
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Novel cyano-activated Cu(II) complexes of arylhydrazones of active methylene nitriles and their catalytic application for azide–alkyne cycloaddition in water and glycerol†
Atash V.
Gurbanov
 *ab,
Abdallah G.
Mahmoud
*ac,
Vusala A.
Aliyeva
a,
M. Fátima C.
Guedes da Silva
ad and
Armando J. L.
Pombeiro
ae
*ab,
Abdallah G.
Mahmoud
*ac,
Vusala A.
Aliyeva
a,
M. Fátima C.
Guedes da Silva
ad and
Armando J. L.
Pombeiro
ae
aCentro de Química Estrutural, Institute of Molecular Sciences, Instituto Superior Técnico, Universidade de Lisboa, Av. Rovisco Pais, 1049-001 Lisbon, Portugal. E-mail: atash.gurbanov@tecnico.ulisboa.pt; abdallah.mahmoud@tecnico.ulisboa.pt
bExcellence Center, Baku State University, Z. Xalilov Str. 23, Az 1148 Baku, Azerbaijan
cDepartment of Chemistry, Faculty of Science, Helwan University, Ain Helwan, Cairo 11795, Egypt
dDepartamento de Engenharia Química, Instituto Superior Técnico, Universidade de Lisboa, Av. Rovisco Pais, 1049-001 Lisboa, Portugal
eResearch institute of chemistry, Peoples’ Friendship University of Russia (RUDN University), 6 Miklukho-Maklaya St., Moscow 117198, Russian Federation
First published on 14th March 2023
Abstract
The reaction of 2-(2-(dicyanomethylene)hydrazineyl)benzoic acid (H2L1) with copper(II) acetate in the absence or presence of imidazole (im), 4,4′-bipyridine (4,4′-bipy) or pyridine (py) leads to the new complexes [Cu2(CH3OH)2(μ-L1a)2] (1), [Cu(L1a)(im)] (2), [Cu(L1a)(H2O)(4,4′-bipy)]·H2O (3) or [Cu(L1a)(py)]n (4), respectively, where (L1a)2− = (Z)-1-(2-carboxylatophenyl)-2-(1-cyano-2-imino-2-methoxyethylidene)hydrazin-1-ide. A one-pot activation of nitrile groups in the reaction of copper(II) acetate monohydrate with 2-(2-(dicyanomethylene)hydrazineyl)terephthalic acid (H3L2) in the presence of pyridine in methanol affords the trinuclear Cu(II) complex [Cu(py)2{Cu(py)(μ-L2a)}2] (5), where (L2a)3− = (Z)-2-((1-cyano-2-imino-2-methoxyethylidene)-1-(2,5-dicarboxylatophenyl)hydrazin-1-ide). Both arylhydrazone ligands and the auxiliary ligands were used to modulate the nuclearity and design of the supramolecular arrangements of the obtained Cu(II) complexes. The complexes were characterized by elemental analyses, electrospray ionization mass spectrometry (ESI-MS), and FT-IR spectroscopy. Their molecular structures were established using single crystal X-ray diffraction (SCXRD) analysis. The catalytic activity of the complexes was investigated for the microwave assisted 1,3-dipolar azide–alkyne cycloaddition reaction using a mixture of water and glycerol as reaction medium. Pre-catalyst 1 was found to be the most efficient one by affording a quantitative conversion to 1,4-disubstituted-1,2,3-triazoles after 30 minutes at 125 °C. The catalytic system proceeds with a broad scope of substrates according to “Click” rules.
1. Introduction
At the beginning of this century, Sharpless1 and Meldal2 independently discovered the copper catalysed azide–alkyne cycloaddition (CuAAC) reaction to selectively obtain 1,4-disubstituted-1,2,3-triazoles, which can be considered as an “ideal” click reaction.3 During the last two decades, CuAAC has become tremendously popular, both at research laboratories and in industry, being the most reliable and efficient synthetic tool to obtain triazole building blocks with applications in several fields including pharmaceuticals, molecular biology, dyes, agricultural chemicals, polymers and materials sciences.4–9 With their discovery of CuAAC, Sharpless and Meldal started a snowball effect that eventually led to them sharing the Nobel Prize in Chemistry 2022 due to the foundation of “click” chemistry, which brought this branch of science into a new era of functionalisation.10Several challenges for the CuAAC reaction still need to be addressed, including the utilization of benign non-toxic solvents. In recent years, there has been growing interest in the use of water as a solvent for catalytic reactions due to its abundance, low cost, and environmentally friendly properties.11–13 In some cases, catalytic conversions are often better in an aqueous medium than in pure organic solvents due to the unique features of water such as high dielectric constant, the ability to act as a nucleophile or a leaving group, and the ability to solubilize polar reactants and catalysts.14–18 Although homogeneous CuAAC can proceed in aqueous medium, the presence of an organic co-solvent (e.g. acetonitrile) is vital to dissolve the organic reactants and increase the efficiency of the catalytic reaction.19–23 Replacing volatile organic and toxic co-solvents with a green organic solvent constitutes one of the main objectives of this study. Utilization of glycerol, which is recognized as an environmentally benign non-toxic solvent,24,25 for CuAAC is still scarce.26,27
A better understanding of the influence of metals and ligands on the mechanistic pathway of the reaction is challenging but will allow the usage of milder conditions ultimately leading to more sustainable transformations. In this context, several metal centres28–30 and ligands including amidophosphines,23,27,31,32 triphenyl phosphine,33,34 polydentate amines,35 tris(pyrazolyl)methanes,20,36 arylhydrazones of β-diketones21 and N-heterocyclic carbenes,37–39 have been explored.
Arylhydrazones of active methylene nitriles (AAMNs, Scheme 1(a)) have been recognized as multifunctional ligands with a rich chemical reactivity.40 The resonance-assisted hydrogen bonding (RAHB, Scheme 1(b)) and/or the presence of a transition metal ion can promote a nucleophilic attack on at least one of the cyano groups affording a variety of new functionalities including amidines, imino ethers and carboxamides.41–43 Besides their role in the formation of the RAHB, the presence of carboxylic or sulfonic groups on the aromatic moiety of the arylhydrazones enhances their ability to act as hydrophilic ligands to obtain water–soluble metal complexes, thereafter applied as catalysts in homogeneous aqueous medium for several organic transformations.44–47
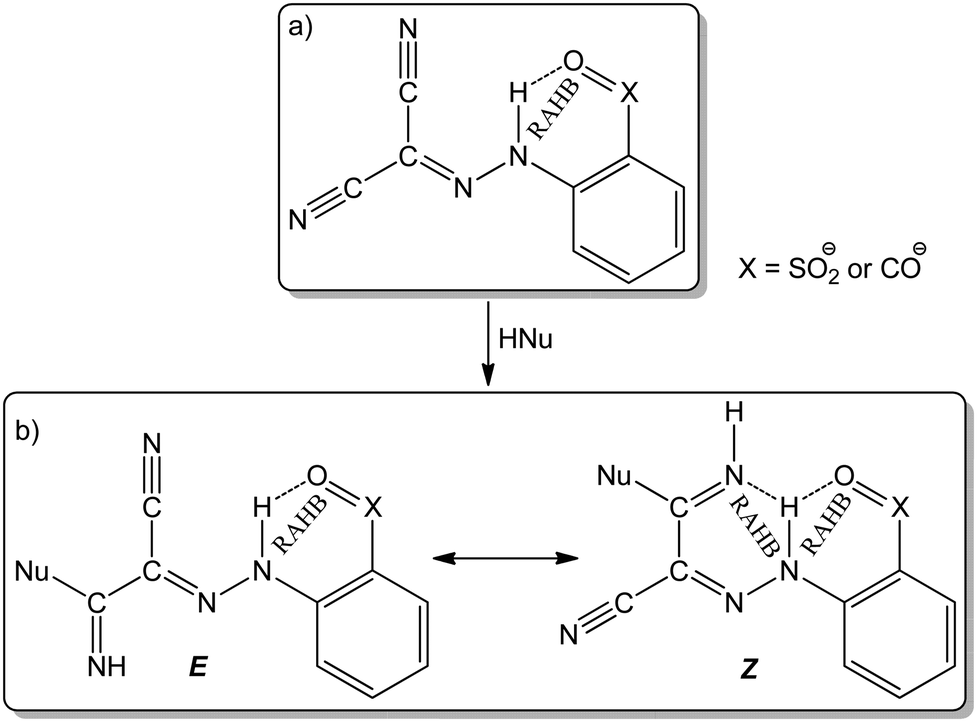 | ||
| Scheme 1 Arylhydrazones of active methylene nitriles with RAHB sites. | ||
Recently, we observed the ability of arylhydrazones of β-diketones to act as Cu-protectors and catalysis promoters for the CuAAC reaction and reported an unprecedent relationship between RAHB of the compounds and their catalytic performances.21 Although arylhydrazones of active methylene compounds are considered as “privileged ligands” due to their high stability and ease of preparation using accessible methods, their application for CuAAC is limited to only that example.21 Utilization of copper catalysts based on AAMNs has not been explored for the CuAAC reaction yet. In this context, this work describes the synthesis and characterization of new five hydrosoluble Cu(II)–AAMN complexes with the hydrazone ligands holding carboxylate groups. Their molecular structures were identified in the solid phase using single crystal X-ray diffraction (SCXRD) analysis. For the first time, complexes based on AAMN are utilized as catalysts for the CuAAC reaction in aqueous medium using glycerol as an organic green co-solvent under microwave irradiation.
2. Results and discussion
2.1. Synthesis and characterization of the complexes
The AAMN 2-(2-(dicyanomethylene)hydrazineyl)benzoic acid (H2L1),42 with a carboxylic group at the ortho-position, was utilized for the template synthesis of the new complexes (Scheme 2(a)). The reaction of copper(II) acetate with H2L1 in methanol afforded the dinuclear complex [Cu2(CH3OH)2(μ-L1a-κNN′:1κO,2κO)2] (1). Performing the reaction in the presence of imidazole (im), 4,4′-bipyridine (4,4′-bipy) or pyridine (py) as auxiliary ligands led to the formation of the complexes [Cu(L1a-κNN′O)(im)] (2), [Cu(L1a-κNN′O)(H2O)(4,4′-bipy)]·H2O (3) or [Cu(μ-L1a-1κNN′O,2κN′′)(py)]n (4), respectively. The structures and topologies of the obtained complexes are dependent on the utilized auxiliary ligand. One of the nitrile groups of H2L1 is activated by the copper ion, undergoes nucleophilic attack by methanol and leads to the iminoester ligand HL1a. The other cyano group remains inactivate, except in 4 where it is linked to a copper centre leading to a coordination polymer. The presence in H2L1 of the carboxylic group in the ortho position promotes the intramolecular N–H⋯O RAHB interaction (Scheme 1), which usually hampers the coordination through this site. The utilization of methanol as a polar solvent with proton-acceptor ability weakened the RAHB, permitting the copper ion to enter the chelating ONN pocket.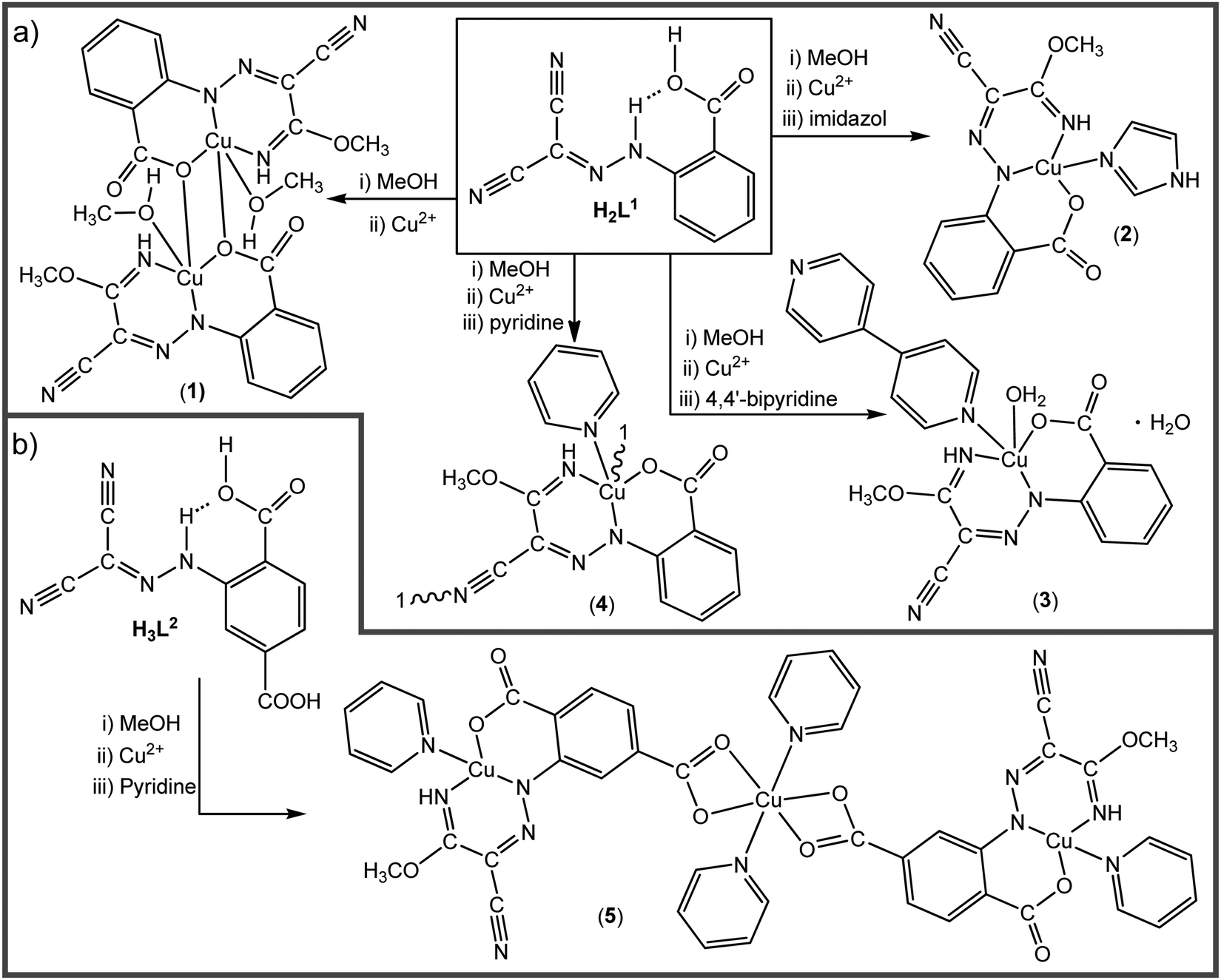 | ||
| Scheme 2 Synthesis of Cu(II)–AAMNs. | ||
The coordinating ability of the carboxylic group facilitates the formation of the copper complexes and affects the type of the obtained structure. Another AAMN compound with two carboxylic groups on the aromatic moiety, namely 2-(2-(dicyanomethylene)hydrazineyl)terephthalic acid (H3L2), was obtained for the first time and reacted with copper acetate in the presence of py as an auxiliary ligand to afford the trinuclear copper complex [Cu(py){Cu(μ-L2a-1κOO′,2κNN′O′′)(py)}2] (5, Scheme 2(b)).
The new hydrosoluble compounds H3L2 and 1–5 were characterized by elemental analysis, ESI-MS and IR spectroscopy. Due to the paramagnetic nature of the copper(II) complexes, only H3L2 was characterized by NMR spectroscopy.
The IR spectra of the compounds show the ν(NH) vibrations in the ∼3313–2948 cm−1 range, while the ν(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) of the unreacted cyano group in 1, 2, 3 and 5 appears in the ∼2212–2221 cm−1 range. The proposed structures of all compounds were confirmed by elemental analysis and ESI-MS. The 1H-NMR spectrum of H3L2 in DMSO-d6 shows a signal at 15.04 ppm assigned to the hydrazone N–H proton, which is attributed to the involvement of that proton in the RAHB system. The molecular structures of complexes 1–5 were established by SCXRD analysis (Fig. 1), selected crystallographic data and structure refinement details are provided in Table S1, ESI† and selected bond distances and angels are given in Table S2 (ESI†).
N) of the unreacted cyano group in 1, 2, 3 and 5 appears in the ∼2212–2221 cm−1 range. The proposed structures of all compounds were confirmed by elemental analysis and ESI-MS. The 1H-NMR spectrum of H3L2 in DMSO-d6 shows a signal at 15.04 ppm assigned to the hydrazone N–H proton, which is attributed to the involvement of that proton in the RAHB system. The molecular structures of complexes 1–5 were established by SCXRD analysis (Fig. 1), selected crystallographic data and structure refinement details are provided in Table S1, ESI† and selected bond distances and angels are given in Table S2 (ESI†).
 | ||
| Fig. 1 Ellipsoid plots of 1–5 (60% probability level) with partial atom numbering schemes. Symmetry operations to generate equivalent atoms: (i) −x, 1 − y, 1 − z (1). (i) −1/2 + x, y, 1/2 − z; (ii) 1/2 + x, y, 1/2 − z (4). (i) 1 − x, 2 − y, 1 − z (5). The crystallization water molecule in 3 is omitted. | ||
The SCXRD shows the diversity in the nuclearity of the compounds and the geometry around the Cu(II) metal centres. The asymmetric unit in 1 contains a Cu(II) cation, a chelating hydrazone and a methanol ligand; symmetry expansion affords the dimeric compound with two bridging oxygen atoms from the symmetry related chelating hydrazone moieties. Thus, each copper centre in 1 possesses a N2O2 square-pyramidal geometry (τ5 = 0.04)48 with the apical site engaged with the O-methanol atom. The Cu–Xequatorial bond distances (average 1.978 Å) are shorter than the Cu–Oaxial one, which assume the value of 2.283(2) Å. The Cu2(μ-O)2 core is planar. The Cu⋯Cu intramolecular distance is of 3.1905(7) Å and the minimum intermolecular one of 6.680(1) Å.
The monomeric compounds 2 crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . It shows a distorted square planar geometry (τ4 = 0.30)49 around the copper centre which is coordinated to a N2O tridentate dianionic L1a ligand (average Cu–X lengths of 1.934 Å) and to an imidazole (Cu–N5 = 1.992(1) Å). The intermolecular Cu⋯Cu distance is as short as 3.919 Å.
. It shows a distorted square planar geometry (τ4 = 0.30)49 around the copper centre which is coordinated to a N2O tridentate dianionic L1a ligand (average Cu–X lengths of 1.934 Å) and to an imidazole (Cu–N5 = 1.992(1) Å). The intermolecular Cu⋯Cu distance is as short as 3.919 Å.
Complexes 3 and 4 present distorted square-pyramidal coordination spheres (τ5 = 0.50 and 0.20, in this order)48 with the chelating hydrazone ligand occupying three equatorial positions and the fourth one being engaged with the nitrogen atom of the auxiliary ligand 4,4′-bipyridine or pyridine, respectively. While the apical site in 3 is filled with an O-water atom at 2.343(2) Å, in 4 it is involved with a N-cyano atom from an adjacent molecule at 2.597(2) Å, thus leading to a 1D coordination polymer that runs along the crystallographic a axis (Fig. S1, ESI†).
Complex 5 crystallizes in the monoclinic space group C2/c, its asymmetric unit including two copper cations (one of them, Cu2, standing in an inversion centre) bridged by the trianionic O2N2O chelating hydrazone moiety, each also coordinated to the N-atom from a pyridine ligand. Symmetry expansion affords a trinuclear entity where the central metal cation stands in a distorted octahedral geometry composed of, in the equatorial plan, four O atoms from two asymmetric bidentate chelating carboxylate groups (Cu2–O of 2.044(4) and 2.411(4) Å), and two Npyridine atoms in the apical sites (Cu2–N = 1.998(5) Å). The copper centre at the chelating ONN hydrazone pocket exhibits a slightly distorted square-planer geometry (τ4 = 0.18)49 with dimensions Cu–O1 1.888(4) Å, Cu–N1 1.947(4) Å and Cu–N3 1.929(4) Å, while the fourth position is completed by the N-pyridine atom at 2.041(4) Å. The smallest intermolecular Cu···Cu distance is of 5.005(1) Å and the intramolecular one is 9.186 Å.
In all cases the copper cations at the ONN hydrazone pocket are involved in two six-membered CuN2C2N and CuNC3O metallacycles, where the average N–N, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and CNH bond lengths of 1.3074, 1.321 and 1.2804 Å, respectively, provide evidence for electron delocalization. The compounds show several intermolecular contacts (Table S3 and Fig. S2–S6, ESI†) resulting in 1D H-bonded aggregates in 1 and 2 (base vectors [100] and [110], respectively), 2D in 4 and 5 (planes (010) and (101), respectively) and 3D in 3 (base vectors [001][010][100]). Only in 1 an intramolecular N–H···Ocarboxylate interaction is recognized (graph set S11(6)50). Since the hydrazone hydrogen was removed and replaced with the metal centre, intramolecular RAHB systems were not observed.
N and CNH bond lengths of 1.3074, 1.321 and 1.2804 Å, respectively, provide evidence for electron delocalization. The compounds show several intermolecular contacts (Table S3 and Fig. S2–S6, ESI†) resulting in 1D H-bonded aggregates in 1 and 2 (base vectors [100] and [110], respectively), 2D in 4 and 5 (planes (010) and (101), respectively) and 3D in 3 (base vectors [001][010][100]). Only in 1 an intramolecular N–H···Ocarboxylate interaction is recognized (graph set S11(6)50). Since the hydrazone hydrogen was removed and replaced with the metal centre, intramolecular RAHB systems were not observed.
2.2. Catalytic activity of 1–5 in the azide–alkyne cycloaddition reaction
The catalytic activities of compounds 1–5 were investigated for the three-component 1,3-dipolar cycloaddition of phenyl acetylene, benzyl bromide and sodium azide under microwave irradiation (Table 1). The initial screening process was performed using 1 mol% of the catalyst in a mixture of H2O/MeCN (v/v, 1/1), which is recognized as one of the most efficient reaction media for CuAAC using hydrosoluble catalysts.19–21,23,27 In the presence of catalysts 1–4, 1-benzyl-4-phenyl-1H-1,2,3-triazole was obtained as unique product after 15 minutes at 125 °C, with complex 1 being the most active catalyst affording 61% of yield. Complex 5 was inactive due to its low solubility in the reaction mixture (Table 1, entries 1–5). Replacing MeCN with glycerol and performing the reactions under the same conditions improved the product yields, with 1 still being recognized as the most active catalyst with the obtained yield of 70% (Table 1, entries 6–9). The improvement of the reaction rate when using glycerol enhances the assumption of its participation in the mechanistic pathway of the CuAAC transformation.27|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Cat. | Cat. loadingb (mol%) | Solvent | Temp. (°C) | Time (min) | Yieldc (%) |
a
Reaction conditions: phenyl acetylene (0.3 mmol, 1.1 equiv.), benzyl bromide (0.3 mmol, 1 equiv.), NaN3 (0.33 mmol, 1.1 equiv.), 0.5 mL of solvent (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volumetric ratio for mixtures), MW irradiation (15 W, 125 °C).
b Calculated based on benzyl bromide.
c Isolated yield.
d Reaction performed in 1 mL of solvent. :1 volumetric ratio for mixtures), MW irradiation (15 W, 125 °C).
b Calculated based on benzyl bromide.
c Isolated yield.
d Reaction performed in 1 mL of solvent.
|
||||||
| 1 | 1 | 1 | Water + MeCN | 125 | 15 | 61.0 |
| 2 | 2 | 1 | Water + MeCN | 125 | 15 | 18.1 |
| 3 | 3 | 1 | Water + MeCN | 125 | 15 | 20.5 |
| 4 | 4 | 1 | Water + MeCN | 125 | 15 | 13.4 |
| 5 | 5 | 1 | Water + MeCN | 125 | 15 | <5% |
| 6 | 1 | 1 | Water + glycerol | 125 | 15 | 69.5 |
| 7 | 2 | 1 | Water + glycerol | 125 | 15 | 22.7 |
| 8 | 3 | 1 | Water + glycerol | 125 | 15 | 27.4 |
| 9 | 4 | 1 | Water + Glycerol | 125 | 15 | 16.1 |
| 10 | 1 | 1 | Water | 125 | 15 | 45.9 |
| 11 | 2 | 1 | Water | 125 | 15 | 15.0 |
| 12 | 3 | 1 | Water | 125 | 15 | 17.6 |
| 13 | 4 | 1 | Water | 125 | 15 | 11.8 |
| 14 | 1 | 1 | Glycerol | 125 | 15 | 35.7 |
| 15 | 2 | 1 | Glycerol | 125 | 15 | 33.3 |
| 16 | 3 | 1 | Glycerol | 125 | 15 | 24.3 |
| 17 | 4 | 1 | Glycerol | 125 | 15 | 15.7 |
| 18 | 1 | 1 | Water + glycerol | 125 | 30 | 87.5 |
| 19 | 1 | 1 | Water + glycerol | 125 | 60 | 74.5 |
| 20 | 1 | 1 | Water + glycerol | 100 | 15 | 45.2 |
| 21 | 1 | 1 | Water + glycerol | 150 | 15 | 56.0 |
| 22 | 1 | 1.5 | Water + glycerol | 125 | 30 | 90.0 |
| 23d | 1 | 1.5 | Water + glycerol | 125 | 30 | 98.9 |
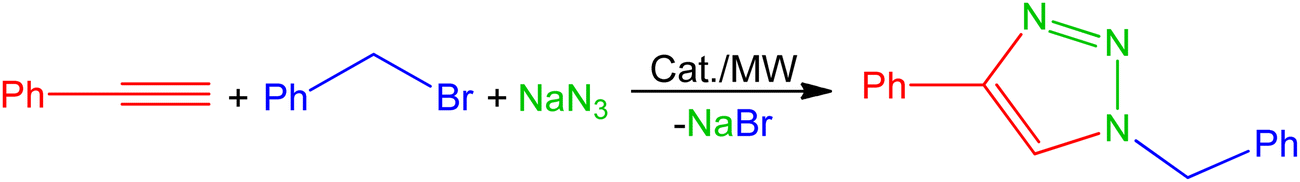
Lower conversions were observed when water (Table 1, entries 10–13) or glycerol (Table 1, entries 14–17) was used as the sole solvent instead of the 1:1 solvent mixture (Table 1, entries 6–9) as a reaction medium. The presence of both water and a miscible organic solvent (glycerol) is vital to dissolve the hydrophilic (the catalyst and NaN3) as well as the hydrophobic (the alkyne and organohalide) reaction components, and to increase the homogeneity of the reaction medium.
As compound 1 revealed the highest activity among the whole set of complexes, it was thus used for the optimization of the reaction conditions. In the presence of 1 mol% of catalyst 1, extending the reaction time from 15 to 30 minutes raised the yield from 70% to 88%, respectively, while a further extension to 90 minutes decreased the product yield to 75% (Table 1, entries 6, 18 and 19).
Decreasing the reaction temperature from 125 to 100 °C led to a drop in yield from 70 to 45% (Table 1, entries 6 and 20); upon raising the temperature to 150 °C, a lower product yield of 56% was observed, which can be attributed to a partial decomposition of the catalyst or the product (Table 1, entries 6 and 21).
Increasing the amount of catalyst 1 to 1.5 mol% afforded the product in 90% yield, at 125 °C after 30 minutes (Table 1, entry 22). Repeating the reaction using a higher amount of solvent mixture, for a better dissolution of the extra catalyst loading, improved the reaction rate and a quantitative conversion was obtained (Table 1, entry 23).
The scope of the reaction was explored with various terminal alkynes and substituted benzyl bromides in the presence of sodium azide, under microwave irradiation with 1 as catalyst (Scheme 3). The corresponding 1,4-disubstituted-1,2,3-triazoles were successfully obtained in excellent yields in the range from 85% to quantitative conversion after 30 minutes; the 1H-NMR spectra of the products show no detectable formation of undesired by-products (Fig. S7–S26, ESI†).
 | ||
| Scheme 3 1,4-disubstituted-1,2,3-triazoles synthesised via a three-component CuAAC reaction catalysed with 1 under microwave irradiation (125 °C, 30 min) in a mixture of water and glycerol (1:1). | ||
Performing the CuAAC reactions in glycerol is limited to a few reports using CuI, Cu(I)-based nanoparticles or Cu(I)–phosphine complexes.26,27 This study constitutes the first example of hydrosoluble well-defined copper(II) complexes being used as catalysts for the CuAAC reaction in glycerol.
While most of the catalytic protocols for the azide–alkyne cycloaddition to obtain 1,4-disubstituted 1,2,3-triazoles use volatile organic solvents (e.g. acetonitrile, tetrahydrofuran and toluene),51 long reaction times,28,52 additional reducing agents,1 costly catalysts based on noble costly metals,28 and may occur in a multistep manner,51,53 our method is straightforward, proceeds in the absence of any toxic organic solvent and uses inexpensive reagents and starting materials. In comparison with the previously reported hydrosoluble copper catalyst based on amidophosphines,23,27,31,32 tris(pyrazolyl)methanes20,36 and arylhydrazones of β-diketones,21 our catalytic system is efficient in aqueous medium in the presence of glycerol as a green organic co-solvent instead of the toxic acetonitrile co-solvent used for the aforementioned catalysts. Moreover, catalyst 1 exhibits a high solubility in water, which simplified its separation from the hydrophobic organic product. This fact represents an important advantage for the large-scale industrial application because an easy way for catalyst separation from the organic product stream can lower the economical and environmental costs, thus leading to a more sustainable chemical process.
Taking into consideration several already reported studies,1,21,54–56 a plausible reaction mechanism is devised in Scheme 4. Cu(I) catalytic species is generated through the alkyne–alkyne homocoupling (also known as Glaser reaction) promoted by the Cu(II) pre-catalyst.21,57 The reaction proceeds in a stepwise manner, beginning with the π-coordination of the alkyne substrate to the Cu(I) metal centre with the increase of the acidity of the alkyne proton. The formation of hydrogen bonding between one hydroxyl group of glycerol and the terminal nitrogen of the azide group would enhance the electrophilicity of the distal nitrogen and thus promote the acetylide–azide coupling.27,58,59 The copper(I) triazolide intermediate was then protonated to afford the favoured regioisomer triazole and regenerates the active copper catalyst.
 | ||
| Scheme 4 Proposed mechanism of the Cu–AAC reaction in glycerol. | ||
3. Conclusions
In this study, five novel copper complexes based on AAMN ligands of various nuclearities were obtained through a simple one-pot template syntheses, which involves the activation of one nitrile group promoted by RAHB and the presence of copper(II). The nuclearity and structures of the prepared complexes are dependent on the auxiliary basic ligands and the carboxylate group at the aromatic moiety of the arylhydrazone ligands.The catalytic activity of the newly synthesized and fully characterized copper(II) complexes was investigated for the microwave assisted CuAAC in glycerol, following the “Click” rules. Among the tested complexes, 1 was found to be the most active pre-catalyst for the regioselective preparation of 1,4-disubstituted-1,2,3-triazoles. Excellent yields were obtained ranging from 85% up to quantitative conversion at 125 °C after 30 min, using a mixture of water and glycerol as a reaction medium.
This protocol constitutes the first example of using copper–AAMN complexes as homogeneous catalysts for the regioselective preparation of 1,4-disubstituted-1,2,3-triazoles in aqueous medium. The conversions obtained are comparable to other related systems based on hydrosoluble catalysts, however, our catalytic system is efficient when glycerol is utilized as an organic green co-solvent instead of acetonitrile. This research direction is worth exploring towards a more sustainable chemical process.
4. Experimental
4.1. Materials and general procedures
All the chemicals were obtained from commercial sources and used as received. The 2-(2-(dicyanomethylene)hydrazineyl)benzoic acid (H2L1) was synthesized according to the reported synthetic procedure.42 The IR spectra (4000–400 cm−1) were recorded on a Bruker Alpha-P ATR-IR spectrometer. The 1H and 13C NMR spectra were obtained at room temperature on a Bruker 400 MHz spectrometer using tetramethylsilane [Si(CH3)4] as the internal reference. Elemental analyses (C, H, and N) were performed on a Finnigan EA 1112 instrument. Electrospray mass spectra (ESI-MS) were run with an ion-trap instrument (Varian 500-MS LC Ion Trap Mass Spectrometer) equipped with an electrospray ion source. For electrospray ionization, the drying gas and flow rate were optimized according to the sample with 35 p.s.i. nebulizer pressure. Scanning was performed from m/z 50 to 1500 in methanol solution. The compounds were observed in the positive or negative ion mode (capillary voltage = 80–105 V).4.2. Preparation of H3L2
N), 1670 ν(CO) and 1621 ν(CN) cm−1. 1H NMR (400.13 MHz) in DMSO-d6, internal TMS, δ (ppm): 7.67–8.40 (3H, Ar–H), 13.15 and 13.52 (s, 1H, O–H), 15.04 (s, 1H, N–H). 13C{1H} NMR (400 MHz, DMSO-d6) δ: 89.6 (CN), 109.1 and 113.4 (CN), 116.0 (Ar–H), 118.8 (Ar–COOH), 125.1 and 132.0 (Ar–H), 136.3 (Ar–COOH), 142.6 (Ar–NHN), 166.1 and 169.0 (Ar–COOH).
4.3. Synthesis of the complexes
N), 1617 ν(CO) and 1591 ν(CN) cm−1.
N), 1643 ν(CO) and 1590 ν(CN) cm−1.
N), 1677 ν(CO) and 1587 ν(CN) cm−1.
N), 1632 ν(CO) and 1580 ν(CN) cm−1.
5: Yield, 54% (based on Cu). Calcd for C44H34Cu3N12O10 (Mr = 1081.47): C 48.87; H 3.17; N 15.54. Found: C, 48.83; H, 3.14; N, 15.49. ESI-MS: m/z: 1082.5 [Mr + H]+. IR (ATR, 298 K): 3313 ν(NH), 2218 ν(CN), 1685 ν(CO) and 1613 ν(CN) cm−1.
4.4. X-ray structure determination
X-ray diffraction intensities of 1–5 were collected using a Bruker SMART APEX-II CCD area detector equipped with graphite-monochromated Mo Kα radiation (λ = 0.71073 Å) at 150 (1, 3 and 4) or 296 K (2 and 5). Absorption corrections were applied by SADABS.60,61 The structure was solved by direct methods and refined on F2 by full-matrix least-squares using Bruker's SHELXTL-97.62 All non-hydrogen atoms were refined anisotropically. The details of the crystallographic data for 1–5 are summarized in Table S1 (ESI†). Crystallographic data for the structural analysis have been deposited to the Cambridge Crystallographic Data Centre [CCDC 2234588 (for 1), 2234589 (for 2), 2234591 (for 3), 2234592 (for 4) and 2234593 (for 5)].†4.5. General procedure for the azide–alkyne cycloaddition
A mixture of substituted benzyl bromide (0.3 mmol), acetylene derivative (0.3 mmol) and sodium azide (0.33 mmol) in 1 mL of solvent (water and glycerol 1:1) was charged into a 4 mL glass vial sealed with an aluminium/Teflon crimp top and equipped with a small magnetic stirring bar. The reaction mixture was then subjected to the microwave irradiation (20 W) under continuous stirring to reach 125 °C for 30 minutes. At the end of the reaction, 10 mL of water were added to produce a precipitate, which was then collected by filtration, washed with petroleum ether and dried in air to furnish the corresponding product triazole. The 1H NMR spectra of all products proved their high purity (ESI,† Section 2 and Fig. S7–S26).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been supported by the Fundação para a Ciência e a Tecnologia (FCT) (Portugal), projects UIDB/00100/2020, UIDP/00100/2020 and LA/P/0056/2020 of Centro de Química Estrutural. AGM and VAA are grateful to Associação do Instituto Superior Técnico para Investigação e Desenvolvimento for their research and post-doctoral fellowships through grants no: BL133/2021-IST-ID and BL110/2022-IST-ID, respectively. A. V. G. thanks to FCT and Instituto Superior Técnico (DL 57/2016, L 57/2017 and CEEC Institutional 2018 Programs, Contract no: IST-ID/110/2018) as well as to the Baku State University (Azerbaijan). Authors thank the Portuguese NMR Network (IST-UL Centre) and the IST Node of the Portuguese Network of mass-spectrometry. This publication is also supported by the RUDN University Strategic Academic Leadership Program (recipient AJLP, preparation).References
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 114, 2708–2711 CrossRef.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- T. Cañeque, S. Müller and R. Rodriguez, Nat. Rev. Chem., 2018, 2, 202–215 CrossRef.
- Y. Zou, L. Zhang, L. Yang, F. Zhu, M. Ding, F. Lin, Z. Wang and Y. Li, J. Controlled Release, 2018, 273, 160–179 CrossRef CAS PubMed.
- P. Z. Li, X. J. Wang and Y. Zhao, Coord. Chem. Rev., 2019, 380, 484–518 CrossRef CAS.
- Y. Dai, X. Chen and X. Zhang, Polym. Chem., 2018, 10, 34–44 RSC.
- M. Arslan, G. Acik and M. A. Tasdelen, Polym. Chem., 2019, 10, 3806–3821 RSC.
- A. K. Agrahari, P. Bose, M. K. Jaiswal, S. Rajkhowa, A. S. Singh, S. Hotha, N. Mishra and V. K. Tiwari, Chem. Rev., 2021, 121, 7638–7956 CrossRef CAS PubMed.
- P. Wu, ACS Chem. Biol., 2022, 2022, 2959–2961 CrossRef PubMed.
- F. Scalambra, P. Lorenzo-Luis, I. de los Rios and A. Romerosa, Coord. Chem. Rev., 2021, 443, 213997 CrossRef CAS.
- I. Borthakur, S. Kumari and S. Kundu, Dalton Trans., 2022, 51, 11987–12020 RSC.
- H. Gröger, F. Gallou and B. H. Lipshutz, Chem. Rev., 2022 DOI:10.1021/ACS.CHEMREV.2C00416.
- F. Scalambra, M. Serrano-Ruiz and A. Romerosa, Dalton Trans., 2017, 46, 5864–5871 RSC.
- G. Hankó, R. Márton, A. Udvardy, M. Purgel, Á. Kathó, F. Joó and G. Papp, Inorg. Chim. Acta, 2021, 522, 120359 CrossRef.
- Á. Kathó, H. H. Horváth, G. Papp and F. Joó, Catalysts, 2022, 12, 518 CrossRef.
- I. L. Librando, A. Paul, A. G. Mahmoud, A. V. Gurbanov, S. A. C. Carabineiro, F. C. Guedes da Silva, C. F. G. C. Geraldes and A. J. L. Pombeiro, RSC Sustain., 2023, 1, 147–158 RSC.
- A. G. Mahmoud, L. M. D. R. S. Martins, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Catalysts, 2019, 9, 611 CrossRef CAS.
- A. G. Mahmoud, M. F. C. Guedes da Silva, J. Sokolnicki, P. Smoleński and A. J. L. Pombeiro, Dalton Trans., 2018, 47, 7290–7299 RSC.
- A. G. Mahmoud, L. M. D. R. S. Martins, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Inorg. Chim. Acta, 2018, 483, 371–378 CrossRef CAS.
- A. G. Mahmoud, M. F. C. Guedes da Silva, K. T. Mahmudov and A. J. L. Pombeiro, Dalton Trans., 2019, 48, 1774–1785 RSC.
- A. G. Mahmoud, P. Smolénski, M. F. C. Guedes Da Silva and A. J. L. Pombeiro, Molecules, 2020, 25, 5479 CrossRef CAS PubMed.
- I. L. Librando, A. G. Mahmoud, S. A. C. Carabineiro, M. F. C. Guedes da Silva, C. F. G. C. Geraldes and A. J. L. Pombeiro, Nanomaterials, 2021, 11, 2702 CrossRef CAS PubMed.
- A. E. Díaz-Álvarez, J. Francos, B. Lastra-Barreira, P. Crochet and V. Cadierno, Chem. Commun., 2011, 47, 6208–6227 RSC.
- A. G. Mahmoud, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Catal. Today, 2023, 114056 CrossRef CAS.
- N. Nebra and J. García-Álvarez, Molecules, 2020, 25, 2015 CrossRef CAS PubMed.
- A. G. Mahmoud, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Dalton Trans., 2021, 50, 6109–6125 RSC.
- C. Wang, D. Ikhlef, S. Kahlal, J.-Y. Saillard and D. Astruc, Coord. Chem. Rev., 2016, 316, 1–20 CrossRef CAS.
- I. L. Librando, A. G. Mahmoud, S. A. C. Carabineiro, M. F. C. G. da Silva, F. J. Maldonado-Hódar, C. F. G. C. Geraldes and A. J. L. Pombeiro, Catalysts, 2022, 12, 45 CrossRef CAS.
- G. Vilé, G. Di Liberto, S. Tosoni, A. Sivo, V. Ruta, M. Nachtegaal, A. H. Clark, S. Agnoli, Y. Zou, A. Savateev, M. Antonietti and G. Pacchioni, ACS Catal., 2022, 12, 2947–2958 CrossRef.
- A. G. Mahmoud, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Coord. Chem. Rev., 2021, 429, 213614 CrossRef CAS.
- I. L. Librando, A. G. Mahmoud, S. A. C. Carabineiro, M. F. C. Guedes da Silva, C. F. G. C. Geraldes and A. J. L. Pombeiro, Catalysts, 2021, 11, 185 CrossRef CAS.
- Z. Gonda and Z. Novák, Dalton Trans., 2010, 39, 726–729 RSC.
- S. Lal and S. Díez-González, J. Org. Chem., 2011, 76, 2367–2373 CrossRef CAS PubMed.
- N. Candelon, D. Lastécouères, A. K. Diallo and J. Ruiz Aranzaes, Chem. Commun., 2008, 741–743 RSC.
- I. Cano, E. Álvarez, M. C. Nicasio and P. J. Pérez, J. Am. Chem. Soc., 2011, 133, 191–193 CrossRef CAS PubMed.
- S. C. Sau, S. R. Roy, T. K. Sen, D. Mullangi and S. K. Mandal, Adv. Synth. Catal., 2013, 355, 2982–2991 CrossRef CAS.
- Y. D. Bidal, M. Lesieur, M. Melaimi, F. Nahra, D. B. Cordes, K. S. Athukorala Arachchige, A. M. Z. Slawin, G. Bertrand and C. S. J. Cazin, Adv. Synth. Catal., 2015, 357, 3155–3161 CrossRef CAS.
- A. Makarem, R. Berg, F. Rominger and B. F. Straub, Angew. Chem., Int. Ed., 2015, 54, 7431–7435 CrossRef CAS PubMed.
- K. T. Mahmudov, M. N. Kopylovich and A. J. L. Pombeiro, Coord. Chem. Rev., 2013, 257, 1244–1281 CrossRef CAS.
- M. N. Kopylovich, K. T. Mahmudov, A. Mizar and A. J. L. Pombeiro, Chem. Commun., 2011, 47, 7248 RSC.
- M. N. Kopylovich, A. Mizar, M. F. C. Guedes da Silva, T. C. O. Mac Leod, K. T. Mahmudov and A. J. L. Pombeiro, Chem. – Eur. J., 2013, 19, 588–600 CrossRef CAS PubMed.
- K. T. Mahmudov, M. N. Kopylovich, A. Sabbatini, M. G. B. Drew, L. M. D. R. S. Martins, C. Pettinari and A. J. L. Pombeiro, Inorg. Chem., 2014, 53, 9946–9958 CrossRef CAS PubMed.
- A. G. Mahmoud, K. T. Mahmudov, M. F. C. Guedes da Silva and A. J. L. Pombeiro, RSC Adv., 2016, 6, 54263–54269 RSC.
- G. A. O. Tiago, K. T. Mahmudov, M. F. C. Guedes da Silva, A. P. C. Ribeiro, F. E. Huseynov, L. C. Branco and A. J. L. Pombeiro, Polyhedron, 2017, 133, 33–39 CrossRef CAS.
- A. V. Gurbanov, F. E. Huseynov, G. Mahmoudi, A. M. Maharramov, F. C. Guedes da Silva, K. T. Mahmudov and A. J. L. Pombeiro, Inorg. Chim. Acta, 2018, 469, 197–201 CrossRef CAS.
- A. V. Gurbanov, L. M. D. R. S. Martins, M. N. Kopylovich, M. Sutradhar, F. I. Zubkov, K. T. Mahmudov and A. J. L. Pombeiro, ChemistrySelect, 2020, 5, 7923–7927 CrossRef CAS.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC.
- M. C. Etter, Acc. Chem. Res., 1990, 23, 120–126 CrossRef CAS.
- E. Haldón, M. C. Nicasio and P. J. Pérez, Org. Biomol. Chem., 2015, 13, 9528–9550 RSC.
- V. Aragão-Leoneti, V. L. Campo, A. S. Gomes, R. A. Field and I. Carvalho, Tetrahedron, 2010, 66, 9475–9492 CrossRef.
- U. Pradere, V. Roy, T. R. McBrayer, R. F. Schinazi and L. A. Agrofoglio, Tetrahedron, 2008, 64, 9044–9051 CrossRef CAS PubMed.
- F. Himo, T. Lovell, R. Hilgraf, V. V. Rostovtsev, L. Noodleman, K. B. Sharpless and V. V. Fokin, J. Am. Chem. Soc., 2005, 127, 210–216 CrossRef CAS PubMed.
- V. D. Bock, H. Hiemstra and J. H. van Maarseveen, Eur. J. Org. Chem., 2006, 51–68 CrossRef CAS.
- B. T. Worrell, J. A. Malik and V. V. Fokin, Science, 2013, 340, 457–460 CrossRef CAS PubMed.
- P. Siemsen, R. C. Livingston and F. Diederich, Angew. Chem., Int. Ed., 2000, 39, 2632–2657 CrossRef CAS PubMed.
- M. Rodríguez-Rodríguez, E. Gras, M. A. Pericàs and M. Gómez, Chem. – Eur. J., 2015, 21, 18706–18710 CrossRef PubMed.
- F. Nemati, M. M. Hosseini and H. Kiani, J. Saudi Chem. Soc., 2016, 20, S503–S508 CrossRef CAS.
- SMART & SAINT Software Reference Manuals, Version 6.22, Bruker AXS Analytic X-ray Systems, Inc., Madison, WI, 2000 Search PubMed.
- G. M. Sheldrick, SADABS. Program for Empirical Absorption Correction, University of Gottingen, Germany. ( 2000) Search PubMed.
- G. M. Sheldrick, SHELXTL V5.1, Software Reference Manual, Bruker AXS Inc., Madison, WI, 1997 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2234588 (for 1), 2234589 (for 2), 2234591 (for 3), 2234592 (for 4) and 2234593 (for 5). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3nj00512g |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2023 |