
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInvestigation of uranium oxide hydrates with barium(II) ions: structural diversity, uranium valences and implications†
Kimbal T.
Lu
ab,
Yingjie
Zhang
 *a,
Tao
Wei
a,
Timothy A.
Ablott
a,
Jakub
Plášil
c,
Inna
Karatchevtseva
a and
Rongkun
Zheng
b
*a,
Tao
Wei
a,
Timothy A.
Ablott
a,
Jakub
Plášil
c,
Inna
Karatchevtseva
a and
Rongkun
Zheng
b
aAustralian Nuclear Science and Technology Organisation, Locked Bag 2001, Kirrawee DC, NSW 2232, Australia. E-mail: yzx@ansto.gov.au
bSchool of Physics and Advanced Materials, University of Sydney, Ultimo, New South Wales 2007, Australia
cInstitute of Physics ASCR, v.v.i., Na Slovance 2, 18221, Praha 8, Czech Republic
First published on 26th June 2023
Abstract
Although barium(II) ions play an important role in the natural weathering of the mineral uraninite (UO2+x) and are expected to therefore do the same for spent nuclear fuel in geological disposal, the detailed structural understanding of this role is rather limited. In this work, we report the synthesis and characterisation of two new uranium oxide hydrate (UOH) phases with barium(II) ions, a layer-structured UOH-Ba1 constructed with undulating uranyl oxide hydroxide layers incorporating hydrated interlayer barium(II) ions; and UOH-Ba2 forming a complex three-dimensional structure by linking six different types of uranium polyhedrons with barium(II) ions located in the narrow channels. Both crystal structures were revealed using synchrotron single crystal X-ray diffraction and confirmed with powder X-ray or electron diffractions. While vibrational modes were elucidated by Raman, electronic structures and the presence of pentavalent uranium were investigated using diffuse reflectance spectroscopy. This work has provided insights into the complexity of uranium crystal chemistry in the presence of barium(II) ions and has implications to both uranium geochemistry in terms of the natural weathering of uraninite and the disposal of spent nuclear fuel in the underground repository.
1. Introduction
Uranium minerals and synthetic compounds have been extensively studied, owing to the revived interest in nuclear energy to mitigate the effects of climate change.1 The in-depth understanding of uranium geochemistry and structural chemistry is directly relevant to the current uranium-based nuclear fuel cycle.2 The uranium oxide mineral uraninite (UO2+x: x = 0–0.25)3 has been used as the natural analogue for the study of spent nuclear fuel (SNF) as the SNF discharged from the nuclear reactor, usually in the form of UO2, is highly radioactive and poses significant risks to the general public.4 As one of the candidate solutions, the direct long-term disposal of this SNF in the geologically stable underground repository is the most attractive SNF management strategy for countries where SNF reprocessing facilities are not available.5–7 As an international pioneer, Finland is on schedule to operate the world first underground repository for SNF in 2024.8 However, there are still concerns on the possible interactions of SNF including fission products and radionuclides from the decay process with the surrounding materials in the presence of underground water.9 While the geological repositories are designed to have a reducing environment where SNF will remain stable, a possible accident could occur where underground water may be in contact with SNF during the long-term storage, leading to the alteration of SNF.10 A 10-year long drip-test conducted in the 1990s has confirmed that SNF undergoes the same oxidation–hydration alteration pattern to that of uraninite.11Comparable to the natural weathering of uraninite, the alteration of SNF can be viewed as an oxidation–hydration process where tetravalent U4+ is progressively converted to hexavalent U6+. The U6+ is prominently present as uranyl ions (UO2)2+,12,13 which are further bonded with O/OH ligands in the equatorial planes to form tetragonal, pentagonal, and hexagonal uranium polyhedra. These uranyl moieties then link together via edge- or corner-sharing to form uranyl oxide hydroxide layers which will incorporate either water molecules (if the layers are charge neutral) or interlayer cations from the nearby environment to form uranium oxide hydrate (UOH) phases/compounds.14,15 So far there are more than two dozen UOH minerals incorporating monovalent (alkali),16,17 divalent (alkaline earth, transition metals and p-block metals),18–20 or a combination of mono- and divalent metal ions21 as interlayer cations for charge compensation. They differ mainly in the secondary metal ions and the O/OH ratios largely dependent on the fluid chemical composition and pH.14 In addition, the uranyl oxide hydroxide layer topologies for various UOH minerals have been comprehensively reviewed.22
Hydrothermal synthesis in the laboratory has been used to assist the formation of the UOH compounds in a simulated natural environment. To date, more than two dozen of synthetic UOH compounds have been made with interlayer cations including alkali,23,24 alkaline earth,25,26 lead,27 limited 3d transition metal28–30 and lanthanide ions.31–34 Complicating these UOH systems are a further number of compounds which have a slightly different structure where the apical oxygen of the uranyl ions on the layer shares the equatorial oxygen with an interlayer uranyl ion acting as a pillar, resulting in an open three-dimensional (3D) framework structure typically incorporating various cations in the framework channels. Synthetic compounds of this type of structure, with various cations such as (NH4)+,35 Sr2+,36 Y3+ and Tb3+,37 Eu3+, Gd3+,38 Sm3+,39 and U4+40 ions have been synthesised and extensively studied, highlighting the complexity and flexibility of the UOH systems. Furthermore, other types of 3D structures with Cs+36 and Pb2+27 are also being revealed.
In examining the SNF environment, barium is anticipated to play an important role during the early alteration of SNF due to its abundance in rocks and groundwater (0.0425% in the Earth crust),41 and more importantly being a fission product from the nuclear fission of 235-uranium (up to 2% in the SNF depending on the fuel burn up).42 Several uranyl minerals containing barium exist in nature including protasite (Ba[(UO2)3O3(OH)2](H2O)3),43 billietite (Ba[(UO2)3O2(OH)3]2(H2O)4),44 bauranoite (Ba[(UO2)(O, OH)](H2O)4.5),45 and in trace amount as in calciouranoite [(Ca, Ba, Pb)U2O7·5(H2O)],46 meta-calciouranoite [(Ca, Na, Ba)U2O7·2H2O],46 and wölsendorfite [(Pb, Ca, Ba)U2O7·2H2O].47 With these minerals making up a reasonable percentage of the UOH minerals in the early stage of the alterations of uraninite, this appears to confirm that barium is likely to be involved in the alteration pathways of SNF. Detailed study of protasite and billietite have found that both minerals form layered UOH structures stabilised by the interlayer barium cations. However, protasite has an α-U3O8 type uranyl layer structure whilst billietite adopts both the α-U3O8 and β-U3O8 uranyl layer structures (Fig. 1), suggesting geological factors influence the preferred formation of one structure type over the other. Limiting the understanding of these factors is the scarcity of structural information, with the crystal structure of bauranoite unknown.45 Interestingly, the significant difference in the charge deficiency per anion (CDA) value between protasite and billietite, a value being used to measure the degree of oxidative alteration with a higher value indicating a later stage of alteration, suggests the possible existence of unexplored intermediate phases that could elucidate the factors governing the transition from β-U3O8 to α-U3O8 type uranyl oxide hydroxide layer in UOH-Ba system. With the difficulties in finding the suitable single crystal in nature and the limited number of minerals of which to study, hindering this progress, the recent success in the hydrothermal synthesis of UOH compounds can ideally overcome this challenge.34
 | ||
| Fig. 1 Polyhedral views of uranyl oxide hydroxide layers: α-U3O8 type (a) and β-U3O8 type (b). | ||
There is only one reported synthetic UOH-Ba phase, made via a hydrothermal reaction of schoepite and barium nitrate at 200 °C for 10 h. The compound was characterised using X-ray diffraction and IR but no detailed structural data were available.48 Thus, there still exists the need to obtain the structural details of these UOH-Ba phases, along with their derived CDA values, in order to provide an insight into their alteration pathways towards the safe management of SNF via long-term disposal in the geological repository.
The search for these intermediate UOH-Ba phases was carried out in this project using hydrothermal synthesis, which resulted in the successful formation of two new UOH-Ba compounds. Importantly, the crystal structures of both compounds were obtained with synchrotron single-crystal X-ray diffraction and verified by powder X-ray and/or electron diffractions. Whereas the UOH-Ba1 compound forms a unique corrugated layered structure with interlayer Ba2+ ions, the UOH-Ba2 is made up of a complex 1D and 2D arrays of both pentagonal and tetragonal uranyl polyhedra forming narrow opened channels which house the Ba2+ ions. Diffuse reflectance spectroscopy further confirmed the existence of pentavalent uranium in an octahedral coordination environment in UOH-Ba2. The results from this work, together with existing literature, provide a foundation to enhance our current understanding in the possible alteration pathway of SNF when in contact with a fluid containing Ba2+ ions under an oxidative environment.
2. Experimental
2.1. Materials synthesis
In the literature, a few UOH minerals are known to contain Ba2+ ions, major in bauranoite, billietite and protasite, and minor in calciouranoite, meta-calciouranoite, and wölsendorfite.43–47 However, the UOH minerals can provide only limited structural information on the role of Ba2+ ions in the natural weathering of uraninite and the subsequent formations of various uranyl minerals. More detailed structural understanding requires further studies through synthetic phases. Unfortunately, only one synthetic compound with the general formula of BaU2O7(H2O)3 (refer to UOH-Ba in Table 1) was made via a hydrothermal reaction of schoepite and barium nitrate solution at 200 °C.48 Although it was characterised by elemental analysis, PXRD and TGA, crystal structure data were not available.| Compound | Precursors | Synthesis conditions | Final product | Ref | |||
|---|---|---|---|---|---|---|---|
| U/Ba | Temp. | Formula | pH | U/Ba | |||
| Time | O/OH | ||||||
| UOH-Ba | (UO2)8O2(OH)12·12H2O | 1/1 | 200 °C | BaU2O7(H2O)3 | — | 2 | 48 |
| Ba(NO3)2 | 10 h | — | |||||
| UOH-Ba1 | U3O8 | 1/2 | 240 °C | Ba(H2O)2[(UO2)4O3(OH)4] | 3.62 | 4 | This work |
| Ba(NO3)2 and Na2CO3 | 24 h | 2.75 | |||||
| UOH-Ba1s | (UO2)8O2(OH)12·12H2O Ba(NO3)2 | 1/2 | 240 °C | Ba(H2O)2[(UO2)4O3(OH)4] | — | 42.75 | This work |
| 72 h | |||||||
| UOH-Ba2 | UO2(NO3)2·6H2O | 1/1 | 240 °C | Ba2[(UO2)10UO12(OH)6 (H2O)2.5] | 3.51 | 5.5 | This work |
| Ba(NO3)2 and NaOH | 24 h | 5.25 | |||||
Schoepite [(UO2)8O2(OH)12·12H2O],49 uranium oxide (U3O8) and uranyl nitrate hexahydrate with natural uranium isotopic abundance were used as the uranium precursors. Compounds containing uranium are radioactive and should be handled in the regulated laboratories. Other chemicals in A. R. grade were purchased from Sigma-Aldrich (Merck).
2.2. Materials characterization
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ba ratios. Transmission electron microscopy (TEM) analysis was carried out with a JEOL 2200FS (JEOL Ltd, Japan) operated at 200 kV, fitted with an Oxford X-Max silicon drift detector for Energy Dispersive X-ray analysis (EDX). TEM specimens were prepared by crushing crystalline compounds in ethanol and dispensing them on holey carbon supported films.
:Ba ratios. Transmission electron microscopy (TEM) analysis was carried out with a JEOL 2200FS (JEOL Ltd, Japan) operated at 200 kV, fitted with an Oxford X-Max silicon drift detector for Energy Dispersive X-ray analysis (EDX). TEM specimens were prepared by crushing crystalline compounds in ethanol and dispensing them on holey carbon supported films.
2.3. Synchrotron single crystal X-ray diffraction
The single crystal data for UOH-Ba1 (CSD-2247026) and UOH-Ba2 (CSD-2247027) were collected at 100(2) K on the MX2 beamline50 at the Australian Synchrotron employing silicon double crystal monochromated synchrotron radiation (λ = 0.71078–0.71093 Å). Data integration and reduction were undertaken with XDS.51 SADABS was applied to the data for absorption corrections.52 The structures were solved by direct methods53 and refined with SHELXL-201454 using the Olex2 graphical user interface.55 All atoms except hydrogen were located on the electron density maps and refined anisotropically. Hydrogen atoms on possible hydroxyl groups and water molecules were unable to be located and thus were omitted in the final structure refinements. Note the diffraction data for UOH-Ba1 could be indexed to either the monoclinic or triclinic space groups. However, the subsequent structure refinement for the monoclinic structure did not make chemical sense. So the final crystal structure was refined in the triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group. The data completeness was a bit low due to only one-circle data collection possible.
space group. The data completeness was a bit low due to only one-circle data collection possible.
3. Results and discussion
3.1. Synthesis and characterization
In this work, two new UOH compounds were successfully synthesised hydrothermally at 240 °C. Compound UOH-Ba1 was synthesised using a mixture of U3O8 and Ba(NO3)2 in water and pH adjusted with Na2CO3. It was also synthesised via a hydrothermal reaction of schoepite and Ba(NO3)2. On the other hand, compound UOH-Ba2 was made by a hydrothermal reaction of UO2(NO3)·6H2O and Ba(NO3)2, with pH adjusted by adding a diluted NaOH solution. Both UOH-Ba1 and UOH-Ba2 formed in solutions with a narrow range of final solution pH 3.62 and 3.51, respectively, underpinning the importance of solution pHs in governing the uranyl hydrolysis and subsequent effects on the formation of synthetic UOH phases.14 However, uranium precursors also affect solution pH and uranium hydrolysis, further influencing the formation and structure of the final UOH phases. The synthetic UOH phases with Ba2+ ions discussed above, together with synthesis conditions are summarised in Table 1.Both compounds were subsequently characterised for structure, microstructure, and elemental ratios. PXRD patterns of both compounds matched the simulated patterns calculated from their single crystal data (Fig. S1, ESI†), confirming that pure phase materials were obtained. The backscattered SEM images (Fig. 2) showed a needle (UOH-Ba1) or a block (UOH-Ba2) crystal morphology, different from the previously reported thin-plate crystal morphology for layer structured UOH compounds.31–33 SEM-EDS multipoint analyses confirmed that they contain U, Ba, and O with the U/Ba ratio of 4 and 5.5 for UOH-Ba1/UOH-Ba1s and UOH-Ba2, respectively (Fig. S2–S4, ESI†).
 | ||
| Fig. 2 Backscattered SEM images of UOH-Ba1 (a) and UOH-Ba2 (b). | ||
3.2. Crystal structures and discussion
The single crystal data and structural refinement details for both compounds are summarised in Table 2, with selected bond lengths and angles listed in Tables S1–S2, ESI.† Compound UOH-Ba1 crystallises in the triclinic P space group. The crystal structure is constructed with undulating uranyl oxide hydroxide layers with interlayer hydrated Ba2+ ions (Fig. 3). There are two unique uranyl moieties (U1 and U2) in pentagonal bipyramidal polyhedrons, with the U![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond lengths ranging from 1.77(3) to 1.79(4) Å and OUO angles from 176.3(18)° to 177.3(17)°. The U–O bond lengths in the equatorial planes range from 2.20(4) to 2.37(4) Å (Table S1, ESI†). These typical UO and U–O bonds for the uranyl moieties are similar to other compounds containing uranyl ions such as UOH minerals and synthetic compounds.56 Either U1 or U2 polyhedrons share edges to form 1D zigzag arrays that are connected to each other repeatedly via corner-sharing between the two types of arrays to form the corrugated uranyl oxide hydroxide layers with interlayer Ba2+ ions (Fig. 3). The Ba2+ ions are 11-fold coordinated with seven uranyl O atoms (O1/O1′/O2/O2′/O4/O4′/O5), two equatorial O atoms (O3 and O6), and two H2O molecules (O9/O9′), with Ba–O bonds ranging from 2.72(3) to 3.08(3) Å for uranyl O atoms, 3.26(4) to 3.29(3) Å for equatorial O atoms and 2.865(5) to 2.869(5) Å for H2O molecules (Table S1, ESI†). Note the relatively longer Ba–O distances for the equatorial O atoms (O3 and O6) are just beyond the normal Ba–O bond range, suggesting weak bonding nature. Such weak bonding between equatorial O atoms and interlayer cations are unique and can exist only in layered UOH compounds with corrugated uranyl oxide hydroxide layers.
O bond lengths ranging from 1.77(3) to 1.79(4) Å and OUO angles from 176.3(18)° to 177.3(17)°. The U–O bond lengths in the equatorial planes range from 2.20(4) to 2.37(4) Å (Table S1, ESI†). These typical UO and U–O bonds for the uranyl moieties are similar to other compounds containing uranyl ions such as UOH minerals and synthetic compounds.56 Either U1 or U2 polyhedrons share edges to form 1D zigzag arrays that are connected to each other repeatedly via corner-sharing between the two types of arrays to form the corrugated uranyl oxide hydroxide layers with interlayer Ba2+ ions (Fig. 3). The Ba2+ ions are 11-fold coordinated with seven uranyl O atoms (O1/O1′/O2/O2′/O4/O4′/O5), two equatorial O atoms (O3 and O6), and two H2O molecules (O9/O9′), with Ba–O bonds ranging from 2.72(3) to 3.08(3) Å for uranyl O atoms, 3.26(4) to 3.29(3) Å for equatorial O atoms and 2.865(5) to 2.869(5) Å for H2O molecules (Table S1, ESI†). Note the relatively longer Ba–O distances for the equatorial O atoms (O3 and O6) are just beyond the normal Ba–O bond range, suggesting weak bonding nature. Such weak bonding between equatorial O atoms and interlayer cations are unique and can exist only in layered UOH compounds with corrugated uranyl oxide hydroxide layers.
| Compounds | UOH-Ba1 | UOH-Ba2 |
|---|---|---|
| a R 1 = Σ‖Fo| − |Fc‖/|Fo|; wR2 = {Σ[w(Fo2 – Fc2)2]/Σ[w(Fo2)2]}1/2. | ||
| Empirical formula | BaO17U4 | Ba2O40.50U11 |
| Formula weight | 1361.46 | 3541.01 |
| Temperature/K | 100(2) | 100(2) |
| Crystal system | Triclinic | Monoclinic |
| Space group |
P |
C2/c |
| a/Å | 4.2320(9) | 13.251(3) |
| b/Å | 8.1990(16) | 10.389(2) |
| c/Å | 10.649(2) | 25.130(5) |
| α/° | 87.40(3) | — |
| β/° | 78.01(3) | 90.33(3) |
| γ/° | 75.01(3) | — |
| Volume/Å3 | 349.79(14) | 3459.5(12) |
| Z | 1 | 8 |
| ρ calc/g cm−3 | 6.463 | 6.799 |
| μ/mm−1 | 49.001 | 53.640 |
| F(000) | 560.0 | 5792.0 |
| Radiation | Synchrotron (λ = 0.71093) | Synchrotron (λ = 0.71078) |
| 2θ range for data collection/° | 3.904 to 49.928 | 3.242 to 51.996 |
| Index ranges | −4 ≤ h ≤ 4, −9 ≤ k ≤ 9, −12 ≤ l ≤ 12 | −15 ≤ h ≤ 15, −12 ≤ k ≤ 12, −29 ≤ l ≤ 29 |
| Reflections collected | 3822 | 21522 |
| Independent reflections | 1079 [Rint = 0.0601, Rsigma = 0.0630] | 3316 [Rint = 0.0516, Rsigma = 0.0325] |
| Data/restraints/parameters | 1079/105/94 | 3316/309/236 |
| Goodness-of-fit on F2 | 1.143 | 1.092 |
| Final R indexes [I > =2σ (I)]a | R 1 = 0.0812, wR2 = 0.2197 | R 1 = 0.0295, wR2 = 0.0751 |
| Final R indexes [all data]a | R 1 = 0.1128, wR2 = 0.2902 | R 1 = 0.0303, wR2 = 0.0757 |
| Largest diff. peak/hole/e Å−3 | 4.84/−4.59 | 3.49/−3.10 |
 | ||
| Fig. 3 Crystal structure of compound UOH-Ba1: a 2D layered structure with interlayer Ba2+ ions (a), and two types of pentagonal bipyramidal uranium polyhedrons (U1 and U2) linking into the corrugated uranyl oxide hydroxide layers (b). | ||
Compound UOH-Ba2 crystallises in the monoclinic C2/c space group. The complex 3D structure comprises of six unique uranium centres, four (U2-U5) in pentagonal bipyramids, one (U1) in a square bipyramid, and one (U6) in an octahedral geometry (Fig. 4). The crystal structure is constructed as stacking of multiple layers of three different building arrays repeatedly, parallel to the (001) plane (Fig. 4a and b). The first 1D chains are formed by edge-sharing of double U1 and U2 polyhedra (Fig. 4c). The second 1D chains are formed by corner-sharing of a repeated edge-sharing U3–U4 pairs (Fig. 4d). The third 2D nets are formed by corner-sharing of U5 and U6 polyhedra at an angle of approximate 76.786° with each U5 linking to two U6 and each U6 linking to four U5 (Fig. 4e and f). The 2D nets are linked to the 1D arrays of U3–U4 pairs on both sides, with one side rotating 90° forming the sandwich-type structural building blocks that are linked between 1D arrays of U3–U4 and U1–U2 (Fig. 4c), via the parallel 1D arrays of U1–U2 by rotating 90° constantly (Fig. 4a and b). The 1D arrays of U3–U4 are connected by the 1D arrays of U1–U2 via uranyl cation–cation interactions resulting in a tilting of approximately 115°. The overall structure could be simplified as layers of U3-U4 and U5-U6 sandwiching layers of U1-U2 to form a 3D open framework, with Ba2+ ions in the narrow channels (Fig. 4a and b).
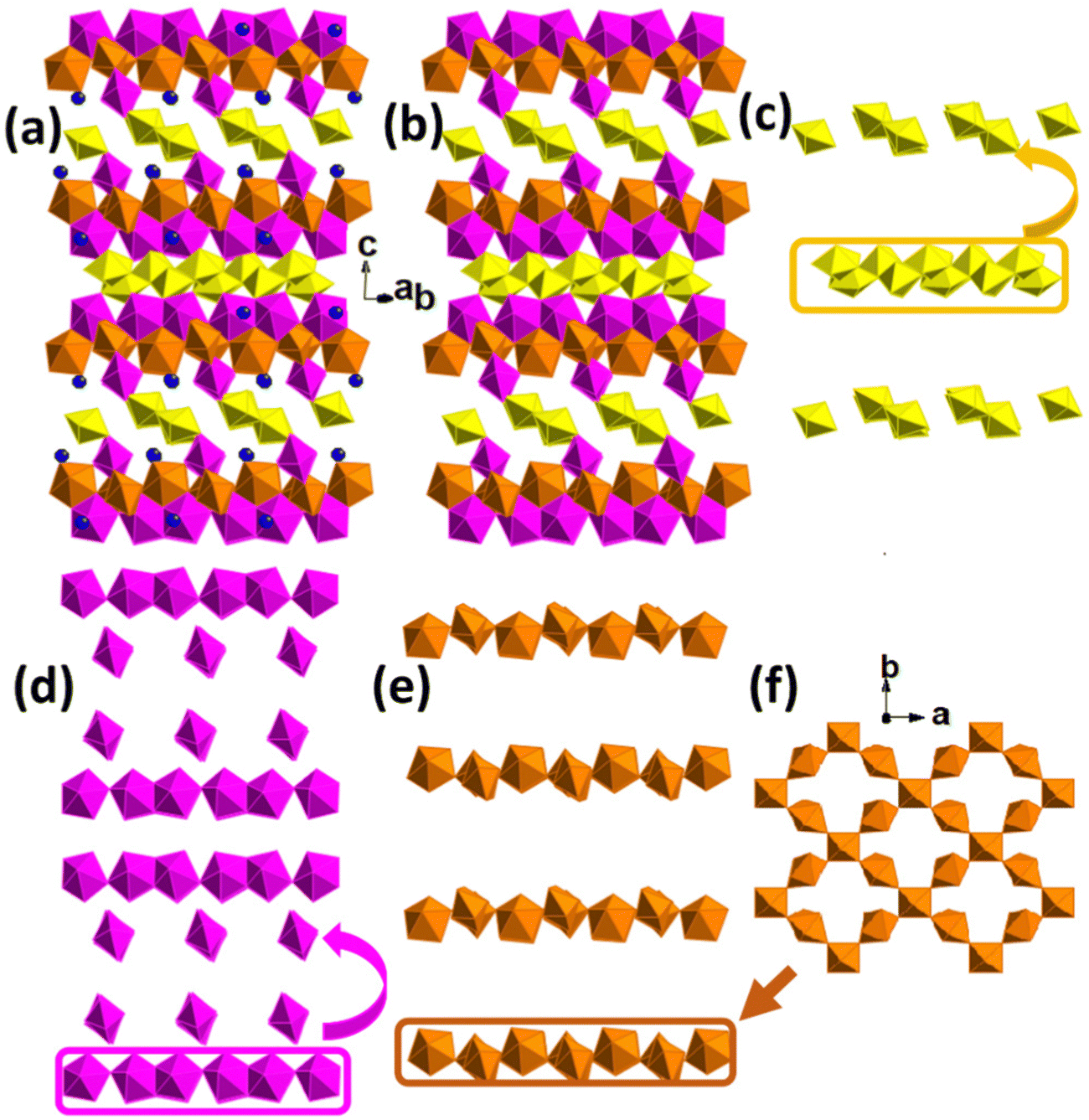 | ||
| Fig. 4 Crystal structure of UOH-Ba2: overall 3D polyhedral crystal structure (a), the same as in (a) but without Ba2+ ions (b), double U1 and U2 edge-sharing to form Zigzag 1D chains (c), pairs of edge-sharing U3 and U4 connected by corner-sharing to form 1D chains (d), U5 and U6 connected by corner-sharing to form 2D nets [(e) and (f)]. Uranium polyhedrons: U1 and U2 are in yellow, U3 and U4 in scarlet, U5 and U6 in orange; Ba in blue spheres. | ||
For uranyl U2–U5, the UO bond lengths range from 1.773(7) to 1.828(7) Å with OUO angles from 176.7(3) to 178.7(3)°, and U–O bond lengths in the equatorial planes ranging from 2.196(7) to 2.583(7) Å (Table S2, ESI†). For U1 a in square bipyramid, UO bond lengths are 1.838(7) and 1.841(7) Å with the OUO angle of 175.9(3)°, and U–O bond lengths in the equatorial plane from 2.139(7) to 2.422(7) Å. The U6 does not show a uranyl centre and it has an octahedral coordination environment with six U–O bonds from 2.052(7) to 2.102(7) Å. The Ba2+ ion is in a 10-fold coordination environment, eight uranyl O atoms with Ba–O bond lengths from 2.717(7) to 2.947(8) Å, and two H2O molecules with Ba–O bond lengths of 3.073(7) and 3.306(6) Å (Table S2, ESI†). The longer than normal Ba–O bond range exists for O21, a disordered H2O molecule in two partially occupied positions.
The bond valence sums (BVS)56,57 were calculated using U6+ parameters (RU–O = 2.051; B = 0.519),57 with BVS results for both compounds listed in Tables S3–S4 (ESI†). The BVS values for UOH-Ba1 (6.60 for U1, 6.59 for U2 and 2.14 for Ba1 in Table S3, ESI†) suggest hexavalent uranium for uranyl groups and Ba2+ ion. In addition, majority of O atoms are O, with OH for O7 and O8, and H2O for O9. The unit cell contains four uranyl units (2U1 and 2U2), one Ba2+ ion, three O (O3 and O6), four OH− (2O7 and 2O8) and two coordinated H2O molecules (2O9), leading to the formula Ba(H2O)2[(UO2)4O3(OH)4]. While four uranium centres (U2-U5) in UOH-Ba2 are indicative of hexavalent, both U1 and U6 in 8-fold coordination may be in pentavalent state with BVS values of 5.66 and 5.78 (Table S4, ESI†). The BVS values also suggest Ba2+ (2.21) and majority of O atoms as O, with OH for O3, O13 and O17, and H2O for O9 and O21. The presence of U(V) in the octahedral environment was confirmed using DRS (Section 3.5 below). The unit cell contains 8 Ba2+ ions, 44 U (40 uranyl units and 4 octahedrons), 164 O (128 O, 24 OH and 12 H2O), leading to the formula Ba2(H2O)[(UO2)10UO12(OH)6 (H2O)2].
So far, three UOH minerals incorporating only Ba2+ ions in their crystal structures have been identified, demonstrating the importance of Ba2+ ions in the natural weathering of uraninite. Protasite and billietite were first reported in 1987,43 with billietite structure being revisited in 2006.44 However, crystal structure of mineral bauranoite (Ba[(UO2)(O,OH)](H2O)4.5) remains unknown.45 While protasite contains the α-U3O8 type uranyl oxide hydroxide layers (Fig. 5a) with interlayer hydrated Ba2+ ions, billietite adopts both the α-U3O8 and β-U3O8 uranyl oxide hydroxide layers (Fig. 5c). In contrast to the two known UOH minerals with Ba2+ ions, the uranyl oxide hydroxide layers for compound UOH-Ba1 contains two types of pentagonal pyramidal uranyl polyhedrons linking parallelly to form opposite distorted triangles between them in zigzag pattern (Fig. 5b). As such, UOH-Ba1 has corrugated uranium oxide hydroxide layers that is not common but observed previously in UOH mineral curite58 and a few synthetic UOH compounds such as Na[(UO2)4O2(OH)5](H2O)259 and Ca(UO2)4O3(OH)4(H2O)2.25
 | ||
| Fig. 5 Polyhedral uranyl oxide hydroxide layers: α-U3O8 type layer in protasite (a), UOH-Ba1 (b), and β-U3O8 layer in billietite (c). | ||
In the literature, two synthetic UOH compounds (UOH-Sr60 and UOH-Pb27) were found to have the similar 3D structures with UOH-Ba2 and their structural similarities and differences are summarised in Table 3. Apart from having the same monoclinic C2/c space group and similar cell parameters, the obvious difference is the smaller β angle for UOH-Ba2 (90.33°) in comparison to around 103.20° for UOH-Pb and UOH-Sr. It is also interesting to note that only 2+ metal ions with relatively large ionic radii (1.35–1.52 Å) such as Pb2+, Sr2+ and Ba2+ can stabilise this structure type. It seems to suggest that the larger 2+ metal ions allow high coordination numbers (9 to 10), which may be the key factor for stabilising such a complex 3D structure, highlighting the complexity and flexibility of UOH systems in the presence of larger 2+ metal ions. As U(V) in the octahedral coordination environment was confirmed for both UOH-Sr and UOH-Ba2, it is very likely that UOH-Pb would have the U(V) centre as well.
| Synthetic phase and formula | Structure | Cell parameters | [n]M | M-ionic radii (Å) | M–O (Å) | U(V) |
|---|---|---|---|---|---|---|
| UOH-Pb 27 | C2/c | a = 13.281(5), b = 10.223(4), c = 26.10(1) Å; β = 103.202(6)° | [9] | 1.35 | 2.53(3) –3.07(3) | — |
| Pb2(H2O)[(UO2)10UO12(OH)6(H2O)2] | ||||||
| UOH-Sr 60 | C2/c | a = 13.289(3), b = 10.170(2), c = 26.139(5) Å; β = 103.04(3)° | [10] | 1.36 | 2.574(18)–3.004(10) | yes |
| Sr2(H2O)[(UO2)10UO12(OH)6(H2O)2] | ||||||
| UOH-Ba2 (this work) | C2/c | a = 13.251(3), b = 10.389(2), c = 25.130(5) Å; β = 90.33(3)° | [10] | 1.52 | 2.717(7)–3.306(6) | yes |
| Ba2(H2O)[(UO2)10UO12(OH)6(H2O)2] |
3.3. Transmission electron microscopy
Compound UOH-Ba1 was further studied by TEM. The key elements of U, Ba and O were confirmed by TEM-EDS. Bright field TEM images of UOH-Ba1 (Fig. 6a and b) showed long needle crystals. Selected area electron diffraction (SAED) patterns in [−1 −1 −1] zone axis (Fig. 6c) and [2 −1 −1] zone axis (Fig. 6d) were both indexed to triclinic P space group, consistent with the results from single crystal and powder XRD.
 | ||
| Fig. 6 TEM of UOH-Ba1: A bright field TEM image (a), a TEM image showing grains in higher magnification (b), SAED patterns from a grain in [−1 −1 −1] zone axis (c) and a grain in [2 −1 −1] zone axis (d) indexed to the P space group. | ||
3.4. Electronic structures and uranium valences
As the calculated BVS values indicating the possible presence of pentavalent uranium in the octahedral coordination site in UOH-Ba2, the electronic structures were investigated by DRS. The DRS spectra of UOH-Ba1 and UOH-Ba2 in the UV-vis region (Fig. 7) showed the similar strong and broad absorption bands reflecting the charge transfer feature for U(VI) ions.61,62 However, the spectra in the near infrared (NIR) region clearly showed the presence of pentavalent U in UOH-Ba2 as the sharp absorption band at 1448 nm due to U(V) ion in 5f1 configuration.63–66 The absorption band (1448 nm) is very close to the value (1445 nm) observed for the non-uranyl U(V) centre in an octahedral coordination environment,64 consistent with the crystallographic result in this work. In contrast, U(V) in an 8-fold coordination environment would give the absorption band at much longer wavelengths (∼1615).67 Note the presence of U(V) in several oxides including minerals and synthetic compounds were reported earlier, but none of them contain pentavalent uranyl ion.68 The weak and broad absorption for UOH-Ba1 in the same area was due to the presence of H2O molecules. As such, UOH-Ba2 is a UOH compound with mixed U valences, major in U(VI) and minor in U(V). Mixed uranium valences are rarely observed in UOH systems,40,69 and a complex 3D UOH structure with mixed-valence uranium in UOH-Ba2 would be rather rare. | ||
| Fig. 7 DR spectra of UOH-Ba1 (a) and UOH-Ba2 (b) with an inset of the spectra in the near infrared regain (1300 to 1600 nm). | ||
3.5. Vibrational modes
Raman spectroscopy was used to probe the vibrational modes. Similar to earlier studies, the signature vibrations for both UOH-Ba1 and UOH-Ba2 (Fig. 8) arise from the uranyl vibrations, with ν1(UO2)2+ in strong and sharp peaks between 700 and 900 cm−1, ν(U3O) and γ[U3(OH)3] in medium and broad peaks at 300 to 600 cm−1, ν2(UO2)2+ in weak and broad peaks at 200 to 300 cm−1 and lattice vibrations at ≤150 cm−1.70–72 It is apparent that ν1(UO2)2+ vibrations split in both compounds reflecting the subtle differences in uranyl moieties. For UOH-Ba1, the three ν1(UO2)2+ vibrations at 779.6 cm−1 (strong), 799.5 cm−1 (weak) and 834.6 cm−1 (strong) correspond to the estimated UO bond lengths of 1.78 to 1.83 Å.71 In contrast, UOH-Ba2 shows multiple splitting bands for ν1(UO2)2+ vibrations, at 740.2 cm−1 (weak), 774.8 cm−1 (strong), 809.4 cm−1 (strong), and 875.0 cm−1 (weak), corresponding to the calculated UO bond lengths of 1.88, 1.84, 1.80 and 1.74 Å, respectively.73 These inferred UO bond values from the Raman vibrations are broadly consistent with the crystal structures with UO bond lengths ranging from 1.773(7) to 1.841(7) Å.
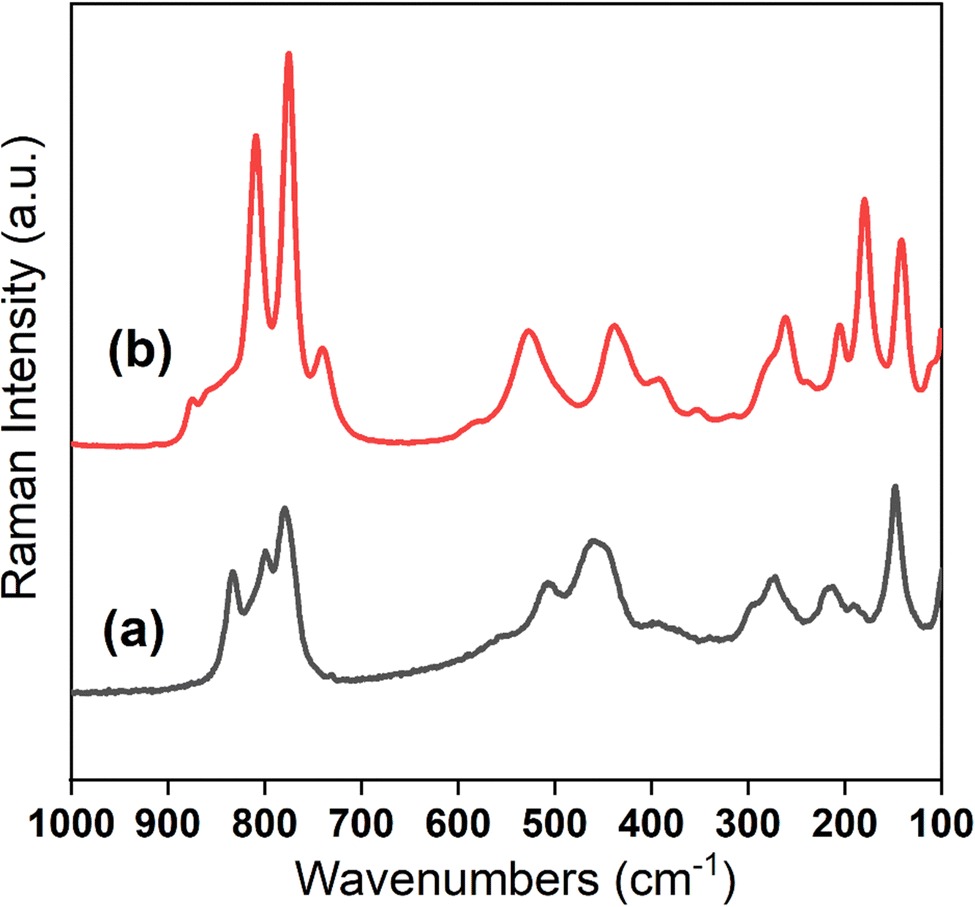 | ||
| Fig. 8 Raman spectra of UOH-Ba1 (a) and UOH-Ba2 (b). | ||
3.6. Thermal stability
The thermal stability of UOH-Ba1 was further investigated. Based on the thermogravimetric and differential thermal analysis results (Fig. S5, ESI†), the thermal decomposition of compound UOH-Ba1 went through a few steps: (1) gradually lost half lattice H2O molecules from 50 to 240 °C with an endotherm at 135 °C (calc. 0.66%, measured 0.6%); (2) lost all H2O molecules and three OH groups from 240 °C to 600 °C in two steps (calc. 5.74%, measured 5.7%); (3) lost final O through UO3 to UO2+x reduction from 800 to 900 °C, with the final residue (BaUO4 + U3O7) (calc. 93.0%, measured 93.5%).3.7. Implications
The charge deficiency per anion (CDA) is a measure of the charge imbalance in the structural units of UOH minerals. As these minerals undergo alteration starting from uraninite, the CDA value increases. The CDA value is closely tied to the average coordination number of oxygens in the structural unit and plays a vital role in determining the crystal-chemical properties of UOH minerals, such as the pH of the liquids they can crystallise in.14 The CDA values, in combination with the U/OH and Me/U ratios, are used to assess the degree of alteration in UOH minerals.Although having more secondary cations in the structure, compound UOH-Ba1 has a CDA value of 0.19, in between the values for billietite (0.15) and protasite (0.22), indicating that it is in between the two minerals in terms of alterations from uraninite. Compound UOH-Ba2 on the other hand has the same CDA value (0.14), the U/OH ratio (1.83), and the Me/U ratio (0.18) to those of compounds UOH-Sr60 and UOH-Pb,27 indicating they could be of the same alteration stage during the oxidative weathering of uraninite.
A weathering path could be seen based on the CDA values of all UOH minerals and synthetic compounds with Ba2+ ions, together with selected UOH minerals (Table 4). An oxidative alteration of SNF starts from UO2 (similar to uraninite) to a mixed valence uranium in UOH-Ba2 with CDA = 0.14 similar to billietite with CDA = 0.15 by gradually taking in Ba2+ ions and balanced by the rearrangement of the pentagonal and square uranyl bipyramidal polyhedra. Up on further alteration, all square bipyramidal uranyl ions transform into pure pentagonal bipyramidal polyhedra while taking in even more interlayer cations resulting in compound UOH-Ba1 with CDA = 0.19. The final stage involves the rearrangement of the pentagonal uranyl polyhedra resulting in a stable α-U3O8 type structure in protasite with CDA = 0.22. However, this is only the observation from the limited information on synthetic UOH materials. The presence of U centres in the square bipyramidal coordination environment among uranyl phosphate hydrate minerals such as autunite (Ca[(UO2)(PO4)]2·10–12H2O)74 clearly highlights the importance of thermodynamic and kinetic factors in controlling the formation of other types of uranyl minerals and synthetic phases.
| UOH minerals and formula14 | Structural unit | CDAa | U/OH | Me/U | [n]U |
|---|---|---|---|---|---|
| a CDA – charge deficiency per anion: calculated as the effective charge of the structural unit divided by the anions in the structural unit. The effective charge is the formal charge plus the charge contributed by the (H)-bonds in the structural unit = n × 0.2. | |||||
| Schoepite | [(UO2)4O(OH)6]0 | 0.08 | 0.67 | 0 | [7] |
| [(UO2)4O(OH)6](H2O)6 | |||||
| Vandendriesscheite | [(UO2)10O6(OH)11]3− | 0.14 | 0.91 | 0.150 | [7] |
| Pb1.5[(UO2)10O6(OH)11](H2O)11 | |||||
| Becquerelite | [(UO2)3O2(OH)3]1− | 0.15 | 1.00 | 0.333 | [7] |
| Ca[(UO2)3O2(OH)3]2(H2O)8 | |||||
| Gauthierite | [(UO2)7O5(OH)7]3− | 0.17 | 1.00 | 0.256 | [7] |
| KPb[(UO2)7O5(OH)7](H2O)8 | |||||
| Fourmarierite | [(UO2)4O3(OH)4]1− | 0.19 | 1.00 | 0.250 | [7] |
| Pb[(UO2)4O3(OH)4](H2O)4 | |||||
| Masuyite | [(UO2)3O3(OH)2]2− | 0.22 | 1.50 | 0.333 | [7] |
| Pb[(UO2)3O3(OH)2](H2O)3 | |||||
| Curite | [(UO2)8O8(OH)6]6− | 0.24 | 1.33 | 0.375 | [6]/[7] |
| Pb3(H2O)2[(UO2)8O8(OH)6](H2O)5 | |||||
| Spriggite | [(UO2)6O8(OH)2]6− | 0.29 | 3.00 | 0.500 | [6]/[7] |
| Pb3[(UO2)6O8(OH)2](H2O)3 | |||||
| Billietite | [(UO2)3O2(OH)3]1− | 0.15 | 1.00 | 0.333 | [7] |
| Ba[(UO2)3O2(OH)3]2(H2O)4 | |||||
| Protasite | [(UO2)3O3(OH)2]2− | 0.22 | 1.50 | 0.333 | [7] |
| Ba[(UO2)3O3(OH)2](H2O)3 | |||||
| Synthetic UOH compounds | Structural unit | CDAa | U/OH | Me/U | [n]U |
|---|---|---|---|---|---|
| UOH-Pb 27 | [(UO2)10UO12(OH)6]4− | 0.14 | 1.83 | 0.18 | [6]/[7] |
| Pb2(H2O)[(UO2)10UO12(OH)6(H2O)2] | |||||
| UOH-Sr 60 | [(UO2)10UO12(OH)6]4− | 0.14 | 1.83 | 0.18 | [6]/[7] |
| Sr2(H2O)[(UO2)10UO12(OH)6(H2O)2] | |||||
| UOH-Ba1 (this work) | [(UO2)4O3(OH)4]2− | 0.19 | 1.00 | 0.25 | [7] |
| Ba[(UO2)4O3(OH)4](H2O)2 | |||||
| UOH-Ba2 (this work) | [(UO2)10UO12(OH)6]4− | 0.14 | 1.83 | 0.18 | [6]/[7] |
| Ba2[(UO2)10UO12(OH)6(H2O)](H2O)1.5 |
Hydrothermal synthesis in the laboratory has shown to be an important method in closing the knowledge gap that is critical to the understanding of the oxidative weathering of UO2 both as mineral uraninite and SNF. The successful synthesis and characterisation of UOH-Ba1 and UOH-Ba2 have filled in the knowledge gaps in the structural understanding on the formation of UOH phases in the presence of Ba2+ ions. This work further reinforces that solution pH plays a vital role in controlling uranium hydrolysis and subsequent formation of UOH phases. Besides, the choice of uranium and secondary metal precursors also affects solution pH value, metal ion hydrolysis speciation and the final product. It is anticipated that a reasonably well designed UOH system can lead to the formation of a variety of UOH products with chosen secondary metal ions, leading to an improved fundamental understanding of potential alteration of SNF under long-term storage/disposal in a geological repository.
4. Conclusions
The primary aim of this work was to reveal the missing structural links in the UOH-Ba system, subsequently to improve our intrinsic understanding the role of Ba2+ ions in the natural weathering and oxidative-hydrolysis alteration of SNF in long-term storage and geological disposal. Consequently, two new synthetic UOH compounds with Ba2+ ions were successfully synthesised hydrothermally and characterised using a variety of structural and spectroscopic techniques. The crystal structures were determined using synchrotron single-crystal XRD and confirmed with PXRD and/or TEM. Compound UOH-Ba1 has an undulating layered structure with interlay Ba2+ ions while UOH-Ba2 has a complex 3D structure built up with several types of uranium polyhedra. The presence of pentavalent U in the octahedral coordination environment in UOH-Ba2 was confirmed with DRS.The successful synthesis and characterisation of these UOH compounds have unravelled the complexity and flexibility of the uranium oxide hydrate system in the presence of Ba2+ ions. The results from the current work, together with the related literature, help not only to improve our fundamental understanding on the possible alteration of SNF but also pave the way for future studies especially on the effect of various synthesis factors such as choices of uranium and secondary metal precursors and controlling of the solution final pH on the formation and structures of UOH phases with other secondary metal ions.
Author contributions
K. T. Lu: data curation, formal analysis, writing – review & editing; Y. Zhang: conceptualization, data curation, formal analysis, project administration, resources, writing – review & editing, supervision; T. Wei: data curation, formal analysis, writing – review & editing; T. A. Ablott: data curation, formal analysis, writing – review & editing; J. Plášil: conceptualization, formal analysis, writing – review & editing; I. Karatchevtseva: data curation, formal analysis, writing – review & editing; R. Zheng: conceptualization, resources, writing – review & editing, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Synthesis and characterisation of materials were performed in the facilities under Nuclear Science and Technology (NST) at ANSTO. The crystallographic data for compounds UOH-Ba1 and UOH-Ba2 were collected on the MX2 beamline at the Australian Synchrotron, ANSTO, and made use of the Australian Cancer Research Foundation (ACRF) detector.References
- V. Popa and O. Cocoş, Cent. Eur. J. Geogr. Sustainable Dev., 2021, 3, 17–25 Search PubMed.
- J. Plášil, J. Geosci., 2014, 59, 99–114 CrossRef.
- J. Janeczek and R. C. Ewing, J. Nucl. Mater., 1992, 190, 128–132 CrossRef CAS.
- S. Spiridonov, A. Perevolotskii, T. Perevolotskaya, R. Aleksakhin and E. Spirin, At. Energy, 2017, 123, 122–126 CrossRef.
- R. J. Baker, Coord. Chem. Rev., 2014, 266, 123–136 CrossRef.
- A. Paulillo, J. M. Dodds, S. J. Palethorpe and P. Lettieri, Sustainable Mater. Technol., 2021, 28, e00278 CrossRef CAS.
- D. Mallants, K. Travis, N. Chapman, P. V. Brady and H. Griffiths, Energies, 2020, 13(4), 833 CrossRef.
- S. El-Showk, Science, 2022, 375, 806–810 CrossRef CAS PubMed.
- R. J. Finch, R. Haddal and G. T. Baldwin, Safeguards Implications for Deep Borehole Disposal of Spent Fuel, SAND2016-4591 report, Sandia National Laboratories, USA, 2016.
- R. C. Ewing, Nat. Mater., 2015, 14, 252–257 CrossRef CAS PubMed.
- D. J. Wronkiewicz, J. K. Bates, S. F. Wolf and E. C. Buck, J. Nucl. Mater., 1996, 238, 78–95 CrossRef CAS.
- R. J. Finch and R. C. Ewing, J. Nucl. Mater., 1992, 190, 133–156 CrossRef CAS.
- J. Janeczek and R. Ewing, J. Nucl. Mater., 1992, 190, 157–173 CrossRef CAS.
- J. Plášil, Eur. J. Mineral., 2017, 29, 1–15 CrossRef.
- P. C. Burns, Hydrated uranium oxides, in Comprehensive Nuclear Materials, ed. R. Konings and R. Stoller, Elsevier, 2nd edn, 2020 Search PubMed.
- T. A. Olds, J. Plasil, A. R. Kampf, T. Spano, P. Haynes, S. M. Carlson, P. C. Burns, A. Simonetti and O. P. Mills, Am. Mineral., 2018, 103, 143–150 CrossRef.
- P. C. Burns, Can. Mineral., 1998, 36, 1061–1067 CAS.
- C. L. Cahill and P. C. Burns, Am. Mineral., 2000, 85, 1294–1297 CrossRef CAS.
- J. Plášil, R. Škoda, J. Čejka, V. Bourgoin and J.-C. Boulliard, Eur. J. Mineral., 2016, 28, 959–967 CrossRef.
- K. A. Hughes, P. C. Burns and U. Kolitsch, Can. Mineral., 2003, 41, 677–685 CrossRef CAS.
- J. Plášil, Eur. J. Mineral., 2018, 30, 237–251 CrossRef.
- M. L. Miller, R. J. Finch, P. C. Burns and R. C. Ewing, J. Mater. Res., 1996, 11(12), 3048–3056 CrossRef CAS.
- P. C. Burns and F. C. Hill, Can. Mineral., 2000, 38, 163–173 CrossRef CAS.
- F. C. Hill and P. C. Burns, Can. Mineral., 1999, 37, 1283–1288 CAS.
- R. E. Glatz, Y. Li, K.-A. Hughes, C. L. Cahill and P. C. Burns, Can. Mineral., 2002, 40, 217–224 CrossRef CAS.
- P. C. Burns and F. C. Hill, Can. Mineral., 2000, 38, 175–181 CrossRef CAS.
- Y. Li and P. C. Burns, Can. Mineral., 2000, 38, 1433–1441 CrossRef CAS.
- M. Rivenet, N. Vigier, P. Roussel and F. Abraham, J. Solid State Chem., 2009, 182, 905–912 CrossRef CAS.
- N. G. Chernorukov, O. V. Nipruk, K. A. Klinshova, O. N. Tumaeva and D. V. Sokolov, New J. Chem., 2021, 45, 9922–9935 RSC.
- T. A. Ablott, K. T. Lu, T. Wei and Y. Zhang, Dalton Trans., 2023, 52, 6629–6640 RSC.
- Y. Zhang, J. Čejka, G. R. Lumpkin, T. Trong Tran, I. Aharonovich, I. Karatchevtseva, J. R. Price, N. Scales and K. Lu, New J. Chem., 2016, 40, 5357 RSC.
- Y. Zhang, R. Aughterson, I. Karatchevtseva, L. Kong, T. T. Tran, J. Čejka, I. Aharonovich and G. R. Lumpkin, New J. Chem., 2018, 42, 12386–12393 RSC.
- Y. Zhang, R. D. Aughterson, Z. Zhang, T. Wei, K. Lu, J. Čejka and I. Karatchevtseva, Inorg. Chem., 2019, 58, 10812–10821 CrossRef CAS PubMed.
- Y. Zhang, K. T. Lu and R. Zheng, Dalton Trans., 2022, 51, 2158–2169 RSC.
- Y. Li, C. L. Cahill and P. C. Burns, Chem. Mater., 2001, 13, 4026–4031 CrossRef CAS.
- K.-A. Kubatko and P. C. Burns, Inorg. Chem., 2006, 45, 10277–10281 CrossRef CAS PubMed.
- T. A. Ablott, K. T. Lu, R. D. Aughterson and Y. Zhang, Dalton Trans., 2022, 51, 15965–15973 RSC.
- K. T. Lu, Y. Zhang, R. D. Aughterson and R. Zheng, Dalton Trans., 2020, 49, 15854–15863 RSC.
- K. T. Lu, Y. Zhang, T. Wei, Z. Wang, D. T. Oldfield and R. Zheng, Inorg. Chem., 2021, 60, 13233–13241 CrossRef CAS PubMed.
- Y. Zhang, T. Wei, T. Trong Tran, K. T. Lu, Z. Zhang, J. R. Price, I. Aharonovich and R. Zheng, Inorg. Chem., 2020, 59, 12166–12175 CrossRef CAS PubMed.
- R. Price, C. Gray, R. Wilson, F. Frey and S. Taylor, Chem. Geol., 1991, 93, 245–265 CrossRef CAS.
- I. Sato, H. Furuya, T. Arima, K. Idemitsu and K. Yamamoto, J. Nucl. Sci. Technol., 1999, 36, 775–780 CrossRef CAS.
- M. K. Pagoaga, D. E. Appleman and J. M. Stewart, Am. Mineral., 1987, 72, 1230–1238 CAS.
- R. J. Finch, P. C. Burns, F. C. Hawthorne and R. C. Ewing, Can. Mineral., 2006, 44, 1197–1205 CrossRef CAS.
- V. Rogova, L. Belova, G. Kiziyarov and N. Kuznetsova, Int. Geol. Rev., 1974, 16, 214–219 CrossRef.
- V. Rogova, L. Belova, G. Kiziyarov and N. Kuznetsova, Int. Geol. Rev., 1974, 16, 1255–1256 CrossRef.
- P. C. Burns, Am. Mineral., 1999, 84, 1661–1673 CrossRef CAS.
- N. G. Chernorukov, O. V. Nipruk, G. N. Chernorukov, R. V. Abrazheev and K. A. Chaplieva, Russ. J. Gen. Chem., 2019, 89, 71–75 CrossRef CAS.
- J. Plášil, J. Geosci., 2018, 63, 65–73 CrossRef.
- D. Aragão, J. Aishima, H. Cherukuvada, R. Clarken, M. Clift, N. P. Cowieson, D. J. Ericsson, C. L. Gee, S. Macedo, N. Mudie, S. Panjikar, J. R. Price, A. Riboldi-Tunnicliffe, R. Rostan, R. Williamson and T. T. Caradoc-Davies, J. Synchrotron Radiat., 2018, 25, 885–891 CrossRef PubMed.
- W. Kabsch, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 133–144 CrossRef CAS PubMed.
- G. M. Sheldrick, SADABS, Empirical Absorption and Correction Software, University of Göttingen, Göttingen, 1996 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- I. D. Brown, Chem. Rev., 2009, 109(12), 6858–6919 CrossRef CAS PubMed.
- P. C. Burns, P. C. Burns, R. C. Ewing and F. C. Hawthorne, Can. Mineral., 1997, 35, 1551–1570 CAS.
- Y. Li and P. C. Burns, Can. Mineral., 2000, 38, 727–735 CrossRef CAS.
- P. C. Burns and K. M. Deely, Can. Mineral., 2002, 40, 1579–1586 CrossRef CAS.
- K. T. Lu, Y. Zhang, T. Wei, T. A. Ablott, T. H. Nguyen and R. Zheng, New J. Chem., 2022, 46, 1371–1380 RSC.
- Y. Zhang, D. J. Fanna, N. D. Shepherd, I. Karatchevtseva, K. Lu, L. Kong and J. R. Price, RSC Adv., 2016, 6, 75045–75053 RSC.
- N. D. Shepherd, Y. Zhang, I. Karatchevtseva, J. R. Price, L. Kong, N. Scales and G. R. Lumpkin, Polyhedron, 2016, 113, 88–95 CrossRef CAS.
- E. R. Vance, Y. Zhang and Z. Zhang, J. Nucl. Mater., 2010, 400(1), 8–14 CrossRef CAS.
- Y. Zhang, T. Wei, Z. Zhang, L. Kong, P. Dayal and D. J. Gregg, J. Am. Ceram. Soc., 2019, 102(12), 7699–7709 CrossRef CAS.
- Y. Zhang, L. Kong, I. Karatchevtseva, R. D. Aughterson, D. J. Gregg and G. Triani, J. Am. Ceram. Soc., 2017, 100(9), 4341–4351 CrossRef CAS.
- Y. Zhang, L. Kong, R. D. Aughterson, I. Karatchevtseva and R. Zheng, J. Am. Ceram. Soc., 2017, 100(11), 5335–5346 CrossRef CAS.
- K. T. Lu, Y. Zhang, T. Wei, Z. Zhang, M. Avdeev and R. Zheng, J. Eur. Ceram. Soc., 2021, 41, 6000–6009 CrossRef CAS.
- P. L. Arnold, J. B. Love and D. Patel, Coord. Chem. Rev., 2009, 253, 1973–1978 CrossRef CAS.
- N. Belai, M. Frisch, E. S. Ilton, B. Ravel and C. L. Cahill, Inorg. Chem., 2008, 47, 10135–10140 CrossRef CAS PubMed.
- R. L. Frost, J. Čejka and M. L. Weier, J. Raman Spectrosc., 2007, 38(4), 460–466 CrossRef CAS.
- Y. Zhang, J. Čejka, I. Karatchevseva, M. Qin, L. Kong, K. Short, S. C. Middleburg and G. R. Lumpkin, J. Nucl. Mater., 2014, 446, 68–72 CrossRef CAS.
- Y. Zhang, I. Karatchevtseva, M. Qin, S. C. Middleburgh and G. R. Lumpkin, J. Nucl. Mater., 2013, 437, 149–153 CrossRef CAS.
- J. R. Bartlett and R. P. Cooney, J. Mol. Struct., 1989, 193, 295–300 CrossRef CAS.
- A. J. Locock and P. C. Burns, Am. Mineral., 2003, 88, 240–244 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Powder XRD, SEM-EDS, supporting tables, check CIFs (PDF), crystallographic information files (CIFs), CSD-2247026 (UOH-Ba1) and 2247027 (UOH-Ba2). CCDC 2247026 and 2247027. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3nj01334k |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2023 |