
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic hydrosilylation using an immobilized Co-terpyridine complex activated by inorganic salts and its application in a flow reactor†
Katsuaki
Kobayashi
 a,
Norihisa
Fukaya
b and
Hiroshi
Nakazawa
*a
a,
Norihisa
Fukaya
b and
Hiroshi
Nakazawa
*a
aDepartment of Chemistry, Graduate School of Science, Osaka Metropolitan University, Sumiyoshi-ku, Osaka 558-8585, Japan. E-mail: nakazawa@omu.ac.jp
bNational Institute of Advanced Industrial Science and Technology (AIST), Tsukuba Central 5, 1-1-1 Higashi, Tsukuba 305-8565, Ibaraki, Japan
First published on 10th May 2023
Abstract
The activation of a dibromo Co-terpyridine complex immobilized on a stationary phase (Co(tpy)Br2@SiO2) as a hydrosilylation catalyst was investigated. Inorganic salts that are sparingly soluble in an organic solvent were examined as activators, and K2CO3 was found to show advantages in activator ability, stability, cost, and ease of handling. Catalytic hydrosilylation using Co(tpy)Br2@SiO2 activated by K2CO3 afforded the products in good yield, in both the reactions of styrene with triethoxysilane (92%) and 1-octene with diphenylsilane (88%). Both Co(tpy)Br2@SiO2 and K2CO3 were easily separable from the hydrosilylated product, which contributed to achieving a reusable hydrosilylation system in both the catalyst and activator. Moreover, the Co(tpy)Br2@SiO2/K2CO3 system was found to be applicable in a continuous flow reactor as a catalyst in the stationary phase.
Introduction
Organosilane compounds are versatile and indispensable chemicals for the silicone industry. Hydrosilylation is one of the most straightforward means of obtaining organosilane compounds that are not produced in natural systems. Hydrosilylation usually requires precious metal catalysts such as Pt complexes.1 However, the problem with precious metals is that there are very few reserves on earth. Recent interest in this research field has brought about the development of base metal complex catalysts.2–6 There are many examples of hydrosilylation catalysts based on Mn,4,7 Fe,3,5,8–10 Co,2,3,9–13 Ni,3,14 or Cu,15 which show high turnover numbers, as well as good stereoselectivity, and chemoselectivity. Most of them have a pincer type ligand. Our group has reported Fe16 and Co17,18 complexes bearing an iminobipyridine ligand. Fe-iminobipyridine complexes catalyze the hydrosilylation of olefins and ketones with good turnover numbers and frequency.16 Co-iminobipyridine complexes show chemoselective hydrosilylation between an olefin and a ketone, which is controlled by the reaction solvent.17 The pincer ligands used in these catalysts have large and complicated substituents. Therefore, the synthesis of these pincer ligands requires considerable effort, time, and cost.Catalyst reuse is one of the ways to make more effective use of specially synthesized catalysts. The immobilization of a catalyst on a stationary phase is a good method for reusing the catalyst because it can be separated readily from the liquid product or product solution. Pt catalysts immobilized on solid supports have been synthesized and their reuse has been reported.19 By contrast, base metal complexes for hydrosilylation that are immobilized on a stationary phase are limited.20,21 Recently, our group developed a Co complex supported on silica gel (Co(tpy)(X)2@SiO2, where X = OH, Br) in which a Co complex with terpyridine (tpy), being one of the simplest pincer ligands, was immobilized on the surface of silica gel or glassware.22Co(tpy)Br2@SiO2 (a rough image of the silica gel surface supporting Co(tpy)Br2 is shown in Fig. 1) exhibited good catalytic activity using NaBHEt3 as the activator for the hydrosilylation of 1-octene with diphenylsilane (Ph2SiH2). This Co-supported silica gel was reusable as a catalyst, but the activity gradually decreased with each reuse. The cause of this decrease in catalytic activity was considered to be the cleavage of the Si–O bond in Co(tpy)Br2@SiO2 by the highly active NaBHEt3. Next, we searched for a system that activates the Co complex without using NaBHEt3.
 | ||
| Fig. 1 Schematic of the silica gel surface supporting Co(tpy)Br2. | ||
Recently, we found that [Co(tpy)Br2] was activated by inorganic salts which are stable under air.23 In particular, K2CO3 is a good activator for the hydrosilylation reaction in terms of its ability, cost and stability. It was interesting that K2CO3 activated [Co(tpy)Br2] even though the salt is sparingly soluble in the reaction system (a mixture of an olefin and hydrosilane).
This paper describes whether or not unprecedented combinations of a Co-complex immobilized on silica gel and activators, which include extremely sparingly soluble inorganic salts, result in catalytic activity for hydrosilylation; in addition, a flow reactor system is reported in which the hydrosilylation product is eluted simply by passing the reactants (the olefin and hydrosilane) through a column packed with this silica gel and K2CO3 powder.
Results and discussion
Catalytic activity of Co(tpy)Br2@SiO2 for hydrosilylation in the presence of an activator
It has been reported that [Co(tpy)Br2] did not show any catalytic activity for hydrosilylation, although it did show activity in the presence of an activator such as KOAc (potassium acetate), KOPv (potassium pivalate), and K2CO3.23 Thus, we examined the catalytic activity of [Co(tpy)Br2] immobilized on silica gel (Co(tpy)Br2@SiO2) in the presence of an activator.KOAc, KOPv, KOtBu, K2CO3, KF, and K3PO4 were selected as the activators, considering our previous report on [Co(tpy)Br2].23 The hydrosilylation of styrene with (EtO)3SiH (eqn (1), Table 1) and that of 1-octene with diphenylsilane (Ph2SiH2) (eqn (2)) were used as model reactions. In the absence of an activator, Co(tpy)Br2@SiO2 did not show any catalytic activity for the hydrosilylation of styrene with (EtO)3SiH (Table 1, entry 1). The hydrosilylation of 1-octene with Ph2SiH2 using Co(tpy)Br2@SiO2 without an activator afforded moderate yields of the products, with the linear one (2l) as the major product and the branched one (2b) as the minor product (Table 1, entry 2).
|
|
||||
|---|---|---|---|---|
| Entry | Olefin | Hydrosilane | Additive | Yielda (%) |
| a Determined via GC. b Not detected. | ||||
| 1 | Styrene | (EtO)3SiH | None | N.D.b |
| 2 | 1-Octene | Ph2SiH2 | None | 2l: 52 (2b: 0.3) |
| 3 | Styrene | (EtO)3SiH | KOAc | 1: 82 |
| 4 | Styrene | (EtO)3SiH | KOPv | 1: 90 |
| 5 | Styrene | (EtO)3SiH | KOtBu | 1: 80 |
| 6 | Styrene | (EtO)3SiH | K2CO3 | 1: 92 |
| 7 | Styrene | (EtO)3SiH | KF | 1: 98 |
| 8 | Styrene | (EtO)3SiH | K3PO4 | 1: 89 |
| 9 | 1-Octene | Ph2SiH2 | KOAc | 2l: 68 (2b: 3.0) |
| 10 | 1-Octene | Ph2SiH2 | KOPv | 2l: 86 (2b: 6.2) |
| 11 | 1-Octene | Ph2SiH2 | KOtBu | 2l: 83 (2b: 2.1) |
| 12 | 1-Octene | Ph2SiH2 | K2CO3 | 2l: 88 (2b: 2.1) |
| 13 | 1-Octene | Ph2SiH2 | KF | 2l: 84 (2b: 3.8) |
| 14 | 1-Octene | Ph2SiH2 | K3PO4 | 2l: 84 (2b: 2.4) |

The addition of KOAc to the reaction mixture of styrene and (EtO)3SiH containing Co(tpy)Br2@SiO2 resulted in a high catalytic activity and the formation of 1 (82%; Table 1, entry 3). Similarly, in the presence of other salts, 1 was produced in high yield. In particular, the addition of KOPv, K2CO3, and KF afforded the corresponding product in more than 90% yield (entries 4, 6, and 7, respectively). The hydrosilylation reaction between 1-octene with Ph2SiH2 was also improved in the presence of inorganic salts: the product yields were more than 80% with the addition of activators such as KOPv, KOtBu, K2CO3, KF, and K3PO4 (entries 10–14, respectively). GC-MS and 1H NMR measurements carried out on the supernatant after the reactions in Table 1 revealed that the hydrosilylation products (i.e., 1 for eqn (1), 2l and 2b for eqn (2)) and a small amount of the starting materials (olefin and hydrosilane) were detected, although no by-products were detected. These results are quite similar to the effect of the inorganic salts on the hydrosilylation of the catalytic system based on [Co(tpy)Br2].23
Among the inorganic additives, K2CO3 afforded good results for both the hydrosilylation of styrene with (EtO)3SiH and that of 1-octene with Ph2SiH2. Both Co(tpy)Br2@SiO2 and K2CO3 were barely soluble in organic solvents, showing the high reuse potential of the system. Thus, optimization of the reaction conditions was carried out for the Co(tpy)Br2@SiO2/K2CO3 system.
Optimization of reaction conditions catalyzed by Co(tpy)Br2@SiO2/K2CO3
Optimization of the amount of Co(tpy)Br2@SiO2 in the presence of 2.5 mol% K2CO3 was conducted. When 5.4 mmol of styrene and (EtO)3SiH were used as substrates, 10 mg of Co(tpy)Br2@SiO2 was enough to obtain a quantitative amount of the corresponding product (Table S1, ESI†). By contrast, the hydrosilylation of 1-octene with Ph2SiH2 (5.4 mmol) required 50 mg Co(tpy)Br2@SiO2 for an 88% yield of the corresponding product (Table S1, ESI†). Thus, 50 mg of Co(tpy)Br2@SiO2 was used for the hydrosilylation of 5.4 mmol of the substrates in the subsequent reactions. The amount of K2CO3 was changed in the range of 0.5–2.5 mol% (eqn (3) and (4), Table 2). In the case of the hydrosilylation of styrene with (EtO)3SiH, the product yields were around 90% (Table 2, entries 1–5). When 1-octene and Ph2SiH2 were used as the substrates, the addition of 0.5 mol% K2CO3 as an activator afforded a 74% yield of 2l as the main product with a small amount of 2b as the minor product (Table 2, entry 6). The yield of 2l increased as the amount of K2CO3 was increased, where an 88% yield was achieved with the addition of 2.5 mol% K2CO3 (Table 2, entry 10).|
|
||||
|---|---|---|---|---|
| Entry | Amount of K2CO3 | Olefin | Hydrosilane | Yielda (%) |
| a Determined via GC. | ||||
| 1 | 0.5 | Styrene | (EtO)3SiH | 1: 87 |
| 2 | 1.0 | Styrene | (EtO)3SiH | 1: 88 |
| 3 | 1.5 | Styrene | (EtO)3SiH | 1: 88 |
| 4 | 2.0 | Styrene | (EtO)3SiH | 1: 91 |
| 5 | 2.5 | Styrene | (EtO)3SiH | 1: 92 |
| 6 | 0.5 | 1-Octene | Ph2SiH2 | 2l: 74 (2b: 1.8) |
| 7 | 1.0 | 1-Octene | Ph2SiH2 | 2l: 80 (2b: 2.3) |
| 8 | 1.5 | 1-Octene | Ph2SiH2 | 2l: 80 (2b: 2.2) |
| 9 | 2.0 | 1-Octene | Ph2SiH2 | 2l: 82 (2b: 2.5) |
| 10 | 2.5 | 1-Octene | Ph2SiH2 | 2l: 88 (2b: 2.1) |

Finally, the temperature dependence in the range of 25–100 °C for the hydrosilylation reaction catalyzed by Co(tpy)Br2@SiO2 was investigated in the presence of 2.5 mol% K2CO3 (eqn (5) and (6), Table 3). The hydrosilylation of styrene with (EtO)3SiH afforded a greater than 86% yield of 1 in the temperature range studied (Table 3, entries 1–5). By contrast, the hydrosilylation of 1-octene with Ph2SiH2 at 25 °C produced a low yield of 2l (14%, entry 6). The yield gradually increased as the reaction temperature was increased (entries 6–10), and reached 88% at 100 °C (2l: 88% and 2b: 2.1%; Table 3, entry 10). As reported before, the [Co(tpy)Br2]/K2CO3 system showed catalytic activity at 25 °C for the hydrosilylation of 1-octene with Ph2SiH2 (99% conversion).23 The low yield of the product for the hydrosilylation of 1-octene with Ph2SiH2 at 25 °C (entry 6) probably comes from the lack of activation energy for the reaction to proceed, since the unreacted substrates were observed via GC analysis without any by-product. Therefore, it seems that a high temperature is required to activate the Co complex supported on silica gel with K2CO3.
|
|
||||
|---|---|---|---|---|
| Entry | Temperature (°C) | Olefin | Hydrosilane | Yielda (%) |
| a Determined via GC. | ||||
| 1 | 25 | Styrene | (EtO)3SiH | 1: 86 |
| 2 | 40 | Styrene | (EtO)3SiH | 1: 88 |
| 3 | 60 | Styrene | (EtO)3SiH | 1: 91 |
| 4 | 80 | Styrene | (EtO)3SiH | 1: 95 |
| 5 | 100 | Styrene | (EtO)3SiH | 1: 92 |
| 6 | 25 | 1-Octene | Ph2SiH2 | 2l: 14 (2b: 0.2) |
| 7 | 40 | 1-Octene | Ph2SiH2 | 2l: 31 (2b: 0.9) |
| 8 | 60 | 1-Octene | Ph2SiH2 | 2l: 76 (2b: 3.5) |
| 9 | 80 | 1-Octene | Ph2SiH2 | 2l: 81 (2b: 2.9) |
| 10 | 100 | 1-Octene | Ph2SiH2 | 2l: 88 (2b: 2.1) |

The hydrosilylation of styrene with (EtO)3SiH catalysed by Co(tpy)Br2@SiO2/K2CO3 (eqn (5), Table 3) was also conducted in air. As a result, the yield of the hydrosilylated product dropped to 45%, which was almost half of that under N2. This result indicates that the catalytically active Co species produced from Co(tpy)Br2@SiO2 and K2CO3 is air-sensitive. The active species has not been identified since it is quite difficult to observe the Co complex on silica gel during the catalytic reaction. By contrast, in our previous report,23 the activation mechanism in the homogeneous system using [Co(tpy)Br2] was proposed in which the carbonate anion undergoes nucleophilic attack at the Si atom of hydrosilane causing the release of a hydride ion as a reductant of the Co complex. Considering the previous report, it can be thought, even in an immobilized catalytic system, that Co(tpy)Br2@SiO2 is activated via hydrosilane activation by K2CO3 to generate the catalytically active Co(I) or Co(0) species.
Turnover frequency (TOF) of the hydrosilylation reaction catalyzed by Co(tpy)Br2@SiO2/K2CO3
To estimate the catalytic activity of the Co complex immobilized on silica gel, it is necessary to determine the amount of the Co complex that is supported on silica gel. Thus, the Co-immobilized silica gel was subjected to energy-dispersive X-ray fluorescence spectrometer (EDXRF) measurements and elemental analysis, and it was found that 1 mg of Co was contained in 1 g of the silica gel (0.1 wt% of Co), and the molar ratio of Co![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Br was 1.0:2.0.
:Br was 1.0:2.0.
A comparison of the catalytic activity for the hydrosilylation reaction derived from discrete [Co(tpy)Br2] with that from immobilized [Co(tpy)Br2] is interesting. In the presence of 0.01 mol% [Co(tpy)Br2] relative to 5.4 mmol of each substrate, and in the presence of 30 mg Co(tpy)Br2@SiO2, which corresponds to 0.01 mol% immobilized-[Co(tpy)Br2], a mixture of the olefin and hydrosilane (both 5.4 mmol) to which 2.5 mol% K2CO3 had been added was heated at 100 °C for 1 h, and the products were examined. The results are summarized in Table 4. In the case of the hydrosilylation of styrene with (EtO)3SiH, the TOF of [Co(tpy)Br2] was 6910 h−1 (6282 h−1 per 0.01 mol% catalyst) and that of Co(tpy)Br2@SiO2 was 9120 h−1 (9702 h−1 per 0.01 mol% catalyst) (Table 4, entries 1 and 2). It is worth noting that our immobilized catalyst showed a 1.5-fold higher activity than the [Co(tpy)Br2] catalyst, even though the immobilization of a catalyst often lowers the catalytic activity. There are two possible reasons for this: (i) the tpy ligand was electronically affected due to the connection of an anchor (a triazole substituent) for immobilization, which enhanced the activity of the Co catalyst; and (ii) [Co(tpy)Br2] is reported to be in equilibrium in solution as shown in eqn (7).24 [Co(tpy)2]Br2 and CoBr2 show no catalytic activity. The immobilization of [Co(tpy)Br2] inhibits the bimolecular association and inhibits the shifting of the equilibrium to the right, resulting in the high catalytic performance. Although there is no experimental evidence, we tentatively think that (ii) is the main reason for the enhanced catalytic activity.
| 2[Co(tpy)Br2] ⇄ [Co(tpy)2]Br2 + CoBr2 | (7) |
| Entry | Catalyst | Co amount (mol%) | Substrates | Yielda (%) | TOF (h−1) (TOF per 0.01 mol%) |
|---|---|---|---|---|---|
| a Determined via GC. b Total yield of 2l and 2b. | |||||
| 1 | [Co(tpy)Br2] | 0.011 | Styrene (EtO)3SiH | 76 | 6910 (6282) |
| 2 | Co(tpy)Br2@SiO2 (30 mg) | 0.0094 | Styrene (EtO)3SiH | 86 | 9120 (9702) |
| 3 | [Co(tpy)Br2] | 0.013 | 1-Octene Ph2SiH2 | 87b | 6690 (5146) |
| 4 | Co(tpy)Br2@SiO2 (30 mg) | 0.0094 | 1-Octene Ph2SiH2 | 34b | 3620 (3851) |
The TOF for the hydrosilylation of 1-octene with Ph2SiH2 using [Co(tpy)Br2] was 6690 h−1 (5146 h−1 per 0.01 mol% catalyst), whereas the TOF using Co(tpy)Br2@SiO2 was 3620 h−1 (3851 h−1 per 0.01 mol% catalyst), showing that immobilization of the Co complex caused a decrease in the catalytic activity, in contrast to the above case. The reason for this is not clear, but one possibility is shown below. First, a comparison of the steric accessibility of the olefin and hydrosilane to the catalytically active Co center was carried out. The olefin substituent is a planar phenyl group in styrene, whereas it is a hexyl group in 1-octene. Therefore, it is sterically more difficult for 1-octene to approach the Co center than it is for styrene. Concerning the hydrosilane substrate, Ph2SiH2 appears to be less able to approach the Co center than (EtO)3SiH, because the Ph group in the former hydrosilane is considered to make the Si environment more crowded than the OEt group in the latter hydrosilane. Therefore, when Co(tpy)Br2 is used as the catalyst, the hydrosilylation of 1-octene with Ph2SiH2 is considered to be sterically less likely than that for styrene with (EtO)3SiH, which is consistent with the results of entries 1 vs. 3 in Table 4. This tendency is more pronounced in the catalytic system with Co(tpy)Br2 immobilized on silica gel. This is because one side of the catalytically active Co species is the silica gel surface, which limits the space that is accessible to the substrates. Therefore, in the Co(tpy)Br2@SiO2 system, the reaction of styrene with (EtO)3SiH, which is less sterically constrained by the immobilization of the complex, can effectively receive the benefit of (ii) shown above (entries 1 vs. 2 in Table 4), whereas the reaction of 1-octene with Ph2SiH2 suffers from more steric constraints due to immobilization of the complex compared with the benefit of (ii) (entries 3 vs. 4 in Table 4).
Scope of substrates
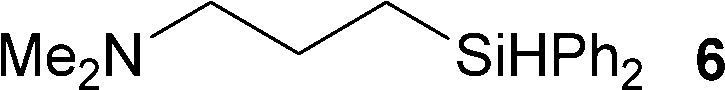
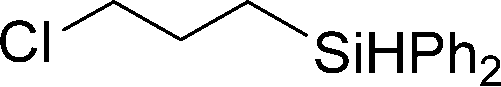
Several hydrosilylation reactions of olefins containing various functional groups were investigated. The mixtures of 5.4 mmol olefin and hydrosilane were treated with 50 mg Co(tpy)Br2@SiO2 activated using 2.5 mol% K2CO3 at 100 °C. Ph2SiH2 was used as the hydrosilane substrate (eqn (8), Table 5). Using Co(tpy)Br2@SiO2/K2CO3, styrene and vinyl cyclohexane underwent hydrosilylation with Ph2SiH2 in good yields (Table 5, entries 1 and 2, respectively). 1,2-Epoxy-5-hexene was converted to the corresponding product with a moderate yield (67%, entry 3). The hydrosilylation of dimethylallylamine with Ph2SiH2 afforded 6 in only 46% yield (entry 4), although [Co(tpy)Br2] produced the same product in 73% yield.23 Allyl chloride, vinyl acetate, and methyl acrylate were not suitable substrates for hydrosilylation using Co(tpy)Br2@SiO2/K2CO3 (entries 5–7, respectively). Methyl acrylate was converted by [Co(tpy)Br2] activated using K2CO3 in 41% yield,23 but Co(tpy)Br2@SiO2/K2CO3 did not afford the product.









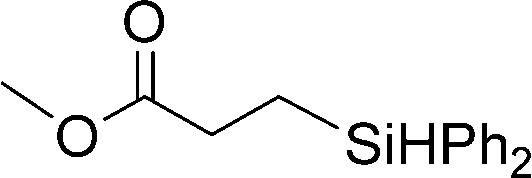
Next, the scope of the hydrosilane and siloxane substrates was investigated. Styrene was employed as the olefin substrate (eqn (9), Table 6), since hydrosilylation using styrene solely produced the anti-Markovnikov type product (3) with Co(tpy)Br2@SiO2/K2CO3 in good yield (Table 5, entry 1). When PhSiH3 was used as the primary silane, the corresponding product (7) was obtained in 90% yield (Table 6, entry 1). In the case of the tertiary silane, hydrosilylation using (EtO)2MeSiH afforded the hydrosilylated product (8) in good yield (84%, entry 2). However, the similar tertiary silane Ph2MeSiH was not reactive at all (entry 3). These three hydrosilanes were quantitatively converted into the corresponding products by [Co(tpy)Br2]/K2CO3.23 This indicates that Co(tpy)Br2@SiO2/K2CO3 is affected by the steric hindrance around the Si atom of the hydrosilane. This tendency was also observed in the hydrosilylation reaction using siloxane substrates with Co(tpy)Br2@SiO2/K2CO3. 1,1,1,3,3-Pentamethyldisiloxane (PMDS) and 1,1,1,3,5,5,5-heptamethyltrisiloxane (MD′M) were employed as the siloxane substrates. When PMDS and MD′M were used as the substrates the products were obtained in 46% (9) and 36% (10) yield, respectively. Thus, the larger framework of MD′M afforded the hydrosilylation product in a lower yield.

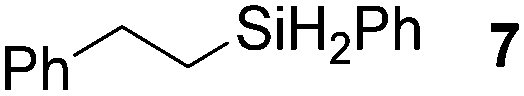




Study on detachment of the Co complex from the silica gel support
It was then examined whether or not the immobilized Co complex moieties detach from the silica gel support during the hydrosilylation reaction in the Co(tpy)Br2@SiO2/K2CO3 catalytic system. The hydrosilylation reaction of 5.4 mmol 1-octene and 5.4 mmol Ph2SiH2 was carried out at 100 °C for 24 h in the presence of 50 mg Co(tpy)Br2@SiO2 and 2.5 mol% K2CO3. After that, the reaction solution was centrifuged to separate the solid portion; the supernatant was subjected to inductively coupled plasma mass spectrometry (ICP-MS), and it was found that Co was not detected. Considering that the detection limit of Co per 1 g of sample in the ICP-MS analysis method is 0.5 μg, it can be said that detachment of the immobilized Co portion from the silica gel support is negligible during this reaction, if it occurs at all.Reusability of Co(tpy)Br2@SiO2/K2CO3
Since Co(tpy)Br2@SiO2 is insoluble and the solubility of K2CO3 is extremely low in an organic solvent, the catalyst and activator can be readily separated via filtration or centrifugation, indicating the possibility of their reusability.A mixture of 5.4 mmol each of 1-octene and Ph2SiH2 containing 50 mg of Co(tpy)Br2@SiO2 and 2.5 mol% K2CO3 was stirred using a mechanical stirrer (not a magnetic stirrer) to avoid crushing the silica gel (Co(tpy)Br2@SiO2). After 2 h of reaction at 100 °C, Co(tpy)Br2@SiO2 and K2CO3 were separated via centrifugation, washed with hexane, dried, and used for the second reaction. These sequential operations were repeated five times, and the yield of 2l was measured for each cycle (the yield of 2b was lower than 2% in each cycle). From the results shown in Fig. 2, it was found that the Co(tpy)Br2@SiO2/K2CO3 system is reusable at least 5 times without any reduction in the catalytic activity.

 | ||
| Fig. 2 Repeated hydrosilylation of 1-octene with Ph2SiH2 catalyzed using the Co(tpy)Br2@SiO2/K2CO3 system. | ||
Application of Co(tpy)Br2@SiO2/K2CO3 to a continuous flow reactor
Since it was shown that the Co(tpy)Br2@SiO2/K2CO3 system can be reused and that the Co complex does not detach from the silica gel support, we tried applying this system to a continuous flow reactor. A powdered mixture of 500 mg Co(tpy)Br2@SiO2 and 200 mg K2CO3 was packed into a stainless-steel tubular reactor (5 mm internal diameter, 100 mm length). This reactor was positioned in the temperature controller. The liquid substrate mixture of 1-octene and Ph2SiH2 was deaerated via N2 bubbling and introduced to the tubular reactor using a pump unit. Shortly after starting, the outflow of the products stopped. Thinking that the reaction tube was clogged due to the high viscosity of the hydrosilylated products (2l and 2b), an organic solvent was added to the substrates and injected into the flow reactor.When toluene was used as the solvent and the flow was carried out at 100 °C, the flow rate was maintained at 0.03 mL min−1 and the conversion rate was around 45% for 24 h (Fig. 3A). When THF was used and the flow was carried out at 60 °C, the conversion rate improved to more than 95% for the first 4 h, which then settled down to ca. 80% after 24 h (Fig. 3B). The real active species is not clear, but it is probably a coordinatively unsaturated Co complex, such as [Co(tpy)], which is air-sensitive. As THF is known to be a coordinative solvent, whereas toluene is not, it is likely that THF coordinates to the real Co active species and prevents decomposition, resulting in the high catalytic activity. Actually, the batch reaction of Co(tpy)Br2@SiO2/K2CO3 in THF was superior to that in toluene (Table S2, ESI†). Thus, the continuous flow reaction in the presence of THF in total afforded 21.6 g of the product (2l) (88% conversion) with 90% purity after removal of the solvent, in which only small amounts of the starting materials and 2b (<2%) were found.
 | ||
| Fig. 3 Time course of the conversion rate of hydrosilylation of 1-octene with Ph2SiH2 by continuous flow reaction using Co(tpy)Br2@SiO2/K2CO3 in the presence of toluene (A) or THF (B) as a solvent. | ||
These results show that a flow reactor using Co(tpy)Br2@SiO2 and K2CO3 can be adapted for hydrosilylation reactions between olefins with hydrosilanes, and the reactor can operate for at least 24 h whilst maintaining a high catalytic activity. Therefore, our reaction system, which combines an air-stable immobilized catalyst and an inorganic salt, is a step towards the practical application of a flow reactor.
Conclusions
The conversion of the dibromo Co-terpyridine complex immobilized on silica gel (Co(tpy)Br2@SiO2) to a catalytically active species by inorganic salts was demonstrated via the hydrosilylation reaction. Among the inorganic salts, K2CO3 was the best in terms of the activator ability, stability, cost, and ease of handling. The insolubility of Co(tpy)Br2@SiO2 and the sparing solubility of K2CO3 in an organic solvent made separation of the silica gel and K2CO3 from the product easy and facilitated the reusability of the Co(tpy)Br2@SiO2/K2CO3 catalytic system. Furthermore, this system was shown to be applicable for a continuous flow reaction system.In many hydrosilylation catalyzed reactions, as the catalytically active species is air-sensitive, it is required to conduct the reaction under an inert atmosphere and to separate the product from the catalyst residues after the reaction has been completed. Since this paper reports that a solid catalyst and a solid activator are packed in a column under air and the deaerated reaction reagents (olefin and hydrosilane) are simply flowed through the column to continuously generate the hydrosilylation product that contains no catalyst residue, our results can be said to be a step towards the realization of an ideal catalytic system. The present research suggests an environmentally friendly catalytic system for hydrosilylation.
Experimental section
Materials
Terpyridine-modified silica gel (tpy@SiO2) and [Co(tpy)Br2] were prepared according to literature methods.13,22 The hydrosilylated products 1,252l,12 and 2b9 were synthesized as authentic samples according to the literature. Silica gel for Co(tpy)Br2@SiO2 preparation was purchased from Nacalai Tesque (Silica Gel 60, spherical, neutral). All other chemicals were purchased from commercial sources and were used as received.Preparation of Co(tpy)Br2@SiO2
2 g of tpy@SiO2 was dispersed in an aqueous solution of CoBr2. The mixture was then stirred at 25 °C for 24 h. The resulting powder (Co(tpy)Br2@SiO2) was collected via centrifugation, washed with water or THF (20 mL), and then dried in vacuo to obtain approximately 1.8 g of Co(tpy)Br2@SiO2.Typical procedure for catalytic hydrosilylation by Co(tpy)Br2@SiO2
Co(tpy)Br2@SiO2 (50 mg) and K2CO3 (19 mg, 2.5 mol%) were placed in a Schlenk tube. The air in the tube was replaced with N2. The olefin (5.4 mmol) and hydrosilane (5.4 mmol) substrates were added to the tube. The suspension was heated at 100 °C and stirred for 24 h. After the reaction, the solution was cooled to room temperature. Co(tpy)Br2@SiO2 and K2CO3 were separated via centrifugation and the supernatant was analyzed using GC.Author contributions
K. K. and H. N. conceived and designed the experiments. K. K. performed the experiments. N. F. measured EDXRF and elemental analysis of Co(tpy)Br2@SiO2. K. K. and H. N. prepared the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the “Development of Innovative Catalytic Processes for Organosilicon Functional Materials” project (PL: K. Sato) of the New Energy and Industrial Technology Development Organization (NEDO).Notes and references
-
(a) D. Troegel and J. Stohrer, Coord. Chem. Rev., 2011, 255, 1440–1459 CrossRef CAS
; (b) B. Marciniec, H. Maciejewski, C. Pietraszuk and P. Pawluć, Hydrosilylation, Springer, Berlin, 2009 Search PubMed
-
(a) J. Sun and L. Deng, ACS Catal., 2016, 6, 290–300 CrossRef CAS
- X. Du and Z. Huang, ACS Catal., 2017, 7, 1227–1243 CrossRef CAS
- X. Yang and C. Wang, Chem. – Asian J., 2018, 13, 2307–2315 CrossRef CAS PubMed
-
(a) D. Wei and C. Darcel, Chem. Rev., 2019, 119, 2550–2610 CrossRef CAS PubMed
- L. D. de Almeida, H. Wang, K. Junge, X. Cui and M. Beller, Angew. Chem., Int. Ed., 2021, 60, 550–565 CrossRef CAS PubMed
-
(a) C. Ghosh, T. K. Mukhopadhyay, M. Flores, T. L. Groy and R. J. Trovitch, Inorg. Chem., 2015, 54, 10398–10406 CrossRef CAS PubMed
-
(a) J. Yang and T. D. Tilley, Angew. Chem., Int. Ed., 2010, 49, 10186–10188 CrossRef CAS PubMed
-
(a) X. Du, Y. Zhang, D. Peng and Z. Huang, Angew. Chem., Int. Ed., 2016, 55, 6671–6675 CrossRef CAS PubMed
- A. Sanagawa and H. Nagashima, Organometallics, 2018, 37, 2859–2871 CrossRef CAS
-
(a) C. C. H. Atienza, T. Diao, K. J. Weller, S. A. Nye, K. M. Lewis, J. G. P. Delis, J. L. Boyer, A. K. Roy and P. J. Chirik, J. Am. Chem. Soc., 2014, 136, 12108–12118 CrossRef CAS PubMed
- A. D. Ibrahim, S. W. Entsminger, L. Zhu and A. R. Fout, ACS Catal., 2016, 6, 3589–3593 CrossRef CAS
- K. Kobayashi and H. Nakazawa, Inorg. Chim. Acta, 2021, 523, 120403 CrossRef CAS
-
(a) G. Vijaykumar, A. Pariyar, J. Ahmed, B. K. Shaw, D. Adhikari and S. K. Mandal, Chem. Sci., 2018, 9, 2817–2825 RSC
-
(a) J. M. W. Gribble, M. T. Pirnot, J. S. Bandar, R. Y. Liu and S. L. Buchwald, J. Am. Chem. Soc., 2017, 139, 2192–2195 CrossRef PubMed
-
(a) K. Hayasaka, K. Kamata and H. Nakazawa, Bull. Chem. Soc. Jpn., 2016, 89, 394–404 CrossRef CAS
- K. Kobayashi, D. Taguchi, T. Moriuchi and H. Nakazawa, ChemCatChem, 2020, 12, 736–739 Search PubMed
- D. Taguchi, K. Kobayashi, T. Moriuchi and H. Nakazawa, Bull. Chem. Soc. Jpn., 2020, 93, 1086–1094 CrossRef CAS
-
(a) J. Zhao, Y. Gui, Y. Liu, G. Wang, H. Zhang, Y. Sun and S. Fang, Catal. Lett., 2017, 147, 1127–1132 CrossRef CAS
- R.-H. Li, X.-M. An, Y. Yang, D.-C. Li, Z.-L. Hu and Z.-P. Zhan, Org. Lett., 2018, 20, 5023–5026 CrossRef CAS PubMed
- Z. Yu, Z. Song, C. Lu, Y. Bai, J. Li, J. Liu, P. Liu and J. Peng, Appl. Organomet. Chem., 2022, 36, e6648 CrossRef CAS
- K. Kobayashi and H. Nakazawa, Chem. – Asian J., 2021, 16, 3695–3701 CrossRef CAS PubMed
- K. Kobayashi and H. Nakazawa, Dalton Trans., 2022, 51, 18685–18692 RSC
-
(a) H. Hogg and R. G. Wilkins, J. Chem. Soc., 1962, 341–350 RSC
- S. Gutiérrez-Tarriño, P. Concepción and P. Oña-Burgos, Eur. J. Inorg. Chem., 2018, 4867–4874 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nj01062g |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2023 |