RHE (Fig. S12, ESI†). The increase in the reduction current after the HER evaluation can be attributed to the desorption of oxygen adsorbed on the Pt-NPs surface during the HER. However, an XPS spectrum of the Pt/LpSi after the HER evaluation shows a doublet peak characteristic of Pt0 with oxygen adsorbed on the surface, which is in contrast to the cyclic voltammogram results (Fig. S13, ESI†). One possible reason for the XPS results is that the Pt-NP surfaces reabsorbed oxygen due to exposure of Pt/LpSi to the atmosphere after the HER evaluation. These results indicate that Pt/LpSi can continuously catalyse electrochemical reactions without any change in crystallinity or electronic state during the reaction.
Conclusion
In this study, we have successfully demonstrated the recovery of Pt ions from aqueous media containing various ionic species using the reducibility of LpSi. The reduction reaction using LpSi proceeds selectively toward Pt ions at normal temperature and pressure and is also effective for the recovery of extremely low concentrations of Pt ions dissolved in ASW. Pt-NPs formed on LpSi were easily isolated by alkali treatment of Pt/LpSi and filtration. The resultant Pt/LpSi also acted as an electrochemical catalyst for the HER by water splitting, and quantitative H2 evolution proceeded on Pt-NPs in response to the charge supplied to Pt/LpSi. Pt-NPs formed on LpSi can be used as an electrochemical catalyst without isolation from LpSi, so that LpSi can serve as not only a recovery agent for Pt ions but also as a catalyst support. Although the electrochemical catalytic activity of Pt/LpSi is not sufficiently high at present, it is expected that it could be improved by further optimization, such as by increasing the electrical conductivity of LpSi. The electrochemical behavior of LpSi changed dramatically following loading of Pt-NPs, and these changes could also be used for monitoring Pt recovery processes or for sensing Pt ions. The unique reduction properties of LpSi are applicable for substances with a higher redox potential than the Si–H group; therefore, LpSi is expected to be applied as a new recovery/recycling and sensing technology for other precious metal ions that have high redox potentials, such as Au, Ag, Cu, Pd, Rh and Ir, dissolved at low concentrations in the natural environment and in industrial wastewater. LpSi is a low environmental impact solid material consisting only of H and Si, which are safe and abundant elements; therefore, it is also expected that LpSi could be applied to the removal of harmful ions, such as Os or Hg, that have leaked into aqueous environments.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to extend special thanks to Mr Naonari Sakamoto (TCRDL) for XPS measurements and Dr Takeo Arai (TCRDL) for helpful discussions.Notes and references
- M. K. Jha, J.-C. Lee, M.-s Kim, J. Jeong, B.-S. Kim and V. Kumar, Hydrometallurgy, 2013, 133, 23–32 CrossRef CAS.
- F. Reith, S. G. Campbell, A. S. Ball, A. Pring and G. Southam, Earth Sci. Rev., 2014, 131, 1–21 CrossRef CAS.
- A.-Q. Zhang, L.-J. Cai, L. Sui, D.-J. Qian and M. Chen, Polym. Rev., 2013, 53, 240–276 CrossRef CAS.
- H. Umeda, A. Sasaki, K. Takahashi, K. Haga, Y. Takasaki and A. Shibayama, Mater. Trans., 2011, 52, 1462–1470 CrossRef CAS.
- M. Moldovan, M. A. Palacios, M. M. Gómez, G. Morrison, S. Rauch, C. McLeod, R. Ma, S. Caroli, A. Alimonti, F. Petrucci, B. Bocca, P. Schramel, M. Zischka, C. Pettersson, U. Wass, M. Luna, J. C. Saenz and J. Santamaría, Sci. Total Environ, 2002, 296, 199–208 CrossRef CAS PubMed.
- V. Queirós, U. M. Azeiteiro, A. M. V. M. Soares and R. Freitas, J. Hazard. Mater., 2021, 412, 125028 CrossRef PubMed.
- N. Vyas, A. Turner and G. Sewell, Sci. Total Environ, 2014, 493, 324–329 CrossRef CAS PubMed.
- A. Suzuki, H. Obata, A. Okubo and T. Gamo, Mar. Chem., 2014, 166, 114–121 CrossRef CAS.
- M. Abdou, J. Schäfer, A. Cobelo-García, P. Neira, J. C. J. Petit, D. Auger, J. F. Chiffoleau and G. Blanc, Mar. Chem., 2016, 185, 104–110 CrossRef CAS.
- H. Nakano and M. Ohashi, in Silicene: Structure, Properties and Applications, ed. M. J. S. Spencer and T. Morishita, Springer International Publishing, Cham, 2016, vol. 235, pp. 85–106 Search PubMed.
- M. Ohashi, R. Yaokawa, Y. Takatani and H. Nakano, ChemNanoMat, 2017, 3, 534–537 CrossRef CAS.
- S. Yamanaka, H. Matsu and M. Ishikawa, Mater. Res. Bull., 1996, 31, 307–316 CrossRef CAS.
- S. Yamanaka, H. Itoh and M. Hattori, in Expanded Clays and Other Microporous Solids, ed. M. L. Occelli and H. E. Robson, Springer Science & Business Media, 1992, ch. 15, pp. 296–317 Search PubMed.
- K. S. Kim, N. Winograd and R. E. Davis, J. Am. Chem. Soc., 1971, 93, 6296–6297 CrossRef CAS.
- A. Cobelo-García, D. E. López-Sánchez, C. Almécija and J. Santos-Echeandía, Mar. Chem., 2013, 150, 11–18 CrossRef.
- J. M. Cosden and R. H. Byrne, Geochim. Cosmochim. Acta, 2003, 67, 1331–1338 CrossRef CAS.
- D. Kobayashi, H. Kobayashi, D. Wu, S. Okazoe, K. Kusada, T. Yamamoto, T. Toriyama, S. Matsumura, S. Kawaguchi, Y. Kubota, S. M. Aspera, H. Nakanishi, S. Arai and H. Kitagawa, J. Am. Chem. Soc., 2020, 142, 17250–17254 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and characterization details, SEM/EDS, XPS, XRD, and time dependence of the cathodic current. See DOI: https://doi.org/10.1039/d3nj01412f |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2023 |