
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, structure and spectroscopic properties of BODIPY dyes incorporating the pentafluorosulfanylphenyl group†
Richard D.
James
,
Fabio
Cucinotta
 ,
Paul G.
Waddell
and
Andrew C.
Benniston
*
,
Paul G.
Waddell
and
Andrew C.
Benniston
*
Chemistry-School of Natural & Environmental Sciences, Newcastle University, Newcastle upon Tyne, NE1 7RU, UK. E-mail: andrew.benniston@newcastle.ac.uk
First published on 31st March 2023
Abstract
Six compounds (BD1–BD6) are discussed based on the borondipyrromethene (BODIPY) core and functionalised in the meso position or the 3,5-positions with pentafluorosulfanylaryl subunits. The SF5 unit in the aryl ring acts an electron-withdrawing group and its effect on the properties of the BODIPY derivatives is described. Two crystal structures were obtained for the simplest compounds in which the meso position of the BODIPY is either pentafluorosulfanylphenyl (BD1) or pentafluorosulfanylphenylethynylphenyl (BD2). The crystal packing features of the two structures were used to rationalise the strong solid-state emission observed for the two compounds in terms of exciton-coupled dimers. Solution phase photophysical properties of the compounds are also described, and trends within them are discussed in terms of the negative hyperconjugation and the “non-bonded” resonance effect of the SF5 group. The long-wavelength absorbing and fluorescing dyes (BD4–BD6) are π-conjugated at the 3,5 positions, but they do not display typical solvatochromic behaviour associated with push–pull chromophores. The alterations in their absorbance and fluorescence maxima with change in solvent are adequately fitted to the solvent polarizability function (f(n2) = (n2 − 1)/(2n2 + 1)), where n is the refractive index. The good linear fit supports no major change in the electronic transition dipole moment between the ground and excited states. Density functional theory (DFT) calculations were consistent with localised low-energy π–π* transitions rather than any charge-transfer transitions despite the existence of the electron-withdrawing SF5 group(s).
Introduction
Since its initial discovery over some fifty years ago, the boron dipyrromethene (BODIPY) group has grown in stature to a point where it is the choice dye for numerous chemical and biophysical applications.1 The popularity of the dye can be attributed to several factors, arising not only from its highly beneficial photophysical properties (e.g., high fluorescence quantum yield), but also to the ease at which it can be functionalised with disparate groups and in multiple positions of the chromophore backbone.2 This versatility has given rise to a library of compounds, tuneable in both their colour and fluorescence signal output.3 In this context, our own work focussed on the “fluorine effect” by incorporation of a fluoroaryl subunit, with different numbers and substituent patterns of fluorines, in the meso position.4 The highly electron-withdrawing nature of the fluoroaryl group afforded strongly fluorescent BODIPY dyes with tuneable redox properties. As a continuation to this work, the highly electron-withdrawing (pentafluorosulfanyl)phenyl (PFSP) unit was targeted, since it has found application in several push–pull structures that display solvatochromic properties.5 The group is also of special interest because of its ability to participate in negative hyperconjugation. To assess the effect of the PFSP group on the BODIPY chromophore, two sets of compounds were pursued based on substitution at the meso site and the 3,5 positions. The examples were developed as highly conjugated systems, in addition bearing a protected carboxylic acid as a masked site for future semiconductor attachment (Scheme 1).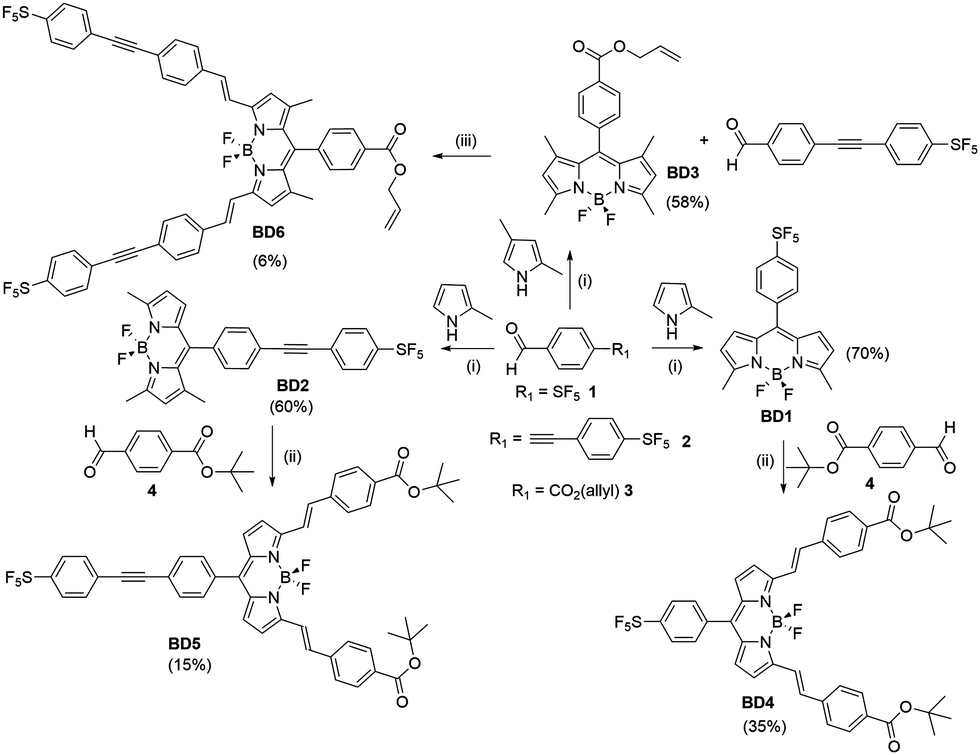 | ||
| Scheme 1 Reagents and Conditions: (i) DCM, TFA, DIPEA, p-chloroanil or DDQ, BF3·Et2O, r.t. (ii) Piperidine, AcOH, MeCN, 3 Å molecular sieves, r.t. (iii) Piperidine, TsOH, toluene, Dean-Stark. Percentage yield given in brackets. | ||
Results and discussion
Synthesis and characterization
The target compounds were prepared using standard methods starting from the commercially available compound 1, or the derivative 3 synthesised using a literature procedure.6 The previously unpublished compound 2 was prepared in 94% yield by Sonogashira coupling of 1-bromo-4-pentafluorosulfanylbenzene with 4-ethynylbenzaldehyde. The preparation of BD1 and BD4 was reported in a prior publication.7 The condensation of an appropriate aldehyde (2 or 3) with either 2-methylpyrrole or 2,4-dimethylpyrrole, coupled with oxidation and chelation to a BF2 unit, afforded the BODIPYs BD2 and BD3 in reasonable yields. These BODIPY compounds along with BD1 were the basis for performing the classic Knoevenagel condensation reaction at the 3,5 methyl groups. The condensation reaction of BD1 with the tBu protected carboxylic acid 4 by simply using MeCN as solvent at room temperature, and driving the reaction using molecular sieves, is known to work well to produce BD4 as a blue solid.7 The Knoevenagel condensation of BD2 with 3 under identical conditions was not successful and heating to 60 °C was required to produce BD5. The same could not be said for the reaction of BD3 with 2 under room temperature conditions, or slight heating, as no product was isolated. The harsher condition of reflux in toluene using a Dean-Stark apparatus to remove the water did succeed, but the yield of the product BD6 was very low. The two 3,5-methyl groups for BD1, and to a lesser extent in BD2, are likely activated to deprotonation by the presence of the electron-withdrawing PFSP unit in the meso position. All compounds were analysed by standard analytical techniques. A typical 19F NMR spectrum for a PFSP containing compound displayed the expected splitting patterns, in addition to the quartet for the BF2 unit, a doublet (equatorial F) and quintet (axial F) for the SF5 group (see ESI† Fig. S25).Further confirmation of the structures of BD1 and BD2 was obtained by single-crystal X-ray crystallography (Fig. 1). The basic dipyrromethene subunit for both structures is essentially identical with bond lengths and angles consistent with previously published structures.8 The first subtle modification in the structures is observed at the dihedral angle relating to the twist of the directly linked meso aryl group. This angle is slightly greater for BD2 (53.7(2)°) when compared to BD1 (49.9(3)°). The small variation is attributed to packing effects, since the crystal packing diagrams for the two compounds are vastly different (see later). The bond lengths and angles for the SF5 are within error the same for both structures. The extra ethynylaryl spacer for BD2 results in a linearly extended structure noting that the two aryl rings are non co-planar.
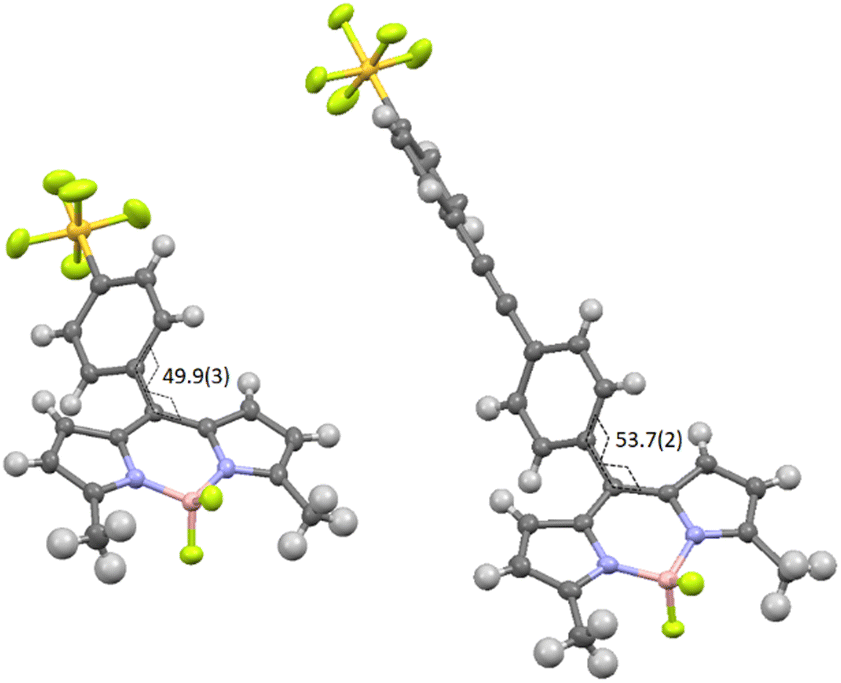 | ||
| Fig. 1 X-Ray determined molecular structures for BD1 (left) and BD2 (right) as thermal ellipsoids (50%). Torsional angle shown in degrees with error in brackets. Sulphur = yellow, fluorine = green, nitrogen = blue, boron = pink, carbon = dark grey and hydrogen = light grey. | ||
The most significant difference between the two structures relates to their respective packing diagrams and the way the two BODIPY subunits align (Fig. 2). A pair of BD1 molecules arrange almost anti-parallel in a cross-like motif which extends in one dimension to give the impression of a wave modulating within the crystal. The SF5 groups of disparate BD1 units are prearranged such that specific F–F intermolecular distances fall within the range 3.7–3.4 Å. In comparison, an offset head-to-tail motif is observed for a pair of BD2 molecules in the unit cell, which gives rise to a crystal packing diagram that can be highlighted as pseudo-dimers and -tetramers of BODIPY moieties. The packing features for both the BODIPYs are considered important in the context of crystal emission as described later.
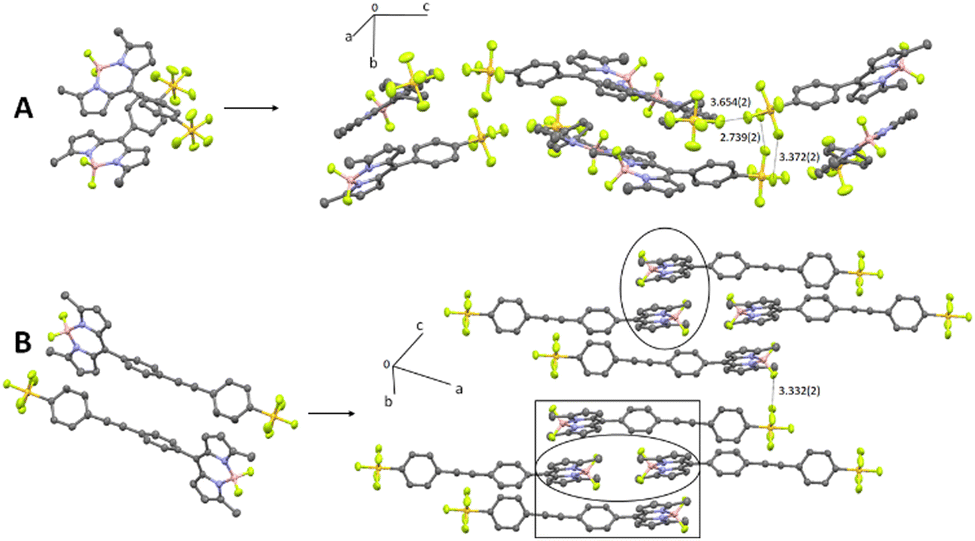 | ||
| Fig. 2 Crystal packing diagrams for BD1 (A) and BD2 (B) based on their simple dimers. Hydrogens are omitted for clarity and distances are given in Ångstroms. The circle highlights a pseudo-dimer and the square a pseudo-tetramer of BODIPYs. | ||
DFT calculations
The energy and spatial location of frontier molecular orbitals for BODIPY derivatives have been used extensively to rationalise their ground- and excited-state properties. Density functional theory (DFT) is commonly applied alongside its time-dependent analogue (TD-DFT) to identify electronic transitions.9 Both these methods, and by employing the B3LYP functional and the 6-311G(d,p) basis set, were applied to the energy-minimised structures of BD1–BD6in vacuo. For comparison purposes, the calculated frontier molecular orbitals of BD1 and BD2 are illustrated in Fig. 3. The HOMOs for both compounds are located on the dipyrromethene backbone and are similar in appearance with a node located at the meso position as observed for other simple BODIPY derivatives. The energy difference (Δ = EBD1HOMO − EBD2HOMO) is −0.14 eV. The LUMOs for BD1 and BD2 are again similar in appearance despite the presence of the additional ethynylaryl group. The energy difference (Δ = EBD1LUMO − EBD2LUMO) is −0.068 eV. Whereas the LUMO+1 for BD1 is exclusively located on the SF5 group, the LUMO+1 for BD2 is very much situated on the second aryl ring; this feature is more associated with the LUMO+2 for BD1. Even the LUMO+2 for BD2 does not exhibit a feature consistent with electron density located on the SF5 group. There is a distinct similarity of HOMO−1 for BD1 with the HOMO−2 for BD2; their difference in energy is 1.79 eV. The HOMO−1 for BD2 is clearly associated with the diarylethynyl subunit. The calculations predict that any electron addition/removal for both compounds will be essentially focussed at the dipyrromethene core. Only for BD1 could electron addition be anticipated to occur at the SF5 unit.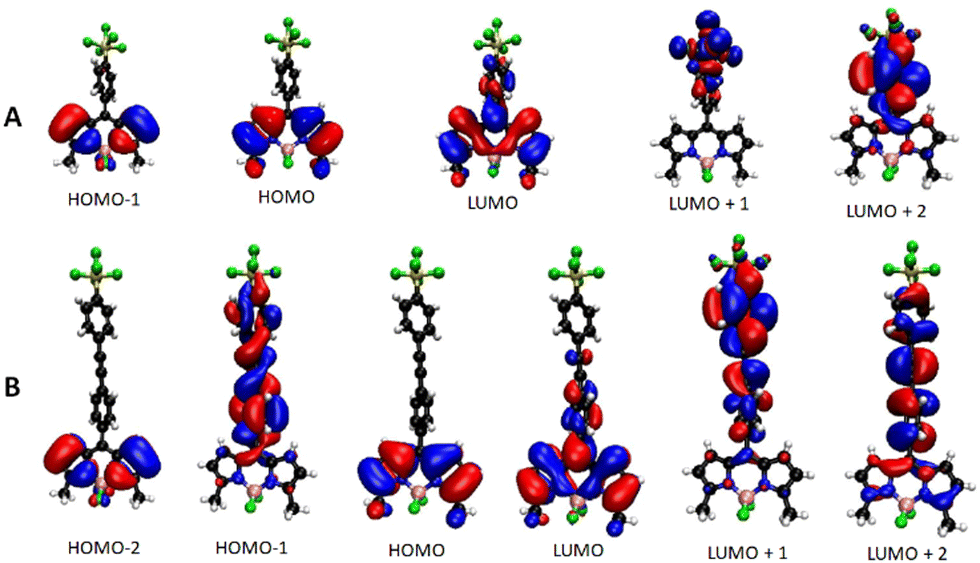 | ||
| Fig. 3 DFT calculated Kohn–Sham molecular orbitals using the B3LYP functional and the 6-311G(d,p) basis set for BD1 (A) and BD2 (B). | ||
The application of TD-DFT to the two compounds identified the first three main electronic transitions, their associated orbital contributions and energies (Table 1). The first point to note is the similarity in the energy for the first electronic transition and orbital contribution when comparing BD1 with BD2. Even though there is a small involvement of a HOMO−2 to LUMO electronic transition for BD2, all the orbitals in both cases are located on the dipyrromethene core consistent with a π–π* absorption. The ratio of oscillator strengths corresponding to BD1/BD2 is 1.34, indicating a slightly more favourable electronic transition for the smaller BODIPY. In contrast, the oscillator strength for the second electronic transition (HOMO−1 to LUMO) of BD1 is calculated to be some six times smaller than the corresponding one for BD2. This difference is explained by the alteration in the molecular orbital shape for the HOMO−1 in BD2 (Fig. 3), which is extended onto the meso diarylethynyl subunit. In fact, the electronic transition would appear to shift electron density from the meso substituent onto the dipyrromethene group more reminiscent of charge-transfer. This charge-transfer character is also observed in the third electronic transition for BD2, along with electron migration from the dipyrromethene unit to the diarylethynyl subunit. A weak third transition with some charge-transfer character is also calculated for BD1.
| Cmp | Electronic transition (oscillator strength) | Orbitals (% contribution) | Energy/eV (nm) |
|---|---|---|---|
| BD1 | 1 (0.4867) | HOMO → LUMO (100) | 2.94 (422) |
| 2 (0.0579) | HOMO−1 → LUMO (98) HOMO → LUMO (2) | 4.02 (308) | |
| 3 (0.0146) | HOMO → LUMO+2 (100) | 4.55 (273) | |
| BD2 | 1 (0.3627) | HOMO → LUMO (94) | 2.97 (417) |
| HOMO−2 → LUMO (6) | |||
| 2 (0.3774) | HOMO−1 → LUMO (100) | 3.44 (360) | |
| 3 (0.0014) | HOMO−2 → LUMO (49) | 3.53 (351) | |
| HOMO → LUMO+1 (46) | |||
| HOMO → LUMO+2 (4) |
Similar calculations to above were performed on the carboxylic acid derivatives for the remaining BODIPY examples and the results are collected in ESI† (Fig S1–S4). The main point to note is the predicted ordering for the energies of the first electronic transition BD5 (2.23 eV) > BD4 (2.12 eV) >BD6 (2.07 eV), corresponding to the BODIPYs with further π-conjugation at the 3,5 positions.
Electrochemistry
Cyclic voltammetry (CV) and square-wave voltammetry (SWV) techniques were used to assess the redox properties of the BODIPY compounds in acetonitrile, probing more directly the HOMO and LUMO orbital energies and assessing any substituent effects. The redox behaviour for BD1 is highlighted as an example, and collected in Table 2 are selected oxidation and reduction potentials for the other compounds. The CV and SWV recorded for BD1 in acetonitrile containing 0.2 M tetrabutylammonium hexafluorophosphate (TBAHFP) versus ferrocene reference are shown in Fig. 4. The oxidative segment of the CV displayed a clear irreversible one-electron wave at +0.89 V (vs. FcH) corresponding to formation of the radical cation. The lack of the reverse wave is consistent with the partially substituted dipyrromethene core and deprotonation of the radical cation. The SWV indicates there are at least two more oxidations. The reductive segment of the CV exhibits a quasi-reversible one-electron wave at −1.26 V (vs. FcH). Upon further scanning to a more negative potential an irreversible wave is observed at −2.04 V (vs. FcH); a partial wave corresponding to oxidation of the initially generated dianion is observed at −1.47 V (vs. FcH). The significant difference between the two waves supports a significant chemical/structural transformation of the dianion initially formed. The irreversible wave is likely a mixture of two one-electron reductions of the initially formed dipyrromethene-based radical anion and the PFSP group.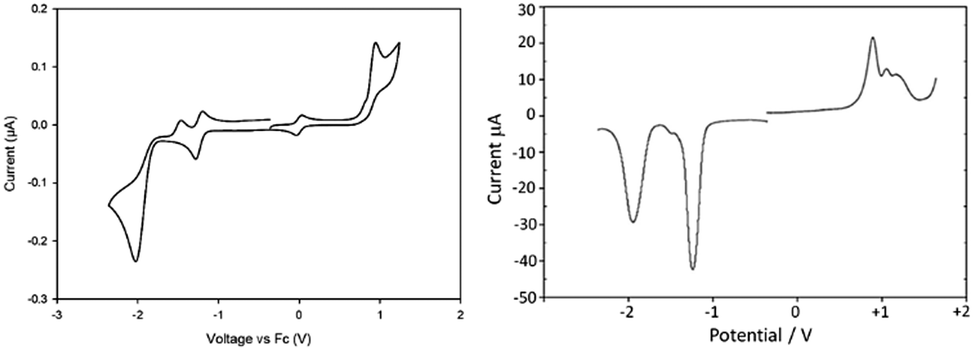 | ||
| Fig. 4 Cyclic voltammogram (left) and square-wave voltammogram (right) recorded for BD1 in a N2-purged acetonitrile solution containing 0.2 M tetrabutylammonium hexafluorophosphate background electrolyte at a glassy carbon working electron. Scan rate = 100 mV s−1. Note: the reversible wave at 0 V is the ferrocene FcH/FcH+ couple. | ||
The electrochemistry response for the remaining derivatives BD2–BD6 was also recorded, and for simplicity only the first oxidation and reduction potentials are highlighted (Table 2). Several points are worth emphasising when comparing the redox values across the series. The greater π-expanded compounds BD4–BD6 are the easiest to reduce; the hardest to reduce compound is BD3 as expected because of the presence of the four electron donating methyl groups. The easiest compound to oxidise is BD6 and coupled to its low reduction potential means that the energy gap Δ is the smallest. This energy difference ordering is BD3 > BD1 > BD2 > BD5 > BD4 > BD6. Such prioritisation is relevant when considering the absorption properties of the compounds.
Photophysical properties
Electronic absorption and fluorescence spectra were recorded for BD1–BD6 in dilute acetonitrile at room temperature. The normalized spectra are shown in Fig. 5 (see also ESI,† Fig. S5–S10) and collected in Table 3 are data for the series. The first point of note is the similarity of the absorption (λABS) and fluorescence (λEM) maxima for BD1 and BD2, despite their obvious structural differences. The similarity of the λABS values was predicted by the DFT calculations, although the absolute numbers were ca. 90 nm too blue shifted. However, it is noted that the DFT calculated ratio for the oscillator strengths corresponding to BD1/BD2 (1.34) is in good agreement with the observed ratio of 1.22 for the molar absorption coefficients (εMAX). The most noteworthy observation is the dramatic 4-fold drop in fluorescence quantum yield when comparing BD2 to BD1.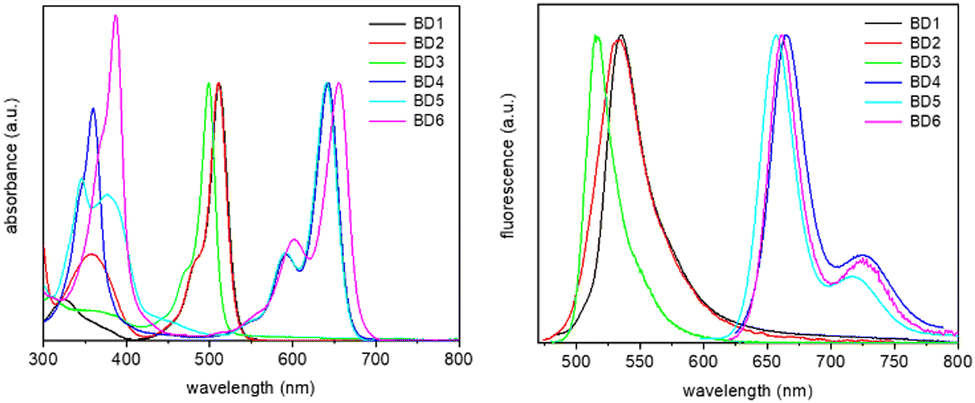 | ||
| Fig. 5 Normalised absorption (left) and fluorescence (right) spectra, recorded for the six dyes in acetonitrile. | ||
| Compound/Data | BD1 | BD2 | BD3 | BD4 | BD5 | BD6 |
|---|---|---|---|---|---|---|
| a Stokes’ shift. b Quantum yield of fluorescence. c First-excited singlet lifetime. d Radiative rate constant. e Non-radiative rate constant. | ||||||
| λ ABS nm | 511 | 511 | 499 | 642 | 641 | 655 |
| λ EM nm | 535 | 535 | 516 | 665 | 657 | 669 |
| ε MAX M−1 cm−1 | 69![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 500 |
57000 |
55000 |
79500 |
43000 |
30000 |
| SSa cm−1 | 878 | 878 | 660 | 539 | 380 | 320 |
| ϕ FLU | 0.30 | 0.07 | 0.63 | 0.57 | 0.66 | 0.25 |
| τ s psc | 487 | 296 | 2100 | 4970 | 4850 | 2130 |
|
k
RAD 108 s−1d |
6.2 | 2.4 | 3.0 | 1.1 | 1.4 | 1.2 |
|
k
NR 109 s−1e |
1.4 | 3.1 | 0.18 | 0.09 | 0.07 | 0.35 |
To try and rationalise the result, the comparison of BD1 with the phenyl (R = H) and cyanobenzene (R = CN) derivatives (Fig. 6A) is made.10 The fluorescence quantum yield for BD1 is more on par with the phenyl derivative, but the λABS and λFLU values are comparable to the cyanobenzene compound; more significantly, the ϕFLU for BD1 is 10-fold greater than this derivative. The Hammett coefficient for cyanide (σ = +0.66)11 is remarkably similar to SF5 (σ = +0.68),12 so a simple electronegativity argument for the change is not convincing. The SF5 group in the para position would appear to enhance the radiative rate constant (kRAD) and reduce the non-radiative rate constant (kNR). The concept of negative hyperconjugation of the SF5 group is used to discuss its effect on push–pull fluorescent compounds.13 The effect arises because of the sufficiently large difference in electronegativity between sulphur and fluorine (valence-bond approach), or alternatively the overall calculated bond order of 3.84 (molecular orbital approach).14 Evidently, the coupling of negative hyperconjugation and “no-bond” resonance (Fig. 6B) would result in enhanced double-bond character at the meso-aryl bond and consequently heightened rigidity in the structure. In the first-excited singlet state, this enriched rigidity would help reduce non-radiative processes. Of course, in the cyanobenzene BODIPY electron delocalization is also feasible, suggesting that the negative hyperconjugation coupled to “non-bonded” resonance has a stronger impact than simple resonance alone. The effect is “switched off” as the SF5 is decoupled from the dipyrromethene core, as evidenced by the dramatic reduction in the ϕFLU for BD2.
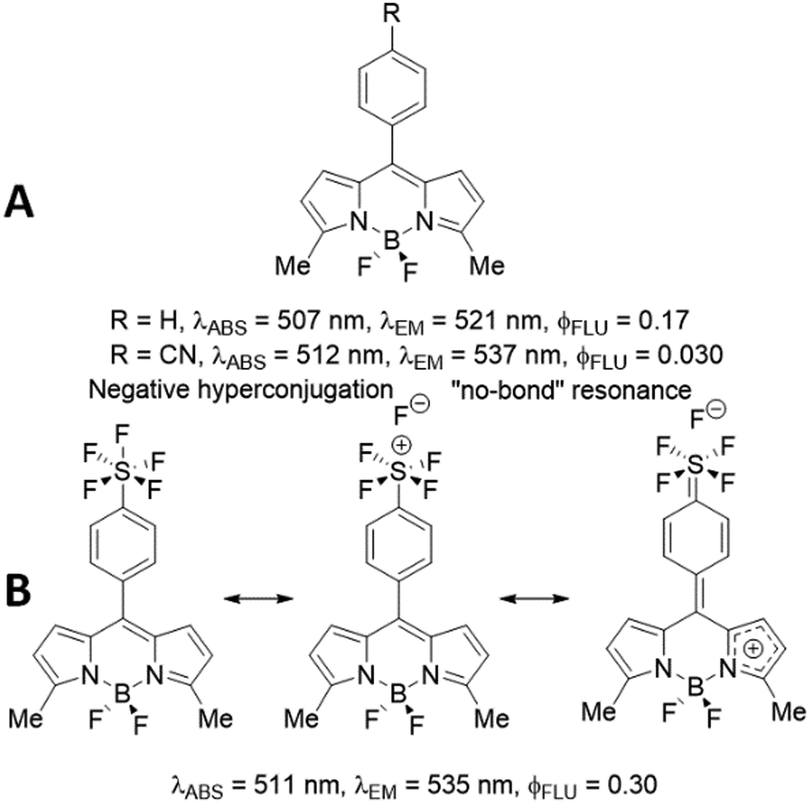 | ||
| Fig. 6 Photophysical parameters for two literature reported BODIPYs (A) and the effect of negative hyperconjugation and “no-bond” resonance on BD1 because of the SF5 unit (B). | ||
The comparison of BD4 with BD5 is highlighted as they represent the π-extended versions of BD1 and BD2, respectively. The absorption maxima, though shifted to the red as expected, for BD4 and BD5 are remarkably similar; a trend also observed for their smaller BODIPY counterparts. Their fluorescence maxima are different and separated by some 8 nm, which is in complete contrast to the trend seen for BD1 and BD2. This difference is likely linked to the alteration of the orbitals associated with the electronic transitions, which for BD4 and BD5 are confined to the two aryl-ethene groups. There is not the same dramatic drop in fluorescence quantum yield comparing BD4 with BD5, and values for ϕFLU are also reasonable for both compounds (∼60%). The SF5 group and its proximity to the dipyrromethene core would appear to be much less important. The longest-wavelength absorbing dye is BD6, with an emission maximum close to that of BD4. As a consequence, the Stokes’ shift is the smallest in the series, supporting a modest structural perturbation upon formation of the first-excited singlet state. Worth noting is the trend in λABS in terms of energy is BD3 > BD1 = BD2 > BD5 > BD4 > BD6, which is very similar to the trend observed in the difference between the oxidation and reduction potentials (Table 2).
Solvatochromic properties
The push–pull character of previously described donor–acceptor compounds incorporating the SF5 unit displayed clear solvatochromic behaviour.5 To ascertain if the same was applicable to BD4–BD6, absorption and fluorescence spectra were recorded in a range of solvents (see ESI† Fig. S11). Definite shifts in both λABS and λFLU were observed. For each compound the difference νABS–νFLU (in cm−1) versus the solvent polarity function (Δf) was analysed in terms of the Lippert-Mataga equation (eqn (1)),15 where ε is the dielectric constant and n is the refractive index.where
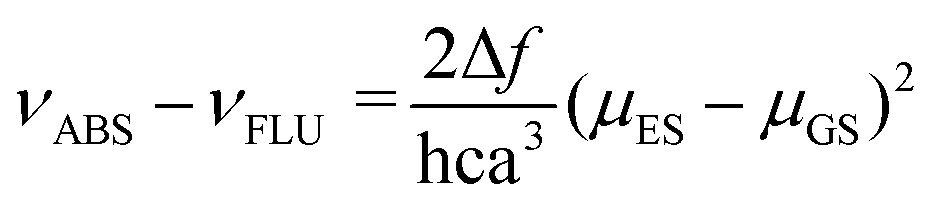
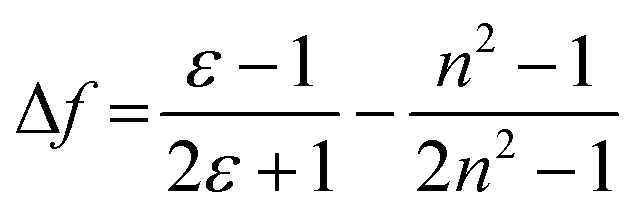
The equation predicts a linear correlation which affords the difference in the dipole moment between the ground and excited state (μES–μGS) within a spherical solvent cavity of radius “a”. There was no discernible linear relationship for any of the compounds, suggesting that the dipole moment in their ground- and excited-states is similar (see ESI,† Fig. S13). The shifts observed for λABS and λFLU are more adequately mapped to the solvent polarizability function f(n2) = (n2 − 1)/(2n2 + 1), which again matches with the concept that permanent dipoles, if they do exist, do not differ between the ground-state and the excited-state (see ESI† Fig. S12). It appears that there is no sign of any enhanced charge-transfer character in the first-excited singlet state because of the electron-withdrawing SF5 group; this tenet is supported by the DFT calculations. It is also noted that strong fluorescence was observed for all the compounds despite the change in solvent polarity.
Solid state emission
Crystals of BD1 and BD2 glowed strongly red when exposed to a UV light source, and their fluorescence optical properties were measured (see Fig. 7 and ESI† Fig. S14 and S15). The emission spectrum of BD1 displayed a clear peak centred at 603 nm which is some 68 nm red shifted with respect to the compound in fluid solution. A broad weaker tail is observed towards much longer wavelength, consistent with emission from a dimer. Excitation spectra collected by monitoring at several wavelengths were similar to one another, but very different to the absorption spectrum in solution. A clear peak is observed at 410 nm with two further broad peaks located at around 500 nm and 570 nm. The emission lifetime measured for the crystalline sample required fitting to three exponentials with lifetimes corresponding to 0.82 ns (31%), 3.2 ns (60%) and 9 ns (9%). The first component is at least comparable with the lifetime measured in solution, supporting that some fraction of the emission is likely from a monomer-like BODIPY, albeit with a non-radiative pathway that is less efficient when compared to the dye in fluid solution. The enhanced rigidity of the dye because of the constraint imposed by crystal packing would contribute to a reduction in the non-radiative rate constant. The emission spectrum for BD2 was significantly more broadened compared to BD1 with a clear maximum at 638 nm and a shoulder located at 750 nm. Again, the features are consistent with emission from a dimer, but the broadness is likely a result of multiple different sites as illustrated by the complexity of the packing diagram. The excitation spectrum in contrast to BD1 showed a maximum at 579 nm, with a much smaller peak located at 425 nm. The emission lifetime measured for the crystalline sample required fitting to only two exponentials with lifetimes corresponding to 3.8 ns (48%) and 9.3 ns (52%). That no short lifetime component was required for the fit would suggest that monomer-like emission is not that significant.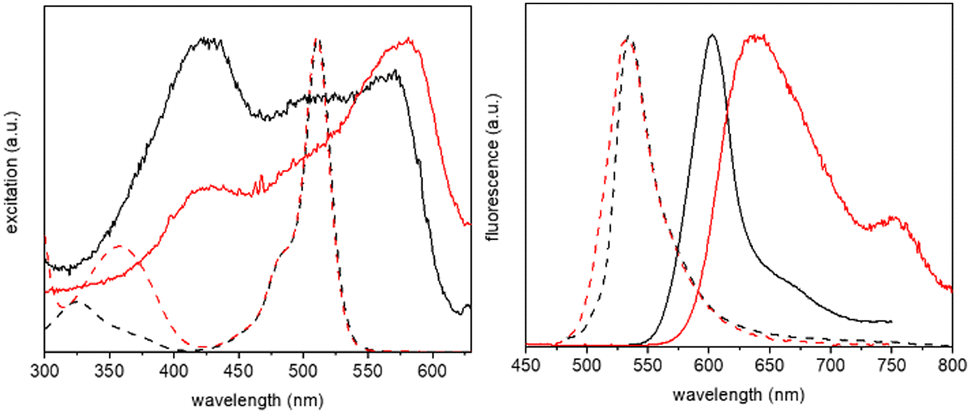 | ||
| Fig. 7 Normalised excitation (left) and fluorescence (right) spectra of BD1 (black, solid line) and BD2 (red, solid line), recorded from crystalline samples, shown in comparison with the spectra from solution samples (dashed lines). | ||
The noticeably clear difference between the two compounds is evidently the result of their disparate crystal packing features as highlighted previously (see ESI† Fig. S16–S22). At a first level of interpretation, exciton coupling16 of dimers would be anticipated to be quite different from inspection of the packing diagrams. For BD1 there is a clear dimer which aligns the electronic transition dipoles at 86° with respect to each other, with a centre-to-centre distance of 5.1 Å. The exciton coupling in this dimer would be weak because of the near perpendicular arrangement of the two chromophores, explaining why some monomer-like emission is observed. A further two dimers are also discernible from the packing diagram, but in these cases the electronic transition dipoles are parallel or more co-linear, with centre-to-centre distances of 7.5 Å and 12.6 Å, respectively. In the classic exciton coupling model,17 the parallel arrangement would result in a short-wavelength absorption band and the co-linear in a long-wavelength absorption band. This interpretation seems reasonable as both the features are observed in the excitation spectrum.
Two distinct dimers are observed for BD2 within the crystal packing diagram, with centre-to-centre distances of 4.4 Å and 8.4 Å, respectively. The short-distance apart dimer represents an offset-stacked arrangement of dipyrromethene subunits. The second dimer places the dipyrromethene subunits in a head-to-head arrangement with parallel electronic transition dipoles. Two more dimer arrangements are observed between stacks in the packing diagram (viewed down c-axis), separated by 9.8 Å and 9.5 Å, respectively, with offset co-linear electronic transition dipoles (see ESI† Fig S21 and S22). The features observed in the excitation spectrum again can be explained by invoking a classic exciton coupling model.
Conclusions
The incorporation of pentafluorosulfanylaryl subunits into a BODIPY core is readily achieved at the meso or 3,5-positions, because of readily available starting materials or easily prepared precursors. Although the SF5 unit within an aryl ring is electron withdrawing, its effect can be adequately discussed in terms of negative hyperconjugation and “non-bonded” resonance. In particular, direct linkage of the PFSP at the meso position of the dipyrromethene core appears to promote structural rigidity in the first-excited singlet state. As the group is decoupled from the dipyrromethene unit, its influence appears to diminish. This “pentafluorosulfanyl effect” may hold for other substitution patterns of BODIPY, or more significantly be universal to other chromophores. Hence, low fluorescence quantum yield flexible dyes may get an enhancement by its presence.It is evident that the PFSP group is not an effectual electron acceptor, as evidenced in the electrochemistry findings and by the fact that no fluorescence quenching is observed even in highly polar solvents. Any charge-transfer state (CTS) associated with electron transfer from the BODIPY core to the PFSP group must be higher in energy than the emitting state; solvent stabilization is not strong enough to bring the CTS into play. In the solid state the situation is likely more complicated since the long lifetime of ca. 9 ns seen for BD1 and BD2 is unusual for J-like dimers. It is possible that a localised CTS associated with a dimer comes into play.
Experimental
Solvents for synthesis were used as received or dried by standard methods. Compounds were characterised by nuclear magnetic resonance (NMR) spectroscopy using Bruker Avance III 300 MHz, Bruker Avance III 400 MHz and Bruker Avance III HD 700 MHz spectrometers. Further analysis was performed using mass spectrometry (MS) and where possible by X-ray crystallography. UV-Vis spectroscopy measurements were conducted on a Shimadzu (UV-1800) UV/Vis spectrometer using 1 cm quartz cuvettes, 1 nm sample interval and ran in air unless otherwise stated. Emission and excitation measurements were run on a Shimadzu (RF-6000) fluorescence spectrometer using 1 cm quartz cuvettes and HPLC grade solvents where appropriate. Scan speed was 600 nm min−1 with slits at 3.0 nm for both source and detector unless otherwise stated. Quantum yield measurements were kept under 0.05 absorbance at the excitation wavelength to eliminate self-quenching and referenced versus either rhodamine 6G (λem = 480 nm, ϕFLU = 0.95 in methanol) or oxazine 1 (λem = 665 nm, ϕFLU = 0.11 in methanol).18 Solid state and lifetime measurements were run using an Edinburgh FLS980 photoluminescence spectrometer, equipped with a 450 W Xenon arc lamp, Czerny Turner excitation and emission monochromators (1.8 nm mm−1 dispersion; 1800 grooves mm−1), time-correlated single photon counting (TCSPC) module and a Hamamatsu R928 P photomultiplier tube (in a fan assisted TE cooled housing, operating temperature −20 °C). For lifetime measurements, samples were excited with an EPL-375 (370.8 nm; 61.1 ps pulse width) and an EPL-475 (471.8 nm; 61.1 ps pulse width) picosecond pulsed diode lasers and data analysis was performed on the F980 software with numerical data reconvolution based on Marquardt–Levenberg algorithm. Electrochemical characterisation used a three-electrode set-up with a platinum counter electrode, either a calomel or silver/silver chloride reference electrode and a glassy carbon working electrode. The background electrolyte was a 0.2 M tetrabutylammonium hexafluorophosphate solution in dry acetonitrile which was degassed before each run. The setting used for DPV were a 0.2 V pulse for 0.02 seconds with a scan rate of 0.01 V s−1. The setting used for square wave voltammetry are 0.01 V step with a 0.1 V amplitude at 20 Hz.Computer calculations were performed using Gaussian 0919 with Gauss view 5.0 to set up calculations and Visual Molecular Dynamics was used to render the images of the orbitals. All calculations where run on the University of Newcastle computing system Rocket HPC. The structures were either optimised before the energy calculations in vacuum or the structure was obtained via X-ray crystallography.
Preparation of 2
4-Ethynyl benzaldehyde (1.0 g, 7.7 mmol), [1,1′-bis(diphenylphosphino) ferrocene] dichloropalladium(II) (280 mg, 0.38 mmol), and copper(I) iodide (73 mg 0.38 mmol) were added to a 100 mL flame dried Schlenk flask under an inert nitrogen atmosphere. The flask was degassed three times. Toluene (30 mL) and diisopropylamine (5 mL) were added to a second flame dried Schlenk flask, the liquids were degassed using freeze pump thaw technique 5 times. The liquids were transferred to the first Schlenk flask via a cannula. The reaction was heated to 80 °C for 16 hours. The reaction was cooled to room temperature and solvents removed under reduced pressure. The crude reaction mixture was purified using silica gel column chromatography, 100% ethyl acetate, to produce 2 as a pale yellow solid (1.1 g, 3.3 mmol, 94%) 1H NMR (300 MHz, CDCl3) δ 10.04 (s, 1H), 7.90 (d, J = 8.5 Hz, 2H), 7.77 (d, J = 9.0 Hz, 2H), 7.70 (d, J = 8.5 Hz, 2H), 7.63 (d, J = 9.1 Hz, 2H). 13C{1H} NMR (176 MHz, CDCl3) δ 191.46, 136.09, 132.49, 132.02, 129.78, 128.64, 126.37, 126.34, 126.32, 91.31, 91.07. 19F NMR (282 MHz, CDCl3) δ 83.67 (quintet), 62.84 (d).Preparation of BD2
Dry DCM (100 mL) containing 2 (1.0 g, 3.0 mmol), 2-methyl-1H-pyrrole (0.57 mL, 6.62 mmol) and three drops of trifluoroacetic acid were added to a 250 mL Schlenk tube under an inert nitrogen atmosphere. The resulting reaction mixture was stirred at room temperature for 3 hours. p-Chloranil (888 mg, 3.61 mmol) was added portion wise over 10 minutes, and the reaction mixture was stirred at room temperature for 3 hours under an inert nitrogen atmosphere. Diisopropylethylamine (3.7 mL, 21 mmol) was added and stirred at room temperature for 15 minutes. Boron trifluoride diethyl etherate (4.0 mL, 33 mmol) was added and the reaction mixture was stirred at room temperature overnight (∼16 h). The reaction mixture was washed with water (5 × 500 mL). The organic layer was separated, dried over Na2SO4 and the solvent was removed under reduced pressure. Following silica gel column chromatography (DCM: petroleum ether 40–60 °C, 2:3) the product was obtained as a red solid (0.90 g, 1.8 mmol, 60%). 1H NMR (300 MHz, CDCl3) δ 7.77 (d, J = 8.9 Hz, 2H), 7.69–7.59 (m, 4H), 7.51 (d, J = 8.5 Hz, 2H), 6.71 (d, J = 4.1 Hz, 2H), 6.29 (d, J = 4.1 Hz, 2H), 2.66 (s, 6H). 13C{1H} NMR (176 MHz, CDCl3) δ 158.20, 141.42, 134.79, 134.39, 131.94, 131.69, 130.64, 130.29, 126.72, 126.31, 124.34, 120.20, 119.82, 91.51, 89.42, 15.10.11B NMR (96 MHz, CDCl3) δ 0.95 (t, J = 30.7 Hz). 19F NMR (282 MHz, CDCl3) δ 83.92 (quintet), 62.68 (d, J = 149.9 Hz), −147.53 (q).
Preparation of BD3
Dry DCM (100 mL) containing allyl 4-formylbenzoate 3 (1.0 g, 5.26 mmol), 2,4-dimethyl-1H-pyrrole (0.92 mL, 11.57 mmol), and three drops of trifluoroacetic acid were added to a 250 mL Schlenk tube under an inert nitrogen atmosphere. The resulting reaction mixture was stirred at room temperature for 3 hours. DDQ (1.43 g, 6.31 mmol) was added portion wise over 10 minutes, and the reaction mixture was stirred at room temperature for 3 hours under an inert nitrogen atmosphere. Diisopropylethylamine (6.41 mL, 36.8 mmol) was added and stirred at room temperature for 15 minutes. Boron trifluoride diethyl etherate (7.14 mL, 36.8 mmol) was added and the reaction mixture was stirred at room temperature overnight (∼16 h). The reaction mixture washed with water (5 × 500 mL). The organic layer was separated, dried over Na2SO4 and the solvent was removed under reduced pressure. Following silica gel column chromatography (DCM: petroleum ether 40–60 °C, 2:3) the product was obtained as a red solid (1.25 g, 3.1 mmol, 58%) 1H NMR (300 MHz, CDCl3) δ 8.20 (d, J = 8.3 Hz, 2H), 7.41 (d, J = 8.3 Hz, 2H), 6.08 (ddt, J = 17.2, 10.4, 5.8 Hz, 1H), 5.99 (s, 2H), 5.46 (dd, J = 17.2, 1.5 Hz, 1H), 5.33 (dd, J = 10.4, 1.3 Hz, 1H), 4.87 (d, J = 5.8 Hz, 2H), 2.55 (s, 6H), 1.36 (s, 6H). 13C{1H} NMR (75 MHz, CDCl3) δ 165.78, 156.15, 143.02, 140.34, 140.08, 132.10, 131.07, 130.95, 130.56, 128.55, 121.63, 118.96, 66.15, 14.67. 11B NMR (96 MHz, CDCl3) δ 0.74 (t, J = 32.3 Hz). 19F NMR (282 MHz, CDCl3) δ −146.23 (quartet, J = 65.5, 32.6 Hz). Mass calc. for C23H24BF2N2O2 409.1899 fnd. m/z = 409.1888 [M + H]+.
Preparation of BD5
BD2 (50 mg, 0.096 mmol), tert-butyl 4-formylbenzoate 4 (79 mg, 0.38 mmol), and 3 Å molecular sieves (1 g) were added to a 100 mL flame dried Schlenk flask under nitrogen. Dry acetonitrile (20 mL) was added. Piperidine (0.19 mL, 1.91 mmol) was added dropwise followed by glacial acetic acid (0.11 mL, 1.91 mmol). The reaction was stirred at 60 °C for 16 hours. The reaction was cooled, filtered and the solvent was removed under reduced pressure. Following silica gel column chromatography (DCM: petroleum ether 40–60 °C, 3:2) the product was isolated as a blue/green solid (10 mg, 0.02 mmol, 19%). 1H NMR (700 MHz, CDCl3) δ 8.04 (d, J = 8.2 Hz, 4H), 7.86 (d, J = 16.3 Hz, 2H), 7.78 (d, J = 8.7 Hz, 2H), 7.73–7.66 (m, 6H), 7.65 (d, J = 8.3 Hz, 2H), 7.56 (d, J = 8.2 Hz, 2H), 7.37 (d, J = 16.3 Hz, 2H), 6.99 (d, J = 4.5 Hz, 2H), 6.84 (d, J = 4.4 Hz, 2H), 1.63 (s, 18H). 13C{1H} NMR (176 MHz, CDCl3) δ 165.48, 154.88, 153.49, 140.23, 139.05, 136.43, 136.12, 134.84, 132.37, 131.96, 131.88, 130.74, 130.12, 129.83, 127.46, 126.69, 126.32, 124.45, 121.24, 117.15, 91.50, 89.62, 81.37, 28.37. 19F NMR (282 MHz, CDCl3) δ 87.39–80.28 (m), 62.67 (d, J = 149.8 Hz), −125.76 to −145.51 (m). 11B NMR (96 MHz, CDCl3) δ 1.30 (t).
Preparation of BD6
Compound 2 (200 mg, 0.49 mmol) and BD3 (208 mg, 1.96 mmol) were added to a 100 mL 3-necked round bottomed flask. Dry toluene (50 mL) was added followed by piperidine (0.97 mL, 9.80 mmol) and p-toluenesulfonic acid (1.69 g, 9.80 mmol). The flask was fitted to a Dean-Stark distillation apparatus. The flask was heated to 150 °C and the water was gradually removed over time. Finally, all the toluene was distilled off and the flask was cooled to room temperature and more dry toluene (50 mL) was added. The flask was heated to 150 °C and the toluene was removed for a second time. The crude reaction mixture was purified using silica gel column chromatography (dichloromethane: petroleum ether 40–60 °C, 3:1). The product was isolated as a blue solid (30 mg, 0.029 mmol, 6%). 1H NMR (700 MHz, CDCl3) δ 8.24 (d, J = 8.3 Hz, 2H), 7.87–7.71 (m, 6H), 7.67–7.61 (m, 8H), 7.58 (d, J = 8.3 Hz, 4H), 7.48 (d, J = 8.2 Hz, 2H), 7.26 (d, J = 16.2 Hz, 2H), 6.68 (s, 2H), 6.25–5.99 (m, 1H), 5.64–5.23 (m, 2H), 4.89 (dd, J = 5.9, 1.5 Hz, 2H), 1.45 (s, 6H). 13C{1H} NMR (176 MHz, CDCl3) δ 165.77, 153.24, 152.84, 142.33, 139.99, 137.95, 137.19, 135.74, 133.45, 132.37, 132.09, 131.83, 131.15, 130.61, 128.95, 127.74, 127.08, 126.37–125.99 (m), 122.85, 120.34, 119.03, 118.55, 92.68, 89.32, 66.22. 11B NMR (96 MHz, CDCl3) δ 1.13 (t, J = 33 Hz). 19F NMR (659 MHz, CDCl3) δ −137.81 (quartet, J = 66 Hz, 33 Hz), −73.20 (s), −72.11 (s). Mass calc. for C53H37BF12N2O2S 1036.2198 fnd. m/z = 1036.6705 [M]+.
X-Ray diffraction
All crystal structure data were collected at 150 K using copper radiation (λCuKα = 1.54184 Å) on an Xcalibur, Atlas, Gemini ultra diffractometer equipped with an Oxford Cryosystems CryostreamPlus open-flow N2 cooling device. The intensities were corrected for absorption empirically using a multifaceted crystal model created by indexing the faces of the crystal for which data were collected.20 Cell refinement, data collection and data reduction were undertaken via the software CrysAlisPro.21All structures were solved using XT22 and refined by XL23 using the Olex2 interface.24 All non-hydrogen atoms were refined anisotropically and hydrogen atoms were positioned with idealised geometry, with the exception of those bound to heteroatoms where the positions could be located using peaks in the Fourier difference map. The displacement parameters of the hydrogen atoms were constrained using a riding model with U(H) set to be an appropriate multiple of the Ueq value of the parent atom.
Author contributions
RDJ carried out all the synthetic work, characterization and spectroscopic work with supervision from FC and ACB. All crystallographic work was performed by PGW. Manuscript preparation was carried out by ACB with contributions from all the other authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the University of Newcastle and the Engineering and Physical Sciences Research Council (EPSRC) (EP/P015395/1) for financial support.Notes and references
-
(a) N. Boens, V. Leen and W. Dehaen, Chem. Soc. Rev., 2012, 41, 1130–1172 RSC
; (b) T. Zhang, C. Ma, T. Sun and Z. Xie, Coord. Chem. Rev., 2019, 390, 76–85 CrossRef CAS
-
(a) A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS PubMed
-
(a) R. Ziessel, G. Ulrich and A. Harriman, New J. Chem., 2007, 31, 496–501 RSC
- M. A. H. Alamiry, A. C. Benniston, J. Hagon, T. P. L. Winstanley, H. Lemmetyinen and N. V. Tkachenko, RSC Adv., 2012, 2, 4944–4950 RSC
- P. Gautam, C. P. Yu, G. Zhang, V. E. Hillier and J. M. W. Chan, J. Org. Chem., 2017, 82, 11008–11020 CrossRef CAS PubMed
- D. Kerwat, S. Grätz, J. Kretz, M. Seidel, M. Kunert, J. B. Weston and R. D. Süssmuth, ChemMedChem, 2016, 11, 1899–1903 CrossRef CAS PubMed
- L. S. Alqahtani, R. D. James, J. Mallows, H. V. Flint, P. G. Waddell, O. Woodford and E. A. Gibson, Sustainable Energy Fuels, 2023, 7, 1494–1501 RSC
- B. Herrado, A. Chana, M. Alonso, F. Amat-Guerria, M. Liras and M. A. Maestro, J. Mol. Struct., 2004, 697, 29–40 CrossRef
- P. Geerlings, F. De Proft and W. Langenaeker, Chem. Rev., 2003, 103, 1793–1873 CrossRef CAS PubMed
- W. Qin, M. Baruah, M. Van der Auweraer, F. C. De Schryver and N. Boens, J. Phys. Chem. A, 2005, 109, 7371–7384 CrossRef CAS PubMed
- D. H. McDaniel and H. C. Brown, J. Org. Chem., 1958, 23, 420–427 CrossRef CAS
- W. A. Sheppard, J. Am. Chem. Soc., 1962, 84, 3072–3076 CrossRef CAS
- P. Gautam, Y. Wang, G. Zhang, H. Sun and J. M. W. Chan, Chem. Mater., 2018, 30, 7055–7066 CrossRef CAS
- O. Exner and S. Böhm, New J. Chem., 2008, 32, 1449–1453 RSC
-
(a) E. Z. Lippert, Z. Naturforsch., A: Astrophys., Phys. Phys. Chem., 1955, 10a, 541–545 CrossRef CAS
- S. G. Telfer, T. M. McLean and M. R. Waterland, Dalton Trans., 2011, 40, 3097–3108 RSC
- M. Kasha, H. R. Rawls and M. A. El-Bayoumi, Pure Appl. Chem., 1965, 11, 371–392 CAS
- A. M. Brouwer, Pure Appl. Chem., 2011, 83, 2213–2228 CrossRef CAS
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2016 Search PubMed
- R. C. Clark and J. S. Reid, Acta Crystallogr., Sect. A: Found. Crystallogr., 1995, 51, 887–897 CrossRef
-
CrysAlisPro, Rigaku Oxford Diffraction, Tokyo, Japan Search PubMed
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2015, 71, 3–8 CrossRef PubMed
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. J. Puschmann, Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Spectroscopic data including NMR spectra, mass spectra, UV-vis spectra and X-ray diagrams; molecular orbital pictures. CCDC 2240726 and 2240727. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3nj00633f |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2023 |