
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The role of carbon dioxide and water in the degradation of zeolite 4A, zeolite 13X and silica gels†
John H.
Jacobs
,
Connor E.
Deering
 ,
Ruohong
Sui
,
Amelia P.
Cann
,
Kevin L.
Lesage
and
Robert A.
Marriott
*
,
Ruohong
Sui
,
Amelia P.
Cann
,
Kevin L.
Lesage
and
Robert A.
Marriott
*
Department of Chemistry, University of Calgary, 2500 University Drive, N.W., Calgary, AB T2N 1N4, Canada. E-mail: rob.marriott@ucalgary.ca
First published on 21st February 2023
Abstract
The degradation of desiccants is important in designing natural gas conditioning processes. Previous studies have focused on the effect of changes in regeneration gas water content, regeneration temperature and number of thermal cycles. However, less is known about how other components impact the lifespan of desiccants over thousands of thermal cycles. Herein we present results on how desiccant degradation is influenced by CO2 in a process fluid. Increasing the CO2 concentration resulted in less degradation across unsupported zeolite 4A, zeolite 13X and silica gels. Additionally, higher water concentrations in the regeneration gas resulted in a decrease in the degradation at the same CO2 concentration. For zeolite 13X, the surface area and pore volumes were larger in the samples subjected to greater CO2 concentrations. For silica gels, a higher capacity for water adsorption after 5000 thermal swing adsorption cycles was observed in samples with a lower concentration of surface silanol groups.
1. Introduction
Natural gas is an important transitional energy resource. The major components of natural gas from a reservoir are methane (CH4), hydrogen sulfide (H2S), carbon dioxide (CO2) and water (H2O); additionally, helium (He) and heavier hydrocarbons (such as ethane, C2H6, propane, C3H8, butane, C4H10, etc.) also can be present in rich natural gas. While the concentrations of natural gas components vary from source to source, the raw natural gas is always saturated with water at the wellhead and after aqueous alkanolamine treatment. This water can lead to pipeline corrosion and possible formation of hydrates during transportation and further processing.1–5 Therefore, water is removed before transportation in sales gas or before cryogenic liquefaction and can be removed before raw gas gathering lines. Industrially, natural gas is dehydrated through either glycol absorption (at plants and for moderate water dew points) or solid adsorption technology (in the field or within liquid natural gas conditioning), with temperature swing adsorption (TSA) processes being the most common adsorption process.6–10 In the TSA process, two or three beds are used. Within a three-bed system, one bed dehydrates the raw natural gas stream and a second bed undergoes thermal regeneration to remove the adsorbed water, while a third bed cools, thus allowing for the continuous operation of this system. These adsorption systems are designed for uninterrupted operation over 3-5 years before the desiccants are replaced due to a loss in adsorption capacity.11–13The decrease in adsorption capacity is often attributed to three factors: (I) the collapse of the materials’ pores;14–16 (II) coking of the desiccant materials, thus effectively limiting the adsorption sites of water;17 and III) the blocking of pores through the condensation of contaminants in the gas stream, such as hydrocarbons.13 The reaction of aluminum and silica with water is believed to be responsible for changes in the porosity of aluminosilicate zeolites.18,19 For silica gels, the loss of porosity is attributed to the restructuring of porous silica materials toward non-porous structures in the presence of water and heat, with reported structural changes occurring at temperatures as low as T = 90 °C.16 Our previous work demonstrated a temperature dependence on the degradation of desiccants, where higher regeneration temperatures result in a greater degree of adsorption capacity loss.20 Our work also showed that utilizing a wet gas during the thermal regeneration of the desiccants mitigated the degradation of the materials. These results indicate that the major degradation mechanisms are most likely due to (i) mechanical stress from the expansion and contraction of the materials during the heating and cooling steps of the TSA process and (ii) overstripping the highest energy adsorption sites, thus increasing the rate of crystalline rearrangement.
When comparing the data of our previous experiments, it was observed that experiments where CO2 was part of the gas mixture, resulted in a lower degree of degradation on zeolite 13X. Thus far, degradation studies on desiccants have been focused on how changes in water concentration and regeneration temperatures impact the degradation of desiccant materials.18,19 There are no published studies on the role of CO2 and desiccant material degradation.
In this work, five desiccant materials (zeolite 4A, zeolite 13X, and 2.2 nm, 3.0 nm and 6.0 nm pore size silica gels) were subjected to 5000 TSA cycles at a regeneration temperature of T = 250 °C. Three feed gas phase compositions were investigated for the TSA systems (pCO2 = 3.49 kPa, pH2O = 2.39 kPa; pCO2 = 3.49 kPa, pH2O = 2.21 kPa; pCO2 = 34.9 kPa, pH2O = 2.21 kPa) and the relationship between the wet or dry CO2 concentrations and amount of degradation was studied.
2. Experimental section
2.1 Materials
The synthesis and characterization of zeolite 4A and 13X materials are reported in our previous publications.8–10 An in-house EMD Millipore system was used to purify double distilled water to an 18 MΩ cm resistivity. Liquid nitrogen (N2, 99.998%), helium (He, 99.9990%, Alphagaz 1) and liquid CO2 (99.8%) were purchased from Air Liquide and used as received. 2.2 nm pore size silica gel (High purity, Davisil Grade 12, 28–200 mesh) and 3.0 nm pore size silica gel (High purity, Davisil Grade 923, 100–200 mesh) were purchased from Sigma Aldrich and used as received. 6.0 nm pore size silica gel (High purity, Davisil Grade 9385, 130–270 mesh) was purchased from Merck and used as received. No binding materials were studied in this work.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.98 sample/KBr mass ratio.
:0.98 sample/KBr mass ratio.
2.2 Water adsorption
Single-point water adsorption measurements were conducted on all materials before and after the TSA cycling experiments using a Thermal Gravimetric Analyser, TGA (TGA 550 Auto apparatus, TA Instruments). The TGA operated by flowing dry He through the balance and the furnace. For the adsorption experiments, the He flowing over the adsorbent was first passed through a water saturator and then diluted by dry He before passing over the adsorbent. The flow rate of He passing through the saturator was controlled using a Brooks SLA5800 mass flow controller. The saturator temperature (T = 15.00 °C) was controlled using a Polyscience chilling circulating bath. During activation, dry He (T = 400 °C, p = 89.05 kPa, at 16 mL min−1) was passed over the adsorbent. The temperature increased at 25 °C min−1, and the sample was held at T = 400 °C for 120 minutes. During the adsorption, 1.4 mole % H2O in He were held at T = 35 °C, pTotal = 89.05 kPa, and 16 mL min−1 for 240–360 minutes until a stable mass reading was obtained.2.3 Rapid thermal swing cycling experiments
To test the materials for their stability in the long-term TSA process, a rapid cycling TSA instrument was used, this instrument was introduced in our previous work21 (Fig. 1). In each adsorbent cell, 25–35 mg of adsorbent material was loaded. For all thermal cycling experiments, the same sequence of events followed: | ||
| Fig. 1 The schematic of the instrument used during the rapid cycling experiments adapted from the our previous literature.21 | ||
(a) The adsorbing gas feed flowed through the adsorbent for 330 seconds and was then replaced with the regeneration gas feed.
(b) The block temperature was increased at a rate of 85 °C min−1. Once the desired regeneration temperature was achieved, the block's heating element was cycled on and off to maintain the temperature for 400 seconds.
(c) After the heating time for the adsorbents concluded, the heating elements were switched off, and the water coolant was flowed until the block temperature reached T = 35 °C.
(d) When the block reached a temperature of T = 80 °C, the regeneration feed was replaced with the adsorbing gas feed.
This procedure was repeated until the desired number of cycles was collected. The details of the adsorption and regeneration feed for the experimental conditions can be found in Table 1. The flow rates in each cell were measured using an Agilent Technologies ADM 2000 universal flow meter. As described in our previous work, a Nafion water saturator was used to introduce water into the adsorbing gas feed.21 The water content of the gas feeds also are present in Table 1.
| Experiment | Inlet water content pH2O/kPa | p CO2/kPab | p total/kPac | Regeneration feed | Regeneration temperature/°C |
|---|---|---|---|---|---|
| a Regeneration feed and adsorption feed are the same. b The CO2 content was the same for the regeneration feed and adsorption feed. c All feeds were balance N2. | |||||
| I | 2.39 ± 0.05 | 3.49 ± 0.03 | 349 ± 60 | N2/CO2 | 246 ± 9 |
| II | 2.39 ± 0.05 | 3.49 ± 0.03 | 338 ± 40 | N2/CO2/H2Oa | 244 ± 8 |
| III | 2.21 ± 0.06 | 34.9 ± 0.3 | 349 ± 60 | N2/CO2 | 246 ± 9 |
| IV | 2.21 ± 0.06 | 34.9 ± 0.3 | 338 ± 40 | N2/CO2/H2Oa | 244 ± 8 |
| V | 2.21 ± 0.06 | 3.49 ± 0.03 | 349 ± 60 | N2/CO2 | 246 ± 9 |
| VI | 2.21 ± 0.06 | 3.49 ± 0.03 | 338 ± 40 | N2/CO2/H2Oa | 244 ± 8 |
2.4 Data analysis
where b0 is the intercept and bi is the slope/correlation corresponding to property xi. A stepwise elimination of the property coefficients was conducted where the coefficient with the lowest magnitude t stat was eliminated until the minimum standard error was achieved. For this analysis, a parameter was only deemed significant if the P-value was below P-value = 0.05.
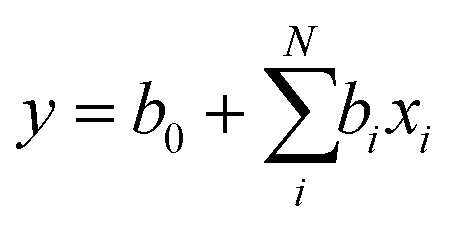
3. Results and discussion
3.1 Material characterization
The characterization of the zeolite 4A and 13X samples were reported in our other work.8,9 Using dynamic light scattering, the average particle diameters of zeolites 4A and 13X were determined as 4.45 μm and 21.41 μm, respectively. Through energy dispersive X-Ray analysis, the Si/Al ratios of the zeolites were determined as 1.14–1.15 for zeolite 4A, and 1.8 for zeolite 13X. The scanning electron microscope images (Fig. 2) of the zeolite show that the 4A has large regular crystallites, whereas the 13X are aggregates of smaller crystallites. The powder X-Ray diffraction patterns of the zeolite 4A and 13X materials are shown in Fig. 2, where the experimental peaks are compared to simulated peaks provided by the international zeolite association from the work of Treacy and Higgins.22 Comparison of the experimental and reference patterns for the two zeolites shows agreement. The diffraction pattern is focused on the range of 10–35 2θ. The significance of this range is it focuses on the HKL peaks used by the ASTM methods to verify the Linde type A,23 and faujasite diffraction patterns.24 Both the comparison of the computational patterns and the ASTM methods with the experimental patterns verify the materials to be zeolites 4A and 13X. The silica gels are amorphous materials, thus PXRD was not used to characterize the silicas. | ||
| Fig. 2 Powder X-Ray diffraction patterns for zeolite 4A (A) and Zeolite 13X (B). Red lines indicate calculated spectra provided by the International Zeolite Association,22 and the HKL indices highlighted by the ASTM methods for identifying Linde Type A23 and Faujasite24 zeolites are indicated. Scanning electron microscope images of zeolite 4A (C) and zeolite 13X (D and E). The white bars indicate a length of 40 μm (C and D) and the white bar in image E represents a length of 500 nm. | ||
The diffuse reflectance infrared Fourier transform (DRIFT) spectra (Fig. 3) of the zeolites showed a strong OH stretching frequency in the range of 2700–3700 cm−1, likely due to the adsorption of water in the zeolite pores. Peaks at ∼1650 cm−1 and ∼1400 cm−1 correspond to water vibration modes.25 On the zeolite 4A spectra, the peaks at ∼983 cm−1 (Si–O–Si and Si–O–Al asymmetric stretch), ∼670 cm−1 (Si–O–Al symmetric stretch), ∼550 cm−1 (complex vibration band of four member rings), and ∼466 cm−1 (O–Si–O bending) were observed, all of which are typical of the Linda type A framework.26 The peaks on zeolite 13X at ∼990 cm−1 (Si–O–Si and Si–O–Al asymmetric stretch), 760–665 cm−1 (complex vibration band of four and six member rings), ∼557 and ∼500 cm−1 (six member ring vibrations), and ∼470 cm−1 (O–Si–O and O–Al–O bending) also were observed.26
 | ||
| Fig. 3 The DRIFT spectra of the five desiccants. | ||
Analysis of the DRIFT spectra (Fig. 3) of the silica gels show isolated silanol peaks on all three silica gels at ∼3700 cm−1. The silica gels all show similar peaks in the 400–1400 cm−1 range with peaks at 1065 cm−1 (Si–O–Si stretch), 940 cm−1 (Si–OH stretch), 790 cm−1 (O–Si–O stretching), 470 cm−1 (O–Si–O bending).27 Comparison of the Si–OH peak at ∼940 cm−1 between the silica gels shows that the 2.2 nm pore silica had the greatest concentration of silanol groups, while the 6.0 pore size had the least.
The results from TGA analysis of the silica gels showed that the concentration of silanol groups on the 2.2 nm pore size silica gel was 2.5 OH nm−2, the 3.0 nm pore size had 2.3 OH nm−2, and the 6.0 nm pore size had 2.1 OH nm−2. This agrees with the results from the DRIFT spectra. Table 2 shows the results from the single point water adsorption experiments for the five desiccants and the BET specific surface area, total pore volume, t-plot micropore volume and mesopore volumes of the three silica gels and zeolite 13X. The Specific surface areas were determined by fitting the BET isotherm equation to the N2 physisorption isotherm at T = 77 K following the procedure of Brunaur, Emmett, and Teller.28 The mesopore volumes were determined using the procedure of Barrett et al.29 The micropore volumes were determined using the t-plot method.30
| Zeolite 4A | Zeolite 13X | 2.2 nm pore silica | 3.0 nm pore silica | 6.0 nm pore silica | |
|---|---|---|---|---|---|
| a The amount of water adsorbed at T = 35 °C and pH2O = 1.25 kPa. | |||||
| n ads H2O /mmol g−1 | 13.5 ± 0.4 | 13.5 ± 1.1 | 6.0 ± 0.7 | 2.7 ± 0.3 | 1.80 ± 0.07 |
| A s,BET/m2 g−1 | — | 589 | 641 | 470 | 453 |
| V pore,total/cm3 g−1 | — | 0.56 | 0.45 | 0.52 | 1.07 |
| V micropore,t-plot/cm3 g−1 | — | 0.27 | 0.10 | 0 | 0 |
| V mesopore/cm3 g−1 | — | 0.02 | 0.16 | 0.37 | 0.57 |
3.2 Material degradation
The five desiccants studied in this work were subject to 5000 TSA cycles at Tregen = 250 °C with the same heating rate across all experiments. All experiments were run in duplicate for zeolite 4A, zeolite 13X, and the 2.2 nm pore size silica gel. In these experiments, the concentrations of H2O and CO2 in the gas feed (adsorption and regeneration) were changed, and the water adsorption capacities after 5000 TSA cycles are shown in Fig. 4. At the same concentration of CO2, a higher water concentration resulted in a higher water adsorption capacity after 5000 TSA cycles, i.e., less degradation. As well, increasing the CO2 concentration increased the water capacity of the desiccants after 5000 TSA cycles. Of these two trends, it is noteworthy that the materials subjected to the gas feed with the highest CO2 concentration had the highest water capacity out of all the experiments. It was observed that for all feed gas streams, zeolite 4A was left with a higher water capacity after 5000 cycles from the wet gas regeneration conditions versus the dry gas regeneration conditions. Whereas zeolite 13X consistently had a lower capacity with the wet gas regeneration experiments, the 3.0 nm pore size silica showed inconsistent trends regarding wet gas and dry gas regeneration. For the 2.2 nm and 6.0 nm pore silicas, there was no statistically significant difference between the dry and wet gas regeneration conditions.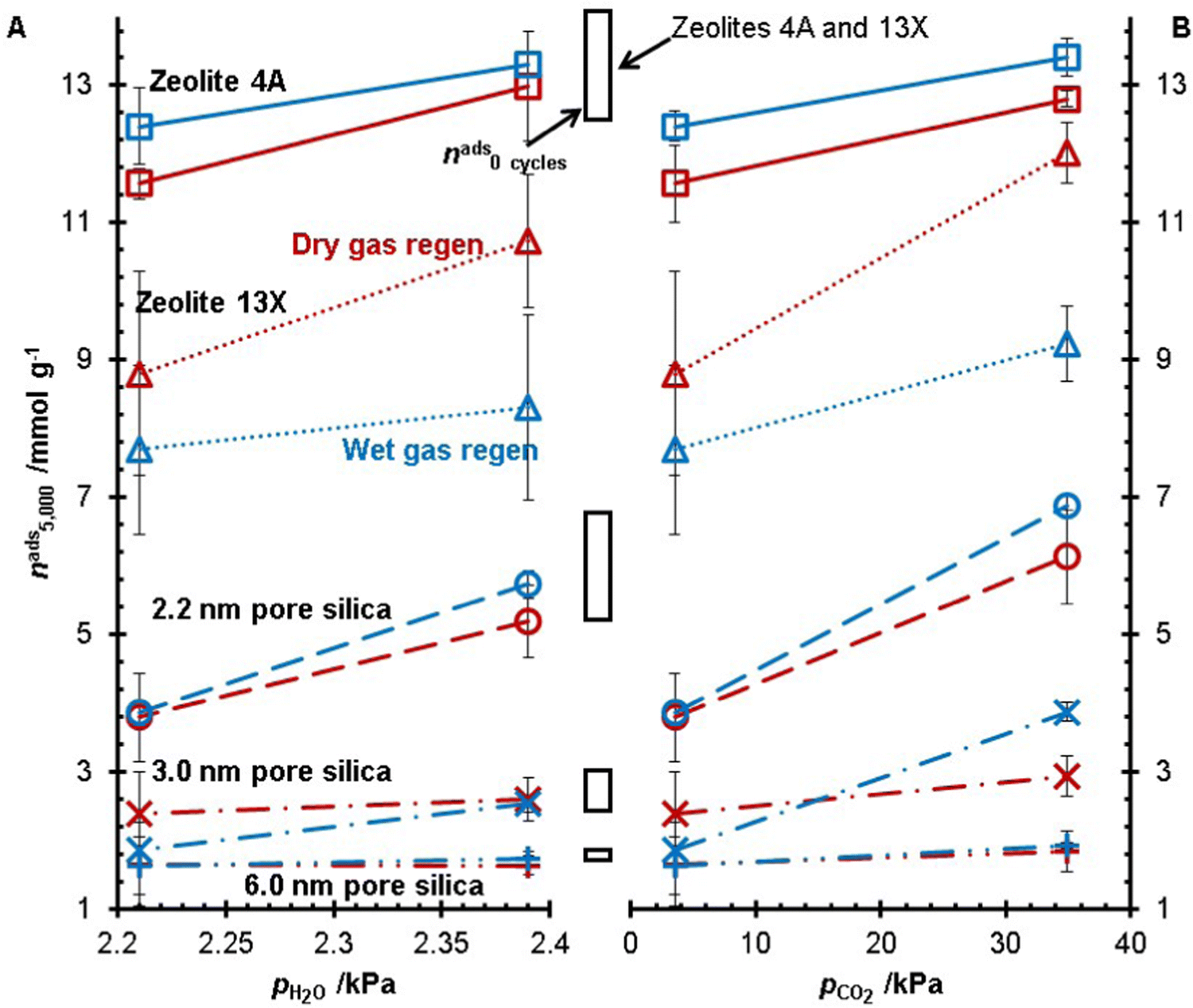 | ||
Fig. 4 The capacity after five thousand cycles of zeolite 4A (□, ![[thick line, graph caption]](https://www.rsc.org/images/entities/char_e117.gif) ), zeolite 13X (Δ, • •), 2.2 nm pore size silica (○, ), 3.0 nm pore size silica (×, •), and 6.0 nm pore size silica (+, • •) compared to different water partial pressures at 1% CO2 (pCO2 = 3.49 kPa) (A) and different CO2 concentrations at pH2O = 2.21 kPa. The colours red and blue represent dry gas and wet gas regeneration conditions, respectively, and the black boxes represent the initial uptake of the materials before cycling. ), zeolite 13X (Δ, • •), 2.2 nm pore size silica (○, ), 3.0 nm pore size silica (×, •), and 6.0 nm pore size silica (+, • •) compared to different water partial pressures at 1% CO2 (pCO2 = 3.49 kPa) (A) and different CO2 concentrations at pH2O = 2.21 kPa. The colours red and blue represent dry gas and wet gas regeneration conditions, respectively, and the black boxes represent the initial uptake of the materials before cycling. | ||
To better visualize the changes in the water adsorption capacity during the TSA experiments, the results for zeolite 13X and the 2.2 nm pore size silica gel are shown in Fig. 5. Zeolite 13X and the 2.2 nm pore size silica gel are presented because these are the two materials with the greatest capacity loss from our previous experiments.20 For the dry gas experiments, the zeolite and the silica gel samples showed greater degradation when no CO2 was present in the adsorption and regeneration gas feed. For the zeolite 13X, in the wet gas regeneration experiments, the experiment VI condition (pCO2 = 3.49 kPa and pH2O = 2.21 kPa) showed the greatest capacity loss, followed by the CO2-free experiment, experiment IV. Finally, experiment II showed the least amount of capacity loss. For the dry regen experiments on the 2.2 nm pore size silica gel, experiments I and V showed similar results during the TSA cycling, but the final measured capacity of the material subjected to experiment V was lower than the results of I. For the dry and wet gas regeneration experiments, capacities during experiments III and IV of the 2.2 nm pore size silica gel increased with the TSA cycling. From this analysis of Fig. 4 and 5, CO2 changes the desiccant materials’ degradation.
 | ||
| Fig. 5 Capacity changes over 5000 TSA cycles for zeolite 13X (A and B) and the 2.2 nm pore size silica gel (C and D). Results for the dry gas regeneration (A and C) and wet gas regeneration (B and D) experiments are shown. For each material, the results of experiments I (blue, ○), II (blue, Δ), III (red, ○), IV (red, Δ), V (purple, ○), and VI (purple, Δ) were compared to the dry (black, ○) and wet (black, Δ) regeneration conditions of the CO2 free experiments.20 For all data sets, solid lines represent empirical polynomial fits to guide the eye. | ||
3.2 Material degradation correlation
The porosity and surface area of the desiccants after 5000 TSA cycles were measured by N2 physisorption (Fig. 6 and 7). The surface area of zeolite 13X decreased less for the three different gas feeds compared to the N2/H2O mixture previously studied.20 The surface area for the 1% CO2 (pCO2 = 3.49 kPa) experiments was similar for the two other water contents, while the 10% CO2 (pCO2 = 3.49 kPa) experiments had the highest surface area for zeolite 13X. This trend was followed for wet and dry gas regeneration conditions, with the wet gas having a lower surface area. The porosity of zeolite 13X showed the same trend as the surface area for the dry gas regeneration conditions. Still, the wet gas conditions showed higher surface areas, and the 1% CO2 (pCO2 = 3.49 kPa) experiment with a higher water content showed the greatest pore volume. The values of BET specific surface area and pore volumes for the three silica geals and zeolite 13X are presented in Table 3.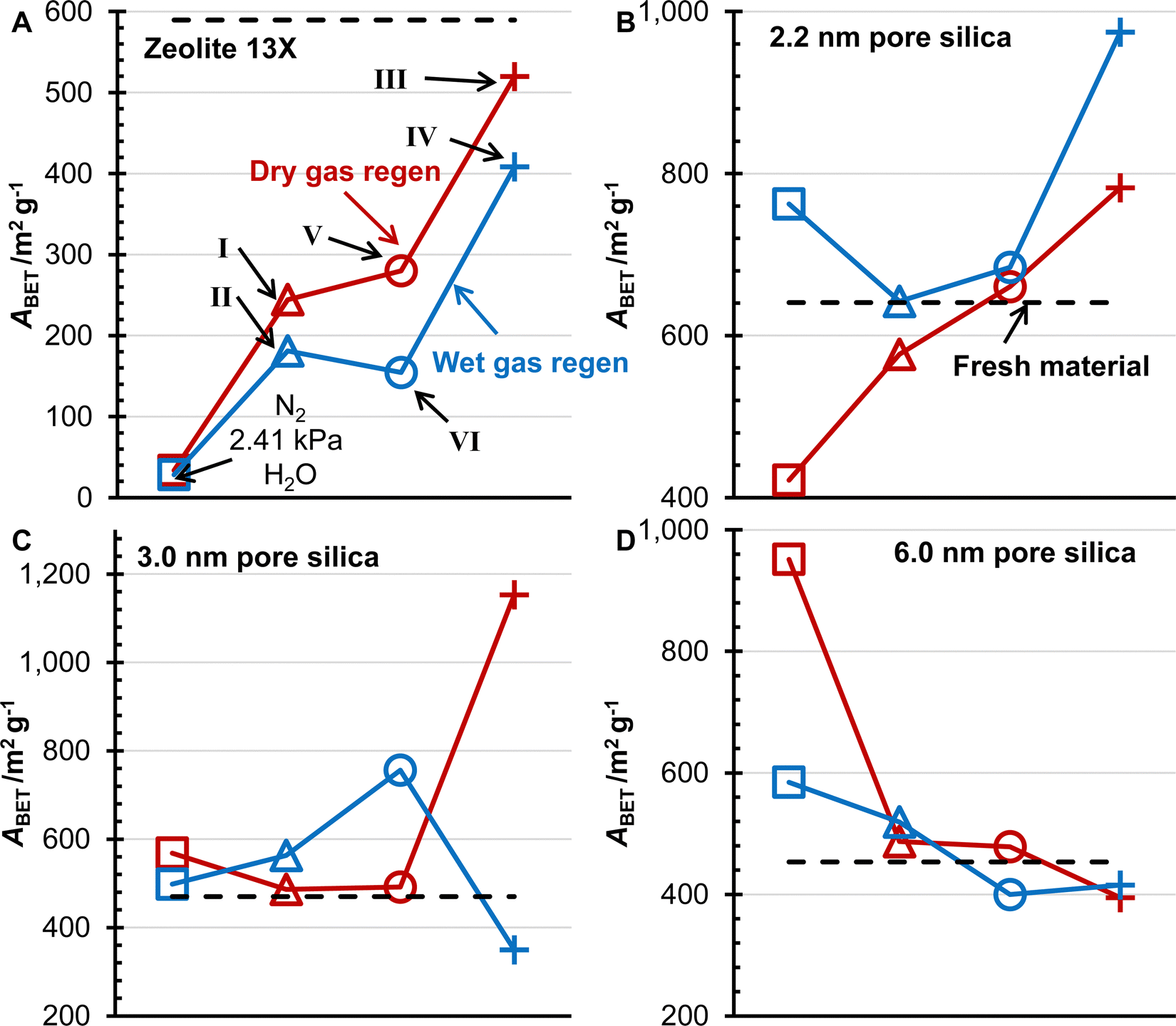 | ||
| Fig. 6 The BET surface area of zeolite 13X (A), 2.2 nm pore size silica (B), 3.0 nm pore size silica (C), and 6.0 nm pore size silica (D) for the three gas mixtures (pCO2 = 3.49 kPa, pH2O = 2.39 kPa, Δ; pCO2 = 3.49 kPa, pH2O = 2.21 kPa, ○; pCO2 = 34.9 kPa, pH2O = 2.21 kPa, +) and the previously published results (pH2O = 2.41 kPa, □) without CO2.20 Results for dry gas (red) and wet gas (blue) regeneration conditions are reported. The solid black line represents the surface area of the fresh material. | ||
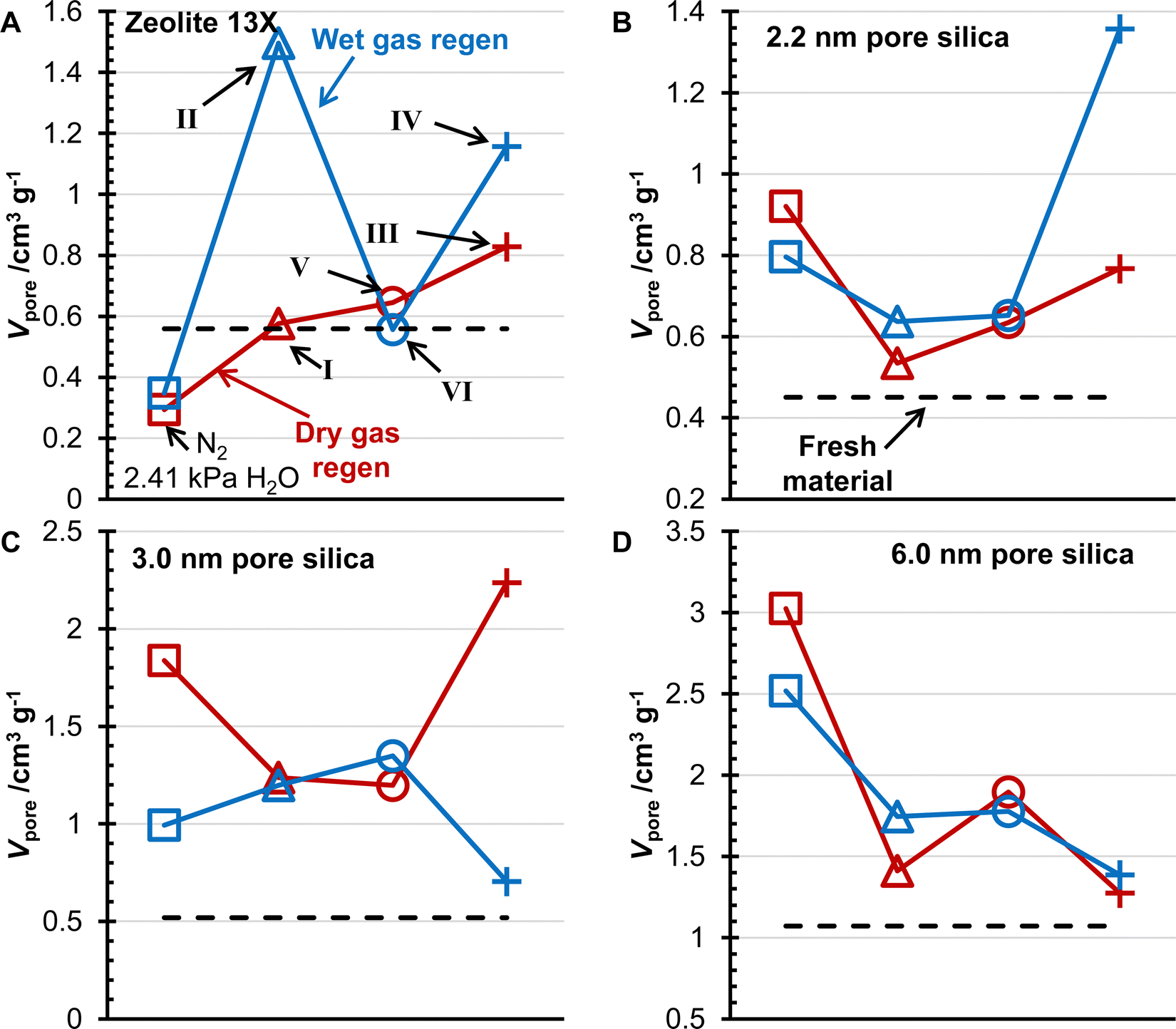 | ||
| Fig. 7 The pore volumes of zeolite 13X (A), •), 2.2 nm pore size silica (B), 3.0 nm pore size silica (C), and 6.0 nm pore size silica (D) for the three gas mixtures (pCO2 = 3.49 kPa, pH2O = 2.39 kPa, Δ; pCO2 = 3.49 kPa, pH2O = 2.21 kPa, ○; pCO2 = 34.9 kPa, pH2O = 2.21 kPa, +) and the previously published results (pH2O = 2.41 kPa, □) without CO2.20 Results for dry gas (red) and wet gas (blue) regeneration conditions are reported. The solid black line represents the surface area of the fresh material. | ||
| Experiment | Zeolite 13X | 2.2 nm pore silica | 3.0 nm pore silica | 6.0 nm pore silica | |
|---|---|---|---|---|---|
| Fresh sample | A BET/m2 g−1 | 589 | 641 | 470 | 951 |
| V pore/cm3 g−1 | 0.56 | 0.45 | 0.52 | 1.07 | |
| I | A BET/m2 g−1 | 245 | 577 | 487 | 478 |
| V pore/cm3 g−1 | 0.58 | 0.53 | 1.24 | 1.41 | |
| II | A BET/m2 g−1 | 181 | 642 | 563 | 519 |
| V pore/cm3 g−1 | 1.50 | 0.64 | 1.20 | 1.75 | |
| III | A BET/m2 g−1 | 520 | 782 | 1153 | 395 |
| V pore/cm3 g−1 | 0.83 | 0.77 | 2.24 | 1.27 | |
| IV | A BET/m2 g−1 | 408 | 975 | 350 | 415 |
| V pore/cm3 g−1 | 1.16 | 1.36 | 0.70 | 1.39 | |
| V | A BET/m2 g−1 | 280 | 660 | 492 | 395 |
| V pore/cm3 g−1 | 0.64 | 0.63 | 1.20 | 1.90 | |
| VI | A BET/m2 g−1 | 155 | 684 | 756 | 400 |
| V pore/cm3 g−1 | 0.56 | 0.65 | 1.53 | 1.78 |
Investigating the changes in surface area of the silica gels, the 2.2 nm pore size silica increased in surface area with a decreased water content in the adsorption gas mixture. In contrast, the 10% CO2(pCO2 = 34.9 kPa) mixture resulted in the highest surface area of the silica gel. The 3.0 nm pore size silica was observed to have similar surface areas between the N2/H2O mixture and the 1% CO2 (pCO2 = 3.49 kPa) mixtures for the dry gas regeneration experiments. The surface area increased with decreasing water content for the wet gas regeneration. Interestingly, the surface area for the 10% CO2 (pCO2 = 34.9 kPa) mixture was the greatest for the dry gas regeneration samples, while the wet gas regeneration showed the lowest surface area. Comparing the N2/H2O mixture results for the 6.0 nm pore size silica with the 1% CO2 (pCO2 = 3.49 kPa) mixtures, it is observed that the surface area decreased with increasing water content for the wet gas regeneration and decreased with increasing CO2 concentration. The changes in porosity of the 6.0 nm pore size silica followed the same trends as the surface area. For the 6.0 nm pore size, there were no qualitative differences in the surface area and porosity trends between the wet and dry gas regeneration experiments.
The ratio of the silanol groups to the siloxane groups (Si–OH/Si–O–Si) was obtained from DRIFT spectra. The ratio of the silica functional groups was compared to the capacity of the samples after 5000 cycles (Fig. 8B). All three silicas showed a decreased capacity with increasing silanol content. This trend is the opposite expected trend but can be related to the changes in surface area. If the accessible surface area decreases due to the collapse of pores, then even if the silanol groups have been preserved, they will not interact with the water molecules. Additionally, it must be mentioned that there was no evidence in any of the DRFIT spectra to indicate the formation of carboxylate groups on the surface of the desiccants.
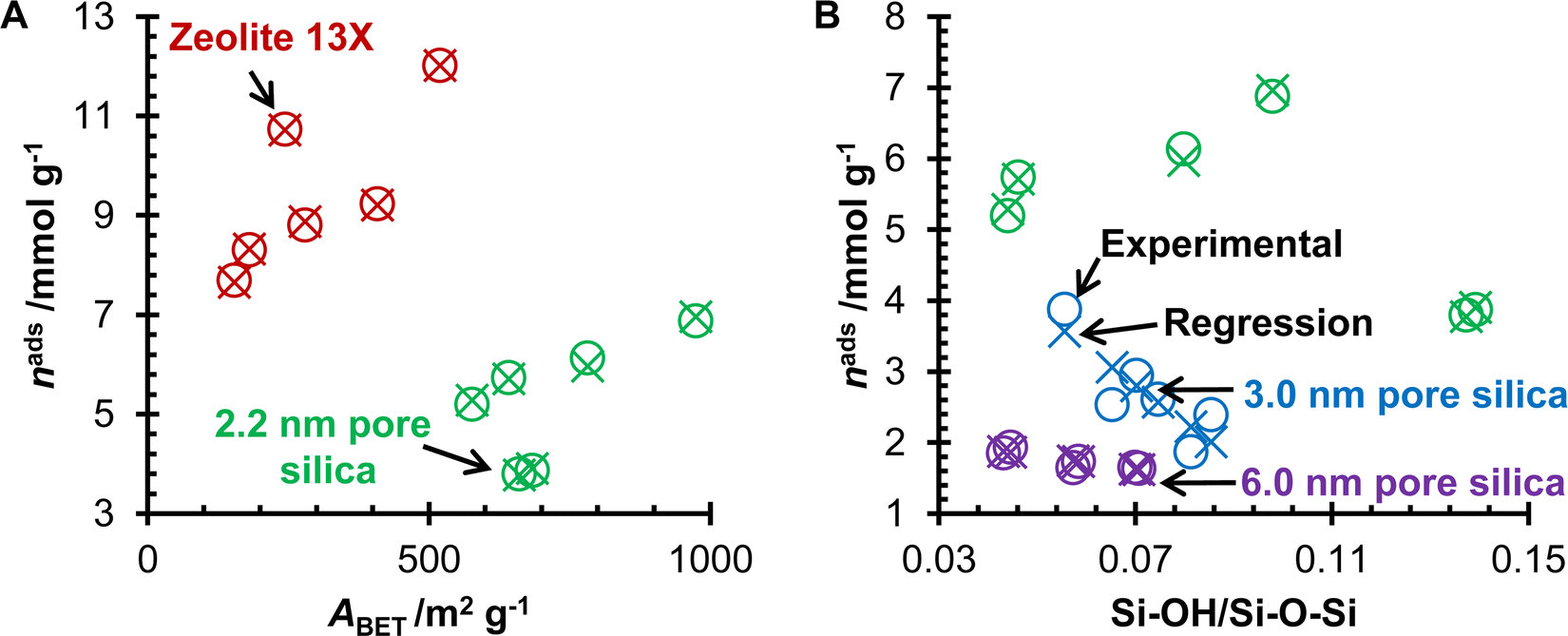 | ||
| Fig. 8 A comparison of the adsorption capacity of zeolite 13X (red) and 2.2 nm pore size silica (green) against the surface area (A) and the adsorption capacity of the silca gels (2.2 nm pore size, green; 3.0 nm pore size, blue; 6.0 nm pore size, purple) against the ratio of silanol to siloxane groups (B). The experimental capacities (○) and the capacities predicted by the regression analysis (×) are presented for both plots. | ||
A multiple parameter regression analysis was conducted for zeolite 13X and the three silica gel materials to investigate the significance of correlation between the changes in material properties and the changes in adsorption capacities. For zeolite 13X, the BET surface area, pore volume, micropore surface area, and micropore volume all showed significant correlations (P-value < 0.05). Of the tested properties, the correlation to BET surface area showed the largest t-statistic. The plot of capacity against BET surface area is shown in Fig. 8A. For the silica gels, the 2.2 nm pore size silica showed significant correlation between both the BET surface area and the silanol/siloxane ratio (P-value < 0.0005), and both parameters had a similar magnitude t-stat. For the 3.0 and 6.0 nm pore size silicas, only the silanol/siloxane ratio showed a significant correlation (P-value < 0.05).
Interestingly, as the silanol/siloxane ratio increased, the adsorption capacity decreased for all three silica gels. It is already established that the water absorption of silica gels depends on the silanol concentration on the surface of the silica gels, so it stands to reason that increasing the silanol concentration would increase water adsorption. One possible explanation would be that changes in the pore structure reduce the accessibility of water to the silanol groups. It should be noted that the regression fitting of the zeolite 13X properties only had one statistical degree of freedom, while the 2.2 nm pore size silica had three statistical degrees of freedom and the 3.0 and 6.0 nm pore size silicas each had four degrees of freedom.
The crystallinity of the zeolite 4A and zeolite 13X samples was investigated by PXRD (Fig. 9). The dry gas and wet gas regeneration conditions for all three gas mixtures showed little change between the samples. Between the gas mixtures, there was a small amount of peak broadening for the zeolite 4A around 30 2θ. It is difficult to attribute this to changes in the material, as sample preparation can also result in such small changes. We note that peak broadening can be caused by changes in crystallite size and changes in crystallinity. Our previous work showed that zeolite 4A crystals became fractured over time, versus loosing crystallinity. The SEM images of the zeolite 4A (Fig. 10) show small particulate around the larger crystallites for all samples, with some of the crystallites showing wear on the surface. However, throughout the imaged samples, there were no indications of fracturing occurring on the crystallites, as was observed in previous experiments without CO2 in the gas mixture.20
 | ||
| Fig. 9 PXRD spectra for zeolite 4A (Left) and zeolite 13X (Right) for the different gas mixtures (A, B: pCO2 = 3.49 kPa, pH2O = 2.39 kPa, N2 balance; C, D: pCO2 = 3.49 kPa, pH2O = 2.21 kPa, N2 balance; E, F: pCO2 = 34.9 kPa, pH2O = 2.21 kPa, N2 balance). Results for dry gas (red) and wet gas (blue) regeneration conditions are reported. | ||
 | ||
| Fig. 10 SEM images of zeolite 4A after 5000 TSA cycles. Images for (A, D: pCO2 = 3.49 kPa, pH2O = 2.39 kPa, N2 balance; B, E: pCO2 = 3.49 kPa, pH2O = 2.21 kPa, N2 balance; C, F: pCO2 = 34.9 kPa, pH2O = 2.21 kPa, N2 balance). Images of the material after dry gas (A–C) and wet gas (D–F) regeneration conditions are presented. The white bars represent a length of four μm. | ||
The PXRD of zeolite 13X (Fig. 9) showed broadening around the 30 2θ region, but this didn’t change between the samples. Unlike the experiments without CO2 in the gas mixture,20 the zeolite generally retained crystallinity without much difference between the dry and wet gas regeneration conditions. The SEM images (Fig. 11) of the zeolite 13X samples after 5000 TSA cycles showed that all six samples retained sharp edges of the crystallites, whereas the images from the experiments without CO2 in the gas mixture20 showed rounding of the crystallite edges.
 | ||
| Fig. 11 SEM images of zeolite 13X after 5000 TSA cycles. Images for (A and D: pCO2 = 3.49 kPa, pH2O = 2.39 kPa, N2 balance; B and E: pCO2 = 3.49 kPa, pH2O = 2.21 kPa, N2 balance; C and F: pCO2 = 34.9 kPa, pH2O = 2.21 kPa, N2 balance). Images of the material after dry gas (A–C) and wet gas (D–F) regeneration conditions are presented. The white bars represent a length of 500 nm. | ||
The adsorption mechanism of CO2 on both zeolite and silica surfaces is believed to be primarily physisorption, with some studies indicating a minor amount of chemisorption occurring on these materials.31,32 The presence of the chemisorbed CO2 could inhibit the degradation of the desiccants during the TSA cycling. For the silica gel materials, the CO2 may interfere with the dehydroxylation/rehydroxylation process during thermal treatment. It is known that silanols on silica surfaces are lost during thermal treatment, and that when water is present, the silanols can be regenerated on the silica surface.33 To better understand the role of CO2 on the surface silanol chemistry, further experiments will need to be conducted.
4. Conclusion
The previous literature18,19 into what factors influence the degradation of desiccants have always focused on changes in water content or regeneration temperature. We present results on how the degradation of desiccants is influenced by the concentration of CO2 in the process fluid. It was observed that increasing the CO2 concentration (in the regeneration gas) resulted in less degradation across all desiccants tested. Additionally, higher water concentrations in the regeneration gas resulted in a decrease in the desiccant degradation at the same CO2 concentration. For zeolite 13X, the surface area and pore volumes were greater in the samples subjected to a 10% CO2 (pCO2 = 34.9 kPa) gas mixture during the TSA process than in the 1% CO2 (pCO2 = 3.49 kPa) mixtures. In the silica gel samples, a higher capacity for water adsorption after 5000 TSA cycles was observed in materials with a lower concentration of surface silanol groups, i.e., silica gel became a better adsorbent upon exposure to hot wet CO2. As expected, higher surface areas were associated with higher adsorption capacities.In terms of looking at the degradation of commercial desiccants, these results are important for materials applications in different process scenarios. For example, dehydration for the purposes of cryogenic liquefaction of natural gas would not benefit from CO2, but could benefit from wet gas regeneration. Alternatively, raw gas conditioning at the wellhead would benefit from the presence of CO2, H2O in the regeneration gas and a lower regeneration temperature (lower dew-point requirements). Future work should consider the effect of binders, which will be challenging for 5000 cycles. Here the rapid cycling is possible due to the small beds explored, whereas larger tests require both large laboratory gas flow and longer experimental time (years versus months).
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The funding for this research was provided through the Natural Science and Engineering Research Council of Canada (NSERC) and Alberta Sulphur Research Ltd. (ASRL) Industrial Research Chair in Applied Sulfur Chemistry. In addition to NSERC, the authors are grateful to the feedback from the member companies of ASRL.References
- A. L. Kohl and R. Nielsen, Gas purification, Gulf Pub, Houston, Tex, 5th edn, 1997 Search PubMed.
- Z. T. Ward, R. A. Marriott, A. K. Sum, E. D. Sloan and C. A. Koh, Equilibrium Data of Gas Hydrates containing Methane, Propane, and Hydrogen Sulfide, J. Chem. Eng. Data, 2015, 60, 424–428 CrossRef CAS.
- S. Zarinabad and A. Samimi, Problems of Hydrate Formation in Oil and Gas Pipes Deals, J. Am. Sci., 2012, 8, 1007–1010 Search PubMed.
- P. Gandhidasan, A. A. Al-Farayedhi and A. A. Al-Mubarak, Dehydration of natural gas using solid desiccants, Energy, 2001, 26, 855–868 CrossRef CAS.
- L. Popoola, A. Grema, G. Latinwo, B. Gutti and A. Balogun, Corrosion problems during oil and gas production and its mitigation, Int. J. Ind. Chem., 2013, 4, 35 CrossRef.
- R. W. Baker and K. Lokhandwala, Natural Gas Processing with Membranes: An Overview, Ind. Eng. Chem. Res., 2008, 47, 2109–2121 CrossRef CAS.
- J. M. Campbell and R. N. Maddox, Gas conditioning and Processing: Gas Treating and Sulfur Recovery, Campbell Petroleum Series, Norman, Okla, 4th edn, 1998 Search PubMed.
- K. G. Wynnyk, B. Hojjati, P. Pirzadeh and R. A. Marriott, High-pressure sour gas adsorption on zeolite 4A, Adsorption, 2017, 23, 149–162 CrossRef CAS.
- K. G. Wynnyk, B. Hojjati and R. A. Marriott, High-Pressure Sour Gas and Water Adsorption on Zeolite 13X, Ind. Eng. Chem. Res., 2018, 57, 15357–15365 CAS.
- K. G. Wynnyk, B. Hojjati and R. A. Marriott, Sour Gas and Water Adsorption on Common High-Pressure Desiccant Materials: Zeolite 3A, Zeolite 4A, and Silica Gel, J. Chem. Eng. Data, 2019, 64, 3156–3163 CrossRef CAS.
- Gas Processors Supplier's Assoc. (GPSA), Engineering Data Book, Tulsa, OK, 1987 Search PubMed.
- R. H. Herold and S. Mokhatab, Optimal design and operation of molecular sieves for gas dehydration-Part 1, Gas Process., 2017, 96, 25–30 CAS.
- R. H. Herold and S. Mokhatab, Optimal design and operation of molecular sieves for gas dehydration-part 2, Gas Process., 2017, 96, 33–36 Search PubMed.
- D. M. Ruthven, Principles of adsorption and adsorption processes, Wiley, New York, 1984 Search PubMed.
- C. Li, W. Jia and X. Wu, Experimental Failure-Mechanism Analysis of 4A Zeolites Used for Natural-Gas Drying, Chem. Technol. Fuels Oils, 2015, 51, 245–251 CrossRef CAS.
- W. Lutz, M. Weber, R. Bertram, R. Kurzhals and G. Kryukova, The Ageing of Silica Gels Affected by Hydrothermal Treatment, Z. Anorg. Allg. Chem., 2011, 637, 421–425 CrossRef CAS.
- R. Gomes Santiago, B. Ferreira dos Santos, I. Gomes Lima, K. Oliveira Moura, D. Carrijo Melo, W. Mantovani Grava, M. Bastos-Neto, S. M. Pereira de Lucena and D. Cristina Silva de Azevedo, Invstigation of premature aging of zeolites used in the drying of gas streams, Chem. Eng. Commun., 2019, 206, 1367–1374 CrossRef.
- M. Suckow, W. Lutz, J. Kornatowski, M. Rozwadowski and M. Wark, Calculation of the hydrothermal long-term stability of zeolites in gas-desulphurization and gas-drying processes, Gas Sep. Purif., 1992, 6, 101–108 CrossRef CAS.
- C. J. Heard, L. Grajciar, F. Uhlík, M. Shamzhy, M. Opanasenko, J. Čejka and P. Nachtigall, Zeolite (In)Stability under Aqueous or Steaming Conditions, Adv. Mater., 2020, 32, 2003264 CrossRef CAS PubMed.
- J. H. Jacobs, C. E. Deering, R. Sui, K. L. Lesage and R. A. Marriott, Degradation of desiccants in temperature swing adsorption processes: The temperature dependent degradation of zeolites 4A, 13X and silica gels, Chem. Eng. J., 2023, 451, 139049 CrossRef CAS.
- J. H. Jacobs, C. E. Deering, K. L. Lesage, M. J. Stashick and R. A. Marriott, Rapid Cycling Thermal Swing Adsorption Apparatus: Commissioning and Data Analyses for Water Adsorption of Zeolites 4A and 13X Over 2000 Cycles, Ind. Eng. Chem. Res., 2021, 60, 7487–7494 CrossRef CAS.
- M. M. J. Treacy and J. B. Higgins, Collection of simulated XRD powder patterns for zeolites, Elsevier, Amsterdam, Boston, 5th rev. edn, 2007 Search PubMed.
- D32 Committee, Test Method for Determination of Relative Crystallinity of Zeolite Sodium A by X-ray Diffraction, ASTM International Search PubMed.
- D32 Committee, Test Method for Determination of Relative X-ray Diffraction Intensities of Faujasite-Type Zeolite-Containing Materials, ASTM International Search PubMed.
- T. Seki, K.-Y. Chiang, C.-C. Yu, X. Yu, M. Okuno, J. Hunger, Y. Nagata and M. Bonn, The Bending Mode of Water: A Powerful Probe for Hydrogen Bond Structure of Aqueous Systems, J. Phys. Chem. Lett., 2020, 11, 8459–8469 CrossRef CAS PubMed.
- W. Mozgawa, M. Król and B. Katarzyna, Chemik, 2011, 65, 667–674 CAS FT-IR studies of zeolites from different structural groups.
- L. B. Cappeletti, E. Moncada, J. Poisson, I. S. Butler and J. H. Z. D. Santos, Determination of the Network Structure of Sensor Materials Prepared by Three Different Sol-Gel Routes Using Fourier Transform Infrared Spectroscopy (FT-IR), Appl. Spectrosc., 2013, 67, 441–447 CrossRef CAS PubMed.
- S. Brunauer, P. H. Emmett and E. Teller, Adsorption of Gases in Multimolecular Layers, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- E. P. Barrett, L. G. Joyner and P. P. Halenda, The Determination of Pore Volume and Area Distributions in Porous Substances. I. Computations from Nitrogen Isotherms, J. Am. Chem. Soc., 1951, 73, 373–380 CrossRef CAS.
- B. Lippens, Studies on pore systems in catalysts V. The t method, J. Catal., 1965, 4, 319–323 CrossRef CAS.
- P. Rzepka, Z. Bacsik, A. J. Pell, N. Hedin and A. Jaworski, Nature of Chemisorbed CO2 in Zeolite A, J. Phys. Chem. C, 2019, 123, 21497–21503 CrossRef CAS.
- R. Roque-Malherbe, R. Polanco-Estrella and F. Marquez-Linares, Study of the Interaction between Silica Surfaces and the Carbon Dioxide Molecule, J. Phys. Chem. C, 2010, 114, 17773–17787 CrossRef CAS.
- L. T. Zhuravlev, The surface chemistry of amorphous silica. Zhuravlev model, Colloids Surf., A, 2000, 173, 1–38 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The tabulated data for the capacities of the desiccants over the continuous cycling, and the N2 physisorption results of the samples. See DOI: https://doi.org/10.1039/d3nj00093a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2023 |