
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and evaluation of new modified diglycolamides with different stereochemistry for extraction of tri- and tetravalent metal ions†
Laura
Diaz Gomez
 a,
Andreas
Wilden
*a,
Dimitri
Schneider
a,
Zaina
Paparigas
a,
Giuseppe
Modolo
a,
Maria Chiara
Gullo
b,
Jurriaan
Huskens
b and
Willem
Verboom
b
a,
Andreas
Wilden
*a,
Dimitri
Schneider
a,
Zaina
Paparigas
a,
Giuseppe
Modolo
a,
Maria Chiara
Gullo
b,
Jurriaan
Huskens
b and
Willem
Verboom
b
aForschungszentrum Jülich GmbH, Institut für Energie und Klimaforschung, Nukleare Entsorgung (IEK-6), 52428 Jülich, Germany. E-mail: a.wilden@fz-juelich.de
bLaboratory of Molecular Nanofabrication, Department for Molecules & Materials, Mesa+ Institute for Nanotechnology, University of Twente, 7500 AE Enschede, The Netherlands
First published on 12th January 2023
Abstract
Americium separation from high level nuclear waste can provide certain benefits such as reducing the long-term heat generation or the possibility of re-use as fuel in innovative reactors. The separation is carried out through solvent extraction. Among commonly used ligands, diglycolamide (DGA) derivatives have shown high distribution ratios for trivalent lanthanides (Ln(III)) and actinides (An(III)). Recently, different diastereomers of DGAs with substitutions in the backbone have shown an inverse selectivity for Am over Cm, in comparison with commonly used DGAs. Hence, we synthesised, characterized, and tested novel syn- and anti-diastereomers of tetra-n-octyl and tetra-n-decyl DGAs with two propyl and ethyl-methyl substituents at the backbone methylene carbon atoms. The distribution ratios for An(III) and Ln(III) were found to be generally lower than for the unsubstituted parent molecules due to steric hindrance. On the other hand, 239Pu was much better extracted. All tested ligands showed the inverse selectivity for Am with an Am/Cm separation factor (SFAm/Cm) around 1.5. Finally, slope analysis confirmed a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 metal:ligand ratio for the DGA complexes.
:3 metal:ligand ratio for the DGA complexes.
Introduction
Numerous processes (e.g., innovative-SANEX,1 AmSel,2 GANEX,3–5etc.6,7) have been developed with the main purpose of minimizing the radiotoxicity of High-Level Waste (HLW) obtained from reprocessing of Spent Nuclear Fuel (SNF). A major focus is the separation of the minor actinides Am(III) and Cm(III) from lanthanides and other fission products with the interest of reducing the long-term heat generation of SNF.8–10 The separation of particular metal ions is achieved by solvent extraction using selective hydrophilic or lipophilic ligands in aqueous and/or organic solutions, or a combination of ligands.11 The main priority nowadays is looking for different ligands which can effectively separate actinides (An) from lanthanides (Ln) and Am from Cm.12 Ideally, the ligands are composed of only C, H, O and N (the so-called CHON principle) which allows incinerating the final waste without generating additional solid waste. Diglycolamides (DGAs, e.g., see Fig. 1) are molecules which contain two amide groups bound by an ether group. Since 2001, they have been tested for their capability to complex actinides and lanthanides in order to achieve the separation of minor An and Ln from HLW.13 However, DGAs usually do not provide the required selectivity for An(III) over Ln(III). Therefore, they are often combined with other ligands in a synergistic system to enable the envisioned selective separation.14,15 | ||
| Fig. 1 Overview of the diglycolamide ligands used in this study with different alkyl side chain lengths (n-octyl and n-decyl), different substituents in the ligand backbone (dipropyl and ethyl-methyl), as well as different stereochemical orientation (syn or anti). | ||
A broad number of DGA derivatives with alkyl substituents on the amide groups have been synthesised and tested to assess their physical and chemical properties, specifically with f-elements.13,16–18 The alkyl chains on the amide groups have a direct influence on the solubility of the ligand, e.g., long alkyl chains (n-octyl and n-decyl) make the compounds lipophilic and present high distribution ratios from process relevant HNO3 concentrations (1–6 mol L−1) with An(III) and Ln(III). However, longer alkyl chains cause reduced distribution ratios due to steric hindrance.13,17,19 On the other hand, short alkyl chains give hydrophilic DGAs. N,N,N′,N′-tetra-n-octyl diglycolamide (TODGA) and N,N,N′,N′-tetra-n-decyl diglycolamide (TDDGA) have been shown to be potential ligands for solvent extraction processes with high lipophilic solubility and high An(III) distribution ratios.1,2,13,19,20 Afterwards, substituents on both methylene carbons in the backbone of the molecule with alkyl groups have been introduced to find that these groups partly reduce radiolysis.16,21–23 Another approach with interesting results concerns diastereomers of the doubly methylated TODGA derivative with methyl substitutions in the backbone.24,25 Usually, DGAs present higher affinity for Cm over Am. For the methyl-substituted derivatives an inverse selectivity of Am over Cm by the Me2-TODGA anti-diastereomer was observed, which was explained by the orientation influencing the interaction between the nitrate ions and the extracted metal complex.23 Generally, the influence of the stereochemistry of extractants has yet only received little attention in the field of spent nuclear fuel treatment, despite the evidence on extraction differences caused by the orientation in ligands.23,26–31 Based on these results, n-octyl and n-decyl DGAs were synthesised with dipropyl and ethyl-methyl substituents in the backbone of the molecules and their respective syn- and anti-isomers were isolated (Fig. 1), and their extraction behaviour and selectivity for An(III) and Ln(III), and specifically for Am(III) and Cm(III) was studied. Fundamental chemistry studies are planned on ligands with wider applicability. Here, the synthesis and first extraction studies were covered.
Results and discussion
The diastereomeric ligands 4, 5 and 9, 10 were prepared in an analogous way as described for Me2-TODGA.23 2-Bromobutanoate was reacted with ethyl (S)-lactate in the presence of NaH as a base to give diastereomeric diesters 1 in a ratio of 3:1, which were separated by flash chromatography. Analogously, 2-bromovalerate was reacted with ethyl (±)-2-hydroxyvalerate to give diastereomeric diesters 6 in a ratio of 5:1. Saponification of the ester groups in 1 and 6 afforded the corresponding dicarboxylic acids 2 and 5 in quantitative yields. Using Schotten-Baumann conditions,322 and 5 were converted into the target compounds 4,5 and 9,10, via the in situ prepared dichlorides, in varying yields.
In our previous study23 we assigned the stereochemistry of the related Me2-TODGA based on the difference in the 1H NMR spectra upon complexation with lanthanum(III) triflate. In the 1H NMR spectrum of the starting diester, the anti-diastereomer showed the C(O)OCH2-signal as a quartet, as expected. However, in case of the syn-diastereomer the corresponding methylene group resides as a complicated multiplet due to steric interaction with the methyl group at the backbone. This characteristic feature is used for the assignment of the stereochemistry of the diesters 1 and 6, showing the same characteristics, and consequently also holds for the ligands 4,5 and 9, 10, respectively.
We expected low distribution ratios (D) associated with high uncertainties due to steric hindrance produced by the long alkyl side chains and the substitution of the methylene carbon atoms in the backbone. However, we are interested in the extraction behaviour and trends of each diastereomer and their affinity for Am(III) over Cm(III).23 Distribution ratios were calculated as the ratio of activity or metal ion (M) concentration in the organic phase vs. the activity or metal ion concentration in the aqueous phase ([M]org/[M]aq) from single measurements. Fig. 2 shows the Am(III) distribution ratios for all synthesized ligands as a function of the nitric acid concentration. The first results of the ligand screening confirmed our predictions for the trivalent metal ions (e.g., Am(III)). Distribution ratios increased with increasing HNO3 concentration.13 Generally, ethyl-methyl substitution for both alkyl side chain lengths (C8 and C10) extracted more Am(III) than for dipropyl substitution and regarding the orientation of the substituents in the backbone syn-diastereomers showed higher D values. The highest Am(III) D value was 7.0 for syn-Et-Me-TDDGA 5A at 9.8 mol L−1 HNO3. The anti-Et-Me-TDDGA 5B diastereomer showed a distribution ratio of 5.5 at the same HNO3 concentration. At lower HNO3 concentrations the distribution ratios were approximately one to two orders of magnitude lower than for the syn diastereomer. Anti-Et-Me-TODGA 4B distribution ratios (Fig. 2, black squares) were close to but slightly lower than the ones of syn-Et-Me-TDDGA 5A (Fig. 2, yellow diamonds). Even higher distribution ratios are expected for the syn-Et-Me-TODGA 4A diastereomer as the syn-diastereomers of these diglycolamide extractants usually give higher distribution ratios than the anti-diastereomer.23,30 However, the syn-Et-Me-TODGA 4A diastereomer was synthetically unavailable and could not be tested in the current study. The differences in extraction by the different diastereomers was explained by the steric hindrance and orientation created by the alkyl groups of the ligand backbone hindering the nitrate ion complexation in the outer sphere of the complexes influencing their selectivity for Am(III) over Cm(III).23,28–30
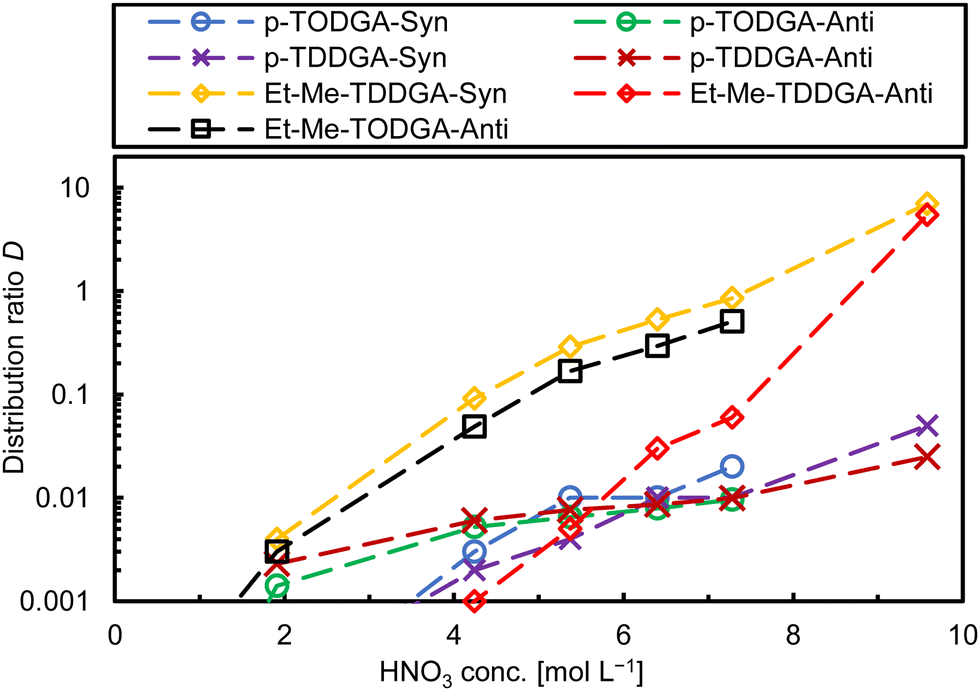 | ||
| Fig. 2 Comparison of Am(III) distribution ratios for the different ligands used in this study. Org.: [Ligands] = 0.1 mol L−1 in n-dodecane; aq.: 10−5 mol L−1 Ln(III) (without Pm) + Y, Sr, Zr, Mo, Pd, Fe, Ru, 152Eu, 239Pu, 241Am, 244Cm; extraction time: 30 min, 22 °C. | ||
The dipropyl substituted DGAs with the same orientation in both analogues showed similar behaviour. Anti-Orientation gave very low D values in a close range (0.005–0.009) slightly above the lower detection limit, without major differences among the HNO3 concentration. Syn-Orientation gave only slightly higher D values at 4 to 7 mol L−1 nitric acid concentration. Hence, dipropyl substitutions present minimal differences on D ratios through the HNO3 concentrations studied here.
The separation factor (SF) between two metal ions was calculated as the ratio of the corresponding distribution ratios (SFM1/M2 = DM1/DM2). As the SF is calculated from two D values with their individual uncertainties, best SF values are obtained where the corresponding D values are close to one. The separation factors reported here were determined accordingly. Concerning Am/Cm selectivity, the studied ligands showed a slight preference for Am(III) over Cm(III) with a separation factor of SFAm/Cm ∼ 1.5 (Fig. S1, ESI†). This finding is different from the behaviour of the two Me2-TODGA diastereomers.23 Our first hypothesis to explain this phenomenon is, that the overall steric hindrance by the longer alky chains of the ligands studied here compared to Me2-TODGA might have a higher influence on Am(III)/Cm(III) selectivity compared to their orientation.
For all the ligands, process-relevant distribution ratios for the trivalent actinides >1 could only be achieved at very high HNO3 concentrations of about 10 mol L−1. Such high HNO3 concentrations can cause splitting of the solvent phase into a light diluent rich phase with reduced extractant concentration, and a heavy phase containing complexes of HNO3 with the extractant.33,34 This 3rd phase formation phenomenon is undesirable for process applications. In other processes using diglycolamides (e.g., TODGA) and high HNO3 concentrations, a phase modifier like 1-octanol is used to mitigate 3rd phase formation.33,35 According to literature, DGAs can form inverse micelles with polar molecules (e.g., HNO3) in long-chain alkane diluents (e.g., n-dodecane or n-octane).33,36,37 Massey et al. found that the addition of n-octanol reduced the degree of aggregation and hence reduced the degree of 3rd phase formation.34 In our experiments, even at 10 mol L−1 HNO3, no 3rd phase formation was observed. The steric hindrance from the substitution in the backbone seems to reduce the probability of increasing aggregate and cluster formation in solution, hence reducing the possibility of third-phase formation at high nitric acid concentrations.33,34,37
Low distribution ratios are not necessarily a disadvantage, as for a process application it must also be possible to back-extract the desired metal ions from the loaded organic phase. Nevertheless, the distribution ratios of the ligands considered here are quite low at a ligand concentration of 0.1 mol L−1 for the trivalent metal ions and associated with high uncertainties. Therefore, it is desirable to increase the distribution ratios maintaining the HNO3 concentrations. Hence, the concentration of syn-p-TODGA 9A was increased to 0.2 mol L−1 for 1.9–9.8 mol L−1 HNO3. Distribution values indeed improved approx. 10-fold. Although, due to the dipropyl substitution, those values are still low compared to the moderate D values required (D ≥ 1). Fig. 3 shows the comparison between 0.1 and 0.2 mol L−1syn-p-TODGA 9A.
 | ||
| Fig. 3 241Am distribution ratios of syn-p-TODGA 9A in two concentrations (exp. conditions see Fig. 2). | ||
Ethyl-methyl substitution in TODGA and TDDGA showed higher extraction for the Ln series (except Pm) and some FP compared to dipropyl substitution (see Fig. S2–S4, ESI†). For the other metal ions, anti-p-TODGA 9B showed a higher extraction for Zr and Mo with values between 0.1 and 1 (Fig. S2–S3, ESI†), in comparison with syn-p-TODGA 9A (D values below 0.1). On the other hand, Ru was not well extracted for most ligands (Fig. S4, ESI†), which is a positive effect since Ru most of the times is co-extracted complicating the system application.17,36 Dipropyl substitution for TODGA and TDDGA syn-diastereomers showed poor extraction for Ln(III), independent of the HNO3 concentration (Fig. 4). All metal ions were tested as radioactive spikes or in low concentrations (1 × 10−5 mol L−1, each) to avoid loading, precipitation or 3rd phase formation. For further process development, these effects, and possible changes in the metal ions speciation (e.g., Zr or Ru) would need to be studied.
 | ||
| Fig. 4 Distribution ratios of Ln (w/o Pm, open symbols), Am and Cm (filled symbols) as a function of the inverse ionic radius for extraction at 7.4 mol L−1 HNO3 (exp. conditions see Fig. 2). | ||
Fig. 4 compares the distribution ratios of Ln(III) (except Pm) as a function of the inverse ionic radius38,39 between ethyl-methyl and dipropyl substitutions. In this figure, the octad-effect is visible for anti-Et-Me-TODGA 4B (Fig. 4, black squares) and syn-Et-Me-TDDGA 5A (Fig. 4, yellow diamonds) contrary to the dipropyl analogues. The octad (2 curves each of 8 elements, including Gd in both) or tetrad-effects (four curves, each made of 4 elements, Gd is include in the 2nd and 3rd curve) observed in solvent extraction processes are due to half-filled f shells resulting in increased electronic stability.40,41
Pu(IV) extraction was tested to study the complexing behaviour of the newly synthesized DGAs with tetravalent actinide ions. Interestingly, in all cases, Pu(IV) was extracted with high distribution ratios. Syn-Et-Me-TDDGA 5A (Fig. 5, yellow diamonds) and anti-Et-Me-TODGA 4BFig. 5, (black squares) gave the highest D values (see Fig. 5). Comparison of the different stereochemical orientation for the Et-Me or dipropyl substitutions gave different results for trivalent and tetravalent metal ions. For ethyl-methyl substitutions, the syn diastereomers showed better extraction, but interestingly, anti-dipropyl substitutions showed higher D values than their syn-analogues for Pu(IV).
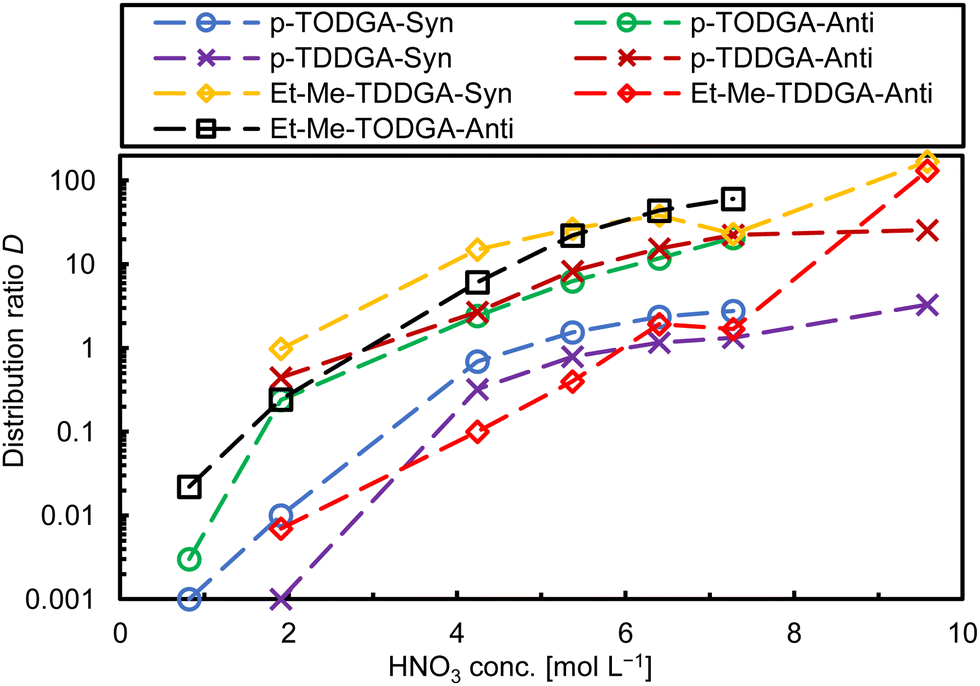 | ||
| Fig. 5 Comparison of the Pu(IV) distribution ratios for the different ligands (exp. conditions see Fig. 2). | ||
The metal:ligand ratio was studied using slope analysis, assuming a solvation mechanism.13 The slope of the log(D) vs. log(ligand concentration) plot then gives the number of ligand molecules per metal ion. For anti-p-TDDGA 10B, we chose a nitric acid concentration of 5.3 mol L−1, for syn-p-TODGA 9A of 7.3 mol L−1 and varied the ligand concentration between 0.1 to 0.5 mol L−1. Fig. 6 shows the logarithms of distribution ratios vs. ligand concentration at the fixed nitric acid concentration, including the results from the first extraction experiments at 0.1 mol L−1. According with the linear trend, the slopes suggest approximately three ligand molecules per metal ion, which agrees well with the formation of 1:3 DGA complexes in the literature.13,17,42,43
 | ||
| Fig. 6 Slope analysis for the extraction of 241Am(III), 244Cm(III) and 239Pu(IV) by anti-p-TDDGA 10B (left; aq. phase: 5.3 mol L−1 HNO3) and syn-p-TODGA 9A (right; aq. phase: 7.4 mol L−1 HNO3). Org. phase: ligand concentrations 0.1–0.5 mol L−1, Extraction time: 30 min, 22 °C. | ||
Conclusions
A series of syn- and anti-diastereomers of novel backbone alkyl-substituted tetra-n-octyl and tetra-n-decyl diglycolamides was prepared and evaluated. Ethyl-methyl substitution for both diglycolamide groups (octyl and decyl side chains) gave the highest D values among the investigated molecules. However, these values were much lower than for their non-substituted parent molecules or for the methyl and dimethyl derivatives. Similarly, the dipropyl substitution for both extractants even further reduced the extraction of trivalent metal ions. Distribution ratios increased with increasing ligand concentration (tested only for the dipropyl substitutions) and slope analysis confirmed a 1:3 metal:ligand ratio in accordance with the literature. However, the dipropyl substituted extractants give only low to moderate D values at high HNO3 concentrations, which limits their possible application. The best results in the first screening were achieved with the ethyl–methyl substituted extractants, although still high HNO3 concentrations were required. For further investigations, both diastereomers of Et-Me-TODGA need to be tested at a higher extractant concentration and reduced HNO3 concentration, including additional spectroscopic and analytical techniques. All the studied diastereomers here showed a preference for Am over Cm, which was only observed for one other diglycolamide before. Although the observed Am/Cm separation factor is quite low here, understanding the fundamental effects causing the different selectivity of diglycolamides is crucial for the development of improved extractants and their application in solvent extraction processes. On the other hand, the ligands showed positive results for Pu(IV) extraction, which opens the possibility to use these ligands for separation of tetravalent ions from Ln(III) and other MA or in group actinide extraction processes.
Experimental
Reagents and solvents
All commercially available chemicals (Sigma-Aldrich, TCI, ChemPur, VWR) used in this study were analytical reagent grade and were used without further purification. All air sensitive reactions were carried out under nitrogen atmosphere using oven-dried glassware. All dry solvents were prepared according to standard procedures and stored over 3 or 4 Å molecular sieves.Instrumentation and techniques
1H NMR (Nuclear Magnetic Resonance) and 13C NMR spectra were recorded on a Bruker AscendeTM 400 MHz NMR spectrometer (observation of 1H at 400 MHz and of 13C at 100 MHz). Partially deuterated solvents were used as internal standards to calculate the chemical shifts (d values in ppm). All 13C NMR spectra were performed with proton decoupling. The used abbreviations in these spectra are singlet (s), doublet (d), triplet (t), quadruplet (q), quintet (quint) and multiplet (m). IR spectra were recorded on an FT-IR spectrometer (Thermo Scientific Nicolet 6700) with a diamond ATR accessory (Thermo Optec Smart Orbit). Electrospray ionization (ESI) mass analyses were performed on a Waters micromass LCT mass spectrometer SQ Detector in positive mode using MeOH or CH3CN as solvents.To monitor the progress of reactions, aluminum sheets covered with silica gel 60 F-254 provided by Merck were used. All aluminum sheets were revealed under an ultraviolet lamp (λ = 254 nm) or using staining reagents.
The characterization data of the individual compounds from 1H and 13C NMR, as well as FT-IR and ESI-MS spectrometry are given in the ESI.†
Procedures
Scheme 1 shows the corresponding structures and synthesis pathway of the tetra n-octyl and n-decyl DGAs with ethyl, methyl substituents in the backbone. During the synthesis several times there was treatment with acid (20% sulfuric acid and oxalyl chloride, the latter one generating HCl). Never any racemization was observed.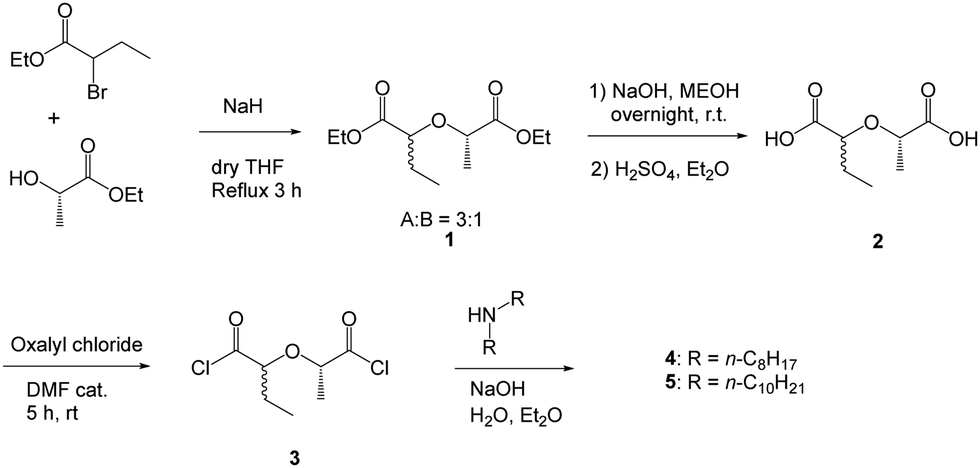 | ||
| Scheme 1 Synthesis of Et-Me-TODGA 4 and Et-Me-TDDGA 5. | ||
:1 afforded diastereomer 1A and diastereomer 1B in 53% and 17% yield, respectively.
General procedure for the in situ preparation of diacyl chlorides 3A,B and 8A,B
To a solution of a substituted dicarboxylic acid (1 mmol) dissolved in pure oxalyl chloride (2 mL), DMF (some drops) was added. The mixture was stirred under nitrogen for 4 hours, whereupon the oxalyl chloride was removed under vacuum. The product was obtained in quantitative yield and immediately dissolved in Et2O to be used for the synthesis of DGAs.:1 as eluent to give the product as a yellowish oil in 42% yield.
:1 as eluent. The product was obtained as a yellowish oil in 30% yield.
Scheme 2 shows the corresponding structures and synthesis pathway scheme of the tetra n-octyl and n-decyl DGAs with n-propyl substituents in the backbone.
 | ||
| Scheme 2 Synthesis of p-TODGA 9 and p-TDDGA 10. | ||
:1 as eluent. Diastereomer A was obtained in 59% yield and diastereomer B in 11% yield.
:2 as eluent. The product was obtained as a yellowish oil in 54% yield.
:1 as eluent. The product was obtained as a yellowish oil in 45% yield.
:2 as eluent to give 10B as a yellowish oil in 50% yield.
Solvent extraction
Nitric acid dilutions (HNO3, Merck AG, 0.82–9.78 mol L−1) were prepared using ultra-pure water (18.2 MΩ cm) which was obtained from an Elga Purelab Ultra water purification system. The radiotracers 241Am, 244Cm and 152Eu were purchased from Isotopendienst M. Blaseg GmbH, Waldburg, Germany, Oak Ridge National Laboratory, Oak Ridge, USA, and Eckert & Ziegler Nuclitec GmbH, Braunschweig, Germany, respectively. 239Pu tracer was used from a laboratory stock solution. The Pu oxidation state in the stock solution was Pu(IV) as confirmed by UV/Vis spectroscopy with ≤5% Pu(VI). The oxidation state was not checked during or after extraction, as the Pu concentration in the extraction samples was too low. Nevertheless, under the applied extraction conditions (high nitric acid concentrations and short extraction time at 22 °C) the formation of other than Pu(IV) oxidation states is highly unlikely.44–47 For the screening experiments diluted metal ion solutions (Fe, Sr, Y, Zr, Mo, Ru, Pd, La, Ce, Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, Yb, Lu) in HNO3 were used with metal ion concentrations of 1 × 10−5 mol L−1, each, to avoid loading, precipitation or 3rd phase formation. Acid concentration was measured by titration against 0.1 mol L−1 NaOH using a Titrando 907, purchased from Metrohm GmbH & Co. KG (Filderstadt, Germany). All chemicals were used without further purification. Organic solutions of 0.1 mol L−1 DGA were prepared using a weighed quantity of ligand in n-dodecane (Sigma Aldrich).Procedures and analytics
Batch solvent extraction experiments were carried out using equal volumes of 500 μL of each phase. The aqueous and organic phases were pipetted into screw-cap glass vials and contacted for a given time on an IKA VIBRAX VXR, IKA®-Werke GmbH & Co. KG (Staufen, Germany) basic automatic shaker at 2,200 rpm and 22 °C. The temperature was controlled by a F25-HE thermostat, JULABO GmbH (Seelbach, Germany). After mixing, the samples were centrifuged with a Hettich EBA 8s centrifuge, Andreas Hettich GmbH & Co. KG (Tuttlingen, Germany), for 5 min. The phases were then separated manually using a fine tipped transfer micropipette. 200 μL of each phase were transferred into new glass vials for further measurements.Gamma measurements of 241Am (60 keV) and 152Eu (122 keV) were carried out using an Eurisys EGC 35-195-R germanium coaxial N-type detector and spectra were evaluated using the GammaVision Software. Samples were measured directly without further treatment of the samples. Alpha measurements were carried out for 239Pu (5157 keV), 241Am (5486 keV) and 244Cm (5805 keV) using an Ortec/Ametek ALPHA-ENSEMBLE-8 eight chamber alpha measurement system equipped with PIPS detectors purchased from Ametek GmbH (Meerbusch, Germany). Sample preparation for alpha measurement was done by homogenizing a 10 μL alpha-spectroscopy sample in 100 μL of a mixture of Zapon varnish and acetone (1:100 v/v). This mixture was distributed over a stainless-steel plate obtained from Berthold, Bad Wildbad, Germany. The sample was dried under a heating lamp and annealed into the stainless-steel plate by a gas-flame burner. For stable elements, Inductively Coupled Plasma Mass Spectrometry (ICP-MS) was applied using a PerkinElmer NexION 2000C. Aqueous samples were measured after dilution in 1% v/v nitric acid solution without further treatment. Organic samples were measured directly in a tenside matrix (Triton-X-100) in 1% v/v HNO3 after dilution.
Distribution ratios (D) were calculated as the ratio of activity or metal ion (M) concentration in the organic phase vs. the activity or metal ion concentration in the aqueous phase ([M]org/[M]aq). The separation factor (SF) between two metal ions was calculated as the ratio of the corresponding distribution ratios (SFM1/M2 = DM1/DM2). Distribution ratios between 0.01 and 100 exhibit an uncertainty of ±5%, while lower/higher values exhibit larger uncertainties, yet they represent the complexing trend of the ligands. Mass balances were calculated as the sum of aqueous and organic concentrations divided by the initial concentration.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding for this research was provided by the German Federal Ministry for the Environment, Nature Conservation, Nuclear Safety and Consumer Protection (BMUV), project SEPAM (02E11921A).References
- A. Wilden, G. Modolo, P. Kaufholz, F. Sadowski, S. Lange, M. Sypula, D. Magnusson, U. Müllich, A. Geist and D. Bosbach, Solvent Extr. Ion Exch., 2015, 33, 91–108 CrossRef CAS.
- C. Wagner, U. Müllich, A. Geist and P. J. Panak, Solvent Extr. Ion Exch., 2016, 34, 103–113 CrossRef CAS.
- M. Miguirditchian, L. Chareyre, X. Heres, C. Hill, P. Baron and M. Masson, Global, 2007, Advanced Nuclear Fuel Cycles and Systems, Boise, ID, United States, 2007 Search PubMed.
- R. Malmbeck, M. Carrott, B. Christiansen, A. Geist, X. Hérès, D. Magnusson, G. Modolo, C. Sorel, R. Taylor and A. Wilden, Sustainable Nuclear Energy Conference, Manchester, UK, 2014 Search PubMed.
- M. Carrott, K. Bell, J. Brown, A. Geist, C. Gregson, X. Hères, C. Maher, R. Malmbeck, C. Mason, G. Modolo, U. Müllich, M. Sarsfield, A. Wilden and R. Taylor, Solvent Extr. Ion Exch., 2014, 32, 447–467 CrossRef CAS.
- A. Geist, J.-M. Adnet, S. Bourg, C. Ekberg, H. Galán, P. Guilbaud, M. Miguirditchian, G. Modolo, C. Rhodes and R. Taylor, Sep. Sci. Technol., 2021, 56, 1866–1881 CrossRef CAS.
- T. L. Authen, J.-M. Adnet, S. Bourg, M. Carrott, C. Ekberg, H. Galán, A. Geist, P. Guilbaud, M. Miguirditchian, G. Modolo, C. Rhodes, A. Wilden and R. Taylor, Sep. Sci. Technol., 2022, 57, 1724–1744 CrossRef.
- E. M. González-Romero, Nucl. Eng. Des., 2011, 241, 3436–3444 CrossRef.
- T. Kooyman, Ann. Nucl. Energy, 2021, 157, 7 CrossRef.
- OECD-NEA, Strategies and Considerations for the Back End of the Fuel Cycle, OECD Nuclear Energy Agency (NEA), Boulogne-Billancourt, France, 2021 Search PubMed.
- Ion Exchange and Solvent Extraction: Changing the Landscape in Solvent Extraction , ed. B. A. Moyer, CRC Press, Taylor & Francis Group, Boca Raton, Fl, USA, 2020 Search PubMed.
- OECD-NEA, Potential Benefits and Impacts of Advanced Nuclear Fuel Cycles with Actinide Partitioning and Transmutation, Report NEA No. 6894, OECD, Nuclear Energy Agency (NEA), 2011 Search PubMed.
- Y. Sasaki, Y. Sugo, S. Suzuki and S. Tachimori, Solvent Extr. Ion Exch., 2001, 19, 91–103 CrossRef CAS.
- P. J. Panak and A. Geist, Chem. Rev., 2013, 113, 1199–1236 CrossRef CAS PubMed.
- P. Matveev, P. K. Mohapatra, S. N. Kalmykov and V. Petrov, Solvent Extr. Ion Exch., 2021, 39, 679–713 CrossRef CAS.
- M. Iqbal, J. Huskens, W. Verboom, M. Sypula and G. Modolo, Supramol. Chem., 2010, 22, 827–837 CrossRef CAS.
- S. A. Ansari, P. Pathak, P. K. Mohapatra and V. K. Manchanda, Chem. Rev., 2012, 112, 1751–1772 CrossRef CAS PubMed.
- Y. Y. Liu, C. Zhao, Z. B. Liu, S. Liu, Y. Zhou, C. S. Jiao, M. Zhang, Y. Gao, H. He and S. W. Zhang, RSC Adv., 2022, 12, 790–797 RSC.
- S. A. Ansari, P. N. Pathak, V. K. Manchanda, M. Husain, A. K. Prasad and V. S. Parmar, Solvent Extr. Ion Exch., 2005, 23, 463–479 CrossRef CAS.
- J. Brown, F. McLachlan, M. J. Sarsfield, R. J. Taylor, G. Modolo and A. Wilden, Solvent Extr. Ion Exch., 2012, 30, 127–141 CrossRef CAS.
- H. Galán, C. A. Zarzana, A. Wilden, A. Núñez, H. Schmidt, R. J. M. Egberink, A. Leoncini, J. Cobos, W. Verboom, G. Modolo, G. S. Groenewold and B. J. Mincher, Dalton Trans., 2015, 44, 18049–18056 RSC.
- V. Hubscher-Bruder, V. Mogilireddy, S. Michel, A. Leoncini, J. Huskens, W. Verboom, H. Galán, A. Núñez, J. Cobos Sabate, G. Modolo, A. Wilden, H. Schmidt, M.-C. Charbonnel, P. Guilbaud and N. Boubals, New J. Chem., 2017, 41, 13700–13711 RSC.
- A. Wilden, P. M. Kowalski, L. Klaß, B. Kraus, F. Kreft, G. Modolo, Y. Li, J. Rothe, K. Dardenne, A. Geist, A. Leoncini, J. Huskens and W. Verboom, Chem. – Eur. J., 2019, 25, 5507–5513 CrossRef CAS PubMed.
- B. Verlinden, K. Van Hecke, A. Wilden, M. Hupert, B. Santiago-Schübel, R. J. M. Egberink, W. Verboom, P. M. Kowalski, G. Modolo, M. Verwerft, K. Binnemans and T. Cardinaels, RSC Adv., 2022, 12, 12416–12426 RSC.
- B. Verlinden, A. Wilden, K. Van Hecke, R. J. M. Egberink, J. Huskens, W. Verboom, M. Hupert, P. Wessling, A. Geist, P. J. Panak, R. Hermans, M. Verwerft, G. Modolo, K. Binnemans and T. Cardinaels, Solvent Extr. Ion Exch., 2023, 41, 59–87 CrossRef CAS.
- A. H. Bond, R. Chiarizia, V. J. Huber, M. L. Dietz, A. W. Herlinger and B. P. Hay, Anal. Chem., 1999, 71, 2757–2765 CrossRef CAS.
- A. M. Costero, J. P. Villarroya, S. Gil, M. J. Aurell and M. C. R. de Arellano, Tetrahedron, 2002, 58, 6729–6734 CrossRef CAS.
- Y. Lin, J. Espinas, S. Pellet-Rostaing, A. Favre-Reguillon and M. Lemaire, New J. Chem., 2010, 34, 388–390 RSC.
- F. L. Guerra Gomez, T. Uehara, T. Rokugawa, Y. Higaki, H. Suzuki, H. Hanaoka, H. Akizawa and Y. Arano, Bioconjugate Chem., 2012, 23, 2229–2238 CrossRef PubMed.
- N. Felines, G. Arrachart, F. Giusti, A. Beillard, C. Marie and S. Pellet-Rostaing, New J. Chem., 2021, 45, 12798–12801 RSC.
- P. Wessling, M. Trumm, T. Sittel, A. Geist and P. J. Panak, Radiochim. Acta, 2022, 110, 291–300 CrossRef CAS.
- A. Leoncini, J. Huskens and W. Verboom, Synlett, 2016, 2463–2466 CAS.
- T. Yaita, A. W. Herlinger, P. Thiyagarajan and M. P. Jensen, Solvent Extr. Ion Exch., 2004, 22, 553–571 CrossRef CAS.
- D. Massey, A. Masters, J. Macdonald-Taylor, D. Woodhead and R. Taylor, J. Phys. Chem. B, 2022, 126, 6290–6300 CrossRef CAS PubMed.
- A. Geist, U. Müllich, D. Magnusson, P. Kaden, G. Modolo, A. Wilden and T. Zevaco, Solvent Extr. Ion Exch., 2012, 30, 433–444 CrossRef CAS.
- J. Ravi, K. A. Venkatesan, M. P. Antony, T. G. Srinivasan and P. R. Vasudeva Rao, Radiochim. Acta, 2013, 101, 301 CrossRef CAS.
- P. Narayanan, K. R. Swami, T. Prathibha and K. A. Venkatesan, ChemistrySelect, 2022, 7, 11 CrossRef.
- P. D'Angelo, A. Zitolo, V. Migliorati, G. Chillemi, M. Duvail, P. Vitorge, S. Abadie and R. Spezia, Inorg. Chem., 2011, 50, 4572–4579 CrossRef PubMed.
- P. D'Angelo, F. Martelli, R. Spezia, A. Filipponi and M. A. Denecke, Inorg. Chem., 2013, 52, 10318–10324 CrossRef PubMed.
- D. F. Peppard, G. W. Mason and S. Lewey, J. Inorg. Nucl. Chem., 1969, 31, 2271–2272 CrossRef CAS.
- T. Monecke, U. Kempe, J. Monecke, M. Sala and D. Wolf, Geochim. Cosmochim. Acta, 2002, 66, 1185–1196 CrossRef CAS.
- Y. Sasaki, P. Rapold, M. Arisaka, M. Hirata, T. Kimura, C. Hill and G. Cote, Solvent Extr. Ion Exch., 2007, 25, 187–204 CrossRef CAS.
- A. Wilden, G. Modolo, S. Lange, F. Sadowski, B. B. Beele, A. Skerencak-Frech, P. J. Panak, M. Iqbal, W. Verboom, A. Geist and D. Bosbach, Solvent Extr. Ion Exch., 2014, 32, 119–137 CrossRef CAS.
- Y. Ban and Y. Morita, Radiochim. Acta, 2012, 100, 879–883 CrossRef CAS.
- M. H. Lee, Y. J. Park and W. H. Kim, J. Radioanal. Nucl. Chem., 2007, 273, 375–382 CrossRef CAS.
- The Chemistry of the Actinide and Transactinide Elements , ed. L. R. Morss, N. M. Edelstein and J.Fuger, Springer, Dordrecht, The Netherlands, 4th edn, 2011 Search PubMed.
- J. M. Cleveland, The Chemistry of plutonium, American Nuclear Society, La Grange Park, IL, Reprint of the 1970 ed. published by Gordon and Breach, New York; with new pref. and foreword edn., 1979 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nj05663a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2023 |