DOI:
10.1039/D3NJ00182B
(Paper)
New J. Chem., 2023,
47, 10296-10308
Synthesis and anticancer properties of dendritic glycoconjugates containing multiple o-carborane clusters†
Received
12th January 2023
, Accepted 1st May 2023
First published on 2nd May 2023
Abstract
Boron-rich glycodendrimers containing three to six peripheral hydrophilic glucose moieties and three ortho-carborane clusters have been synthesized. Adding more number of glucose moieties compensated the hydrophobicity of ortho-carboranes and made the glycodendrimers completely water soluble. In vitro cytotoxicity of the synthesized glycodendrimers 16, 18 and 20 has been evaluated against MCF-7 breast cancer cell lines and compared with that of the core compound 7 without any peripheral glucose moieties. The glycodendrimer 20 having nine glucose moieties has shown the highest cell death potential with an IC50 value of 41.35 ng mL−1 (0.01 μM) while lower cell death potential with an IC50 value of 822.33 ng mL−1 (0.8 μM) has been observed in the case of compound 7. The cytotoxicity of glycodendrimer 20 was found to be 600 times higher than the cytotoxicity of the chemotherapeutic drug cisplatin (IC50 6 μM). The study of caspase-3 activity reveals that compounds 7, 16, 18 and 20 induce cell death in MCF-7 cell lines via apoptosis. The results indicate that the glycodendrimers containing multiple o-carborane clusters could be useful tumor specific anticancer agents.
Introduction
Dendrimer based drug delivery systems show enhanced cellular uptake because of their unique branching architecture and a large number of peripheral functional groups.1 Macromolecular and dendrimer based drug delivery systems selectively carry and accumulate drugs inside tumors because of the leaky vasculature of tumor tissues in a method known to be the enhanced permeability and retention (EPR) effect.2–5 Icosahedral carboranes possess remarkable anticancer properties. Incorporation of multiple carborane clusters into the dendritic platform makes it boron-rich for effective cancer treatment.6,7 Dendritic molecules containing multiple o-carborane clusters showed higher cytotoxicity towards cancer cells than the commonly used anticancer agent cisplatin.8 Glycoconjugates of carboranes have been synthesized for selective boron delivery to cancer cells. Hydrophilic carrier compounds such as sugar moieties compensate for the hydrophobicity of carborane clusters and enhance their selectivity towards tumor cells.9–11 Carbohydrate–protein interactions facilitate many biological processes and cellular adhesion at the molecular level. Due to the overexpression of lectins in tumor tissues polyglycosylated derivatives selectively target cancer cells.12 Glycodendrimers have been used as selective delivery agents for cancer cells.13 We reported dendritic glycoconjugates containing one o-carborane moiety and evaluated anticancer efficacy in vitro against two cancer cell lines (MCF-7 and A-431) and one normal skin epidermal cell line (HaCaT). The results of the study revealed that the synthesized compounds were less toxic towards HaCaT normal cell lines while more cytotoxic towards cancer cells. However, they showed lower anticancer activity when more sugar moieties were added to enhance water solubility.14 These findings inspired us to synthesize dendritic molecules containing multiple o-carborane clusters with peripheral hydrophilic carbohydrate moieties. Herein, we report the preparation of a series of dendritic carboranyl glycoconjugates containing multiple o-carbore clusters in the core. The core has been appended to three, six and nine peripheral glucose moieties using Cu(I) catalyzed azide–alkyne click cycloaddition reactions.7,8,14 The anticancer efficacy of the synthesized glycoconjugates has been investigated against MCF-7 breast carcinoma cells through in vitro cytotoxicity assay. Caspase-3 activity assay was carried out to explore the mode of cell death i.e. necrosis or apoptosis.
Experimental
General methods
All the chemicals used in this work were purchased from Sigma-Aldrich/Merck India Pvt. Ltd/Spectrochem India and were used without any further purification. 3-(4,5-Dimethylthiazol-2-yl)-2,5 di-phenyl-tetrazolium bromide (MTT), Dulbecco's Modified Eagle's Medium (DMEM), fetal bovine serum (FBS), antibiotic solution (penicillin–streptomycin), caspase assay buffer and caspase-3 colorimetric substrate were purchased from Sigma Aldrich and 96 wells plates were purchased from Tarsons, India. All experiments were carried out in oven-dried glassware under dry argon/nitrogen. Solvents were distilled under nitrogen from an appropriate drying agent. Reagents were purchased and were used without further purification. All compounds were purified by column chromatography on silica gel/activated neutral alumina (60–120 mesh, Spectrochem, India). The yield of the products refers to analytically pure samples. 1H and 13C NMR spectra were recorded on a Fourier Transform multinuclear NMR spectrometer at 400 MHz and 100 MHz respectively. Chemical shifts were reported relative to TMS (1H: δ = 0.00 ppm), CDCl3 (13C: δ = 77.0 ppm) and coupling constants are given in Hz. All 13C spectra are proton-decoupled. 11B NMR spectra are proton-decoupled and were recorded at 128 MHz relative to BF3·Et2O. Accurate mass measurements were performed for compounds using high resolution instruments (in the positive or negative mode) and a MALDI-TOF mass spectrometer.
Cell culture
The MCF-7 cell line was obtained from the National Center for Cell Science, Pune, India, and was maintained in Dulbecco's minimal essential medium containing 10% fetal bovine serum, 1% L-glutamine, and 1% penicillin–streptomycin at 37 °C in a humidified incubator with 5% CO2/95% air. MCF-7 cells at a concentration of 20 × 103 cells in 200 μL per well were seeded into 96-well culture plates and kept for 24 h. Then compounds 7, 16, 18 and 20 were added separately and kept for 48 h. After the incubation period, cell survival was monitored using MTT assay. The concentrations used were calibrated after a pilot experiment taking the test compounds on a log scale (1–1000 ng mL−1).
Assay of cytotoxicity
After incubation for 48 h, the media were removed carefully from all wells, and the cells were washed in phosphate buffer saline (PBS) two times and used for MTT assay. Then, 100 μL of 10% (v/v) MTT solution (5 mg mL−1 in PBS) in medium was added. After incubation for 3 h, the MTT solution was replaced with 100 μL of 20% (w/v) sodium dodecyl sulfate solution and 0.6% (v/v) of 37% HCl in dimethyl sulfoxide, which acted as cell lysis buffer, breaking the cells and allowing the purple crystals to be released into the solution. The 96-well plates were then shaken in a multi-microplate shaker to dissolve the MTT crystals, resulting in a homogeneous purple solution and the absorbance was measured at 570 nm using a plate reader. The darker the solution, the higher is the number of viable and metabolically active cells. The percentage of viable cells is calculated using the following formula:
| % Viable cell = (abssample − absblank/abscontrol − absblank) × 100 |
Measurement of apoptosis from caspase-3 activity
Caspase-3, which is an effector caspase in apoptosis, is the most studied mammalian caspase. It plays a central role in nuclear apoptosis which leads to cell death. The caspase-3 colorimetric assay is based on the hydrolysis of the peptide substrate acetyl-Asp-Glu-Val-Asp p-nitroanilide by caspase-3, resulting in the release of the p-nitroaniline (pNA) moiety, the concentration of which is calculated from the absorbance value at 405 nm. The caspase-3 activity was determined as per the reported procedure.15 After replacing media with caspase assay buffer (50 mM HEPES, pH 7.4, 100 mM NaCl, 0.1% CHAPS, 1 mM EDTA, 10% glycerol and 10 mM DTT), cell lysates were incubated with a caspase-3 colorimetric substrate acetyl-Asp-Glu-Val-Asp-p-nitroanilide-7-amino-4-methylcoumarin. Reaction mixtures were incubated at 37 °C for 15 min to 1 hour, and the absorbance value was measured at 405 nm using an ELISA microplate reader.
Live dead assay
To observe the morphological changes incurred upon MCF-7 cells after treatment with the compounds 7, 16, 18, and 20, the cells were seeded in culture dishes for 48 h at 37 °C. The cells were then stained with Hoechst 33342 and PI solution at room temperature for 10 min in the dark. The cellular morphology was observed under an inverted fluorescence microscope.
Statistical analysis
All statistical analyses were performed using the SPSS Version 20.0 for Windows (significance was established at P < 0.05). Data have been expressed as mean ± SD and statistical significance was determined by one-way analysis of variance (ANOVA) combined with Duncan's multiple range tests. All experiments were performed in triplicate, unless otherwise indicated.
Preparation and analytical data of compounds
Compound 3.
To a solution of compound 1 (1.60 g, 12.11 mmol) in 30 mL of Et3N and 10 mL of acetonitrile, p-bromo acetophenone (2) (1.68 g, 8.48 mmol) was added under an argon atmosphere. To the reaction mixture, Pd(PPh3)2Cl2 (85 mg, 0.12 mmol) was added followed by CuI (46 mg, 0.24 mmol) and refluxed at 90 °C for 8 hours. The progress of the reaction was monitored by TLC. After completion of the reaction, the reaction mixture was cooled to room temperature and filtered using short silica pads and the solvent was removed under reduced pressure. The crude product was purified by silica gel chromatography using 10% ethyl acetate in hexane. Pure product: 1.30 g. Yield: 61%. Colourless solid. MP: 137 °C. 1H NMR (400 MHz, CDCl3, δ ppm): 7.91 (d, 2H, J = 8 Hz, Ar-H), 7.57 (d, 2H, J = 8 Hz, Ar-H), 7.48 (d, 2H, J = 8 Hz, Ar-H), 6.88 (d, 2H, J = 8 Hz, Ar-H), 3.82 (s, 3H, OCH3-H), 2.59 (s, 3H, COCH3-H); 13C {1H} NMR (100 MHz, CDCl3, δ ppm): 197.4 (carbonyl-C), 160.1 (Ar-C), 135.9 (Ar-C), 133.3 (Ar-C), 131.5 (Ar-C), 128.6 (Ar-C), 128.3 (Ar-C), 114.7 (Ar-C), 114.2 (Ar-C), 93.0 (alkyne-C), 87.6 (alkyne-C), 55.4 (OCH3-C), 26.6 (COCH3-C); IR (KBr): 2954, 2033, 1672, 1509, 1274, 1078 cm−1; ESI-MS: calcd for C17H14O2: 250.29; found: 251.10 [M + H]+.
Compound 4.
Decaborane (878 mg, 7.19 mmol, 1.2 equiv.) was refluxed at 95 °C in 5 mL of dry acetonitrile for 2 hours under an argon atmosphere. To this, compound 3 (1.5 g, 5.99 mmol, 1 equiv.) dissolved in 5 mL of dry toluene was added at room temperature and then refluxed at 95 °C for 17 hours. The progress of the reaction was monitored by TLC. Excess decaborane was quenched using 2 mL of methanol and then the resulting mixture was concentrated. The crude product obtained was purified by silica gel chromatography using 25% ethyl acetate in hexane. Pure product: 800 mg. Yield: 36%. Colourless solid. MP: 123 °C. 1H NMR (400 MHz, CDCl3, δ ppm): 7.39 (d, 2H, J = 8 Hz, Ar-H), 7.33 (d, 2H, J = 8 Hz, Ar-H), 7.13 (d, 2H, J = 8 Hz, Ar-H), 6.61 (d, 2H, J = 8 Hz, Ar-H), 4.77 (s, 1H, OH-H), 3.69 (s, 3H, OCH3-H), 1.91 (br s, 1H, tertiary CH-H), 1.38 (s, 3H, CH3-H); 13C {1H} NMR (100 MHz, CDCl3, δ ppm): 160.8 (Ar-C), 147.8 (Ar-C), 132.1 (Ar-C), 130.9 (Ar-C), 129.9 (Ar-C), 125.3 (Ar-C), 123.0 (Ar-C), 113.6 (Ar-C), 85.8 (cage-C), 85.3 (cage-C), 69.5 (tertiary CH-C), 55.3 (OCH3-C), 25.2 (CH3-C); 11B {1H} NMR: −2.82, −9.14, −10.82 ppm; IR (KBr): 3445, 3029, 2945, 2592 (B–H), 1608, 1516, 1274, 1078 cm−1; ESI-MS: calcd for C17H26B10O2: 370.49; found: 370.48 [M]+.
Compound 5.
Compound 4 (800 mg, 2.16 mmol) was added to a solution of PCC (1.39 g, 6.48 mmol) in 30 mL of dry dichloromethane at 0 °C and stirred at room temperature for 2 hours. The progress of the reaction was monitored by TLC. After completion of the reaction, the reaction mixture was filtered over a short silica pad and concentrated. The crude product was purified by silica gel chromatography using 8% ethyl acetate in hexane. Pure product: 500 mg. Yield: 83%. Colourless solid. MP: 137 °C. 1H NMR (400 MHz, CDCl3, δ ppm): 7.63 (d, 2H, J = 12 Hz, Ar-H), 7.45 (d, 2H, J = 8 Hz, Ar-H), 7.26 (d, 2H, J = 8 Hz, Ar-H), 6.55 (d, 2H, J = 12 Hz, Ar-H), 3.61 (s, 3H, OCH3-H), 2.43 (s, 3H, COCH3-H); 13C {1H} NMR (100 MHz, CDCl3, δ ppm): 197.1 (Carbonyl-C), 161.1 (Ar-C), 137.9 (Ar-C), 135.2 (Ar-C), 132.1 (Ar-C), 131.0 (Ar-C), 128.1 (Ar-C), 122.6 (Ar-C), 113.8 (Ar-C), 86.0 (cage-C), 84.1 (cage-C), 55.3 (OCH3-C), 26.7 (CH3-C); 11B {1H} NMR: −3.28, −10.70, −11.82 ppm; IR (KBr): 3034, 2933, 2594 (B–H), 1656, 1523, 1259 cm−1; ESI-MS: calcd for C17H24B10O2; 368.48 found: 369.47 [M + H]+.
Compound 6.
To a solution of compound 5 (450 mg, 1.22 mmol) in a mixed solvent (18 mL ethanol and 6 mL toluene) system at 0 °C, 3 mL of SiCl4 was added carefully and stirred at room temperature for 15 hours. The progress of the reaction was monitored by TLC. Upon completion of reaction, it was quenched carefully by adding distilled water; the product was extracted using dichloromethane and concentrated to get the crude product. The crude product was washed carefully with cold dichloromethane to get the pure product. Column chromatography was not required. Pure product: 350 mg. Yield: 27%. Colourless solid. MP: >250 °C. 1H NMR (400 MHz, CDCl3, δ ppm): 7.52 (s, 3H, Ar-H), 7.50 (d, 6H, J = 8 Hz, Ar-H), 7.39 (d, 6H, J = 4 Hz, Ar-H), 7.37 (d, 6H, J = 4 Hz, Ar-H), 6.63 (d, 6H, J = 8 Hz, Ar-H), 3.69 (s, 9H, OCH3-H); 13C NMR {1H} (100 MHz, CDCl3, δ ppm): 160.9 (Ar-C), 142.0 (Ar-C), 140.8 (Ar-C), 132.2 (Ar-C), 131.2 (Ar-C), 130.5 (Ar-C), 127.0 (Ar-C), 125.5 (Ar-C), 123.1 (Ar-C), 113.6 (Ar-C), 85.9 (cage-C), 85.0 (cage-C), 55.3 (OCH3-C); 11B {1H} NMR: −3.24, −4.21, −10.93, −11.75 ppm; IR (KBr): 3036, 2942, 2589 (B–H), 1612, 1523, 1246 cm−1; ESI-MS: calc. for C51H66B30O3: 1056.78; found: 1057.92 [M + H]+.
Compound 7.
BBr3 (2.8 mL, 2.8 mmol, 1 M in CH2Cl2, 10.5 equiv.) was added dropwise to a solution of compound 6 (280 mg, 0.266 mmol, 1 equiv.) in 30 mL of CH2Cl2 at 0 °C and stirred at room temperature for 20 hours. The progress of the reaction was monitored by TLC. After completion, the reaction mixture was quenched with distilled water (20 mL) at 0 °C. The excess CH2Cl2 was evaporated using a rotary evaporator. The crude product was then extracted using ethyl acetate (3 × 20 mL), and combined organic layers were dried over anhydrous sodium sulfate and filtered. Removal of the solvent afforded the crude product, which was purified carefully by washing with cold dichloromethane. Pure product: 240 mg. Yield: 89%. Colourless solid. MP: >250 °C. 1H NMR (400 MHz, CD3OD, δ ppm): 7.45 (d, 6H, J = 8 Hz, Ar-H), 7.35 (d, 6H, J = 8 Hz, Ar-H), 7.11 (s, 3H, Ar-H), 7.06 (d, 6H, J = 8 Hz, Ar-H), 6.50 (d, 6H, J = 8 Hz, Ar-H); 13C {1H} NMR (100 MHz, CD3OD, δ ppm): 159.4 (Ar-C), 141.7 (Ar-C), 139.3 (Ar-C), 132.2 (Ar-C), 131.1 (Ar-C), 130.2 (Ar-C), 126.6 (Ar-C), 124.5 (Ar-C), 121.4 (Ar-C), 115.0 (Ar-C), 86.8 (cage-C), 85.4 (cage-C); 11B {1H} NMR: −3.24, −4.21, −10.92, −11.75 ppm; IR (KBr): 3415, 3024, 2936, 2587 (B–H), 1607, 1518, 1256 cm−1; ESI-MS: calcd for C48H60B30O3: 1009.31; found: 1010.77 [M + H]+.
Compound 11.
To a solution of compound 7 (140 mg, 0.138 mmol, 1 equiv.) and compound 8 (113 mg, 0.496 mmol, 3.6 equiv.) in 30 mL of dry acetone under an argon atmosphere, K2CO3 (171 mg, 1.242 mmol, 9 equiv.) was added and refluxed at 70 °C for 17 hours. The progress of the reaction was monitored by TLC. After completion of the reaction, the reaction mixture was filtered through a short silica pad and concentrated. The crude product was purified by silica gel chromatography using 25% ethyl acetate in hexane. Pure product: 145 mg. Yield: 73%. Colourless solid. MP: 151 °C. 1H NMR (400 MHz, CDCl3, δ ppm): 7.55 (s, 3H, Ar-H), 7.50 (d, 6H, J = 8 Hz, Ar-H), 7.40–7.36 (m, 12H, Ar-H), 7.23 (d, 6H, J = 8 Hz, Ar-H), 6.91 (d, 6H, J = 8 Hz, Ar-H), 6.69 (d, 6H, J = 8 Hz, Ar-H), 4.84 (s, 6H, OCH2-H), 4.62 (s, 6H, OCH2-H), 2.16 (s, 3H, alkyne-H); 13C {1H} NMR (100 MHz, CDCl3, δ ppm): 160.1 (Ar-C), 157.6 (Ar-C), 142.0 (Ar-C), 140.9 (Ar-C), 132.2 (Ar-C), 131.3 (Ar-C), 130.5 (Ar-C), 129.2 (Ar-C), 129.0 (Ar-C), 127.0 (Ar-C), 125.5 (Ar-C), 123.2 (Ar-C), 115.1 (Ar-C), 114.5 (Ar-C), 85.9 (cage-C), 85.0 (cage-C), 78.4 (alkyne-C), 75.7 (alkyne-C), 69.8 (OCH2-C), 55.8 (OCH2-C); 11B {1H} NMR: −3.84, −10.74 ppm; IR (KBr): 3207, 3026, 2924, 2591 (B–H), 2401, 2125, 1612, 1514, 1225 cm−1; ESI-MS: calcd for C78H84B30O6: 1441.82; found: 1442.02 [M]+.
Compound 12.
To a solution of compound 7 (170 mg, 0.168 mmol, 1 equiv.) and compound 9 (169 mg, 0.607 mmol, 3.6 equiv.) in 30 mL of dry acetone under an argon atmosphere, K2CO3 (209 mg, 1.51 mmol, 9 equiv.) was added and refluxed at 70 °C for 17 hours. The progress of the reaction was monitored by TLC. After completion of reaction, the reaction mixture was filtered through a short silica pad and concentrated. The crude product was purified by silica gel chromatography using 25% ethyl acetate in hexane. Pure product: 175 mg. Yield: 64%. Colourless solid. MP: 121 °C. 1H NMR (400 MHz, CDCl3, δ ppm): 7.55 (s, 3H, Ar-H), 7.50 (d, 6H, J = 8 Hz, Ar-H), 7.40–7.36 (m, 12H, Ar-H), 6.68 (d, 6H, J = 8 Hz, Ar-H), 6.56–6.53 (m, 6H, Ar-H), 6.50–6.47 (m, 3H, Ar-H), 4.86 (s, 6H, OCH2-H), 4.58–4.56 (m, 12H, OCH2-H), 2.42–2.40 (m, 6H, alkyne-H); 13C {1H} NMR (100 MHz, CDCl3, δ ppm): 159.9 (Ar-C), 158.9 (Ar-C), 142.0 (Ar-C), 140.8 (Ar-C), 138.7 (Ar-C), 132.2 (Ar-C), 131.3 (Ar-C), 130.5 (Ar-C), 127.0 (Ar-C), 125.5 (Ar-C), 123.4 (Ar-C), 114.6 (Ar-C), 101.7 (Ar-C), 85.8 (cage-C), 85.0 (cage-C), 78.2 (alkyne-C), 75.9 (alkyne-C), 69.6 (OCH2-C), 55.9 (OCH2-C); 11B {1H} NMR: −2.94, −10.75 ppm; IR (KBr): 3203, 3026, 2924, 2593 (B–H), 2402, 2123, 1606, 1514, 1218 cm−1; ESI-MS: calcd for C87H90B30O9: 1606.52; found: 1606.89 [M]+.
Compound 13.
To a solution of compound 7 (100 mg, 0.099 mmol, 1 equiv.) and compound 10 (118 mg, 0.357 mmol, 3.6 equiv.) in 30 mL of dry acetone under an argon atmosphere, K2CO3 (123 mg, 0.891 mmol, 9 equiv.) was added and refluxed at 70 °C for 17 hours. The progress of the reaction was monitored by TLC. After completion of reaction, the reaction mixture was filtered through a short silica pad and concentrated. The crude product was purified by silica gel chromatography using 30% ethyl acetate in hexane. Pure product: 120 mg. Yield: 68%. Colourless solid. MP: 115 °C. 1H NMR (400 MHz, CDCl3, δ ppm): 7.56 (s, 3H, Ar-H), 7.52 (d, 6H, J = 8 Hz, Ar-H), 7.42–7.38 (m, 12H, Ar-H), 6.71 (d, 12H, J = 8 Hz, Ar-H), 4.87 (s, 6H, OCH2-H), 4.67–4.65 (m, 18H, OCH2-H), 2.42 (t, J = 4 MHz, 3H, alkyne-H), 2.33 (t, J = 4 Hz, 6H, alkyne-H); 13C {1H} NMR (100 MHz, CDCl3, δ ppm): 159.9 (Ar-C), 151.8 (Ar-C), 142.0 (Ar-C), 140.8 (Ar-C), 137.0 (Ar-C), 132.29 (Ar-C), 132.26 (Ar-C), 131.3 (Ar-C), 130.6 (Ar-C), 127.0 (Ar-C), 125.5 (Ar-C), 123.5 (Ar-C), 114.6 (Ar-C), 107.7 (Ar-C), 107.0 (Ar-C), 85.8 (cage-C), 85.0 (cage-C), 79.1 (alkyne-C), 78.3 (alkyne-C), 76.1 (alkyne-C), 75.4 (alkyne-C), 69.8 (OCH2-C), 60.4 (OCH2-C), 57.1 (OCH2-C); 11B {1H} NMR: −3.67, −10.78 ppm; IR (KBr): 3201, 3018, 2931, 2592 (B–H), 2402, 2126, 1610, 1516, 1217 cm−1; ESI-MS: calcd for C87H90B30O9: 1770.96; found: 1770.80 [M]+.
Compound 15.
To a solution of alkynyl core 11 (150 mg, 0.104 mmol, 1 equiv.) and glucosyl azide 14 (159 mg, 0.374 mmol, 3.6 equiv.) in 5 mL of dry THF, 3 mL of distilled water was added. To this suspension, a solution of sodium ascorbate (19 mg, 0.093 mmol, 0.9 equiv.) and CuSO4·5H2O (24 mg, 0.093 mmol, 0.9 equiv.) in 2 mL of water was added and the reaction mixture was stirred at room temperature for 24 hours. After completion of reaction, the crude product was extracted using ethyl acetate (3 × 20 mL), dried over sodium sulfate and concentrated. The crude product was purified by neutral alumina column chromatography using 2% methanol in ethyl acetate. Pure product: 192 mg. Yield: 68%. Colourless solid. MP: 95 °C. 1H NMR (400 MHz, CDCl3, δ ppm): 7.67 (s, 3H, triazole-H), 7.55 (s, 3H, Ar-H), 7.50 (d, 6H, J = 8 Hz, Ar-H), 7.40–7.36 (t, 12H, J = 8 Hz, Ar-H), 7.24 (d, 6H, J = 8 Hz, Ar-H), 6.95 (d, 6H, J = 8 Hz, Ar-H), 6.69 (d, 6H, J = 8 Hz, Ar-H), 5.19–5.13 (3H (Glu-H); 6H (OCH2-H)), 5.07–4.95 (m, 6H, OCH2-H), 4.83 (s, 3H, OCH2-H), 4.62–4.57 (m, 3H, OCH2-H), 4.52–4.43 (m, 6H, OCH2-H), 4.24–4.20 (m, 6H, OCH2-H), 4.13–4.06 (m, 6H, Glu-H), 3.93–3.87 (m, 3H, Glu-H), 3.69–3.65 (m, 3H, Glu-H), 2.06–1.92 (4s, 36H, 12CH3); 13C {1H} NMR (100 MHz, CDCl3, δ ppm): 170.6, 170.2, 169.5, 169.4 (4 × OAc-C), 160.2 (Ar-C), 158.3 (Ar-C), 143.7 (Ar-C), 142.0 (Ar-C), 140.9 (Ar-C), 132.4 (Ar-C), 132.2 (Ar-C), 131.3 (Ar-C), 131.0 (Ar-C), 130.5 (Ar-C), 129.3 (Ar-C), 128.9 (Ar-C), 128.6 (Ar-C), 127.0 (Ar-C), 125.5 (Ar-C), 124.3 (Ar-C), 123.2 (Ar-C), 114.9 (Ar-C), 114.5 (Ar-C), 100.5 (Glu-C), 85.9 (cage-C), 85.0 (cage-C), 72.5 (OCH2-C), 72.0 (Glu-C), 71.8 (Glu-C), 71.0 (OCH2-C), 69.8 (Glu-C), 68.2 (Glu-C), 67.8 (Glu-C), 62.9 (CH2-C), 61.8 (glucose-C), 50.1 (CH2-C), 27.8, 20.8, 20.6, 19.2 (4 × CH3-C); 11B {1H} NMR: −2.92, −10.78 ppm; IR (KBr): 3029, 2924, 2589 (B–H), 1751 (ester-CO), 1660, 1593, 1512, 1228, 1047 cm−1; ESI-MS: calcd for C126H155B30N9O36Na: 2723.32; found: 2723.76 [M + Na]+.
Compound 16.
To the acetylated derivative 15 (100 mg, 0.037 mmol, 1 equiv.) dissolved in 10 mL of dry methanol, a solution of sodium methoxide (2 mg, 0.009 mmol, 1 equiv.) in 2 mL of dry methanol was added and stirred at room temperature for 45 minutes. After completion of the reaction monitored by TLC, the reaction mixture was neutralized by the addition of Amberlite IR-120 (H+-form) ion exchange resin (pre-washed with methanol), filtered, and concentrated under reduced pressure to afford the desired hydroxylated product 16. Pure product: 72 mg. Yield: 88%. Colourless solid. MP: 111 °C. 1H NMR (400 MHz, DMSO-d6, δ ppm): 8.22 (s, 3H, triazole-H), 7.74 (s, 3H, Ar-H), 7.70–7.67 (m, 6H, Ar-H), 7.57 (d, 6H, J = 8 Hz, Ar-H), 7.46 (d, 6H, J = 8 Hz, Ar-H), 7.23 (d, 6H, J = 8 Hz, Ar-H), 6.94 (d, 6H, J = 8 Hz, Ar-H), 6.81 (d, 6H, J = 8 Hz, Ar-H), 5.03 (s, 9H, OCH2-H), 4.93 (dd, 6H, J = 8 Hz, J = 8 Hz, OCH2-H), 4.84 (s, 6H, OCH2-H), 4.55–4.50 (m, 9H, OCH2-H, Glu-H), 4.19 (d, J = 8 Hz, 3H, Glu-H), 4.07–4.02 (m, 3H, Glu-H), 3.97 (d, J = 12 Hz, 6H, Glu-H), 3.89–3.84 (m, 3H, Glu-H), 3.66–3.62 (m, 3H, Glu-H), 3.11–2.90 (m, 12H, Glu-H); 13C {1H} NMR (100 MHz, DMSO-d6, δ ppm): 160.5 (Ar-C), 158.4 (Ar-C), 142.8 (Ar-C), 141.9 (Ar-C), 140.2 (Ar-C), 132.8 (Ar-C), 132.2 (Ar-C), 132.1 (Ar-C), 131.7 (Ar-C), 130.2 (Ar-C), 129.9 (Ar-C), 129.2 (Ar-C), 128.9 (Ar-C), 125.9 (Ar-C), 115.2 (Ar-C), 115.0 (Ar-C), 103.4 (Ar-C), 87.1 (cage-C), 86.3 (cage-C), 77.5 (CH2-C), 77.1 (CH2-C), 73.8 (OCH2-C), 71.6 (OCH2-C), 70.4 (OCH2-C), 69.7 (Glu-C), 68.3 (Glu-C), 67.8 (Glu-C), 61.6 (Glu-C), 61.5 (Glu-C), 50.2 (CH2-C); 11B {1H} NMR: −5.52, −13.13 ppm; IR (KBr): 3405, 2929, 2857, 2592 (B–H), 1627, 1607, 1512, 1387, 1247, 1065 cm−1; ESI-MS: calcd for C102H129B30N9O2 + Na: 2217.19; found: 2218.09 [M + H + Na]+.
Compound 17.
To a solution of alkynyl core 12 (200 mg, 0.128 mmol, 1 equiv.) and glucosyl azide 14 (385 mg, 0.923 mmol, 7.2 equiv.) in 5 mL of dry THF, 3 mL of distilled water was added. To this suspension, sodium ascorbate (45 mg, 0.23 mmol, 1.8 equiv.) and CuSO4·5H2O (57 mg, 0.23 mmol, 1.8 equiv.) in 2 mL of water were added and stirred at room temperature for 24 hours. After completion of the reaction, the crude product was extracted using ethyl acetate (3 × 20 mL), dried over sodium sulfate and concentrated. The crude product was purified by neutral alumina column chromatography using 5% methanol in ethyl acetate. Pure product: 320 mg. Yield: 60%. Colourless solid. MP: 95 °C. 1H NMR (400 MHz, CDCl3, δ ppm): 7.71–7.66 (m, 6H, triazole-H), 7.54–7.48 (m, 12H, Ar-H), 7.41–7.35 (m, 12H, Ar-H), 6.69 (d, 6H, J = 8 Hz, Ar-H), 6.57 (dd, 6H, J = 4 Hz, Ar-H), 5.17–4.94 (m, 30H, OCH2-H), 4.84 (s, 6H, OCH2-H), 4.61–4.55 (m, 6H, OCH2-H), 4.50–4.45 (m, 6H, Glu-H), 4.24–4.19 (m, 12H, Glu-H), 4.12–4.05 (m, 12H, Glu-CH2), 3.93–3.87 (m, 6H, Glu-H), 7.70–3.65 (m, 6H, Glu-H), 2.04, 1.99, 1.96, 1.90 (4s, 72H); 13C {1H} NMR (100 MHz, CDCl3, δ ppm): 170.6, 170.1, 169.5, 169.4 (4 × OAc-C), 159.9 (Ar-C), 159.6 (Ar-C), 143.5 (Ar-C), 142.0 (Ar-C), 140.8 (Ar-C), 138.8 (Ar-C), 132.4 (Ar-C), 131.0 (Ar-C), 128.9 (Ar-C), 127.1 (Ar-C), 124.4 (Ar-C), 123.3 (Ar-C), 114.6 (Ar-C), 106.5 (Ar-C), 101.5 (Ar-C), 100.5 (Glu-C), 85.9 (cage-C), 85.1 (cage-C), 72.5 (OCH2-C), 72.0 (Glu-C), 71.8 (Glu-C), 71.0 (OCH2-C), 69.7 (Glu-C), 68.2 (Glu-C), 67.7 (Glu-C), 61.9 (CH2-C), 61.8, 50.1 (CH2-C), 27.7, 20.8, 20.6, 19.24 (4 × CH3-C); 11B {1H} NMR: −4.53, −10.77 ppm; IR (KBr): 3026, 2924, 2584 (B–H), 1757 (ester-CO), 1665, 1597, 1509, 1223, 1037 cm−1; ESI-MS: calcd for C183H288B30N18O69 + 2H2O: 4144.17; found: 4144.12 [M + 2H2O]+.
Compound 18.
To the acetylated derivative 17 (200 mg, 0.048 mmol, 1 equiv.) dissolved in 5 mL of dry methanol, a solution of sodium methoxide (5 mg, 0.097 mmol, 2 equiv.) in 2 mL of dry methanol was added and the reaction mixture was stirred at room temperature for 45 minutes. After completion of the reaction, the reaction mixture was neutralized by the addition of Amberlite IR-120 (H+-form) ion exchange resin (pre-washed with methanol), filtered, and concentrated under reduced pressure to afford the desired hydroxylated product 18. Pure product: 160 mg. Yield: 60%. Colourless solid. MP: 105 °C. 1H NMR (400 MHz, DMSO-d6, δ ppm): 8.21 (s, 6H, triazole-H), 7.76 (s, 6H, Ar-H), 7.71–7.67 (m, 12H, Ar-H), 7.65–7.46 (m, 6H, Ar-H), 6.83 (d, 3H, J = 8 Hz, Ar-H), 6.61 (d, 9H, J = 8 Hz, Ar-H), 5.02 (s, 18H, OCH2), 4.89 (s, 3H, OCH2), 4.53 (t, J = 4 Hz, 18H, OCH2), 4.21 (d, 12H, Glu-H), 4.07–4.03 (m, 9H, Glu-H), 3.97 (d, J = 12 Hz, 12H, Glu-H), 3.89–3.84 (m, 12H, Glu-H), 3.11–2.97 (m, 24H, Glu-H); 13C {1H} NMR (100 MHz, DMSO-d6, δ ppm): 160.3 (Ar-C), 159.7 (Ar-C), 144.7 (Ar-C), 142.7 (Ar-C), 141.9 (Ar-C), 139.2 (Ar-C), 132.2 (Ar-C), 132.1 (Ar-C), 129.9 (Ar-C), 129.2 (Ar-C), 125.9 (Ar-C), 122.7 (Ar-C), 115.3 (Ar-C), 107.1 (Ar-C), 103.5 (Ar-C), 103.4 (Ar-C), 86.3 (cage-C), 77.4 (CH2-C), 77.0 (CH2-C), 73.8 (OCH2-C), 71.6 (OCH2-C), 70.6 (OCH2-C), 67.9 (Glu-C), 67.8 (Glu-C), 61.6 (Glu-C), 61.5 (Glu-C), 50.2 (CH2-C); 11B {1H} NMR: −5.63, −13.24 ppm; IR (KBr): 3417, 2937, 2842, 2593 (B–H), 1621, 1602, 1509 1379, 1244, 1045 cm−1; ESI-MS: calcd for C135H180B30N18O45: 3103.51; found: 3157.54 [M + 3H2O]+.
Compound 19.
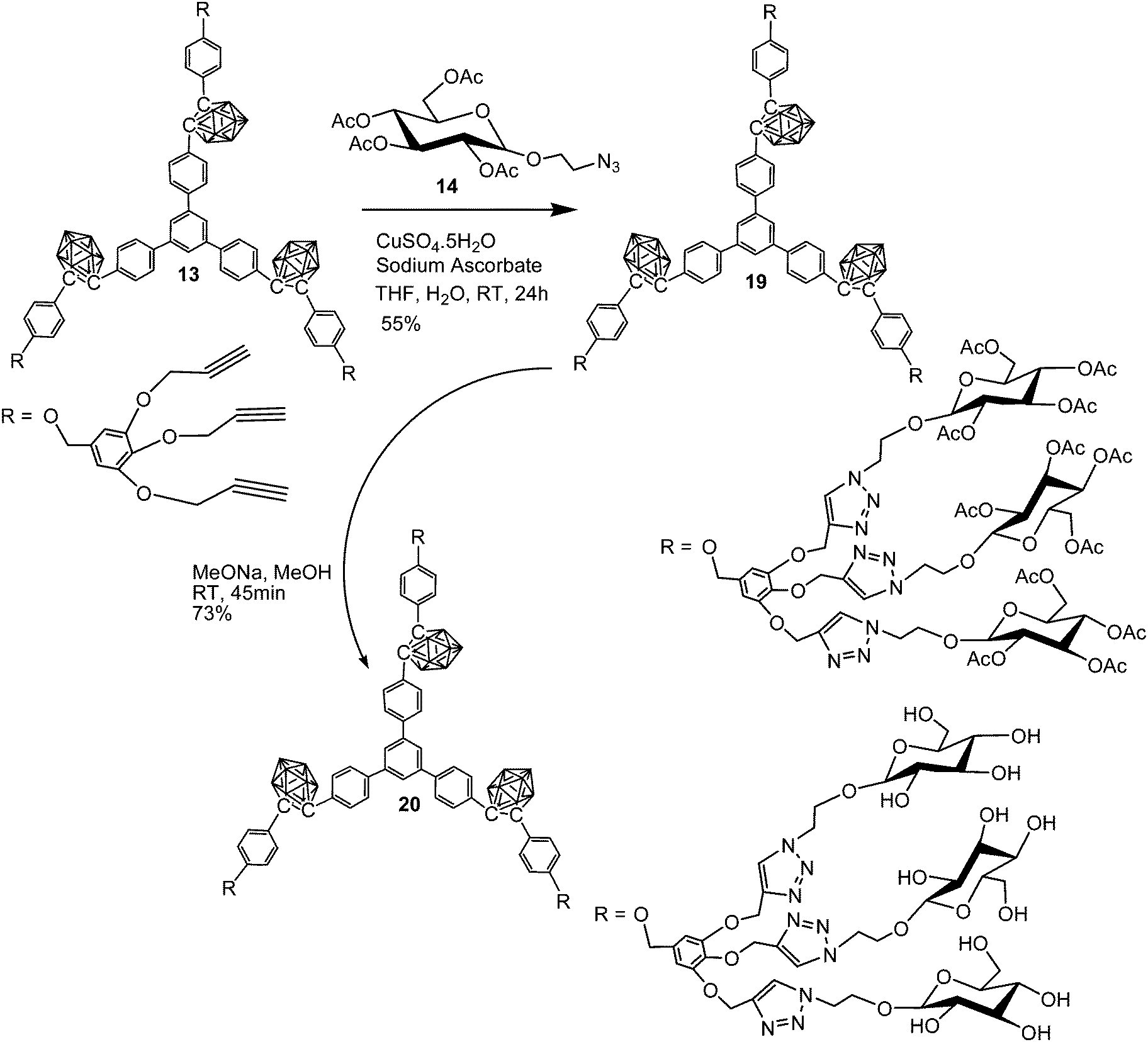
To a solution of alkynyl core 13 (200 mg, 0.113 mmol, 1 equiv.) and glucosyl azide 14 (548 mg, 1.315 mmol, 11.6 equiv.) in 5 mL of dry THF, 3 mL of distilled water was added. To this suspension, sodium ascorbate (60 mg, 0.306 mmol, 2.7 equiv.) and CuSO4·5H2O (76 mg, 0.306 mmol, 2.7 equiv.) in 2 mL of water were added and stirred at room temperature for 24 hours. After completion of the reaction monitored by TLC, the crude product was extracted using ethyl acetate (3 × 20 mL), dried over sodium sulfate and concentrated. The crude product was purified by neutral alumina column chromatography using 6% methanol in ethyl acetate. Pure product: 345 mg. Yield: 55%. Colourless solid. MP: 94 °C. 1H NMR (400 MHz, CDCl3, δ ppm): 7.87–7.70 (m, 9H, triazole-H), 7.57 (s, 3H, Ar-H), 7.53–7.50 (m, 6H, Ar-H), 7.43, 7.41, 7.39 (t, J = 8 Hz, 12H, Ar-H), 6.74 (t, J = 8 Hz, 12H, Ar-H), 5.17–5.01 (m, 36H, OCH2-H), 4.97–4.92 (m, 9H, OCH2-H), 4.85 (s, 6H, OCH2-H), 4.61–4.51 (m, 24H, OCH2-H, Glu-H), 4.26–4.21 (m, 18H, Glu-H), 4.12–4.06 (m, 12H, Glu-H), 4.01–3.91 (m, 9H, Gluc-H), 3.72–3.68 (m, 9H, Gluc-H), 2.05, 1.99, 1.95, 1.88 (4s, 108H); 13C {1H} NMR (100 MHz, CDCl3, δ ppm): 170.6, 170.1, 169.5, 167.7 (4 × OAc-C), 159.9 (Ar-C), 152.4 (Ar-C), 144.3 (Ar-C), 143.6 (Ar-C), 142.1 (Ar-C), 140.8 (Ar-C), 137.5 (Ar-C), 132.4 (Ar-C), 132.2 (Ar-C), 131.3 (Ar-C), 131.0 (Ar-C), 130.4 (Ar-C), 128.9 (Ar-C), 127.1 (Ar-C), 125.0 (Ar-C), 124.6 (Ar-C), 123.3 (Ar-C), 114.6 (Ar-C), 107.2 (Ar-C), 100.5 (Ar-C), 100.6 (Glu-C), 85.9 (cage-C), 85.2 (cage-C), 72.5 (OCH2-C), 71.8 (Glu-C), 70.9 (OCH2-C), 68.3 (Glu-C), 67.7 (Glu-C), 63.0 (CH2-C), 61.8 (Glu-C), 60.4 (Glu-C), 50.0 (CH2-C), 27.7, 20.8, 20.6, 19.2 (4 × CH3-C); 11B {1H} NMR: −5.34, −10.75 ppm; IR (KBr): 3021, 2928, 2586 (B–H), 1754 (ester-CO), 1659, 1591, 1512, 1207, 1021, cm−1; ESI-MS: calcd for C240H303B30N27O102: 5521.42; found: 5520.58 [M-H]+.
Compound 20.
To the acetylated derivative 19 (200 mg, 0.036 mmol, 1 equiv.) dissolved in 5 mL of dry methanol, a solution of sodium methoxide (6 mg, 0.018 mmol, 3 equiv.) in dry methanol was added and stirred at room temperature for 45 minutes. After completion of the reaction, the reaction mixture was neutralized by the addition of Amberlite IR-120 (H+-form) ion exchange resin (pre-washed with methanol), filtered, and concentrated under reduced pressure to afford the desired hydroxylated product 20 as white foam. Pure product: 106 mg. Yield: 73%. Colourless solid. MP: 105 °C. 1H NMR (400 MHz, D2O, δ ppm): 8.21–8.01 (m, 30H, triazole-H, Ar-H), 6.77 (br s, 12H, Ar-H), 4.78–4.63 (m, 54H, OCH2-H), 4.41 (s, 12H, Glu-H), 4.20 (s, 12H, Glu-H), 4.07 (s, 15H, Glu-H), 3.88 (s, 18H, Glu-H), 3.68 (d, 12H, J = 12 Hz, Glu-H), 3.58 (d, 24H, J = 8 Hz, Glu-H), 3.46 (s, 12H, Glu-H); 13C {1H} NMR (100 MHz, D2O, δ ppm): 160.2 (Ar-C), 151.7 (Ar-C), 143.3 (Ar-C), 142.7 (Ar-C), 138.0 (Ar-C), 135.8 (Ar-C), 132.7 (Ar-C), 132.3 (Ar-C), 128.0 (Ar-C), 125.3 (Ar-C), 125.1 (Ar-C), 120.5 (Ar-C), 110.1 (Ar-C), 107.4 (Ar-C), 102.4 (Ar-C), 87.4 (cage-C), 82.8 (cage-C), 75.9 (CH2-C), 75.6 (CH2-C), 73.0 (OCH2-C), 72.9 (OCH2-C), 69.5 (OCH2-C), 68.4 (Glu-C), 67.8 (Glu-C), 62.6 (Glu-C), 60.7 (Glu-C), 50.2 (CH2-C); 11B {1H} NMR: −6.23, −14.52 ppm; IR (KBr): 3425, 2954, 2834, 2589 (B–H), 1633, 1606, 1519, 1387, 1237, 1054 cm−1; ESI-MS: calcd for C168H231B30N27O66 + 2H2O: 4042.86; found: 4042.80 [M + 2H2O]+.
Results and discussion
The dendritic carboranyl glycosides (16, 18 and 20) were synthesized from compound 1 following a sequence of reaction conditions. The synthetic method includes Sonogashira cross-coupling reaction, decaborane insertion and oxidation with PCC followed by a cyclotrimerization reaction mediated by silicon tetrachloride to produce a symmetrical phenylene-carboranyl compound containing three o-carborane clusters. Finally, BBr3 promoted demethylation reaction yielded the symmetrical hydroxylated compound 7 (Scheme 1). The core compounds 11, 12 and 13 were prepared in good yield from nucleophilic substitution reaction of compound 7 with different alkyne dendrons in the presence of K2CO3 in dry acetone. These core compounds further underwent a Cu(I) catalyzed azide–alkyne [3+2] cycloaddition reaction with glucosyl azide followed by Zemplén deacetylation reaction to furnish the desired boron rich dendritic glycoconjugates 16, 18 and 20 in good to moderate yields.14,16–19
 |
| | Scheme 1 Synthesis of dendritic core 7 containing three o-carborane moieties. | |
Synthesis of the hydroxylated core 7
Synthesis of the compound 7 was initiated from the Sonogashira cross-coupling reaction as illustrated in Scheme 1. Compound 1 was prepared as per the reported procedures.14 The Sonogashira cross-coupling reaction of compound 1 with compound 2 (4-bromoacetophenone) produced the unsymmetrical alkyne 3 in 61% yield.16 The alkynyl compound 3 was refluxed with decaborane to form compound 4.17 The 1H and 13C NMR data indicated the reduction of the sensitive carbonyl group of compound 3 to secondary alcohol. Thus, a pyridinium chloro chromate (PCC) mediated oxidation reaction of compound 4 was necessary to obtain the required carbonyl compound 5. Subsequently silicon tetrachloride mediated cyclotrimerization reaction of compound 5 provided the symmetrical phenylene cored methoxy compound 6 containing three o-carborane clusters.18,19 Disappearance of the carbonyl peak in the 13C NMR spectrum along with other NMR and mass spectral evidences confirmed the formation of compound 6. The boron tribromide promoted demethylation of compound 6 led to the formation of the hydroxylated core 7 containing three hydroxyl groups and three o-carborane clusters in good yield.17 The disappearance of the peak for –OCH3-H at δ 3.69 ppm in 1H NMR and OCH3-C at δ 55.3 ppm in 13C NMR along with mass spectral evidence confirmed the formation of compound 7 (see ESI†).
The alkynyl dendrons 8, 9 and 10 used for the preparation of the alkynyl core compounds were synthesized from commercially available methyl-4-hydroxy benzoate, methyl-3,5-dihydroxy benzoate and methyl-3,4,5-trihydroxy benzoate respectively in accordance with the reported procedure (Scheme 2).20 The dendritic alkynyl core compounds 11, 12 and 13 were prepared from the hydroxylated compound 7 in quantitative yield as illustrated in Scheme 2. The hydroxylated compound 7 with compound 8 under reflux conditions in the presence of K2CO3 in dry acetone produced alkynyl core 11 in 73% yield. Through a similar approach, the core compounds 12 and 13 were synthesized in good yields upon treatment of compound 7 with compounds 9 and 10, respectively.8 FT-IR analysis showed the presence of an alkyne C–C triple bond stretching band at 2125 cm−1 and a terminal alkyne C–H stretching band at 3200 cm−1 in all the three synthesized core compounds,. The existence of the characteristic peaks for alkyne C–H protons in the range δ 2.16–2.42 ppm in 1H NMR along with the mass spectral analysis confirmed the synthesis of the alkynyl core compounds (see ESI†).
 |
| | Scheme 2 Synthesis of alkynyl core compounds 11, 12 and 13. | |
Synthesis of the glycodendrimers 16, 18 and 20
The glucosyl azide 14 was prepared from commercially available D-glucose in accordance with the literature procedure.21,22 Preparation of the desired glycodendrimers was accomplished in two steps. Treatment of alkynyl cores with glucosyl azide using Huisgen-type Cu(I) catalyzed azide−alkyne “click” cycloaddition reaction yielded acetylated derivatives, which upon subsequent sodium methoxide promoted trans-acetylation reactions in dry methanol yielded the desired final compounds.7,21,22 At each step of the reactions, the formation of the anticipated products was confirmed by 1H, 13C and 11B NMR data. The disappearance of alkyne C–H protons at around δ 2.5 ppm, the appearance of four singlets for the acetyl protons around δ 2 ppm in 1H NMR and peaks for cage-C and carbonyl-C in the appropriate regions in 13C NMR corroborate the formation of click reaction products. The complete disappearance of the peak for ester-H in 1H NMR and carbonyl-C in 13C NMR spectra respectively along with 11B NMR and FT-IR data confirmed the formation of the hydroxylated carborane–glucose conjugates. Formation of the desired compounds was further confirmed from their mass spectral analysis.
The reaction of the core 11 with the glucosyl azide 14 led to the formation of the acetylated derivative 15 in 68% yield, which upon deacetylation produced dendrimer 16 having three peripheral glucose moieties in 88% yield (Scheme 3). For the click cycloaddition product 151H NMR shows the presence of a singlet for the triazole-H at δ 7.67 ppm and four singlets for ester-H in the range δ 2.06–1.92 ppm. 13C NMR shows four peaks for the ester carbons in the range δ 170.6–169.4 ppm and two peaks for the cage-C at δ 85.9 ppm and 85.0 ppm respectively. FT-IR analysis shows a band at 2589 cm−1 corresponding to B–H stretching of o-carborane clusters. The disappearance of the peaks for ester-H in 1H NMR and peaks for ester-C in 13C NMR along with mass spectral data confirms the formation of the fully hydroxylated product 16 containing three glucose moieties at the periphery.
 |
| | Scheme 3 Synthesis of carboranyl dendrimer 16 containing three glucose moieties. | |
The acetylated dendritic compound 17 was prepared in 60% yield upon reaction of core 12 with glucosyl azide 14. A subsequent deacetylation reaction in the presence of sodium methoxide in dry methanol produced hydroxylated glycodendrimer 18 containing six peripheral glucose moieties in 80% yield (Scheme 4). 1H NMR for the click cycloaddition product 17 shows four singlets for ester-H in the range δ 2.04–1.90 ppm. 13C NMR shows four peaks for the ester carbons in the range δ 170.6–169.4 ppm and two peaks for the cage-C at δ 85.9 ppm and 85.1 ppm. FT-IR shows the presence of a B–H stretching band of o-carborane clusters at 2584 cm−1 and an ester-CO stretching band at 1757 cm−1. The disappearance of the peaks for ester-H in 1H NMR, peaks for ester-C in 13C NMR and mass spectral analysis confirmed the formation of hydroxylated dendrimer 18.
 |
| | Scheme 4 Synthesis of carboranyl dendrimer 18 containing six glucose moieties. | |
Through a similar approach using click chemistry, the reaction of core 13 and glucosyl azide 14 produced acetylated glycodendrimer 19 in 55% yield. The trans-acetylation was carried out in the presence of sodium methoxide in dry methanol to obtain the desired dendrimer 20 in 73% yield containing nine peripheral glucose moieties (Scheme 5). The formation of acetylated dendrimer 19 was evident from the presence of four singlets for ester-H in the range δ 2.05 ppm–1.88 ppm. 13C NMR shows four peaks for the ester-C in the range δ 170.1 ppm–167.7 ppm and two peaks for the cage-C at δ 85.9 ppm and 85.2 ppm. FT-IR shows the presence of a B–H stretching band of o-carborane clusters at 2586 cm−1 and an ester CO stretching band at 1754 cm−1. The formation of the hydroxylated water-soluble glycodendrimer 20 was evident from the absence of peaks for ester-H in 1H NMR and peaks for ester-Cs in 13C NMR in appropriate regions (see ESI†).
 |
| | Scheme 5 Synthesis of carboranyl dendrimer 20 containing nine glucose moieties. | |
Water solubility
The o-carboranyl core 7 with three hydroxyl groups and the glycodendrimer 16 containing three peripheral glucose moieties and twelve hydroxyl groups were completely insoluble in water. But the glycodendrimer 18 containing six glucose moieties and twenty-four hydroxyl groups at the periphery was sparingly water soluble up to 20 mg mL−1. With an increase in the number of peripheral glucose moieties to nine, the glycodendrimer 20 with thirty-six hydroxyl groups was found to be completely water soluble. The water solubility of 20 was enhanced to 150 mg mL−1 (Table 1). There are only a few reports on the preparation of water soluble carboranyl glycoconjugates.11,14m- and p-carborane containing bis(glycophosphonates) with one and two galactosyl moieties showed very high water solubility, even higher than that of galactose.23
Table 1 Water solubility and IC50 values of the carboranyl glycoconjugates against MCF-7
| Compound |
Water solubility (mg mL−1) |
Regression equation |
IC50 (ng mL−1) |
IC50 (μM) |
|
7
|
— |
y = 97.3199 + −0.05955x |
823 |
0.8 |
|
16
|
— |
y = 92.9639 + −0.2233x |
216 |
0.1 |
|
18
|
20 ± 10 |
y = 96.1886 + −0.4750x |
104 |
0.03 |
|
20
|
150 ± 10 |
y = 97.9492 + −1.2318x |
41 |
0.01 |
In vitro cytotoxicity, live dead and apoptosis assay
In vitro cytotoxicity and caspase-3 activity studies of the synthesized glycodendrimers 16, 18 and 20 were carried out against the MCF-7 breast cancer cell line to explore their ability to treat cancer as chemotherapeutic agents and promising boron neutron capture therapy (BNCT) drugs as well. MTT assay employs a colorimetric method for determining the percentage of viable cells based on mitochondrial dehydrogenase activity measurement at 595 nm. It is considered as an economic, convenient and rapid move toward the detection of viable and dead cells. To test the cytotoxic effect of the glucose containing dendritic carboranyl glycoconjugates, a cell viability study was carried out with the conventional MTT reduction assay. The IC50 value is a quantitative measure of the substance required to inhibit, in vitro, a given biological process or a biological component by 50%. Here, we measured the IC50 values of the newly synthesized compounds 7, 16, 18 and 20 by evaluating the concentration of the compounds required to inhibit the survival of MCF-7 cancer cells by 50%. All the four compounds under study showed remarkable dose-dependent cytotoxicity toward the MCF-7 breast cancer cell line as shown in Fig. 1. The results of the in vitro cytotoxicity studies are shown in Table 1. Fig. 1 shows the % cell survivability of the cell line MCF-7 after treatment with the dendritic carboranyl compounds 7, 16, 18 and 20 containing zero, three, six and nine peripheral glucose moieties, respectively. The IC50 value for the dendritic compound 20 containing nine glucose units at the periphery was found to be 41.35 ng mL−1 (0.01 μM), whereas, it was found to be 822.53 ng mL−1 (0.8 μM) for compound 7 without a glucose moiety at the periphery. The IC50 value for the dendritic compound 16 containing three peripheral glucose units was found to be 215.65 ng mL−1 (0.1 μM) and 104.47 ng mL−1 (0.03 μM) in the case of dendritic compound 18 containing six glucose units at the periphery. These results indicate that the water soluble glycodendrimer 20 is most cytotoxic while the core compound 7 is least cytotoxic towards MCF-7. These results were compared with those of the commonly used cisplatin drug which has an IC50 value of 6 μM towards the MCF-7 breast cancer cell line.24 From the comparison of the IC50 value (Table 1), it was estimated that the water-soluble glycodendrimer 20 was 600-fold more cytotoxic than the chemotherapeutic drug cisplatin towards the MCF-7 breast cancer cell line. However, the core compound 7 was estimated to be more than 7-fold cytotoxic than cisplatin. Water insoluble glycodendrimer 16 and partially water soluble glycodendrimer 18 were found to be 60 and 200-fold more cytotoxic towards MCF-7 cancer cell lines than cisplatin, respectively. The cytotoxicity of the water-soluble carboranyl glycodendrimer 20 was comparable with that of the clinically used cancer drug paclitaxel. Its IC50 value towards the MCF-7 cancer cell line has been found to be between 8–54 ng mL−1 (0.009–0.06 μM).25 The cytotoxicity of the partially water-soluble carboranyl glycodendrimer 18 was comparable with that of the anticancer agent raloxifene (IC50 0.025 μM towards MCF-7).26 These preliminary observations indicate that the dendritic molecules under study could be used as better anticancer therapeutics than cisplatin. The trend of cytotoxicity indicates that cytotoxicity increases with an increase in the number of peripheral glucose moieties and increasing water solubility. This result might be attributed to peripheral glucose moieties, which enhance the uptake of the carboranyl glycodendrimers into the cancer cells for the cell killing action.
 |
| | Fig. 1
In vitro cytotoxic effect of the carboranyl glycoconjugates at different concentrations (ng mL−1) on MCF-7 cells (breast cancer cell line) determined using MTT assay (data are expressed as mean ± SD, n = 10 with respect to control). | |
Hoechst-propidium iodide (PI) staining assay is generally performed to detect apoptotic and necrotic cells by fluorescence microscopy.27 Typically, the nuclei of healthy cells are spherical and the DNA is evenly distributed. However, the DNA becomes condensed during apoptosis but remains unchanged during necrosis. Thus, nuclear condensation can be used to distinguish between apoptotic and necrotic cells. Hoechst 33342 is a remarkable dye that binds to DNA, and is employed to observe nuclear condensation. For a normal cell, the nucleus is stained bright blue. The dead cells are characterized by red spots in propidium iodide staining. Usually, PI cannot pass through intact cell membranes but penetrates dead or damaged cells to bind with DNA and RNA by intercalating between the bases. In the present study, the cells stained with PI showed no fluorescence in the control indicating no dead cells in the range. On the other hand, compounds 7, 16, 18 and 20 displayed red fluorescence entirely in their vicinity, thereby validating cell death at their IC50 value (Fig. 2).
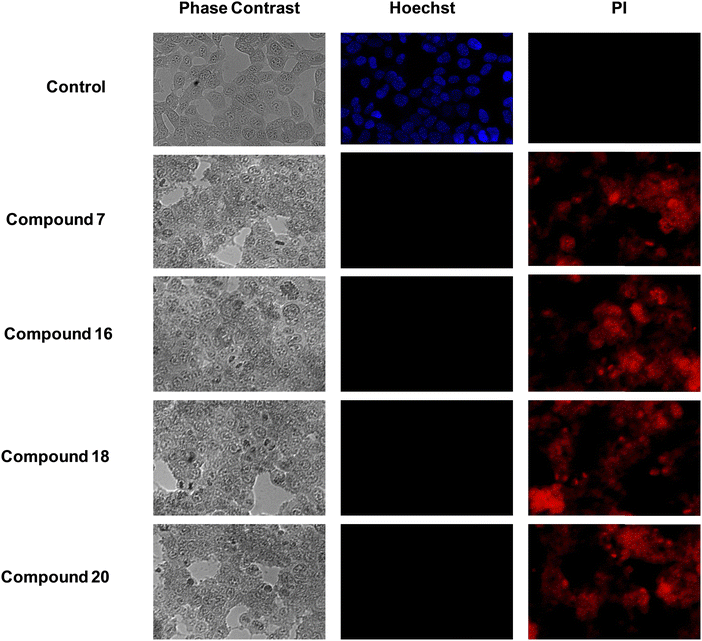 |
| | Fig. 2 Representative bright-field (phase contrast) and live/dead fluorescent images of MCF-7 cells treated with compound 7, 16, 18 and 20. The cells were stained with Hoechst-PI to demonstrate the cellular viability after 48 h. | |
After evaluating the IC50 values of glycodendrimers against MCF-7 cancer cell lines, the mode of cell death was studied through caspase-3 activity assay (Fig. 3). Apoptosis or programmed cell death involves controlled dismantling of intracellular components while avoiding inflammation and damage to surrounding cells. This mode of cell death is coordinated by members of the caspase family of cysteine proteases and 14 different caspases are reported in mammalian cells. Out of different caspases, caspase-3 is essential in the execution phase of cell apoptosis.28 It was found from caspase-3 activity assays (Fig. 3) that the non-glycoconjugated compound 7 and all the three dendritic glycoconjugated o-carboranyl dendrimers 16, 18 and 20 induced caspase-3 activity in a dose-dependent manner, suggesting the induction of cell death via apoptosis. It was found that the water soluble carboranyl glycoside 20 containing nine peripheral glucose moieties showed the maximum induction in the caspase-3 activity at a lower concentration. In hormone responsive breast cancers, the steroid hormone estrogen plays a crucial role in its progression. It elicits its action by binding to the ligand binding domain of the estrogen receptor (ER, a protein), leading to its dimerization which in turn binds to DNA promoter elements, initiating gene transcription.29 Through genome-wide approaches, Bailey et al. reported that ER prevents p53-dependent apoptosis, promoting cell proliferation and can be treated with ER antagonists such as fulvestrant.29 ER agonists or antagonists are compounds that can bind with it to either promote or inhibit estrogenic activity, respectively. Based on the inhibition of ERα, Endo et al. have shown that a carborane derivative has higher anti-estrogen response that is comparable to tamoxifen.30 Therefore, it may be stated that o-carborane induces p53-dependent apoptosis by inhibiting ER as evidenced by induction of caspase-3 activity. Furthermore, the augmentation in the apoptosis process in glycoconjugated carboranes may be attributed to their enhanced bio-availability and enhanced cellular uptake.
 |
| | Fig. 3 Effect of different concentrations (in ng mL−1) of synthesized dendritic compounds 7, 16, 18 and 20 on caspase-3 activity in MCF-7 cancer cells. | |
Conclusions
In summary, we have synthesized boron-rich glycodendrimers containing three o-carborane moieties. The glycodendrimer 16 contains three, 18 contains six and 20 possesses nine peripheral glucose moieties. Compound 20 contains thirty-six hydroxyl groups at the periphery and was found to be completely water soluble while partial water solubility was observed for dendrimers 18 with twenty-four hydroxy groups. The glycodendrimer 16 with twelve hydroxyl groups was not water soluble. The in vitro cytotoxicity test of dendritic compounds 7, 16, 18 and 20 against the MCF-7 breast cancer cell line revealed that all these compounds show stronger anticancer activity in comparison to the commonly used chemotherapeutic drug cisplatin. The water-soluble dendrimer 20 was found to be 600-fold more cytotoxic than the chemotherapeutic drug cisplatin whereas 16 and 18 were more than 60 and 200-fold cytotoxic compared to cisplatin. The least cytotoxicity was observed for the core 7 without any glucose moieties. The high cytotoxicity of glycodendrimers 18 and 20 can be compared with that of clinically used anticancer agents paclitaxel and raloxifene. Caspase-3 activity assay suggested that the mode of cell death is via apoptosis. Preliminary experimental and comparison results suggest that the high boron content dendrimers containing three polyhedral o-carborane cages in a single molecule could act as effective anticancer agents for cancer therapy. The high boron content symmetrical dendrimer 20 might be able to incorporate a therapeutic dosage of boron atoms into malignant tissues at a lower concentration due to the enhanced permeability and retention (EPR) effect.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
Authors thank Science & Engineering Research Board, India (grant numbers: CRG/2018/002635, SB/FT/CS-028/2012, and SB/FT/CS-058/2012), the Council of Scientific & Industrial Research, India (grant number: 02(0308)/17/EMR-II), the University Grants Commission, India, grant number: MRP-MAJOR-CHEM-2013-11320 and the Higher Education Department, Government of Odisha, grant number: 26913/HED/HEPTC-WB-02-17 (OHEPEE) for financial support.
Notes and references
- A.-M. Caminade and C.-O. Turrin, J. Mater. Chem. B, 2014, 2, 4055–4066 RSC.
- S. H. Medina and M. E. El-Sayed, Chem. Rev., 2009, 109, 3141–3157 CrossRef CAS PubMed.
- M. R. Carvalho, R. L. Reis and J. M. Oliveira, J. Mater. Chem. B, 2020, 8, 1128–1138 RSC.
- H. Maeda, J. Wu, T. Sawa, Y. Matsumura and K. Hori, J. Controlled Release, 2000, 65, 271–284 CrossRef CAS PubMed.
- J. Zhu and X. Shi, J. Mater. Chem. B, 2013, 1, 4199–4211 RSC.
- M. C. Parrott, E. B. Marchington, J. F. Valliant and A. Adronov, J. Am. Chem. Soc., 2005, 127, 12081–12089 CrossRef CAS.
- B. P. Dash, R. Satapathy, B. P. Bode, C. T. Reidl, J. W. Sawicki, A. J. Mason, J. A. Maguire and N. S. Hosmane, Organometallics, 2012, 31, 2931–2935 CrossRef CAS.
- B. B. Jena, S. R. Jena, B. R. Swain, C. S. Mahanta, L. Samanta, B. P. Dash and R. Satapathy, Appl. Organomet. Chem., 2020, 34, e5754 CrossRef CAS.
- L. F. Tietze, U. Griesbach, I. Schuberth, U. Bothe, A. Marra and A. Dondoni, Chem. – Eur. J., 2003, 9, 1296–1302 CrossRef CAS PubMed.
- S. Stadlbauer, P. Wetzel and E. Hey-Hawkins, Inorg. Chem., 2009, 48, 5005–5010 CrossRef CAS PubMed.
- R. Satapathy, B. P. Dash, C. S. Mahanta, B. R. Swain, B. B. Jena and N. S. Hosmane, J. Organomet. Chem., 2015, 798, 13–23 CrossRef CAS.
- H. Lis and N. Sharon, Chem. Rev., 1998, 98, 637–674 CrossRef CAS PubMed.
- D. Goyard, P. I. Diriwari and N. Berthet, RSC Med. Chem., 2022, 13, 72–78 RSC.
- B. R. Swain, C. S. Mahanta, B. B. Jena, S. K. Beriha, B. Nayak, R. Satapathy and B. P. Dash, RSC Adv., 2020, 10, 34764–34774 RSC.
- J. O. Suberu, I. Romero-Canelon, N. Sullivan, A. A. Lapkin and G. C. Barker, ChemMedChem, 2014, 9, 2791–2797 CrossRef CAS.
- B. P. Dash, R. Satapathy, J. A. Maguire and N. S. Hosmane, Chem. Commun., 2009, 3267–3269 RSC.
- B. P. Dash, R. Satapathy, E. R. Gaillard, K. M. Norton, J. A. Maguire, N. Chug and N. S. Hosmane, Inorg. Chem., 2011, 50, 5485–5493 CrossRef CAS PubMed.
- B. P. Dash, R. Satapathy, J. A. Maguire and N. S. Hosmane, Org. Lett., 2008, 10, 2247–2250 CrossRef CAS.
- B. P. Dash, R. Satapathy, J. A. Maguire, E. R. Gaillard and N. S. Hosmane, J. Am. Chem. Soc., 2010, 132, 6578–6587 CrossRef CAS PubMed.
- J. Camponovo, J. Ruiz, E. Cloutet and D. Astruc, Chem. – Eur. J., 2009, 15, 2990–3002 CrossRef CAS.
- Y. M. Chabre, P. P. Brisebois, L. Abbassi, S. C. Kerr, J. V. Marcotte, I. Fahy and R. Roy, J. Org. Chem., 2011, 76, 724–727 CrossRef CAS.
- N. P. Bizier, S. R. Atkins, L. C. Helland, S. F. Colvin, J. R. Twitchell and M. J. Cloninger, Carbohydr. Res., 2008, 343, 1814–1818 CrossRef CAS PubMed.
- S. Stadlbauer, P. Lonnecke, P. Welzel and E. Hey-Hawkins, Eur. J. Org. Chem., 2009, 6301–6310 CrossRef CAS.
- J. O. Suberu, I. Romero-Canelon, N. Sullivan, A. A. Lapkin and G. C. Barker, ChemMedChem, 2014, 9, 2791–2797 CrossRef CAS PubMed.
- H. Onyüksel, E. Jeon and I. Rubinstein, Cancer Lett., 2009, 274, 327–330 CrossRef.
- Y. Okamoto, X. Liu, N. Suzuki, K. Okamoto, M. Sekimoto, Y. R. S. Laxmi and S. Shibutani, Int. J. Cancer, 2008, 122, 2142–2147 CrossRef CAS PubMed.
- I. Vermes, C. Haanen and C. Reutelingsperger, J. Immunol. Methods, 2000, 243, 167–190 CrossRef CAS.
- L. M. Bomfim, F. A. de Araujo, R. B. Dias, C. Sales, C. A. G. Rocha, R. S. Correa, M. B. Soares, A. A. Batista and D. P. Bezerra, Sci. Rep., 2019, 9, 11483 CrossRef.
- S. T. Bailey, H. Shin, T. Westerling, X. S. Liu and M. Brown, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 18060–18065 CrossRef CAS.
- Y. Endo, T. Yoshimi and C. Miyaura, Pure Appl. Chem., 2003, 75, 1197–1205 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b
*b