
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFrom CO2 to CS2: a theoretical investigation of the cycloaddition to aziridines mediated by metal-free porphyrin-based catalytic systems†
Caterina
Damiano
 *a,
Nicola
Panza
a,
Jakub
Nagy
b,
Emma
Gallo
a and
Gabriele
Manca
*c
*a,
Nicola
Panza
a,
Jakub
Nagy
b,
Emma
Gallo
a and
Gabriele
Manca
*c
aDepartment of Chemistry, University of Milan, Via Golgi 19, I-20133, Milan, Italy. E-mail: caterina.damiano@unimi.it
bFaculty of Science in Department of Chemistry, Masaryk University, Czech Republic
cIstituto di Chimica dei Composti OrganoMetallici, CNR-ICCOM, Via Madonna del Piano 10, I-50019, Sesto Fiorentino, Italy. E-mail: gabriele.manca@iccom.cnr.it
First published on 19th January 2023
Abstract
A detailed computational analysis of the synthesis of thiazolidine-2-thiones by CS2 insertion into three membered aziridine rings is here reported. DFT studies highlighted the feasibility of the atom-efficient process by employing either the metal-free binary TPPH2/TBACl system or bifunctional TPPH4Cl2 molecules as the catalytic species. Detailed investigations into the mechanism promoted by the TPPH2/TBACl system indicated, as in the analogous cycloaddition of CO2, the pivotal role of the adduct between TPPH2 and TBACl salts in the synthesis of both N-aryl and N-alkyl thiazolidine-2-thiones. The latter species can be also obtained by using the eco-compatible TPPH4Cl2 catalyst, which is able to simultaneously activate CS2 and aziridine molecules. The comparison of the overall energy gains of the reaction between aziridines and CS2 with respect to the same process involving CO2 revealed that the formation of thiazolidine-2-thiones is more favoured than the synthesis of oxazolidin-2-ones paving the way to develop efficient and eco-compatible synthesis of thiazolidine-2-thiones.
Introduction
The synthesis of medium size N-heterocycles is one of the most dynamic and constantly evolving areas of chemistry in view of the widespread occurrence of N-heterocyclic fragments in various biologically and pharmaceutically active molecules. Among all the naturally and synthetically derived N-heterocycles, increasing interest has been devoted to the sulphur containing ones, which have proved to be very promising bioactive spiecies1 and chiral auxiliaries in organic synthesis.2 In particular, the five-membered thiazolidine-2-thione scaffold has aroused great attention due to its activity as a regulator of cancer cells apoptosis,3 inhibitor of the aldose reductase and xanthine oxidase4 and anti-inflammatory and antifungal agents.5Considering the potential applicability of thiazolidine-2-thiones, many efforts have been devoted to developing synthetic methodologies that are more efficient and sustainable than the conventional ones. Traditionally, thiazolidine-2-thiones, which are the sulphur analogous of oxazolidin-2-ones, can be synthesised by reacting amino alcohols with high concentration of carbon disulphide (CS2) under harsh reaction conditions (high temperatures, strong alkaline solutions, and extended reaction times).6 In order to overcome these experimental drawbacks and increase the process sustainability, microwave-assisted syntheses7 and various multicomponent reactions have been developed. The latter include: (i) the iodocyclization of allyl amines, carbon disulphide and iodine;8 (ii) the three-component coupling of amines, alkynes and aldehydes followed by the cyclization with CS2;9 (iii) the three-component one-pot reaction of amines, CS2 and either α-bromoketones10 or α-chloro-β,γ-alkenoate esters11 and (iv) the phosphine-catalysed one-pot reaction between dithiocarbamates (prepared in situ from amines and CS2) and arylpropiolates.12 However, one of the most attractive procedures in terms of atom-economy is the CS2 cycloaddition to aziridine rings. It is important to note that this approach allows the combination of a 100% atom-efficient process with an alternative disposal of carbon disulphide. In fact, CS2 plays a central role in many industrial sectors, such as chemical fiber plants (e.g. viscose production) or petroleum processing, but it is particularly harmful13 and its common disposal requires an incineration with the consequent emission in the atmosphere of pollutant gases (e.g. CO2, SO2 or H2S) with many environmental negative effects.14
Even if the CS2 cycloaddition to aziridines offers several advantages and shows analogous characteristics to the synthesis of oxazolidin-2-ones from aziridines and CO2, it has been much less studied both from the experimental and theoretical point of view. To date, only a few examples of organo- and metal-based catalytic systems capable of promoting this reaction have been reported in the literature and they usually require high catalytic loadings of alkali metal halides,15 tributylphosphine,16 pyrrolidine,17 pyridinecarboxaldehyde oxime,18 amidato divalent lanthanide complexes19 or MOFs based on the trivalent dysprosium center.20
Considering the analogy between CO2 and CS2, it is reasonable to think that catalytic systems, optimised for the synthesis of oxazolidin-2-ones, could be equally efficient in the synthesis of thiazolidine-2-thiones from aziridines and CS2.
In the recent years we have investigated the catalytic activity of porphyrin-based systems in the CO2 cycloaddition to aziridines21–24 and the symbiotic computational/experimental approach has highlighted the high efficiency of the metal-free TPPH2/TBACl binary catalyst (TPP = dianion of tetraphenyl porphyrin, TBACl = tetrabutyl ammonium chloride) in promoting the synthesis of N-alkyl and N-aryl oxazolidine-2-ones under both homogenous and heterogeneous conditions.25 DFT calculations pointed out the formation of the active adduct 1 (Scheme 1) where the butyl substituents of the TBA+ cation are perfectly hosted in the crib formed by the peripheral meso substituents of the porphyrin macrocycle. Adduct 1 has been estimated to be more stable by ca. −7.5 kcal mol−1 in free energy than the separated components due to the cooperative effects of the dispersion and weak forces between the alkyl chains of TBA+ and the aromatic framework of TPPH2. Adduct 1 is the real catalyst of the process by weakening the ionic interactions between TBA+ and the chloride anion, which makes the latter more prone to attack the aziridine ring with the following formation of intermediate 3.
 | ||
| Scheme 1 Catalytic activity of the TPPH2/TBACl adduct (1) and the bifunctional TPPH4Cl2 catalyst in the reaction of aziridines with either CO2 or CS2. | ||
At this point, the former aziridine nitrogen atom has acquired a sufficient basic character to activate carbon dioxide with the consequent synthesis of N-alkyl and N-aryl oxazolidin-2-ones.
The nature of the R substituents (alkyl or aryl) does not affect the reaction mechanism and only small variations in the height of the barriers or in the energy stabilization of the encountered intermediates were observed.
It is worth noting that additional experimental studies revealed the possibility of replacing the binary TPPH2/TBACl system with the more convenient bifunctional TPPH4Cl2 catalyst to promote CO2 cycloaddition (Scheme 1).26 The very cheap and eco-compatible latter catalyst, simply obtained by treating TPPH2 with HCl, efficiently mediated the synthesis of N-alkyl oxazolidin-2-ones in the absence of any Lewis acid or additive. DFT studies of the reaction mechanism revealed that the TPPH4Cl2 catalyst allows the synthesis of N-alkyl oxazolidin-2-ones through the simultaneous activation of aziridine and CO2 molecules by forming an adduct between all the three species involved in the reaction (TPPH4Cl2, aziridine and CO2). This synergic activation can occur thanks to the co-presence of the porphyrin skeleton and the nucleophilic chloride anion, which are responsible for the reaction regioselectivity and the ring-opening of the aziridine moiety, respectively.
The intriguing results that were obtained on CO2 cycloadditions pushed us to investigate, from the computational point of view, the feasibility of the CS2 cycloaddition to N-alkyl and N-aryl aziridines in the presence of either the binary TPPH2/TBACl system or the bifunctional TPPH4Cl2 catalyst (Scheme 1) in order to pave the way for future experimental studies.
According to the reported studies of the CO2 cycloaddition to aziridines, the catalytic activity of the binary TPPH2/TBACl system was theoretically evaluated in the synthesis of both N-alkyl and N-aryl aziridines while the catalytic performance of the bifunctional TPPH4Cl2 catalyst was assessed in the sole CS2 cycloaddition to N-alkyl aziridines.
Results and discussion
Binary TPPH2/TBACl system for the CS2 cycloaddition to aziridines
The precedent analysis of cycloaddition reactions involving CO2 revealed that no productive reactions occurred between CO2 and aziridine or TPPH2 and that the first part of the mechanism is the formation of intermediate 3 (with R = alkyl or aryl substituent) that is responsible for the CO2 activation. Thus, in order to confirm that this intermediate is the key-species also in the reaction between CS2 and aziridines, preliminary studies were carried out on the reactivity of CS2 towards either aziridine or TPPH2.The quasi null interaction between aziridine and CS2, confirmed by the Naz–C(CS2) distance longer than 3.2 Å, excluded any possible reaction between these two species in the early stage of the process. The formation of a possible adduct between porphyrin and the incoming CS2 (Fig. 1) was also discharged for both structural (too long distance, 3.6 Å, between one N–H moiety and the carbon atom of CS2) and disfavouring energy features (free energy cost of +3.4 kcal mol−1). As shown in Fig. 1, CS2 maintains the starting linear arrangement and any kind of perturbations are highlighted.
 | ||
| Fig. 1 Optimised structure of the hypothetical adduct between TPPH2 and CS2. | ||
Once the possibility of a preliminary activation of CS2 through an interaction with either aziridine or TPPH2 macrocycle was discarded, we assumed that, as occurred in the case of CO2, the catalytic cycle should evolve through the formation of the adduct 1 that interacts with the aziridine cycle forming the active species 3. As pointed out in the previous articles on CO2 activation, the encountered free energy barriers are high up to 40 kcal mol−1, in agreement with the requested experimental conditions (T > 80 °C). Two different typologies of N-substituted-2-phenylaziridines were modelled, which show on the nitrogen atom either the nbutyl or the 3,5(CF3)2C6H3 aromatic substituent. The electrophilicity of the two aziridines was estimated by applying the procedure developed by Domingo et al.27–30 In particular, the electrophilicity of N-aryl aziridine was estimated to be particularly greater than that of N-butyl one, with the electrophilicity index ω value of 2.28 vs. 1.3 eV that was calculated for N-butyl aziridine. This trend is in accordance with the higher free energy barrier (by ca. 10 kcal mol−1) associated with the nucleophilic attack of chloride to N-butyl aziridine compared to that of Cl− to the aryl one. In both cases, the local electrophilicity index, ω+K, of the two aziridinic carbon atoms was estimated and it was found that the C1 electrophilicity was double than that of C2. In particular, while the ω+C1 and ω+C2 values of the N-aryl aziridine are 0.53 and 0.22, these values of N-butyl one are 0.31 and 0.14, respectively. These theoretical results are in agreement with the observed stereoselectivity as well as with the computed lower free energy barrier (ca. 8.5 kcal mol−1) that was obtained for the chloride attack to C1 rather than to C2 atom.
Considering that the formation of intermediate 3 is not affected by the nature of the other involved reagent (CO2 or CS2) the theoretical investigation of the formation of 3Bu (TPPH2/TBACl + N-butyl-2-phenylaziridine) and 3Ar (TPPH2/TBACl + N-aryl-2-phenylaziridine) has not been reported again but only indicated in the final free energy pathways of Fig. 4 and 6. All the calculations were performed by considering the tetrahydrofuran (THF) as the reaction solvent, according to the experiments already performed for the CO2 activation process.
The direct interaction of the out-pointing lobe of Naz with the carbon atom of CS2, named C3, is responsible for the desymmetrisation of carbon disulphide and the consequent formation of intermediate 4 (Fig. 2) with an energy gain of −17.1 kcal mol−1.
 | ||
| Fig. 2 Optimised structure of adduct 4 and the Transition State 4TS. Hydrogen atoms were omitted for clarity. | ||
It is important to note that the free energy gain estimated for the formation of 4 is comparable to that obtained for the CO2 activation (estimated to be exergonic by −20.3 kcal mol−1) and it suggests the suitability of the CS2 cycloaddition to N-butyl-2-phenylaziridine. As reported in Fig. 3, intermediate 4 presents an already formed Naz–C3 bond of 1.39 Å thanks to which the non-linear CS2 moiety reaches a new SCS angle of 121°. The lack of linearity causes the appearance of an IR band at 552 cm−1 associated with the in phase C–S stretching, which is 100 cm−1ca. red shifted with respect to the value computationally predicted for the free CS2.
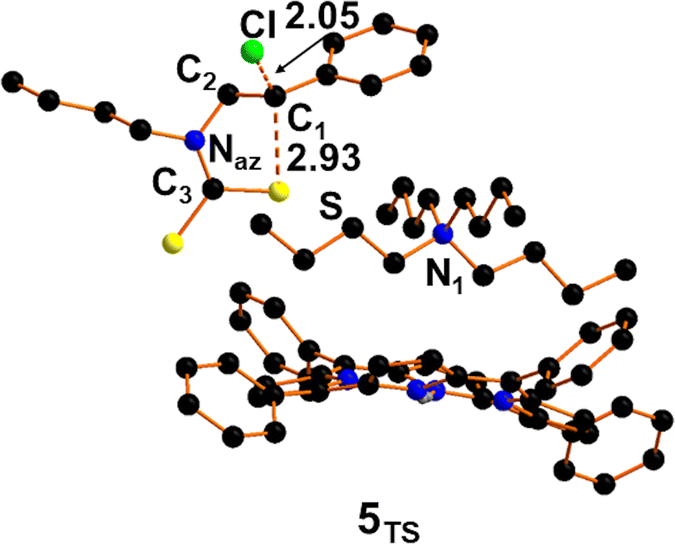 | ||
| Fig. 3 Optimised structure of transition state 5TS. | ||
A Transition State, namely 4TS shown in Fig. 2 with a free energy barrier of +8.4 kcal mol−1 was isolated for the approaching of the CS2 moiety to 3Bu species. The structure features the formation of the Naz–C3 bonding with a distance of 2.26 Å together with a corresponding 31° bent of the firstly linear CS2. After the Transition State, the complete formation of the Naz–C3 bonding in 4 occurred with a large free energy gain of −25.5 kcal mol−1.
Due to the loss of symmetry, the original neutral CS2 moiety acquires a net charge of ca. −1 e− on the sulphur atoms, which are enough electron-rich to perform a nucleophilic attack at the C1 atom of the opened aziridine of 3Bu. This process occurs by-passing a very small free energy barrier of +3.7 kcal mol−1 that allows achieving the Transition State 5TS (Fig. 3).
Even if the sulphur is particularly distant from C1 (the S–C1 distance is 2.93 Å) in the transition state 5TS, it can exert its nucleophilic action toward the C1 centre in a SN2 fashion forming an S–C1–Cl angle of 156°. The approach of the sulphur atom to the C1 centre allows a weakening of the corresponding C1–Cl distance by ca. 0.2 Å, which elongates up to 2.05 Å. The Transition State nature of 5TS was confirmed by the detection of a single imaginary frequency at −60.3 cm−1 associated with the approach of the sulphur atom to C1 and the cleavage of the C1–Cl bond. It is noteworthy that the resulting energy barrier required for the formation of 5TS is quasi three times smaller than that calculated in the same process involving CO2 (3.7 vs. 9.4 kcal mol−1). After the accomplishment of 5TS, the system evolves forming the final thiazolidine-2-thione 5 with a free energy gain of −39.2 kcal mol−1 and releasing the chloride anion and the adduct 1, which can restart the catalytic cycle.
The overall free energy gain for the cycloaddition reaction of CS2 to N-butyl-2-phenylaziridine forming 5 is of −14.5 kcal mol−1. A complete schematic representation of the overall mechanism with all the main intermediates and Transition States that are involved in the CS2 cycloaddition to N-butyl-2-phenylaziridine is shown in Fig. 4.
 | ||
| Fig. 4 Free energy profile (kcal mol−1) associated with the synthesis of 5 catalysed by the TPPH2/TBACl system. | ||
Analogously to what was discussed above for the reaction involving N-butyl-2-phenylaziridine, the starting point of the computational analysis is the analysis of the structure of intermediate 3Ar.
As a general consideration for all the steps precedent to the formation of 3Ar, the encountered free energy barrier associated with the achievement of 3ArTS (the rate determining step of the reaction) is lower than that found for the formation of 3BuTS due to the presence of CF3 substituents, which render the aziridine ring very electron-poor, as also confirmed by the calculated electrophilicity index. The EWGs placed onto the aromatic substituent of the aziridine nitrogen atom (called Naz) are also responsible for a limited availability of its electron pair in the activation of inert substrates such as CO2 and CS2. The nucleophilicity index for the species 3Ar was evaluated, revealing a nucleophilicity Nu index of 1.2 eV and a local nucleophilic character for the nitrogen NuNaz of 0.43 eV smaller with respect to that obtained by modelling N-butyl species. This assumption is confirmed by the higher encountered free energy barrier for the very limited free energy gain associated with the formation of the corresponding 3Ar/CO2 (1.7 kcal mol−1) and 3Ar/CS2 (−1.0 kcal mol−1) adducts.
The optimised structure of the adduct 6 between 3Ar and CS2 (Fig. 5) shows a bent alignment of the CS2 moiety and a new Naz–C3 bond of 1.46 Å, which results in an increase in the length by 0.07 Å with respect to that calculated by modelling N-butyl-2-phenylaziridine, suggesting again the weaker donor power of the lone pair placed on the Naz centre.
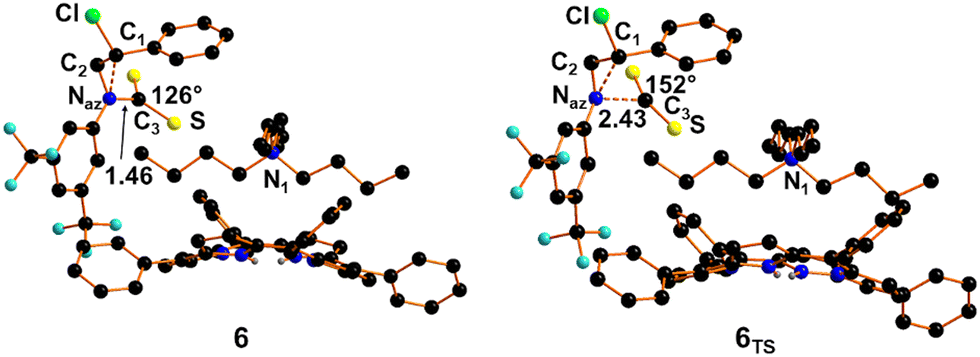 | ||
| Fig. 5 Optimised structure of the adduct 6 and the transition state 6TS. | ||
As occurred for the reaction involving N-butyl-2-phenylaziridine, also in this case an IR peak related to the symmetric stretching of the CS2 appears at 582 cm−1, 30 cm−1 blue shifted with respect to that observed for adduct 4, to confirm the less donor power of Naz when linked to aryl CF3-substituted groups. As stated for 4, also for the formation of the intermediate 6 we isolated a Transition State, namely 6TS, (Fig. 5) with a free energy barrier of +12.4 kcal mol−1. After that, the system gains −13.4 kcal mol−1 for the complete formation of the C3–Naz bonding in 6. In 6TS, the CS2 approaching Naz becomes bent by ca. 28° already at a long distance of 2.43 Å. The Transition State nature was confirmed by the detachment of a single imaginary frequency at −181.7 cm−1.
In 6 the electron donation from the nitrogen centre confers a sufficient nucleophilicity to sulphur atoms for attacking C1 in a SN2 mechanism. The so-formed Transition State 7TS (Fig. 6) shows a quasi-linear S–C1–Cl bond with an angle of 166°. The free energy barrier required for the achievement of 7TS is estimated to be +7.8 kcal mol−1 and it is associated with an imaginary IR frequency at −40 cm−1. At this point, N-aryl-thiazolidine-2-thione 7 is formed and the adduct 1 regenerated with a free energy gain of −30.9 kcal mol−1.
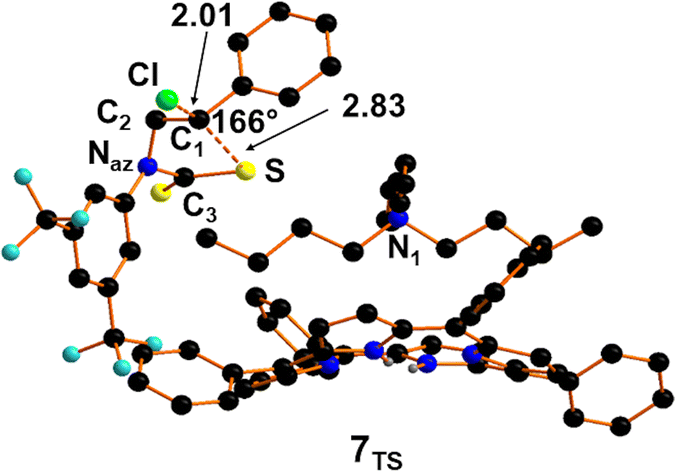 | ||
| Fig. 6 Optimised structure of transition state 7TS. | ||
A detailed free energy pathway for the synthesis of N-aryl-thiazolidine-2-thione 7 catalysed by the TPPH2/TBACl system is depicted in Fig. 7. The estimated free energy associated with the formation of 7, starting from CS2 and the model N-aryl-2-phenylaziridine, is −7.8 kcal mol−1.
 | ||
| Fig. 7 Free energy profile (kcal mol−1) associated with the synthesis of 7 catalysed by adduct 1. | ||
Bifunctional TPPH4Cl2 catalyst for the CS2 cycloaddition to N-alkyl aziridines
In view of the already reported DFT studies describing the CO2 activation by the bifunctional TPPH4Cl2 catalysts,26 the CS2 cycloaddition to N-alkyl aziridines in the presence of the same catalyst was theoretically investigated.The chlorohydrate TPPH4Cl2 porphyrin, whose formation by the protonation of TPPH2 in THF has been estimated to be exergonic of −31.5 kcal mol−1, can interact with N-butyl-2-phenylaziridine and CS2 forming adduct 8 (Fig. 8) with a free energy cost of +29.6 kcal mol−1. This high energy barrier is mainly due to entropic factors since the enthalpy cost is only +4.3 kcal mol−1.
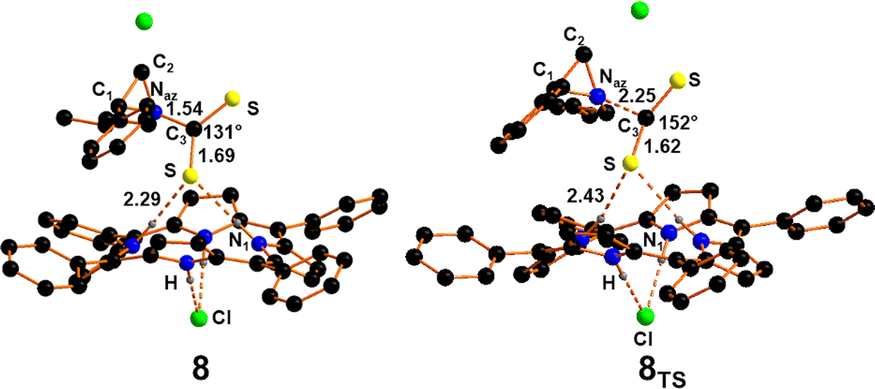 | ||
| Fig. 8 Optimised structure of adduct 8 and the Transition State 8TS. Hydrogen atoms were omitted for clarity. | ||
As shown in Fig. 8, the structure of adduct 8 shows a non-linear CS2 molecule with a S–C–S angle of 131° and a new Naz–C3 bond of 1.54 Å is formed. The transition state 8TS for the obtainment of intermediate 8 was also isolated with a free energy barrier of 33.7 kcal mol−1 after which the system gains −4.3 kcal mol−1. Even if the energy barrier was particularly high, the formation of 8 could reasonably occur by employing the same experimental conditions (temperatures higher than 80 °C) required for CO2 activation.21–26 The high temperature reasonably allows overcoming the high estimated energy barrier.
As shown in Fig. 8, in the Transition State 8TS the CS2 moiety is approaching the aziridine nitrogen at a N2⋯C3 distance of 2.25 Å. The CS2 in 8TS appears already bent by ca. 30° compared to the starting linear arrangement. The Transition State nature of 8TS has been confirmed by the detection of a unique imaginary frequency at −144 cm−1 associated with the CS2 approaching as well as the bending of the triatomics. The interaction between the Naz centre and the CS2 moiety is also responsible for the electronic depletion of the aziridine ring that makes more favourable the nucleophilic attack of the chloride anion on the more substituted carbon (C1) of the three-membered ring. The less rich electron feature of the aziridine ring after the interaction with CS2 is confirmed by the detection of the transition state 9TS (Fig. 9) in which the chloride anion can approach the C1 centre overcoming the low free energy barrier of 5.9 kcal mol−1. Comparing the structure of 9TS with a similar Transition State observed in the process involving CO2, the chloride anion seems to be a less efficient nucleophile due to the presence of a Cl–C1 distance shorter by 0.2 Å with respect to that detected in the case of CO2 (1.88 Å). These data suggest a lower electron transfer from the aziridine Naz to the CS2 moiety, as also confirmed by the higher free energy barrier required for the formation of 9TS (5.9 vs. 1.5 kcal mol−1 for CS2 and CO2 cases, respectively). After the obtainment of the Transition State 9TS, the system evolves to the intermediate 9 (Fig. 9) with a free energy gain of −25.1 kcal mol−1. The process is associated with the formation of a new Cl–C1 bond and the complete cleavage of the Naz–C1 linkage. At this point the sulphur atoms of intermediate 9 are nucleophilic enough to attack the C1 atom allowing the ring-closing reaction required for the formation of product 5.
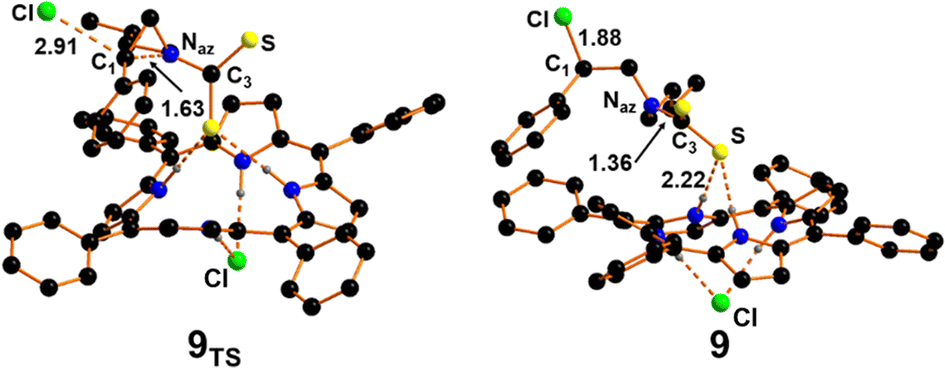 | ||
| Fig. 9 Optimised structures of transition state 9TS and intermediate 9. Hydrogen atoms are omitted for clarity. | ||
The ring-closing reaction and the release of the chloride anion occur through the formation of transition state 10TS (Fig. 10) reached by-passing a low free energy barrier of 2.8 kcal mol−1.
 | ||
| Fig. 10 Optimised structure of transition state 10TS. | ||
In the transition state 10TS the C1 centre achieves a bipyramidal trigonal arrangement with the chloride and one sulphur atom lying quasi co-linear (the S–C1–Cl angle is 162°) at the two apical positions.
The free energy gain from 10TS to achieve the N-butyl-thiazolidine-2-thione 5, together with the restoration of TPPH4Cl2, is of −28.9 kcal mol−1. A detailed free energy reaction pathway for the CS2 cycloaddition to N-butyl-2-phenylaziridine catalysed by the bifunctional TPPH4Cl2 molecule is summarised in Fig. 10.
It is interesting to note that despite the use of different catalytic systems (the binary TPPH2/TBACl or the bifunctional TPPH4Cl2 catalyst), the overall free energy gain to provide thiazolidine-2-thione 5 is comparable. In fact, the energy gain to achieve 5 is −14.7 kcal mol−1 when TPPH4Cl2 is employed as the catalyst (Fig. 11) and −14.5 kcal mol−1 if the binary TPPH2/TBACl system is used (Fig. 4). The energetic similarity between the two catalytic processes is also in agreement with the values previously estimated for the synthesis of N-alkyl oxazolidin-2-ones in which the resulting energy gain for the CO2 cycloaddition to the N-butyl-2-phenylaziridine is equal to 4.7 kcal mol−1 using either the TPPH4Cl226 or the TPPH2/TBACl system.22
 | ||
| Fig. 11 Free energy profile (in kcal mol−1) associated with the synthesis of 5, catalysed by the bifunctional TPPH4Cl2. | ||
Experimental
Computational details
All the compounds along the reaction pathway were isolated at the B97D-DFT level of theory34 within Gaussian 16 package.35 All the obtained structures were validated as minima or transition states by the vibrational frequencies calculations. The experimental solvent, tetrahydrofuran, has been considered within the CPCM model.36 The 6-31G basis set, with the addition of the polarization functions (d,p), was adopted. The coordinates of all the optimised structures as well as their main energetic features are reported in the ESI.†Conclusions
The present manuscript describes a detailed theoretical investigation on the feasibility of the CS2 cycloaddition to N-alkyl and N-aryl aziridines promoted by metal-free porphyrin-based systems. The performed DFT studies highlighted that either the binary TPPH2/TBACl system or the bifunctional TPPH4Cl2 catalyst could be competent for the synthesis of thiazolidine-2-thiones. The exhaustive study of the possible involved intermediates and Transition States has shown that thiazolidine-2-thiones can be synthesised through reaction mechanisms quite similar to those already suggested in the case of CO2 cycloaddition to aziridines. The TPPH2/TBACl system can promote the synthesis of both N-alkyl and N-aryl thiazolidine-2-thiones with an overall energy gain of −14.5 kcal mol−1 and −7.8 kcal mol−1, respectively. The acquired data are in accordance with the lower reactivity of N-aryl aziridines with respect to the N-alkyl ones.In view of the possibility of replacing the TPPH2/TBACl binary system with a cheaper system, eco-compatible bifunctional TPPH4Cl2 species, the CS2 cycloaddition to N-alkyl aziridines in the presence of this catalyst was also theoretically investigated. The obtained data revealed the formation of an active intermediate in which the protonated porphyrin can simultaneously activate CS2 and N-alkyl aziridine. The overall energy gain of the process was estimated to be 14.7 kcal mol−1. This value is comparable to that calculated for the reaction catalysed by the TPPH2/TBACl binary system to confirm the possibility of employing either the binary porphyrin-based system or the bifunctional porphyrin-based catalyst for the synthesis of N-alkyl thiazolidine-2-thiones.
In all the investigated cases, the CS2 cycloaddition to the aziridine rings yielded better results than the same process involving carbon dioxide suggesting the synthetic applicability of the procedure. Future efforts need to be focused on testing the TPPH2/TBACl system and the TPPH4Cl2 catalyst as promoters for the synthesis of thiazolidine-2-thiones.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is part of the project “One Health Action Hub: University Task Force for the resilience of territorial ecosystems”, Supported by Università degli Studi di Milano – PSR 2021 – GSA – Linea 6. We thank the University of Milan (PSR 2020 – financed project “Catalytic strategies for the synthesis of high added-value molecules from bio-based starting materials”) for financial support.References
- P. K. Sharma, A. Amin and M. Kumar, Open Med. Chem. J., 2020, 14, 49–64 CrossRef CAS.
- F. Velazquez and H. Olivo, Curr. Org. Chem., 2002, 6, 303–340 CrossRef CAS.
- A. Degterev, A. Lugovskoy, M. Cardone, B. Mulley, G. Wagner, T. Mitchison and J. Yuan, Nat. Cell Biol., 2001, 3, 173–182 CrossRef CAS PubMed.
- M. X. Wang, H. W. Qin, C. Liu, S. M. Lv, J. S. Chen, C. G. Wang, Y. Y. Chen, J. W. Wang, J. Y. Sun and Z. X. Liao, PLoS One, 2022, 17, 2–13 Search PubMed.
- Y. S. Prabhakar, V. R. Solomon, M. K. Gupta and S. B. Katti, Top. Heterocycl. Chem., 2006, 4, 161–249 CrossRef CAS.
- N. Chen, W. Jia and J. Xu, Eur. J. Org. Chem., 2009, 5841–5846 CrossRef CAS.
- R. Morales-Nava, M. Fernández-Zertuche and M. Ordóñez, Molecules, 2011, 16, 8803–8814 CrossRef CAS.
- A. Ziyaei-Halimehjani, K. Marjani and A. Ashouri, Tetrahedron Lett., 2012, 53, 3490–3492 CrossRef CAS.
- A. A. Nechaev, A. A. Peshkov, K. Van Hecke, V. A. Peshkov and E. V. Van der Eycken, Eur. J. Org. Chem., 2017, 1063–1069 CrossRef CAS.
- S. F. Gan, J. P. Wan, Y. J. Pan and C. R. Sun, Synlett, 2010, 973–975 CAS.
- A. M. Jacobine and G. H. Posner, J. Org. Chem., 2011, 76, 8121–8125 CrossRef CAS PubMed.
- S. Gabillet, D. Lecerclé, O. Loreau, M. Carboni, S. Dézard, J. M. Gomis and F. Taran, Org. Lett., 2007, 9, 3925–3927 CrossRef CAS PubMed.
- A. W. Demartino, D. F. Zigler, J. M. Fukuto and P. C. Ford, Chem. Soc. Rev., 2017, 46, 21–39 RSC.
- X. F. Jiang, H. Huang, Y. F. Chai, T. L. Lohr, S. Y. Yu, W. Lai, Y. J. Pan, M. Delferro and T. J. Marks, Nat. Chem., 2017, 9, 188–193 CrossRef CAS PubMed.
- A. Sudo, Y. Morioka, E. Koizumi, F. Sanda and T. Endo, Tetrahedron Lett., 2003, 44, 7889–7891 CrossRef CAS.
- T. Able, J. Org. Chem., 2008, 9137–9139 Search PubMed.
- M. Sengoden, M. Vijay, E. Balakumar and T. Punniyamurthy, RSC Adv., 2014, 4, 54149–54157 RSC.
- A. Khalaj and M. Khalaj, J. Chem. Res., 2016, 40, 445–448 CrossRef CAS.
- Y. Xie, C. Lu, B. Zhao, Q. Wang and Y. Yao, J. Org. Chem., 2019, 1951–1958 CrossRef CAS PubMed.
- Y. Shi, B. Tang, X.-L. Jiang, Y.-E. Jiao, H. Xu and B. Zhao, J. Mater. Chem. A, 2022, 10, 4889–4894 RSC.
- D. Carminati, E. Gallo, C. Damiano, A. Caselli and D. Intrieri, Eur. J. Inorg. Chem., 2018, 5258–5262 CrossRef CAS.
- C. Damiano, P. Sonzini, G. Manca and E. Gallo, Eur. J. Org. Chem., 2021, 2807–2814 CrossRef CAS.
- P. Sonzini, C. Damiano, D. Intrieri, G. Manca and E. Gallo, Adv. Synth. Catal., 2020, 362, 2961–2969 CrossRef CAS.
- C. Damiano, P. Sonzini, M. Cavalleri, G. Manca and E. Gallo, Inorg. Chim. Acta, 2022, 540, 121065 CrossRef CAS.
- P. Sonzini, N. Berthet, C. Damiano, V. Dufaud and E. Gallo, J. Catal., 2022, 414, 143–154 CrossRef CAS.
- A. M. Cavalleri, C. Damiano, G. Manca and E. Gallo, Chem. – Eur. J., 2023, 29, e202202729 CrossRef PubMed.
- P. K. Chattaraj, S. Duley and L. R. Domingo, Org. Biomol. Chem., 2012, 10, 2855–2861 RSC.
- L. R. Domingo, M. J. Aurell, P. Pérez and J. A. Saez, RSC Adv., 2012, 2, 1334–1342 RSC.
- L. R. Domingo, M. Rios-Gutierrez and P. Pérez, Molecules, 2016, 21, 748–760 CrossRef PubMed.
- L. R. Domingo, P. Perez and J. S. Saez, RSC Adv., 2013, 3, 1486–1494 RSC.
- A. Z. Bialvaei, M. Rahbar, M. Yousefi, M. Asgharzadeh and H. S. Kafil, J. Antimicrob. Chemother., 2017, 72, 354–364 CrossRef CAS PubMed.
- D. McBride, T. Krekel, K. Hsueh and M. J. Durkin, Expert Opin. Drug Metab. Toxicol., 2017, 13, 331–337 CrossRef CAS PubMed.
- F. Moureau, J. Wouters, D. Vercauteren, S. Collin, G. Evrard, F. Durant, F. Ducrey, J. Koenig and F. Jarreau, Eur. J. Med. Chem., 1992, 27, 939–948 CrossRef CAS.
- S. Grimme, J. Chem. Phys., 2006, 124, 034108 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Gaussian, Inc., Wallingford CT, 2016, 1 Search PubMed.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nj05479e |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2023 |