
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNitrogen-doped reduced graphene oxide-polyaniline composite materials: hydrothermal treatment, characterisation and supercapacitive properties†
Edwin T.
Mombeshora‡
a,
Edigar
Muchuweni§
 a,
Matthew L.
Davies
*ab,
Bice S.
Martincigh
a and
Vincent O.
Nyamori
*a
a,
Matthew L.
Davies
*ab,
Bice S.
Martincigh
a and
Vincent O.
Nyamori
*a
aSchool of Chemistry and Physics, University of KwaZulu-Natal, Westville Campus, Private Bag X54001, Durban, 4000, South Africa. E-mail: nyamori@ukzn.ac.za
bSPECIFIC IKC, Materials Science and Engineering, Faculty of Science and Engineering, Swansea University, UK
First published on 17th January 2023
Abstract
Among other factors, the electrochemical capacitor (EC) properties of graphene oxide (GO) are limited by conductivity issues emanating from high oxygen concentrations. Hence, innovative GO modifications, such as derivatisation and synthesising composites with conducting polymers, are required to boost the EC properties. This allows the exploitation of the low material cost associated with carbonaceous materials. Herein, the effect of temperature (70, 130 and 190 °C) on the hydrothermal treatment of a GO/urea mixture (to form nitrogen-doped reduced GO samples U70, U130 and U190 (N-RGO), respectively) was investigated in order to study the resulting physicochemical and EC properties of N-RGO in a K2SO4 electrolyte. Subsequently, the EC properties of the N-RGO obtained from the optimum temperature were investigated in composites with polyaniline (PANI). The functionality of the composites in the electrolytes, namely, K2SO4 (SO42− radius: 258 pm) versus KOH (OH− radius: 133 pm), was compared. The nitrogen at% in GO and N-RGO (U190) was 0.39 and 6.74%, respectively, with corresponding conductivities of 4.61 × 10−7 and 4.17 × 10−1 S cm−1. Compared with GO, U130 achieved the highest increase in specific capacitance (Cs) of 200 times at 10 mV s−1. The 5 wt% PANI composite displayed the highest enhancement of Cs of 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 032% at 50 mV s−1 and 4749% at 5 mV s−1 relative to N-RGO and PANI in K2SO4, respectively. The highest Cs values for NRGO-PANI composites in KOH were more than double those of K2SO4. This work demonstrates that the hydrothermal treatment temperature tailors the physicochemical properties of the doped GO and, together with the anion size of the electrolyte and PANI wt%, synergistically tunes the EC properties of the ultimate PANI-N-RGO composite.
032% at 50 mV s−1 and 4749% at 5 mV s−1 relative to N-RGO and PANI in K2SO4, respectively. The highest Cs values for NRGO-PANI composites in KOH were more than double those of K2SO4. This work demonstrates that the hydrothermal treatment temperature tailors the physicochemical properties of the doped GO and, together with the anion size of the electrolyte and PANI wt%, synergistically tunes the EC properties of the ultimate PANI-N-RGO composite.
Introduction
Energy-related problems, such as depletion of non-renewable resources, pollution, supply and demand, have been growing global concerns for decades. This has driven researchers to devote their focus to strategies that mitigate these energy-related concerns. Developing alternative clean and renewable energy resources to replace fossil fuels is central to this cause. However, renewables as alternative energy resources still need effective energy storage technologies. Hence, electrical energy storage is fundamental in shifting to clean, sustainable and dependable renewable energy resources.Of all the electrical energy storage systems, capacitors have achieved higher power densities (∼5 kW kg−1) and longer cycle lives (>106 charge–discharge cycles) than batteries and fuel cells.1–3 On the other hand, electrochemical capacitors (ECs) are lightweight and have higher energy densities than conventional capacitors.1,4 Hence, ECs are attractive for further developments toward high energy densities since they can be viewed as the bridge between high energy density devices (Li-ion batteries ∼300 W h kg−1)3 and high-power density devices (conventional capacitors).
One key aspect influencing the performance of ECs is their electrode materials. Owing to high surface area and mechanical strength, ease of manufacturing, non-toxicity, relatively low costs, and abundance; carbonaceous materials are promising contenders for electrode materials.1,5,6 Among the carbonaceous materials, graphene-based materials have been widely studied in constructing EC electrodes because of their large ion-accessible surface areas. Graphene forms the backbone of most carbonaceous materials and consists of a one-carbon atom thick sheet of hexagonally arranged sp2-hybridised carbon atoms.7 However, it tends to agglomerate, resulting in poor dispersion in liquid media. In such cases, its derivative graphene oxide (GO) is preferred. GO with randomly distributed sp3-hybridised carbons contains a high concentration of oxygen functionalities attached to the sp2 framework and instils a highly exfoliated state.8,9 However, GO on its own is a poor electrode material for ECs due to limited electro-active sites and its insulating nature.10,11 For example, a high amount of GO in a Ni–metal–organic framework-GO-graphene nanoplatelet EC electrode was reported to lower electric energy storage capabilities due to the increased insulating effect from GO.12 The conducting properties of graphene can be restored in GO by reducing it through chemical, thermal, and other methods. In this alternative form of GO, known as reduced graphene oxide (RGO), the oxygen content is decreased, and the aromatic structure of graphene is restored. This process does not entirely convert GO to graphene since some oxygen content remains, but these defects can be utilised for functionalisation to enhance the properties of RGO.
A wide range of studies involving graphene derivatives, such as GO and RGO in particular, in ECs have been reported.5,6,13–15 Some current research efforts aim to further enhance their performance through composite synthesis,16 surface functionalisation,17 and heteroatom doping.18 Chemical doping with effective atoms, such as nitrogen, phosphorus, sulfur, and boron, improves the charge density and electrical conductivity.17,19–21 Incorporating nitrogen moieties into/onto the graphitic carbon framework has been reported to enhance several physicochemical properties, such as carrier concentrations, conductivity, and chemical reactivity, as detected by content and species. Muangrat et al.22 synthesised a composite of nitrogen-doped graphene and double-walled carbon nanotubes through a chemical vapour deposition method and reported a 162% improvement in the specific capacitance (Cs) due to N-doping. Among other methods used to incorporate nitrogen moieties, hydrothermal treatment is a suitable nitrogen doping method because it facilitates the retention of substantial oxygen functionalities that can aid wettability in ECs. From a study undertaken to improve understanding of the effect of N-doping levels in RGO, done by employing the hydrothermal method at 150 °C, a higher amount of urea dopant raised the nitrogen content and lowered oxygen moieties, and the resulting Cs decline was attributed to aggregation of sheets.17 Studies have also deduced the dependency of electric charge storage processes on the electronic structure of the electrode materials, which is, in turn, influenced by their bond characteristics.23 For example, from a study of the effect of the type of nitrogen species in N-RGO on Cs, measured in Na2SO4 electrolyte, EC electrodes with pyrrolic-N-doped RGO were deduced to be more effective than those with pyridinic-N-doped-RGO.18 Hence, since doping and reduction of GO introduce heteroatoms and lower oxygen concentrations of GO, methodical approaches are needed that can suitably alter the electronic characteristics and tune the electrochemical energy storage characteristics for EC applications.
In order to provide both physical and chemical charging storage mechanisms in one electrode, carbonaceous materials are often combined with another carbon-based material, a metal oxide or a conducting polymer, to form a nanocomposite. In particular, adding conducting polymers, such as polythiophene, polypyrrole, polyethylene or polyaniline (PANI) in composites, can play an important role in improving the performance of carbonaceous-based EC electrodes through the delivery of high specific energy (Es) from faradaic processes.14,24–26 Among these, PANI has high potential in ECs because of its low relative costs, elongated conjugated chain structure, high electrical conductivity, variable oxidation state and excellent doping/de-doping nature.27 Despite being a promising polymer material for ECs, the poor cycle stability and rate capability of PANI restrict its applicability in ECs.26,28,29 However, the synthesis of graphene derivative-PANI composites that proved to be functional EC materials has been reported.30 For illustration, a PANI–MnO2–GO ternary composite enhanced the electrochemical properties and achieved the highest Cs of ∼150 F g−1.15 Thus, creating hybrid materials can enhance the synergistic properties of the individual components, but at an extra cost.
In this work, we investigated the suitability of a PANI-N-RGO composite for use as an EC electrode material. The rationale for developing this composite was to positively enhance the capacitive properties of GO by augmenting the favourable effects of reduction, N-doping and composite synthesis with PANI. The N-RGO was produced by hydrothermal treatment of GO in urea. Since the synthesis method can alter the energy storage characteristics of the resulting material, the effect of three doping temperatures, namely, 70, 130 and 190 °C, on the physicochemical properties of the resulting N-RGO was studied. The material with the best storage characteristics was then formed into a hybrid material with different amounts of PANI and further characterised. In addition, the effect of the size of the electrolyte anion on the performance of the PANI-N-RGO composites was elucidated by changing the electrolyte from K2SO4 to KOH. This research illustrates that systematic tailoring of the physicochemical properties of the hybrid PANI-N-RGO material can boost the electrochemical energy storage capabilities of the individual components through synergistic effects and arrive at an optimum choice of EC electrode material.
Experimental section
The N-RGO samples were synthesised and first characterised by various physicochemical and electrochemical techniques.Synthesis of GO and nitrogen-doped reduced GO
The GO was synthesized using a modified Tour's method.31,32 In short, 3 g of graphite (Sigma Aldrich, <150 μm, 99.9%) was added to 400 mL of an orthophosphoric acid (Labchem, 85% AR) and sulfuric acid (Promark Chemicals, 98%) mixture in a 1:9 (v/v) ratio. Thereafter, 9 g of potassium permanganate (Promark Chemicals, 99.4%) was added to the mixture contained in an ice water bath to ensure a temperature of <10 °C throughout the addition. Subsequently, the reaction temperature was increased to 50 °C and maintained for 3 h. A volume of 30 mL of hydrogen peroxide (Promark Chemicals, 30–35%) was added to terminate the reaction. The sample was decanted, and the supernatant was discarded. Decanting was repeated, and washing was done consecutively with 240 mL of each double-distilled water (DI, Glass chem water distiller model Ws4lcd), hydrochloric acid (Sigma Aldrich, 32%) and ethanol (crude). The sample was then dialysed through a dialysis tubing cellulose membrane (Merck, D9652-100FT) until the pH of the dialysate was neutral. The resulting GO was dried to constant mass in an oven at 50 °C.
The GO was simultaneously reduced and doped with nitrogen by hydrothermal treatment in urea (CO(NH2)2, Saarchem, 99%). In brief, a mass of 0.1 g of GO was added to 10 g of urea dissolved in 44 mL DI (GO:urea weight ratio of 1:100). The reagents were loaded into an autoclave, and the thermal treatment was carried out at three different temperatures in an oven at 70, 130 and 190 °C for 8 h to synthesise the samples named as U70, U130 and U190, respectively. After cooling, the supernatant was decanted, and the samples were washed several times with DI before they were dried at 60 °C in an oven for 48 h.
Physicochemical characterisation
The materials were imaged with a JEOL JEM 2100 transmission electron microscope. Elemental analyses were conducted with an Elementar vario El cube elemental analyser on two replicates for each sample. X-ray photoelectron spectroscopy (XPS) analysis was performed by means of a Kratos Axis Supra instrument with a charge neutralizer and monochromatic Al Kα source. The data was collected with a pass energy of 20 eV, and the peaks were deconvoluted using the Casa XPS software. For Fourier transform infrared spectroscopy (FTIR) analysis, samples were diluted 300 times with KBr before pelletising with a 10 ton press and left to dry overnight in an oven. FTIR spectra were acquired with a PerkinElmer Spectrum RXI spectrophotometer. Raman spectra were acquired with a 100 mW Delta Nu Advantage 532™ spectrometer at a resolution of 10 cm−1 and excitation wavelength of 532 nm from a Nd: YAG laser with a 2D CCD detector and grating lines of 1800 mm−1. The powder X-ray diffraction data were collected with a Bruker D8-Advance multi-purpose X-ray diffractometer operated in a continuous J–J scan in locked coupled mode with Cu Ka radiation (λ = 1.5406 Å). The EVA software from Bruker was used for data evaluation. A position-sensitive detector, Lyn-Eye, was used to record diffraction data at a typical speed of 0.5 s per step (equivalent to an effective time of 92 s per step for a scintillation counter). For nitrogen sorption analysis, samples were degassed at 160 °C for 18 h in a Micromeritics Vacprep 061 sample degassing system. Thereafter, analyses were done using a Micromeritics TriStar 3020 (V1.03) surface area and porosity analyser at 77 K with nitrogen as the sorbent. Thermal stability studies were carried out with a TA Instruments SDT Q600 thermal analyser in air flowing at 100 mL min−1 from 25 to 1000 °C at a heating rate of 10 °C min−1. The electrical conductivity characterisations were performed on 0.02 mm pellets of samples by means of a Jandel four-point probe connected to a Keithley 2450 source-meter. The Hall effect measurements were done using a DHE-21 Hall effect set-up connected to a DPS-50 constant power supply and DGM-202 digital gaussmeter at a constant magnetic field of 1377 G (all from Scientific Equipment, Roorkee).Electrochemical measurements
The N-RGO samples were mixed thoroughly with Nafion perfluorinated resin (Sigma, ion-exchange resin, 20 wt%) in a 95:5 wt% ratio, respectively, to form a uniform slurry. The slurry was then drop-cast onto a clean, glassy carbon electrode and dried under a sodium lamp. The mass loading was determined by subtracting the mass of the glassy carbon electrode from the mass of the dried casted electrode. All electrochemical tests were carried out with a Princeton Applied Research VersaSTAT 3F, model-500, potentiostat/galvanostat, electrochemical workstation, while the VersaStudio software (version 2.60.6.0) was used for data acquisition. The three-electrode cyclic voltammetry (CV) system utilised in the current work consisted of the test material, Pt wire, Ag/AgCl and 0.5 M K2SO4 as the active electrode, counter electrode, reference electrode and electrolyte system, respectively. The CV scan speeds (v) were from 5 to 200 mV s−1 in the potential range (ΔV) of 0 to 0.8 V. Electrochemical properties were also investigated by means of the electrochemical impedance spectroscopy (EIS) technique with an amplitude of 5 mV in the 100000–1 Hz frequency range.
After optimisation, to establish the N-RGO sample with the best Cs, a second set of electrochemical analyses was carried out with the addition of PANI (synthesised via oxidative polymerisation of aniline (Merck, 98%), as reported elsewhere)33 at wt% ratios of 5, 10, 20, 50, 80 and 100% in 0.5 M K2SO4 with samples drop-cast as discussed earlier. This step was repeated using 0.5 M KOH as the electrolyte (as opposed to 0.5 M K2SO4) to study the effect of different electrolytes and the anion size.
Given the current, i, and mass, m, the specific capacitance (Cs) was calculated from eqn (1):17,34
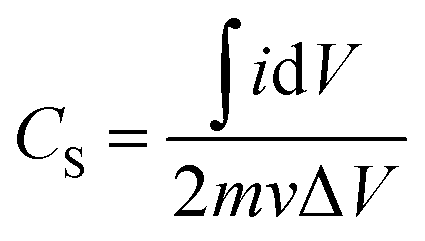 | (1) |
Results and discussion
Firstly, the effects of temperature on the simultaneous reduction and N-doping of GO in a urea solution using a hydrothermal method are discussed. Secondly, details of the electrochemical energy storage capabilities of N-RGO samples to establish the optimum sample are presented. Hence, the third section discusses the effect of different wt% of PANI in the PANI-N-RGO composites (PANI–U130 since U130 was the optimum) when either K2SO4 or KOH are used as electrolytes.Effect of hydrothermal temperature on physicochemical and electronic properties of N-RGO
 | ||
| Fig. 1 TEM micrographs of (a) GO, (b) U70, (c) U130 and (d) U190 for pristine and hydrothermally treated graphene oxide at 70, 130 and 190 °C, respectively. | ||
Elemental composition and surface functional group analysis
To establish the effect of temperature of the hydrothermal treatment in urea solution on the chemical composition of N-RGO, elemental (EA) and X-ray photoelectron spectroscopy (XPS) analyses were carried out (Table 1 and ESI,† Table S1). Both EA and XPS showed the presence of N and S atoms. The XPS binding energies at ca. 532, 401, 399 and 168 eV were assigned to oxygen-containing moieties (O1s), pyrrolic N1s (N2), pyridinic N1s (N1) and S2p, respectively (Fig. 2(a)–(c)).24,35,36 The small component of sulfur found in the samples was attributed to H2SO4 used in the oxidation reaction of graphite to produce GO. This agrees with previous reports on the formation of sulfur functionalities/adsorbed species under similar experimental conditions.32,37 The poor signal-to-noise ratios in the spectra for N1s and S2p were due to respective low elemental concentrations and inelastic electron scattering in the samples causing a related rise in the background (Fig. 2(b) and (c)).38| Sample | Elemental composition | X-ray photoelectron spectroscopy | |||||
|---|---|---|---|---|---|---|---|
| S (%) | N (%) | C/N | S (%) | N (%) | C/N | C/O | |
| GO | 0.33 | 0.00 | — | 0.59 | 0.39 | 180.28 | 2.48 |
| U70 | 0.32 | 3.46 | 15.79 | 0.14 | 2.70 | 26.93 | 2.97 |
| U130 | 0.32 | 2.52 | 22.48 | 0.15 | 1.73 | 42.03 | 2.86 |
| U190 | 0.21 | 6.89 | 7.99 | 0.00 | 6.74 | 12.39 | 8.55 |
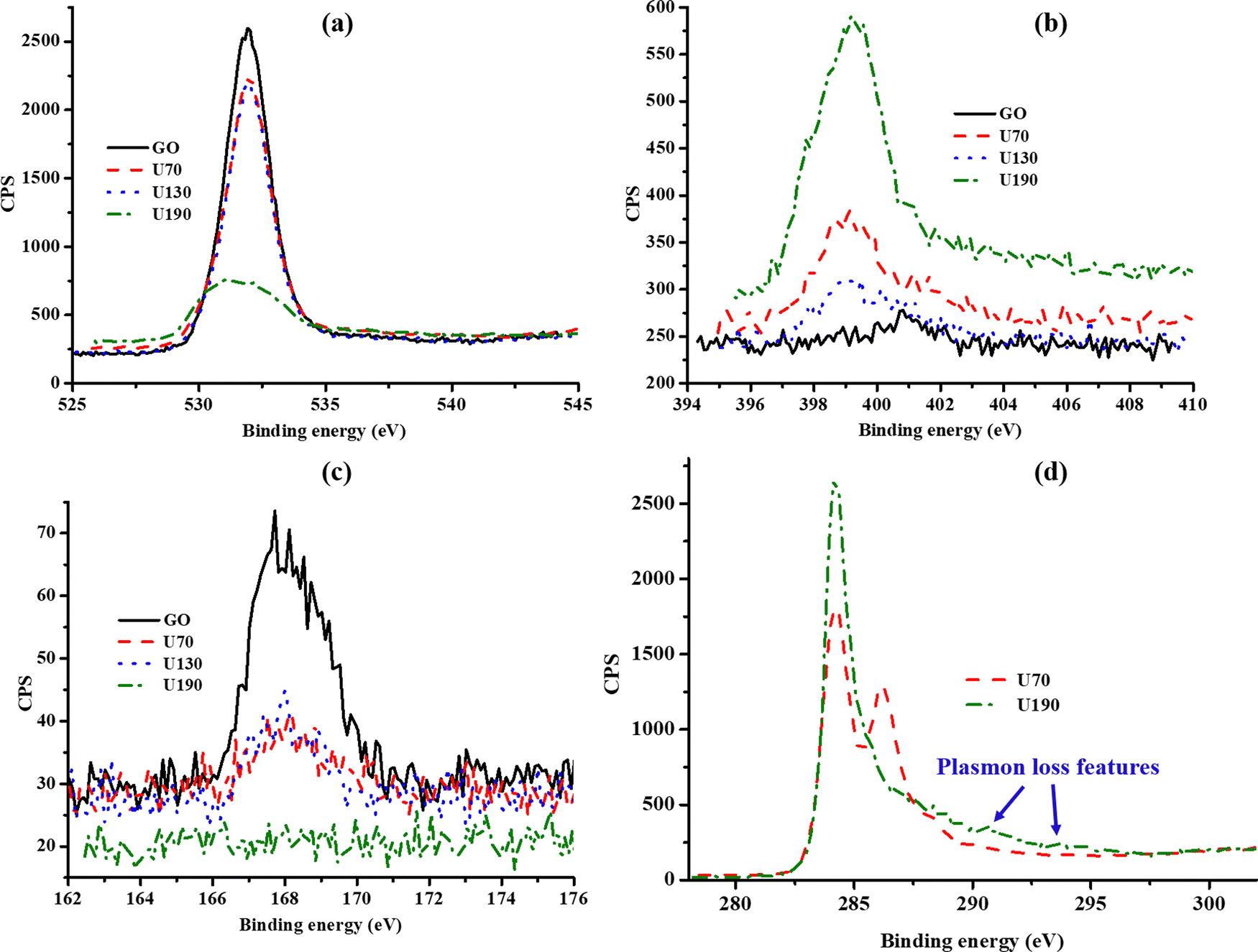 | ||
| Fig. 2 The (a) O1s, (b) N1s and (c) S2p spectra from X-ray photoelectron spectroscopy, and (d) illustration of removal of oxygen through C1s upon raising the solvothermal temperature. | ||
U190 contained the largest amount of N (smallest C/N ratio) and the least O content (highest C/O ratio) (Table 1). The lowest O content at the highest temperature investigated (190 °C) is an indication of the highest degree of reduction of GO at this temperature (loss of gaseous COx). Reduction causes a decline in repulsions between oxygen functionalities on sheet surfaces; hence, restacking of sheets was observed in the TEM micrographs (Fig. 1). Since N could either be incorporated on surface functionalities or doped in the carbon framework, the decline observed between U70 and U130 can be attributed to the decomposition of unstable surface nitrogen species to form NOx gases due to a higher temperature.8 The fact that U190 contains the highest N at% (XPS) and elemental composition (∼7%) probably infer N-doping since considerably more energy is required to force the replacement of carbon with nitrogen, and this would have been provided by the higher temperature (Table 1 and Fig. 2(b)).
In addition, the increase of the strong and sharp peak at 284.5 eV and decline of the weaker peak at 286.5 eV, ascribed to sp2 C–C and C–O–C/C–OH interactions,35,39,40 respectively, upon raising the temperature of solvothermal treatment up to 190 °C relative to lower temperatures (70 and 130 °C) illustrate the increase in graphitisation and reduction, respectively (Fig. 2(d)). The subtle “bumps” (blue arrows in Fig. 2(d)) that are often described as ‘plasmon loss features’ emanate from the transitions to π anti-bonding orbitals (electrons in the highest occupied molecular orbitals are promoted into the lowest unoccupied molecular orbitals).40 This indicates a high degree of sp2 hybridisation in U190.
To study the chemical functionalities present, FTIR analysis was performed over the 500–3500 cm−1 range (Fig. 3). The peaks at ca. 3220, 2912, 2346, 1704, 1600, 1450, 1250 and 1060 cm−1 were assigned to the O–H vibrations of hydroxyl and carboxyl, dangling C–H vibrations, dangling C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bonds, C
N bonds, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of carbonyl, in-plane CC, C–N (sp3 N) stretching vibrations, C–O–C of carbonyl and C–O stretching, respectively.14,41,42 Relative to GO, for the thermally treated samples, the C–H and C–OH (also less broad) bond vibrations were red-shifted, while the CC bond intensity increased (Fig. 3), which may infer modified chemical interactions and an increase in the graphitic carbon lattice upon hydrothermal treatment from reduction/N-doping. The presence of C–N and CN peaks after the urea hydrothermal treatment indicates N-doping/N functionalities in the samples. As in the work of Ombaka et al.,43 the broadening of the peak at ca. 1600 cm−1 can also be interpreted to indicate that CC and CN (due to the introduction and incorporation of nitrogen in the carbon backbone) peaks overlap in this absorption region. Also, the peak at 1060 cm−1 increased after hydrothermal treatment, possibly due to peak overlap due to C–N bonds in the carbon framework. These results corroborate previous work that suggests that oxygen functionalities are active sites for nitration.8
O of carbonyl, in-plane CC, C–N (sp3 N) stretching vibrations, C–O–C of carbonyl and C–O stretching, respectively.14,41,42 Relative to GO, for the thermally treated samples, the C–H and C–OH (also less broad) bond vibrations were red-shifted, while the CC bond intensity increased (Fig. 3), which may infer modified chemical interactions and an increase in the graphitic carbon lattice upon hydrothermal treatment from reduction/N-doping. The presence of C–N and CN peaks after the urea hydrothermal treatment indicates N-doping/N functionalities in the samples. As in the work of Ombaka et al.,43 the broadening of the peak at ca. 1600 cm−1 can also be interpreted to indicate that CC and CN (due to the introduction and incorporation of nitrogen in the carbon backbone) peaks overlap in this absorption region. Also, the peak at 1060 cm−1 increased after hydrothermal treatment, possibly due to peak overlap due to C–N bonds in the carbon framework. These results corroborate previous work that suggests that oxygen functionalities are active sites for nitration.8
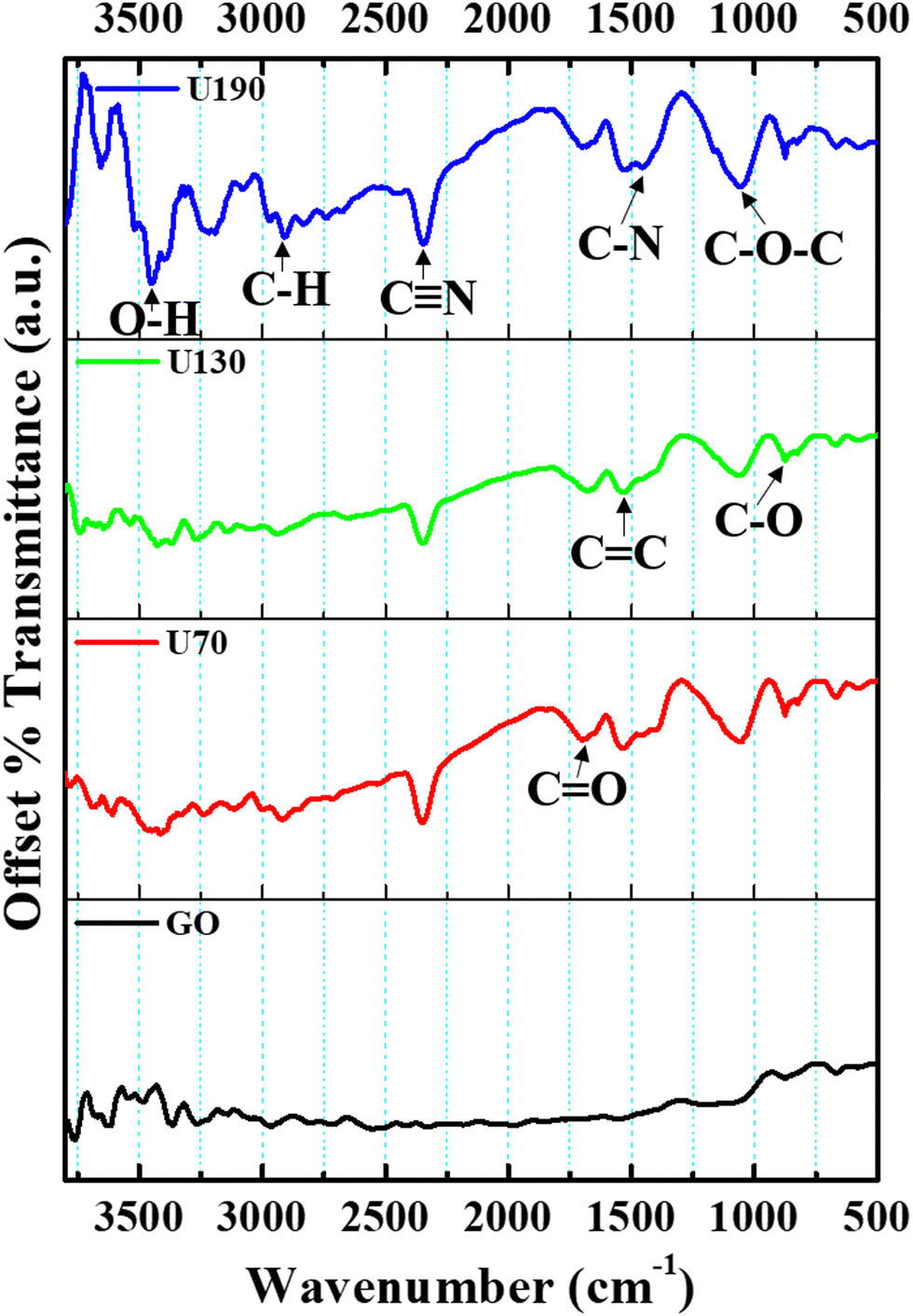 | ||
| Fig. 3 FTIR spectra for GO and N-RGO samples recorded as KBr discs. | ||
Vibrational and crystallinity studies
Raman spectroscopy was used to determine crystallinity and structural properties. The peaks at ca. 1350 and 1600 cm−1 were attributed to the D- and G-bands, respectively (Fig. 4 and Table 2).13,44 Whilst the D-band implies the existence of sp3-hybridized carbon and disordered carbon structures, the G-band signals the presence of a graphitic structure from sp2 carbon stretching vibrations. The decreased intensity of both peaks as the reduction temperature was raised agrees with work reported by Kavinkumar et al.45 upon GO reduction. The red shifting of both peaks, relative to GO, indicates an n-type doping phenomenon that induces a strong electron-donating character (Fig. 4 and Table 2).37 The defect intensity was calculated by dividing the area under the D-band by that under the G-band. | ||
| Fig. 4 Raman spectra of GO and N-RGO. | ||
| Sample | D-band position (cm−1) | G-band position (cm−1) | I D/IG | L a (nm) |
|---|---|---|---|---|
| GO | 1351 | 1597 | 0.64 | 30.20 |
| U70 | 1347 | 1586 | 1.05 | 18.31 |
| U130 | 1342 | 1588 | 0.86 | 22.35 |
| U190 | 1345 | 1586 | 0.98 | 19.62 |
The increase in defect intensity in the RGO samples relative to GO agreed with earlier reports17,46,47 and was ascribed to the generation of more local defects and structural disorders from the hydrothermal reduction/N-doping process (Table 2). Unlike the rest of the samples that contained more sp2 than sp3 carbons, it is noteworthy that the concentration of sp3 hybridized carbons in U70 is almost equal to that of the sp2 species (ID/IG ≈ 1).44,48 The ID/IG trend was independent of the reaction temperature and was most likely influenced by the nitrogen composition in two ways (Tables 1 and 2). Firstly, at 130 °C, most of the unstable surface nitrogen species had been removed without substantial N-doping, hence, a decline in ID/IG (relative to 70 °C). Secondly, N-doping in the carbon framework was enhanced at 190 °C leading to an increase in ID/IG (relative to 130 °C), and this is also a possible reason for the poor crystallinity of U190.
The RGO domain size (La) was calculated from eqn (2):32,49
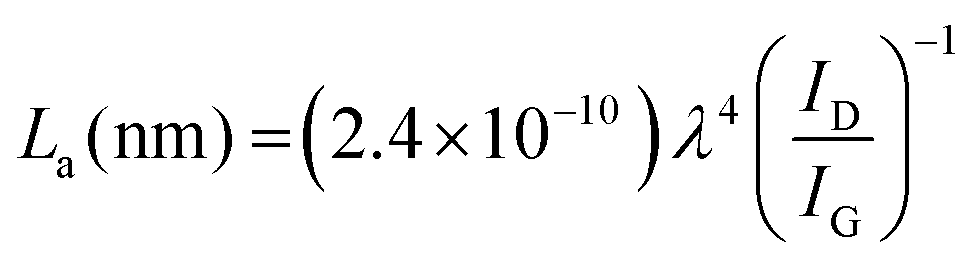 | (2) |
The GO nanosheets were cut into smaller sizes during the reduction/N-doping process, and the least tearing effect was observed on U130 (Table 2). The sheet disintegration during reduction can be attributed to the eradication of bridged oxygen molecules during reduction.50 The tearing effect on the sheets is a probable reason for the defect intensification traits since the ascending order of defect intensities (GO < U130 < U190 < U70) was supported by the descending order of sheet sizes (GO > U130 > U190 > U70).32
The X-ray diffraction peaks at 2θ values of 11.5°, 25° (broad) and 43° were due to the (002) plane of GO, shifted (002) plane in RGO and (001) crystal plane of GO/RGO, respectively (Fig. 5).45,46,51
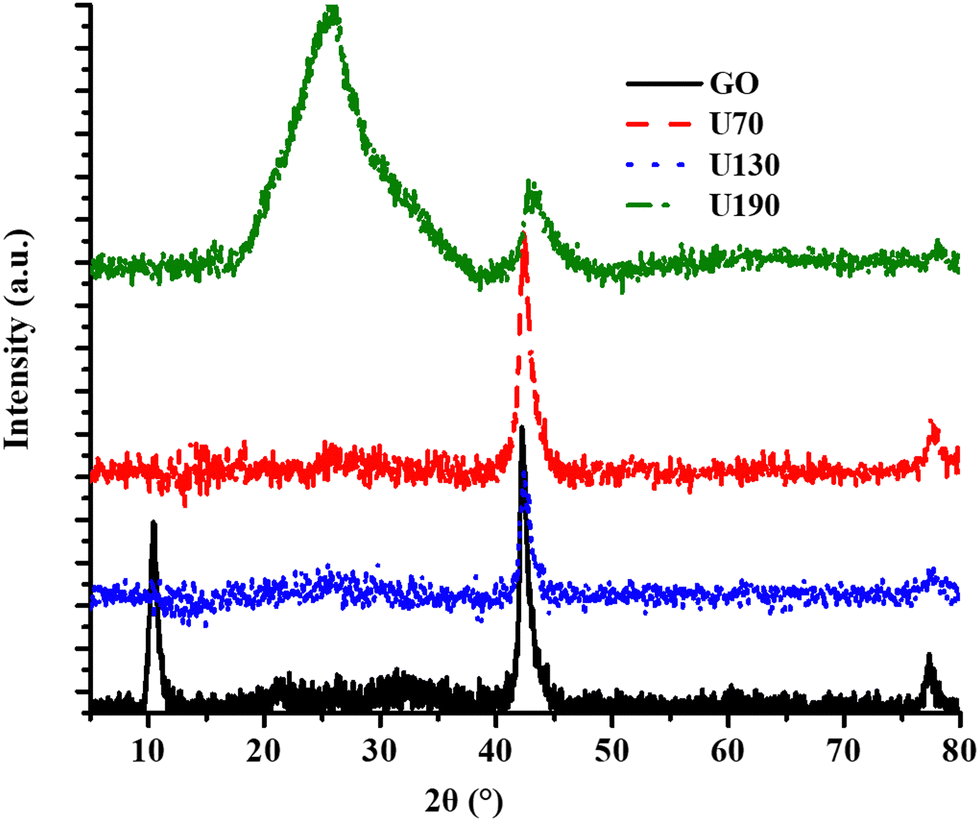 | ||
| Fig. 5 The effect of hydrothermal treatment in urea at different temperatures on crystallinity. | ||
The disappearance/shifting of the peak at 2θ of 11.5° upon hydrothermal treatment is an indication of crystallinity transformation due to the doping effect and reduction process.18 In addition, the intensity of the peak due to the (001) crystal plane of GO decreased as the oxidation/reduction temperature was raised (Fig. 5), and this was ascribed to structural transformations due to the increased reduction effect (Table 1). The broad peak from reduction/N-doping at 2θ of 25° for U190 (Fig. 5) suggests a large number of structural transformations on the N-RGO surface, possibly from the most effective removal of oxygen-containing moieties.8,45,46 The most intense 002 peak of U190 is a signal for improved crystallinity and agrees with XPS data (Fig. 2(d) and 5).52 Significant structural transformations among the N-RGO samples were possible since the difference in hydrothermal treatment temperature was 60 °C in each case (Fig. 5). Hence, different hydrothermal temperatures induced different reduction/doping efficacy that culminated in various levels of oxygen vacancies and lattice fragmentation, among others (Fig. 2, 3 and Table 2).
Textural characterization
All the samples displayed type IV nitrogen adsorption isotherms with H3 type hysteresis loops (ESI,† Fig. S2). This signifies a mesoporous character that could aid the rapid infiltration of electrolyte ions into the electrode.36,53,54 The N2 sorption studies show that the hydrothermal reduction/N-doping process in urea solution negatively affected the textural properties (Table 3). This agrees with agglomerated morphologies, as observed on TEM micrographs (Fig. 1), which limits exposed surfaces and the participation of graphene sheet interspaces in N2 sorption. Since the order for surface area, pore volume, and ID/IG are the same: U130 < U190 < U70, while for La, it is the inverse U130 > U190 > U70 (Tables 2 and 3), the trends probably mean that the relative higher defect intensities, large pore volume and surface area, were caused by disintegration of sheets.| Sample | Specific surface area (m2 g−1) | Pore volume (cm3 g−1) | Pore size (nm) |
|---|---|---|---|
| GO | 302 | 1.25 | 19.84 |
| U70 | 82 | 0.42 | 7.14 |
| U130 | 20 | 0.04 | 8.75 |
| U190 | 66 | 0.23 | 7.05 |
Thermal stability and compositional studies
The thermograms obtained for GO and the three N-RGO samples in air are shown in Fig. 6. Similar to earlier reports,42,47 the weight loss at 200 °C was assigned to the loss due to the decomposition of labile oxygen-containing moieties (Fig. 5). The thermograms show that the descending order of the losses at ca. 200 °C were GO (∼74%), U70 (∼20%), U130 (∼14%) and U190 (∼4%), and this agreed with the ascending weight loss order due to the carbon framework at temperatures >600 °C (U70, U130 and U190 losses were 33, 38 and 66%, respectively) (Fig. 6(a)). This probably infers that the large surface area and high oxygen concentration of GO (Tables 1 and 3) facilitated with ease the thermal decomposition of the carbon framework to CO2 at 200 °C,32,55 unlike in the rest of the samples, and that the composition of the oxygen-containing moieties decreased as the reduction/N-doping temperature (in the hydrothermal treatment) was raised (Fig. 6). A low content of oxygen-containing moieties lowers hydrophilicity; hence, the descending order of weight loss due to adsorbed water at <100 °C for RGO was U70 (∼40%), U130 (∼34%) and U190 (∼12%) (Fig. 6). Thus, the thermal analysis infers that the increased reduction that occurs at higher reduction/N-doping temperatures in the hydrothermal treatment lowers the wettability/hydrophilicity of the materials. | ||
| Fig. 6 The thermograms of GO and N-RGO samples recorded in an atmosphere of air. | ||
Electrical conductivity and Hall measurements
The simultaneous reduction and N-doping process improved the conductivity of GO due to changes in bond configurations and the availability of a lone pair of electrons from the N atom; also, the conductivity increased as the reduction temperature was raised (Table 4). A further rise of reduction/N-doping temperature beyond 200 °C to establish the limit was not feasible due to the thermal stability limit of the autoclave. These conductivity traits are attributed to the synergistic effect of both the restoration of aromaticity arising from reduction and N-doping of pristine GO that increased the graphitic carbon lattice content (decrease in C/N and increase in C/O ratios in Table 1 and increase in CC peak intensity in Fig. 3).8 Possibly, this is through the graphitic-N species, which adopt the sp2 planar structure of the carbon framework to facilitate electron transfer (enhanced carrier mobility) and improve electrical conductivity and reduce bulk resistivity (Tables 4 and 5).5,22,26 The rationale for the decrease in sheet resistance with an increase in reduction/N-doping temperature is possibly the increase in the C/O ratios (Tables 1 and 4).
| Sample | Sheet resistance (Ω sq−1) | Electrical conductivity (S cm−1) |
|---|---|---|
| GO | 2.15 × 107 | 4.61 × 10−7 |
| U70 | 5.90 × 102 | 3.85 × 10−4 |
| U130 | 1.30 × 102 | 1.70 × 10−3 |
| U190 | 6.00 × 102 | 4.17 × 10−1 |
| Sample | Bulk resistivity (Ω cm) | Hall coefficient (cm3 C−1) | Carrier density (cm−3) | Carrier mobility (cm2 V−1 s−1) |
|---|---|---|---|---|
| LOD – limit of detection | ||||
| GO | <LOD | <LOD | <LOD | <LOD |
| U70 | 2.60 × 103 | −18.17 | 3.44 × 1017 | 0.03 |
| U130 | 5.90 × 102 | −1.21 | 5.16 × 1018 | 0.50 |
| U190 | 2.40 | −3.24 | 1.93 × 1018 | 1.35 |
The high oxygen concentration of GO was the main cause of the high sheet resistance and low electrical conductivity (Tables 1 and 4);12 hence, the current driven through the sheets was below the limit of detection (LOD), and this made it impossible to carry out Hall measurements (the instrument required current ≥mA range). The increase in carrier mobility for N-RGO was ascribed to the decrease in bulk resistivity as the synthesis temperature was raised, due to reduction/N-doping of the graphene framework since reduction restores the conjugated structure and nitrogen has an electron-donating nature (n-type). These results corroborate the Raman analysis and EA and XPS data (Tables 1, 2 and 5). The carrier mobility and carrier density in the current work were comparable to 1.61 cm2 V−1 s−1 and 3.90 × 1020 cm−3 for N-doped graphene from a chemical vapour deposition method.22
Electrochemical characterization
CV and EIS were used to investigate the effect of hydrothermal temperature on the electrochemical properties of N-RGO in 0.5 M K2SO4. Changes in local acidity due to the removal of oxygen-containing moieties, such as carboxylic acid and hydroxyl groups, on the electrode materials (induced by hydrothermal treatment), caused slight variations in the operational voltage window in the K2SO4 electrolyte (Fig. 7).34,56,57 On the one hand, the CV technique showed that the Cs of U130 was the highest, followed by U190, at low v of 10 and 25 mV s−1 (Fig. 7(a)–(f)). On the other hand, U190 was highest at higher v, whilst the U130 was second (Fig. 7(a)–(f)). The improved electrochemical properties of U130 and U190, relative to GO, can be attributed to the decrease in bulk resistivity and improved conductivity, carrier density and mobility (Fig. 7(a)–(e) and Table 5). Another possible reason is the tailored lattice electronic and physicochemical properties emanating from the pyridinic- and pyrrolic-N doping effect that induced pseudocapacitive contributions and enhanced electron transfer from graphitic-N.22,37 Despite lower bulk resistivity, and higher electrical conductivity, carrier mobility, elemental N and N at% (Tables 1 and 5), U190 had lower Cs than U130 at slower v. This could be because of greater wettability (deductions from Fig. 6(a)) and higher carrier density effects of U130 (Table 5).17 Additionally, the decrease in Cs as v was raised was ascribed to the minimal contributions from the electro-active area of the inner pores at higher v (Fig. 7(f)).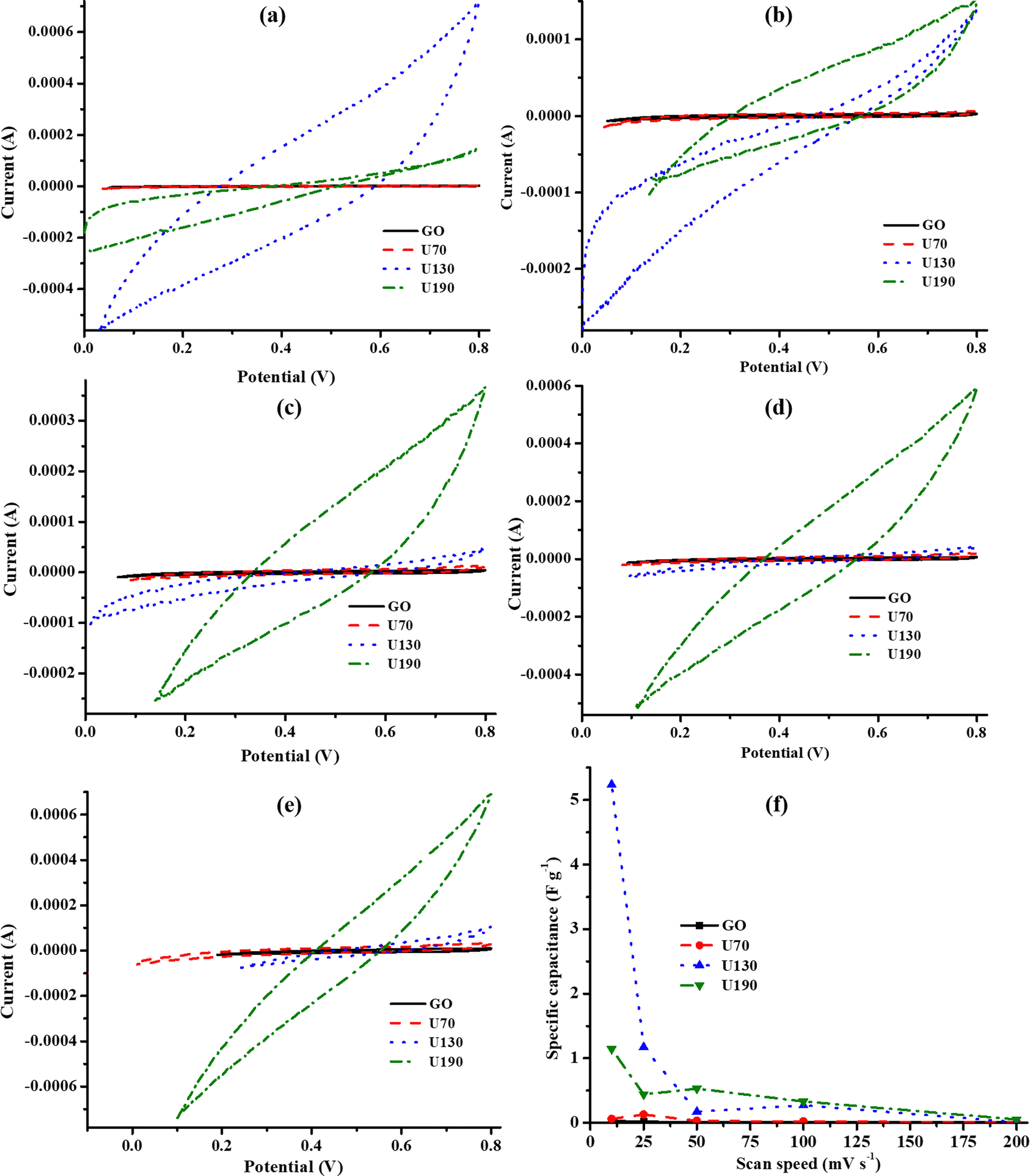 | ||
| Fig. 7 Cyclic voltammograms of GO and N-doped RGO at different v (a) 10, (b) 25, (c) 50, (d) 100, (e) 200 mV s−1 and (f) Cs in 0.5 M K2SO4 electrolyte. | ||
The curve inclination exhibited by GO and U70 in the low-frequency region of the Nyquist plot (Fig. 8) supported their more quasi-rectangular cyclic voltammograms (Fig. 7(a)–(e)); hence, the data suggest that the electric double layer capacitor (EDLC) quality deteriorated with hydrothermal treatment temperature. In addition, Fig. 7(a)–(e) shows that U130 exhibited a better EDLC character (a more rectangular CV curve) than U190. This possibly arises because nitrogen functionalities introduce some pseudocapacitive characteristics on carbonaceous materials that can cause deviations from EDLC behaviour. The EIS spectra were analysed with the aid of a modelled equivalent circuit, which comprised Rs (intrinsic material strength, mass of the electrolyte, and the electrolyte ionic impedance between electrodes), EDLC at the electrode/electrolyte interface, RCT (electrode/electrolyte contact impedance), and W (Warburg impedance) (inset in Fig. 8).58,59
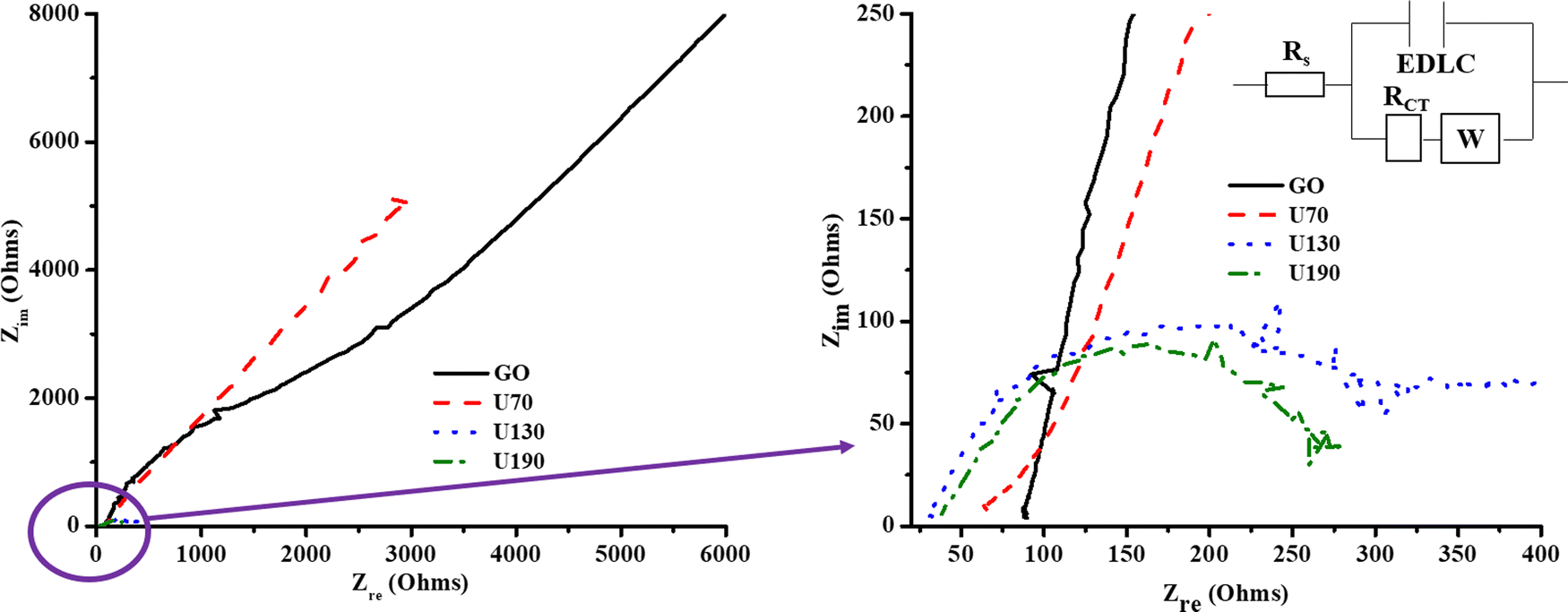 | ||
| Fig. 8 The influence of hydrothermal temperature on the electrochemical impedance of N-RGO. | ||
Tailoring of electrochemical properties by altering the reduction/N-doping temperature was displayed by the EIS spectra, which showed clear semi-circles in the high-frequency region for U130 and U190, and linear lines in the low-frequency region for the pristine GO and U70 samples (Fig. 8). The x-intercept and diameter of the semi-circle in the high-frequency region of a Nyquist plot are reported methods to determine RS and RCT, respectively.44,60,61 The RS for GO, U70, U130 and U190 were estimated to be 88, 63, 32 and 38 Ω, respectively (Fig. 8). Possibly, an increase in graphitisation via the presence/content of graphitic-N is a suitable justification for the decrease in RS with the increase in reduction/oxidation temperature. Since the electrolyte factor was constant, the RS declining trend agreed with the increase in carrier density of the electrode materials (Table 5), hence, ultimately enhancing the electrochemical energy storage of U130 and U190 in K2SO4 (Fig. 7(f)). In addition, the highest current responses for U190 can be associated with the higher conductivity (Fig. 7(b)–(e) and Table 4). On the other hand, the RCT of GO, U70, U130 and U190 was estimated to be 0 (absence of small circle), 10, 269 and 231 Ω, respectively (Fig. 8). This data shows that samples with higher RCT are associated with higher electric energy storage capabilities (Fig. 8 and 7(e)). This occurrence concurs with an earlier report by Muangrat et al.22 that higher RCT implies slow charge transfer at the electrode/electrolyte interface62 and, hence, instils good electrochemical energy storage. Also, since the length of the Warburg curve in the Nyquist plot signifies the diffusion path-length,34,48 the ascending diffusion path-length of U130 < U190 < U70 < GO (Fig. 8) is a feasible contributing factor to the Cs trend in the samples (Fig. 7(f)).
Electrochemical properties of PANI-N-RGO hybrid materials
Since hydrothermal treatment of GO at 130 °C achieved the highest energy density (11.57 W h kg−1) and power density (41.67 kW kg−1) (ESI,† Table S2) and Cs (Fig. 7(f)), the sample U130 was thereafter used to investigate the influence of PANI wt% in the functionality of N-RGO as electrodes for ECs in 0.5 M K2SO4. In addition to the highest Cs at all v (uppermost% improvement, ESI,† Table S3), the Cs of 5 wt% PANI PANI–U130 composite increased with a raised v (Fig. 9(a)). Also, the composites with smaller amounts of PANI (5–20 wt%) recorded higher Cs values than both pristine U130 and PANI; hence, this implies that the synergistic effect of the composite components is dependent on the mixing ratio, and the composite synthesis is a potential route to boost the Cs of both N-RGO (highest Cs boost of 14032% at 50 mV s−1 for 5 wt% PANI composite) and PANI (highest Cs boost of 4749% at 5 mV s−1 for 5 wt% PANI composite) (ESI,† Table S3). Interestingly, the highest Cs boost relative to U130 was at higher v, while with reference to PANI, it occurred at lower v for all PANI–U130 composites (ESI,† Table S3). In addition, the Cs improvement with reference to U130 was relatively more than that of PANI (Fig. 9(a) and ESI,† Table S3).
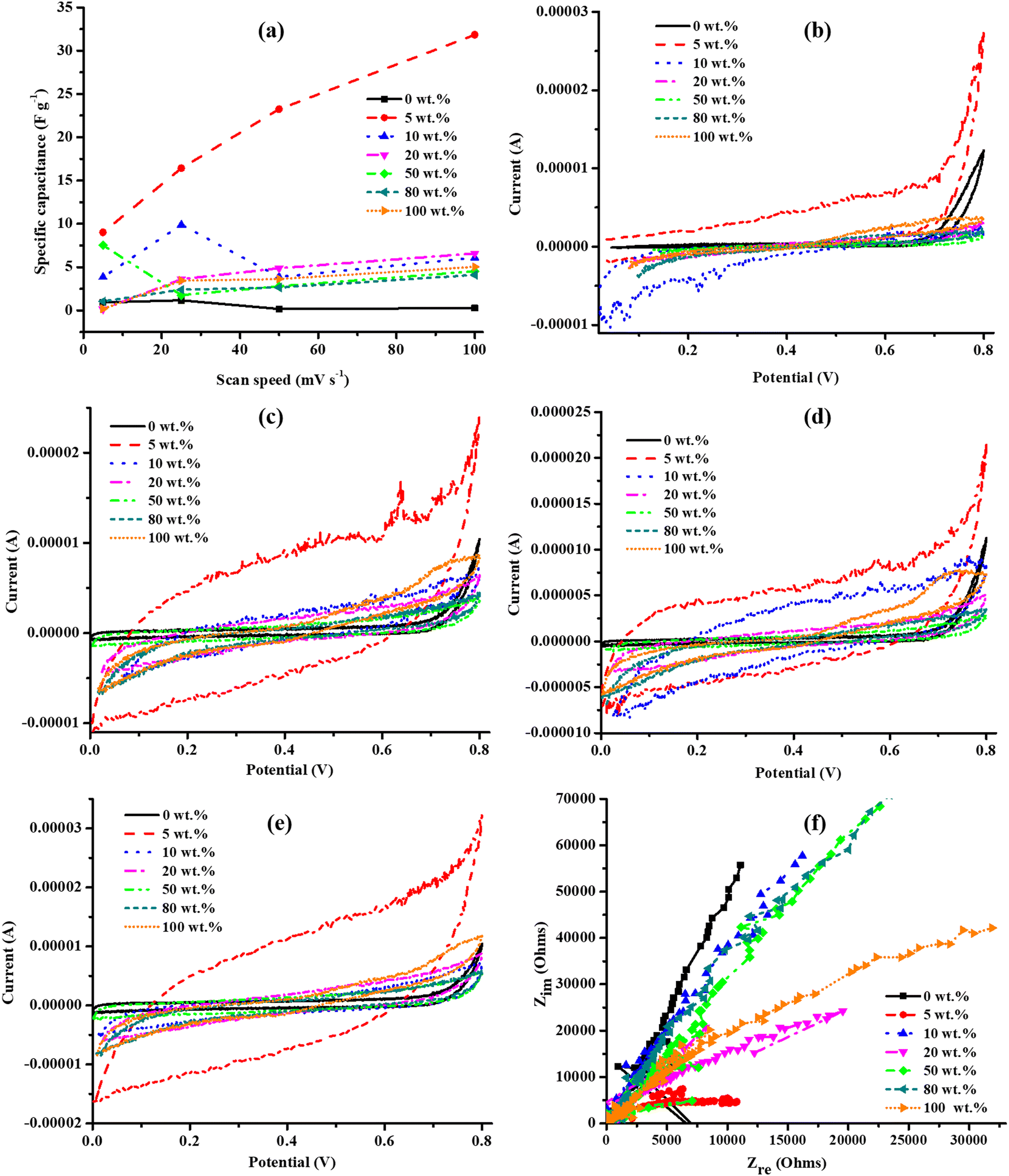 | ||
| Fig. 9 The (a) Cs and cyclic voltammograms for PANI–U130 composites at a v of (b) 5, (c) 25, (d) 50 and (e) 100 mV s−1, and (f) electrochemical impedance spectroscopy studies in K2SO4 electrolyte. | ||
The EDLC capacitive quality of electrodes from all composites was similar (Fig. 9(b)–(f)); this could be because this phenomenon depends mostly on the carbonaceous framework, which was a constant factor in the current study. Similar to current work, Lee et al.17 attributed the absence of redox peaks and quasi-rectangular shapes from N-RGO, doped from urea as the nitrogen source, to the predominant contributions of nitrogen functionalities to Cs. This can further be understood to mean that N-doping of RGO may indirectly affect Cs by altering conductivity and Hall effect parameters (Tables 4 and 5). All the composites exhibited good rate capability since the quasi-rectangular shape of the cyclic voltammograms was still retained at a high v of 100 mV s−1 (Fig. 9(b)–(e)).48,63
The RS values were approximated as 32, 76, 108, 136, 185, 143 and 114 Ω for 0 (U130); 5, 10, 20, 50, 80 and 100 (PANI) wt% of PANI, respectively, and only 0 (269 Ω) and 5 (9924 Ω) wt% of PANI displayed RCT. Hence, these traits support the observed electrochemical energy storage capabilities of the composites, as discussed earlier. The highest Cs from 5 wt% PANI composites could also be attributed to the shortest diffusion path-length; therefore, Cs increased as v was raised because time limitations of the electrolyte diffusion process into electrode pores were minimal at higher v (Fig. 9(a) and (f)).
The electrolyte in the cell was changed from 0.5 M K2SO4 to 0.5 M KOH to investigate the effect of a smaller anionic size of 133 pm for the OH−,64 relative to 258 pm for SO42−,65 on the electrochemical properties of the PANI–U130 composites. The narrower potential windows of the PANI–U130 composites relative to that of U130 (widest) and pristine PANI were similarly ascribed to changes in the local acidity of composites (Fig. 10(a)–(d)). Again, in KOH, the quasi-rectangular cyclic voltammograms (Fig. 10(a)–(d)) and the curve inclinations in the low-frequency region of the Nyquist plot (Fig. 10(f)) for PANI–U130 composites imply a similar EDLC capacitive quality due to reasons elaborated in the K2SO4 study. Unlike the earlier deductions from the K2SO4 study, the 10 wt% PANI composites displayed the best electrochemical activities and highest Cs (Fig. 10(a)–(e)). Similar to the electrochemical studies in K2SO4, the composite materials in the 5–20 wt% range of PANI were highly enhanced than both U130 (17733% Cs boost for the 10 wt% PANI composite at 5 mV s−1) and PANI (1248% Cs improvement for the 10 wt% PANI composite at 50 mV s−1) (ESI,† Table S4). Notably, the Cs from the 5–20 wt% range of PANI composites approach a constant value at higher v (Fig. 10(e)). This could be attributed to the relative declining contribution of the inner pores to electrochemical processes; hence, the physicochemical variations due to wt% ratios become less significant. The 50 and 80 wt% PANI loadings were not beneficial to the electrical energy storage capabilities of U130.
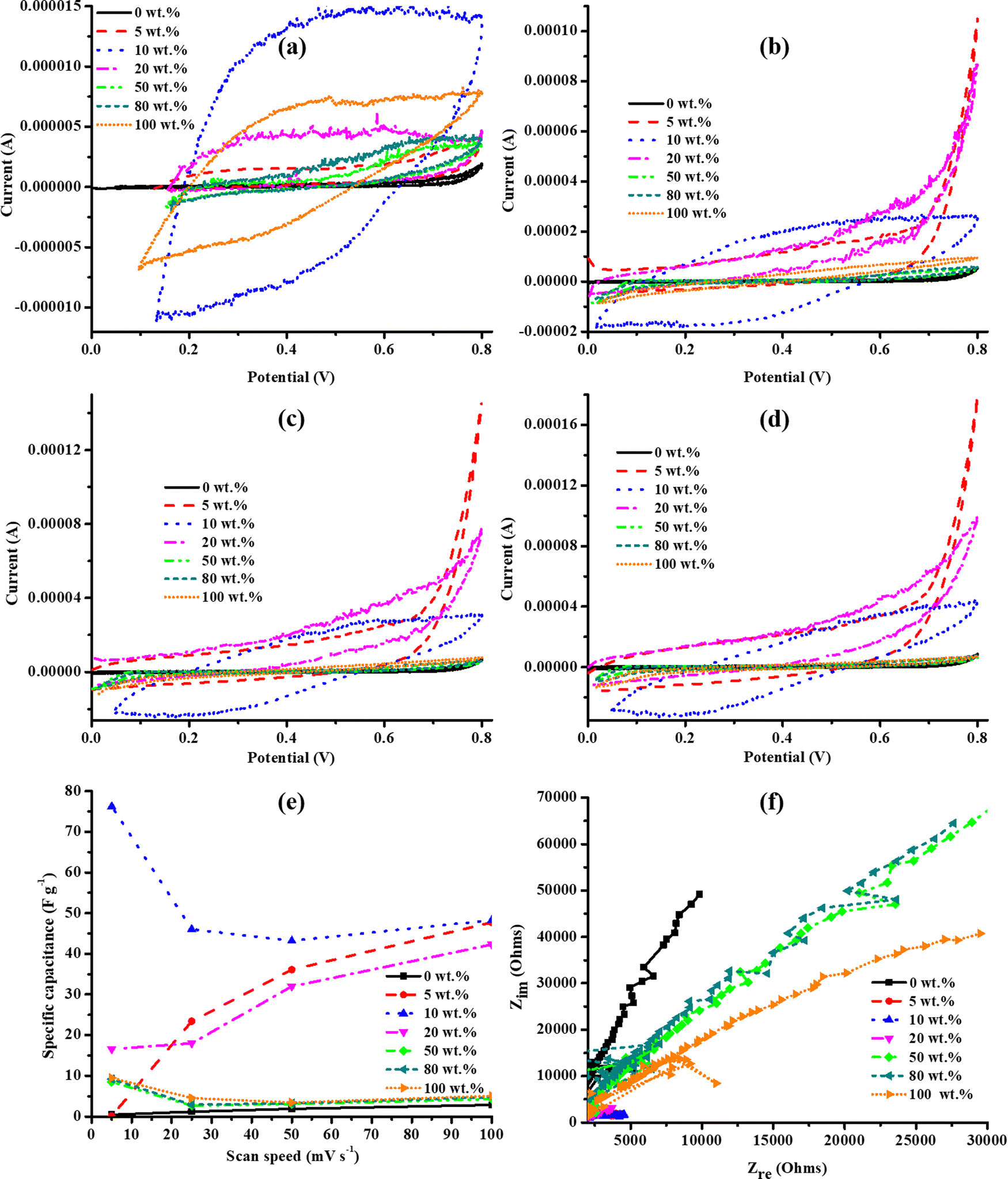 | ||
| Fig. 10 Cyclic voltammograms for PANI–U130 composites at v of (a) 5, (b) 25, (c) 50 and (d) 100 mV s−1, (e) Cs and (f) electrochemical impedance spectroscopy study in KOH electrolyte. | ||
The noise observed in EIS spectra may be due to the low alternating current amplitude used in the current study, defects (instability), surface area, and formation of gas bubbles on the electrode surface (Fig. 9(f) and 10(f)).66–68 The approximate RS values of PANI–U130 composites in KOH were 133, 102, 69, 95, 148, 191 and 118 Ω for 0 (U130), 5, 10, 20, 50, 80 and 100 (PANI) wt% of PANI, respectively. It was only possible to estimate RCT for the 5 and 10 wt% of PANI composites as 3532 and 3898 Ω, respectively. The noisier EIS spectra for the PANI–U130 composite-based electrodes studied in KOH than those in the K2SO4 electrolyte possibly arise due to the observed higher impedance in the KOH electrolyte (Fig. 9(f) and 10(f)).67 Furthermore, the shorter diffusion path-length in the 5–20 wt% range of PANI of the PANI–U130 composites implies an effective diffusion process (Fig. 10(f)). Therefore, the high Cs values from electrodes of the 5–20 wt% PANI range of composites in KOH electrolyte was attributed to synergistic contributions from effective diffusion, shorter diffusion path-length, lower RS and higher RCT than the rest of the samples (Fig. 10(e) and (f)). In addition, the steeper curve in the high-frequency region of the Nyquist plot for 0 wt% of PANI composite suggests that PANI induces a deviation from the EDLC capacitive character (Fig. 10(f)). This is because PANI induces pseudocapacitive characteristics through the benzenoid/quinoid rings interconversion induced by an electron transfer.24,27
From Fig. 9(a) and 10(e), it can be deduced that the composite materials had better electrochemical energy storage capabilities in KOH (anionic radius: 133 pm and champion Cs: 76.25 F g−1) than in K2SO4 (anionic radius: 258 pm and champion Cs: 31.84 F g−1) electrolyte. The maximum Cs enhancement from PANI addition to U130, was 14032% and 17733% in K2SO4 and KOH electrolytes, respectively (ESI,† Tables S3 and S4). Since RS is the sum of the internal resistance of the electrode material, the electrolyte resistance, and the interfacial contact resistance between the electrode and current collector materials,48,69 it means that the internal resistance contributions of the electrode material per given PANI wt% were constant. Hence, the RS and Cs variations can be assigned to the electrolyte and interfacial contact resistances, originating from differences in the chemical nature and size of the electrolyte anion.
The current response (i) depends on either diffusion of ions into electrode pores or electrochemical adsorption/desorption according to eqn (3):70,71
| i = avb | (3) |
i against logv at a fixed potential. If b is 0.5, 1 and 0.5 < b < 1, then the electrochemical charge storage will be characterised by a battery-like and diffusion-controlled process, a surface-controlled kinetics process (capacitive mechanism), and mixed processes, respectively.23 From the values of b (ESI,† Table S5), it is clear that the electrochemical energy storage processes in the K2SO4 electrolyte for both U70 and U130 were based on surface-controlled kinetics. The mixed electrochemical energy storage processes in both GO and U190 (ESI,† Table S5) originate from the high content of oxygen and nitrogen, respectively (Table 1). The oxygen- and nitrogen-containing species induce pseudocapacitive characteristics in addition to the intrinsic EDLC traits.
The values of b agree with the EIS data in that adding PANI in the PANI–U130 composites transformed the electrochemical energy storage processes from capacitive to battery-like and diffusion-controlled nature processes in both electrolytes (ESI,† Table S5, Fig. 8(f) and 9(f)). This is also a possible reason for the improved electrochemical energy storage capabilities of the composites induced by PANI. For example, the Cs trends of the composites can be interpreted to mean that at lower PANI wt%, diffusion-controlled electrochemical processes allowed participation of both inner and outer pores as v was raised (Fig. 8(a) and 9(e)).
Conclusion
The study highlighted that the temperature of hydrothermal treatment of GO in urea positively tuned physicochemical properties, such as lowering the oxygen concentration, diffusion path-length, sheet resistance, RS, and bulk resistivity, and increasing the nitrogen content, electrical conductivity, carrier density and mobility towards improved Cs. The highest N at% of 6.74% was achieved at 190 °C. From Hall measurements, the highest carrier mobility of 1.35 cm2 V−1 s−1 and lowest bulk resistivity of 2.40 Ω cm was displayed by U190; hence, reduction/N-doping positively tuned the electrical properties. In this study, the optimum reduction/N-doping temperature of GO with synergistic properties, from a GO:urea ratio of 1:100, towards a high Cs was 130 °C.
Synergistic properties induced by the anionic size of the electrolytes used and low PANI wt% (5–20 wt%) in PANI–U130, together with the intrinsically tailored physicochemical and electronic properties of U130, improved the capacitive properties of composites relative to that of the individual components. PANI transformed the electrochemical energy storage processes of U130 from capacitive to battery-like and diffusion-controlled activities. The highest Cs of the PANI–U130 composite was almost double that of an electrolyte with an anionic radius of 258 pm (Cs: 31.84 F g−1) in an electrolyte with approximately half the anionic radius of 133 pm (Cs: 76.25 F g−1). The current work validates that the hydrothermal treatment temperature controls the physicochemical and electrochemical properties of nitrogen-doped GO, the anion size of the electrolyte influences the electrochemical capacitance, and the synthesis of composite materials from N-RGO and PANI can enhance the electrochemical energy storage capabilities of both individual components through synergistic effects.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is based on the research supported mainly by funding through the EPSRC GCRF SUNRISE project (grant number: EP/P032591/1) and partly by the National Research Foundation (NRF) of South Africa. MLD is grateful for the financial support of the EPSRC (EP/S001336/1) and the funding of the SPECIFIC Innovation and Knowledge Centre by the EPSRC [EP/N020863/1], Innovate UK [920036], and the European Regional Development Fund [c80892] through the Welsh Government. The authors wish to thank the University of KwaZulu-Natal (UKZN) and the UKZN Nanotechnology Platform for supporting this work by providing the necessary research infrastructure. The authors are also grateful to James Mcgettrick for assistance in X-ray photoelectron spectroscopy analysis.References
- Y. Wang, L. Zhang, H. Hou, W. Xu, G. Duan, S. He, K. Liu and S. Jiang, J. Mater. Sci., 2020, 56, 173–200 CrossRef CAS.
- Q. Abbas, R. Raza, I. Shabbir and A. G. Olabi, J. Sci.: Adv. Mater. Devices, 2019, 4, 341–352 Search PubMed.
- P. Simon and Y. Gogotsi, Nat. Mater., 2020, 19, 1151–1163 CrossRef CAS PubMed.
- E. T. Mombeshora and V. O. Nyamori, Int. J. Energy Res., 2015, 39, 1955–1980 CrossRef CAS.
- A. Kumar, C.-S. Tan, N. Kumar, P. Singh, Y. Sharma, J. Leu, E.-W. Huang, T. Winie, K.-H. Wei and T. Y. Tseng, RSC Adv., 2021, 11, 26892–26907 RSC.
- M. Zhang, Y. Shan, Q. Kong and H. Pang, FlatChem, 2022, 32, 100332 CrossRef CAS.
- E. T. Mombeshora and A. Stark, Biomass Convers. Biorefin., 2021 DOI:10.1007/s13399-021-01499-6.
- J. S. Roh, H. W. Yoon, L. Zhang, J.-Y. Kim, J. Guo and H. W. Kim, ACS Appl. Nano Mater., 2021, 4, 7897–7904 CrossRef CAS.
- Q. Zhou, G. Yuan, K. Guo, S. Li, M. Lin, J. Hong and Y. Huang, FlatChem, 2021, 30, 100303 CrossRef CAS.
- K. Adaikalam, S. Ramesh, P. Santhoshkumar, H. S. Kim, H.-C. Park and H.-S. Kim, Int. J. Energy Res., 2022, 46(4), 4494–4505 CrossRef CAS.
- E. T. Mombeshora, E. Muchuweni, R. Garcia-Rodriguez, M. L. Davies, V. O. Nyamori and B. S. Martincigh, Nanoscale Adv., 2022, 4, 2057–2076 RSC.
- I. Ibrahim, S. Zheng, C. Y. Foo, N. M. Huang and H. N. Lim, J. Energy Storage, 2021, 43, 103304 CrossRef.
- E. T. Mombeshora and V. O. Nyamori, J. Mater. Sci.: Mater. Electron., 2017, 28, 18715–18734 CrossRef CAS.
- J. Yang, J. Cao, Y. Peng, M. Bissett, I. A. Kinloch and R. A. W. Dryfe, J. Power Sources, 2021, 516, 230663 CrossRef CAS.
- G. Zeplin and E. G. C. Neiva, J. Electroanal. Chem., 2021, 902, 115776 CrossRef CAS.
- E. T. Mombeshora, P. G. Ndungu, A. L. L. Jarvis and V. O. Nyamori, Mater. Chem. Phys., 2018, 213, 102–112 CrossRef CAS.
- H.-J. Lee, A. Abdellah, F. M. Ismail, C. Gumeci, N. Dale, J. Parrondo and D. C. Higgins, Electrochim. Acta, 2021, 397, 139241 CrossRef CAS.
- E. T. Mombeshora, P. G. Ndungu and V. O. Nyamori, Electrochim. Acta, 2017, 258, 467–476 CrossRef CAS.
- D. J. Tarimo, K. O. Oyedotun, A. A. Mirghni and N. Manyala, Int. J. Hydrogen Energy, 2020, 45, 13189–13201 CrossRef CAS.
- N. P. D. Ngidi, M. A. Ollengo and V. O. Nyamori, New J. Chem., 2020, 44, 16864–16876 RSC.
- P.-J. Yen, C.-C. Ting, Y.-C. Chiu, T.-Y. Tseng, Y.-J. Hsu, W.-W. Wu and K.-H. Wei, J. Mater. Chem. C, 2017, 5, 2597–2602 RSC.
- W. Muangrat, M. Obata, M. T. Htay, M. Fujishige, P. Dulyaseree, W. Wongwiriyapan and Y. Hashimoto, FlatChem, 2021, 29, 100292 CrossRef CAS.
- Y. Sim, S. Surendran, H. Cha, H. Choi, M. Je, S. Yoo, D. Chan Seok, Y. Ho Jung, C. Jeon, D. Jin Kim, M.-K. Han, H. Choi, U. Sim and J. Moon, Chem. Eng. J., 2022, 428, 132086 CrossRef CAS.
- J. Vigneshwaran, S. Abraham, B. Muniyandi, T. Prasankumar, J.-T. Li and S. Jose, Surf. Interfaces, 2021, 27, 101572 CrossRef CAS.
- R. B. Choudhary and S. Ansari, J. Energy Storage, 2022, 46, 103912 CrossRef.
- Y. Chen, L. Fang, Y. Hu, Y. Lu, J. He, S. Wang, Q. Yang, Z. Shi and C. Xiong, Diamond Relat. Mater., 2022, 122, 108813 CrossRef CAS.
- A. K. Thakur, M. Majumder, R. B. Choudhary, S. B. Singh, S. P. Patole and D. Dubal, Mater. Today Commun., 2021, 29, 102915 CrossRef CAS.
- A. Dang, T. Li, C. Fang, C. Tang, T. Zhao, X. Chen, C. Xiong and Q. Zhuang, Fullerenes, Nanotubes, Carbon Nanostruct., 2019, 27, 440–445 CrossRef CAS.
- Y. Wang, Y. Ding, X. Guo and G. Yu, Nano Res., 2019, 12, 1978–1987 CrossRef CAS.
- F. Movassagh-Alanagh, A. Bordbar-Khiabani and A. Ahangari-Asl, Int. J. Hydrogen Energy, 2019, 44, 26794–26806 CrossRef CAS.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
- E. T. Mombeshora and A. Stark, Mater. Chem. Phys., 2022, 277, 125535 CrossRef CAS.
- E. C. Gomes and M. A. S. Oliveira, Am. J. Polym. Sci., 2012, 2, 5–13 CrossRef.
- K. Mugadza, E. T. Mombeshora, A. Stark, P. G. Ndungu and V. O. Nyamori, J. Mater. Sci.: Mater. Electron., 2021, 32, 27923–27936 CrossRef CAS.
- N. Huu Hieu, H. Huu Dat, L. Tran Trung Nghia, N. Minh Dat, L. Nam Phat, N. Thi Tinh, P. Tan Khang, N. Thai Hoang and M. Thanh Phong, FlatChem, 2022, 31, 100318 CrossRef CAS.
- X. Hou, P. Ren, Z. Dai, Z. Guo, Z. Zhang, A. Sun, W. He, F. Ren and Y. Jin, Energy Technol., 2021, 9, 2100743 CrossRef CAS.
- N. H. A. Rosli, K. S. Lau, T. Winie, S. X. Chin and C. H. Chia, Diamond Relat. Mater., 2021, 120, 108696 CrossRef CAS.
- T. G. Avval, E. F. Smith, N. Fairley and M. R. Linford, Vac. Technol. Coating, 2019, 1–3 Search PubMed.
- J. Vigneshwaran, S. Abraham, B. Muniyandi, T. Prasankumar, J.-T. Li and S. Jos, Surf. Interfaces, 2021, 27, 101572 CrossRef CAS.
- T. R. Gengenbach, G. H. Major, M. R. Linford and C. D. Easton, J. Vac. Sci. Technol., A, 2021, 39, 013204 CrossRef CAS.
- A. Mohsenipour, M. Mozaffarian, G. Pazuki and L. Naji, J. Alloys Compd., 2021, 892, 162093 CrossRef.
- S. Ayyaru and Y.-H. Ahn, J. Membr. Sci., 2017, 525, 210–219 CrossRef CAS.
- L. M. Ombaka, P. G. Ndungu, B. Omondi, J. D. McGettrick, M. L. Davies and V. O. Nyamori, J. Solid State Chem., 2016, 235, 202–211 CrossRef CAS.
- M. Vandana, S. Veeresh, H. Ganesh, Y. S. Nagaraju, H. Vijeth, M. Basappa and H. Devendrappa, J. Energy Storage, 2022, 46, 103904 CrossRef.
- T. Kavinkumar, N. Naresh, G. Mathew and B. Neppolian, J. Alloys Compd., 2022, 891, 162052 CrossRef CAS.
- S. W. Bokhari, S. Wei and W. Gao, Electrochim. Acta, 2021, 398, 139300 CrossRef CAS.
- Y. Chen, P. Lian, J. Feng, Y. Liu, L. Wang, J. Liu and X. Shi, Chem. Eng. J., 2022, 429, 132274 CrossRef CAS.
- D. Mohanadas, M. A. A. Mohd Abdah, N. H. N. Azman, J. Abdullah and Y. Sulaiman, Int. J. Hydrogen Energy, 2021, 46, 35385–35396 CrossRef CAS.
- A. Capasso, T. Dikonimos, F. Sarto, A. Tamburrano, G. D. Bellis, M. S. Sarto, G. Faggio, A. Malara, G. Messina and N. Lisi, Beilstein J. Nanotechnol., 2015, 6, 2028–2038 CrossRef CAS PubMed.
- H. Wang, H. Tian, S. Wang, W. Zheng and Y. Liu, Mater. Lett., 2012, 78, 170–173 CrossRef CAS.
- J. Xu, J. Liang, Y. Zou, F. Xu, Q. Chen, C. Xiang, J. Zhang and L. Sun, J. Energy Storage, 2021, 43, 103195 CrossRef.
- A. S. Al-Sherbini, M. Bakr, I. Ghoneim and M. Saad, J. Adv. Res., 2017, 8, 209–215 CrossRef CAS PubMed.
- E. T. Mombeshora, R. Simoyi, V. O. Nyamori and P. G. Ndungu, S. Afr. J. Chem., 2015, 68, 153–164 CrossRef CAS.
- X. Liu, Z. Lu, X. Huang, J. Bai, C. Li, C. Tu and X. Chen, J. Power Sources, 2021, 516, 230682 CrossRef CAS.
- M. A. M. Motchelaho, H. Xiong, M. Moyo, L. L. Jewell and N. J. Coville, J. Mol. Catal. A: Chem., 2011, 335, 189–198 CrossRef CAS.
- H. Tomiyasu, H. Shikata, K. Takao, N. Asanuma, S. Taruta and Y. Y. Park, Sci. Rep., 2017, 7, 45048 CrossRef CAS PubMed.
- M. S. Hong, S. H. Lee and S. W. Kim, Electrochem. Solid-State Lett., 2002, 5, A227 CrossRef CAS.
- H. Ahmadi, V. Haddadi-Asla, E. Kowsari and N. Mohammadi, J. Alloys Compd., 2022, 927, 167021 CrossRef CAS.
- S. R. Charandabinezha, H. Asgharzadeh and N. Arsalani, J. Mater. Sci.: Mater. Electron., 2021, 32, 1864–1876 CrossRef.
- W. Chen, Y. Peng, Z. Qiu, X. Zhang and H. Xu, J. Alloys Compd., 2022, 901, 163614 CrossRef CAS.
- Y. Yao, G. Huang, Y. Liu, Y. Liu, Y. Li, G. Han, B. Xing, Q. Liu, J. Jia and C. Zhang, Appl. Surf. Sci., 2022, 580, 152236 CrossRef CAS.
- C. Xu, W. Yang, J. Zhao, J. Ma and M. Wu, ACS Omega, 2021, 6, 24931–24939 CrossRef CAS PubMed.
- E. O. Oseghe, S. O. Akpotu, E. T. Mombeshora, A. O. Oladipo, L. M. Ombaka, B. B. Maria, A. O. Idris, G. Mamba, L. Ndlwana, O. S. Ayanda, A. E. Ofomaja, V. O. Nyamori, U. Feleni, T. T. I. Nkambule, T. A. M. Msagati, B. B. Mamba and D. W. Bahnemann, J. Mol. Liq., 2021, 344, 117820 CrossRef CAS.
- Y. Marcus, J. Chem. Phys., 2012, 137, 154501 CrossRef PubMed.
- B. Wu, X. J. Yang, C. Janiak and P. G. Lassahn, Chem. Commun., 2003, 902–903 RSC.
- K. Elsøe, M. R. Kraglund, L. Grahl-Madsen, G. G. Scherer, J. Hjelm, S. H. Jensen, T. Jacobsen and M. B. Mogensen, Fuel Cells, 2018, 18, 640–648 CrossRef.
- D.-H. Xia, S. Song, Y. Behnamian, W. Hu, Y. F. Cheng, J.-L. Luo and F. Huet, J. Electrochem. Soc., 2020, 167, 081507 CrossRef CAS.
- M. Tretjak, E. Palaimiene, S. Pralgauskaitė, J. Matukas, J. Banys, J. Macutkevič, V. Fierro, S. Schaefer and A. Celzard, Polymers, 2021, 13, 997 CrossRef CAS PubMed.
- Y. Du, D. Cao, B. Li, H. Lü and Y. Shen, Electrochim. Acta, 2021, 395, 139201 CrossRef CAS.
- H. Li and H. Xuan, Int. J. Hydrogen Energy, 2021, 46, 38198–38211 CrossRef CAS.
- X. Huang, R. Sun, Y. Li, J. Jiang, M. Li, W. Xu, Y. Wang, H. Cong, J. Tang and S. Han, Electrochim. Acta, 2022, 403, 139680 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nj05112e |
| ‡ Present address: Department of Chemistry and Earth Sciences, University of Zimbabwe, Post Office Box MP167, Mount Pleasant, Harare, Zimbabwe. |
| § Permanent address: Bindura University of Science Education, Department of Engineering and Physics, Private Bag 1020, Bindura, Zimbabwe. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2023 |