
Gelation-induced enhanced emission active stimuli-responsive low-molecular-weight organogelator: dual-channel recognition of cyanide from water and food samples with superhydrophobic surface formation†
Kingshuk
Debsharma
 *a,
Sunanda
Dey
bc,
Jyothibabu Sajila
Arya
a,
Krishna Sundar
Das
d,
Chittaranjan
Sinha
b and
Edamana
Prasad
*a
*a,
Sunanda
Dey
bc,
Jyothibabu Sajila
Arya
a,
Krishna Sundar
Das
d,
Chittaranjan
Sinha
b and
Edamana
Prasad
*a
aDepartment of Chemistry, Indian Institute of Technology Madras, Chennai 600036, India. E-mail: ju34kingshukdbs@gmail.com; cy16d041@smail.iitm.ac.in
bDepartment of Chemistry, Jadavpur University, Kolkata 700032, India
cDepartment of Chemistry, Mrinalini Datta Mahavidyapith, Birati, Kolkata, 700051, India
dDepartment of Chemistry, Indian Association for the Cultivation of Science (IACS), Jadavpur, 700032, West Bengal, India
First published on 8th February 2023
Abstract
A pyrene-appended low-molecular-weight organogelator (LMWG) [2-(4-nitrophenyl)-3-(pyren-1-yl)acrylonitrile] (CN1) is presented which exhibits mechano–thermo-responsive gelation, superhydrophobic surface formation and naked-eye colorimetric and fluorogenic dual-channel cyanide sensing from different real-world water and food samples based on gelation-induced enhanced emission (GIEE). Attaching pyrene and 4-nitrophenyl acetonitrile enables CN1 to orient into a J-type aggregate, where restricted intramolecular rotation (RIR) results in GIEE without supportive gelling units. In the presence of cyanide ions (CN−), the cyanovinyl unit in the GIEEgen triggers a selective Michael-type nucleophilic addition, leading to qualitative and quantitative sensing both in solutions [detection limit (DL): 5.42 ± 0.256 nM], gels (DL = 0.48 ± 0.01 μM) and thin films (134.24 pg cm−2). Importantly, in the absence of self-assembly, the complete hindrance of optical sensing is noticed for the CN1 congener [2-(4-fluorophenyl)-3-(pyren-1-yl)acrylonitrile] (F1), which emphasizes the importance of the functional supramolecular material for achieving a dual-channel sensory response towards GIEE-driven day-to-day practical applications.
Introduction
The gelation propensity in π-conjugated low-molecular-weight organogelators (LMWGs, molar mass ≤3000) has led to the development of well-ordered supramolecular arrays, guided by multiple weak non-covalent interactions (π-interactions, hydrogen bonding interactions and different van der Waals forces).1–3 In contrast to gel formation in polymeric and inorganic counterparts, the evolved ‘softness’ in the structural framework, which is due to malleable intermolecular forces, enables the low-molecular-weight organogels to respond sensitively to various environmental stimuli such as temperature,4 shearing forces,5 various analytes (cations, anions),6 biomolecules and so on.7 By virtue of π-electron delocalization, intrinsic electronic properties like luminescence can be greatly modulated via the molecular packing in such molecular systems.8,9 However, most of the LMWGs are known to emit brightly only from dilute solutions, and their luminescence efficiency is diminished dramatically upon self-assembly where severe co-facial dispositions amongst the flat planar polycyclic molecules cause π-electron delocalization, which weaken the light emission.10 This detrimental effect is termed aggregation-caused quenching (ACQ), which seriously obstructs the real-world applications of self-assembled organic molecules.11,12Aggregation-induced enhanced emission (AIEE) in low-molecular-weight organogels enables researchers to counter the effect of ACQ by developing LMWGs with typical AIEE units (AIEEgens) along with various molecular groups [steroidal units, urea, nucleobases, surfactants, long alkyl chains, poly(aryl ether) dendrons]. The combined effect from the particular AIEEgen and the molecular interactions, assisted by the tethered substituents, is found to be beneficial for gelation and gelation-induced enhanced emission (GIEE).10,13 Such LMWGs can be utilized to fabricate stimuli-responsive materials, where ‘smart’ alteration of the GIEE enables the materials to act as fluorescence-based sensors towards toxic analytes such as cyanide (CN−), which is a potential threat to human life and the environment.14,15 In spite of its tremendous applications in industrial fields (viz., metallurgy, tanning, herbicides, gold extraction, electroplating and so on), the accidental release of CN−, mostly via industrial waste water followed by its interaction with mammalian bodies via oral exposure, dermal exposure or inhalation, can cause serious signs of damage in the form of vomiting, convulsions, and disruption of the respiratory system owing to its binding with iron (Fe3+) in metalloenzymes, which ultimately leads to death, even within a few minutes.16–18 In addition, more than 1000 kinds of plants, food crop species (sprouted potatoes, apples and bitter seeds, almonds, etc.) are known to contain CN− in the form of cyanogenic glycoside, where their direct consumption results severe neurological diseases as a result of the toxification of HCN through hydrolysis of the cyanogenic glycosides.18–20 Therefore, it is of the utmost importance to develop sensitive and selective CN− sensors with efficient and fast responsive activities in the contemporary arena of green chemistry.21 Upon comparing various analytical techniques (electrochemistry, flow injection, chromatography),22 naked eye dual-channel (colorimetric and fluorogenic) sensing has emerged as superior owing to the sensitivity of its visual output during real-time detection.23,24 Chemodosimetric sensing, initiated via the irreversible nucleophilic reaction of CN− with the active functional group (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, CN, CO) in the probe is found to be dominant over other reported mechanisms (viz., hydrogen bonding,25 and displacement strategies26) on the basis of former's sensitivity towards CN−.27,28 However, most CN− sensors are known to suffer from the drawbacks of requiring tedious synthesis steps, which include adverse reaction conditions (high temperatures, long reaction times, multistep derivatization processes), low sensitivity and high detection limits (DL), where some of them provide the sensory response only in the presence of additional supportive analytes, which sometimes limits their sensory activity for practical applications.14,16
C, CN, CO) in the probe is found to be dominant over other reported mechanisms (viz., hydrogen bonding,25 and displacement strategies26) on the basis of former's sensitivity towards CN−.27,28 However, most CN− sensors are known to suffer from the drawbacks of requiring tedious synthesis steps, which include adverse reaction conditions (high temperatures, long reaction times, multistep derivatization processes), low sensitivity and high detection limits (DL), where some of them provide the sensory response only in the presence of additional supportive analytes, which sometimes limits their sensory activity for practical applications.14,16
Thus, innovating an LMWG through a single step, which exhibits sensitive stimulus-responsive behavior towards CN− ions through a dual-channel pathway is highly desirable. In the present study, a pyrene-based LMWG (CN1) is synthesized via a single-step Knoevenagel condensation reaction.29,30 It is hypothesized that, attaching a known ACQphore (pyrene) with a twisted functional core might produce GIEEgenic behavior, even in the absence of typical AIEEgens. From the design of the LMWG it is also predicted that the aggregated (solid and gel) state should disclose the stimuli responsiveness and sensitive recognition towards CN− through multiple avenues. Moreover, the enhanced tendency for aggregation in CN1 is utilized to develop superhydrophobic surfaces without incorporating any additional hydrophobic core units.
Results and discussion
Rationale for the design
The pyrene-based LMWG, CN1 [2-(4-nitrophenyl)-3-(pyren-1-yl)acrylonitrile], was synthesized based on a single-step Knoevenagel condensation reaction between 1-pyrene carboxaldehyde and 4-nitrophenyl acetonitrile.29,30 The detailed synthetic route is mentioned in the (Scheme S1, ESI†). The formation of the product was confirmed via spectroscopic and high-resolution mass spectrometry (HRMS) techniques. The 1H NMR spectrum of CN1 in DMSO-d6 showed a sharp characteristics peak at 9.30 (s, 1H), and other signals at 8.67 (d, 1H), 8.61 (d, 1H), 8.38–8.47 (m, 5H), 8.36 (d, 1H), 8.25–8.29 (m, 4H), and 8.19 (t, 1H), which supported the formation of the desired compound (Fig. S1, ESI†). The ESI-MS+ spectrum also showed an ion peak at m/z 374.1091, consistent with the formation of [CN1 + H]+, and at m/z 392.1393 for [CN1 + NH4]+ (Fig. S2, ESI†).
Fig. 1(a) depicts the molecular structure of CN1. Designing such a molecular system relied on the following considerations: (1) the flat, planar, polycyclic architecture of pyrene will offer intermolecular π-interactions, responsible for one of the driving forces behind gelation. (2) Even though pyrene has been considered to be a typical ACQphore,31 attaching it to a flexible functional unit may result in specific J-type self-assembly, where RIR behavior will instigate enhanced emission from the system. It is expected that the unfavorable co-facial arrangement of native pyrene will be prevented in the present case, even in the absence of well-known AIEE units. (3) The presence of the anion-responsive cyanovinyl unit in CN1 might furnish a Michael type reaction exclusively in the presence of nucleophilic CN−, which will result in the desired sensory response. The dihedral angles of pyrene and the nitrophenyl unit with the cyanovinyl moiety were confirmed to be 46° and 30°, respectively, from the energy optimized structure of CN1 (Fig. 1b) calculated at the B3LYP/6-311(+)G level of theory. The higher magnitudes of the dihedral angles produced definite twisting in CN1, activated by the intramolecular steric repulsion between the bulky cyano moiety and ortho hydrogen atoms, which results in torsional-driven non-radiative spoiling of the excitation energy.32 Interestingly, in the condensed phase, the presence of different intermolecular interactions between two pyrene units [optimum distance = 3.78 Å (i)], cyano groups [optimum distance = 3.04 Å (ii)], and pyrene and phenyl units [optimum distance = 3.45–3.73 Å (i)], together with cyano and hydrogen atoms [optimum distance = 4.83 Å (iii)] rigidifies the molecular structure in a particular head-to-tail conformation, as illustrated by the energy-optimized dimer structure of CN1 (Fig. 1c). The desired structural stability is sufficient to freeze the torsional motions in the dimeric structure by overcoming low rotational energy losses at the C–C bonds of pyrene and phenyl units with the central double bond of the cyanovinyl motif, which significantly suppresses the non-radiative relaxation pathways when avoiding the co-facial arrangement of luminogens in the aggregated state.33 Moreover, the spontaneous tendency towards head-to-tail arrangement might develop constructive J-type self-assembly when a greater number of molecules are allowed to participate in aggregation, where the augmented rigidity will lead to tight molecular packing which results in enhanced PL. Accordingly, the sliding angle between the line joining the centres of two adjacent molecules and the long molecular axis of any one of the participating molecules in the dimeric structure is found to be 34.31° [Fig. 1c, (iv)] which determines the J-aggregation.34,35 The electrostatic potential landscape of CN1 is given in (Fig. 1d). The π-rich character of aromatic pyrene is made available for a charge transfer (CT) interaction with the π-inferior nitrophenyl unit due to the strong electron-withdrawing nature of the nitro group (–NO2) group. However, the substantial electropositive potential of the CC bond (indicated by the blue arrow), inspired by the concomitant electronegative potential of the cyano group (indicated by the red arrow), manifests the nucleophilic addition of anions on the cyanovinyl unit in CN1, which might cause the real-time sensing event by disturbing the initial CT effect in the donor–acceptor (D–A) architecture that can conveniently be perceived via dual-channel recognition from the solution and emissive aggregated states.
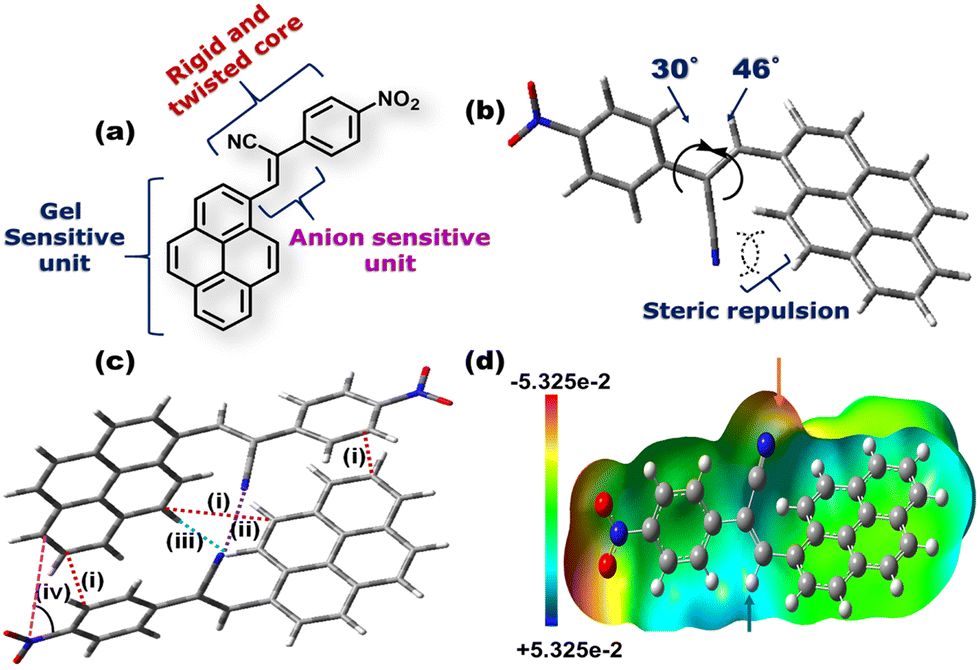 | ||
Fig. 1 (a) Molecular structure of CN1. (b) Geometry optimized simulated structure of CN1 calculated at the B3LYP/6311(+)G level of theory. (c) MM+ optimized dimer structure of CN1 with intermolecular π⋯π (i), C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N⋯NC (ii) and CN⋯H interactions (iii), and sliding angles (iv) between two molecules. (d) Electrostatic potential map [B3LYP/6311(+)G] depicting the electropositive and electronegative potentials of the CC and CN units in CN1, as indicated by the blue and red arrow, respectively (scale values are in kcal mol−1). N⋯NC (ii) and CN⋯H interactions (iii), and sliding angles (iv) between two molecules. (d) Electrostatic potential map [B3LYP/6311(+)G] depicting the electropositive and electronegative potentials of the CC and CN units in CN1, as indicated by the blue and red arrow, respectively (scale values are in kcal mol−1). | ||
Self-assembly features of CN1: investigation of gelation behavior
As anticipated, the prevalence of different non-covalent interactions might promote the present system to construct supramolecular architecture, and the gelation behavior of CN1 was examined by mixing the luminogen in a series of solvents and solvent mixtures with heating, followed by slow cooling at room temperature. To our surprise, CN1 was able to develop a self-supporting gel by hindering the free-flow of solvent molecules in DMSO, DMF, decanol, o-dichlorobenzene, mesitylene, and anisole as well as in DMSO/water, DMF/water, anisole/methanol, anisole/hexane and anisole/chloroform (Table S1, ESI†). The ‘stable to inversion of the vial’ technique36,37 confirmed the reluctance of the gel to exhibit any gravitational flow, confirming that CN1 acted as a non-traditional π-conjugated LMWG, which endured ambient conditions for several weeks even in the absence of any classical gelling units. Subsequently, the gelation propensity of CN1 was characterized via XRD, microscopic and rheological techniques. The PXRD pattern of the CN1 xerogel from DMSO (Fig. 2a) exhibited several well-resolved diffraction peaks, at 2θ = 5.30°, 8.31° and 10.71° in the low-diffraction-angle regime (2θ < 15°), which indicated d-spacing values of 16.65 Å (d100), 10.63 Å (d110) and 8.25 Å (d010), respectively, in the ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1/√3:1/√4 and accounted for the hexagonal columnar assembly (Colh) with an intercolumnar distance of 19.23 Å (Scheme 1).35,37 Also, the wide-angle XRD region (2θ > 20°) indexed the ‘d’ value of 3.56 Å (2θ = 24.94°), which is attributed the π-interaction between two pyrene units,38 and agreed well the theoretically obtained intermolecular π-interaction based on the energy-optimized dimer structure of CN1 (inset of Fig. 2a). Moreover, the calculated molecular length obtained from the fully optimized geometry of CN1 was found to be 13.68 Å, which matched closely with the d-spacing of 16.65 Å (2θ = 5.3°) (Fig. 2a, including inset). Since the molecular length obtained via both experiment and theory did not differ significantly, the first peak indeed originated from a head-to-tail orientation via the inclination of monolayer species whose optical properties should be consistent with that of a typical J-type self-assembly, where the steric requirement of the cyano group favoured a somewhat tilted stacking instead of assembling molecules in the face-to-face orientation.39
:1/√3:1/√4 and accounted for the hexagonal columnar assembly (Colh) with an intercolumnar distance of 19.23 Å (Scheme 1).35,37 Also, the wide-angle XRD region (2θ > 20°) indexed the ‘d’ value of 3.56 Å (2θ = 24.94°), which is attributed the π-interaction between two pyrene units,38 and agreed well the theoretically obtained intermolecular π-interaction based on the energy-optimized dimer structure of CN1 (inset of Fig. 2a). Moreover, the calculated molecular length obtained from the fully optimized geometry of CN1 was found to be 13.68 Å, which matched closely with the d-spacing of 16.65 Å (2θ = 5.3°) (Fig. 2a, including inset). Since the molecular length obtained via both experiment and theory did not differ significantly, the first peak indeed originated from a head-to-tail orientation via the inclination of monolayer species whose optical properties should be consistent with that of a typical J-type self-assembly, where the steric requirement of the cyano group favoured a somewhat tilted stacking instead of assembling molecules in the face-to-face orientation.39
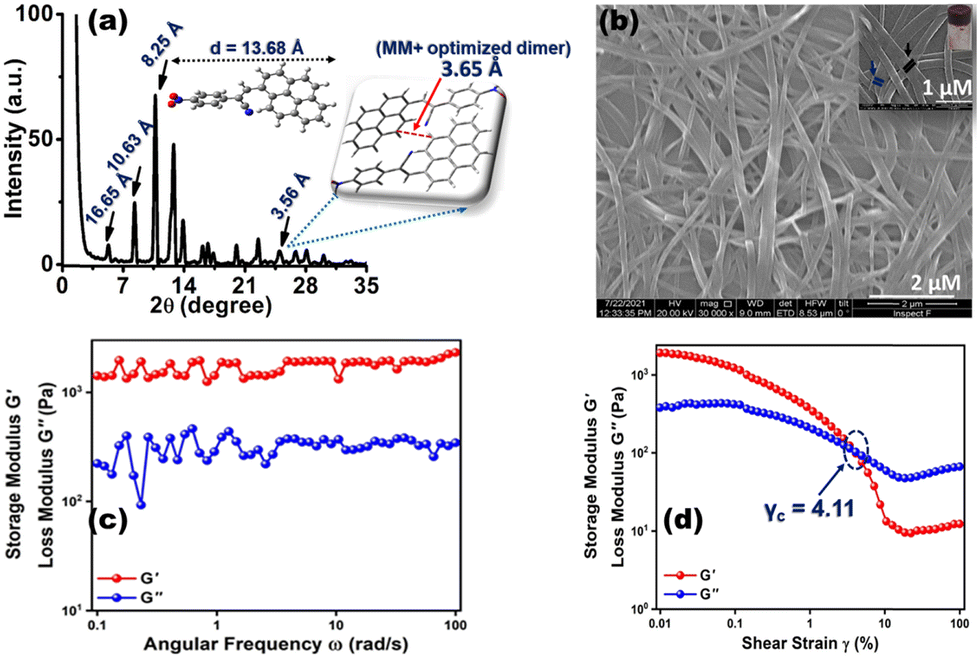 | ||
| Fig. 2 (a) PXRD pattern of the CN1 xerogel (1 wt%, DMSO). The corresponding inset shows the calculated molecular length [B3LYP/6311(+)G] of simulated CN1 and the π-interaction between two pyrene units of the MM+-optimized CN1 dimer. (b) SEM image of the CN1 xerogel (1 wt%, DMSO) is showing the entangled and robust fibrous morphology. The fibres are interpenetrated and knotted along the same nodes as represented by inset of (b). (c) Oscillatory frequency sweep and (d) amplitude sweep rheological studies for the CN1 gel (1 wt%, DMSO). | ||
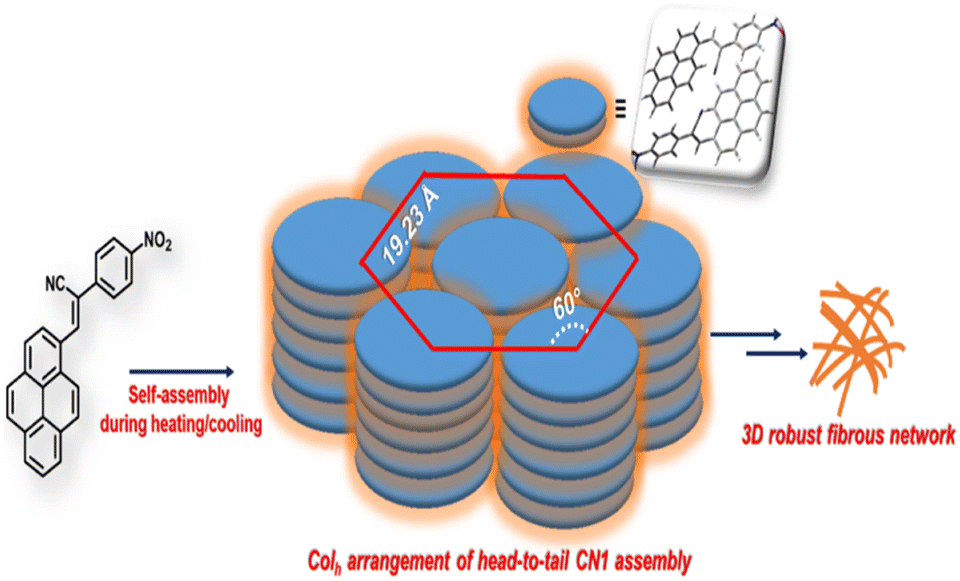 | ||
| Scheme 1 Plausible self-assembly pathway of CN1 during the heating/cooling event. The packing arrangement is believed to consist of a slipped head-to-tail ensemble of CN1, as illustrated by the optimized dimer structure in the inset. | ||
In order to observe the visual impact of the gel morphology, FE-SEM (field-emission scanning electron microscopy) images of the CN1 xerogel obtained from DMSO were examined. Subsequently, the opaque gel formed at 10 mg mL−1 (1 wt%) revealed a hierarchically oriented interpenetrating three-dimensional (3D) network, comprised of intertwined fibrous aggregates (Fig. 2b). Closer inspection of the entangled network suggested that the existing fibres were tightly bundled and adjoined at the same node accompanied with the voids (inset of Fig. 2b), which was responsible for entrapping and immobilizing the solvent molecules and ultimately leading to gelation. To achieve solid-like behavior, the dynamic storage modulus (G′) of a gel, which represents its elastic nature, should be higher than the loss modulus (G′′), which represents the flow behavior of a gel.40,41Fig. 2c shows the results of a frequency sweep experiment for the DMSO gel at room temperature, monitored within the frequency range from 0.1 to 100 rad s−1 at a constant strain of 0.01%. These results demonstrated that G′ remains more or less independent of the applied oscillatory frequency, and that its magnitude is higher than G′′ over the entire tested frequency range, indicated the typical solid-like nature of the gel.42 Again, in the amplitude sweep experiment carried out for the same gel (Fig. 2d), which monitored the strain (γ) region of 0.01–100% at a constant angular frequency of 10 rad s−1, it was demonstrated that, in the initial stage, G′ was higher than G′′ and the elastic nature of the gel overruled its flow behavior. Subsequently, with increasing strain%, both G′ and G′′ were decreased, indicating partial breakage of the gel structure; even though the gel successfully retained its solid structure with G′ > G′′.43 However, as the stain% was increased further, the gel lost its elastic/solid character, as indicated by the cross-over of G′ and G′′ at a particular strain%. Beyond that point, the flow behavior of the gel exceeded that if its solid character (G′′ > G′). The cross-over point (γc) of the DMSO gel was found to be at 4.11% of the strain amplitude or 5.83 Pa of shear stress as obtained from the stress sweep test (Fig. S3, ESI†).
Photophysical features of CN1: investigation of the GIEE and solid-state PL characteristics
Initially, the photophysical properties of CN1 were probed by monitoring the UV/Vis absorption and emission characteristics in dimethyl sulfoxide (DMSO). The absorption spectrum of CN1 (Fig. S4, left, ESI†) exhibited a peak maximum (λa) at 415 nm along with a shoulder band at 390 nm. The lower energy absorbance was attributed to the charge transfer transition between the pyrene and nitrophenyl units, and the higher energy absorption was attributed to the π–π* transition of the aromatic segments.44 The emission spectrum of CN1 in DMSO (Fig. S4, right, ESI†) exhibited a maximum (λf) at 600 nm with poor magnitudes of both the fluorescence lifetime (τf) and the emission quantum yield (ϕf) (Table 1). Evidently, the typical non-luminescent features of CN1 indicated the presence of different conformations in the excited-state lifetime of the luminogen, where the desired twisting in the structural framework resulted in either a quinoidal resonance structure or twisted intramolecular charge transfer species in the molecularly dissolved state of CN1.45 Subsequently, taking the magnitudes of τavg and ϕf, the radiative (kr) and non-radiative (knr) relaxation constants were calculated for the luminogen, where the knr values were found to be higher than those of kr (Table 1). The above observation clearly supports the prevailing non-radiative relaxation of the excitation energy as described previously.18 Interestingly, conspicuous variations in the photophysical characteristics of CN1 were noticed in the self-assembled states (the gel and the as-synthesized powder). From Fig. 3a it is shown that, in contrast to the absorption spectrum in dilute solution, the absorption spectra of CN1 in the xerogel and solid-state forms broadened significantly in the higher wavelength region, with the appearance of red-shifted structureless bands around 480 nm for the xerogel and at 450 nm for the powder. For both the cases, the tailing in the absorption spectrum in the higher wavelength region (Fig. S5, ESI†) signifies scattering from the aggregates.46 Moreover, the apparent similarity between the xerogel and the solid-state absorbance certainly suggest the identical packing arrangements in aggregated states. Also, the analogous PXRD patterns of both the CN1 powder and xerogel (Fig. S6 and Table S2, ESI†) further establish the similar Colh assembly.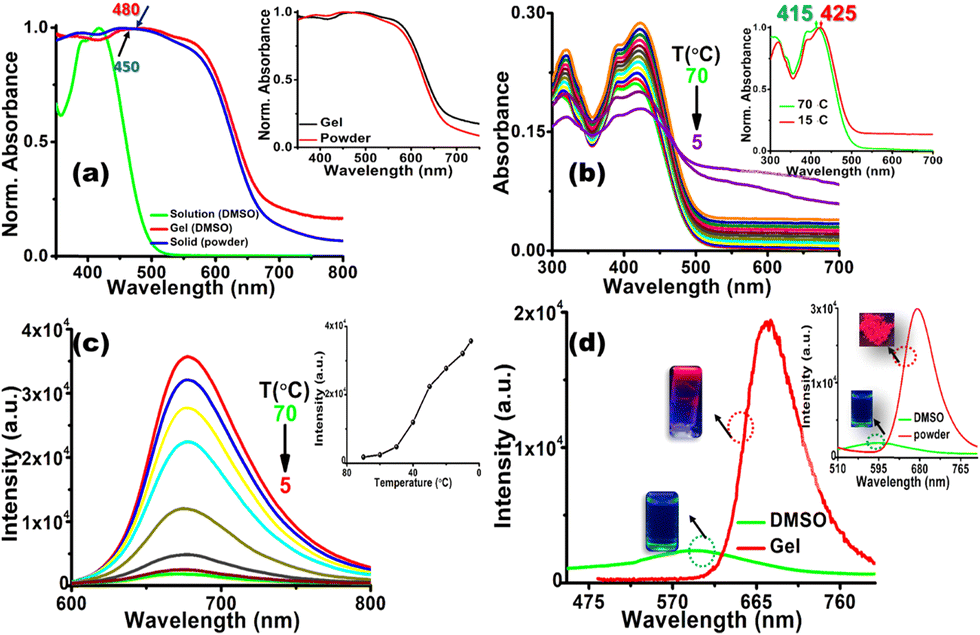 | ||
| Fig. 3 (a) Normalized absorption spectrum of CN1 in solution (DMSO, 10 μM), as a xerogel (DMSO, 1 wt%) and in the solid (as-synthesized powder) state. The inset shows the similarity between the absorption spectra of CN1 in the xerogel (DMSO, 1 wt%) and the as-synthesized powder forms. (b) Variation in absorption spectra of CN1 (concentration ∼1 wt%) with decreasing temperature along with the bathochromic shift in the peak position (inset). Herein, DMF was used as the solvent medium instead of DMSO due to the lower freezing temperature of DMF compared with DMSO. (c) Variation in emission spectra of CN1 (concentration ∼1 wt%, λex = 420 nm) with decreasing temperature, together with change in emission intensity versus temperature (inset). (d) Augmented emission spectral response of the CN1 gel (1 wt%, DMSO; λex = 450 nm) and solid powder (inset; λex = 450 nm) compared with that in DMSO solution (λex = 420 nm). The respective photographs were obtained under UV light irradiation (λex = 365 nm). | ||
Presumably, the intermolecular interactions in CN1 caused the conformational planarization in the aggregated states, where the red-shifted broad and structureless absorption spectra confirmed the tight molecular array of CN1 molecules in an ordered head-to-tail fashion, which ultimately gives rise to J-aggregation by increasing the effective conjugation length of the self-assembled molecules.33,39Fig. 3b shows the variation in the absorption spectrum of CN1 when a hot solution (70 °C) of the gelator (concentration ∼1 wt%) is allowed to cool to 5 °C. Initially, cooling the solution from 70 °C to 15 °C caused a red-shift in the absorption band from 415 to 425 nm (inset of Fig. 3b) along with appearance of intensified tailing in the higher wavelength region (Fig. S7a, ESI†). Interestingly, further cooling of the solution caused a decrease in the absorbance, with broad spectral features (Fig. S7b, ESI†) which denoted J-aggregate formation.47,48 As expected, the hot solution of CN1 (concentration ∼1 wt%) was non-emissive under UV light irradiation (inset of Fig. 3d); however, the emission intensity increased during cooling of the solution (Fig. 3c) with a bathochromic shift in the peak position (Fig. S8, ESI†) from 670 nm at 70 °C to 680 nm at 5 °C, which discloses a particular signature of J-type self-assembly.34 Consequently, the prevailing J-aggregation caused the CN1 gel and powder samples to reveal notable far-red PL characteristics (λf = 680 nm) with more than ∼8- and ∼10-fold enhancements in the emission intensity compared with that of the monomeric state (Fig. 3d and its inset), which indicates striking GIEE and solid-state PL properties. The superior J-type self-assembly amongst the molecules generated spatial constraints, driven by π-interactions and other van der Waals forces (CN⋯NC and CN⋯H), causing sufficient rigidification of the molecular framework by arresting all possible intramolecular motions that are present in the molecularly dissolved states of CN1. Accordingly, to understand the involvement of the non-covalent interactions behind the construction of emissive supramolecular self-assembly, a temperature-dependent 1H NMR study of CN1 in DMSO-d6 was carried out. The 1H signals corresponding to the aromatic protons showed a considerable downfield shift at 70 °C upon increasing the solution temperature gradually from 25 to 70 °C (Fig. S9 and Table S3, ESI†). This observation clearly indicates the presence of intermolecular interactions amongst the molecules which suffered considerable disruption at higher temperature, resulting in the observed variation of the NMR signals.49 In order to obtain the further information on the different intermolecular interactions, a concentration-dependent 1H NMR study of CN1 was carried out (Fig. S10, ESI†). Concurrently, increasing the concentration from ∼3 mM to ∼15 mM caused a significant upfield shift for most of the aromatic as well as the vinyl protons of CN1 (Table S4, ESI†) due to intermolecular π-interactions (π⋯π and C–H⋯π), which resulted in shielding of the aromatic ring over the respective protons.22,34 Conversely, the downfield shift of some of the aromatic protons (Fig. S11 and Table S4, ESI†) specified weak intermolecular CN⋯H interactions between neighbouring molecules, where sufficient polarity enables the cyano group to interact with adjacent aromatic protons.50 Also, to demonstrate the role of the cyano interaction (CN⋯NC), FT-IR spectroscopic measurements were carried out (Fig. S12, ESI†). A dilute solution of CN1 exhibited a characteristic cyano stretching frequency at 2275 cm−1. Upon aggregation, a noticeable variation in the frequency from 2275 to 2210 cm−1 in the powder and 2208 cm−1 in the xerogel clearly showed the existing intermolecular cyano interactions in the self-assembled state of CN1.50 Consequently, those intermolecular interactions caused RIR upon aggregation, which in association with the prevailing J-type aggregation opened up the radiative channel in a dominating way and caused the clear enhancement of the emission. Evidently, ϕf (Table 1) and τf (Fig. S13, ESI† and Table 1) were improved significantly from 0.60% and 0.38 ns, respectively, in DMSO to 28.00% and 4.06 ns for the gel and 31.00% and 4.33 ns for the solid, quantitatively demonstrating the momentous GIEE (αGIEE, ϕf,xerogel/ϕf,DMSO = 46.67 s−1) and AIEE (αAIEE, ϕf,powder/ϕf,DMSO = 51.67 s−1) effects from the gel and solid states, respectively.
Finally, to affirm RIR as one of the primary mechanisms for the enhanced PL characteristics, the emission behavior of CN1 was monitored in DMSO with an increasing fraction of glycerol added [fgly(%)s]. Subsequently, emission spectrum of CN1 was found to be enhanced significantly with increasing fgly(%)s, where at fgly = 90%, a 2.4-fold enhancement in the emission intensity was noted compared with that in pure DMSO (fgly = 0%) (Fig. S14a and its inset, ESI†) along with a hypsochromic shift in the peak position from 598 nm to 538 nm (Fig. S14b, ESI†). The observed spectral features presumed that the bridging interactions of the glycol units to bring the molecules closer to each other suppressed the free rotation in CN1, resulting in the blue-shifted enhanced PL spectra due to RIR.51
Stimuli-responsive behavior of the CN1 gel
It is noteworthy that, the GIEE feature produced in the CN1 gel, impregnated with dynamic non-covalent interactions, benefited us for monitoring the variation in the self-assembly in real-time by looking for reversible fluorescence modulation through a tunable and repeated gel-to-sol phase transition in the presence of different external stimuli. Therefore, when the gel was shaken manually, its 3D architecture collapsed immediately to a quasi-liquid sol upon turning the vial upside down (Fig. 4a). Interestingly, the gel structure was restored by keeping the vial undisturbed for 10 minutes. From this observation, it was anticipated that the low-molecular-weight organogel based on CN1 can exhibit reversible thixotropic behavior during alternating shaking/resting cycles. Meanwhile, the bright far-red emission underwent considerable quenching (‘OFF’) once the as-synthesized gel had suffered mechanical shearing. The emission intensity was revived (‘ON’) once the agitation was discontinued (Fig. 4b). This reversible OFF–ON switching could be repeated for several cycles without any significant fatigue of the gel emission (inset of Fig. 4b). Furthermore, these isothermal thixotropic features were inspected by disrupting the gel structure after being submitted to a higher strain (30%) in a time sweep experiment followed by reducing the strain values to 0.01% in the subsequent steps, separated by reasonable time gaps (Fig. 4c). The CN1 self-assembly disclosed complete gel-to-sol (G′ > G′′) and sol-to-gel (G′′ > G′) switching that occurred repeatedly during the sequential application of high and low strains, which confirmed its reversible stimulus-responsive and self-healing nature with a profound tendency of recovery, suitable for practical applications.37,52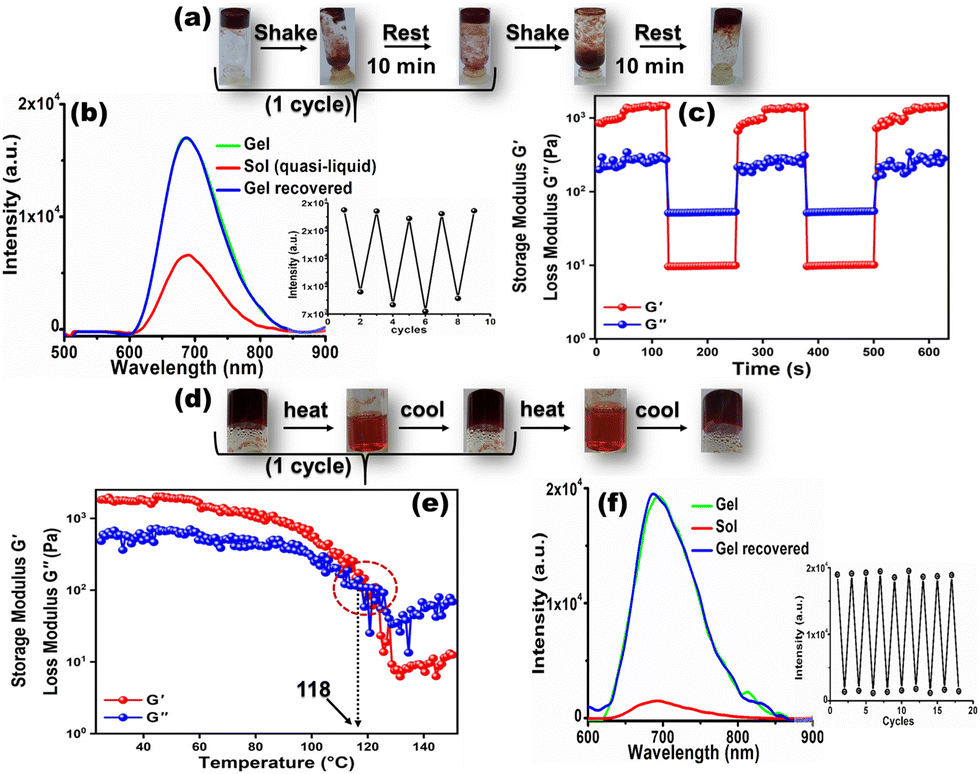 | ||
| Fig. 4 (a) Photographs of the reversible integration/disintegration of the gel structure of CN1 during alternating shaking/resting events. (b) Variation in the GIEE characteristics of the CN1 gel (DMSO; 1 wt%) when subjected to 1 cycle of shaking and resting. The inset shows the ON–OFF PL switching during the reversible thixotropic cycles (λex = 450 nm). (c) Time-sweep rheological profile of the CN1 gel (DMSO; 1 wt%) subjected to repeated higher (30%) and lower (0.01%) strains. (d) Photographs showing the reversible thermochromic characteristics of the CN1 gel (DMSO; 1 wt%) during alternating heating/cooling events. (e) Temperature-sweep rheological profile of the CN1 gel (DMSO; 1 wt%). (f) Variation in the GIEE characteristics of the CN1 gel (DMSO; 1 wt%) when subjected to 1 cycle of heating and cooling. The inset denotes the ON–OFF PL switching of the CN1 gel during reversible thermochromic cycles (λex = 450 nm). | ||
Again, the evolved self-assembled CN1 gel was found to exhibit completely thermo-reversible characteristics (Fig. 4d). Initially, the gel-to-sol phase-transition temperature (Tg) was determined via a temperature-sweep experiment where the variation in G' and G′′ was evaluated as a function of the temperature (Fig. 4e). The solid-like structural stability of the CN1 gel enabled G′ to be independent of temperature up to 100 °C, where the elasticity of the gel network was due to the predominant enthalpic contribution. When the temperature was higher than 100 °C, G′ dropped rapidly and crossed over G′′ at 118 °C, which directed the melting of the gel network. Consequently, by heating the assembly above 118 °C, the gel was turned completely into a sol, where the phase transition was accompanied by drastic quenching of the gel emission at 680 nm (OFF) (Fig. 4f). However, instantaneous re-gelation was noticed when the sol was kept at room temperature, with complete restoration of the PL intensity (ON). Subsequently, the reversible sol–gel transformation could be carried out many times without any perturbation in the emission intensity (inset of Fig. 4f). Again, the existence of stimuli-responsiveness via reversible the gel/sol transition was substantiated via the diffuse reflectance UV/Vis spectral responses during shaking/resting and heating/cooling events. Subsequently, the reflectance (%R) of the as-synthesized gel was seen to reduce drastically (Fig. S15a and b, ESI†) after shaking and/or heating, which indicates the collapse of its solid-like 3D architecture to a quasi-liquid sol state (insets of Fig. S15a and b, ESI†) with disrupted non-covalent interactions. Interestingly, once the shaken/heated sol was kept undisturbed the gel architecture was reinstated with the appearance of the original broad reflectance (Fig. S15a and b, and insets, ESI†) due to the restored non-covalent interactions.
Dual-channel spectral responses to CN−
Since the electron deficiency of the CC bond facilitates nucleophilic reactions with anions, the sensory behavior of CN1 was first monitored in the presence of aqueous solutions of different anions, i.e., Br−, I−, Cl−, F−, OAc−, SCN−, AsO3−, H2PO4−, N3−, S2O52−, CO32−, HCO3−, SO42−, HSO4−, S2O32−, NO2−, NO3− and CN−. Amongst the different anions tested herein, CN1 demonstrated a prompt colorimetric response only for CN−, which resulted in a change of the solution color from straw yellow to violet in a selective manner, as observed with the naked eye (inset above Fig. 5a and b, and inset of Fig. 5b).
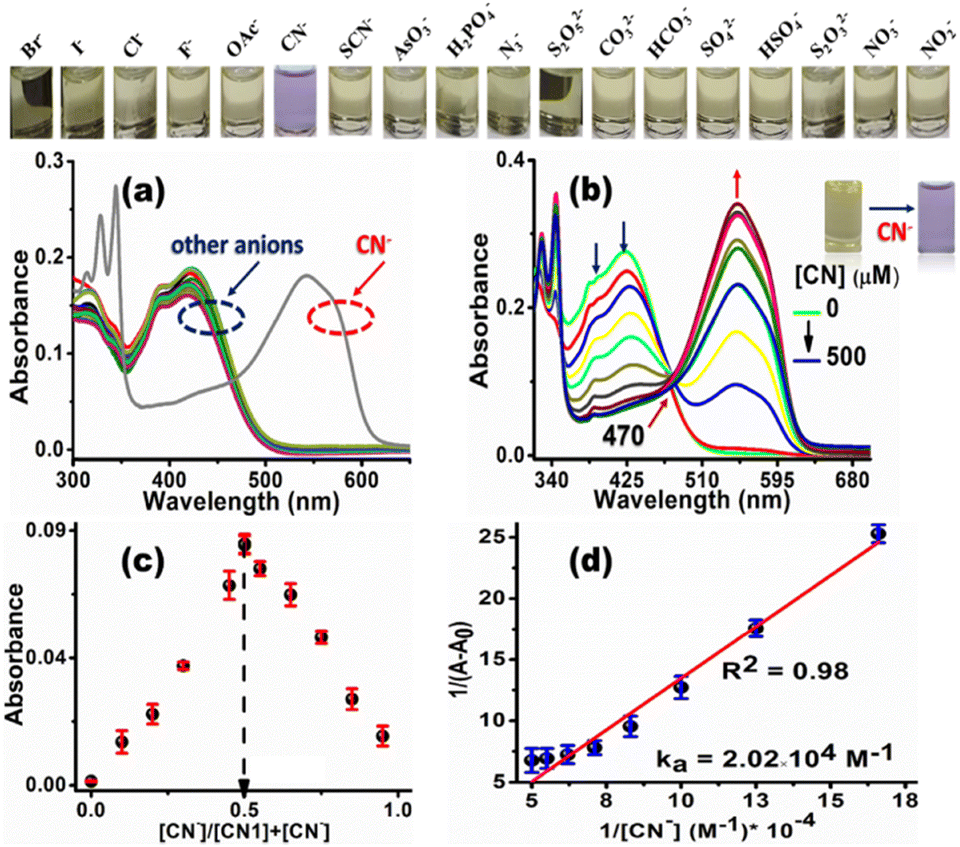 | ||
| Fig. 5 (a) UV/Vis absorption spectra of CN1 (10 μM) in an 8:2 (v/v) DMSO/water mixture in the presence of different aqueous anionic solutions (5 equiv.), where the inset above (a and b) shows the naked eye colorimetric response of the CN1 solution in the presence of each anion. (b) Variation in the UV/Vis absorption spectra of CN1 (10 μM) in an 8:2 (v/v) DMSO/water mixture with increasing aqueous [CN−], where the inset shows the naked eye colorimetric response of the CN1 solution in the presence of CN−. (c) Job's continuous variation plot and (d) Benesi–Hildebrand plot of CN1 (10 μM) with increasing [CN−]. | ||
Concurrently, the absorbance at ∼390 and ∼415 nm in native CN1 decreased dramatically in the presence of CN−, accompanied by the evolution of a new red-shifted absorption peak at 545 nm (Fig. 5a), where the other anions exerted practically no effect on the absorption spectrum of CN1. In addition, the variation in the absorption spectrum, induced by CN− remained more or less unaltered, even with an excess concentration of the other anions (Fig. S16, ESI†), which showed that the selective colorimetric response of CN1 to CN− was scarcely interfered with by the competitive anions. Fascinated by this selective spectral disclosure, the variation in the absorbance was monitored with a successively increasing concentration of aqueous CN− added (i.e., [CN−], 0–50 equiv.) to a 20% aqueous DMSO solution of CN1 via a titration experiment (Fig. 5b). A gradual enhancement in the absorption band at ∼545 nm was noted with increasing [CN−] along with a decrease in the absorbance at ∼390 and ∼415 nm, which indicated the formation of a [CN1–CN−] adduct. Evidently, the isosbestic point that appeared at 470 nm designated the dynamic equilibrium between CN1 and the adduct.53 Consequently, the successively decreasing absorbance at 415 nm was found to be dependent upon the [CN−]. Therefore, to gain a better understanding of the sensitivity of the probe, the concentration-dependent variation of the absorption [(1 − A/A0) (%)] of CN1 was plotted with respect to [CN−] (Fig. S17a, ESI†). The change in the absorbance increased nonlinearly with the increasing addition of CN− and was almost saturated when the analyte concentration was above 350 μM. A linear relationship between the absorbance and [CN−] was observed below 350 μM (Fig. S17b, ESI†), and the detection limit (DL) of CN1 towards aqueous CN− was found to be 5.42 ± 0.256 nM. Through the variation in the absorbance, the colorimetric response was then utilized to determine the mode of binding between the analyte and the probe. From Job's continuous variation method, the maximum absorbance was noted at the mole fraction of ∼0.5 (Fig. 5c), which indicated a 1:1 binding stoichiometry between CN− and CN1.28 Furthermore, the nature of the binding interaction was evaluated from the Benesi–Hildebrand equation (eqn (S1), ESI†),54 where the slope and intercept of the straight line obtained from the double-reciprocal plot between 1/[A − A0] and 1/[CN−] gave rise to a reasonable binding constant of (2.02 × 104) ± 0.045 M−1 (Fig. 5d), which illustrated the strong bonding between CN− and CN1. Meanwhile to affirm the working window of cyanide sensing, the variation in the absorption spectral response of CN1 was first monitored at different pH values. From Fig. S18a, ESI,† it was observed that the absorption spectrum of CN1 was unaffected upon variation of the pH from 2 to 12. In the presence of aqueous CN− in the acidic regime (pH 2–5), the absorption spectral response of CN1 was found to be insensitive towards the analyte as there occurred hardly any evolution of the absorption peak at 545 nm, presumably due to ineffectiveness of the nucleophilic addition of cyanide under acidic conditions (Fig. S18b, ESI†). However, upon increasing the pH (to >5), the peak at 545 nm started to increase, approaching a maximum value at pH 7 and/or 8 before decreasing when under extreme basic conditions (pH > 9) (Fig. S18b and c, ESI†). From this observation, it can be inferred that the working zone for cyanide sensing lies in the pH range between 6 and 8, which matches closely with physiological conditions.55
Encouraged by the selective and sensitive colorimetric outcomes from the solution state, the enriched PL feature of the CN1 self-assembly was subsequently utilized for the fluorogenic sensing of CN−. For this purpose, primarily, a self-assembled portable thin film was fabricated via drop casting of a chloroform solution of CN1 (∼13 mM) onto a glass slide followed by drying under open air. As expected, the aggregated CN1 exhibited a strong far-red emission (λf = 680 nm) from the thin film after solvent evaporation, as observed under UV light (Fig. S19, ESI†). When the thin film was treated using a small cotton swab loaded with solid tetrabutylammonium cyanide (TBACN) to write ‘CN’ on it, the bright red luminescence was found to be quenched drastically on the written part (Fig. 6a), which demonstrated that the nucleophilic reaction of CN− led to the formation of the non-emissive [CN1–CN−] adduct.
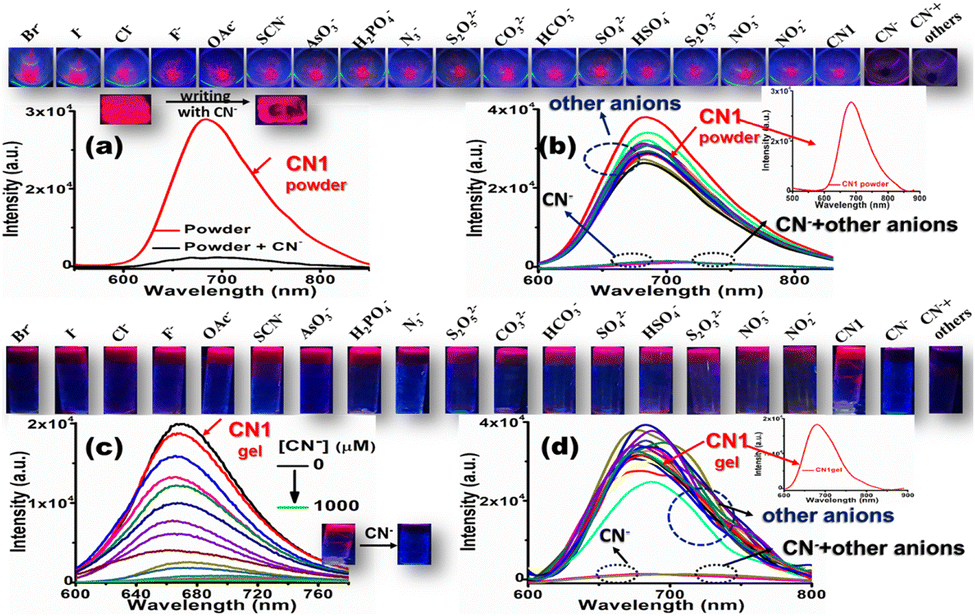 | ||
| Fig. 6 (a) Emission spectral response of CN1 powder upon grinding with solid TBACN (λex = 450 nm). The inset above the spectra shows photographs under UV irradiation of the CN1-coated film before and after writing with a CN−-loaded cotton swab. (b) Variation of the solid-state PL spectrum of CN1 in the presence of different anionic salts (λex = 450 nm), where the inset also shows the initial emission spectrum of the CN1 powder. (c) Variation in the PL spectrum of CN1 gel [8:2(v/v) DMSO/water] in the presence of increasing aqueous [CN−] (λex = 450 nm). (d) Change in the emission spectrum of CN1 gel [8:2 (v/v) DMSO/water] in presence of aqueous solutions of different anions, where the inset also shows the initial emission spectrum of the CN1 gel. The insets above (a and b) and above (c and d) show photographs of ground CN1 solid and CN1 gel respectively in the presence of added solid salts and aqueous solutions of various anions taken under UV light. | ||
Henceforth, when the as-synthesized powder was ground with a trace amount of solid TBACN using a mortar and pestle, the peak at 680 nm underwent notable fluorescence quenching (Fig. 6a). Interestingly, solid salts of other anions, viz., Br−, I−, Cl−, F−, OAc−, SCN−, AsO3−, H2PO4−, N3−, S2O52−, CO32−, HCO3−, SO42−, HSO4−, S2O32−, NO2−, and NO3−, brought about neither a significant change in the fluorescence spectrum nor interference in the selective quenching of CN−, which further demonstrated the excellent selectivity of CN1 for CN− in the solid state (Fig. 6b, the inset above Fig. 6a and b and Fig. S20, ESI†]. Again, the entangled porous and interlinked nanofibrous morphology of the CN1 gel enabled the effective accumulation of CN− as the electron and/or energy trap, followed by its rapid diffusion via interpenetrating nanofibers, which resulted in long-range exciton migration throughout the network, leading to the amplified fluorescence quenching based on the surface-adsorbed analytes. Therefore, when an aqueous solution of CN− was allowed to diffuse successively into the hot DMSO/water [8:2(v/v)] solution of CN1 to develop analyte-containing gels of various strengths, its native bright fluorescence underwent successive quenching (Fig. 6c and its upper panel) with increasing [CN−], with a 30 nm red-shift in the peak position (Fig. S21, ESI†). Even a low [CN−] (<50 μM) induced a significant QE of 33.67 ± 1.780%, which indicated the high sensitivity of CN1 towards CN− in the gel state. Based on the linear fitted curve of the QE (%) with respect to [CN−] (Fig. S22a and its inset, ESI†), the DL was found to be 0.48 ± 0.01 μM. Interestingly, the fibre matrix remained unchanged after treatment with CN−, where the retained 3D aggregated network, as confirmed from the SEM image of the cyanide-treated gel (Fig. S23, ESI†) favoured the self-assembled molecules for the swift detection of CN− without hampering the non-covalent interactions via the competitive binding of CN− in the aggregated structure.56 The kinetics of the fluorescence intensity of the gel, monitored by fitting the intensity decay to a single-exponential equation,57 indicated a rapid response time of 3.33 s in the presence of CN− (Fig. S22b, ESI†) for its detection. Moreover, the gel emission showed negligible variation in the presence of other anions (Fig. 6d, Fig. S24, ESI†], which confirmed the desired selectivity for CN−. Accordingly, the emission intensity of CN1 [8:2 (v/v) DMSO/water] in the presence of aqueous CN− was analysed using the Stern–Volmer equation (eqn (S2), ESI†), which disclosed upward trend in the emission intensity (Fig. S25a, ESI†), indicating a reasonable interaction between the analyte and probe. Incidentally, the linear relationship of the quenched intensity with respect to the analyte concentration ([CN−] < 1.6 equiv./50 μM) gave rise to a quenching constant (KSV) of 26212.86 M−1 (Fig. S25b, ESI†), which was high enough to impart strong binding of the gelator with cyanide. Subsequently, to test the practical applicability, xerogel films of CN1 (∼10 mg mL−1) from an 8:2 (v/v) DMSO/water mixture were utilized for a dual-channel sensing event by simply pressing a swab loaded with various cyanide concentrations on the self-assembled surface (∼1 cm2). From this, CN1 can detect CN− at a concentration of 10−7 M via a visual color change as well as via the quenched emission, monitored under normal and UV light, respectively, without the need for any sophisticated instrumentation (Fig. S26, ESI†). Subsequently, the contact-mode detection of CN− by CN1 was calculated to be as low as 134.24 pg cm−2 (calculation of the DL is mentioned in the ESI†). Furthermore, to confirm the response time of the thin film, the fabricated test-kit was dip-coated using an aqueous CN− solution and the time-dependent variation in the emission spectrum of the film was monitored. After fitting the PL decay kinetics of the CN1 film at 680 nm according to an exponential decay equation (Fig. S27, ESI†), the response time was calculated as 0.30 s. In addition, the sensory response of CN1 was screened in HEPES buffer (pH = 7.4) as well as in a series of mixed buffer solvents with an increasing buffer fraction [fbuffer(%)]. Accordingly, the probe exhibited a colorimetric and fluorogenic dual-channel response only in 8:2 (v/v) DMSO/HEPES and 8:2 (v/v) DMF/HEPES solvent mixtures in the presence of CN−, with the appearance of a red-shifted absorption maximum at 545 nm (Fig. S28a and c, ESI†) accompanied by a change in the solution color from straw yellow to violet (inset of Fig. S28a and c, ESI†) as well as considerable quenching of the emission spectra from both the 8:2 (v/v) DMSO/HEPES and 8:2 (v/v) DMF/HEPES gel states (Fig. S28b and d, ESI† and the respective insets). However, in mixed buffer solutions with fbuffer > 20%, CN1 was unable to produce any colorimetric response (Fig. S29, ESI†), presumably due to the higher hydration energy of CN− with the elevated buffer content. Also, in those solvent systems, CN1 scarcely showed any gelation properties to generate a fluorogenic response towards CN−. Table S5, ESI† presents a comparison of the chemical sensing properties of CN1 with previously reported CN− sensors.
Meanwhile, to check the reversibility of the system, the UV/Vis absorption and emission spectral responses of CN1 were recorded from the simultaneous addition of cyanide (CN−) and trifluoroacetic acid (TFA). Accordingly, it was hypothesized that the addition of TFA to the [CN1–CN−] adduct may result in the acid-induced loss of cyanide for reversible alterations in the optical features of the sensor.58 Interestingly, the addition of TFA tended to restore the original absorbance of the sensor at ∼390 and ∼415 nm at the expense of diminished absorbance at λa = 545 nm (Fig. S30a, ESI†) with the repaired gel emission at λf = 680 nm (Fig. S30b, ESI†), as shown by reversible variations in the solution and gel colors (insets of Fig. S30a and b, ESI†), due to the acid-induced loss of cyanide. These outcomes indicate the reversible sensory response of CN1, which augments its practical applicability as a desirable dual-channel sensor.
The photostability of a sensor is considered to be one of the pivotal factors for ascertaining its practical applicability.59,60 Therefore, to inspect the photostability, CN1 in 8:2 (v/v) DMSO/water, as a thin film and as the synthesized gel was subjected to UV-irradiation for 2 hours and the time-dependent variation in its UV/Vis absorption and emission spectral responses was recorded. From Fig. S31a and (c and e), ESI,† it can be seen that both the UV/Vis absorption and emission spectra of CN1 scarcely show any significant alteration during UV-irradiation and remained more-or-less unchanged, without any fatigue being shown (Fig. S31b, d and f, ESI†). Hence, CN1 exhibits considerable photostability both in the solution and self-assembled states.
Elucidation of the dual-channel sensing mechanism
The Michael-type nucleophilic reaction of CN− was studied by monitoring the variation of the 1H NMR spectrum in the presence of the analyte (Fig. S32, ESI†). The singlet resonance signal of the vinylic proton that appeared at δ = 9.304 ppm in pristine CN1 underwent progressive disappearance upon increasing the [CN−] (0–1 equiv.), with the simultaneous appearance of a gradually increasing upfield-shifted NMR peak at δ = 6.294 ppm (Table S6, ESI†). The above observation showed the nucleophilic association of the analyte with the CC bond of CN1 followed by the formation of the [CN1–CN−] adduct, which resulted in obvious breakage of the initial π-electron delocalization in the D–A architecture. In addition to checking the binding site in the nucleophilic addition of cyanide to CN1, natural bond orbital (NBO) charge calculations were performed. The charge distributions on some important selected atoms as labelled are shown in Fig. S33 and Table S7, ESI.† The relatively low negative charge on the C2 atom of CN1 indicated its faintly electropositive character compared with the other carbon atoms, which made it susceptible to nucleophilic attack by the cyanide anion, and it is clearly evident in Table S7, ESI,† that the charge was decreased on this atom in the [CN1–CN−] adduct. Consequently, the pyrene unit in [CN1–CN−] lost its electron-donating character, which led to the gradual upfield shift of the aromatic protons (Table S6, ESI†) at elevated [CN−].17,61 The formation of the [CN1–CN−] adduct was further supported from the appearance of a resonance signal at 51.93 ppm (Fig. S34, ESI†), as confirmed from the 13C NMR study of CN1 in the presence of CN−, and the appearance of the peak at m/z 400.0940 from the HRMS results (Fig. S35, ESI†).61 The FTIR spectrum of free CN1 (Fig. S36, ESI†) displayed vibration bands at 3107, 2275 and 1661 cm−1, which corresponded to the stretching frequencies of the unsaturated hydrocarbon (C–H), cyano and vinyl CC units, respectively. Evidently, in presence of CN− (1 equiv.), the disappearance of the CC stretch at 1661 cm−1 and the appearance of characteristic vibrational bands at 2927, 2268 and 2112 cm−1 indicated the stretching vibrations of the saturated hydrocarbon group (C–H) and two cyano units, which clearly demonstrated that the nucleophilic addition of CN− to the vinyl CC linkage caused the observed changes in the IR spectral response of CN1.62 Therefore, the addition of CN− essentially blocked the primary ICT nature of CN1 by generating a negatively charged species of D′–A character (Fig. 7a), which creates a ‘dark’ charge transfer state via the further channelization of negative charge through the formation of a non-emissive cyclohexadienylidene-type species.
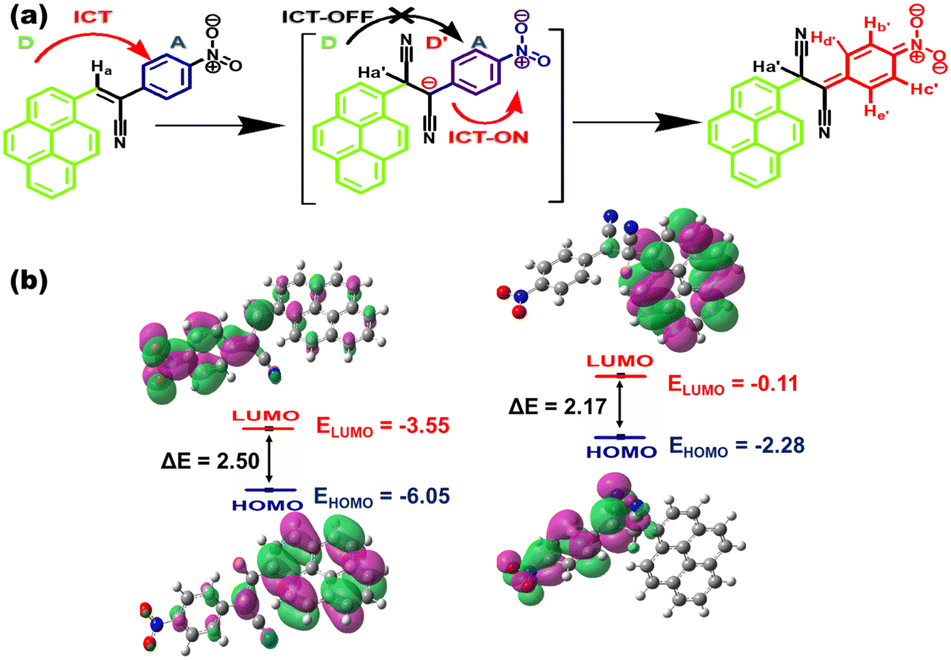 | ||
| Fig. 7 (a) Mechanistic pathway of the naked-eye colorimetric and fluorogenic dual-channel response of CN1 towards CN−. (b) HOMO–LUMO energy profiles of CN1 and the [CN1–CN−] adduct. | ||
Fig. S37, ESI† shows the electrostatic potential map of the [CN1–CN−] adduct based on the B3LYP/6311(+)G level of theory. From this figure it is shown that, after CN− addition, there is clear localization of the negative charge on the nitrophenyl unit, as confirmed from its higher electronegativity, which supports the desired alteration in the charge transfer pathway of CN1 before and after CN− addition. Also, two pairs of doublets (Hb′/Hc′ and Hd′/He′) appeared gradually in the region at (7.717, 7.689)/(7.612/7.583) and (6.836, 6.820)/(6.520, 6.488) ppm, respectively, as noted from the 1H NMR spectrum of CN1 with the subsequent addition of CN− (1 equivalent) indicated the formation of such species (Fig. S38 and Table S6, ESI†). As a result, there occurred red-shifting in absorption band (λa = 545 nm) with desired color-switching and severe fluorescence quenching response in the presence of CN−.
In order to understand the nature of the ground and excited states of the cyano complex, DFT and TDDFT calculations were carried out on the [CN1–CN−] adduct to provide detailed information on the participating molecular orbitals in the spectral transitions (Fig. S39 and Table S8, ESI†).23,55 The simulated absorption spectrum, obtained at the TD-CAM-B3LYP/cc-pVDZ level for calculation of the energy optimized geometry of [CN1–CN−] indicated the appearance of a red-shifted absorbance peak (λa,theo = 507.2 nm), which validated the experimental observation (Fig. S40, ESI†). Subsequently, breakage of the π-electron delocalization in the [CN1–CN−] adduct resulted higher HOMO (EHOMO = −2.28 eV) and LUMO (ELUMO = −0.11 eV) energy levels compared with those of the original species (EHOMO = −6.05 eV; ELUMO = 3.55 eV), where a complete reversal of the electron cloud distribution was observed between pyrene and the nitrophenyl unit in going from the HOMO to the LUMO (Fig. 7b) due to the alteration in the D–A architecture.23,53,63 The lowering of the magnitude of the HOMO–LUMO energy from ΔE = 2.5 eV in CN1 to ΔE = 2.17 eV in [CN1–CN−] clearly supports a new CT pathway which leads to the decreased PL characteristics in CN1 with the red-shifted absorption spectrum at high [CN−].18 Moreover, negligible changes in the time resolved fluorescence decay kinetics of CN1 in the presence of CN− (Fig. S41 and Table S9, ESI†) signified that the fluorescence quenching followed the static mood, which was primarily initiated by the ground state reaction between the analyte and the probe causing a change in both the absorption and emission characteristics of the receptor to execute the dual-channel sensory response.64
Evidently, to confirm the importance of the supramolecular structure, a CN1 congener, i.e., F1 [2-(4-fluorophenyl)-3-(pyren-1-yl)acrylonitrile], was synthesized based on a similar Knoevenagel condensation reaction (Scheme S2, ESI†)29 and characterized accordingly (Fig. S42 and S43, ESI†). In the design aspect of F1, the π-inferior nitrophenyl unit was replaced with a π-superior fluorophenyl core, which may alter the self-assembly pathway and subsequently affect the chemical sensing towards the CN− ion. Accordingly, heating/cooling treatment of F1 (concentration ∼1 wt%) led only precipitation (P) from the 8:2 (v/v) DMSO/water mixture (inset of Fig. S44a, ESI†). Henceforth, the SEM image of the extracted solid F1 barely revealed the formation of an entangled 3D network (Fig. S44a, ESI†) for the effective accumulation and diffusion of the analyte, and this its barely disclosed fluorogenic response towards CN−. By contrast, the variation in UV/Vis absorption spectrum of F1 towards aqueous CN− was also checked under experimental conditions identical to those described for CN1. From the inset of Fig. S44b, ESI,† it can be observed that F1 did not reveal any discrete colorimetric response to CN−, where the UV/Vis absorption spectrum did not shown any new band formation under an increased [CN−] (50 equiv.) in the 8:2 (v/v) DMSO/water mixture (Fig. S44b, ESI†). Even though the existing cyanovinyl unit in F1 will invite a Michael-type nucleophilic reaction, the developed negative charge was presumably not engaged in any further charge transfer interaction with the immediate fluorophenyl core due to the high electron density of the fluorine (–F) unit (Scheme S3, ESI†). Hence, unlike CN1, there was no room for F1 to exhibit any naked eye dual-channel response towards CN−, which demonstrated that the ingenious design strategy is certain to promote GIEE-triggered colorimetric and fluorogenic dual-channel chemical sensing. Furthermore, for elucidating the performance of CN1, the sensing efficiency of individual molecules containing 1-pyrene carboxaldehyde (PYCHO) and 4-nitrophenyl acetonitrile (4-ACN) were screened separately under identical experimental conditions. Accordingly, PYCHO hardly disclosed any colorimetric and fluorogenic signals as observed under daylight and UV light (insets of Fig. S45a and b, ESI†), where both the absorption (Fig. S45a, ESI†) and emission spectra (Fig. S45b, ESI†) neither evolved a new peak nor showed any fluorescence variation, even in the presence of 50 equiv./500 μM CN− in 8:2 (v/v) DMSO/water. The above observations indicate that PYCHO alone is not efficient as an optical sensor towards CN−. Although the addition of excess CN− (1100 μM/110 equiv.) did alter the UV/Vis absorption spectrum of 4-ACN by increasing the absorbance at λa ≈ ∼540 nm (Fig. S45c, ESI†) in 8:2 (v/v) DMSO/water, the above variation was not sufficient to show any noticeable change in the solution color in the presence of the analyte (inset of Fig. S45c, ESI†), which makes it a poorly efficient sensor towards CN−. However, monitoring the change in absorbance at λa = 540 nm with respect to the increased [CN−] (Fig. S45d, ESI†), the detection limit (DL) was calculated to be as high as 62.08 ± 0.09 μM upon considering its linear variation under lower analyte concentrations (0 ≤ [CN−] ≤ 200 μM) (inset of Fig. S45d, ESI†), which was significantly higher than CN1 both in the gel (DL = 0.48 ± 0.01 μM) and solution (DL = 5.42 ± 0.256 nM) forms (Table S10, ESI†). By contrast, the typical non-luminescent feature of 4-ACN (inset of Fig. S45d, ESI†) hindered us in obtaining a proper emission spectrum in solution before and after CN− addition, which weakened it as a fluorogenic sensor for the analyte. The above outcomes clearly indicate the importance of the present low-molecular-weight organogelator (CN1), which reveals a distinct naked-eye colorimetric [straw yellow to violet in solution and red to brownish-red in the gel with the evolution of a red-shifted absorption band at λa = 510 nm (Inset of Fig. S45c, ESI†)] and fluorogenic (notable quenching of gel fluorescence) dual-channel sensing pathway towards trace quantities of CN− for fruitful real-world applications.
Real-world applications in cyanide ion sensing
Encouraged by the rapid sensitivity of CN1 towards CN− with excellent selectivity, the dual-channel colorimetric and fluorogenic sensing event was explored for practical applications in real water samples including tap water (Kolkata Municipal supplied drinking water in the Jadavpur University campus), drinking water and distilled water, preserved freshly in specially designed PVC bottles, and recovery experiments were performed using the standard addition method. Evidently, when such real water samples, with CN− (0.5–2 μM) were be treated with a DMSO/water [8:2 (v/v)] solution (10 μM) and a gel [DMSO/water = 8:2(v/v)] of CN1 (1 wt%), an instant change in solution color and considerable fluorescence quenching of gel were obtained, respectively. Considering the linear regression plot from the variation in the absorbance and fluorescence spectra with respect to increasing [CN−], the percentage recovery (%R) values were evaluated and are tabulated accordingly in Table 2. In all cases, the [CN−] recovered by CN1 was very close to that of added concentration. Therefore, the outstanding sensitivity of CN1 stimulated our use of the present luminogen to detect as well as recover CN− precisely from real water samples with excellent efficiency.
| Sample | Dual-channel recovery of CN− | |||||||
|---|---|---|---|---|---|---|---|---|
| Colorimetric [8:2 (v/v) DMSO/water] |
Fluorogenic [8:2 (v/v) DMSO/water gel] |
|||||||
| [CN−]a (μM) | [CN−]d (μM) | %R | %E | [CN−]a (μM) | [CN−]d (μM) | %R | %E | |
| [CN−]a = analyte added; [CN−]d = analyte detected; %R = percentage recovery; %E = percentage error. For all the sets, the recovery experiments were repeated thrice to include standard errors. | ||||||||
| Distilled water | 0.5 | 0.496 | 99.32 | 0.68 | 0.5 | 0.496 | 99.30 | 0.70 |
| 1 | 0.944 | 94.40 | 5.60 | 1 | 0.974 | 97.40 | 2.60 | |
| 2 | 1.971 | 98.55 | 1.45 | 2 | 1.970 | 98.33 | 1.65 | |
| Drinking water | 0.5 | 0.498 | 99.75 | 0.25 | 0.5 | 0.483 | 96.61 | 3.40 |
| 1 | 0.974 | 97.43 | 2.57 | 1 | 0.991 | 99.14 | 0.86 | |
| 2 | 1.884 | 94.20 | 5.80 | 2 | 1.977 | 98.85 | 1.15 | |
| Tap water | 0.5 | 0.482 | 96.34 | 3.66 | 0.5 | 0.470 | 93.82 | 6.00 |
| 1 | 0.990 | 98.64 | 1.00 | 1 | 0.963 | 96.32 | 3.68 | |
| 2 | 1.992 | 99.62 | 0.38 | 2 | 1.904 | 95.20 | 4.80 | |
In addition, practical applications of the sensor CN1 were further investigated in our daily life by checking the quality of different food consumables that contain trace amounts of cyanide. Quantitative analytical experiments were carried out to examine the capability of CN1 to detect endogenous cyanide from sprouted potatoes, bitter seeds, apple seeds and Sudan grasses (the detailed procedure for this is given in the ESI†).
Afterwards, when the cyanide-containing solutions extracted from the respective food samples were added to the DMSO/water [8:2(v/v)] solution and gel [DMSO/water = 8:2(v/v)] of CN1, an abrupt change in the solution color was noticed via the naked eye, and the dramatic fluorescence quenching of the gel emission was obtained by surveying the red shifted absorption (λa = 545 nm) and the decreased emission (λf = 680 nm) (Fig. 8 and insets). In all cases, the respective changes in fluorescence were applied to the linear regression plot to evaluate the [CN−]. From Table 3 and Fig. S46, ESI,† it was confirmed that CN1 was proficient for the quantitative detection of cyanide in food samples. Therefore, the inspected results confirmed that CN1 was capable of achieving a convenient, cost-effective and efficient sensing performance through a rapid dual-channel spectral pathway for the sensitive detection of cyanide in both real water samples and food samples.
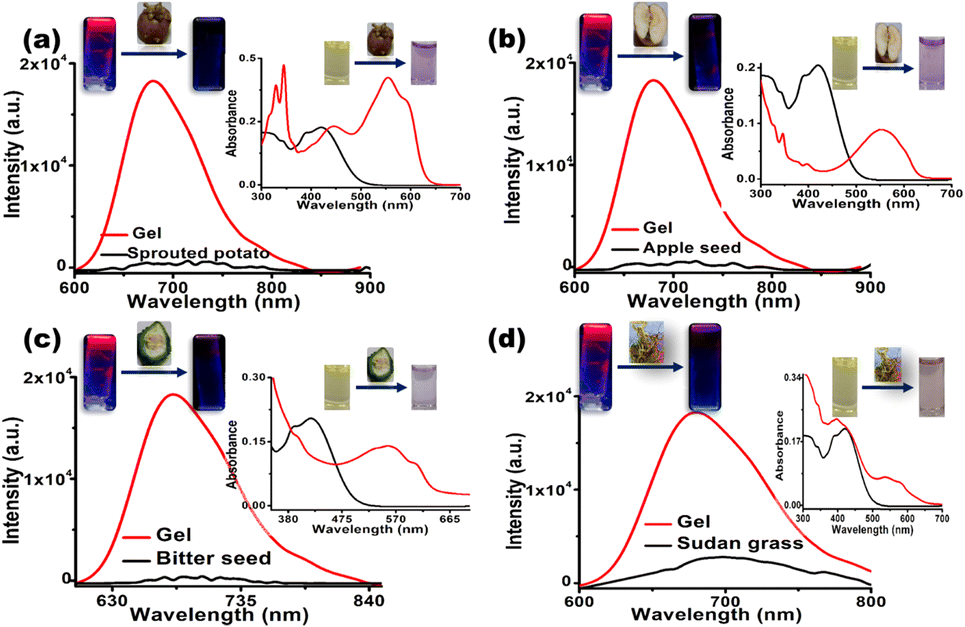 | ||
| Fig. 8 Fluorogenic response of the CN1 gel [8:2 (v/v) DMSO/water (v/v); 1 wt%] upon treatment with food extractions obtained from (a) sprouted potato, (b) apple seed, (c) bitter seed and (d) Sudan grass (λex = 450 nm) samples. The corresponding insets show the naked-eye colorimetric changes of the CN1 solution (10 μM) in DMSO/water [8:2(v/v)], monitored from the variations in UV/Vis absorption spectra upon treatment with the respective food extraction. | ||
| Food sample | No. | QE (%) | [CN−]d (μM) | [CN−]d,avg (μM) |
|---|---|---|---|---|
| QE (%) = (1 − I/I0) × 100; [CN−]d = concentration of estimated CN−; [CN−]d,avg = average concentration of estimated CN−. Individual measurements were repeated to include the error values. | ||||
| Sprouted potato | 1 | 73.87 | 6.19 | 5.89 ± 0.201 |
| 2 | 72.64 | 5.98 | ||
| 3 | 69.68 | 5.51 | ||
| Apple seed | 1 | 74.16 | 6.25 | 6.18 ± 0.043 |
| 2 | 73.80 | 6.18 | ||
| 3 | 73.32 | 6.10 | ||
| Bitter seed | 1 | 72.83 | 6.02 | 6.06 ± 0.106 |
| 2 | 74.31 | 6.27 | ||
| 3 | 72.22 | 5.91 | ||
| Sudan grass | 1 | 69.51 | 5.48 | 5.19 ± 0.148 |
| 2 | 66.83 | 5.10 | ||
| 3 | 66.02 | 4.99 | ||
Self-assembly leading to superhydrophobic surface formation
Superhydrophobic materials with a surface coating ability are candidates of paramount importance for a wide range of applications owing to their tendency to protect surfaces from wetting.65 The vigorous tendency to aggregate with spontaneous self-wrapping among the molecules may prompt CN1 to show water-repellence properties for the creation of a superhydrophobic surface by utilizing different non-covalent interactions in the self-assembled architecture. To authenticate the above assumption, drop-cast thin film and xerogel surfaces of CN1 (∼27 mM in both cases) were constructed and dried properly under high-vacuum conditions.Initially, a water droplet was seen to spread and soak completely on an CN1-un-coated glass slide or filter paper, indicating the inherent hydrophilic nature of these surfaces. However, when the water droplet was just placed on the xerogel or a dip-coated glass slide or filter paper, it is interesting to note that, rather than spreading over the surface, the droplet appeared as a spherical pearl-like structure [Fig. 9(a)–(d), inset]. The mean contact angles were calculated to be as high as 158.74 and 155.48° for the xerogel-coated glass slide [Fig. 9(a)] and filter paper [Fig. 9(b)], respectively. The CN1 film exhibited a mean contact angle of 154.53° for both the dip-coated glass slide [Fig. 9(c)] and dip-coated filter paper [Fig. 9(d)]. In all the cases, self-assembled surfaces of CN1 exhibited reasonable super-hydrophobicity without tethering any additional surface modifying hydrophobic agents [e.g., long alkyl chains, poly(aryl ether)dendrons]65,66 to the luminogen structure. Moreover, thin films constructed using only native pyrene on a glass slide or filter paper showed insignificant hydrophobicity, with immediate spreading of the water droplet on the respective substrate, which clearly demonstrated importance of definite supramolecular packing to create a self-assembled superhydrophobic surface where the cooperative non-covalent interactions undeniably play a decisive role in bringing about the water repellence phenomenon.
 | ||
| Fig. 9 Mean water contact angles and (insets) photographs of water droplets on (a and b) CN1 xerogel and (c and d) CN1 dip-coated glass slides (a and c) and filter papers (b and d). | ||
Although the CN1 self-assembly is hydrophobic in nature, indicating water repellence, it does not interfere with the sensory response from a purely aqueous medium as various concentrations of aqueous CN− were added in situ into the hot solution of CN1 (10 mg mL−1) in order to develop analyte-containing gels of different strengths, where the aqueous conditions triggered CN1 to develop an entangled porous and interlinked nanofibrous morphology utilizing different hydrophobic forces for gel-to-gel amplified fluorescence quenching based on the surface-adsorbed analytes. Furthermore, unlike just placing the water droplet on different CN1-coated surfaces during the mean water contact angle measurements, swabs loaded with various strengths the aqueous CN− analyte were pressed onto the xerogel film to facilitate the analyte–probe interaction for demonstrating the contact mode detection of CN− in the solid state.
Conclusion
The present study introduces an atypical GIEEgenic LMWG, CN1, where prolific mechano–thermo-responsive self-assembly behavior as well as qualitative and quantitative dual-channel cyanide sensing was achieved in real time from the single molecular architecture. The twisted structural framework of CN1 from attaching the typical ACQphore pyrene with a nitrophenyl acetonitrile core enabled the corresponding luminogen for constructive head-to-tail packing upon aggregation, where the occurrence of several non-covalent interactions led to the establishment of J-type self-assembly. Accordingly, the effect of RIR in an ordered array for the supramolecular framework enabled CN1 to show enhanced far-red PL characteristics from both the as-synthesized powder and the gel state without the assistance of any auxiliary substituents, in spite of being completely non-emissive in the molecularly dissolved state due to the torsional-induced rotational motion. Owing to the flexibility in the constituent non-covalent interactions, the CN1 gel responded rapidly to externally applied force and heat via disintegration and reintegration of the gel structure to realize ‘ON–OFF’ fluorescence switching over several cycles without any loss of PL intensity. The electropositive nature of the existing C bond in CN1 induced a Michael-type nucleophilic reaction of CN−, which disturbed the initial π-electron delocalization in D–A architecture of the luminogen via forming a CT active non-emissive ground-state cyano adduct. Subsequently, the selective color switching and the drastic fluorescence quenching response of CN1 towards CN− could commendably be monitored via naked-eye colorimetric and fluorogenic pathways from both the solution and self-assembled states, which were utilized further for the rapid and cost-effective quantitative detection of CN− in real water samples and food consumables with the desired accuracy. Moreover, the robust ambience of the aggregation enables CN1 self-assembly to engineer superhydrophobic surfaces without the need for any additional hydrophobic motifs. These findings established a detailed understanding into the structural strategy and photophysical aspects of a GIEEgen, which will not only enrich the library of LMWGs but also indorse supramolecular self-assembly to disclose multifunctional sensing features for productive real-world applications.
Experimental section
Experimental methodologies, instrumentation, computational details, respective synthesis schemes of the LMWG, preparation of food extractions for analytical studies, figures and tables regarding the photophysics, solid-state emission, gel formation, superhydrophobic surface formation together with the sensitive and selective dual-channel sensory responses are available in the ESI.†Author contributions
Kingshuk Debsharma: conceptualization, data curation, formal analysis, methodology, software, supervision, validation, visualization, writing – original draft, writing – review and editing; Sunanda Dey: data curation, formal analysis, software, validation, visualization, writing – original draft; Jyothibabu Sajila Arya: data curation, formal analysis, software; Krishna Sundar Das: data curation, formal analysis, software; Chittaranjan Sinha: funding acquisition, investigation, resources, visualization, writing – review and editing; Edamana Prasad: conceptualization, funding acquisition, investigation, project administration, resources, supervision, visualization, writing – review and editing.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors thank Dr Raju Mondal, Department of Chemistry, Indian Association for the Cultivation of Sciences (IACS) for rheology measurements. The authors acknowledge the Department of Chemical Engineering, IIT Madras for SEM facilities. The authors acknowledge Prof. Abhijit Deshpande for providing instrumentation for the water contact angle measurements. The authors are grateful to the P.G. Senapathy Centre for computing resources, IIT Madras and Department of Chemistry Jadavpur University for providing the opportunity to utilize their supercomputing features for DFT calculations. We are thankful to the Department of Chemistry, IIT Madras and Department of Chemistry, Jadavpur University for NMR, and HRMS facilities. K. D. personally acknowledges Prof. A. K. Mishra, Prof. Sujay Baitalik and their labs for providing the TCSPC instrumentation facilities. K. D. is thankful to the Ministry of Human Resource and Development (MHRD) for fellowship.References
- A. Dawn, T. Shiraki, S. Haraguchi, S.-I. Tamaru and S. Shinkai, Chem. – Asian J., 2011, 6, 266–282 CrossRef CAS PubMed.
- X. Cao, A. Gao, J.-T. Hou and T. Yi, Coord. Chem. Rev., 2021, 434, 213792 CrossRef CAS.
- S. Panja, A. Panjab and K. Ghosh, Mater. Chem. Front., 2021, 5, 584–602 RSC.
- C. Wang, Z. Wang, D. Zhang and D. Zhu, Chem. Phys. Lett., 2006, 428, 130–133 CrossRef CAS.
- S. Suganya, K. Debsharma, E. Ravindran, M. K. Mahato and E. Prasad, ACS Appl. Polym. Mater., 2020, 2, 1222–1233 CrossRef CAS.
- Q. Lin, T.-T. Lu, X. Zhu, T.-B. Wei, H. Li and Y.-M. Zhang, Chem. Sci., 2016, 7, 5341–5346 RSC.
- J. Y. C. Lim, S. S. Goh and X. J. Loh, ACS Mater. Lett., 2020, 2, 918–950 CrossRef CAS.
- V. K. Praveen, B. Vedhanarayanan, A. Mal, R. K. Mishra and A. Ajayaghosh, Acc. Chem. Res., 2020, 53, 496–507 CrossRef CAS PubMed.
- S. S. Babu, K. K. Kartha and A. Ajayaghosh, J. Phys. Chem. Lett., 2010, 1, 3413–3424 CrossRef CAS.
- Z. Zhao, J. W. Y. Lam and B. Z. Tang, Soft Matter, 2013, 9, 4564–4579 RSC.
- Q. Zhao and J. Z. Sun, J. Mater. Chem. C, 2016, 4, 10588–10609 RSC.
- R. Das, S. Bej, H. Hirani and P. Banerjee, ACS Omega, 2021, 6, 14104–14121 CrossRef CAS PubMed.
- S. S. Babu, V. K. Praveen and A. Ajayaghosh, Chem. Rev., 2014, 114, 1973–2129 CrossRef CAS.
- H.-W. Zhao, G. Wu, X.-Y. Sun, J.-B. Chao, Y. Q. Li, L. Jiang and H. Han, J. Lumin., 2018, 201, 474–478 CrossRef CAS.
- S. K. Samanta, N. Dey, N. Kumari, D. Biswakarma and S. Bhattacharya, ACS Sustain. Chem. Eng., 2019, 7, 12304–12314 CAS.
- Q. Niu, L. Lan, T. Li, Z. Guo, T. Jiang, Z. Zhao, Z. Feng and J. Xi, Sens. Actuators, B, 2018, 276, 13–22 CrossRef CAS.
- F. N. Moghadam, M. Amirnasr, K. Eskandari and S. Meghdadi, New J. Chem., 2019, 43, 13536–13544 RSC.
- R. Das, S. Paul, S. Bej, M. Ghosh, J. C. Bose and P. Banerjee, Colloids Surf., A, 2022, 653, 130022 CrossRef CAS.
- P. K. Dhas, P. Chitra, S. Jayakumar and A. R. Mary, Indian J. Occup. Health, 2011, 15, 133–136 Search PubMed.
- M. Hou, Y.-C. Liu, W. Zhou, J.-D. Zhang, F.-D. Yu, Y. Zhang, G.-J. Liu and G.-W. Xing, Chem. – Asian J., 2021, 16, 2014–2017 CrossRef CAS.
- S. Bej, R. Das, H. Hirani, S. Ghosh and P. Banerjee, New J. Chem., 2019, 43, 18098–18109 RSC.
- W.-J. Qu, H.-H. Yang, J.-P. Hu, P. Qin, X.-X. Zhao, Q. Lin, H. Yao, Y.-M. Zhang and T.-B. Wei, Dyes Pigm., 2021, 186, 108949 CrossRef CAS.
- S. Khatua, D. Samanta, J. W. Bats and M. Schmittel, Inorg. Chem., 2012, 51, 7075–7086 CrossRef CAS PubMed.
- R. Das, S. Bej, N. C. Murmu and P. Banerjee, Anal. Chim. Acta, 2022, 1202, 339597 CrossRef CAS PubMed.
- Z. Xu, X. Chen, H. N. Kim and J. Yoon, Chem. Soc. Rev., 2010, 39, 127–137 RSC.
- X. Lou, D. Ou, Q. Li and Z. Li, Chem. Commun., 2012, 48, 8462–8477 RSC.
- S. Pramanik, V. Bhalla and M. Kumar, ACS Appl. Mater. Interfaces, 2014, 6, 5930–5939 CrossRef CAS PubMed.
- S. D. Padghan, C.-Y. Wang, W.-C. Liu, S.-S. Sun, K.-M. Liu and K.-Y. Chen, Dyes Pigm., 2020, 183, 108724 CrossRef CAS.
- G. Paramaguru, R. V. Solomon, S. Jagadeeswari, P. Venuvanalingam and R. Renganathan, J. Photochem. Photobiol., A, 2013, 271, 31–44 CrossRef.
- S. Bej, R. Das, A. Mondal, R. Saha, K. Sarkar and P. Banerjee, Spectrochim. Acta A, 2022, 273, 120989 CrossRef CAS PubMed.
- J. B. Birks, Photophysics of aromatic molecules, Wiley-Interscience, London, UK, 1970 Search PubMed.
- D. Oelkrug, A. Tompert, J. Gierschner, H.-J. Egelhaaf, M. Hanack, M. Hohloch and E. Steinhuber, J. Phys. Chem. B, 1998, 102, 1902–1907 CrossRef CAS.
- B.-K. An, J. Gierschner and S. Y. Park, Acc. Chem. Res., 2012, 45, 544–554 CrossRef CAS.
- P. Xue, B. Yao, J. Sun, Z. Zhang and R. Lu, Chem. Commun., 2014, 50, 10284–10286 RSC.
- M. K. Nayak, B.-H. Kim, J. E. Kwon, S. Park, J. Seo, J. W. Chung and S. Y. Park, Chem. – Eur. J., 2010, 16, 7437–7447 CrossRef CAS PubMed.
- B.-K. An, D.-S. Lee, J.-S. Lee, Y.-S. Park, H.-S. Song and S. Y. Park, J. Am. Chem. Soc., 2004, 126, 10232–10233 CrossRef CAS.
- S. Satapathy, P. Prabakaran and E. Prasad, Chem. – Eur. J., 2018, 24, 6217–6230 CrossRef CAS.
- S. Sivalingam, K. Debsharma, A. Dasgupta, S. Sankararaman and E. Prasad, ChemPlusChem, 2019, 84, 392–402 CrossRef CAS PubMed.
- J. Seo, J. W. Chung, E.-H. Jo and S. Y. Park, Chem. Commun., 2008, 2794–2796 RSC.
- D. López and E. M. García-Frutos, Langmuir, 2015, 31, 8697–8702 CrossRef PubMed.
- J. M. Guenet, Thermoreversible gelation of polymers and biopolymers, Academic Press, London, 1992 Search PubMed.
- G. M. Kavanagh and S. B. Ross-Murphy, Prog. Polym. Sci., 1998, 23, 533–562 CrossRef CAS.
- T. G. Mezger, The rheology handbook: for users of rotational and oscillatory rheometers, Vincentz Network GmbH & Co KG, 2006 Search PubMed.
- A. Baheti, C. H. Lee, K. R. J. Thomas and K. C. Ho, Phy. Chem. Chem. Phy., 2011, 13, 17210–17221 RSC.
- A. Hawe, M. Sutter and W. Jiskootm, Pharm. Res., 2008, 25, 1487–1499 CrossRef CAS PubMed.
- G. S. Thool, K. Narayanaswamy, A. Venkateswararao, S. Naqvi, V. Gupta, S. Chand, V. Vivekananthan, R. R. Koner, V. Krishnan and S. P. Singh, Langmuir, 2016, 32, 4346–4351 CrossRef CAS.
- P. Xue, R. Lu, G. Chen, Y. Zhang, H. Nomoto, M. Takafuji and H. Ihara, Chem. – Eur. J., 2007, 13, 8231–8239 CrossRef CAS PubMed.
- Y. Xu, P. Xue, D. Xu, X. Zhang, X. Liu, H. Zhou, J. Jia, X. Yang, F. Wanga and R. Lu, Org. Biomol. Chem., 2010, 8, 4289–4296 RSC.
- S. J. George and A. Ajayaghosh, Chem. – Eur. J., 2005, 11, 3217–3227 CrossRef CAS PubMed.
- Y. Zhang, C. Liang, H. Shang, Y. Ma and S. Jiang, J. Mater. Chem. C, 2013, 1, 4472–4480 RSC.
- L. Zhu, C. Yang and J. Qin, Chem. Commun., 2008, 6303–6305 RSC.
- A. Adhikary, K. S. Das, S. Saha, M. Roy and R. Mondal, Dalton Trans., 2020, 49, 13487–13495 RSC.
- S. D. Padghan, L.-C. Wang, W.-C. Lin, J.-W. Hu, W.-C. Liu and K.-Y. Chen, ACS Omega, 2021, 6, 5287–5296 CrossRef CAS PubMed.
- T. M. Elmorsi, T. S. Aysha, O. Machalicky, M. B. I. Mohamed and A. H. Bedair, Sens. Actuators, B, 2017, 253, 437–450 CrossRef CAS.
- Y.-Q. Xie, Y.-M. Zhang, Z.-H. Li, X.-N. Qi, H. Yao, B.-B. Shi, W.-J. Qu, T.-B. Wei and Q. Lin, New J. Chem., 2021, 45, 18421–18432 RSC.
- Q. Lin, X. Zhu, Y.-P. Fu, Y.-M. Zhang, R. Fang, L.-Z. Yang and T.-B. Wei, Soft Matter, 2014, 10, 5715–5723 RSC.
- Y. Che, D. E. Gross, H. Huang, D. Yang, X. Yang, E. Discekici, Z. Xue, H. Zhao, J. S. Moore and L. Zang, J. Am. Chem. Soc., 2012, 134, 4978–4982 CrossRef CAS PubMed.
- B. H. Shankar, D. T. Jayaram and D. Ramaiah, Chem. – Asian J., 2014, 9, 1636–1642 CrossRef CAS PubMed.
- P. Xue, B. Yao, P. Wang, P. Gong, Z. Zhang and R. Lu, Chem. – Eur. J., 2015, 21, 17508–17515 CrossRef CAS PubMed.
- K. Debsharma, J. Santhi, B. Baire and E. Prasad, ACS Appl. Mater. Interfaces, 2019, 11, 48249–48260 CrossRef CAS PubMed.
- Z. M. Dong, H. Ren, J. N. Wang and Y. Wang, Microchem. J., 2020, 155, 104676 CrossRef CAS.
- T. Sun, Q. Niu, Y. Li, T. Li, T. Hu, E. Wang and H. Liu, Sens. Actuators, B, 2018, 258, 64–71 CrossRef CAS.
- J. Jin, J. Zhang, L. Zou and H. Tian, Analyst, 2013, 138, 1641–1644 RSC.
- S. Suganya, E. Ravindran, M. K. Mahato and E. Prasad, Sens. Actuators, B, 2019, 91, 426–432 CrossRef.
- T. Nakanishi, Y. Shen, J. Wang, H. Li, P. Fernandes, K. Yoshida, S. Yagai, M. Takeuchi, K. Ariga, D. G. Kurth and H. Möhwald, J. Mater. Chem., 2010, 20, 1253–1260 RSC.
- N. V. Lakshmi, T. M. Babu and E. Prasad, Chem. Commun., 2016, 52, 617–620 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental methods, relevant tables and figures, characterization, theoretical calculation parameters and other additional required experiments. See DOI: https://doi.org/10.1039/d2nj06220h |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2023 |