
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAssessing the role of substituents in ferrocene acylphosphines and their impact on gold-catalysed reactions†
Petr
Vosáhlo
 and
Petr
Štěpnička
*
and
Petr
Štěpnička
*
Department of Inorganic Chemistry, Faculty of Science, Charles University, Hlavova, 2030, 128 40 Prague, Czech Republic. E-mail: petr.stepnicka@natur.cuni.cz
First published on 3rd February 2023
Abstract
Substituents can be used to efficiently modify the coordination and catalytic behaviour of phosphine ligands. In this article, we analyse how substituents affect the properties of ferrocene acylphosphines FcC(O)PR2 (1a–d), where PR2 is PPh2 (1a), PCy2 (1b), PAd2 (1c) and PCg (1d; Fc = ferrocenyl, Cy = cyclohexyl, Ad = 1-adamantyl, and PCg = 1,3,5,7-tetramethyl-2,4,6-trioxa-8-phosphaadamantane-8-yl). The 31P–77Se scalar coupling constants (1JPSe), determined for the corresponding phosphine selenides FcC(O)P(Se)R2 (2a–d), suggest that the basicity of the phosphine groups increases with the donor ability of the substituents R, as expected. Au(I) complexes [AuCl(1-κP)] (3a–d), obtained by replacing the dimethylsulfide ligand in [AuCl(SMe2)] with acylphosphines 1a–d, were tested in Au-catalysed alkyne hydration and intramolecular cyclisation of N-propargyl benzamide to yield 5-methylene-2-phenyl-4,5-dihydrooxazole. The collected results indicate that the highest reaction yields were generally obtained using catalysts derived from acylphosphines bearing the electron-donating aliphatic substituents 1b and 1c. From a wider perspective, the carbonyl moiety in the acylphosphines FcC(O)PR2 appears to lower steric crowding around the phosphorus atom (especially for compounds with bulky R substituents) and counterbalances the electron-donating effect of the ferrocenyl moiety.
Introduction
Tertiary acylphosphines, RC(O)PR′2, have emerged as alternatives to conventional triorganophosphine ligands.1,2 However, such compounds remain rare among the widely studied ferrocene phosphines.3 Thus, the triafulvene derivative A (Scheme 1), a rather obscure ferrocene acylphosphine, was reported in 1991.4 The more conventional compounds, B and C, accessible from alkylidenephosphines FcP![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CR(OSiMe3), followed in 1997.5 More recently,6 we reported the synthesis of ferrocene acylphosphine FcC(O)PPh2 (1a; Scheme 1) and its facile orthopalladation, which differentiates this compound from its conventional counterpart, (diphenylphosphino)ferrocene (FcPPh2). Subsequently, we also prepared a series of monoacyl diphosphines, R2PfcC(O)PR′2 (A; R/R′ = Ph and cyclohexyl (Cy); all possible combinations),7 which are analogues of the widely studied 1,1′-(diphenylphosphino)ferrocene (dppf),8 and compared the coordination and catalytic properties of these compounds.
CR(OSiMe3), followed in 1997.5 More recently,6 we reported the synthesis of ferrocene acylphosphine FcC(O)PPh2 (1a; Scheme 1) and its facile orthopalladation, which differentiates this compound from its conventional counterpart, (diphenylphosphino)ferrocene (FcPPh2). Subsequently, we also prepared a series of monoacyl diphosphines, R2PfcC(O)PR′2 (A; R/R′ = Ph and cyclohexyl (Cy); all possible combinations),7 which are analogues of the widely studied 1,1′-(diphenylphosphino)ferrocene (dppf),8 and compared the coordination and catalytic properties of these compounds.
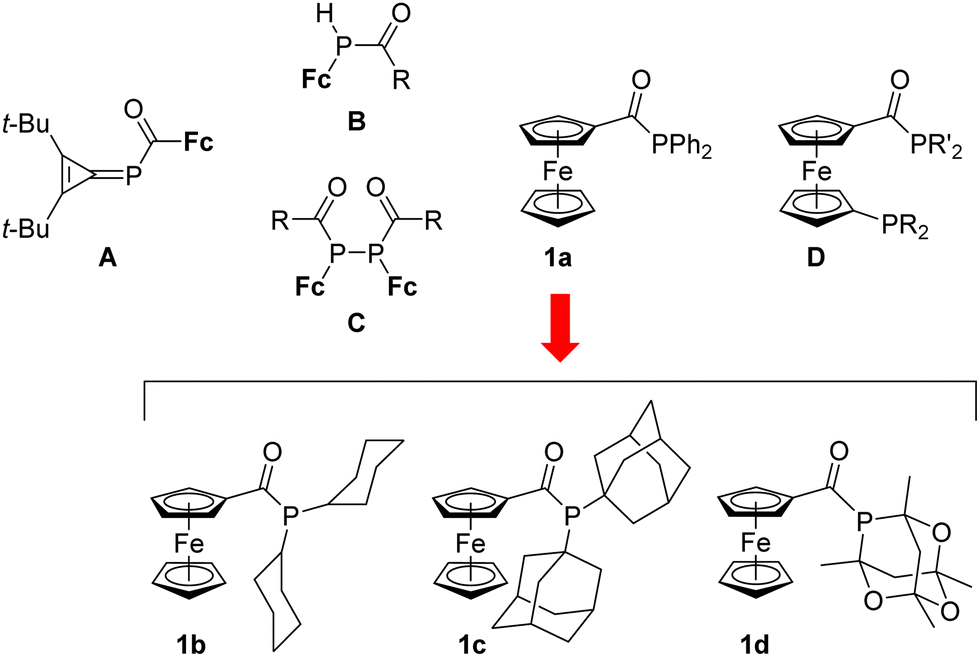 | ||
| Scheme 1 Ferrocene acylphosphines 1a, A–D (in B and C: R = Fc, Ph, and t-Bu; Fc = ferrocenyl) and newly prepared compounds 1b–d. | ||
In continuation of these studies, we are currently investigating the properties and catalytic behaviour of ligands related to compound 1a. In particular, we report the syntheses of three additional compounds of this type bearing different substituents at the phosphorus atom, viz. FcC(O)PCy2 (1b; Cy = cyclohexyl), FcC(O)PAd2 (1c, Ad = 1-adamantyl) and the “cage” phosphine,9,10 FcC(O)PCg (1c, PCg = 1,3,5,7-tetramethyl-2,4,6-trioxa-8-phosphaadamantane-8-yl). These compounds were used to prepare corresponding phosphine selenides and Au(I) complexes [AuCl(L-κP)] (L = 1a–d). The gold complexes were further studied as defined precatalysts in two model gold-catalysed reactions, namely, the hydration of terminal alkynes and cyclisation of N-propargylbenzamide to 2-phenyl-5-methyleneoxazoline, and the catalytic results were analysed in terms of the ligand properties.
Results and discussion
Syntheses and characterisation
The acylphosphines 1b–d (Scheme 2) were obtained similarly to 1a6 using the following one-pot procedure: ferrocenecarboxylic acid was initially reacted with oxalyl chloride to provide ferrocenecarbonyl chloride, and the formed acyl chloride was directly reacted with the respective secondary phosphine in the presence of triethylamine as an HCl scavenger. The reaction with dicyclohexylphosphine was clean and rapid and afforded 1b in a 93% yield after simple chromatographic purification. An analogous reaction employing di(1-adamantyl)phosphine required a longer reaction time and a modified workup procedure because both the secondary phosphine and 1c were only sparingly soluble in diethyl ether used as a solvent during the second reaction step, and the product separated out of the reaction mixture along with (Et3NH)Cl. Therefore, the reaction mixture was evaporated, and the residue was partitioned between dichloromethane and water. Subsequent chromatography afforded pure 1c in an average 54% yield. Longer reaction times were also required to prepare compound 1d bearing 1,3,5,7-tetramethyl-2,4,6-trioxa-8-phosphaadamantane-8-yl group. Purification by chromatography and crystallisation from pentane afforded 1d in a 17% yield. Such a low isolated yield reflects low solubility and reactivity of the electron-poor phosphine HPCg and high solubility of the acylphosphine. | ||
| Scheme 2 Preparation of 1b–d (b: PR2 = PCy2, c: PR2 = PAd2, d: PR2 = PCg; Cy = cyclohexyl, Ad = 1-adamantyl, and PCg = 1,3,5,7-tetramethyl-2,4,6-trioxa-8-phosphaadamantane-8-yl; see Scheme 1). | ||
Compounds 1b–d were obtained as burgundy red solids, which could be stored in air for months without any observable decomposition and were characterised by NMR and IR spectroscopy, ESI MS and elemental analysis. In addition, the structure of 1c was determined by single-crystal X-ray diffraction analysis.
The 31P chemical shifts of the acylphosphines are given in Table 1. The 1H and 13C NMR spectra of the compounds exhibited the characteristic signals of phosphine substituents and the ferrocene unit, namely a C5H5 singlet and two multiplets due to C5H4 in the 1H NMR spectra and resonances from three CHs and one Cipso in the 13C NMR spectra. The signals of the carbonyl group were observed as phosphorus-coupled doublets at δC 210–220. A downfield shift of the CO resonances with respect to ferrocenecarboxylic acid (δC 177.6)11 and analogous amides (cf. δC 160.5 for FcC(O)NCy2 and 171.2 for FcC(O)NPh2)12 suggests a lower carbonyl-heteroatom conjugation in 1b–d. Carbonyl stretching bands in the IR spectra of 1b–d appeared in the 1606–1616 cm−1 range (cf. νCO 1609 cm−1 for 1a). These values are difficult to compare with the data for the corresponding amides because IR spectra may change with the condensation state and experimental setup (neat or dispersed samples).
| Compound | δ P [1JPSe] | ||
|---|---|---|---|
| L (1a–d) | LSe (2a–d) |
[AuCl(L-κP)] (3a–d) | |
| a The spectra were recorded in CDCl3 at 25 °C. The chemical shifts are reported in ppm with respect to 85% H3PO4, and the coupling constants are given in Hz. b Data from ref. 6. | |||
| FcC(O)PPh2 (a) | 11.0b | 28.2 [749]b | 29.3 |
| FcC(O)PCy2 (b) | 17.8 | 52.3 [718] | 45.0 |
| FcC(O)PAd2 (c) | 45.3 | 68.2 [709] | 64.1 |
| FcC(O)PCg (d) | −14.5 | 24.2 [761] | 16.0 |
The molecular structure of 1c and selected geometric data are presented in Fig. 1. The structure comprises unperturbed ferrocene unit. The CO bond is twisted by 9.5(5)° from the plane of its parent cyclopentadienyl ring C(1–5), which is further manifested by the torsion angles C2–C1–C11–O and C5–C1–C11–O of −168.3(5) and 6.0(8)°, respectively. The CO bond length in 1c (1.228(7) Å) is practically identical to that in 1a (1.225(2) Å).6 However, 1c has longer P–C(Ad) bonds than the mentioned reference compound, which is in line with the lack of conjugation and higher steric demands of the adamantyl substituents. Correspondingly, the C12–P–C22 angle is the largest of the C–P–C angles.
 | ||
| Fig. 1 Structure diagram for 1c. Selected distances and angles (in Å and deg): Fe–C(1–10) 2.029(7)–2.053(5), dihedral angle of the cyclopentadienyl planes 2.5(3), C11O 1.228(7), P–C11 1.882(6), P–C12 1.895(5), P–C22 1.884(6), C1–C11–P 115.3(4), C11–P–C12 97.4(2), C11–P–C22 102.3(2), and C12–P–C22 112.4(2). | ||
To compare the “electronic” properties (basicity) of the prepared ligands,13 the acylphosphines 1b–d were treated with grey selenium in refluxing chloroform to give the respective phosphine selenides 2b–d (Scheme 3). The selenation reactions proceed cleanly and with full conversions. Thus, the isolated yields mainly reflected the efficacy of the crystallisation procedure used to isolate the compounds (yields: 66% for 2b, 85% for 2c, and 53% for 2d).
 | ||
| Scheme 3 Synthesis of the phosphine selenides 2 and Au(I) complexes 3. | ||
The selenylation of the phosphines resulted in a low-field shift of the 31P NMR signals and the appearance of characteristic 77Se satellites (I = 1/2, natural abundance: 7.6%). The 13C NMR resonances due to the carbonyl groups were also downfield-shifted (ΔδC ≈ 12 ppm), reflecting the high electron-withdrawing abilities of the P(Se)R2 substituents.
The 1JPSe scalar coupling constants of phosphine selenides are directly proportional to the pKB of the parent phosphines,14 such that larger 1JPSe values indicate a lower Brønsted basicity. However, steric factors must also be considered,14,15 especially for bulky phosphines, because the s-character of the P–Se bond that determines the 1JPSe coupling constant changes with the C–P–C angles. In the present case, however, steric factors appear to play a minor role due to the presence of the CO linker.
The 1JPSe values of 2a–d, which are compiled in Table 1 and compared in Fig. 2, suggest that compounds 1a–d cover a wide basicity range, whereas the trend in the 1JPSe values of 2c < 2b < 2a < 2d reflects the electron-donating ability of the phosphine substituents.16 Furthermore, the increase in 1JPSe from the “simple” phosphine FcPR2 (R = Ph, 1JPSe = 735 Hz; R = Cy, 1JPSe = 700 Hz)17 to the corresponding acylphosphine FcC(O)PR2 (Δ1JPSe ≈ 17 Hz) indicates that the carbonyl group inserted between the phosphine moiety and the electron-donating ferrocenyl substituent significantly lowers the basicity (σ-donor ability) of the phosphorus atom.
 | ||
| Fig. 2 Graphical comparison of the 1JPSe values for phosphine selenides derived from 1a–d and related compounds.18 | ||
Fig. 2 presents a wider comparison, showing that the introduced carbonyl moiety neutralises (at least partially) the electron-donating effect of the ferrocenyl group (cf. the 1JPSe values for FcPPh2 and FcPCy2). As such, FcC(O) and additional substituents allow modification of the ligand basicity in both directions from that of the archetypal phosphine PPh3.
The solid-state structures of the selenides 2b–d are shown in Fig. 3, and selected geometric data are presented in Table 2. All structures contain regular ferrocene units with negligibly tilted cyclopentadienyl rings (≤2°). The C1–C11–P angles are close to 120°, as expected for an sp2 carbon (120–123° in the series), and the acylphosphine fragment {C11,P,O1} is tilted with respect to the bonding cyclopentadienyl ring C(1–5) by 3.5(1)° in 2b, 1.4(2)° in 2c, and 9.3(1)° in 2d. The PSe distances in 2b–d are mutually similar and compare well with the value for 2a (2.1035(7) Å).6 Finally, a comparison of the parameters determined for the pairs 1a–2a and 1c–2c reveals a slight opening of the C–P–C angles upon selenylation.
 | ||
| Fig. 3 Molecular structures of 2b–d (the displacement ellipsoid plots are presented in the ESI†). | ||
| Parametera | 2b | 2c | 2d |
|---|---|---|---|
a The tilt is the dihedral angle of the least-squares planes of the cyclopentadienyl rings C(1–5) and C(6–10), and CR denotes the pivotal carbon atoms of the phosphine substituents for 2b and 2c and the C12/C15 atoms for 2d.
b The C3PSe moiety in the structure of 2d is disordered, with the PSe bond exhibiting approximately mirror orientations (97![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3). Only data for the more populated orientation are provided. :3). Only data for the more populated orientation are provided.
|
|||
| Fe–C | 2.027(1)–2.059(1) | 2.034(2)–2.059(2) | 2.024(1)–2.066(2) |
| Tilt | 1.45(7) | 2.0(1) | 1.92(9) |
| C11–O | 1.222(1) | 1.223(2) | 1.218(2) |
| C11–P | 1.892(1) | 1.908(2) | 1.898(1) |
| P–CR | 1.840(1)/1.836(1) | 1.887(2)/1.890(2) | 1.883(1)/1.872(1) |
| PSe |
2.1124(4) | 2.1172(5) | 2.0981(5) |
| C1–C11–P | 122.69(8) | 122.3(1) | 119.56(9) |
| CR–P–CR | 107.27(5) | 113.70(8) | 95.34(6) |
| CR–P–C11 | 102.98(5)/101.54(5) | 101.70(8)/104.13(8) | 106.52(6)/105.16(6) |
Compounds 1a–d were treated with stoichiometric quantities of [AuCl(SMe2)] to produce the corresponding gold(I)–phosphine complexes [AuCl(1-κP)] (3a–d) (Scheme 3), which were isolated as deep burgundy red solids in good yields (74–85%) after crystallisation or flash chromatography. While stable as solids, the compounds slowly decomposed in solution upon exposure to daylight, depositing a violet precipitate and gold mirror. Coordination to the AuCl fragment resulted in a downfield shift of the 31P NMR signals (by 18–30 ppm in the series) and the resonances due to the carbonyl moiety and the ferrocene Cipso (by approximately 12 and 4 ppm, respectively) but only marginally affected the νCO frequencies (cf. 1612 cm−1 for 3c and 1606 cm−1 for 1c).
The crystal structure of 3c·1.5C6H12 (Fig. 4) comprises a linear P–Au–Cl moiety (177°) in which the Au–P and Au–Cl distances are similar to those in [AuCl(FcPPh2)] (Au–P 2.228(2), Au–Cl 2.280(2) Å)19 and slightly shorter than those in [AuCl(PAd3)] (Au–P 2.2570(7) Å, Au–Cl 2.2955(7) Å).20 The ferrocene cyclopentadienyls in 3c are tilted by 4.0(3)°, and the {C11,O,P} moiety deviates from the plane of its bonding cyclopentadienyl ring by 16.0(4)°. Compared to the free ligand 1c, the P–C(Ad) bonds in the structure of 3c are 0.01–0.02 Å shorter (the P–C11 bond length remains virtually the same), and the C–P–C angles are 2–3° wider.
 | ||
| Fig. 4 View of the complex molecule in the structure of 3c·1.5C6H12. Selected distances and angles (in Å and deg): Au–P 2.244(1), Au–Cl 2.285(1), P–Au–Cl 177.38(4), Fe–C(1–10) 2.034(4)–2.061(5), C11–O 1.214(6), C1–C11–P 120.7(3), P–C11 1.885(4), P–C12 1.875(4), P–C22 1.871(4), C11–P–C12 100.2(2), C11–P–C22 104.2(2), and C12–P–C22 114.3(2). | ||
Catalytic testing
The complexes 3a–d were studied as defined precatalysts for gold-mediated reactions.21 We initially investigated alkyne hydration, for which gold compounds successfully replaced conventional catalysts (e.g., toxic mercury salts).22,23 The hydration reaction is advantageously performed in methanol, which probably first adds to the triple bond to produce a ketal (vinyl ether) intermediate that subsequently hydrolyses to afford the target ketone.24 In our experiments, we also used methanol as the reaction solvent and performed the hydration reaction at a relatively low temperature (40 °C) to effectively differentiate among the performances of the tested catalysts (the typical reaction temperatures are 60–70 °C). Initial screening reactions were performed for the hydration of 4-ethynyltoluene (4a) in the presence of complex 3a (Scheme 4). | ||
| Scheme 4 Model Au-catalysed alkyne hydration (the reaction conditions are presented in Table 3). | ||
Considering the complex role played by Ag(I) ions in hydration catalysis,25 we first attempted to “activate” complex 3a with sodium salts (4 equiv.). Regrettably, using the catalysts generated from 3a/Na[BF4] and 3a/NaBARF resulted in only poor conversions (at 40 °C for 16 h; see Table 3), presumably due to inefficient halogen removal and, hence, a low concentration of Au(1a)+ as the plausible active species. Next, we applied silver(I) salts as activating agents. To minimise the influence of these compounds on the reaction course, we used 1 equiv. of a silver salt and activated the gold catalyst separately by mixing with the silver salt and filtering the reaction mixture through a Celite pad to remove the formed AgCl. The use of Ag[BF4] resulted in a 26% yield of 4-methylacetophenone (5a), which further improved upon replacing the silver salt with AgNTf2 (35%; AgNTf2 by itself did not catalyse the reaction). Complete conversion was achieved by increasing the reaction temperature to 60 °C. Halving the reaction time (60 °C/8 h) decreased the yield of 5a to 72%. No other products were detected in the crude reaction mixture.
| Entry | Catalyst | Additive | T [°C] | t [h] | Yield of 5a [%] |
|---|---|---|---|---|---|
| a Conditions: substrate 4a (0.5 mmol) and water (50 μL) were reacted in methanol (1 mL) in the presence of a preformed catalyst (1 mol% Au; see ESI). The yields were determined by integration of 1H NMR spectra using 4-anisaldehyde as an internal standard and represent the average of two independent runs. NaBARF = Na[B{C6H3(CF3)2-3,5}4]. b 5 mol% of AgNTf2 were used. | |||||
| 1 | 3a | Na[BF4] | 40 | 16 | 5 |
| 2 | 3a | NaBARF | 40 | 16 | 6 |
| 3 | 3a | Ag[BF4] | 40 | 16 | 26 |
| 4 | 3a | AgNTf2 | 40 | 16 | 35 |
| 5 | None | AgNTf2b | 40 | 16 | 0 |
| 6 | 3a | AgNTf2 | 60 | 16 | 100 |
| 7 | 3a | AgNTf2 | 60 | 8 | 72 |
| 8 | 3b | AgNTf2 | 40 | 16 | 77 |
| 9 | 3c | AgNTf2 | 40 | 16 | 94 |
| 10 | 3d | AgNTf2 | 40 | 16 | 13 |
| 11 | [AuCl(PPh3)] | AgNTf2 | 40 | 16 | 97 |
| 12 | [AuCl(FcPPh2)] | AgNTf2 | 40 | 16 | 97 |
When using the remaining complexes 3 under the optimised conditions, the best yield of 5a was achieved with 3c as the most electron-rich and bulky ligand, whereas the lowest performance was observed for the complex 3d featuring the electron-poor, cage phosphine ligand. Notably, all catalysts 3 were outperformed by their conventional analogues, [AuCl(PPh3)] and [AuCl(FcPPh2)], which resulted in nearly complete conversion under the same conditions.
The most active complex 3c was subsequently evaluated in reactions of different substrates (Scheme 5). Replacing the methyl group with an electron-withdrawing trifluoromethyl substituent (substrate 4b) had a detrimental effect on the reaction yield, affording only 6% of the hydration product 5b. By contrast, 1-octyne (4c) and ethynylferrocene (4d) yielded 100 and 65% of the hydration products, respectively. Last, tolane hydration produced ketone 5e in an 11% yield, in line with the lower reactivity of internal alkynes compared to those of the aforementioned substrates with terminal triple bonds.
 | ||
| Scheme 5 Au-catalysed hydration of different alkynes (the reaction conditions are presented in the Experimental section and the footnote to Table 3; the reported yields are the average value from two different runs). | ||
The second test reaction was the intramolecular cyclisation of N-propargyl benzamide (6) to 5-methylene-2-phenyl-4,5-dihydrooxazole (7, Scheme 6).26 This reaction was performed under previously reported conditions27 with 1 mol% of the in situ-activated Au catalyst in CD2Cl2 at 25 °C and monitored by 1H NMR spectroscopy (Fig. 5). The cyclisation proceeded cleanly and selectively, thereby enabling the yield of 7 to be determined simply by integration of the NMR spectra.
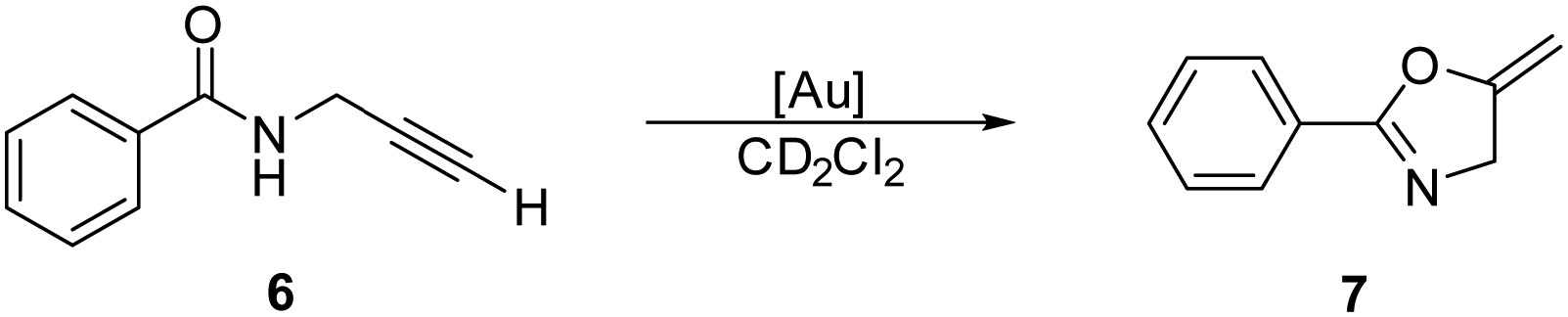 | ||
| Scheme 6 Gold-catalysed cyclisation of propargyl amide 6. | ||
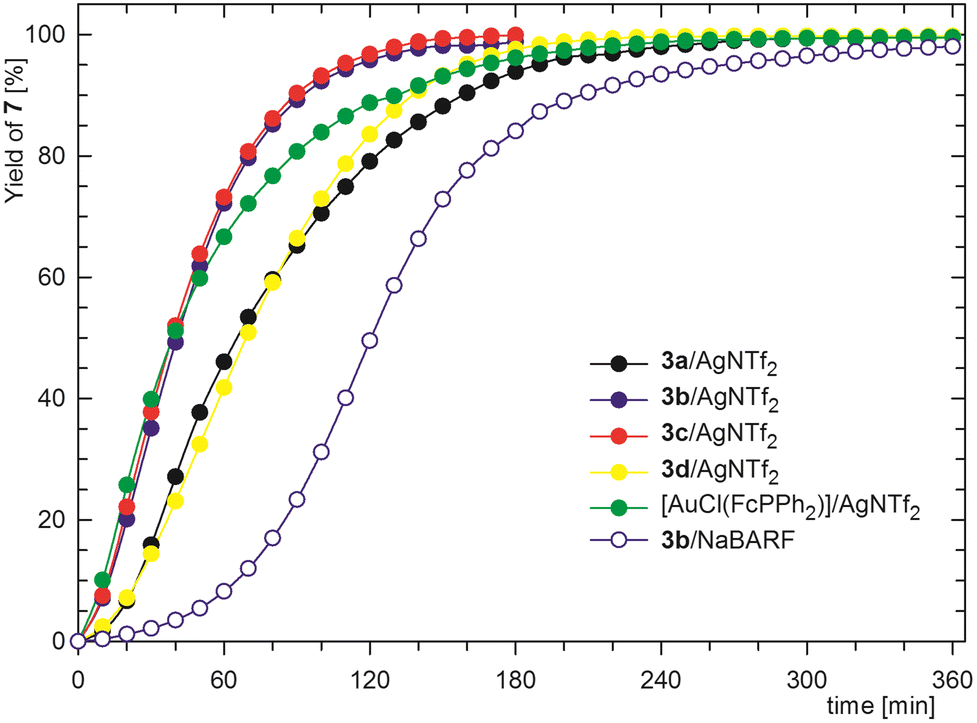 | ||
| Fig. 5 Kinetic profiles for the Au-catalysed cyclisation of amide 6 (the reaction was performed at 25 °C in CD2Cl2 in the presence of 1 mol% Au catalyst; the data are reported as the average of two independent runs). | ||
Among the catalysts generated from complexes 3 and AgNTf2 (1:1), the highest yields of 72 and 73% after 1 h reaction time (Table 4) were obtained using catalysts based on 3b and 3c, respectively, that contained the most electron-rich ligands, whereas yields of 46% and 42% were obtained using catalysts based on 3a and 3d. Notably, the kinetic profiles for the catalysts based on 3b and 3c were very similar but differed from those for the catalysts containing ligands with lower electron-donating abilities. The yields achieved using the catalysts based on 3a and 3d were 79 and 84%, respectively, after 2 h (i.e., the order was inverted from that for the 1 h reaction time) and 94 and 98% for a 3 h reaction time (N.B. the reactions with catalysts generated from 3b and 3c achieved nearly complete conversions within 2 h). This result can be ascribed to the varying stability of Au(1)+ species formed by halide abstraction (reflecting the different abilities of ligands 1 to stabilise such species) and the ease of catalyst activation (the reaction profiles for individual catalysts differ during the first 30 min, showing a gradual reaction acceleration for 3a and 3d). The activation of precatalysts 3 appears to be facilitated by phosphine ligands with high electron-donating ability due to their large trans influence,28 whereas steric and the electronic characteristics of the auxiliary phosphine ligands appear to control the overall catalyst stability (the differences among the steric properties of 1a, b, c, and d are reflected in the estimated buried volume (Vbur)29 values of 34, 31, 38 and 30%, respectively; see the ESI†).
| Catalyst | Yield of 7 after 1 h [%] | Yield of 7 after 3 h [%] | Time for >95% conversion [min] |
|---|---|---|---|
|
a Au catalyst (1 mol%) generated in situ from 3 and AgNTf2 (1:1) was used. The reaction was performed in CD2Cl2 (c(6) = 0.25 M) at 25 °C.
b NaBARF (2 equiv.) was used instead of the silver salt (as described in the main text).
|
|||
| 3a | 46 | 94 | 190 |
| 3b | 72 | 99 | 120 |
| 3c | 73 | 100 | 110 |
| 3d | 42 | 98 | 160 |
| [AuCl(FcPPh2)] | 67 | 96 | 170 |
| 3b | 8 | 84 | 260 |
In agreement with the basicity trend (vide supra), the reaction profile obtained using the catalyst produced from [AuCl(FcPPh2)] fell in between those of 3b/3c and 3a/3d. Even in this case, the catalytic species was formed quickly and was highly reactive (a 67% yield was achieved in 1 h) but degraded over time, as shown by the change in the slope of the kinetic profile.
As illustrated for 3b, using NaBARF to remove the Au-bound chloride instead of the silver salt resulted in significantly slower catalyst activation and slower reaction (Fig. 5). The sigmoidal shape of the kinetic profile during the initial 90 min indicates slow and gradual chloride removal and, together with a slower reaction rate, may suggest equilibria, e.g., between 3a/NaBARF and Au(1b)+BARF−/NaCl.
Conclusions
In summary, we demonstrated that ferrocene acylphosphines FcC(O)PR2 are reasonably stable phosphine derivatives that are readily accessible from ferrocenecarboxylic acid and various secondary phosphines R2PH. Insertion of a carbonyl spacer between the ferrocene unit and the phosphine moiety increases the steric freedom of these compounds and can reduce steric strain in compounds bearing bulky substituents at the phosphorus atom. In addition, the carbonyl group modulates the electronic properties of the terminal phosphine group by counterbalancing the electron-donating properties of the ferrocenyl substituent. These changes in the steric and electronic properties are reflected in the catalytic properties of gold(I) complexes supported with these ligands: substituents with good electron-donating ability and bulky structures may facilitate catalyst activation through halide loss and increase stability of the formed cationic species.Experimental
Materials and methods
Unless noted otherwise, all syntheses were performed under a nitrogen atmosphere using standard Schlenk techniques. Ferrocenecarboxylic acid,30 1,3,5,7-tetramethyl-2,4,6-trioxa-8-phosphaadamantane (CgPH),16b1a6 and 627 were prepared according to procedures reported in the literature. Other chemicals were purchased from Sigma-Aldrich or TCI and were used as received. Anhydrous dichloromethane, diethyl ether and tetrahydrofuran were obtained using a PureSolv MD5 solvent purification system (Innovative Technology, USA). Triethylamine was distilled from sodium metal and stored under nitrogen. Solvents used during workup and for crystallisation were purchased from Lach-Ner (analytical grade) and used without additional purification.NMR spectra were recorded at 25 °C on a Varian UNITY Inova 400 spectrometer operating at 399.95, 100.58 and 161.92 MHz for 1H, 13C and 31P, respectively. Chemical shifts (δ/ppm) are reported relative to internal SiMe4 (1H and 13C NMR) and external 85% aqueous H3PO4 (31P NMR). FTIR spectra were measured on a Thermo Scientific IS50 instrument in the range of 400–4000 cm−1. Electrospray ionisation mass spectra (ESI MS) were recorded with a Compact QTOF-MS spectrometer (Bruker Daltonics) for samples dissolved in methanol. Elemental analyses were performed on a PE 2400 Series II CHNS/O Elemental Analyser (PerkinElmer).
Synthesis of the acylphosphines 1b–d
:1) and eluted with the same solvent mixture. The first red band was collected and evaporated under vacuum to produce pure phosphine 1b as an amorphous red solid. Yield: 2.67 g (93%).
1H NMR (CDCl3, 400 MHz): δ 1.10–1.36 (br m, 10 H, Cy), 1.62–1.86 (br m, 10 H, Cy), 1.94–2.05 (m, 2 H, Cy), 4.23 (s, 5 H, C5H5), 4.54 (vt of d, J′ = 2.0, 1.3 Hz, 2 H, C5H4), 4.93 (vt of d, J′ = 2.0, 0.9 Hz, 2 H, C5H4). 13C{1H} NMR (CDCl3, 101 MHz): δ 26.33 (d, JPC = 1 Hz, Cy), 27.20 (d, JPC = 9 Hz, Cy), 27.34 (d, JPC = 10 Hz, Cy), 29.83 (d, JPC = 11 Hz, Cy), 30.81 (d, JPC = 10 Hz, Cy), 32.37 (d, JPC = 13 Hz, Cy), 69.65 (d, JPC = 8 Hz, CH of C5H4), 69.91 (C5H5), 72.37 (d, JPC = 2 Hz, CH of C5H4), 85.05 (d, 2JPC = 39 Hz, Cipso of C5H4), 217.19 (d, 1JPC = 41 Hz, CO). 31P{1H} NMR (CDCl3, 162 MHz): δ 17.8 (s). IR (DRIFTS, KBr): νmax 3102 w, 3082 w, 2927 s, 2851 m, 1613 s (νCO), 1441 m, 1370 m, 1338 w, 1293 w, 1243 s, 1199 w, 1181 w, 1108 w, 1056 w, 1037 m, 1027 m, 1001 w, 949 w, 873 w, 850 w, 838 m, 818 m, 744 w, 692 w, 585 w, 549 w, 490 m, 478 m, 444 w cm−1. ESI MS: m/z 433 ([M + Na]+), 449 ([M + K]+). Anal. calcd for C23H31FeOP (410.3): C 67.33, H 7.62%. Found: C 67.09, H 7.60%.
:1) and hexane-dichloromethane (1:1) to remove unreacted di(1-adamantyl)phosphine and, finally, with dichloromethane to elute the product as the only red band, which was collected and evaporated to afford pure 1c as an orange powdery solid. Yield: 562 mg (54%).
1H NMR (CDCl3, 400 MHz): δ 1.65–1.76 (m, 12 H, CH2 of Ad), 1.93 (br s, 6 H, CH of Ad), 2.00–2.10 (m, 12 H, CH2 of Ad), 4.27 (s, 5 H, C5H5), 4.52 (vt of d, J′ = 2.0, 1.3 Hz, 2 H, C5H4), 4.95 (vt of d, J′ = 2.0, 1.1 Hz, 2 H, C5H4). 13C{1H} NMR (CDCl3, 101 MHz): δ 28.91 (d, 3JPC = 8 Hz, CH of Ad), 36.89 (CH2 of Ad), 38.31 (d, 1JPC = 22 Hz, Cipso of Ad), 41.69 (d, JPC = 9 Hz, CH2 of Ad), 69.72 (C5H5), 70.39 (d, JPC = 10 Hz, CH of C5H4), 72.06 (d, JPC = 2 Hz, CH of C5H4), 87.57 (d, 2JPC = 42 Hz, Cipso of C5H4), 219.05 (d, 1JPC = 46 Hz, CO). 31P{1H} NMR (CDCl3, 162 MHz): δ 45.3 (s). IR (DRIFTS, KBr): νmax 3096 w, 2926 m, 2898 s, 2844 m, 1606 s (νCO), 1452 m, 1434 m, 1393 w, 1370 m, 1352 m, 1342 m, 1302 w, 1238 s, 1106 m, 1037 m, 1025 m, 1000 w, 970 w, 941 w, 837 w, 822 m, 815 m, 698 m, 588 w, 553 m, 496 m, 487 m, 479 m, 432 w cm−1. ESI MS: m/z 515 ([M + H]+), 537 ([M + Na]+). Anal. calcd for C31H39FeOP (514.5): C 72.37, H 7.64%. Found: C 72.26, H 7.53%.
:1), transferred to the top of a chromatographic column and eluted with the same solvent. The first red band was collected and evaporated to give an oily residue, which was diluted with pentane (≈5 mL) and stored in a refrigerator. The separated crystals were decanted, washed with cold pentane and dried under vacuum to give 1d as a red microcrystalline solid. Yield: 146 mg (17%).
1H NMR (CDCl3, 400 MHz): δ 1.35 (s, 3 H, CH3), 1.39 (s, 3 H, CH3), 1.47 (d, 3JPH = 12.2 Hz, 3 H, CH3), 1.51 (d, 3JPH = 13.4 Hz, 3 H, CH3), 1.64 (dd, J = 13.2, 4.9 Hz, 1 H, CH2), 1.86 (dd, J = 24.8, 13.0 Hz, 1 H, CH2), 2.05 (dd, J = 13.0, 6.5 Hz, 1 H, CH2), 2.61 (d, J = 13.2, 1 H, CH2), 4.24 (s, 5 H, C5H5), 4.58–4.63 (br m, 2 H, C5H4), 4.95–4.98 (m, 2 H, C5H4). 13C{1H} NMR (CDCl3, 101 MHz): δ 27.78 (CH3), 27.82 (CH3), 28.56 (d, 2JPC = 10 Hz, CH3), 28.90 (d, 2JPC = 20 Hz, CH3), 37.78 (d, 2JPC = 1 Hz, CH2), 45.49 (d, 2JPC = 16 Hz, CH2), 69.48 (d, JPC = 7 Hz, CH of C5H4), 70.34 (C5H5), 70.35 (d, JPC = 10 Hz, CH of C5H4), 72.92 (d, JPC = 1 Hz, CH of C5H4), 72.94 (CH of C5H4), 73.08 (d, 1JPC = 8 Hz, Cipso of PCg), 74.50 (d, 1JPC = 26 Hz, Cipso of PCg), 85.46 (d, 2JPC = 39 Hz, Cipso of C5H4), 96.38 (Cipso of PCg), 96.73 (Cipso of PCg), 213.33 (d, 1JPC = 53 Hz, CO). 31P{1H} NMR (CDCl3, 162 MHz): δ –14.5 (s). IR (DRIFTS, KBr): νmax 3101 w, 3085 w, 2987 w, 2975 w, 2962 w, 2936 w, 2920 w, 2910 w, 1631 m (νCO), 1615 m (νCO), 1430 m, 1418 w, 1390 w, 1377 m, 1364 m, 1339 m, 1266 w, 1237 s, 1213 s, 1189 s, 1133 s, 1106 w, 1088 m, 1068 w, 1045 m, 1026 m, 1001 w, 981 s, 965 m, 950 w, 938 m, 897 s, 852 m, 825 s, 814 s, 791 m, 699 m, 686 m, 662 w, 608 w, 574 m, 549 w, 536 m, 498 s, 477 m, 451 w, 437 w cm−1. ESI MS: m/z 451 ([M + Na]+), 467 ([M + K]+). Anal. calcd for C21H25FeO4P (428.2): C 58.90, H 5.88%. Found: C 58.79, H 5.62%.
Preparation of phosphine selenides
1H NMR (CDCl3, 400 MHz): δ 1.12–1.53 (br m, 10 H, Cy), 1.65–2.04 (br m, 10 H, Cy), 2.23–2.35 (m, 2 H, Cy), 4.36 (s, 5 H, C5H5), 4.68–4.70 (m, 2 H, C5H4), 5.51 (vt, J′ = 1.8 Hz, 2 H, C5H4). 13C{1H} NMR (CDCl3, 101 MHz): δ 25.75 (d, JPC = 2 Hz, Cy), 25.99 (d, JPC = 1 Hz, Cy), 26.38 (d, JPC = 5 Hz, Cy), 26.51 (d, JPC = 6 Hz, Cy), 27.17 (d, JPC = 3 Hz, Cy), 36.36 (d, JPC = 37 Hz, Cy), 70.96 (C5H5), 71.90 (CH of C5H4), 73.73 (CH of C5H4), 80.73 (d, 2JPC = 50 Hz, Cipso of C5H4), 203.05 (d, 1JPC = 35 Hz, CO). 31P{1H} NMR (CDCl3, 162 MHz): δ 52.3 (s with 77Se satellites, 1JSeP = 718 Hz). IR (DRIFTS, KBr): νmax 2930 s, 2852 m, 1605 s (νCO), 1450 m, 1435 w, 1410 w, 1371 w, 1350 w, 1258 m, 1107 w, 1052 m, 1030 w, 1002 w, 839 m, 829 m, 746 w, 732 w, 693 w, 587 m, 560 s, 527 w, 499 m, 484 m, 468 m, 448 w cm−1. ESI MS: m/z 513 ([M + Na]+). Anal. calcd for C23H31FeOPSe (489.3): C 56.46, H 6.39%. Found: C 56.24, H 6.27%.
:1) to give a purple amorphous solid after evaporation. Crystallization by liquid-phase diffusion of hexane into the solution of crude selenide in chloroform produced pure 2c as purple crystals. Yield: 50 mg (85%).
1H NMR (CDCl3, 400 MHz): δ 1.65–1.78 (m, 12 H, CH2 of Ad), 1.95–2.06 (m, 6 H, CH of Ad), 2.18–2.31 (m, 12 H, CH2 of Ad), 4.36 (s, 5 H, C5H5), 4.64 (vt of d, J′ = 2.0, 0.9 Hz, 2 H, C5H4), 5.63 (vt, J′ = 2.0 Hz, 2 H, C5H4). 13C{1H} NMR (CDCl3, 101 MHz): δ 28.70 (d, 3JPC = 9 Hz, CH of Ad), 36.44 (d, JPC = 2 Hz, CH2 of Ad), 38.49 (d, JPC = 2 Hz, CH2 of Ad), 43.36 (d, 1JPC = 23 Hz, Cipso of Ad), 70.72 (C5H5), 72.69 (CH of C5H4), 73.23 (CH of C5H4), 82.77 (d, 2JPC = 49 Hz, Cipso of C5H4), 205.79 (d, 1JPC = 31 Hz, CO). 31P{1H} NMR (CDCl3, 162 MHz): δ 68.2 (s with 77Se satellites, 1JSeP = 709 Hz). IR (DRIFTS, KBr): νmax 2902 m, 2850 m, 1604 s (νCO), 1451 w, 1424 w, 1407 w, 1374 w, 1353 w, 1341 w, 1315 w, 1302 w, 1247 m, 1182 w, 1107 w, 1051 m, 1044 m, 1026 w, 1003 w, 973 m, 831 m, 819 m, 696 w, 583 m, 567 m, 532 w, 505 w, 489 m, 478 s, 469 m, 451 m, 430 m cm−1. ESI MS: m/z 617 ([M + Na]+). Anal. calcd for C31H39FeOPSe (593.4): C 62.74, H 6.62%. Found: C 62.55, H 6.92%.
:1) to provide a purple amorphous solid after evaporation. The solid was dissolved in chloroform (≈1 mL), and the resulting solution was layered with hexane in a test tube. Crystals that formed during several days were decanted, washed with cold pentane and dried under vacuum. Yield of 2d: 27 mg (53%), purple crystals.
1H NMR (CDCl3, 400 MHz): δ 1.36 (s, 3 H, CH3), 1.44 (s, 3 H, CH3), 1.52 (d, 3JPH = 15.3 Hz, 3 H, CH3), 1.58 (d, 3JPH = 14.5 Hz, 3 H, CH3), 1.68 (dd, J = 24.2, 13.7 Hz, 1 H, CH2), 2.04 (dd, J = 20.9, 13.8 Hz, 1 H, CH2), 2.57 (dd, J = 13.8, 1.1 Hz, 1 H, CH2), 3.04 (dd, J = 13.3, 3.6 Hz, 1 H, CH2), 4.33 (s, 5 H, C5H5), 4.68–4.72 (br m, 2 H, C5H4), 5.30 (dtd, J = 2.6, 1.3, 0.6 Hz, 1 H, C5H4), 5.49 (dt, J = 2.6, 1.3 Hz, 1 H, C5H4). 13C{1H} NMR (CDCl3, 101 MHz): δ 23.97 (d, 2JPC = 2 Hz, CH3), 24.08 (d, 2JPC = 2 Hz, CH3), 27.31 (CH3), 27.34 (CH3), 39.36 (d, 2JPC = 5 Hz, CH2), 40.96 (d, 2JPC = 2 Hz, CH2), 71.74 (C5H5), 72.32 (d, JPC = 2 Hz, CH of C5H4), 72.51 (CH of C5H4), 73.27 (d, 1JPC = 31 Hz, Cipso of PCg), 73.54 (CH of C5H4), 73.69 (CH of C5H4), 76.73 (d, 1JPC = 29 Hz, Cipso of PCg), 79.59 (d, 2JPC = 57 Hz, Cipso of C5H4), 96.57 (d, 3JPC = 2 Hz, Cipso of PCg), 96.63 (d, 3JPC = 1 Hz, Cipso of PCg), 201.61 (d, 1JPC = 27 Hz, CO). 31P{1H} NMR (CDCl3, 162 MHz): δ 24.2 (s with 77Se satellites, 1JSeP = 761 Hz). IR (DRIFTS, KBr): νmax 2967 w, 2925 w, 1637 w (νCO), 1620 m (νCO), 1453 w, 1431 m, 1412 w, 1394 w, 1381 m, 1373 m, 1364 w, 1345 m, 1323 w, 1262 w, 1248 m, 1213 m, 1202 m, 1183 m, 1136 m, 1106 w, 1090 m, 1047 m, 1034 w, 1006 w, 998 w, 983 s, 959 w, 895 m, 852 m, 839 m, 819 s, 795 m, 702 m, 680 w, 661 w, 635 w, 610 m, 582 m, 568 w, 546 m, 528 w, 503 m, 483 m, 471 m, 445 s, 421 m cm−1. ESI MS: m/z 451 ([M – Se + Na]+), 531 ([M + Na]+). Anal. calcd for C21H25FeO4PSe (507.2): C 49.73, H 4.97%. Found: C 49.66, H 4.71%.
Synthesis of gold(I) complexes
1H NMR (CDCl3, 400 MHz): δ 4.45 (s, 5 H, C5H5), 4.70–4.72 (m, 2 H, C5H4), 5.15–5.17 (m, 2 H, C5H4), 7.46–7.58 (m, 6 H, PPh2), 7.64–7.72 (m, 4 H, PPh2). 13C{1H} NMR (CDCl3, 101 MHz): δ 70.98 (d, JPC = 4 Hz, CH of C5H4), 71.30 (C5H5), 74.48 (d, JPC = 1 Hz, CH of C5H4), 78.81 (d, 2JPC = 55 Hz, Cipso of C5H4), 126.36 (d, 1JPC = 60 Hz, Cipso of PPh2), 129.26 (d, JPC = 12 Hz, CH of PPh2), 132.35 (d, 4JPC = 3 Hz, CHpara of PPh2), 134.89 (d, JPC = 13 Hz, CH of PPh2), 199.78 (d, 1JPC = 33 Hz, CO). 31P{1H} NMR (CDCl3, 162 MHz): δ 29.3 (s). IR (DRIFTS, KBr): νmax 3097 w, 3085 w, 3051 w, 1637 m (νCO), 1626 s (νCO), 1571 w, 1479 w, 1436 s, 1410 m, 1372 w, 1350 w, 1328 w, 1314 w, 1252 s, 1186 w, 1103 m, 1050 m, 1028 m, 999 m, 945 w, 870 w, 836 m, 817 m, 750 m, 743 m, 718 w, 691 m, 591 m, 563 m, 515 m, 497 s, 472 m, 456 m cm−1. ESI MS: m/z 653 ([M + Na]+). Anal. calcd for C23H19AuClFeOP (630.6): C 43.80, H 3.04%. Found: C 43.57, H 2.94%.
1H NMR (CDCl3, 400 MHz): δ 1.14–1.52 (br m, 10 H, Cy), 1.66–2.04 (br m, 10 H, Cy), 2.24–2.36 (m, 2 H, Cy), 4.46 (s, 5 H, C5H5), 4.76–4.78 (m, 2 H, C5H4), 5.30 (vt, J′ = 1.8 Hz, 2 H, C5H4).13C{1H} NMR (CDCl3, 101 MHz): δ 25.60 (d, JPC = 2 Hz, Cy), 26.52 (d, JPC = 12 Hz, Cy), 26.64 (d, JPC = 13 Hz, Cy), 29.48 (d, JPC = 2 Hz, Cy), 29.88 (s, Cy), 33.23 (d, JPC = 31 Hz, Cy), 70.81 (C5H5), 71.18 (d, JPC = 3 Hz, CH of C5H4), 74.64 (CH of C5H4), 81.68 (d, 2JPC = 48 Hz, Cipso of C5H4), 202.05 (d, 1JPC = 27 Hz, CO). 31P{1H} NMR (CDCl3, 162 MHz): δ 45.0 (s). IR (DRIFTS, KBr): νmax 3202 w, 3087 w, 2927 s, 2850 m, 1614 s (νCO), 1447 m, 1436 m, 1411 w, 1373 m, 1352 w, 1329 w, 1292 w, 1258 s, 1199 w, 1180 w, 1171 w, 1106 w, 1052 m, 1031 m, 1003 m, 948 w, 915 w, 888 w, 851 m, 838 m, 823 m, 752 w, 730 w, 693 w, 584 w, 561 m, 500 s, 486 m, 468 m, 444 m, 429 w cm−1. ESI MS: m/z 665 ([M + Na]+). Anal. calcd for C23H31AuClFeOP (642.7): C 42.98, H 4.86%. Found: C 43.02, H 4.49%.
1H NMR (CDCl3, 400 MHz): δ 1.68–1.80 (m, 12 H, CH2 of Ad), 2.00–2.07 (m, 6 H, CH of Ad), 2.18–2.32 (m, 12 H, CH2 of Ad), 4.46 (s, 5 H, C5H5), 4.74 (vt of d, J′ = 2.0 Hz, 1.2 Hz, 2 H, C5H4), 5.39 (vt, J′ = 1.8 Hz, 2 H, C5H4). 13C{1H} NMR (CDCl3, 101 MHz): δ 28.58 (d, 3JPC = 9 Hz, CH of Ad), 36.26 (d, JPC = 1 Hz, CH2 of Ad), 41.63 (CH2 of Ad), 42.65 (d, 1JPC = 20 Hz, Cipso of Ad), 70.65 (C5H5), 71.86 (d, JPC = 3 Hz, CH of C5H4), 74.26 (CH of C5H4), 83.78 (d, 2JPC = 46 Hz, Cipso of C5H4), 204.96 (d, 1JPC = 21 Hz, CO). 31P{1H} NMR (CDCl3, 162 MHz): δ 64.1 (s). IR (DRIFTS, KBr): νmax 2905 s, 2850 m, 1612 s (νCO), 1449 m, 1429 m, 1371 w, 1353 w, 1343 m, 1300 w, 1249 m, 1107 w, 1047 m, 1026 w, 1000 w, 972 w, 837 m, 826 m, 696 w, 587 w, 565 m, 521 w, 506 w, 489 m, 479 s, 464 m, 451 m, 430 m cm−1. ESI MS: m/z 769 ([M + Na]+). Anal. calcd for C31H39AuClFeOP·C6H12 (831.0): C 53.47, H 6.19%. Found: C 53.42, H 5.97%.
1H NMR (CDCl3, 400 MHz): δ 1.38 (s, 3 H, CH3), 1.44 (s, 3 H, CH3), 1.58 (d, 3JPH = 8.1 Hz, 3 H, CH3), 1.62 (d, 3JPH = 9.6 Hz, 3 H, CH3), 1.84 (dd, J = 25.5 Hz, 13.6 Hz, 1 H, CH2), 1.96 (dd, J = 16.9 Hz, 13.8 Hz, 1 H, CH2), 2.50 (dd, J = 13.7 Hz, 4.5 Hz, 1 H, CH2), 2.51 (dd, J = 13.7 Hz, 0.9 Hz, 1 H, CH2), 4.47 (s, 5 H, C5H5), 4.79–4.85 (br m, 2 H, C5H4), 5.21–5.24 (m, 1 H, C5H4), 5.38–5.40 (m, 1 H, C5H4). 13C{1H} NMR (CDCl3, 101 MHz): δ 27.07 (d, 2JPC = 4 Hz, CH3), 27.27 (CH3), 27.30 (CH3), 28.05 (d, 2JPC = 7 Hz, CH3), 38.60 (d, 2JPC = 1 Hz, CH2), 44.68 (d, 2JPC = 8 Hz, CH2), 70.99 (d, JPC = 4 Hz, CH of C5H4), 71.40 (C5H5), 71.54 (d, JPC = 3 Hz, CH of C5H4), 74.25 (d, 1JPC = 30 Hz, Cipso of PCg), 74.78 (d, JPC = 2 Hz, CH of C5H4), 75.10 (CH of C5H4), 75.11 (d, 1JPC = 23 Hz, Cipso of PCg), 81.37 (d, 2JPC = 52 Hz, Cipso of C5H4), 96.66 (d, 3JPC = 2 Hz, Cipso of PCg), 97.05 (Cipso of PCg), 199.05 (d, 1JPC = 12 Hz, CO). 31P{1H} NMR (CDCl3, 162 MHz): δ 16.0 (s). IR (DRIFTS, KBr): νmax 3086 w, 2999 w, 2974 w, 2963 w, 2921 w, 1632 m (νCO), 1617 (νCO), 1450 w, 1436 m, 1394 w, 1385 w, 1373 w, 1346 w, 1249 m, 1231 w, 1216 m, 1205 m, 1187 m, 1135 m, 1109 w, 1088 m, 1048 m, 1035 w, 1004 w, 983 s, 961 w, 940 w, 893 m, 855 m, 836 s, 813 m, 792 m, 703 w, 683 m, 605 m, 579 m, 546 m, 516 w, 495 s, 487 m, 463 m, 443 s cm−1. ESI MS: m/z 683 ([M + Na]+). Anal. calcd for C21H25AuClFeO4P (660.7): C 38.18, H 3.81%. Found: C 37.89, H 3.58%.
Catalytic experiments
X-ray crystallography
Diffraction data were recorded at 120 or 150 K with a Bruker D8 VENTURE Kappa Duo diffractometer with a PHOTON III detector and a Cryostream Cooler (Oxford Cryosystems) using Cu Kα (λ = 1.54178 Å for 1b) or Mo Kα radiation (λ = 0.71073 Å for all other compounds). The structures were solved by direct methods (SHELXT, recent version36) and subsequently refined by a full-matrix least-squares routine based on F2 (SHELXL-2014/201737). All nonhydrogen atoms were refined with anisotropic displacement parameters, and the hydrogen atoms were placed in their theoretical positions and refined as riding atoms using the standard parameters implemented in SHELXL.Compound 2c formed partially disordered crystals with the PSe bonds occupying approximately mirror-image positions within the space defined by the bulky adamantyl substituents. The refined occupancies for the two orientations were 97:3. Furthermore, the crystals of 1b and 3c·1.5C6H12 were nonmerohedral, two-component twins. The refined contributions from the two crystal domains were approximately 70:30 for 1b and 51:49 for 3c·1.5C6H12.
Selected crystallographic data and structure refinement parameters are available in the ESI† (Table S1). The geometric data and structural diagrams were obtained using a recent version of the PLATON program.38 All numerical values were rounded to one decimal place with respect to their estimated standard deviations.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Czech Science Foundation is acknowledged for financial support (project No. 19-09334S). The authors thank Dr Ivana Císařová of the Department of Inorganic Chemistry, Faculty of Science, Charles University, for recording the X-ray diffraction data.References
- P. Kumar, U. Sharma and G. S. Ananthnag, Appl. Organomet. Chem., 2022, 36, e6672 CAS.
- For examples, see: (a) J. Li, C. Yang, Y. Bai, X. Yang, Y. Liu and J. Peng, J. Organomet. Chem., 2018, 855, 7 CrossRef CAS; (b) J. Yang, X. Chen and Z. Wang, Tetrahedron Lett., 2015, 56, 5673 CrossRef CAS; (c) P. Kumar, M. M. Siddiqui, Y. Reddi, J. T. Mague, R. B. Sunoj and M. S. Balakrishna, Dalton Trans., 2013, 42, 11385 RSC; (d) S. Gowrisankar, H. Neumann, A. Spannenberg and M. Beller, Organometallics, 2014, 33, 94 CrossRef CAS; (e) S. Gowrisankar, C. Federsel, H. Neumann, C. Ziebart, R. Jackstell, A. Spannenberg and M. Beller, ChemSusChem, 2013, 6, 85 CrossRef CAS PubMed; (f) R. A. Baber, M. L. Clarke, A. G. Orpen and D. A. Ratcliffe, J. Organomet. Chem., 2003, 667, 112 CrossRef.
- (a) In Ferrocenes: Homogeneous Catalysis, Organic Synthesis, Materials Science, ed. A. Togni and T. Hayashi, VCH, Weinheim, 1995 Search PubMed; (b) In Ferrocenes: Ligands, Materials and Biomolecules, ed. P. Štěpnička, Wiley, Chichester, 2008 Search PubMed; (c) R. C. J. Atkinson, V. C. Gibson and N. J. Long, Chem. Soc. Rev., 2004, 33, 313 RSC; (d) R. Gómez Arrayás, J. Adrio and J. C. Carretero, Angew. Chem., Int. Ed., 2006, 45, 7674 CrossRef PubMed; (e) L. Cunningham, A. Benson and P. J. Guiry, Org. Biomol. Chem., 2020, 18, 9329 RSC; (f) P. Štěpnička, Dalton Trans., 2022, 51, 8085 RSC.
- E. Fuchs, B. Breit, H. Heydt, M. Regitz, W. Schoeller, T. Busch, C. Krüger and P. Betz, Chem. Ber., 1991, 124, 2843 CrossRef CAS.
- R. Pietschnig, E. Niecke, M. Nieger and K. Airola, J. Organomet. Chem., 1997, 529, 127 CrossRef CAS.
- P. Vosáhlo, J. Schulz, I. Císařová and P. Štěpnička, Dalton Trans., 2021, 50, 6232 RSC.
- P. Vosáhlo, I. Císařová and P. Štěpnička, New J. Chem., 2022, 46, 21536 RSC.
- (a) K.-S. Gan and T. S. A. Hor, in Ferrocenes: Homogeneous Catalysis, Organic Synthesis Materials Science, ed. A. Togni and T. Hayashi, Wiley-VCH, Weinheim, Germany, 1995, ch. 1, pp. 3–104 Search PubMed; (b) S. W. Chien and T. S. A. Hor, in Ferrocenes: Ligands, Materials and Biomolecules, ed. P. Štěpnička, Wiley, Chichester, UK, 2008, ch. 2, pp. 33–116 Search PubMed; (c) T. J. Colacot and S. Parisel, in Ferrocenes: Ligands, Materials and Biomolecules, ed. P. Štěpnička, Wiley, Chichester, UK, 2008, ch. 3, pp. 117–140 Search PubMed; (d) G. Bandoli and A. Dolmella, Coord. Chem. Rev., 2000, 209, 161 CrossRef CAS; (e) S. Dey and R. Pietschnig, Coord. Chem. Rev., 2021, 437, 213850 CrossRef CAS.
- H. Shet, U. Parmar, S. Bhilare and A. R. Kapdi, Org. Chem. Front., 2021, 8, 1599 RSC.
- (a) F. Horký, I. Císařová and P. Štěpnička, ChemCatChem, 2021, 13, 4848 CrossRef; (b) F. Horký, I. Císařová and P. Štěpnička, J. Organomet. Chem., 2022, 957, 122145 CrossRef.
- B. Breit and D. Breuninger, Synthesis, 2005, 2782 CrossRef CAS.
- (a) Z. Szarka, R. Skoda-Földes, Á. Kuik, Z. Berente and L. Kollár, Synthesis, 2003, 545 CAS; (b) C. Lu, X. Wang, Y. Yang and X. Liu, Inorg. Chim. Acta, 2016, 447, 121 CrossRef CAS.
- C. A. Tolman, Chem. Rev., 1977, 77, 313 CrossRef CAS.
- U. Beckmann, D. Süslüyan and P. C. Kunz, Phosphorus, Sulfur Silicon Relat. Elem., 2011, 186, 2061 CrossRef CAS.
- R. P. Pinnell, C. A. Megerle, S. L. Manatt and P. A. Kroon, J. Am. Chem. Soc., 1973, 95, 977 CrossRef CAS.
- (a) C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165 CrossRef CAS; (b) J. H. Downing, J. Floure, K. Heslop, M. F. Haddow, J. Hopewell, M. Lusi, H. Phetmung, A. G. Orpen, P. G. Pringle, R. I. Pugh and D. Zambrano-Williams, Organometallics, 2008, 27, 3216 CrossRef CAS.
- B. Milde, M. Lohan, C. Schreiner, T. Rüffer and H. Lang, Eur. J. Inorg. Chem., 2011, 5437 CrossRef CAS.
- The data were taken from: (a) A. Muller, S. Otto and A. Roodt, Dalton Trans., 2008, 650 RSC; (b) M. L. Kelty, A. J. McNeece, J. W. Kurutz, A. S. Filatov and J. S. Anderson, Chem. Sci., 2022, 13, 4377 RSC and refs. 14 and 17.
- K. Rössler, T. Rüffer, B. Walfort, R. Packheiser, R. Holze, M. Zharnikov and H. Lang, J. Organomet. Chem., 2007, 692, 1530 CrossRef.
- L. Chen, P. Ren and B. P. Carrow, J. Am. Chem. Soc., 2016, 138, 6392 CrossRef CAS PubMed.
- (a) A. S. K. Hashmi, Chem. Rev., 2007, 107, 3180 CrossRef CAS PubMed; (b) R. Dorel and A. M. Echavarren, Chem. Rev., 2015, 115, 9028 CrossRef CAS PubMed.
- L. Hintermann and A. Labonne, Synthesis, 2007, 1121 CrossRef CAS.
- For the first report of applications of Au(I) compounds in alkyne hydration, see: E. Mizushima, K. Sato, T. Hayashi and M. Tanaka, Angew. Chem., Int. Ed., 2002, 41, 4563 CrossRef CAS PubMed.
- A. Leyva and A. Corma, J. Org. Chem., 2009, 74, 2067 CrossRef CAS PubMed.
- On the one hand, silver(I) salts are commonly used to generate active species of the type LAu+ from stable precursors [LAuCl]. On the other hand, formation of relatively stable, geminally dimetallated intermediates, R2C = C(AuL)(AgLn), can slow the Au-mediated reaction: (a) D. Weber and M. R. Gagné, Org. Lett., 2009, 11, 4962 CrossRef CAS PubMed; (b) D. Wang, R. Cai, S. Sharma, J. Jirak, S. K. Thummanapelli, N. G. Akhmedov, H. Zhang, X. Liu, J. L. Petersen and X. Shi, J. Am. Chem. Soc., 2012, 134, 9012 CrossRef CAS PubMed.
- (a) A. S. K. Hasmi, J. P. Weyrauch, W. Frey and J. W. Bats, Org. Lett., 2004, 6, 4391 CrossRef PubMed; (b) J. P. Weyrauch, A. S. K. Hashmi, A. Schuster, T. Hengst, S. Schetter, A. Littmann, M. Rudolph, M. Hamzic, J. Visus, F. Rominger, W. Frei and J. W. Bats, Chem. – Eur. J., 2010, 16, 956 CrossRef CAS PubMed.
- O. Bárta, I. Císařová, J. Schulz and P. Štěpnička, New J. Chem., 2019, 43, 11258 RSC.
- (a) P. G. Jones and A. F. Williams, J. Chem. Soc., Dalton Trans., 1977, 1430 RSC; (b) P. G. Jones, A. G. Maddock, M. J. Mays, M. M. Muir and A. F. Williams, J. Chem. Soc., Dalton Trans., 1977, 1434 RSC.
- (a) L. Falivene, Z. Cao, A. Petta, L. Serra, A. Poater, R. Oliva, V. Scarano and L. Cavallo, Nat. Chem., 2019, 11, 872 CrossRef CAS PubMed; (b) L. Falivene, R. Cudendino, A. Poater, A. Petta, L. Serra, R. Oliva, V. Scarano and L. Cavallo, Organometallics, 2016, 35, 2286 CrossRef CAS.
- P. C. Reeves, J. J. Mrowca, M. M. Borecki and W. A. Sheppard, Org. Synth., 1977, 56, 28 CrossRef CAS.
- Y. Yang, A. Qin, K. Zhao, D. Wang and X. Shi, Adv. Synth. Catal., 2016, 358, 1433 CrossRef CAS.
- H. Morimoto, T. Tsubogo, N. D. Litvinas and J. F. Hartwig, Angew. Chem., Int. Ed., 2011, 50, 3793 CrossRef CAS PubMed.
- R. E. Ebule, D. Malhotra, G. B. Hammond and B. Xu, Adv. Synth. Catal., 2016, 358, 1478 CrossRef CAS.
- V. A. Darin, A. F. Neto, J. Miller, M. M. F. Afonso, H. C. Fonsatti and Á. D. L. Borges, J. Prakt. Chem., 1999, 341, 588 CrossRef CAS.
- A. Rühling, H.-J. Galla and F. Glorius, Chem. – Eur. J., 2015, 21, 12291 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct, Chem., 2015, 71, 3 Search PubMed.
- (a) A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS; (b) A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional structural diagrams, summary of the crystallographic parameters and copies of the NMR spectra. CCDC 2235366–2235370. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3nj00201b |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2023 |