
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of aromatic 1,2-disulfonimides and their prospects as monomers†
Denis Andzheevich
Sapegin
 *ab,
Danila Alexandrovich
Kuznetsov
b and
Joseph C.
Bear
*a
*ab,
Danila Alexandrovich
Kuznetsov
b and
Joseph C.
Bear
*a
aDepartment of Chemical and Pharmaceutical Sciences, Kingston University, Penrhyn Rd, Kingston upon Thames KT1 2EE, UK. E-mail: J.Bear@kingston.ac.uk
bQuantori, 625 Massachusetts Ave, Cambridge, MA 02139, USA
First published on 10th February 2023
Abstract
The correlation between chemical structure and selective transport properties in polyimides led to the suggestion of poly-1,2-disulfonimides as prospective materials for high-performance diffusion membranes. Higher polarity along with steric features of the 1,2-disulfonimide fragment in comparison with the imide suggests a superior rate of inter-chain interactions along with presumably higher inter-planar distances in poly-1,2-disulfonimides. A structure of a novel 1,2-disulfonimide-containing dicarboxylchloride is proposed for the obtainment of high-molecular-weight polymers analogous to the routes utilised for the synthesis of poly(amide-imide)s. This article aims at the investigation of approaches for the synthesis of target 1,2-disulfonimide-fragment-containing monomers and the estimation of their properties using analytical and computational techniques. This article discloses the peculiarities of 1,2 disulfobenzoic acid chlorination, which leads to the substitution in the aromatic ring instead of the expected disulfonyl-chloride and instability of 1,2-disulfonimides during oxidation with potassium permanganate in various media.
1. Introduction
The most recognised and commonly used approaches for the description of low-molecular-weight penetrant transport through dense polymeric films are based on the solution-diffusion model.1 The diffusion of a penetrant through the polymer matrix is considered to be the limiting step of the mass transfer in such types of processes.2 The crucial role in the diffusion of such penetrants through the bulk of the polymer is played by the segmentation mobility of the macromolecular chains, which regulates openings of the quasi-stationary free volume voids available for penetrant molecules. Penetrants are hypothesised to be transported through and between such voids driven by the Brownian motion in the direction of lower chemical potential.3 Hence, the segmentation mobility, free volume, inter and intra-chain interactions, and supramolecular chain packaging are considered the main parameters that define the polymer's selective transport properties in gas separation processes. High mobility of the polymer chains along with high free volume values corresponds to the high overall permeability of the material; however, it is also commonly associated with low selectivity values.4 The search for structures which provide the optimal compromise between selectivity and permeability is considered one of the most important challenges in the area of polymeric gas separation membrane development.4,5 Such a combination of properties is found in numerous polymers; however, polyimides rank among the best.Polyimides are considered one of the most promising polymers in the area of gas separation membranes. They are often characterized by high values of selectivity along with relatively low permeability values, which are both caused by the presence of strong interchain interactions.5,6 It was not until the late 1980s that special polyimides which combined high selectivity along with high permeability were developed.7 The so-called first-generation polyimides for gas separation applications based on the 4,4′-(hexafluoroisopropylidene)diphthalic anhydride (6FDA) were designed guided by the understanding of diffusion transport mechanisms and known structure–property correlations.8,9 Introduction of bulky perfluorinated methyl fragments that hinder chain packaging achieved high free volume values and, hence, permeability. This was in combination with high selectivity values, a characteristic of most polyimides.6 Representatives of such high-permeable polyimides may be found in the centre and to the left near the Robeson upper bound.4 Today, the understanding of correlations between the chemical structure of monomers, which are used for the obtainment of various polyimides, and polymers' transport properties is significantly improved.5 However, basic approaches are similar to those reported 50 years ago:10
• Increasing the free volume value by hindering the chain packaging.
• Introduction of bulky groups while controlling the values of cohesive energy density.
• Introduction of crosslinks in order to assure selectivity.
It should be noted that the presence of the imide group in the main chain of the polymer assures the occurrence of inter-chain interactions in polyimides, which are believed to be responsible for their superior thermal, mechanical, dielectric and transport properties.6,7,11 However, these interactions usually also lead to a decrease in the polymer's free volume. The nature of the interactions between the polyimide macromolecules is believed to be defined by the presence of the charge-transfer complexes, which are formed due to the electron-withdrawing effects of the imide ring.11 Additionally, the imide group is believed to be almost flat with the oxygen atoms of the carbonyls12 being in the plane of the imide ring, which along with the strong inter-chain interactions gives relatively low values of inter-planar distances characteristic for most polyimides.6 Following these observations, it may be suggested that substituting a bulkier fragment into the monomer's structure may significantly increase the selective transport performance of the polymer. One such fragment which could be considered a suitable competitor for the imide ring is the benzene-1,2-disulfonimide fragment.13 Possession of a higher electron-withdrawing ability than the imide fragment along with a non-planar structure13 makes such fragments extremely promising in the area of high-performance gas separation polymer development.
The aim of the present research is to investigate a synthetic approach that could be used to synthesize high-molecular-weight poly-1,2-disulfonimides and develop synthetic procedures for the obtainment of monomers for the implementation of the proposed approach. The structural and physical properties and their potential for application in diffusion transport of the polymers which may be synthesized using the obtained monomers were estimated using computational and theoretical approaches on the basis of physical and chemical properties of the obtained compounds.
2. Results and discussion
2.1 Synthesis of 1,2 disulfonimides and their precursors
The published procedures for the successful synthesis of 1,2-disulfonimide compounds in considerable yields usually employ the reaction between the corresponding 1,2-disulfonyl chloride and amine.13–16 Having two bulky sulfonyl chloride groups in the neighbouring positions of the benzene ring 1,2-disulfonyl chlorides are quite challenging to obtain. Most approaches for their synthesis reported in the literature are based on the diazotization of corresponding aromatic 1,2-amino acids.13–15 A number of approaches where diazotization of 1,2-amino sulfonic acid is followed by nucleophilic substitution of the diazonium fragment with SO2 to form a sulfonic acid group followed by the chlorination of obtained 1,2-disulfonic acid are reported.15 Another recently reported approach13 involves the diazotization of 1,2-amino carboxylic acid followed by its decomposition with the formation of an aryne, which then reacts with carbon disulfide to form 1,2-dithiol, which is isolated from the reaction mixture and converted into corresponding 1,2-disulfonyl chloride by oxidation in the presence of chlorine. While the second approach is undoubtedly less time-consuming, it is very sensitive to the presence of the electron-withdrawing groups in the precursor. These hamper the formation of the aryne, negatively affecting the yields and making it impossible to obtain certain disulfonyl chlorides. In view of this and also the cost-effectiveness of the reagents necessary for its implementation, the first approach, involving the substitution of the diazonium group with SO2, was considered the most suitable for the task.2.2 Design of the monomer structure
Taking into account, the specifics of chemical reactions involved in the formation of 1,2-disulfonimides, polycondensation techniques seem the most suitable for the target polymers. The strategies which could be used to obtain corresponding macromolecular compounds may be divided into two principally different groups: first group, strategies in which a 1,2-disulfonimide fragment is formed directly during the polycondensation, such as the case of polyimides;6,7,9 and a second group, strategies in which a monomer containing 1,2-disulfonimide fragment is introduced into the polycondensation reaction, such as in methods described for the obtainment of some poly(amide-imide)s.17 Considering the low selectivity of the 1,2-disulfonimide formation (yield 34–45%),13–15 the nature of polycondensation18 and the data reported on the direct obtainment of polysulfonamides by reactions between disulfonyl chlorides and diamines,19 strategies of the second group are the most suitable for the synthesis of high-molecular-weight polymers. In this vein, the synthesis of a dicarboxylic chloride, containing a 1,2-disulfonimide fragment was suggested to be the optimal approach for the obtainment of target polymers. The reaction between carbonyl chlorides and amines is highly selective, well studied and is commonly used in polycondensation procedures to obtain high-molecular-weight polyamides and poly(amide-imide)s.20 Given the reported data on the synthesis, structural, morphological, physical and selective transport features of various poly(amide-imide)s20,21 4-chloroformyl-N-(p-chloroformylphenyl)-benzene-1,2-disulfonimide (DSI-DA-Cl) (Chart 1) was proposed as the most prospective monomer structure, analogous to 4-chloroformyl-N-(p-chloroformylphenyl)-phthalimide (CFPI) (Chart 1), which was used to obtain various high-performance poly(amide-imide)s.17,20,21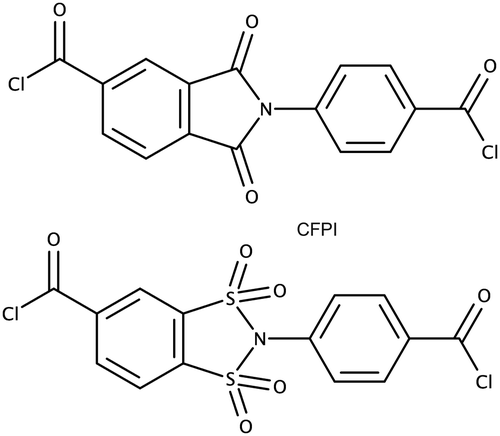 | ||
| Chart 1 Chemical structures of prospective imide and 1,2-disulfonimide dicarbonyl chlorides. DSI-DA-Cl-4-chloroformyl-N-(p-chloroformylphenyl)-benzene-1,2-disulfonimide; CFPI-4-chloroformyl-N-(p-chloroformylphenyl)-phthalimide. | ||
2.3 Synthesis of target 1,2-disulfonimides
The approach proposed for the synthesis of a target 1,2-disulfonimide-dicarbonyl chloride is illustrated in Scheme 1a. Both routes start with the diazotization reaction of 2-amino-5-methyl-benzenesulfonic acid (AMBSA) followed by nucleophilic substitution of a diazo group with SO2 leading to 4-methyl-1,2-benzenedisulfonic acid (Scheme 1, BMDSA, I).15 Broad availability of the reagents and relative simplicity of the procedure made this reaction attractive for the formation of 1,2-disulfonic acids in the present project. | ||
| Scheme 1 Illustration of the routes for the synthesis of DSI-DA-Cl-a-planned pathway, b-updated pathway. | ||
An interesting observation was that, unlike many diazo compounds, 2-diazonium-5-methyl-sulfonate showed outstanding stability during storage and was able to withstand prolonged heating up to 60 °C. This behaviour may be attributed to the high electron-withdrawing character of the sulfonic acid group coupled with the compound's ability to form an intramolecular salt. The isolated diazo-sulfonic acid was characterised by means of FTIR and NMR spectroscopy (1H: 2.6 s; 7.78 m; 7.96 d; 8.6 d; 13C: 155.00; 150.30; 134.82; 132.51; 130.00; 108.36; 22.76), the FTIR spectrum is illustrated in Fig. 1.
 | ||
| Fig. 1 FTIR spectrum of 2-diazonium-5-methylsulfonate. | ||
The characteristic band at 2287 cm−1 corresponds to the stretching vibrations of the diazonium group.23 The medium peak at 3078 cm−1 corresponds to the C–H vibrations of the methyl group, while the presence of sulfonic acid is indicated by the band at 1219 cm−1 corresponding to the S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations.24 The 1H NMR spectrum of the obtained crude disodium 5-methyl-1,2-benzenedisulfonate is presented in Fig. 2. Along with the major substitution product, there are shifts corresponding to the by-product, which as a fraction in the crude residue is not more than 9 mol%, according to the ratio of shifts’ areas corresponding to the protons of the methyl group (2.59 and 2.31 ppm). Accounting for the nature of the diazonium group substitution and the reaction conditions, most likely, the by-product is 2-chloro-5-methyl-benzenesulfonic acid. The superposition of the by-product spectra with the modelled shifts of 2-chloro-5-methyl-benzenesulfonic acid (1H NMR: δ 2.45 (3H, s), 7.58–7.74 (2H, 7.64 (dd), 7.68 (dd)), 7.90 (1H, dd))25 shows a noticeable similarity which acts in favour of the suggested by-product structure.
O stretching vibrations.24 The 1H NMR spectrum of the obtained crude disodium 5-methyl-1,2-benzenedisulfonate is presented in Fig. 2. Along with the major substitution product, there are shifts corresponding to the by-product, which as a fraction in the crude residue is not more than 9 mol%, according to the ratio of shifts’ areas corresponding to the protons of the methyl group (2.59 and 2.31 ppm). Accounting for the nature of the diazonium group substitution and the reaction conditions, most likely, the by-product is 2-chloro-5-methyl-benzenesulfonic acid. The superposition of the by-product spectra with the modelled shifts of 2-chloro-5-methyl-benzenesulfonic acid (1H NMR: δ 2.45 (3H, s), 7.58–7.74 (2H, 7.64 (dd), 7.68 (dd)), 7.90 (1H, dd))25 shows a noticeable similarity which acts in favour of the suggested by-product structure.
 | ||
| Fig. 2 1H NMR spectrum of unpurified BMDSA in D2O. | ||
Synthesis of the dicarboxylic acid precursor of DSI-DA-Cl was initially suggested to be performed through the oxidation of 4-methyl-N-(p-methyl)-benzene-1,2-disulfonimide (DSI-Me Route IV.a, Scheme 1). It was expected that the 1,2-disulfonimide cycle should be able to withstand the conditions of methyl groups’ direct oxidation with potassium permanganate.14,22 Synthesis of corresponding disulfonyl chloride (BMDSA-Cl, II.a, Scheme 1) and its condensation with p-toluidine (III.a, Scheme 1) is simple and demonstrates good yields, allowing the isolation of pure DSI-Me from the reaction mixture by flash chromatography. Chlorination of BMDSA with POCl3 leads to the corresponding disulfonyl chloride (BMDSA-Cl), which after purification does not show traces of the diazotization by-product. The total yield of the BMDSA-Cl from reactions I and II.a calculated from AMBSA was 15.4%. The 1H NMR spectrum of BMDSA-Cl matches the one reported in the literature.13 DSI-Me was characterised by NMR and FTIR spectroscopy. Both spectra are presented in Fig. 3.
 | ||
| Fig. 3 1H NMR in the range 5.5–8.4 ppm in CDCl3 (a) and FTIR (b), spectrum of DSI-Me. | ||
The labelled 1H NMR spectrum of DSI-Me is presented in Fig. 3a. The 1H NMR spectrum allowed the suggested structure of DSI-Me to be confirmed and correlates well with the ones reported for other 1,2-disulfonimides.13 The characteristic bands of FTIR spectra illustrated in Fig. 3b in the range from 800–1600 cm−1 correspond well with the ones reported for 4-methyl-1,2-benzenedisulfonimide.13 The band at 825 cm−1 may be attributed to the aromatic C–H bending vibrations.26 The peaks in the range 1100–1400 cm−1 correspond to the vibrations of sulfone fragments of the cycle.27 The characteristic peak at 1506 cm−1 most likely relates to the deformational vibrations of a sulfonimide cycle, analogous to the case of carboxyl imides.28 The bands in the range 2800–3100 cm−1 may be attributed to C–H vibrations in the methyl groups of DSI-Me.
The melting point of DSI-Me was measured to be 198.6 °C with an interval of 0.6 °C, which is significantly higher than that of CFPI (137.5 °C). While having a similar molecular weight (CFPI −348.14 g mol−1, DSI-Me −323.38 g mol−1) and accounting for the difference in the character of terminal groups in the considered compounds, higher melting points of disulfonimide may indicate higher levels of sterical hindrance and/or intermolecular interaction presumably caused by the formation of a stronger charge-transfer complex.
Contrary to the initial suggestion, DSI-Me could not withstand direct oxidation with potassium permanganate, the disulfonimide was observed to have extremely low solubility in water, which prevented its oxidation in aqueous alkaline and acidic solutions. For these reasons, the oxidation was attempted to be carried out in the pyridine and acetone solutions. The 1H NMR spectra of the crude oxidation products showed the absence of carboxylic acid protons shift along with a number of shifts in the range of 10–11 ppm, which were attributed to the protons in the sulfonamide group and indicated the hydrolysis of the disulfonimide ring.
For these reasons, it was proposed to obtain the target dicarboxy-terminated 1,2-disulfonimide by the condensation of 3,4-dichlorosulfonyl-benzoic acid (DSBA-Cl) and p-aminobenzoic acid (IV.b, Scheme 1). In order to accomplish that the methyl group of BMDSA was successfully oxidised in alkaline potassium permanganate to obtain 3,4-disulfobenzoic acid (DSBA) (II.b, Scheme 1). The 1H NMR (DMSO d6) spectrum of DSBA is illustrated in Fig. 4a. Along with the shifts attributed to the protons of the main oxidised product—the shifts of the oxidised diazotization (I) by-product (d 7.52, dd 7.86, d 8.46 ppm) were also present. To verify the suggested structure of the by-product, 4-chloro-3-sulfobenzoic (CSBA) acid was synthesised separately and characterised with 1H NMR spectroscopy. The superposition of DSBA—by-product and CSBA acid spectra is illustrated in Fig. 4b. Identical shifts from both spectra in Fig. 4b allowed the suggested structure of the by-product to be proved.
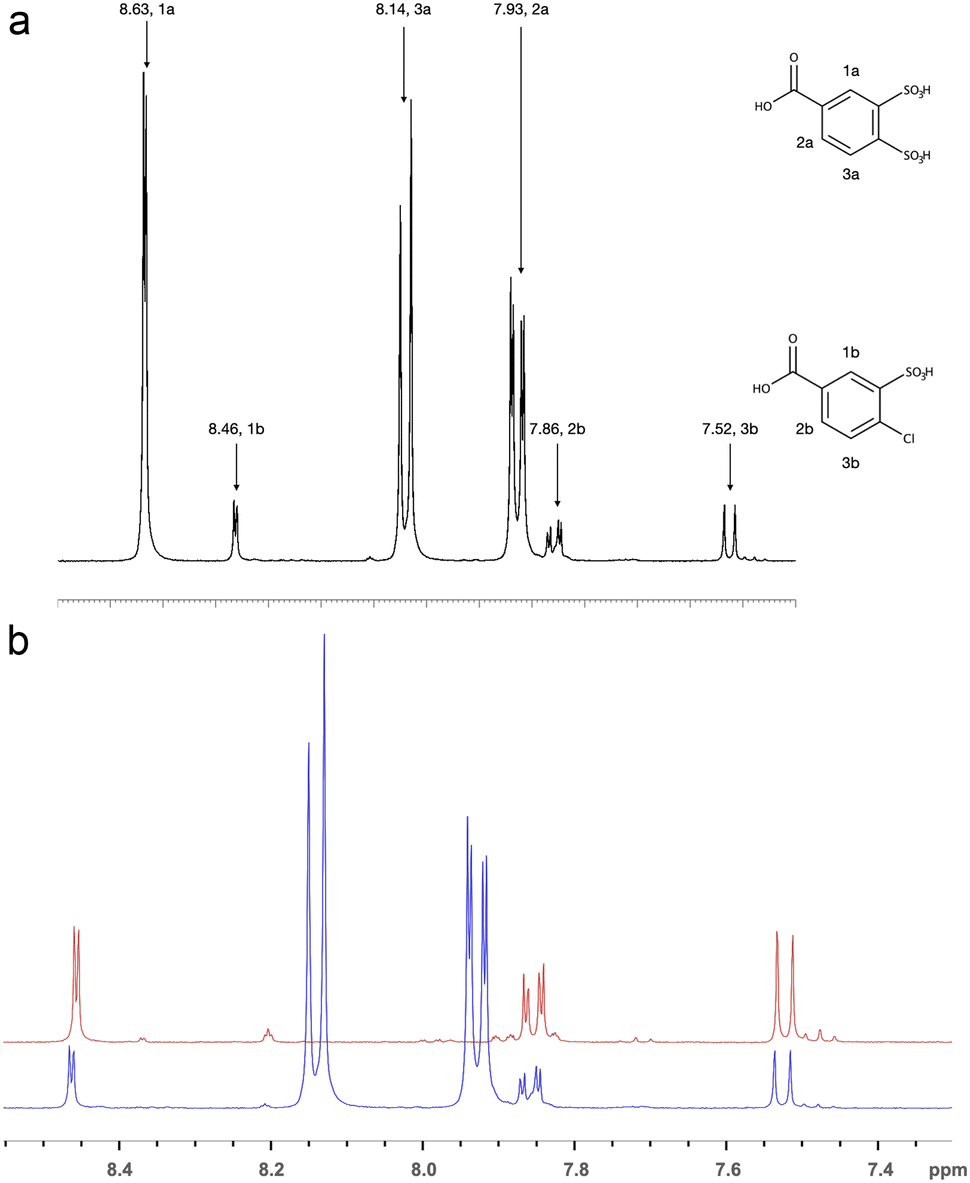 | ||
| Fig. 4 1H NMR spectrum in DMSO d6 of crude DSBA (a), and superposition of spectra of the byproduct and 4-chloro-3-sulfobenzoic acid (CSBA) (b). | ||
Chlorination of DSBA, contrary to expectations formed on the basis of information on similar reactions,15 was not straightforward. The isolation of target disulfonyl chloride from reactions of DSBA with phosphorus oxychloride, similar to II (Scheme 1) was unsuccessful. For these reasons, the peculiarities of DSBA chlorination were studied in more detail. The reaction of DSBA with thionyl chloride in mild conditions (40–45 °C, DMFA cat.) was conducted as an alternative. The 1H NMR spectrum of the hydrolysed product of the DSBA reaction with thionyl chloride in CDCl3 (P1) is illustrated in Fig. 5. The shift at 13.64 ppm corresponds to an acid proton of the carbonyl group, while shifts in the range of 7.7–8.9 ppm relate to the protons in the substituted aromatic ring.
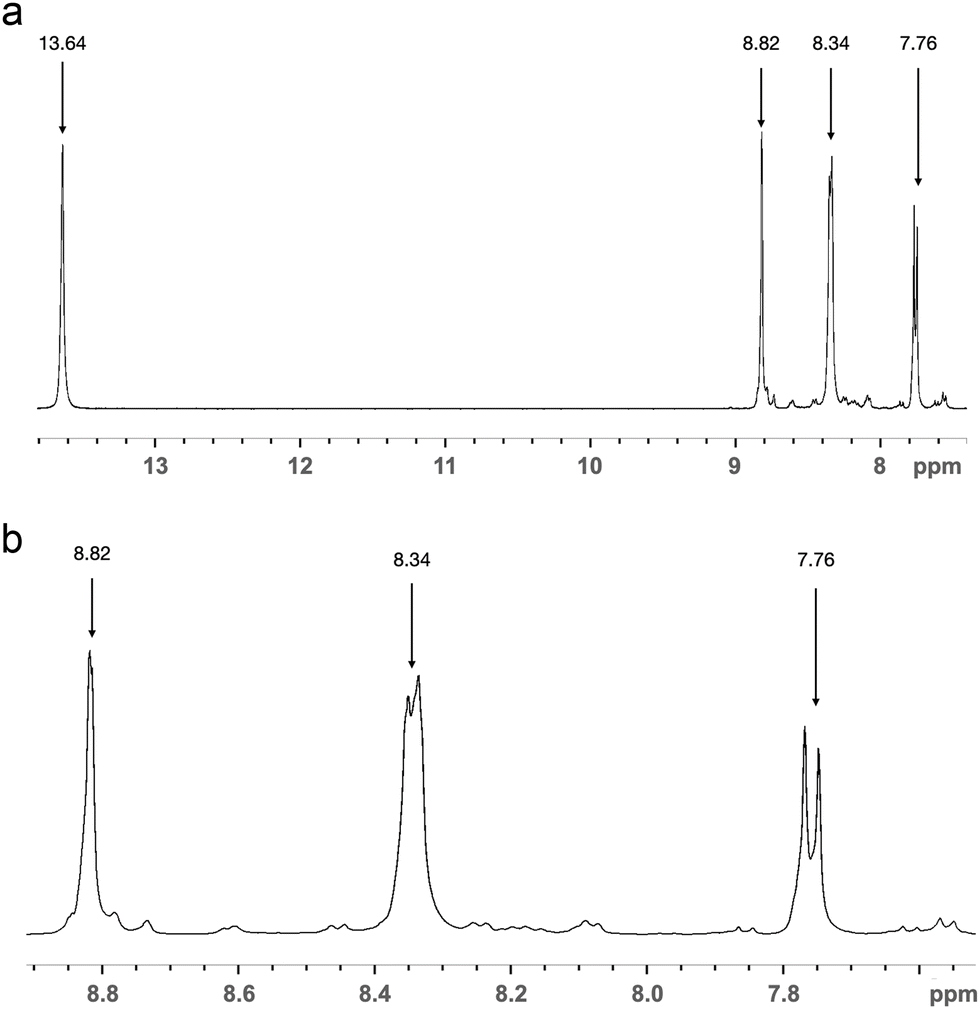 | ||
| Fig. 5 1H NMR spectra in CDCl3 of a hydrolyzed and isolated product of DSBA chlorination with thionyl chloride; (a) full spectrum, (b) fragment with signals of aromatic protons enlarged. | ||
Contrary to expectations, the isolated and purified P1 did not form target disulfonimide in the condensation reaction with p-aminobenzoic acid. The spectra of the crude condensation product were not informative, so it was attempted to isolate individual compounds by means of flash chromatography. P1, unlike DSBA, or CSBA acid showed excellent solubility in CHCl3 and methylene chloride which along with the spectral data indicated the change in chemical structure. However, the distribution of the proton signals along with their multiplicity showed great similarity with the P1 and CSBA spectra which may indicate the same substituents in the aromatic ring.
Judging by the multiplicity of the protons on the P1 spectra (Fig. 5), it was suggested that, accounting for the highly electron-poor aromatic ring, the substitution of the sulfonic acid group in the para position to the carboxylic group may take place, along with the chlorination. The purification of the crude condensation product allowed us to isolate p-aminobenzoic acid and 4-chloro-3-sulfobenzoic acid in the form of triethylammonium salts, supporting our deduction. The chlorination of DSBA was subsequently repeated with a stronger chlorinating agent – PCl5. The 1H NMR spectra of DSBA chlorination with PCl5 hydrolysed products (P2) with reaction times ranging from 2 to 20 hours are presented in Fig. 6.
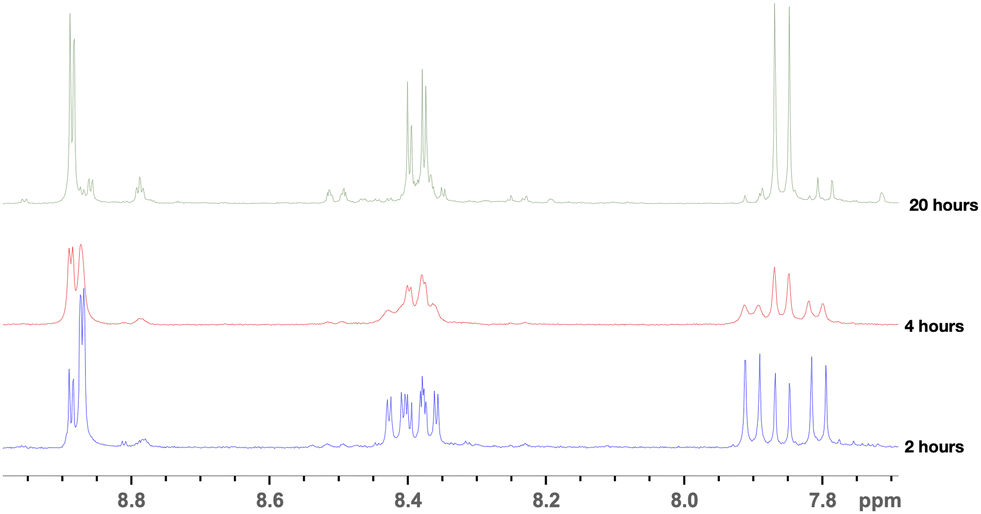 | ||
| Fig. 6 1H NMR spectra of DSBA chlorination products with PCl5 in POCl3 after isolation via hydrolysis, obtained during different reaction times. | ||
The NMR spectra of products isolated from the reactions conducted for 2 and 4 hours showed that the shifts of the reagents and intermediates fade, while the reaction is almost complete after 20 hours leading to a sole major product. The spectra of P1 and P2 products show significant similarity, which attests that P1 and P2 (after 20-hour reaction and hydrolysis) are the same product. As P1, the product of the 20-hour process did not react with p-aminobenzoic acid in any other unexpected way, it was suggested that both chlorination reactions led to the formation of 4-chloro-3-chlorsulfonylbenzoyl chloride, which, during treatment with ice water, hydrolyzed to 4-chloro-3-chlorsulfonylbenzoic acid, which, after further hydrolysis, converted to 4-chloro-3-sulfobenzoic acid. The 3 highly electron-withdrawing substituents leave an electron-poor aromatic ring. Therefore, it is difficult to obtain disulfonylchloride-benzoic acid by direct chlorination of the corresponding disulfonic acid. The nucleophilic substitution occurs in the position para to the carboxyl group directed by the electron-withdrawing substituents. The suggested reaction equation is presented in Scheme 2, the updated reaction scheme is presented in Scheme 1b, while the updated dicarboxy terminated precursor synthesis route, which is planned to be studied in more detail, is illustrated in Scheme 3. The nature of the substituents in the ring means the reaction most likely occurs through an SNAr mechanism.
 | ||
| Scheme 2 Suggested DSBA reactions with chlorinating agents. | ||
 | ||
| Scheme 3 Prospective synthetic path for further development of the DSI-DA synthesis. | ||
Due to the difficulties in the obtainment of carboxy substituted 1,2 disulfonyl chloride through direct chlorination, route b was considered non-prospective. Based on these observations, modification of DSI-Me oxidation along with different thiol-based synthetic approaches13 seem to be the most promising paths to develop.
2.4 Computational studies
In order to evaluate the prospects of the considered 1,2-disulfonimide for the synthesis of novel polymers, the difference in CFPI and DSI-DA-Cl chemical structures was analysed. The correlation between CFPI chemical structure and the properties of polymers based on it is well studied,17,20,21 making it a perfect candidate for the basis of analytical comparison. Conformational and electrostatic differences between the compounds were compared using computational methods in order to form a basis for the suggestion on the properties of the polymers DSI-DA-Cl may form.The intermolecular interactions in polyimides are believed to be caused by the formation of charge-transfer complexes.29 An increase in the electron deficiency of the corresponding links of the macromolecule should increase the charge difference, and by so lead to the increase in electrostatic force responsible for strong intermolecular interactions in the polymer. The difference in the electronic density between DSI-DA-Cl and CFPI was assessed using computational methods. Both structures were optimised (100-step optimisation) using the N-31 (6-31G) basis in GAMESS software. The illustration of calculated electron density maps of both components is illustrated in Fig. 7.
 | ||
| Fig. 7 Chemical structures (a); electron density maps (b) and interplanar angles of interest (c) of DSI-DA-Cl and CFPI obtained using the N-31 (6-31G) basis in GAMESS software. | ||
The dihedral angle between the imide ring and benzene plains in the DSI-DA-Cl molecule (56.14°) is significantly higher than that in CFPI (26.33°). Along with the non-planar arrangement of oxygen atoms, these structural features may introduce steric hindrance during the packing of the chains and lead to an increase of the inter-planar distance in the polymer. Higher electron deficiency of the 1,2-disulfonimide group along with its steric features should lead to an increase in the rate of inter-chain interactions along with the free volume in the corresponding polymers. Such features demonstrate that poly(1,2-disulfonimides) may be considered extremely promising in the area of gas-separation membranes’ development.
Materials and methods
General
2-Amino-5-methyl-benzenesulfonic acid (99%), p-toluidine (99%), N(Et)3 (99%) NaNO2 (99.8%), and CuSO4·5H2O (99.5%) were purchased from Sigma-Aldrich and used without further purification. Thionyl chloride (≥ 99%) and phosphorus oxychloride ((≥ 99%)) were also supplied by Sigma–Aldrich and were purified by vacuum distillation prior to the reactions. SO2 gas was obtained by acid hydrolysis of saturated sodium sulphite solution by dropwise addition of sulfuric acid and bubbled through ice-acetic acid to obtain a 30 mol% solution. All other chemicals and solvents were of reagent grade and were used without further purification unless otherwise stated. 1H and 13C NMR spectra were recorded on a Bruker AVANCE II −500 WB at 400 and 100 MHz, respectively. Attenuated total reflection (ATR) infrared spectra were recorded on a Bruker Vertex 70 using a Pike FTIR attachment with a ZnSe working element in the mode of Frustrated transition [ATR Fourier transform infrared (FTIR)]. The spectra were recorded with a resolution of 4 cm−1 with 60 scans. During processing, a correction was introduced to take into account the dependence of the radiation penetration depth on the wavelength. The elemental composition was determined using the LECO TruSpec Micro CHNS analyzer.Synthesis of 4-methyl-1,2-benzenedisulfonic acid (BMDSA)
2-Amino-5-methyl-benzenesulfonic acid (AMBSA 60.0 mmol, 11.233 g) was dissolved in 2.4 g of NaOH solution in 133 ml of water. After the full dissolution of AMBSA, 32.2 ml of 10 mol% (0.24 mol) HCl water solution was added to the reaction, and the precipitation of AMBSA intramolecular salt was observed. The reaction mixture was cooled using an ice bath to 2–4 °C and NaNO2 (60.0 mmol, 4.148 g) dissolved in 13 ml of water was added dropwise, while the reaction temperature was maintained below 4 °C. After all the NaNO2 solution was added, the reaction was left for an hour in a cooling bath. The formation of pink benzene-diazonium sulfonate crystals was observed. The crystals were filtered and dispersed in 75 ml of concentrated aqueous HCl solution containing a Cu (I) catalyst in a 500 ml flask. The HCl solution with a catalyst was prepared in advance by reduction of dissolved 4.37 g of CuSO4·5H2O by the addition of 1.2 g of sodium sulphite. A freshly prepared 30 mol% solution of SO2 in 118 ml of ice acetic acid was added to the reaction with intense stirring. The reaction temperature was slowly raised to 42 °C and maintained for 6 hours, while SO2 was constantly bubbled through the reaction solution. The mixture was then thoroughly degassed using a water-jet pump, and the solvents were evaporated in vacuo.The solid residue was washed with 10 ml of saturated NaCl solution at 60 °C, the green paste obtained was then cooled for 10–12 hours at 4 °C, filtered and washed with 5 ml of methanol. The solvents from the filtered solution were removed using a rotary evaporator and the solid residue was dried in vacuum at 60 °C until a constant weight was reached. 9.581 g of a powder containing 4-methyl-1,2-benzenedisulfonic acid (BMDSA) in the form of a sodium salt was obtained. The presence of BMDSA in the residue was confirmed with NMR spectroscopy (1H (400 MHz, D2O): δ, ppm −2.59 (s, 3H), 7.79 (dd, 1H, J1 = 8.47 Hz, J2 = 1.4 Hz), 8.05 (d, 1H, J = 1.11 Hz), 8.49 (d, 1H, J = 8.53 Hz) (Fig. 2); 13C (100 MHz, D2O): δ, ppm −20.4, 129.7, 130.0, 133.3, 137.3, 139.9, 142.7).
Synthesis of 4-methyl-1,2-dichlorosulfonylbenzene (BMDSA-Cl)
5 g of dried powder containing BMDSA was dispersed in 50 ml (0.32 mol) of POCl3. The mixture was refluxed for 12 hours. The reaction solution was filtered, and POCl3 was removed completely from the filtered solution under vacuum at 100 °C. The oil obtained after the removal of POCl3 was dissolved in the 100 ml 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 vol.p. water/chloroform mixture. After the full dissolution of the oil, the phases were separated. The organic phase was washed twice with 50 ml of water, twice with a 5 wt% solution of sodium carbonate and again, twice with 50 ml of water. The water phase was extracted with 50 ml of chloroform. Organic phases were combined, dried with anhydrous sodium sulphate and concentrated on a rotary evaporator. The obtained concentrate was dried in vacuo at 60 °C to give 2.68 g of pale brown crystals. BMDSA-Cl was isolated from the crude product by means of flash chromatography using hexane: ethyl acetate (7:3) as the eluent (TFC hexane: ethyl acetate Rf = 0.46). 1.88 g of purified BMDSA-Cl was isolated (1H (400 MHz, CDCl3): δ, ppm – 2.47 (s, 3H), 7.47 (dd, 1H, J1 = 8.20 Hz, J2 = 2.13 Hz), 7.54 (d, 1H, J = 8.24 Hz), 7.97 (d, 1H, J = 1.92 Hz); 13C (100 MHz, CDCl3): δ, ppm – 20.9, 129.7, 130.6, 132.6, 136.7, 138.2, 140.8), which corresponds to the overall yield of 15.4% for 2 stages, calculated from the amount of AMBSA.
:1 vol.p. water/chloroform mixture. After the full dissolution of the oil, the phases were separated. The organic phase was washed twice with 50 ml of water, twice with a 5 wt% solution of sodium carbonate and again, twice with 50 ml of water. The water phase was extracted with 50 ml of chloroform. Organic phases were combined, dried with anhydrous sodium sulphate and concentrated on a rotary evaporator. The obtained concentrate was dried in vacuo at 60 °C to give 2.68 g of pale brown crystals. BMDSA-Cl was isolated from the crude product by means of flash chromatography using hexane: ethyl acetate (7:3) as the eluent (TFC hexane: ethyl acetate Rf = 0.46). 1.88 g of purified BMDSA-Cl was isolated (1H (400 MHz, CDCl3): δ, ppm – 2.47 (s, 3H), 7.47 (dd, 1H, J1 = 8.20 Hz, J2 = 2.13 Hz), 7.54 (d, 1H, J = 8.24 Hz), 7.97 (d, 1H, J = 1.92 Hz); 13C (100 MHz, CDCl3): δ, ppm – 20.9, 129.7, 130.6, 132.6, 136.7, 138.2, 140.8), which corresponds to the overall yield of 15.4% for 2 stages, calculated from the amount of AMBSA.
Synthesis of 4-methyl-N-(p-methyl)-benzene-1,2-disulfonimide (DSI-Me)
BMDSA-Cl (7.00 mmol, 1.88 g) was dissolved in a two-neck flask in 34 ml of methylene chloride. The reaction mixture was cooled using an ice bath to 0–5 °C, a freshly prepared solution of 0 (8.5 mmol, 0.910 g). p-Toluidine in 15 ml of methylene chloride with 3 ml of (20 mmol) N(Et)3 was added with vigorous stirring. The completion of a reaction was monitored by means of TFC (hexane: ethyl acetate 7:3), when the spot of BMDSA-Cl disappeared, after approximately 4 hours, the reaction was complete. The reaction mixture was washed twice with 50 ml of 0.1 M aqueous H2SO4 solution, three times with 50 ml of water, by separating the phases after each addition. The aqueous extracts were combined and washed two times with 50 ml of methylene chloride. The combined organic phase was dried with anhydrous sodium sulphate and concentrated using a rotary evaporator. The obtained powder was dried in vacuum at 60 °C until a constant weight was reached. 0.7 g of pale-yellow pure DSI-Me crystalline powder (yield 40%) (NMR:1H (400 MHz, CDCl3): δ, ppm −2.46 (s, 3H), 2.62 (s, 3H), 7.39 (dd, 2H, J1 = 8.13 Hz, J2 = 2.20 Hz), 7.55 (dd, 2H, J1 = 8.24 Hz, J2 = 1.67 Hz), 7.75 (d, 1H, J = 8.24 Hz), 7.90 (d, 1H, J = 0.93 Hz), 7.98 (d, 1H, J = 7.85 Hz); 13C (100 MHz, CDCl3): δ, ppm −21.5, 22.0, 121.8, 122.4, 122.6, 130.9 (2C), 132.1 (2C), 132.4, 135.3, 135.4, 142.3, 146.7; m.p. = 198.6 °C; calcd for C14H13NO4S2: C 52.00%, H 4.05%, N 4.33%, S 19.83%, found: C 52.06%, H 4.08%, N 4.38%, S 19.80%) were isolated from the crude product by means of flash chromatography (hexane: ethyl acetate 5:2, TLC Rf = 0.34).
Oxidation of DSI-Me
Synthesis of 3,4-disulfobenzoic acid (DSBA)
1 g of unpurified BMDSA powder was dissolved in 12 ml of water. The solid sodium hydroxide in small portions was added with stirring until a pH of 7–8 was reached. KMnO4 (9.66 mol, 1.526 g) was added to the reaction, the mixture was refluxed with constant stirring at 90 °C for 2 hours. The excess of potassium permanganate was reduced by adding a saturated sodium sulfite solution dropwise until the purple colour disappeared. The mixture was filtered to remove MnO2, which was then washed with 20 ml of hot water. Combined liquid residues were neutralised by adding concentrated aqueous HCl dropwise, concentrated on a rotary evaporator and dried to a constant weight in a vacuum oven at 60 °C to obtain 1.61 g of powder containing DSBA. The presence of DSBA in the crude product was verified by means of NMR spectroscopy (1H (400 MHz, DMSO d6): δ, ppm – 7.93 (dd, 1H, J1 = 8.43 Hz, J2 = 2.00 Hz), 8.14 (d, 1H, J1 = 8.03 Hz), 8.63 (d, 1H, J = 2.03 Hz), 13.13 (s, broad, 1H); 13C (100 MHz, D2O): δ, ppm −129.9, 131.5, 132.8, 139.3, 140.0, 141.8, 173.6).Chlorination of DSBA
Condensation of P1 and P2 with p-aminobenzoic acid
0.500 g of p-aminobenzoic acid were dissolved in 10 ml of chloroform with 2 ml of N(Et)3 and added to 0.430 g of DSBA chlorination product (P1 or P2) dissolved in 20 ml of chloroform. The reaction mixture was stirred at room temperature for 24 hours, refluxed for additional 3 hours and solvents were removed on a rotary evaporator. The crude residue was dissolved in 5 ml of water with 0.25 ml of 20% sodium hydroxide and washed through a column filled with ion-exchange resin (H+ form), the fractions were collected until the neutral pH was reached. Solvents were removed from the obtained solution, and this was dried to obtain 0.36 g of crude condensation products. The crude condensation products were purified by means of flash chromatography (hexane/acetone (1 → 10%) 0.1% acetic acid), the major products isolated are p-aminobenzoic acid and 4-chloro-3-sulfobenzoic acid in triethylammonium salt forms.Synthesis of 4-chloro-3-sulfobenzoic acid
The benzene diazonium sulfonate was obtained from AMBSA as described in the synthesis of 4-methyl-1,2-benzenedisulfonic acid (BMDSA) section. The diazo-compound (1 g) was suspended in 10 ml of HCl with 0.1 g of Cu2Cl2, the mixture was stirred at 40 °C for 2 hours and left overnight at room temperature. The reaction solution was neutralised with a 20% solution of sodium hydroxide until a pH of 8–9 was reached. The precipitate was filtered before the filtrate was then concentrated on a rotary evaporator. The solid residue was oxidised with potassium permanganate as described in the synthesis of 3,4-disulfobenzoic acid (DSBA) section to obtain 0.64 g of 4-chloro-3-sulfobenzoic acid (1H (400 MHz, DMSO d6): δ, ppm – 7.52 (d, 1H, J1 = 8.52 Hz), 7.85 (dd, 1H, J1 = 8.43 Hz J2 = 2.44 Hz), 8.46 (d, 1H, J = 2.50 Hz))Conclusions
The structure and an approach for the synthesis of novel 1,2-disulfonimide monomers were proposed and discussed in detail. Based on the state-of-the-art understanding of the correlation between the polymer's structure and its transport properties, poly-1,2-disulfonimides are suggested to be a promising material for gas-separation membranes. Novel 1,2-disulfonimide (4-methyl-N-(p-methylphenyl)-benzene-1,2-disulfonimide) was obtained and characterised. The chlorination of 1,2-disulfobenzoic acid, contrary to expectations, has led to the 4-chloro-3-sulfobenzoic acid instead of 1,2-disulfonylchloride-benzoic acid. The tendency of the reaction towards the nucleophilic substitution in the aromatic ring was explained by the presence of three highly electron-withdrawing substituents in the ring, which drastically decrease the electron density around the carbons of the ring.The attempts at the oxidation of model dimethyl-terminated 1,2-disulfonimide in various media showed the inability of the 1,2-disulfonimide cycle to withstand the oxidation conditions. Due to the complexities encountered in the synthesis of 1,2-disulfonylchloridebenzoic acid from 1,2-disulfobenzoic acid and the low stability of the 1,2-disulfonimide cycle in the oxidation reactions, the most promising approach for the synthesis of 4-chloroformyl-N-(p-chloroformylphenyl)-benzene-1,2-disulfonimide is considered to be through the thiol-based synthetic pathway.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors express their deepest gratitude to Elena Nikolaevna Vlasova for the conduction of FTIR measurements and the NMR department of the Institute of Macromolecular Compounds Russian Academy of Science for the obtainment of NMR spectra.References
- A. Heintz and W. Stephan, J. Membr. Sci., 1994, 89, 153–169, DOI:10.1016/0376-7388(93)E0223-7.
- R. W. Baker, Membrane technology and applications, John Wiley & Sons Ltd., Chichester, 3rd edn, 2012 Search PubMed.
- A. Y. Alentiev and Yu. P. Yampolskii, J. Membr. Sci., 2002, 206, 291–306, DOI:10.1016/s0376-7388(01)00777-3.
- L. M. Robeson, J. Membr. Sci., 2008, 320, 390–400, DOI:10.1016/j.memsci.2008.04.030.
- H. Sanaeepur, A. Ebadi Amooghin, S. Bandehali, A. Moghadassi, T. Matsuura and B. Van der Bruggen, Prog. Polym. Sci., 2019, 91, 80–125, DOI:10.1016/j.progpolymsci.2019.02.001.
- H. Ohya, V. V. Kudryavtsev and S. I. Semenova, Polyimide Membranes: Applications, Fabrications and Properties, CRC Press, Florida, 1st edn, 1997 Search PubMed.
- X.-H. Ma and S.-Y. Yang, Polyimide Gas Separation Membranes. Advanced Polyimide Materials, Elsevier, Amsterdam, 2018, ch. 6, pp. 257–322 DOI:10.1016/b978-0-12-812640-0.00006-8.
- M. R. Coleman and W. J. Koros, J. Membr. Sci., 1990, 50, 285–297, DOI:10.1016/s0376-7388(00)80626-2.
- K. Tanaka, M. Okano, H. Toshino, H. Kita and K.-I. Okamoto, J. Polym. Sci., Part B: Polym. Phys., 1992, 30, 907–914, DOI:10.1002/polb.1992.090300813.
- C. Nagel, K. Günther-Schade, D. Fritsch, T. Strunskus and F. Faupel, Macromolecules, 2002, 35, 2071–2077, DOI:10.1021/ma011028d.
- Y. Jung, Y. Yang, S. Kim, H.-S. Kim, T. Park and B. W. Yoo, Eur. Polym. J., 2013, 49, 3642–3650, DOI:10.1016/j.eurpolymj.2013.07.034.
- T. Okada and S. Ando, Polymer, 2016, 86, 83–90, DOI:10.1016/j.polymer.2016.01.037.
- M. Barbero, S. Berto, S. Cadamuro, P. G. Daniele, S. Dughera and G. Ghigo, Tetrahedron, 2013, 69, 3212–3217, DOI:10.1016/j.tet.2013.02.053.
- Y. Bekdemir, B. Eren and H. Kütük, Phosphorus, Sulfur Silicon Relat. Elem., 2012, 2012(187), 689–696, DOI:10.1080/10426507.2011.642431.
- K. Sørbye, C. Tautermann, P. Carlsen and A. Fiksdahl, Tetrahedron: Asymmetry, 1998, 9, 681–689, DOI:10.1016/s0957-4166(98)00033-0.
- W. H. Mills and R. E. D. Clark, J. Chem. Soc., 1936, 35, 175–181, 10.1039/jr9360000175.
- S. V. Kononova, D. A. Sapegin, E. V. Kruchinina, G. N. Gubanova, K. A. Romashkova, A. L. Didenko, V. E. Smirnova, E. N. Popova, N. N. Saprykina, E. N. Vlasova and V. M. Svetlichnyi, J. Appl. Polym. Sci., 2019, 136, 48197, DOI:10.1002/app.48197.
- S. Kuchanov, H. Slot and A. Stroeks, Prog. Polym. Sci., 2004, 29, 563–633, DOI:10.1016/j.progpolymsci.2004.02.001.
- E. P. Mel’nikova, B. A. Tagiev and A. A. Vansheidt, Vysokomol. Soed., 1969, 11, 1209–1213 Search PubMed.
- D. A. Sapegin, G. N. Gubanova, E. N. Popova and S. V. Kononova, J. Appl. Polym. Sci., 2020, 138, 49982, DOI:10.1002/app.49982.
- V. A. Gusinskaya, M. M. Koton, T. V. Batrakova and K. A. Romashkova, Vysokomol. Soedin., Ser. A, 1976, 18, 2677–2680 Search PubMed.
- C. Fahlberg and I. Remsen, Chem. Ges., 1879, 12, 469–473, DOI:10.1002/cber.187901201135.
- R. H. Nuttall, E. R. Roberts and D. W. A. Sharp, Spectrochim. Acta, 1961, 17, 947–952, DOI:10.1016/0371-1951(61)80030-1.
- P. D. S. Babu, S. Periandy, S. Mohan, S. Ramalingam and B. G. Jayaprakash, Spectrochim. Acta, Part A, 2011, 78, 168–178, DOI:10.1016/j.saa.2010.09.017.
- A. M. Castillo, L. Patiny and J. Wist, J. Magn. Reson., 2011, 209, 123–130, DOI:10.1016/j.jmr.2010.12.008.
- R. D. Kross, V. A. Fassel and M. Margoshes, J. Am. Chem. Soc., 1956, 78, 1332–1335, DOI:10.1021/ja01588a019.
- K. C. Schreiber, Anal. Chem., 1949, 21, 1168–1172, DOI:10.1021/ac60034a005.
- T. Uno and K. Machida, Bull. Chem. Soc. Jpn., 1961, 34, 551–556, DOI:10.1246/bcsj.34.551.
- B. V. Kotov, T. A. Gordina, V. S. Voishchev, O. V. Kolninov and A. N. Pravednikov, Polym. Sci. U.S.S.R., 1977, 19, 711–716 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nj06005a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2023 |