
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of 1,1′-diaryl-4,4′-bibenzo[c]thiophene derivatives with aryl substituents on the thiophene rings by Stille or Suzuki coupling reaction†
Taiki
Higashino
a,
Yasuto
Hara
a,
Keiichi
Imato
 a,
Seiji
Akiyama
b,
Mio
Ishida
b and
Yousuke
Ooyama
*a
a,
Seiji
Akiyama
b,
Mio
Ishida
b and
Yousuke
Ooyama
*a
aApplied Chemistry Program, Graduate School of Advanced Science and Engineering, Hiroshima University, 1-4-1 Kagamiyama, Higashi-Hiroshima 739-8527, Japan. E-mail: yooyama@hiroshima-u.ac.jp
bScience & Innovation Center, Mitsubishi Chemical Corporation, 1000 Kamoshida-cho, Aoba-ku, Yokohama-shi, Kanagawa 227-8502, Japan
First published on 2nd May 2023
Abstract
Benzo[c]thiophene and its derivatives are attractive chromophores or π-building blocks for emitters, photosensitizers, and semiconductors used in organic optoelectronic devices. Herein, we provide facile synthetic methods of 1,1′-diaryl-4,4′-bibenzo[c]thiophene derivatives by Stille or Suzuki coupling reaction and their photophysical properties in the solution and the solid state, electrochemical properties, and X-ray crystal structures.
Benzo[c]thiophene and its derivatives have attracted not only growing scientific interest in synthetic organic chemistry, polymer chemistry, electrochemistry, photochemistry, and theoretical chemistry,1 but also considerable attention as a promising chromophore or π-building block of emitters, photosensitizers, and semiconductors for organic optoelectronic devices, including organic light-emitting diodes (OLEDs),2 organic photovoltaics (OPVs),3 dye-sensitized solar cells (DSSCs),4 and organic field-effect transistors (OFETs).5 Accordingly, some benzo[c]thiophene derivatives with substituents on the thiophene ring and/or the benzene ring such as 1,3- or 5,6-disubstituted benzo[c]thiophenes6 and 1,3,4,7- or 1,3,5,6-tetrasubstituted benzo[c]thiophenes7 have been synthesized (Fig. 1a). Moreover, a few 3,3′-disubstituted-1,1′-bibenzo[c]thiophenes as the 1,1′-dimer of bibenzo[c]thiophene (abbr. as BBT) have been developed by Cava,8 Ono,4,9 and Mohanakrishnan et al.,10 and they offered their synthetic methods and photophysical and electrochemical properties. On the other hand, in our previous work,11a,b we have developed 1,1′-disubstituted 4,4′-BBT derivatives (BBT-Si, BBT-Sn, and BBT-CHO) with various substituents (R = Si(CH3)2C(CH3)3, Sn(CH3)3, and CHO, Fig. 1a) as well as unsubstituted 4,4′-BBT (BBT-H) by a straightforward reaction method from 1,1′,3,3′-tetrahydro-[4,4′-bibenzo[c]thiophene] 2,2′-dioxide (SO-BBT-H4, Scheme 1), and revealed their photophysical and electrochemical properties. Furthermore, we achieved the development of 1,1′-diaryl-4,4′-BBT by Stille coupling reaction of 1,1′-distannyl-4,4′-BBT (BBT-Sn) with aryl halide (Method A in Scheme 1).11c For example, the Stille coupling reaction of BBT-Sn with 1-bromo-4-tert-butylbenzene gave BBT-PhtBu in a moderate yield (Fig. 1b). Indeed, the expansion of the π-conjugated system by the introduction of aryl substituents on chromophores is an effective way to adjust the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energy levels, that is, the photophysical and electrochemical properties for the development of high-performance optoelectronic devices. However, 1,1′-diaryl-4,4′-BBT could be obtained only by Stille coupling reaction using BBT-Sn so far. Therefore, for this purpose it is necessary to develop facile synthetic methods for 1,1′-diaryl-4,4′-BBT derivatives.
 | ||
| Fig. 1 (a) Chemical structures of benzo[c]thiophenes, 1,1′-bibenzo[c]thiophenes, and 4, 4′-bibenzo[c]thiophenes. (b) Previous work (ref. 11c) and this work for the synthesis of 1,1′-diaryl-4,4′-BBT. | ||
 | ||
| Scheme 1 Synthetic routes to 1,1′-diaryl-4,4′-bibenzo[c]thiophene derivatives BBT-PhtBu and BBT-PhCN by Stille coupling reaction (Method A) and Suzuki coupling reaction (Method B). | ||
In this work, with the aim of providing a new synthetic method of 1,1′-diaryl-4,4′-BBT derivatives and their photophysical and electrochemical properties, we successfully synthesized 1,1′-dibromo-4,4′-BBT (BBT-Br) as an intermediate in the synthesis of the BBT derivative form SO-BBT-H4 (Fig. 1b); BBT-Br as a synthetic building block enables us to develop various BBT series for optoelectronic materials by a variety of synthetic approaches, including homo- and cross-coupling reactions, nucleophilic aromatic substitution, and organometallic reagent formation. Indeed, we have achieved the synthesis of BBT-PhCN with an electron-withdrawing cyanophenyl group using BBT-Br and evaluation of its photophysical and electrochemical properties, as well as BBT-PhtBu with an electron-donating tert-butylphenyl group. Herein, we provide facile methods of 1,1′-diaryl-4,4′-BBT derivatives by Stille coupling reaction of BBT-Sn with aryl halide and Suzuki coupling reaction of BBT-Br with arylboronic acid and their photophysical properties in the solution and the solid state, electrochemical properties and X-ray crystal structures.
As previously indicated, we have demonstrated that BBT-Sn is a useful intermediate in the synthesis of 1,1′-diaryl-4,4′-BBT derivatives by Stille coupling reaction with aryl halides (Method A in Scheme 1); in fact, the reaction gave BBT-PhtBu in a moderate yield (44%).11c Likewise, the Stille coupling reaction of BBT-Sn with 4-bromobenzonitrile afforded BBT-PhCN with a 4-cyanophenyl group on each thiophene ring in a yield of 49%. Meanwhile, for Suzuki coupling reaction, we prepared BBT-Br by the dehydration of SO-BBT-H4 using tetramethylethylenediamine (TMEDA) and then n-BuLi, followed by treatment with carbon tetrabromide (CBr4). Thus, we conducted the Suzuki coupling reaction of BBT-Br with 4-tert-butylphenylboronic acid or 4-cyanophenylboronic acid at room temperature (25 °C) due to the thermal instability of BBT-Br (Method B in Scheme 1). As a result, BBT-PhtBu and BBT-PhCN were successfully prepared by the Suzuki coupling reactions even at a low temperature, although the yields of the two BBT derivatives by Suzuki coupling reactions were lower than those by the Stille coupling reactions. Therefore, this result provides facile synthetic methods for the introduction of aryl substituents into the 1,1′-positions on the thiophene rings of the 4,4′-BBT skeleton, that is, 1,1′-diaryl-4,4′-BBT derivatives by Stille and Suzuki coupling reactions.
A single-crystal X-ray structural analysis was successfully performed for BBT-PhtBu, but unfortunately we could not obtain single crystals of BBT-PhCN with a sufficient size to make the X-ray structural analysis possible. (Fig. 2). The crystal structure of BBT-PhtBu has two crystallographically independent molecules in which the dihedral angles between the two benzo[c]thiophene units are 127.61 and 126.90°, respectively (Fig. 2a). There are no short π–π contacts of less than 3.60 Å between the neighboring molecules (Fig. S3, ESI†), which indicates the absence of π–π stacking between the molecules. On the other hand, the formation of a one-dimensional continuous molecular chain by the intermolecular CH⋯S hydrogen bonding interactions12 between the thiophene rings of neighboring molecules was observed in the crystal structure of BBT-PhtBu (C(1)H(1)⋯S(3) angle = 162.85°, C(1)⋯S(3) distance = 3.833 Å, and H(1)⋯S(3) distance = 2.915 Å and C(55)H(55)⋯S(2) angle = 153.77°, C(55)⋯S(2) distance = 3.668 Å, and H(55)⋯S(2) distance = 2.792 Å, Fig. 2b). Meanwhile, the X-ray powder diffraction (XRD) patterns for BBT-PhCN as well as BBT-PhtBu did not show diffraction peaks over 2θ = 20°, suggesting the absence of π–π stacking. On the other hand, the XRD patterns for BBT-H exhibited diffraction peaks at around 2θ = 22°, 26°, and 28°, suggesting the presence of π–π stacking (Fig. S4 in ESI†).
 | ||
| Fig. 2 Crystal structure of BBT-PhtBu: (a) molecular structure and (b) intermolecular interactions. | ||
The photoabsorption and fluorescence spectra of BBT-H, BBT-PhtBu, and BBT-PhCN in toluene are shown in Fig. 3a and their photophysical data are summarized in Table 1. BBT-PhtBu and BBT-PhCN show intense photoabsorption bands (λmaxabs = 386 nm and 399 nm, respectively) with relatively high molar extinction coefficients (εmax = 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700 M−1 cm−1 and 27000 M−1 cm−1, respectively) in longer wavelength regions by 27 nm and 40 nm, respectively, in comparison with BBT-H (λmaxabs = 359 nm, εmax = 7500 M−1 cm−1), due to the expansion of the π-conjugated system by the introduction of an aryl substituent on the benzo[c]thiophene chromophore. The corresponding fluorescence bands (λmaxfl = 469 nm and 472 nm) of BBT-PhtBu and BBT-PhCN also appear in longer wavelength regions by 59 nm and 62 nm, respectively, in comparison with that (λmaxfl = 410 nm) of BBT-H. Therefore, the Stokes shift (SS) values of BBT-PhtBu and BBT-PhCN are 4585 cm−1 and 3876 cm−1, respectively, which are larger than that (3465 cm−1) of BBT-H. Meanwhile, the fluorescence quantum yield (Φfl = 0.27) of BBT-PhCN is lower than those (Φfl = 0.41 and 0.39, respectively) of BBT-H and BBT-PhtBu. Time-resolved fluorescence spectroscopy demonstrated that the fluorescence lifetime (τfl = 1.27 ns) of BBT-PhCN is shorter than those (3.46 ns and 2.33 ns, respectively) of BBT-H and BBT-PhtBu. The radiative rate constant (kr = 2.13 × 108 s−1) for BBT-PhCN is somewhat larger than those (1.18 × 108 s−1 and 1.63× 108 s−1, respectively) for BBT-H and BBT-PhtBu, but the nonradiative rate constant (knr = 5.75 × 108 s−1) for BBT-PhCN is two to three times higher than those (2.62 × 108 s−1 and 1.70 × 108 s−1, respectively) for BBT-PhtBu and BBT-H. Consequently, the ratio of nonradiative constant to radiative constant (knr/kr = 2.70) for BBT-PhCN is larger than those (1.44 and 1.56, respectively) for BBT-H and BBT-PhtBu, indicating that the lower Φfl value of BBT-PhCN is mainly due to the larger knr value compared to those of BBT-H and BBT-PhtBu. The larger knr value of BBT-PhCN may be induced by rotation between the electron-withdrawing cyanophenyl group and BBT fluorophore, leading to excited-state intramolecular charge transfer (ICT)-based fluorescence quenching.13
700 M−1 cm−1 and 27000 M−1 cm−1, respectively) in longer wavelength regions by 27 nm and 40 nm, respectively, in comparison with BBT-H (λmaxabs = 359 nm, εmax = 7500 M−1 cm−1), due to the expansion of the π-conjugated system by the introduction of an aryl substituent on the benzo[c]thiophene chromophore. The corresponding fluorescence bands (λmaxfl = 469 nm and 472 nm) of BBT-PhtBu and BBT-PhCN also appear in longer wavelength regions by 59 nm and 62 nm, respectively, in comparison with that (λmaxfl = 410 nm) of BBT-H. Therefore, the Stokes shift (SS) values of BBT-PhtBu and BBT-PhCN are 4585 cm−1 and 3876 cm−1, respectively, which are larger than that (3465 cm−1) of BBT-H. Meanwhile, the fluorescence quantum yield (Φfl = 0.27) of BBT-PhCN is lower than those (Φfl = 0.41 and 0.39, respectively) of BBT-H and BBT-PhtBu. Time-resolved fluorescence spectroscopy demonstrated that the fluorescence lifetime (τfl = 1.27 ns) of BBT-PhCN is shorter than those (3.46 ns and 2.33 ns, respectively) of BBT-H and BBT-PhtBu. The radiative rate constant (kr = 2.13 × 108 s−1) for BBT-PhCN is somewhat larger than those (1.18 × 108 s−1 and 1.63× 108 s−1, respectively) for BBT-H and BBT-PhtBu, but the nonradiative rate constant (knr = 5.75 × 108 s−1) for BBT-PhCN is two to three times higher than those (2.62 × 108 s−1 and 1.70 × 108 s−1, respectively) for BBT-PhtBu and BBT-H. Consequently, the ratio of nonradiative constant to radiative constant (knr/kr = 2.70) for BBT-PhCN is larger than those (1.44 and 1.56, respectively) for BBT-H and BBT-PhtBu, indicating that the lower Φfl value of BBT-PhCN is mainly due to the larger knr value compared to those of BBT-H and BBT-PhtBu. The larger knr value of BBT-PhCN may be induced by rotation between the electron-withdrawing cyanophenyl group and BBT fluorophore, leading to excited-state intramolecular charge transfer (ICT)-based fluorescence quenching.13
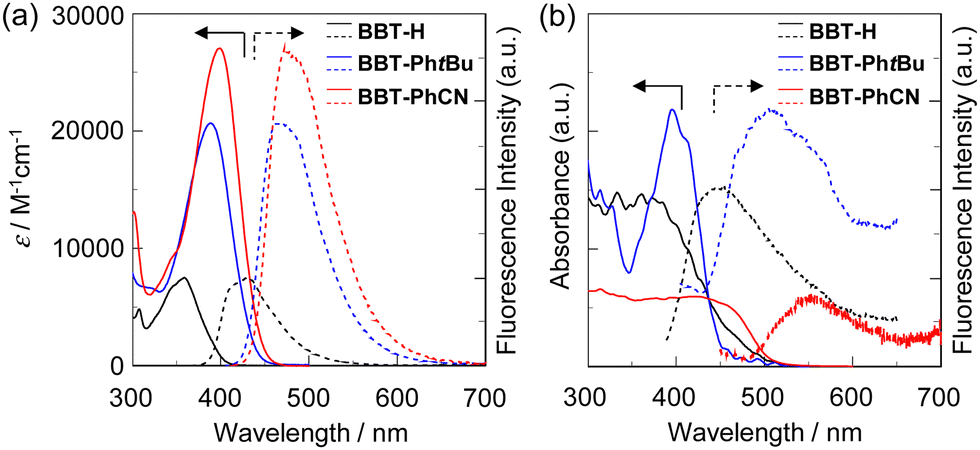 | ||
| Fig. 3 (a) Photoabsorption (solid line) and fluorescence (dotted line) spectra of BBT-H, BBT-PhtBu and BBT-PhCN (3.0 × 10−5 M) in toluene. (b) Solid-state UV-vis diffuse reflection–absorption (solid line) and fluorescence (dotted line) spectra (λex = 360 nm for BBT-H and BBT-PhtBu and 422 nm for BBT-PhCN) of the as-recrystallized BBT-H, BBT-PhtBu and BBT-PhCN. | ||
| Dye | λ max abs/nm (εmax/M−1 cm−1)a | λ max fl/nm (Φfl)b | τ fl/nsc | k r/s−1d | k nr/s−1e | k nr /k r | E onset ox/Vf | E g opt/eVg | HOMO/eVh | LUMO/eVh |
|---|---|---|---|---|---|---|---|---|---|---|
| a In toluene. b In toluene. Fluorescence quantum yields (Φfl) were determined by using a calibrated integrating sphere system (λex = 359 nm, 386 nm and 399 nm for BBT-H, BBT-PhtBu, and BBT-PhCN, respectively). c Fluorescence lifetime. d Radiative rate constant (kr = Φfl/τfl). e Nonradiative rate constant (knr = (1 − Φfl)/τfl). f Onset (Eonsetox) versus Fc/Fc+ of the oxidation potential. g Optical energy gaps (Egopt) were determined from the intersection (393 nm, 432 nm, and 439 nm for BBT-H, BBT-PhtBu, and BBT-PhCN, respectively) of the photoabsorption and fluorescence spectra in toluene. h Versus vacuum level. i Previous work (ref. 11c). | ||||||||||
| BBT-H | 359 (7500)i | 410 (0.41)i | 3.46i | 1.18 × 108i | 1.70 × 108i | 1.44i | 0.75i | 3.16i | −5.55i | −2.39i |
| BBT-PhtBu | 386 (20 700)i | 469 (0.39)i | 2.33i | 1.68 × 108i | 2.62 × 108i | 1.56i | 0.42i | 2.87i | −5.22i | −2.35i |
| BBT-PhCN | 399 (27 000) | 472 (0.27) | 1.27 | 2.13 × 108 | 5.75 × 108 | 2.70 | 0.59 | 2.82 | −5.39 | −2.57 |
Moreover, the solid-state photophysical properties of BBT-H, BBT-PhtBu, and BBT-PhCN have been investigated by using the solid-state UV-Vis diffuse reflection–photoabsorption and fluorescence spectral measurements (Fig. 3b) and their photophysical data are summarized in Table 2. BBT-PhtBu in the solid state exhibits a photoabsorption maximum (λmaxabs-solid) at 400 nm, which is similar to the photoabsorption spectrum in toluene (Fig. 3a). On the other hand, the photoabsorption bands of BBT-H and BBT-PhCN in the solid state are broadened in longer wavelength regions with an onset of ca. 500 nm, compared to those in toluene. In the corresponding solid-state fluorescence spectra, BBT-H, BBT-PhtBu, and BBT-PhCN show fluorescence bands (λmaxfl-solid = 455 nm, 510 nm and 548 nm, respectively) in longer wavelength regions by 45 nm, 41 nm, and 76 nm, respectively, compared to those in toluene. The Φfl-solid values of BBT-H, BBT-PhtBu, and BBT-PhCN in the solid state are <0.02, 0.04, and <0.02, respectively, which are significantly lower than those in toluene (Table 1). Thus, the precise evaluations of the τfl-solid values for the three derivatives were difficult due to their feeble solid-state fluorescence properties. In general, the bathochromic shifts of λmaxabs and λmaxfl and the lowering of the Φfl value by changing from the solution to the solid state is quite common and explained in terms of the formation of intermolecular π–π interactions or continuous intermolecular hydrogen bonding between the fluorophores in the solid state and consequent delocalization of excitons or excimers.14 Thus, based on the XRD patterns for BBT-H, the bathochromic shift of λmaxfl and the lowering of the Φfl value of BBT-H by changing from the solution to the solid state would be attributed to the formation of intermolecular π–π interactions between the molecules in the solid state. Meanwhile, based on the XRD patterns for BBT-PhtBu and BBT-PhCN as well as the crystal structure of BBT-PhtBu, the continuous intermolecular CH⋯S hydrogen bonding interactions between the neighboring molecules might be responsible for the aggregation-caused quenching (ACQ) of BBT-PhtBu and BBT-PhCN.
| Dye |
λ
max
bs-solid/nm |
λ max fl-solid/nm (Φfl-solid)a |
|---|---|---|
| a Fluorescence quantum yields (Φfl-solid) were determined by using a calibrated integrating sphere system (λex = 360 nm for BBT-H and BBT-PhtBu and 422 nm for BBT-PhCN). b Previous work (ref. 11c). | ||
| BBT-H | 360b | 455 (< 0.02)b |
| BBT-PhtBu | 400b | 510 (0.04)b |
| BBT-PhCN | 422 | 548 (< 0.02) |
The electrochemical properties of BBT-H, BBT-PhtBu, and BBT-PhCN were investigated by cyclic voltammetry (CV) in acetonitrile or DMF containing 0.1 M tetrabutylammonium perchlorate (Bu4NClO4). The potentials were internally referenced to ferrocene/ferrocenium (Fc/Fc+). The cyclic voltammograms of the three derivatives showed an irreversible oxidation at 0.88 V for BBT-H, 0.55 V for BBT-PhtBu, and 0.73 V for BBT-PhCN, while any obvious reduction wave did not appear within the potential window (Fig. S5, ESI†). The oxidation waves for BBT-PhtBu and BBT-PhCN were cathodically shifted by 0.33 V and 0.15 V, respectively, compared to that for BBT-H. This result indicates that the introduction of the electron-donating tert-butylphenyl group and the electron-withdrawing cyanophenyl group into the benzo[c]thiophene skeleton causes the lowering of the oxidation potential. The HOMO energy levels (−[Eonsetox + 4.8] eV) versus vacuum level were estimated from the onset potentials (Eonsetox = 0.75 V for BBT-H, 0.42 V for BBT-PhtBu, and 0.59 V for BBT-PhCN) of the oxidation waves and the LUMO energy levels were estimated from the Eonsetox and intersections (optical energy gap: Egopt = 3.16 eV for BBT-H, 2.87 eV for BBT-PhtBu, and 2.82 eV for BBT-PhCN) of the photoabsorption and fluorescence spectra in toluene. The HOMO energy levels (−5.22 eV and −5.39 eV, respectively), of BBT-PhtBu and BBT-PhCN are higher than that (−5.55 eV) of BBT-H. On the other hand, the LUMO energy levels (−2.39 eV and −2.35 eV, respectively) of BBT-H and BBT-PhtBu are similar to each other, but the LUMO energy level (−2.57 eV) of BBT-PhCN is lower than that of BBT-H. Thus, this fact reveals that the bathochromic shift of the photoabsorption band from BBT-H to BBT-PhtBu and BBT-PhCN is mainly attributed to the destabilization of the HOMO energy level for BBT-PhtBu and the destabilization of the HOMO energy level and the stabilization of the LUMO energy level for BBT-PhCN through the introduction of the electron-donating tert-butylphenyl groups and the electron-withdrawing cyanophenyl groups, respectively, into the benzo[c]thiophene skeleton, leading to a decrease in the HOMO–LUMO band gap.
The electronic structures and molecular orbitals of BBT-H, BBT-PhtBu, and BBT-PhCN were calculated by using DFT at the B3LYP/6-31G(d,p) level (Fig. S6, ESI†). The DFT calculations demonstrate that the HOMO for BBT-H is delocalized on each benzo[c]thiophene unit, but the HOMOs for BBT-PhtBu and BBT-PhCN are delocalized on each benzo[c]thiophene unit and the aryl substituent. The LUMOs for BBT-H and BBT-PhtBu are mainly delocalized over the whole benzo[c]thiophene skeleton through the 4,4′-positions, but the LUMO for BBT-PhCN is delocalized over the whole molecule containing cyanophenyl groups through the 4,4′-positions. It was found that the HOMO energy level (−5.06 eV) of BBT-PhtBu is higher than that (−5.29 eV) of BBT-H, but the LUMO energy levels (−1.55 eV and −1.58 eV, respectively) of BBT-H and BBT-PhtBu are similar to each other. The HOMO and LUMO energy levels (−5.64 eV and −2.32 eV, respectively) of BBT-PhCN are significantly lower than those of BBT-H, but the lowering in the LUMO energy level is larger than that in the HOMO energy level, although CV showed that the HOMO energy level of BBT-PhCN is higher than that of BBT-H. Nevertheless, the DFT calculations suggested that compared to BBT-H, the rise of the HOMO energy level for BBT-PhtBu and the lowering of the LUMO energy level for BBT-PhCN result in a decrease in the HOMO–LUMO band gap (3.74 eV, 3.56 eV, and 3.32 eV, respectively), from BBT-H to BBT-PhtBu and BBT-PhCN. Furthermore, time-dependent density functional theory (TD-DFT) calculations were performed to elucidate the photophysical properties of the three derivatives (Fig. S7, ESI†). The calculated λmaxabs-calcd and εcalcd values of BBT-H, BBT-PhtBu, and BBT-PhCN are 345 nm and 7800 M−1 cm−1, 361 nm and 44300 M−1 cm−1, and 403 nm and 38300 M−1 cm−1, respectively. The S0 → S1 transitions are mainly attributed to the transitions from the HOMO to the LUMO (67% for BBT-H, 88% for BBT-PhtBu, and 93% for BBT-PhCN). Indeed, the TD-DFT calculations are in good agreement with the experimental results about bathochromic shifts of the photoabsorption bands from BBT-H to BBT-PhtBu and BBT-PhCN, although the εcalcd value of BBT-PhCN is lower than that of BBT-PhtBu, which is opposite to the experimental results.
In conclusion, we developed facile synthetic methods of 1,1′-diaryl-4,4′-bibenzo[c]thiophene (-BBT) derivatives by Stille or Suzuki coupling reaction and their photophysical and electrochemical properties and X-ray crystal structures were elucidated sufficiently by an experimental approach and DFT calculations. Further studies on the synthetic routes from 1,1′-diaryl-4,4′-BBT derivatives to 1,1′,3,3′-tetraaryl-4,4′-BBT derivatives are now in progress to gain insight into the effect of aryl substituents on the photophysical and electrochemical properties of 4,4′-BBT derivatives.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Grants-in-Aid for Scientific Research (B) from the Japan Society for the Promotion of Science (JSPS) KAKENHI Grant Number 22H02123.Notes and references
- (a) F. Wudl, M. Kobayashi and A. J. Heeger, J. Org. Chem., 1984, 49, 3382–3384 CrossRef CAS; (b) O. D. Abrbanel, J. Rozon and G. R. Hutchison, J. Phys. Chem. Lett., 2022, 13, 2158–2164 CrossRef PubMed; (c) G. Grover, J. D. Tovar and M. Kertesz, J. Phys. Chem. C, 2022, 126, 5302–5310 CrossRef CAS.
- Y.-C. Hu, Z.-L. Lin, T.-C. Huang, J.-W. Lee, W.-C. Wei, T.-Y. Ko, C.-Y. Lo, D.-G. Chen, P.-T. Chou, W.-Y. Hung and K.-T. Wong, Mater. Chem. Front., 2020, 4, 2029–2039 RSC.
- J. D. Douglas, G. Griffini, T. W. Holcombe, E. P. Young, O. P. Lee, M. S. Chen and J. M. J. Fréchet, Macromolecules, 2012, 45, 4069–4074 CrossRef CAS.
- Q. Liu, Q.-Y. Feng, H. Yamada, Z.-S. Wang, N. Ono, X.-Z. You and Z. Shen, Chem. – Asian J., 2012, 7, 1312–1319 CrossRef CAS PubMed.
- X. Chen, D. Zhang, Y. He, M. U. Ali, Y. Wu, C. Zhao, P. Wu, C. Yan, F. Wudl and H. Meng, Mater. Chem. Front., 2020, 4, 3578–3584 RSC.
- (a) S. T. Meek, E. E. Nesterov and T. M. Swager, Org. Lett., 2008, 10, 2991–2993 CrossRef CAS PubMed; (b) A. K. Mohanakrishnan and P. Amaladass, Tetrahedron Lett., 2005, 46, 4225–4229 CrossRef CAS; (c) K. Kawabata and H. Goto, J. Mater. Chem., 2012, 22, 23514–23524 RSC; (d) U. Mitschke and P. Bäuerle, J. Chem. Soc., Perkin Trans. 1, 2001, 740–753 RSC; (e) J. W. Terpstra and A. M. van Leusen, J. Org. Chem., 1986, 51, 230–238 CrossRef CAS; (f) R. M. El-Shishtawy, K. Fukunishi and S. Miki, Tetrahedron Lett., 1995, 36, 3177–3180 CrossRef CAS.
- (a) D.-T. Hsu and C.-H. Lin, J. Org. Chem., 2009, 74, 9180–9187 CrossRef CAS PubMed; (b) A. Ishii, J. Nakayama, J. Kazami, Y. Ida, T. Nakamura and M. Hoshino, J. Org. Chem., 1991, 56, 78–82 CrossRef CAS; (c) R. R. Amaresh, M. V. Lakshmikantham, J. W. Baldwin, M. P. Cava, R. M. Metzger and R. D. Rogers, J. Org. Chem., 2002, 67, 2453–2458 CrossRef CAS PubMed.
- E. Aqad, M. V. Lakshmikantham and M. P. Cava, Org. Lett., 2004, 6, 3039–3041 CrossRef CAS PubMed.
- Y. Shimizu, Z. Shen, S. Ito, H. Uno, J. Daub and N. Ono, Tetrahedron Lett., 2002, 43, 8485–8488 CrossRef CAS.
- P. Amaladass, J. A. Clement and A. K. Mohanakrishnan, Eur. J. Org. Chem., 2008, 3798–3810 CrossRef CAS.
- (a) K. Obayashi, T. Higashino, K. Imato and Y. Ooyama, New J. Chem., 2021, 45, 13258–13261 RSC; (b) K. Obayashi, K. Imato, S. Aoyama, T. Enoki, S. Akiyama, M. Ishida, S. Suga, K. Mitsudo and Y. Ooyama, RSC Adv., 2021, 11, 18870–18880 RSC; (c) K. Obayashi, S. Miho, M. Yasui, K. Imato, S. Akiyama, M. Ishida and Y. Ooyama, New J. Chem., 2021, 45, 17085–17094 RSC.
- (a) H. A. Fargher, T. J. Sherbow, M. M. Haley, D. W. Johnson and M. D. Pluth, Chem. Soc. Rev., 2022, 51, 1454–1469 RSC; (b) H. S. Karmakar, V. S. Kadam, A. L. Patel and S. S. Zade, ChemistrySelect, 2020, 5, 5776–5780 CrossRef CAS; (c) M. Domagała, S. J. Grabowski, K. Urbaniak and G. Mlostoń, J. Phys. Chem. A, 2003, 107, 2730–2736 CrossRef.
- (a) D. Liese and G. Haberhauer, Isr. J. Chem., 2018, 58, 813–826 CrossRef CAS; (b) C. Yan, Z. Guo, W. Chi, W. Fu, S. A. A. Abedi, X. Liu, H. Tian and W.-H. Zhu, Nat. Commun., 2021, 12, 3869 CrossRef CAS PubMed.
- (a) H. Langhals, T. Potrawa, H. Nöth and G. Linti, Angew. Chem., Int. Ed. Engl., 1989, 28, 478–480 CrossRef; (b) H.-C. Yeh, W.-C. Wu, Y.-S. Wen, D.-C. Dai, J.-K. Wang and C.-T. Chen, J. Org. Chem., 2004, 69, 6455–6462 CrossRef CAS PubMed; (c) Y. Ooyama, T. Nakamura and K. Yoshida, New J. Chem., 2005, 29, 447–456 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2248179. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3nj01299a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2023 |