
Shape control with atomic precision: anisotropic nanoclusters of noble metals
Yingwei
Li†
 * and
Rongchao
Jin
*
* and
Rongchao
Jin
*
Department of Chemistry, Carnegie Mellon University, Pittsburgh, PA 15213, USA. E-mail: yingweil@alumni.cmu.edu; rongchao@andrew.cmu.edu
First published on 12th June 2023
Abstract
When plasmonic metal nanoparticles become smaller and smaller, a new class of nanomaterials—metal nanoclusters of atomic precision—comes to light and has become an attractive research topic in recent years. These ultrasmall nanoparticles (or nanoclusters) are unique in that they are molecularly uniform and pure, often possess a quantized electronic structure, and can grow into single crystals as do protein molecules. Exciting achievements have been made by correlating their properties with the precise structures at the atomic level, which has provided a profound understanding of some mysteries that could not be elucidated in the studies on conventional nanoparticles, such as the critical size at which plasmons are emergent. While most of the reported nanoclusters are spherical or quasi-spherical owing to the reduced surface energies (and hence stability), some anisotropic nanoclusters of high stability have also been obtained. Compared to the anisotropic plasmonic nanoparticles, the nanocluster counterparts such as rod-shaped nanoclusters can provide insights into the growth mechanisms of plasmonic nanoparticles at the early stage (i.e., nucleation), reveal the evolution of properties (e.g., optical), and offer new opportunities in catalysis, assembly, and other themes. In this Review, we highlight the anisotropic nanoclusters of atomic precision obtained so far, primarily gold, silver, and bimetallic ones. We focus on several aspects, including how such nanoclusters can be achieved by kinetic control, and how the anisotropy gives rise to new properties over the isotropic ones. The anisotropic nanoclusters are categorized into three types, (i) dimeric, (ii) rod-shaped, and (iii) oblate-shaped nanoclusters. For future research, we expect that anisotropic nanoclusters will provide exciting opportunities for tailoring the physicochemical properties and thus lead to new developments in applications.
Introduction
One of the fundamental goals of nanoscience is to design and synthesize size- and shape-controllable materials at the nanometer scale.1,2 It has been widely accepted that the physicochemical properties of metal nanoparticles (NPs) are highly dependent on their size and shape.3–7Starting from Faraday's time and later developments by Turkevich, Frens, Schmid,8–10et al., the preparation of colloidal gold NPs of monodispersity has long been of major interest to researchers.11 Ordered monolayers of alkanethiol-stabilized gold NPs with tunable interparticle spacing (by different lengths of alkane chains) were reported by Mulvaney and co-wokers.12 Gold NPs functionalized by thiol-terminated DNA led to major research in nanobiomedicine.13 A two-phase method was established by Brust et al., which stimulated interest in organic soluble Au NPs.14 The Murray group demonstrated that the mean size of Au core can be adjusted by Au![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :thiolate ratio as well as temperature and the reduction rate.15 In the meantime, Whetten et al. isolated a series of ultrasmall Au NPs with tight distributions and observed the molecular nature in the smaller cores of Au NPs.16,17 In 2004, the Tsukuda group successfully separated different Au clusters (∼1 nm, ranging from Au10 to Au39) protected by glutathione via polyacrylamide gel electrophoresis, and their chemical compositions were precisely determined by electrospray ionization mass spectrometry (ESI-MS).18,19 About three years later, crystallization was worked out and the total structures of Au102(SR)44 and [Au25(SR)18]− (SR = thiolate) nanoclusters (NCs) were solved by X-ray crystallography,20–22 thus, a new age began, so-called atomically precise nanochemistry.23–26 Since then, the research thrust on Au-SR NCs by the Jin group and others24,27–30 has led to the creation of a series of noble metal NCs of atomic precision.
:thiolate ratio as well as temperature and the reduction rate.15 In the meantime, Whetten et al. isolated a series of ultrasmall Au NPs with tight distributions and observed the molecular nature in the smaller cores of Au NPs.16,17 In 2004, the Tsukuda group successfully separated different Au clusters (∼1 nm, ranging from Au10 to Au39) protected by glutathione via polyacrylamide gel electrophoresis, and their chemical compositions were precisely determined by electrospray ionization mass spectrometry (ESI-MS).18,19 About three years later, crystallization was worked out and the total structures of Au102(SR)44 and [Au25(SR)18]− (SR = thiolate) nanoclusters (NCs) were solved by X-ray crystallography,20–22 thus, a new age began, so-called atomically precise nanochemistry.23–26 Since then, the research thrust on Au-SR NCs by the Jin group and others24,27–30 has led to the creation of a series of noble metal NCs of atomic precision.
Evolution from complexes to nanoclusters to nanoparticles
The metal (e.g., Au) NCs occupy a unique position in bridging the metal–organic complexes and regular metal NPs, revealing the important molecule-to-metal transition (Scheme 1). A model binuclear Au(I)–L complex—Au2(S2PH2)2—and its orbital interaction diagram for Au22+ with two S2PH2− ligands (no d orbitals are included) are illustrated in Scheme 1A (simplified from the original work).31 Substantial s and p mixing into the d block gives aurophilic interactions,34 which is responsible for the tendency of Au(I) ions to clustering. Note that there is no metal–metal (M–M) bond in these Au(I)–L complexes. As the Au(I)–L precursor is reduced, Au–Au bonds form and give rise to a Au(0) kernel, which is the nucleation process at the initial stage of NP growth. To the surprise of nanochemists, at such an early stage, NCs with tens to hundreds of metal atoms (ultrasmall NPs) can also be very stable due to the passivation by ligands which are strongly bonded to the metal surface, as well as their close-shelled geometric structures (e.g., magic numbers) and/or electronic structures (e.g., superatoms).35,36 Taking the most thoroughly studied Au25(SR)18− NC as example, it has an icosahedral (Ih) Au13 kernel and six Au2(SR)3 staple-like motifs at the interface (Scheme 1B, bottom), and the metal core (Aukernel + Aumotif) is then wrapped inside an organic ligand shell. Almost all of the Au–thiolate NCs have such a kernel-motif structure, but for NCs without any surface motif (e.g., Au–phosphine NCs), only a metal core is inside the ligand shell. Note that the oxidation state of the Aukernel is not exactly 0, but between 0 and 1 with more Au(0) character. In addition, NCs have discrete energy levels with a distinct HOMO–LUMO gap (Eg > 0, HOMO = highest occupied molecular orbital, LUMO = lowest unoccupied molecular orbital) that are akin to molecules (Scheme 1B, top). A distinctive feature of NCs compared to complexes is that they have multiple free valence electrons to show unique optical properties with rich features (see Fig. 1).32 For example, isomeric Au30(SR)18 NCs (SR = adamantanethiolate) with face-centered-cubic (fcc) and hexagonal-close-packed (hcp) kernels have been obtained by the Jin group,37,38 and their absorption spectra are very different (Scheme 1C); note that the discussion on the fcc and hcp kernel structures is given in the section of “Rod-shaped nanoclusters”. Remarkably, a series of Au-SR NCs protected by the same adamantanethiol ligand have been achieved, which exhibit an atom-by-atom addition evolution and vivid colors, including Au16(SR)12 (orange), Au16Ag1(SR)13 (light green), Au21(SR)15 (red), Au22(SR)16 (light brown), Au22Cd1(SR)16 (turquoise), Au24(SR)16 (greenish brown), Au29(SR)19 (brown), Au30(SR)18-fcc (green), Au30(SR)18-hcp (magenta), and Au36Ag2(SR)18 (brown), although their sizes are very close (Scheme 1D). This phenomenon is different from the chalcogen-bridged silver or copper clusters (Ag(I) or Cu(I/II) sulfides/selenides) by Fenske et al., which often show less features in absorption,39,40 or the chalcogenide quantum dots with tunable band gaps by changing the composition,33,41,42 or the metal NPs with surface plasmon resonance which, for example, shifts from 520 to 570 nm as the diameter of spherical Au NPs increases from ∼10 to ∼100 nm (Scheme 1G).24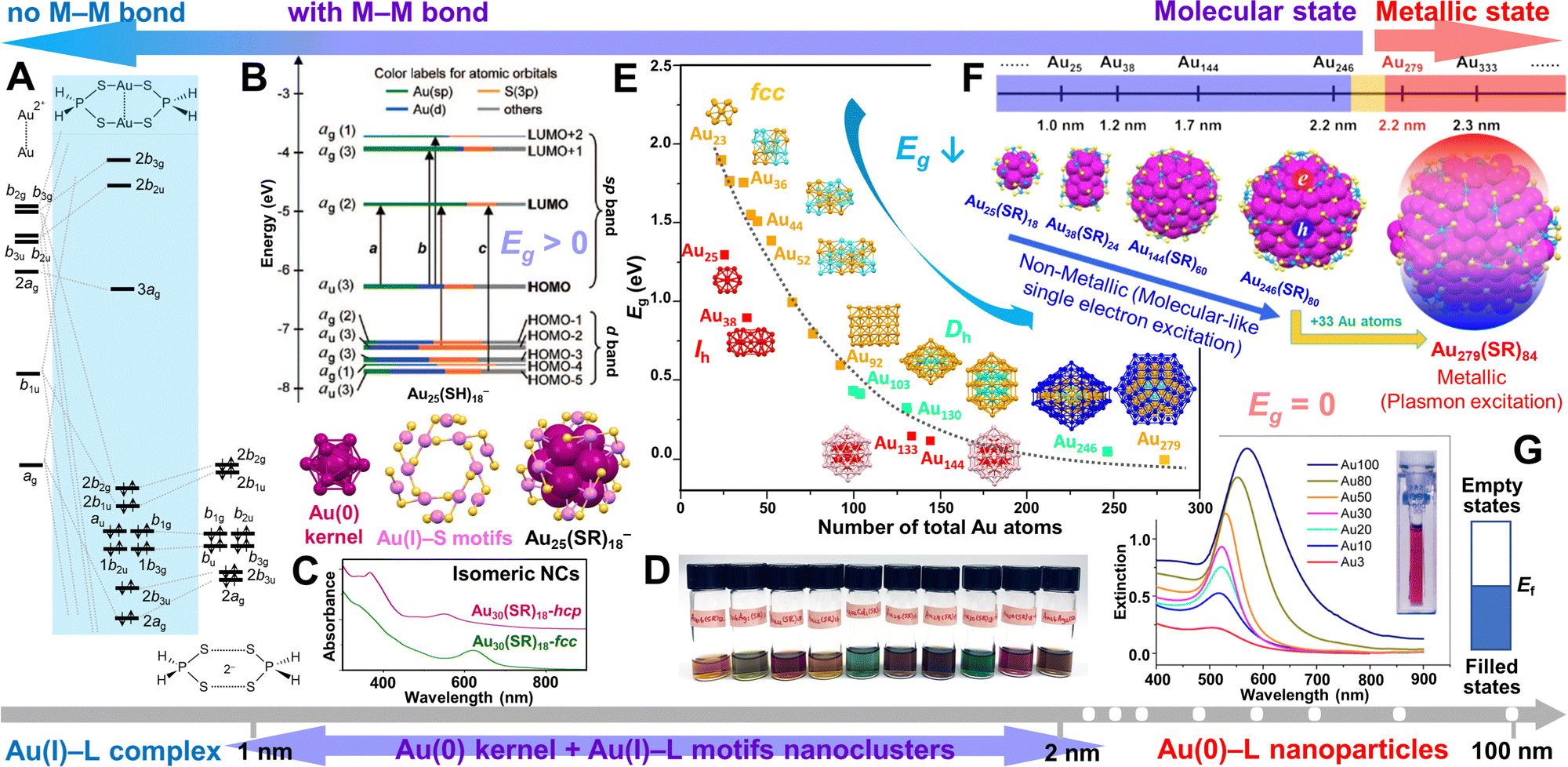 | ||
| Scheme 1 The size evolution from Au(I)–L complexes, to Au(0/I)–L NCs, then to Au(0)–L NPs. (A) Binuclear Au(I)–L complex Au2(S2PH2)2 and its orbital diagram.31 Redrawn by the authors. (B) The crystal structure of Au25(SR)18− NC and its molecular-like electronic structure with HOMO–LUMO gap (Eg) > 0. Reproduced with permission.21 Copyright 2008, American Chemical Society. (C) The optical absorption spectra of isomeric Au30(SR)18-hcp and Au30(SR)18-fcc (the spectra are offset to avoid overlap). (D) The photographs of a series of adamantanethiol-protected NCs in CH2Cl2, from left to right: Au16(SR)12, Au16Ag1(SR)13, Au21(SR)15, Au22(SR)16, Au22Cd1(SR)16, Au24(SR)16, Au29(SR)19, Au30(SR)18-fcc, Au30(SR)18-hcp, and Au36Ag2(SR)18. (C/D) Data collection performed by Y. L. (E) Eg of Au-SR NCs with Ihversus fcc and decahedral kernels as a function of the total number of Au atoms in NC. Reproduced with permission.32 Copyright 2021 Wiley-VCH GmbH. (F) The size dependence of the electronic properties in atomically precise gold nanoparticles with a sharp transition from non-metallic Au246(SR)80 (Eg > 0) to metallic Au279(SR)84 (Eg = 0). Reproduced with permission.33 Copyright 2021 The Author(s). (G) Optical extinction spectra of gold colloids (3–100 nm diameters), inset: photograph of 10 nm Au colloid, and the continuous band electronic structure of metallic-state NPs (Ef = Fermi level). Reproduced with permission.24 Copyright 2016 American Chemical Society. | ||
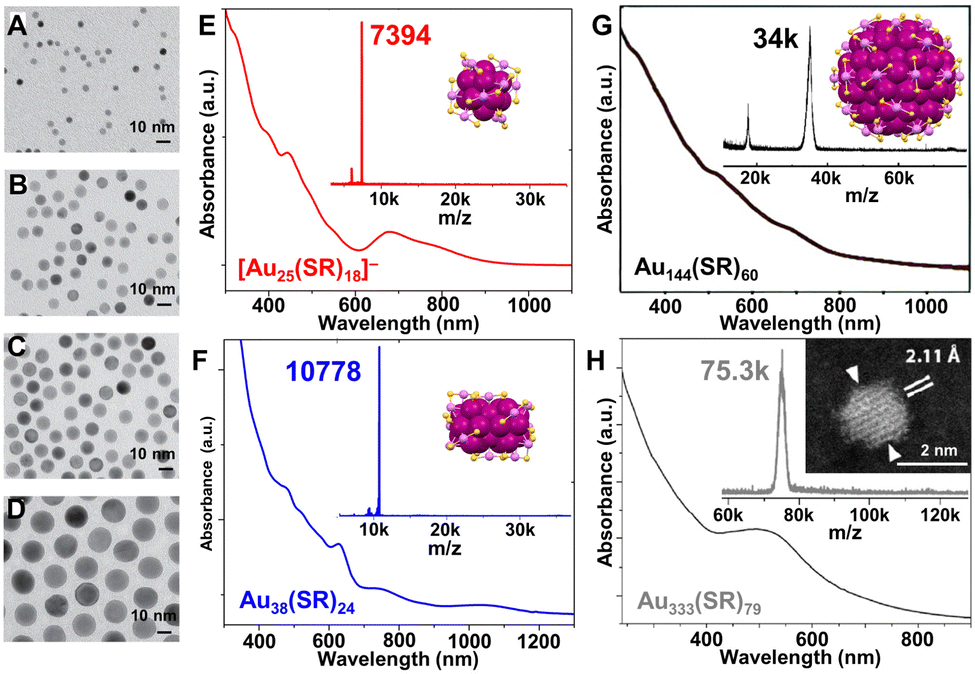 | ||
| Fig. 1 TEM images of single-crystalline Au nanospheres with controlled average diameters (A–D) 5, 8, 10, and 16 nm, by adding (A–D) 1000, 500, 50, and 20 μL Au seed solution, respectively. In Au seed solution preparation, the molar ratio of Au:ligand:reductant = 1:400:2.4. Reproduced with permission.67 Copyright 2013, WILEY-VCH GmbH. UV-vis spectra, MALDI-MS spectra, and corresponding crystal structures or scanning-TEM image of (E) [Au25(SR)18]−; (F) Au38(SR)24; (G) Au144(SR)60; and (H) Au333(SR)79 NCs. (E and F) Data collected by Y. L.; (G) reproduced with permission.69 Copyright 2022, The Authors, published by the Royal Society of Chemistry. (H) Reproduced with permission.70,71 Copyright 2012/2019, The Authors, published by the National Academy of Sciences USA. Color codes: magenta/violet = Au, yellow = S, R groups are omitted for clarity. | ||
Noble metal NCs show multiple absorption peaks and their energy gaps are very sensitive to and highly tunable by size (i.e., the number of metal atoms), composition (e.g., doping or alloying) and structure (e.g., shape).24–29 In general, the energy gap of atomically precise NCs decreases when the size of the NCs increase (Scheme 1E).43 As the Au(0) kernel grows larger, however, the number of Au atoms in the Au(I)–S motifs does not change much: there are 12 Aumotif atoms in Au25(SR)18, and still only 40 Aumotif atoms in the much larger Au246(SR)80, indicating the Au(0)/Au(I) ratio increases when the NC becomes larger. Moreover, with atomically precise nanochemistry, a sharp transition from semiconducting Au246(SR)80 to metallic-state Au279(SR)84 with only 33 Au atoms in the growth (Scheme 1F) has been observed.33,44 The Au279(SR)84 NC shows a quasicontinuous band electronic structure as in metallic-state NPs. With the enlightenment by atomically precise NCs, we believe that the regular Au NPs should also have staple-like Au(I)–L surface structures although they cannot be fully resolved by transmission electron microscopy due to insufficient resolution and image contrast. But since the number of motif Au(I) atoms are quite small compared to the large Au(0) kernel, regular Au NPs are regarded as all Au(0) atoms of metallic state (Eg = 0, no energy gap between the conduction band and valence band), with tunable plasmon resonances as the size changes (Scheme 1G).
The advantages of achieving NCs at the atomically precise level are obvious: (1) although transmission electron microscopy (TEM) can easily determine the size and shape of NPs, analyze their chemical compositions, crystalline structures, and valence states, insufficient image contrast limits the identification of organic monolayer on the particle surface. By contrast, the inter- or intra-particle interactions between ligands—which are especially important in elucidating the self-assembly in supercrystals—can be clearly determined by X-ray crystallography for atomically precise NCs;38,45–48 (2) for both quantification of adsorption and understanding of the catalytic mechanism, revealing the nature and structure of the active site(s) is critical since the interactions of active sites with substrates (i.e. reactants) can only be understood when the active site structure is known;30,49–52 (3) the organic–inorganic interface of the nanostructure which is crucial for resolving the surface science can be fully unveiled with the total structure (metal core + ligand shell) solved, showing that the intrinsic origin of chirality of metal NCs is the chiral arrangements of interface atoms and motifs;53–56 (4) not only a sharp transition from non-metallic Au246(SR)80 to metallic Au279(SR′)84 is demonstrated,44 but also the energy gaps of NCs are found to be related to different atomic packing modes (e.g. Ihvs. fcc or decahedral (Dh) kernels) due to the different distribution of electron wavefunctions in the NCs (Scheme 1E).32,43
A brief introduction concerning the synthetic evolution from size controllable Au NPs to Au NCs is given. Note that there have been excellent reviews on noble metal NP syntheses,57–62 thus in this Review, only selected works relating the size control of Au NPs are highlighted in order to introduce how atomically precise Au-SR NCs of different sizes are produced. For Au NPs larger than 5 nm, a digestive ripening process in excess thiol developed by Lin et al. resulted in greatly narrowed size distribution and long-range-ordered superlattices.63 Based on the seed-growth procedure—which has been a popular technique for decades,64,65 Jana and Murphy prepared Au NPs in the range of 5–40 nm, of which the size can be manipulated by varying the ratio of seed to metal salt.66 The size of single crystalline Au nanospheres could also be controlled by altering the size or volume of the seeds.67 As shown in Fig. 1A–D, the diameter of the Au nanospheres could be varied conveniently by changing the volume of the seed solution added to the reaction mixture. The precursor ratio to make size-controlled Au NPs9 gives an opportunity to develop an advanced kinetic control for making atomically precise NCs.24 In the typical Brust–Schiffrin method for Au–thiolate NPs, the Au:ligand:reductant molar ratio was 1:1.1:0.1.14 By contrast, in the one-pot method to produce the atomically precise Au25(SR)18 NCs (SR = C2H4Ph), the molar ratio of Au:PhC2H4SH:reductant was 1:5:10.68 The optical absorption spectrum, MALDI-mass spectrometry (MS) spectrum, and the corresponding single crystal structure of anionic Au25(SR)18 NC are shown in Fig. 1E.21
As indicated by the above comparison, the successful synthesis of atomically precise Au-SR NCs usually go through two steps: (1) excess thiol is used to convert AuIII into AuI-SR complexes (or polymers), and (2) a great excess of NaBH4 is used to reduce AuI-SR to Au(0). Moreover, the size control of atomically precise NCs is no longer dependent on the seed size or its concentration since they are at the scale of seed size (i.e., ∼1–3 nm in diameter).72,73 Then, how can one obtain NCs of different sizes in a controllable manner just like what was achieved in the plasmonic NPs? The Jin group provided an early strategy—size-focusing—to create a chemical environment for the formation of an exclusive size of Au-SR NC while suppressing other sizes.74 Specifically, the mass range of the initial product (i.e., Aux(SR)y mixture) was first kinetically controlled by Au:thiol ratio, temperature, reducing rate, etc., and then the Aux(SR)y nanocluster mixture was etched at elevated temperatures and with excess thiol (i.e., thermodynamic selection for the most stable size), and finally a specific size such as Au38(SR)24 of molecular purity was achieved (Fig. 1F).75,76 The Au144(SR)60 NCs were obtained by etching for 24 h to eliminate impurities of small sizes (Fig. 1G),77 and the total structure of Au144(SR)60 was later solved by using HSCH2Ph instead of HSC2H4Ph in the initial synthesis (Fig. 1G, inset).78 When adjusting the Au:thiol molar ratio from 1:3 (for the preparation of Au144(SR)60) to 1:2, larger sized Au333(SR)79 was made (Fig. 1H).70,71 Other methodologies, including ligand-exchange-induced size and/or structure transformation,79–84 ligand substituent group hindrance induced size selection,85–88 heteroatom doping induced size/structure transformation, metal exchange or anti-galvanic reduction,89–92 have been developed to enrich the library of noble metal NCs during the past two decades.
The unique chemical and physical properties, especially the optical properties, of plasmonic NPs are not only determined by the size, but also by the shape of the core. Shape-controlled syntheses of NPs,93–97 especially the anisotropic structures with gold nanorods as the most thoroughly studied one98–102 were obtained in a convenient way around 2000. Different from the less tunable plasmon peak wavelength range of isotropic Au NPs of 4–200 nm, anisotropic Au nanorods permit one to drastically tune the longitudinal plasmon wavelength from the visible to near-infrared range by controlling the aspect ratio (AR) of rods. Over the past two decades, researchers have frequently taken advantages of such specific phenomena to enable sensing applications,103–105 plasmon-enhanced spectroscopies,93,106–109 bio-imaging,110,111 and photothermal therapy.112–117
The same trend has also been seen in the relatively short history of atomically precise metal–thiolate NCs, in which isotropic structures were obtained at the beginning of research since the spherical structures have less surface energy, that is, higher stability.20,21,77,118 Fortunately, with the advances in synthetic methods, we have witnessed more and more cases on non-spherical NCs. Spheroidal shapes (Scheme 2) are found in large-sized NCs with more than 100 metal atoms. For example, Au130(SR)50 is the first reported NC to have multiple concentric shells with a barrel-shaped structure,88 but only with a small aspect ratio (AR = 1.13). The Ag@Au17@Ag27@Au17(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CR)34 structure exhibits a spheroidal shape with AR = 1.26, and this Au34Ag28(CCR)34 NC shows much better catalytic performance in its intact form than the partially or completely ligand-removed NCs.119 AR of 1.17 is found in the Au156(CCR)60 NC, which is in the transition region from molecular to metallic state.120 These spheroidal shapes are at the borderline between isotropic and anisotropic structures, and will not be the focus in this review. Larger AR values are highly desirable from the shape effect point of view.
CR)34 structure exhibits a spheroidal shape with AR = 1.26, and this Au34Ag28(CCR)34 NC shows much better catalytic performance in its intact form than the partially or completely ligand-removed NCs.119 AR of 1.17 is found in the Au156(CCR)60 NC, which is in the transition region from molecular to metallic state.120 These spheroidal shapes are at the borderline between isotropic and anisotropic structures, and will not be the focus in this review. Larger AR values are highly desirable from the shape effect point of view.
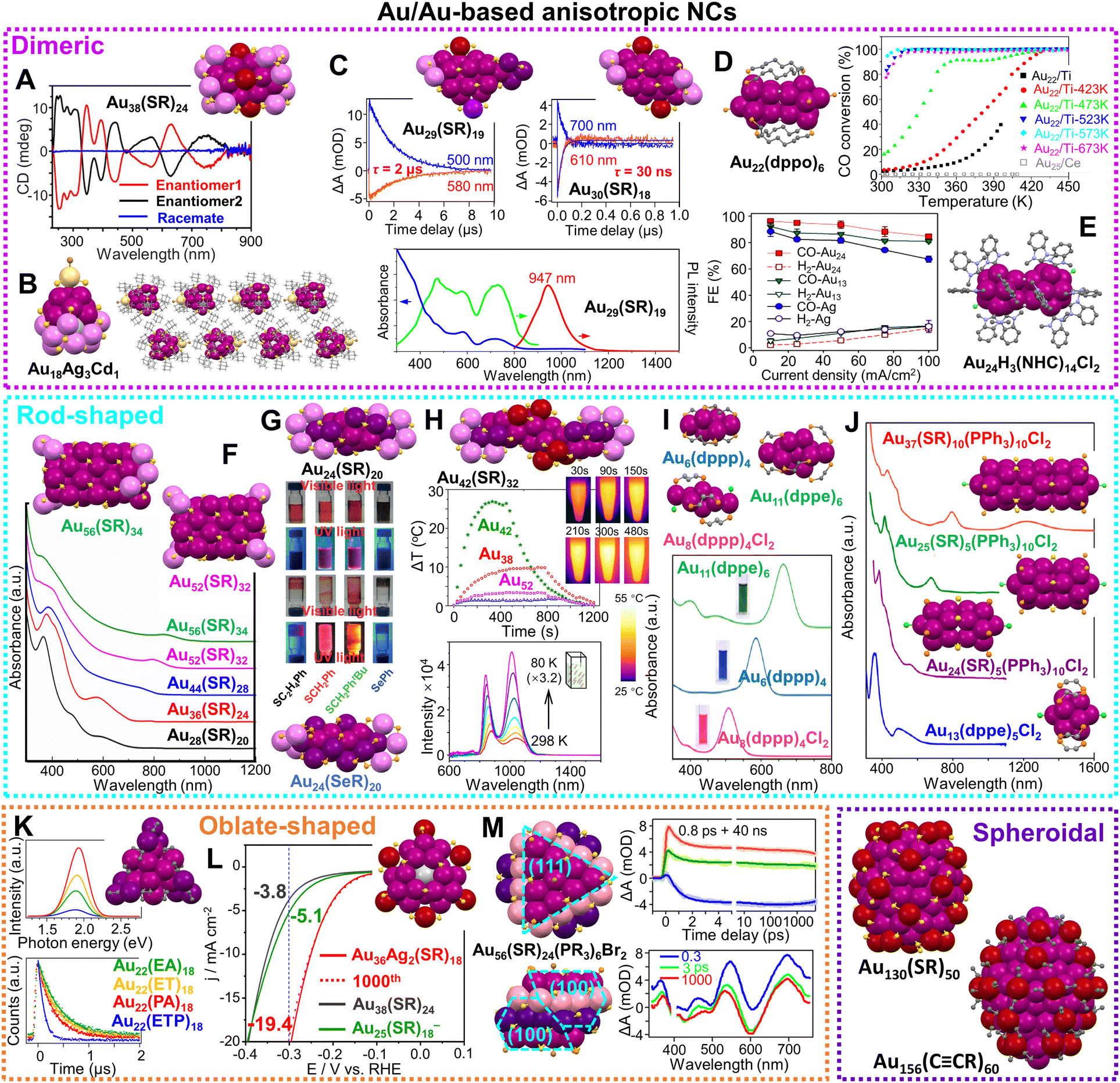 | ||
| Scheme 2 Selected anisotropic Au and Au-based NCs of atomic precision and their corresponding properties. Dimeric structures include: (A) Au38(SR)24 and the circular dichroism spectra of its two enantiomers. Reproduced with permission.121 Copyright 2013 Wiley-VCH GmbH; (B) Au18Ag3Cd1(SR)15Br and its assembly in supercrystals.122 Redrawn by the authors; (C) kinetic traces of nanosecond transient absorption for Au29(SR)19 and Au30(SR)18, and spectra of absorbance, emission and excitation for Au29(SR)19. Reproduced with permission.38 Copyright 2021 Springer Nature Limited; (D) Au22(dppo)6 and the CO oxidation of Au22(dppo)6/TiO2 catalyst pretreated in O2 at different temperatures. Reproduced with permission.123 Copyright 2016 American Chemical Society; (E) Au24H3(NHC)14Cl2 and its electrochemical CO2 reduction performance at different current densities. Reproduced with permission.124 Copyright 2022 American Chemical Society; rod-shaped structures include: (F) the fcc series and absorption spectra. Reproduced with permission.125 Copyright 2020 Wiley-VCH GmbH; (G) Au24(SR)20 and Au24(SeR)20 and the photographs of their solutions or solids under visible and UV light irradiation. Reproduced with permission.126 Copyright 2016 Wiley-VCH GmbH; (H) Au42(SR)32 and its temperature changes upon irradiation compared to Au38(SR)24, Au52(SR)32, Au24(SR)20, and time-dependent images of Au42(SR)32 solution upon irradiation; temperature-dependent PL spectra of Au42(SR)32 solution. Reproduced with permission.127,128 Copyright 2022 American Chemical Society; (I) Au6(dppp)4, Au8(dppp)4Cl2, Au11(dppe)6 and their absorption spectra. Reproduced with permission.129 Copyright 2013 American Chemical Society; (J) Au37(SR)10(PPh3)10Cl2, Au25(SR)5(PPh3)10Cl2, Au24(SR)5(PPh3)10Cl2, Au13(dppe)5Cl2, and their absorption spectra. Reproduced with permission.130 Copyright 2017 National Academy of Science. Oblate-shaped structures include: (K) Au22(CCR)18 and its PL spectra and lifetimes with different ligands. Reproduced with permission.131 Copyright 2019 American Chemical Society; (L) Au36Ag2(SR)18 and its HER electocatalytic activity compared to Au38(SR)24 and Au25(SR)18−. Reproduced with permission.132 Copyright 2021 American Chemical Society; (M) Au56(SR)24(PR3)6Br2 and the kinetic traces of its transient absorption, and corresponding fits at selected wavelengths, and spectra at selected time-delays. Reproduced with permission.133 Copyright 2022 The Author(s). Spheroidal structures include: Au130(SR)50 and Au156(CCR)60. The structures are redrawn from the cif in ref. 88 and 120. | ||
Herein, we focus on the work on noble metal NCs of atomic precision with anisotropic structures in the past 15 years to highlight their distinctive chemical and physical properties. Some illustrations are collected in Scheme 2. We divide the anisotropic NCs into three categories—dimeric structures, rod shapes and oblate (or disk-shaped) structures. Homoleptic Au and Au-based NCs (ligands including thiol, phosphine, and alkynyl) will be our primary coverage, whereas multileptic NCs, and Ag- and Cu-based NCs will be selectively discussed as supplementary cases. Note that we eliminate the charges and counter ions on most of the NCs in the Review for the simplicity of the formulas, unless those specifics become necessary in the discussion of electronic properties of the anisotropic NCs.
Dimeric nanoclusters
Plasmonic Au NP dimers and trimers can be constructed and separated in high purity,134 and such nanostructures have been used for surface-enhanced Raman spectroscopy.135 In terms of structural control, such dimers and trimers are formed by NPs attracting each other without any bond (i.e., with a subnanometer or larger gap between NPs). By contrast, when dimerization or trimerization occurs in atomically precise NCs, monomeric units are linked by metal–metal bonds inside a single particle. Thus, NC dimers can be regarded as the simplest anisotropic structures.Y. Zhu et al. found that two Au25(SR)18 (SR = SC2H4Ph) monomers could be linked by two Ag atoms to form a Au50Ag2(SR)36 dimer (Fig. 2A, left).136 Although the optical absorption spectrum does not show distinct differences when two monomers are bridged (Fig. 2A, right), normalized partial density of states diagrams indicate that AgI has an influence on the HOMO of the dimer, causing band broadening. Moreover, the Au50Ag2(SR)36/CeO2 catalyst exhibited enhanced activity for CO oxidation compared to Au25(SR)18/CeO2, while the catalyst based on Au25−xAgx(SR)18 NCs had much less activity (Fig. 2B).136
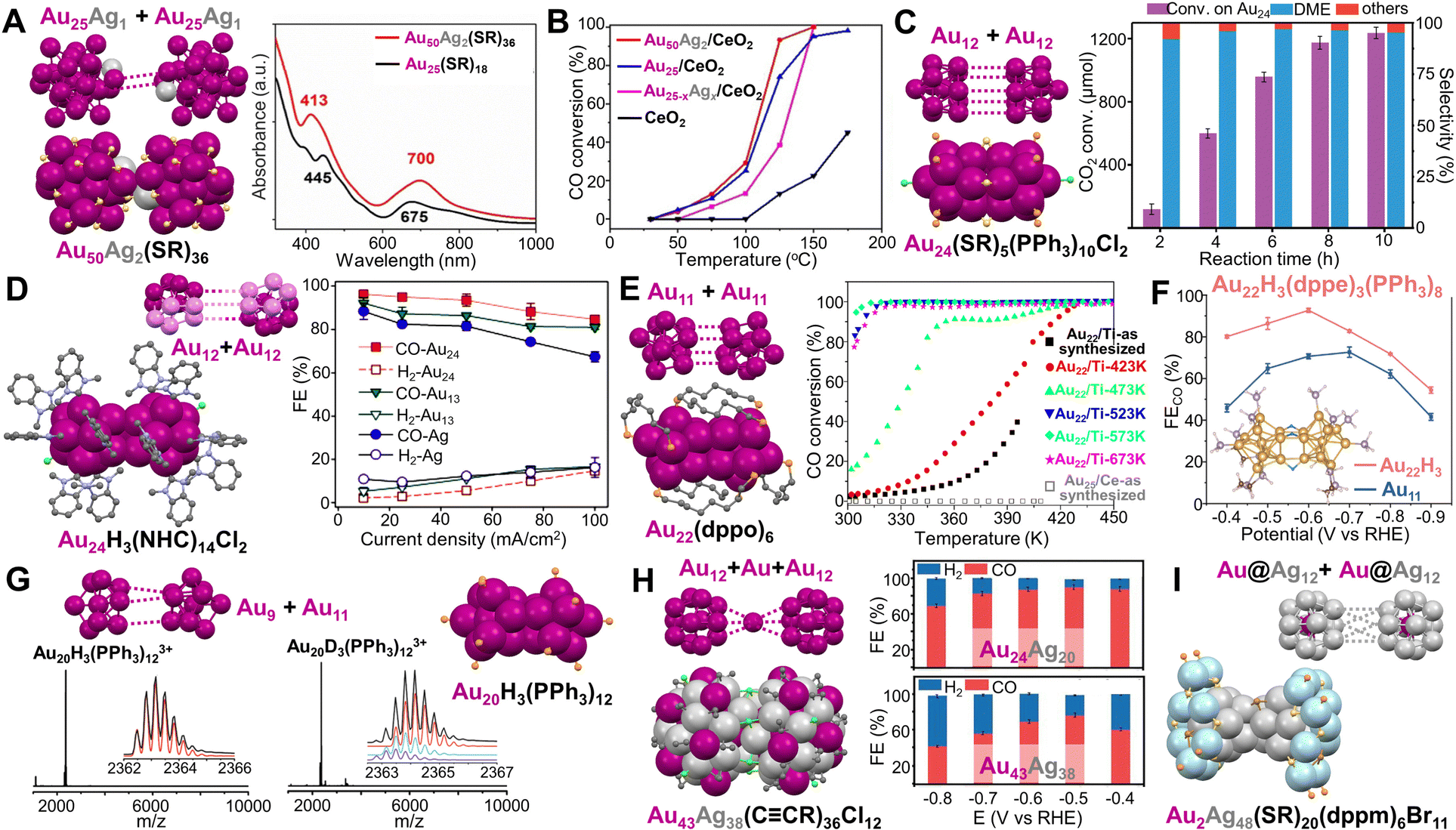 | ||
| Fig. 2 (A) Dimeric Au50Ag2(SR)36 and its UV-vis spectrum compared to monomeric Au25(SR)18. (B) CO oxidation profiles of Au50Ag2(SR)36, Au25(SR)18, and Au25−xAgx(SR)18 catalysts supported on CeO2. Reproduced with permission.136 Copyright 2020, Wiley-VCH GmbH. (C) Au24(SR)5(PPh3)10Cl2 and the CO2 hydrogenation on Au24/SiO2 catalysts with respect to the reaction time. Reaction conditions: Au24/SiO2 (0.5% Au) 100 mg, 2 MPa reaction gas (CO2:H2 = 1:3) with 5% N2, H2O 15 mL, 130 °C. Reproduced with permission.137 Copyright 2020, American Chemical Society. (D) Au24H3(NHC)14Cl2 and its electrochemical CO2 reduction performance compared to Au13(NHC)9Cl3 NCs and Ag NPs at different current densities. Reproduced with permission.124 Copyright 2022, American Chemical Society. (E) Au22(dppo)6 and the CO oxidation curves of the Au22(dppo)6/TiO2 catalysts pretreated in O2 at different temperatures. Reproduced with permission.123 Copyright 2016, American Chemical Society. (F) Electrocatalytic reduction of CO2 to CO based on Au22H3(dppe)3(PPh3)8 catalyst compared to Au11(dppe)5, inset: the theoretical structure of Au22H3(dppe)3(PPh3)8. Reproduced with permission.138 Copyright 2022, American Chemical Society. (G) The structure and mass spectra of Au20H3(PPh3)12 and Au20D3(PPh3)12. Reproduced with permission.139 Copyright 2020, The Royal Society of Chemistry. (H) Au43Ag38(CCR)36Cl12 and its CO2 reduction performance compared to Au24Ag20(CCR)24Cl2. Reproduced with permission.140 Copyright 2022, The Author(s). (I) Au2Ag48(SR)20(dppm)6Br11. Color codes: magenta/violet = Au, light grey/light blue = Ag, light orange = P, yellow = S, green = Cl, brown = Br, light purple = N, and grey = C, R groups are omitted for clarity. | ||
Heteroligand-protected NCs composed of two connected monomeric units without staple motifs have been studied as well, and such NCs also show interesting catalytic properties. In the process of making rod-shaped Au25(SR)5(PPh3)10Cl2 (SR = SC2H4Ph), adding excess PPh3 resulted in Au24(SR)5(PPh3)10Cl2 consisting of two Au12 units (one vertex missing) joined together in an eclipsed manner (Fig. 2C, left).141 Further work indicated that the internal vacancy (i.e., the missing vertex) could provide Au24(SR)5(PPh3)10Cl2 with more structural flexibility and mitigate the deactivation of the Au24/SiO2 catalysts which efficiently converted CO2 to dimethyl ether (DME) (Fig. 2C, right).137 When the open pentagonal faces of the two incomplete Ih Au12 units rotated by ∼120° with respect to each other, an N-heterocyclic carbene (NHC) stabilized NC—Au24H3(NHC)14Cl2—was obtained by the Crudden group (Fig. 2D, left).124 Three bridging hydrides were not resolved by X-ray analysis but were predicted to be located at the connection points by density-functional theory (DFT). Using a membrane electrode assembly electrochemical cell in which the cathode and anode were pressed against an anion exchange membrane, Au24H3(NHC)14Cl2 was tested for CO2 electroreduction to CO, and the dimeric NC-based catalyst exhibited higher selectivity (i.e., faradaic efficiency) than the catalysts of monomeric Au13(NHC)9Cl3 and benchmark Ag NPs at all tested current densities (Fig. 2D, right).124
L.-S. Wang et al. reported a Au22(dppo)6 (dppo = Ph2P(CH2)8PPh2) NC with two Au11 units clipped together by four dppo ligands (Fig. 2E, left).142 Low-temperature CO oxidation activity was observed for the Au22(dppo)6/TiO2 catalyst pretreated in O2 at different temperatures without ligand removal (Fig. 2E, right).123 Recently, the group found that Au22(dppo)6 gradually transformed via H evolution from Au22H4(dppo)6 of which the tetrahydrides were confirmed by electrospray ionization MS and DFT calculations.143 In another work, three hydrides were determined on heteroleptic Au22H3(dppe)3(PPh3)8 (dppe = Ph2P(CH2)2PPh2) by ESI-MS and 1H-NMR, and the structure was proposed to have two Au11 units connected at six Au sites and bridged by three H (Fig. 2F, inset). The six bridged Au atoms in the trihydrido-NC were found to be critical in catalyzing electrochemical CO2 reduction to CO, and a 92.7% faradaic efficiency at −0.6 V vs. RHE and high activity (134 A/gAu mass activity) were obtained (Fig. 2F).138 Beside the homodimeric Au NCs, the Q.-M. Wang group demonstrated an Au20H3(PPh3)12 NC with a heterodimeric core, i.e., an Au9 unit was connected to an Au11 unit (Fig. 2G). The presence of trihydrides on Au was probed by ESI-MS and 1H-NMR, and the hydrido-Au NC showed good stability for long-time storage.139
When two hollow Ih Au12 units are linked by one additional Au atom to form a dimeric kernel which is enclosed by a bimetallic shell, an Au43Ag38(CCR)36Cl12 NC is obtained (Fig. 2H, left). Compared to Au24Ag20(CCR)24Cl2 with a monomeric kernel and a superatomic electronic configuration, the Au43Ag38(CCR)36Cl12 NC shows less activity in catalytic reduction of CO2 to CO (Fig. 2H, right).140 This is in contrast to the observations in the hyrido-Au cases, i.e., dimeric Au22H3(dppe)3(PPh3)8 and Au24H3(NHC)14Cl2 are better than their monomeric counterparts (Fig. 2D and F). We suppose that the hydrides and chlorides at the bridging sites are responsible for the catalytic difference. Besides, two Au@Au12 superatomic units (1S21P6) can also be combined into a dimeric kernel which is protected by different surface Ag motifs to form [Au2Ag42(SR)27]+ and [Au2Ag48(SR)20(dppm)6Br11]3+, respectively (dppm = Ph2PCH2PPh2, Fig. 2I).144 DFT indicates the valence electronic structures of both NCs are analogous to that of Ne2.
Another category of dimeric NCs has kernels formed by fusion of two Ih monomeric units. Jin et al. first solved the total structure of Au38(SR)24 (SR = SC2H4Ph) with a dimeric kernel as another benchmark NC76 (in addition to the Au25(SR)18 in earlier work). Since then, Au38(SR)24 remains actively studied to establish extensive structure–property correlations in atomically precise nanomaterials. The dimeric Au23 kernel is composed of two Ih Au13 units fused together via sharing a common Au3 face, i.e., Au13 + Au13 – Au3. The Au23 kernel is protected by six Au2(SR)3 and three Au3(SR)4 staples in a chiral configuration (Fig. 3A),76 consistent with the DFT calculations by Aikens et al.145 Compared to the superatom model of (1S)2(1P)6(1D)6 for a 14e− NC, a theoretical study based on supervalence bond (SVB) model does a better job in demonstrating that the electronic configuration of the Au23 kernel is (1Σ)2(1Σ*)2(1Π)4(2Σ)2(1Π*)4, where each orbital is created by the bonding and antibonding interactions between the 1S and 1P superatomic orbitals of the two Ih Au13 units.146,147 In this way, the SVB model relates the Au23(14e−) kernel to an isoelectronic analog of the F2 molecule (Fig. 3B). Based on the structure of Au38(SR)24, a systematic ligand-exchange study revealed that ortho-substituted benzenethiols could preserve the Au38(SR)24 structure, while para- or non-substituted benzenethiols would cause the transformation of Au38(SR)24 into Au36(SR′′)24.148 In Au38(SC2H4Ph)24, the six Au2(SR)3 motifs resemble two tri-blade fans at the top and bottom, being arranged in a staggered conformation by ∼60° to each other (Fig. 3C, left);76 by contrast, when applying an ortho-substituted aromatic thiol, the crystal structure of Au38(SR′)24 (SR′ = SPh-2,4(CH3)2) shows that one fan rotates only by ∼45° relative to the other along the C3 axis (Fig. 3C, right). As a result, each Au(SR′)2 motif at the waist is parallel to the two adjacent Au2(SR′)3 motifs of the Au38(SR′)24 structure (Fig. 3C, indicated by blue lines).148 The dimeric Au38(SR)24 NC shows a series of absorption peaks, with the first band (i.e., the lowest energy) centered at 1050 nm (see Fig. 1F),75 which is significantly red-shifted from that of Au25(SR)18 with a mono-Ih Au13 kernel (its first absorption band at 780 nm).149
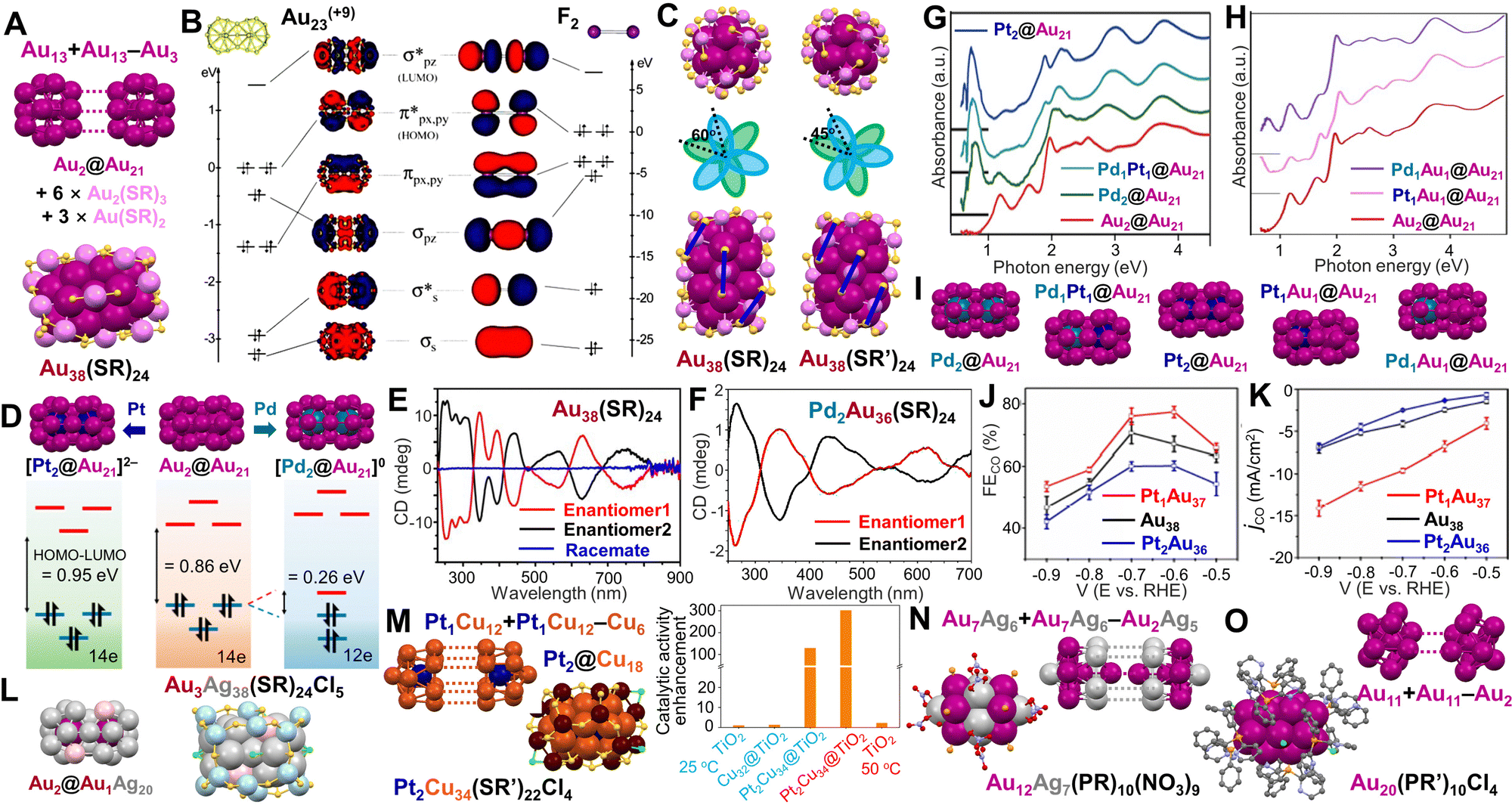 | ||
| Fig. 3 (A) The face-fused Au2@Au21 kernel in Au38(SR)24. (B) Comparison of the Kohn–Sham orbital diagrams between Au23(+9) (left) and F2 (right). Reproduced with permission.146 Copyright 2013, The Royal Society of Chemistry. (C) Top and side views, and the schematic diagram of two tri-blade ‘fans’ of Au38(SR)24 (left) and Au38(SR′)24 (right). Reproduced with permission.148 Copyright 2020, The Royal Society of Chemistry. (D) DFT-calculated electronic energy levels of [Pt2Au36(SCH3)24]2−, Au38(SCH3)24, and [Pd2Au36(SCH3)24]0. Reproduced with permission.154 Copyright 2018, American Chemical Society. (E) Circular dichroism spectra of two enantiomers and the racemic Au38(SR)24. Reproduced with permission.121 Copyright 2012, Macmillan Publishers Limited. (F) Circular dichroism spectra of two enantiomers of Pd2Au36(SR)24. Reproduced with permission.156 Copyright 2014, American Chemical Society. (G) Absorption spectra of Pd2Au36(SR)24, PdPtAu36(SR)24, Pt2Au36(SR)24 and Au38(SR)24 as a function of photon energy. Reproduced with permission.157 Copyright 2020 Wiley-VCH GmbH. (H) Absorption spectra of Pd1Au37(SR)24, Pt1Au37(SR)24 and Au38(SR)24 as a function of photon energy. Reproduced with permission.158 Copyright 2022, The Authors. (I) The 23-metal kernels with different dopants. (J) CO faradaic efficiency and (K) CO partial current density for Pt1Au37(SR)24, Au38(SR)24, and Pt2Au36(SR)24 catalysts at different applied potentials. Reproduced with permission.159 Copyright 2022, Wiley-VCH GmbH. (L) The Au2@Au1Ag20 kernel of Au3Ag38(SR)24X5.163 (M) The Pt2@Cu18 kernel in Pt2Cu34(SR′)22Cl4, and the enhanced catalytic activity of Pt2Cu34@TiO2 for silane oxidation to silanol. Reproduced with permission.160 Copyright 2021, American Chemical Society. (N) The fusion of Au12Ag7 core in Au12Ag7(PR)10(NO3)9. (O) The fusion of Au20 core in Au20(PR’)10Cl4. Color codes: magenta/violet = Au, dark cyan = Pd, dark blue = Pt, light grey/light blue = Ag, pink = Au/Ag, orange/brown = Cu, yellow = S, light orange = P, green = Cl, red = O, light purple = N, and grey = C. R groups are omitted or simplified for clarity. | ||
In light of the successful synthesis and crystallization of Au38(SR)24, heteroatom doping was investigated subsequently. Co-reducing AuI-SR and AgI-SR, followed by thermo-etching with excess thiol, Au38−xAgx(SR)24 was resulted (x = 1–6, SR = SC2H4Ph) with the 1050 and 750 nm absorption bands blue-shifted and diminished at higher Ag dopants.150 Crystallography shows that the doped Ag atoms are distributed in the kernel on the middle fusion plane and the two ends.151 By contrast, Au38−xCux(SR′)24 (x = 0–6, SR′ = SPh–2,4(CH3)2) obtained by a ligand exchange method demonstrates that all Cu atoms are selectively doped in the trimeric motifs, and the optical absorption spectrum remains the same as that of the homogold counterpart. This is reasonable since Cu dopants contribute little to the frontier orbitals.152
Doping the group 10 elements (i.e., Pd, Pt) into Au38(SR)24 provides a more interesting story. In 2011, Negishi et al. used size exclusion chromatography to separate PdxAu38−x(SR)24 (x = 1, 2), which was then heated at 60 °C for 8 days to preferentially decompose the singly doped NC.153 The resulted pure Pd2Au36(SR)24 NC was found to be more stable than Au38(SR)24 against degradation.153 Lee et al. used a similar method to produce Au38(SR′′)24, [Pd2Au36(SR′′)24]0 and [Pt2Au36(SR′′)24]2− (SR′′ = SC6H13), and studied their optical and electronic properties.154 X-ray photoelectron spectroscopy (XPS) shows the Pd0 and Pt0 state, indicating that both dopants are located at the central positions of the bi-Ih kernel, in contrast to the observation of Pd–S bonds in the K-edge extended X-ray absorption fine-structure data.155 Moreover, the Pt-doped NC is dianionic as two associated cations are observed by NMR analysis.154 Voltametric investigations show that the HOMO–LUMO gap of [Pt2Au36(SR′′)24]2− (14e−) is 0.95 eV, slightly larger than the 0.86 eV gap of Au38(SR′′)24 (14e−); whereas [Pd2Au36(SR′′)24]0 shows a drastically decreased gap (0.26 eV) due to the Jahn–Teller distortion of the 12e− NC (Fig. 3D).
As mentioned above, the chiral arrangement of Au3(SR)4 motifs on the surface of Au23 kernel gives rise to the chirality of Au38(SR)24. Bürgi et al. first separated the two enantiomers by chiral high-performance liquid chromatography, showing mirror-image circular dichroism (CD) responses (Fig. 3E) of Au38(SR)24 with large anisotropy factors (up to 4 × 10−3).121 The enantiomers with two doped Pd atoms show significantly changed CD spectra compared to their parent NCs (Fig. 3F), although the anisotropy factors are of similar magnitude. However, the doped 38-metal NCs undergo racemization at a much lower temperature.156
Syntheses other than the original size-focusing method have also been developed to understand the formation of dimeric Au38(SR)24 NCs. Maran et al. found that the charge-neutral Au25(SR)18 NCs could undergo a spontaneous bimolecular fusion to form the dimeric Au38(SR)24 NC during a period of two weeks.161 The Tuskuda group developed a hydride-mediated transformation approach by reacting equivalent amounts of [HM1Au8(PPh3)8]+ (8e) and [M1Au24(SR)18]− (M = Pd/Pt), resulting in homodimeric Pd2Au36(SR)24 and Pt2Au36(SR)24 as well as heterodimeric Pd1Pt1Au36(SR)24 with crystal structures solved (Fig. 3G), proving that Pd or Pt atoms are doped at the centers of the individual icosahedra. The lowest-energy absorption bands of M2Au36(SR)24 (12e−) are determined to be ∼0.7 eV, and the electron configuration of (1Σ)2(1Σ*)2(1Π)4(2Σ)2(1Π*)2 corresponds to the O2 electron configuration.157 Very recently, the authors modified the method by mixing equivalent [HAu9(PPh3)8]2+ (8e) and [M1Au24(SR)18]− with excess Au(I)-SR oligomer, and after chromatography separation, heterodimeric M1Au37(SR)24 (13e−) was obtained.158 The optical absorption spectra of M1Au37(SR)24 show an additional band below 1 eV compared to that of Au38(SR)24 (Fig. 3H), which can be assigned to the electronic transition to a singly occupied molecular orbital. The 23-metal-atom kernel structures with different dopant(s) are shown in Fig. 3I. In the meantime, Y. Zhu et al. independently reported the same 13e− Pt1Au37(SR′′′)24 NC (SR′′′ = SCH2PhtBu) by traditional co-reduction and separation methods. More interestingly, Pt1Au37(SR′′′)24 with an unpaired electron displays a higher catalytic activity than that of Pt2Au36(SR′′′)24 in electroreduction of CO2, whereas Au38(SR′′′)24 is in between (Fig. 3J and K).159
Moreover, Au38(SR)24 has a structural isomer, denoted Au38(SR)24′ which possesses an Au23 kernel composed of one Ih Au13 and one Au12 cap fused together by sharing two Au atoms.162 Although Au38(SR)24′ is thermally less stable than the homodimeric Au38(SR)24, it shows much higher catalytic activity in reducing 4-nitrophenol to 4-aminophenol at 0 °C.
In addition to the NCs with Au-based bi-Ih kernels, Au3Ag38(SR)24X5 (X = Cl or Br, SR = SCH2Ph) with an Ag-based bi-Ih kernel was also reported.163 The two icosahedrons centrally doped with Au atoms are face-fused into a dimeric Au2@Au1Ag20 kernel (one Au is distributed at two sites (∼50%) on the surface of the kernel, Fig. 3L, left), which is further capped by two halogens at two ends and an outer Ag18(SR)24X3 shell (Fig. 3L, right). The NC also has the same 14e− configuration as the Au38(SR)24 system, and shows an emission peak at 827 nm and a shoulder band at 851 nm in solid state at room temperature. In another work, due to the distinct chemical reactivity of [Cu32H8(SR′)24Cl2]2− (SR′ = SC2H4Ph), Hyeon et al. reacted it with Na2PtCl6·6H2O under an Ar atmosphere to produce a [Pt2Cu34(SR′)22Cl4]2− (10e−) NC. Two Pt1Cu12 units are fused at the Cu6 hexagon, resulting in a rod-shaped Pt2Cu18 kernel inside a Cu16(SR′)22Cl4 shell (Fig. 3M). The synergistic effect of the bimetallic NC was found to enhance the catalytic activity by ∼300 fold in silane-to-silanol conversion under mild conditions.160
For phosphine-protected NCs, when two Ih Au7Ag6 units are fused by sharing an Au2Ag5 part, Au12Ag7(PR)10(NO3)9 (PR = PPh(CH3)2) with a nearly staggered Au5–Ag5–Au5 configuration is obtained (Fig. 3N).164 Another Au20(PR′)10Cl4 NC (PR′ = bis(2-pyridyl)phenyl-phosphine) has an Au20 core generated from the fusion of two Au11 units (incomplete icosahedron) via sharing two vertices (Fig. 3O).165
The formation of dimeric structures is elusive due to their ultrasmall sizes. But we noticed that many of the dimeric Ih NCs also have their corresponding monomeric Ih structures that can be separately synthesized to have superatomic character. Thus, the formation mechanism of dimeric NCs could be referred to the model of diatomic molecules or fusion growth.
All the above dimeric structures are formed by two Ih or quasi-Ih units which are easy to be identified. By contrast, understanding the formation of fcc nanostructures at the atomic level remains a major task. The Jin group used an atom-tracing strategy by heteroatom doping into Au30(SR)18 (SR = StBu) to label the specific positions in it, which is consistent with the Ag-doping site in Au20Ag1(SR)15, indicating that the 30-metal-atom NC is a homodimeric structure (Fig. 4A). Electronic orbital analysis by DFT also shows the intrinsic orbital localization at the two specific positions in M30(SR)18 (M = Au/Ag), regardless of Au or Ag occupancy. The mechanism is proposed as follows: first, Au16Ag1(SR)11 intermediates (its structure proposed from Au20Ag1(SR)15 by losing Au4(SR)4) are formed at the initial stage; then, two of them are fused in a face-to-face manner; after eliminating another Au4(SR)4 unit from the adduct, the dimeric Au28Ag2(SR)18 is achieved (Fig. 4A).166
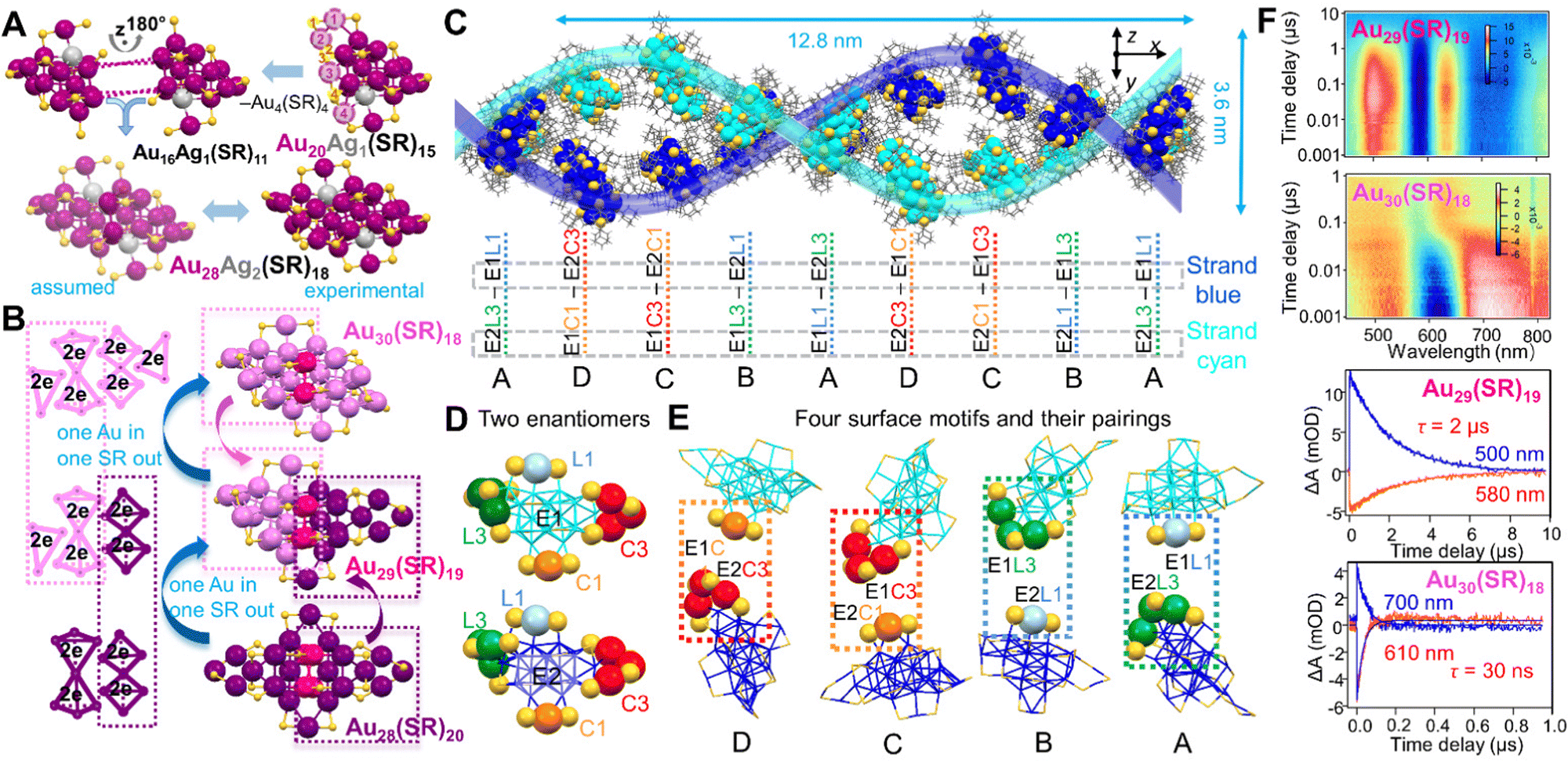 | ||
| Fig. 4 (A) Proposed formation of dimeric Au28Ag2(SR)18. Reproduced with permission.166 Copyright 2020, American Chemical Society. (B) Geometric and electronic structural evolutions achieved by the ‘one gold in, one thiolate out’ approach, from Au28(SR)20 to Au29(SR)19, then to Au30(SR)18. Color codes in (A/B): magenta/violet/dark pink/purple = Au, light grey = Ag, yellow = S, R groups are omitted for clarity. (C) Self-assembly of Au29(SR)19 NCs into a double-helical structure; (D) two enantiomers (E1 and E2) of Au29(SR)19 in the supercrystal; and (E) four types of motifs matching between neighboring enantiomers: E1C1–E2C3 (D type), E1C3–E2C1 (C type), E1L3–E2L1 (B type) and E1L1–E2L3 (A type). Color code in C/D/E: blue and cyan = Au in the different strands; yellow = S; grey = C. The four different staple motifs of Au29(SR)19 are marked with larger spheres of different colors. (F) Nanosecond transient absorption data and kinetic traces for Au29(SR)19, and Au30(SR)18. λex = 400 nm. Reproduced with permission.38 Copyright 2021 Springer Nature Limited. | ||
The homodimeric structure of fcc Au NCs has long been overlooked until the report on heterodimeric fcc NCs. Before being experimentally proved, Pei et al. proposed in 2017 a “Au insertion, SR elimination” mechanism based on the structures of Au30(SR)18 and Au28(SR)20, and predicted the atomic structure of Au29(SR)19 with 10e−.167 The crystal structure of heterodimeric Au29(SR)19 was finally obtained in 2021 by applying adamantanethiol (SR = SC5H10).38 Indeed, half of the Au29(SR)19 structure is the same as that of Au30(SR)18, and the other half is identical to that of Au28(SR′)20 (SR′ = S-c-C6H11) reported before.168 In addition to their geometric structure relationship, Au29(SR)19 is also heterodimeric in terms of its electronic structure (4e− + 6e−), whereas the two parent homodimeric NCs show 6e− + 6e− for Au30(SR)18, and 4e− + 4e− for Au28(SR′)20 (Fig. 4B).38 With respect to the NP assembly behavior, isotropic NPs lacking directional interactions usually adopt basic packing rules in their self-assembly, whereas remarkable physicochemical properties in metallic NPs are usually originated from the symmetry breaking in the particles (i.e., anisotropic NPs). Thus, we are also expecting some special properties resulted from the heterodimeric structure of the atomically precise Au29(SR)19 NC. Suprisingly, these heterodimeric Au29(SR)19 NCs self-assemble into double- and quadruple-helical superstructures, whereas the homodimeric Au30(SR)18 NCs only form layer-by-layer assemblies.38 Each pitch of the double helix contains 16 NCs (pitch length 12.8 nm and width 3.6 nm), in which 8 NCs in each strand adopt different rotation angles in the assembly (Fig. 4C).38 Moreover, the great advantage of atomically precise nanochemistry is that it reveals how such complex arrangements are formed, while this cannot be achieved in conventional NPs due to imperfections. With atomically precise NCs, two enantiomers (E1 and E2) are identified, and the heterodimeric Au29(SR)19 has four types of staple motifs: one monomeric Au(SR)2 motif (denoted L1) and one trimeric Au3(SR)4 (denoted L3) inherited from Au30(SR)18; and one monomeric Au(SR)2 (denoted C1) and one trimeric Au3(SR)4 (denoted C3) inherited from Au28(SR)20 (Fig. 4D). When assembling into a supercrystal, the two neighboring enantiomers approach each other by matching their C1 and C3 staple motifs, or their L1 and L3 staple motifs (Fig. 4E), resulting in four types of interactions: E1C1–E2C3 (denoted D), E1C3–E2C1 (denoted C), E1L3–E2L1 (denoted B) and E1L1–E2L3 (denoted A).38 Specifically, when the C1 and C3 staple motifs of two enantiomers pair up (Fig. 4E, via D and C interactions), the ligands surrounding them are triply paired (resembling base pairing of C and G in DNA); when the L1 and L3 staple motifs of two enantiomers pair up (Fig. 4E, via B and A interactions), the ligands surrounding them are doubly paired (resembling base paring of A and T in DNA). It is noteworthy that the helical assembly is driven by van der Waals interactions through particle rotation and conformational matching, as opposed to hydrogen-bonding and π–π stacking in DNA. Furthermore, the heterodimeric Au29(SR)19 has a photoexcited carrier lifetime that is ∼65 times longer than that of the homodimeric Au30(SR)18 (Fig. 4F), resulting in the NIR photoluminescence in Au29(SR)19 (see Scheme 2C).38 An Au29(SAdm)19 film in which nanoclusters are randomly packed enhances the photoluminescence by suppressing non-radiative decays. Supercrystals in which nanoclusters are self-assembled into double-helixes lead to further photoluminescence enhancement, arising from the collective properties of assembly and ordered anisotropic interactions.38
Additionally, M. Zhu et al. reported a halogen-induced anisotropic growth of Ag40(C6H5COO)13(SR)19(CH3CN) into Ag45(C6H5COO)13(SR)22Cl2 (SR = SCH2PhtBu), in which the adsorption of halogen ions might lead to the formation of defected intermediates for epitaxial growth at both ends of the kernel.169 In another work, Ag48Cl14(SR′)30 and Ag50Cl16(SR′)28(dppp)2 (SR′ = SC5H10, dppp = Ph2P(CH2)3PPh2) share the same framework containing a prolate Ag8 kernel (an Ag6 octahedron with two polar Ag atoms), except that the Ag2(SR′)3 active units at the two ends of Ag48Cl14(SR′)30 are replaced with Ag3(SR′)2(dppp)Cl units in Ag50Cl16(SR′)28(dppp)2 with a more anisotropic shape.170 In Au7Cu12(PR)6(SR′′)6Br4 (PR = diphenylphosphinopyridine, SR′′ = SPhtBu), the central Au is capped by an Au3Cu3 ring and further by a Cu3 motif from either side.171 The bimetallic NC demonstrates strong emission with 13.2% quantum yield (QY), which can be further improved by exchanging the aromatic thiolates with non-aromatic ones to break the intramolecular π⋯π interactions.171
Jin et al. also introduced a Cd–Br bond into Au NCs, resulting in bimetallic Au22Cd1(SR)15Br (see Scheme 2B), and trimetallic Au22−xAgxCd1(SR)15Br (avg. x = 1.87, SR = SC5H10).122 Half of the Ih Au13 or Au13−xAgx kernel is capped by three Au3(SR)4 motifs, whereas the other half has only a (SR)3CdBr motif on it.122 Due to the presence of the special Cd–Br on the surface, the NC with metal core radius of 5 Å shows a giant dipole (∼18 D), falling in the experimental trend of II–VI quantum dots. The strong dipole–dipole interactions lead to a “head-to-tail” alignment of the Au22−xAgxCd1(SR)15Br NCs in the crystalline state.122 In another case resulted from surface reconstruction due to doping, the cuboctahedral Au13 kernel is capped by two Cd3[Au(SR′)2]3 (SR′ = S-c-C6H11) motifs on each side, resulting in a Au19Cd2(SR′)16 NC.172 Compared to Au23(SR′)16 prior to surface reconstruction, the Cd-substituted NC greatly enhances the CO2 electroreduction selectivity to 90–95% at applied potential between −0.5 to −0.9 V vs. RHE.173
Rod-shaped nanoclusters
The Konishi group has done extensive works on diphosphine coordinated [core + exo]-type Au NCs (nuclearity = 6, 7, 8, and 11) featuring a polyhedral core with exo one or two gold atom(s),174–176 resembling ultrasmall Au nanorods. The Au6(dppp)4, Au8(dppp)4Cl2, and Au11(dppe)6 solutions (dppp = Ph2P(CH2)3PPh2, dppe = Ph2P(CH2)2PPh2) show vivid colors corresponding to the intense single band for each NC in the visible region, well-separated from the absorption at higher energy (Fig. 5A).129 By contrast, the core-only isomers of the same nuclearity show overlapped bands from UV to visible region. DFT calculations revealed that the exo Au atoms are highly involved in the frontier molecular orbitals and HOMO–LUMO transition, giving rise to the isolated single absorption band of the [core + exo]-type Au NCs.177 By replacing dppp with dppmb (dppmb = Ph2P-Ph-PPh2), Au6(dppmb)4 has its Au6 framework proximate to the phenylene bridges, and hydrogen bonding interaction can be identified and a large red shift (45 nm) of the visible absorption band is observed by spectroscopy.178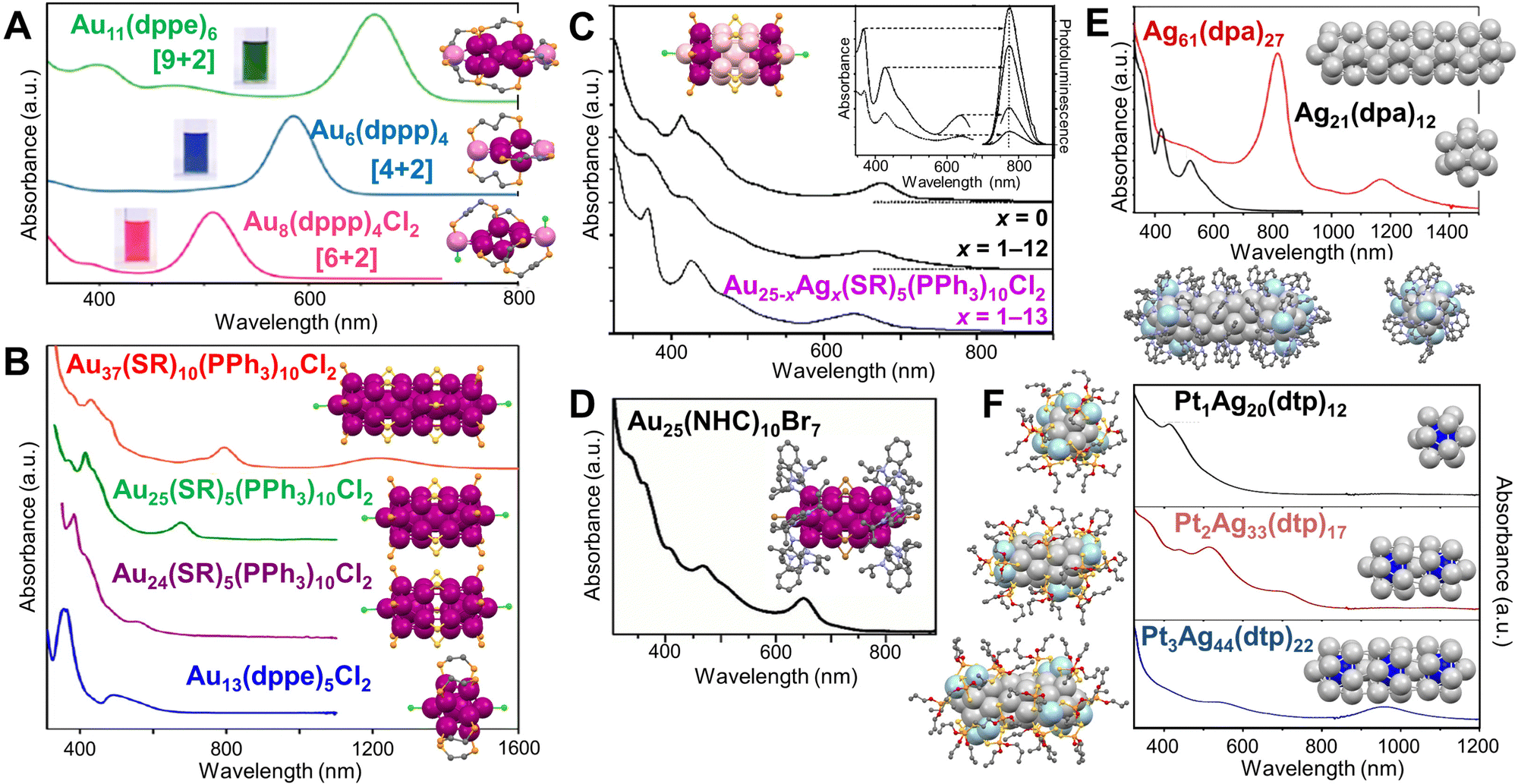 | ||
| Fig. 5 (A) Absorption spectra of Au6(dppp)4, Au8(dppp)4Cl2, Au11(dppe)6, and corresponding structures. Reproduced with permission.129 Copyright 2013, American Chemical Society. (B) Absorption spectra of Au37(SR)10(PPh3)10Cl2, Au25(SR)5(PPh3)10Cl2, Au24(SR)5(PPh3)10Cl2, Au13(dppe)5Cl2, and corresponding structures. Reproduced with permission.130 Copyright 2017, The Authors, published by National Academy of Sciences. (C) Absorption spectra of Au25−xAgx(SR)5(PPh3)10Cl2 (x = 0, 1–12, or 1–13), and corresponding structure. Inset, absorption and excitation spectra (left), and emission spectrum (right) at different excitation wavelengths. Reproduced with permission.179 Copyright 2014, Wiley-VCH GmbH. (D) Absorption spectra of Au25(NHC)10Br7, and corresponding structure. Reproduced with permission.180 Copyright 2019, Wiley-VCH GmbH. (E) Optical absorption spectra of Ag61(dpa)27 and Ag21(dpa)12 NCs, and corresponding total structures and Ih cores. Reproduced with permission.181 Copyright 2021, American Chemical Society. (F) Optical absorption spectra of Pt1Ag20(dtp)12, Pt2Ag33(dtp)17, Pt3Ag44(dtp)22, and corresponding total structures and Ih cores. Reproduced with permission.182 Copyright 2019, American Chemical Society. Color labels: magenta/violet = Au, pink = Au/Ag, light gray/light blue = Ag, blue = Pt, yellow = S, light orange = P, green = Cl, brown = Br, purple = N, red = O, grey = C. R groups are omitted or simplified for clarity. | ||
Using bimetallic Ih building blocks to form cluster of clusters was reported by the Teo group in the 1980s, and the M13 (M = Au/Ag) units are connected by vertex sharing.183–185 In 2005, when thiolate was introduced to the solution of Au11(PPh3)8Cl2, Au25(SR)5(PPh3)10Cl2 (SR = SCnH2n+1, n = 2–18) NCs with two vertex-sharing Ih Au13 units (aspect ratio of 2.3) was produced.186,187 Later, the Jin group reported another bi-Ih Au24(SR)5(PPh3)10Cl2 NC (Fig. 5B, purple line) with the shared vertex eliminated by adding excess PPh3, which is in contrast to the 25-metal-atom rod counterpart (Fig. 5B, green line). DFT calculations indicate that the broad band at ∼560 nm arises from the HOMO−1 to LUMO+2 transition.141 Excitingly, a tri-Ih Au37(SR)10(PPh3)10X2 (X = Cl/Br) with AR = 3.5 was prepared by reducing AuI(PPh3)X to clusters, then etching the clusters with excess thiol (Fig. 5B, red line).188 Along with the Au13(dppe)5Cl2 NC (Fig. 5B, blue line) with a mono-Ih core reported by Konishi and coworkers in 2010,189 the first rod-shaped series of Au NCs is achieved, and the mono-Ih, bi-Ih, and tri-Ih NCs possess 8e−, 16e−, and 24e−, respectively, according to Mingo's electron-counting rule.190 Moreover, the series shows an interesting trend in the optical absorption spectra (Fig. 5B): the high-energy peaks are similar for the series of NCs since the electronic transitions occur within the individual Au13;130,191 whereas the features of dimerization or trimerization due to the interaction between neighboring Au13 units are reflected in the red-shifted peaks in the visible range.32 It is noteworthy that the peak significantly shifts to 1230 nm for tri-Ih Au37 due to the trimeric interaction (Fig. 5B, red line). Moreover, an excited-state exciton localization was observed in the Au37 NC which exhibits strong coupling between Au13 units after photoexcitation, and kinetic traces indicate the existence of both radial and axial vibration modes.130 Femtosecond spectroscopic investigations on Au25(SR)5(PPh3)10Cl2 show overlapped excited state absorption and ground state bleach signals. The 0.8 ps component of the lifetime is attributed to the fast internal conversion process from LUMO+n to LUMO, whereas the 2.4 μs long component is due to electron relaxation to the ground state.192
The interesting stories of rod-shaped Au25 are also highlighted by heteroatom doping. Wang et al. continued to illustrate that substituting Au with Ag not only changed the absorption profile (Fig. 5C), but also drastically enhanced the photoluminescence (PL) to as high as QY = 40.1% for Au25−xAgx(SR)5(PPh3)10Cl2 (x = 1–13, Fig. 5C, inset),179 in striking contrast to the weakly luminescent species (x = 1–12, QY = 0.21%) and the homogold counterpart.193 In other words, the 13th Ag atom is critical in enhancing PL. Based on the highly photoluminescent rod-shaped Au25−xAgx(SR)5(PPh3)10Cl2, (x = 13) the self-annihilation electrogenerated chemiluminescence (ECL) was found to be 10 times higher than the standard tris(bipyridine)–ruthenium(ii) complex, and co-reactant (tripropylamine) ECL was about 400 times stronger.194
By replacing the 10 phosphine ligands with N-heterocyclic carbene ligands and the 5 thiolates with halides, a rod-shaped Au25(NHC)10Br7 NC (NHC = 1,3-diisopropylbenzimidazolin-2-ylidene) was resulted,180 showing the first absorption peak at ∼650 nm, similar to the case of Au25(SR)5(PPh3)10Cl2 (Fig. 5D). The high thermal and air stabilities of this NC are ascribed to the stronger Au–carbene bond than the Au–phosphine bond (difference up to 0.9 eV) as indicated by DFT. The NC also shows high catalytic activity in homogenous cycloisomerization of alkynyl amines to indoles compared to the trace activity of Au25(SR)5(PPh3)10Cl2.180 With the presence of Pd2+, the assembly of two Ih Pd@Au12 units into rod-shaped Pd2Au23(PPh3)10Br7 was also achieved.195 Hetero-bi-Ih Pd1Au24(SR)5(PPh3)10Cl2, i.e., only one Ih unit is central-doped with Pd, was synthesized by etching the crude product (made by co-reducing AuCl(PPh3) and Pd(PPh3)4) with thiol in refluxing chloroform.196 Compared to the rod-shaped Au25 NC, obtaining the rod-shaped Ag25 NC (i.e., the silver counterpart) of the same structure is challenging. However, Pt or Pd doping at the centers of the two Ih units could improve the stability of the NCs, resulting in M2Ag23(PPh3)10Cl7 (M = Pt/Pd).197,198
Very recently, the first supercluster with four Ih Ag13 units linearly connected was reported by Q.-M. Wang and Xu.181 The resulted Ag61(dpa)27 (dpa = dipyridylamine) was produced by reducing AgI-dpa in the presence of bidentate phosphine, AgSbF6, and CH3ONa in dichloromethane. The 30e− of Ag61 makes it isolobal to the linear tetraiodide anion I42−.199 Compared with Ag21(dpa)12 of which the large energy gap is mainly decided by the superatomic Ag135+ (8e−) unit,200 Ag61(dpa)27 with AR = 4.2 shows an intense absorption peak extending into NIR (819 nm) and the first absorption band at 1170 nm (Fig. 5E).181 The authors attributed the loss of the optical features of mono-Ih Ag21 in the spectrum of tetra-Ih Ag61 to the stronger electron couplings between the Ih Ag13 units, in contrast to the case of Au13vs. Au37 (Fig. 5B). Besides, with the help of Pt doping, Liu et al. reported another series of NCs containing one, two or three Ih Pt1@Ag12 units (Fig. 5F) by co-reducing AgI and PtII with LiBH4 and chromatography separation. Similar to the Au series, the lowest-energy absorption bands of the Ag series display a red-shift from 412 nm in mono-Ih Pt1Ag20(dtp)12 to 710 nm in bi-Ih Pt2Ag33(dtp)17, then to 956 nm in tri-Ih Pt3Ag44(dtp)22 (dtp: dipropyl dithiophosphate). Although the first two alloy NCs follow the superatomic electron counts of 8e− and 16e−, respectively, the tri-Ih NC bears only 22e−, representing the first case in NCs that is isoelectric to linear triiodide anion I3−.182 Note that in heteroleptic rod-shaped 25-metal or 37-metal NCs,82,187,188,196–198 the two or three M13 (M = Au/Ag) units are aligned in an eclipsed conformation; whereas in the homoleptic rod-shaped Pt2Ag33, Pt3Ag44 as well as Ag61 NCs,181,182 a gauche rotational conformation is adopted in the connection of multiple Ih units.
Compared to the rod-shaped NCs composed of more commonly observed Ih units, anisotropic growth of fcc structures into nanorods (bulk Au also adopts fcc packing) was also reported for Au NCs of atomic precision. It was surprising when an fcc kernel was first discovered in Au36(SR)24 (SR = SPhtBu) in 2012.79 The close-packed atomic planes show an a–b–c–a stacking sequence (Fig. 6A), resulting in the cubic and cuboid shapes of NCs of fcc structures. The numbers (Fig. 6A) indicate how many metal atoms are in each (001) layer. After continuous successes in synthesizing and characterizing Au28(SR)20,80 and Au52(SR)32,87 the Jin group completed a magic series with a unified formular of Au8n+4(SR)4n+8 (n = 3–6) in 2016 thanks for the structural determination of Au44(SR)28.201 The progression of NCs from small to large is neatly shown in the mass spectra of the four NCs (Fig. 6B). In a concise layer-by-layer mode, all six surfaces of the series of NCs are exclusively terminated by {100} facets, and the anisotropic growth of the magic series can be viewed as subsequently adding an additional 8-gold-atom (001) layer along the z direction, with n representing the number of (001) layers (Fig. 6A). That is, n = 3 for Au28(SR)20, n = 4 for Au36(SR)24, n = 5 for Au44(SR)28, and n = 6 for Au52(SR)32.201 In 2019, Wu et al. reported that a half (001) layer composed of 4 Au atoms could also be stacked on the full 8-gold-atom (001) layer, resulting in Au56(SR)34.125 Both works show the evolution of optical properties, which is indicated by the red-shift of the lowest energy peak (assigned to sp → sp transition) as the kernel grows, and their optical energy gaps are estimated to be 1.76, 1.75, 1.50, 1.39, and 1.30 eV, respectively (Fig. 6C, from Au28 to Au56). Based on the observation of a top half layer, the theoretical structures of Au32(SH)22, Au40(SH)26, and Au48(SH)30 are predicted as well.202 Additionally, when the (001) layers composed of 6 Au atoms and 8 Au atoms are stacked alternatively (total 6 layers), another Au42(SR)26 NC can be obtained (Fig. 6A).203
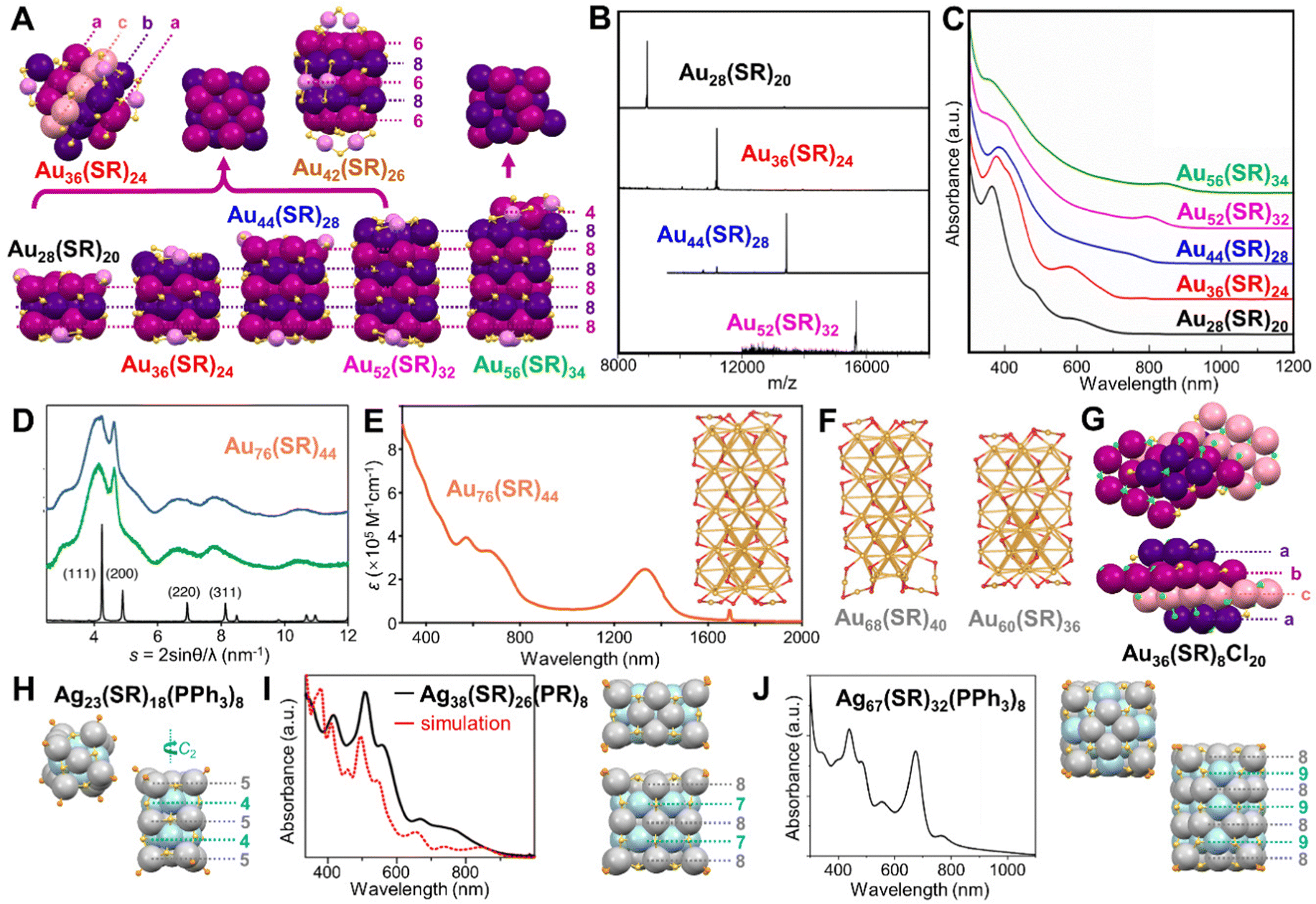 | ||
| Fig. 6 (A) The anisotropic growth of the fcc lattice of Au28(SR)20, Au36(SR)24, Au44(SR)28, Au52(SR)32, and Au56(SR)34 NCs with top views of the Au8n+4(SR)4n+8 magic series compared to Au56(SR)34, and the structure of Au42(SR)26. (B) ESI-MS spectra of the Au8n+4(SR)4n+8 series, n = 3–6. The m/z = [Aun(SR)m + Cs]+. Reproduced with permission.201 Copyright 2016, American Chemical Society. (C) Absorption spectra of the fcc series. Reproduced with permission.125 Copyright 2020, Wiley-VCH GmbH. (D) Powder XRD patterns of Au76(SR)44 before (green) and after (blue) amidation reaction, and bulk gold (black). (E) UV-vis-NIR absorption spectrum of Au76(SR)44 solution. Reproduced with permission.204 Copyright 2015, American Chemical Society. (F) The optimized theoretical structures of Au60(SR)36, Au68(SR)40, and Au76(SR)44. Reproduced with permission.205 Copyright 2016, The Royal Society of Chemistry. (G) Top and side views of Au36(SR)8Cl20. (H) Top and side views of Ag23(SR)18(PPh3)8. (I) Experimental and calculated absorption spectra of Ag38(SR)26(PR)8, and corresponding top and side views of the structure. Reproduced with permission.210 Copyright 2016, American Chemical Society. (J) Absorption spectra of Ag67(SR)32(PPh3)8, and corresponding top and side views of the structure. Reproduced with permission.211 Copyright 2016, American Chemical Society. Color codes: magenta/purple/pink/violet = Au, light grey/light blue = Ag, yellow = S, green = Cl, light orange = P. R groups are omitted for clarity. | ||
In the meantime that the fcc magic series was achieved, the Tsukuda group also synthesized a water soluble Au76(SR)44 NC (SR = SC2H4PhCOOH) by slow reduction,204 and the powder X-ray diffraction indicated a fcc kernel structure elongated along the {100} direction (Fig. 6D). Interestingly, the proposed rod-shaped fcc Au76 NC exhibited a strong NIR absorption band centered at 1340 nm with molar absorption coefficient ε of 3 × 105 M−1 cm−1 (Fig. 6E), much larger than that of the coefficient of HOMO–LUMO transition in most of other gold–thiolate NCs (∼1 × 104 M−1 cm−1).19 The large value of ε was tentatively ascribed to the large electronic–transition moment arising from the one-dimensional structure.204 Since Au76(SR)44 could be fitted in the Au8n+4(SR)4n+8 series with n = 8, theoretical calculations were conducted to predict its structure. Zeng et al. found highly negative values of the nucleus-independent chemical shift in the tetrahedral Au4 units, explaining the overall stability of the series of fcc NCs, and the structures of Au60(SR)36, Au68(SR)40, and Au76(SR)44 were simulated (Fig. 6E inset and Fig. 6F).205 Infinite extending of the lattice showed a band gap of 0.78 eV for the gold–thiolate nanowire.205 At the same time, the Pei group also obtained the same simulated structure of Au76(SR)44 and assigned its NIR absorption to the anisotropic Au kernel which enhanced the longitudinal orbital transition and the ligand passivation, resulting in the red shift of the absorption peak.206 In another fcc NC, Au–Cl–Au motif was first observed in Au36(SR)8Cl20 (SR = SCH2PhtBu), and the total structure can be regarded as four stacking layers in a–b–c–a sequence (Fig. 6G).207
In 2013, Zheng et al. reported a cubic fcc Ag building block —Ag14(SR)12(PPh3)8 (SR = SPhF2) NC.208 Later, by stacking multiple of such building blocks together, a series of cuboid fcc Ag NCs were achieved with six faces and twelve edges capped by thiolates and eight corners terminated by phosphine ligands. In the Ag23(SR′)18(PPh3)8 (SR′ = SC2H4Ph), its structure is a combination of two cubic units with some twist along the C2 axis, endowing chirality to the NC (Fig. 6H).209 Ag38(SR)26(PR′)8 (PR′ = tributylphosphine) is composed of four of such cubes, and its optical spectrum shows three peaks at 413, 507, 563 nm, with some weak absorption bands as well (Fig. 6I),210 while the Ag14 building block NC only shows two shoulder bands.208 Linear-response time-dependent DFT indicates that the Ag38 is rather electronically stable, with an energy gap of 0.67 eV. A larger cubic Ag63(SR)38(PR′)8 of 2 × 2 × 2 size was also reported in the same work and a theoretical 3 × 3 × 3 Ag172(SR)72(PR′)8 was predicted.210 Additionally, the Bakr group revealed an Ag NC built up with 12 units, i.e., Ag67(SR′′)32(PPh3)8 (SR′′ = SPh(CH3)2). Its UV-vis spectrum shows similar strong absorption between 400 to 600 nm as Ag38(SR)26(PR′)8, but with an additional peak at ∼680 nm (Fig. 6J).211
So far, NCs with Ih, fcc, or decahedral kernels constitute the majority of atomically precise noble metal NCs.32,48,212–214 By comparison, Au NCs with hexagonal-close-packed (hcp) kernels are very rare, and no hcp structure is ever found in Ag NCs. We start from Au24(SR)20, although it does not have an hcp kernel. Based on the formular of Au24(SR)20 obtained in experiment,215 two isomeric structures with low energies were predicted by Pei et al. in 2011,216 and both of them were proved by crystallography later. The total structure of Au24(SR)20 was first solved by applying SR = SCH2PhtBu.82 Two tetrahedral Au4 units are joined at their triangle faces by 60° rotation to form a bi-tetrahedral Au8 kernel. One pair of Au4(SR)5 staples is assembled onto the two ends of the Au8 kernel via bi-dentate bonding; and a second pair of the tetrameric staples is further attached to the Au8 kernel by rotating ∼90° relative to the first pair (Fig. 7A, inset). Other thiols were also used to prepare the same 24-gold-atom NC, and the fluorescence QY decreased from 2% for Au24(SCH2PhtBu)20, to 1.5% for Au24(SCH2Ph)20, then to 0.3% for Au24(SC2H4Ph)20, with the emission peak slightly blue-shifted (Fig. 7B and C), correspondence to the electron-donor strength of the ligand.126 By contrast, selenolate-protected Au24(SePh)20 shows an isomeric structure in which two tetrahedral Au4 units cross-join together at the edges (rather than the faces), and the new Au8 kernel is protected by a pair of Au5(SePh)6 motifs as well as a pair of Au3(SePh)4 motifs (Fig. 7D, left).217 The Au24(SePh)20 NC is not luminescent in the detected range (Fig. 7B and C).126 In another case, the Au9 kernel of Au18(SR)14 (SR = S-c-C6H11) can be viewed as three Au3 layers arranged in an a–b–a manner (Fig. 7D, middle),218,219 and the middle Au3 layer can be replaced by an Ag3 layer without changing the structure.220 It is until the report of Au30(SR)18 (SR = adamantanethiolate, SC5H10) that a complete hcp kernel was identified since four layers of Au3–Au6–Au6–Au3 are packed in an a–b–a–b manner, and the total structure has an unprecedentedly high S6 symmetry (Fig. 7D, right), resulting in the special solubility of the NC, i.e., only soluble in benzene.221
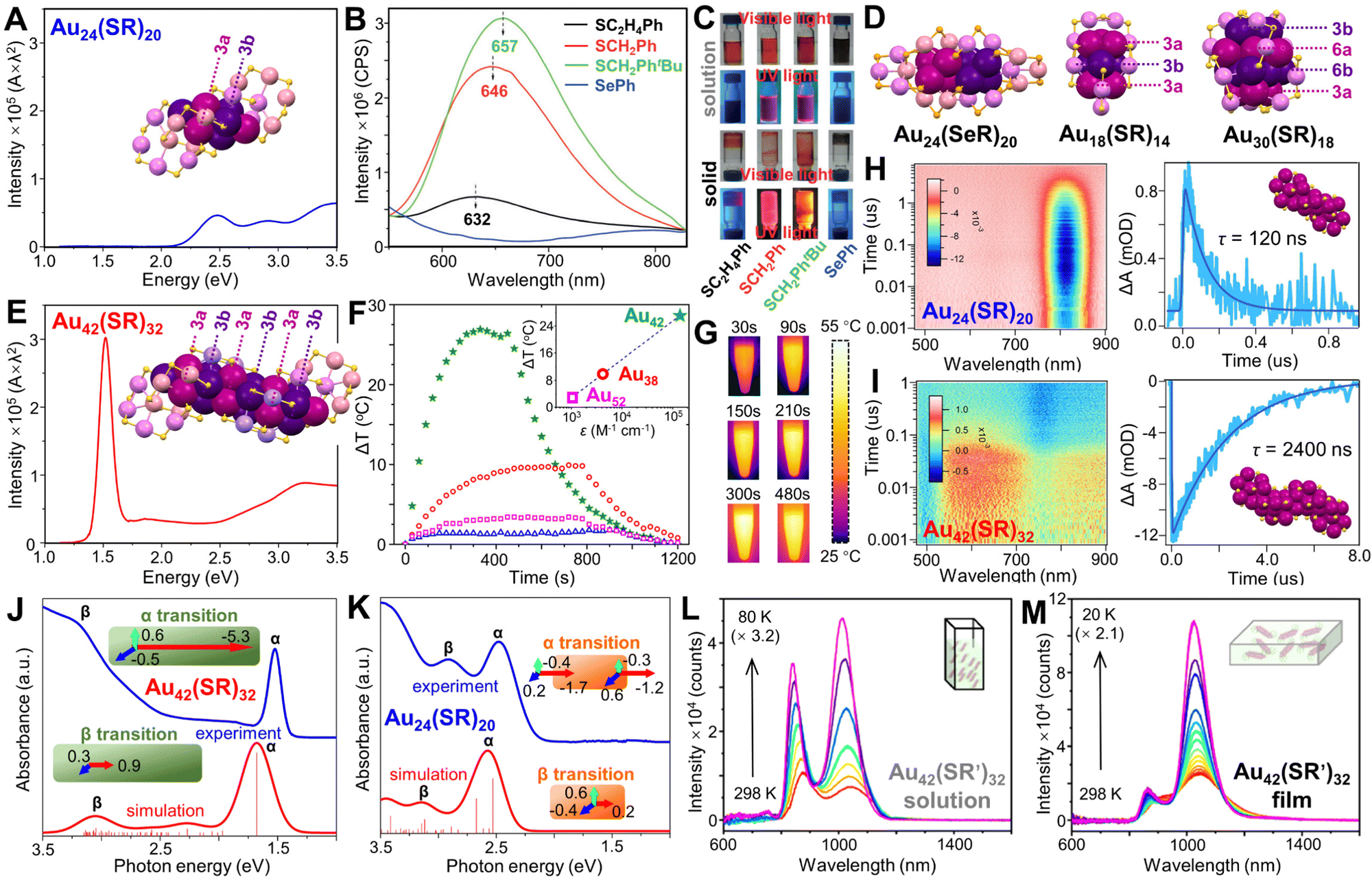 | ||
| Fig. 7 (A) Absorption spectrum (photon energy scale) of Au24(SR)20 with corresponding structure shown in the inset. (B) Fluorescence spectra of Au24(SR)20 NCs with different thiolate ligands and Au24(SeR)20 NCs in CH2Cl2, and (C) digital photographs of samples in solution or solid state under visible and 365 nm UV light irradiation. Reproduced with permission.126 Copyright 2016, Wiley-VCH GmbH. (D) Structures of Au24(SeR)20, Au18(SR)14 and Au30(SR)18. (E) Absorption spectra (photon energy scale) of Au42(SR)32 with corresponding structure shown in the inset. (F) Temperature changes of Au42(SR)32, Au38(SR)24, Au52(SR)32, and Au24(SR)20 in toluene (50 μg mL−1) upon irradiation at 808 nm, 1 W cm−2, inset: the relationship between ΔT and molar absorption coefficients of NC solutions at irradiation wavelength. (G) Time-dependent images of Au42(SR)32 solution upon irradiation. (H) ns-TA data map of Au24(SR)20 and its TA kinetic traces and fitting (probed at 580 nm). (I) ns-TA data map of Au42(SR)32 and its TA kinetic traces and fitting (probed at 813 nm). λex = 400 nm. (J) Experimental absorption spectrum (blue) and TDDFT-simulated absorption spectrum (red) of (J) Au42(SR)32 and (K) Au24(SR)20, insets, schematic illustrations of α and β electronic transitions. (A and E–K) Reproduced with permission.127 Copyright 2022, American Chemical Society. Temperature-dependent PL spectra of (L) Au42(SR′)32 in 2-methyltetrahydrofuran, and (M) Au42(SR′)32/PS film. Reproduced with permission.128 Copyright 2022, American Chemical Society. Color codes: magenta/purple = kernel Au, violet/pink = motif Au, yellow = S, light orange = Se. Organic ligands are omitted for clarity. | ||
A new breakthrough was achieved when the total structure of a rod-shaped Au42(SR)32 (SR = SCH2Ph) NC with a long hcp kernel was resolved very recently.127 When comparing the optical absorption spectra, the lowest energy absorption peak of Au24(SR)20 is at 2.5 eV (Fig. 7A), whereas that of Au42(SR)32 red-shifts to 1.5 eV (Fig. 7E) with 6.5 times stronger intensity. The hcp Au20 kernel of Au42(SR)32 comprises six Au3 layers with two capping Au atoms on opposite ends of the rod, i.e., Au1–Au3–Au3–Au3–Au3–Au3–Au3–Au1, compared to the Au8 kernel (Au1–Au3–Au3–Au1) in Au24(SR)20. Two pairs of Au4(SR)5 staples are attached to the two ends of the Au20 kernel, similar to what is observed in Au24(SR)20, but the much longer kernel further requires six short Au(SR)2 motifs to protect its body (Fig. 7E, inset). The aspect ratio of the long hcp Au20 kernel is as large as 6.2, providing a great opportunity to study new properties, since the confinement of excitons within the long but narrow kernel leads to strong photothermal conversion upon photoexcitation. The molar absorption coefficient ε of Au42(SR)32 is 1.4 × 105 M−1 cm−1 at 815 nm, and upon 808 nm excitation (the most commonly used laser for photothermal therapy with minimum extinction by human tissues),222,223 very rapid heating was observed as the temperature change (ΔT) reached ∼27 °C within only ∼5 min (Fig. 7F, green stars/Fig. 7G).127 By comparison, other anisotropic Au-SR NCs, including Au38(SR)24 with dimeric Au23 kernel (see Fig. 3A), and Au52(SR)32 with a layer-by-layer fcc Au48 kernel (see Fig. 6A), increased only by ∼10 and ∼3 °C, respectively (Fig. 7F, red circles and magenta squares). ΔT is almost linear with respect to the exponential scale of ε at the irradiation wavelength. In transient absorption (TA) analysis, although almost the same fast decay was observed in both Au42(SR)32 and Au24(SR)20 (long and short rods), the nanosecond TA showed the decay lifetimes (τ) of Au24(SR)20 was 120 ns (Fig. 7H), much shorter than the 2400 ns of Au42(SR)32 (Fig. 7I). Moreover, TDDFT calculations (Fig. 7J) found that the transition dipole moment of peak α (corresponding to HOMO to LUMO transition) of Au42(SCH3)32 is ∼10 times stronger along the longitudinal transition than along the transverse transition, indicating the strong NIR absorption is a longitudinal transition, reminiscent of the NIR band of plasmonic Au nanorods. It is a surprise to see that longitudinal excitation can be inherited by NCs of molecular state. For Au24(SCH3)20 with a much shorter kernel, the longitudinal transition for the first peak is not as strong as the case of Au42(SCH3)32, but still stronger than the transverse transitions (Fig. 7K).127
In addition to the unprecedented photothermal conversion properties, a photoluminescence study on the same Au42(SR′)32 NC (SR′ = SC2H4Ph) found a dual NIR emission at 875 and 1040 nm, corresponding to fluorescence and phosphorescence, respectively (Fig. 7L).128 The QY of Au42(SR′)32 was measured to be 11.9% in ambient CH2Cl2 solution at room temperature. Furthermore, when the NCs were embedded in polystyrene (PS) films (solid state), the fluorescence was dramatically suppressed while the phosphorescence was significantly enhanced (Fig. 7M), which was ascribed to the largely enhanced intersystem crossing from singlet to triplet excited state induced by dipolar interaction.128 On a note, Wu et al. independently reported the same Au42(SR)32 NC obtained by a thermal and ligand exchange induced fcc-to-hcp structural transformation, and time-dependent DFT was performed to interpret the Au42 dual emission.224
Finally, some clues can be drawn from the synthesis to understand how the ultrasmall Au42(SR)32 nanorod was achieved. In the synthesis reported by the Jin group,127 Au42(SR)32 was separated from Au24(SR)20 by thin layer chromatography, and the as-synthesized mixture of NCs also contains Au25(SR)18 and Au38(SR)24. Since Au38(SR)24 is a dimeric structure of two Au25(SR)18 NCs through fusion (see Fig. 3 and discussion therein), Au42(SR)32 might also be formed from the anisotropic growth of Au24(SR)20. In the synthesis by the Wu group, the Au42(SR)32 nanocluster was obtained by a thermally induced ligand-exchange reaction from Au28(SPhtBu)20, and then purified by thin layer chromatography,224 indicating that the anisotropic growth occurs simultaneously with fcc-to-hcp structural transformation.
Among the bimetallic NCs, Au10Ag2(CCR)3(dppy)6 (CCR = 2-pyridylethynyl, dppy = 2-pyridyldiphenylphosphine) has a trigonal bipyramidal Au10Ag2 core with Ag atoms at the two ends. The dual emission of the NC in solution includes (i) the visible emission originating from metal-to-ligand charge transfer from Ag atoms to phosphine ligands and (ii) the intense NIR emission which is associated with the participation of 2-pyridylethynyl in the frontier orbitals of the NC.225 The Pt1Ag26(SR)18(PPh3)6 (SR = SPh(CH3)2) NC has an Ih Pt@Ag12 kernel with two more Ag atoms at the two ends. With the Ag12(SR)18(PPh3)6 shell wrapping on the surface, the bimetallic NC shows an overall rod shape. Compared to Pt1Ag24(SR)18 with a Pt@Ag12 kernel, the optical absorption spectrum of Pt1Ag26(SR)18 is almost the same, but their electrochemical gaps are quite different (1.48 V vs. 1.89 V) due to the charge influence.226 When replacing the central Pt with Au, the authors co-crystallized [Au1Ag26(SR′)18(PPh3)6]+ and [Au1Ag24(SR′)18]− (SR′ = SPhC2H5) into alternating stacking array along the [001] direction via weak C–H⋯π and π⋯π interactions and strong electrostatic interactions.227
Additionally, Sun et al. prepared quite many polyoxometalate-templated Ag NCs under solvothermal conditions, and some of the NCs possess rod-shaped structures. Selected works include Ag72 (a long Ag shell wrapping two linearly aligned [EuW10O36]9− anions);228 Ag76 (two Mo6O228− templates encaged in an Ag72 shell);229 Ag80 (the inside Ag10 kernel is locked by a pair of Mo7O2610− anions and further encapsulated by an outer Ag70 shell);230 and Ag90 (the dumbbell-shaped Ag90 cage covers two smaller SO42− and two larger W5O198− anions inside).231 Compared to the linear Au coordination to two S atoms, a complete cage usually forms due to higher Ag–S coordination,55 which is more flexible to accommodate the inner kernel shape. Note that these AgI NCs have no free valent electron as we discussed above for Au NCs.
Oblate nanoclusters
Heteroleptic Au19(CCPh)9(dppa)3 (dppa = N,N-bis-(diphenyl-phosphino)amine) has an Ih Au13 kernel, and three V-shaped Au2(CCPh)3 motifs along the C3 axis.232 The alkynyl ligands are anchored on surface Au atoms via both σ and π bondings, which is different from the bonding mode of thiolate on Au. The NC shows two major absorption peaks at 1.25 and 2.25 eV, and orbital analysis indicates that the PhCC– groups actively participate in the frontier orbitals of the whole NC (Fig. 8A). Although not mentioned in the original work, we here point out that the metal part of the NC resembles a tiny triangular disk despite that no (111) facet is found on the surface. Another alkynyl-protected Au22(CCR)18 NC (R = tBu) also has a triangular shape in which a flattened cuboctahedral Au13 kernel is surrounded by three Au3(CCR)4 motifs.233 The Au22(CCR)18 NC required a relatively weak reduction environment in the synthesis. Although the QY of the NC solution (in CH2Cl2) was only 0.4%, the solid-state emission yield of Au22(CCR)18 increased to 15% with a predominant lifetime of 1.37 μs.234 The same Au22 NC could also be prepared using triethylamine as a mild reductant, and different CCR′, including 3-ethynylthiophene (ETP-H), phenylacetylene (PA-H), 3-ethynyltoluene (ET-H), and 3-ethynylanisole (EA-H) were adopted.131 The PL QY was found to be dependent on the R group, and the highest QY = 4.6% (solution) was obtained when R = Ph (Fig. 8C).131
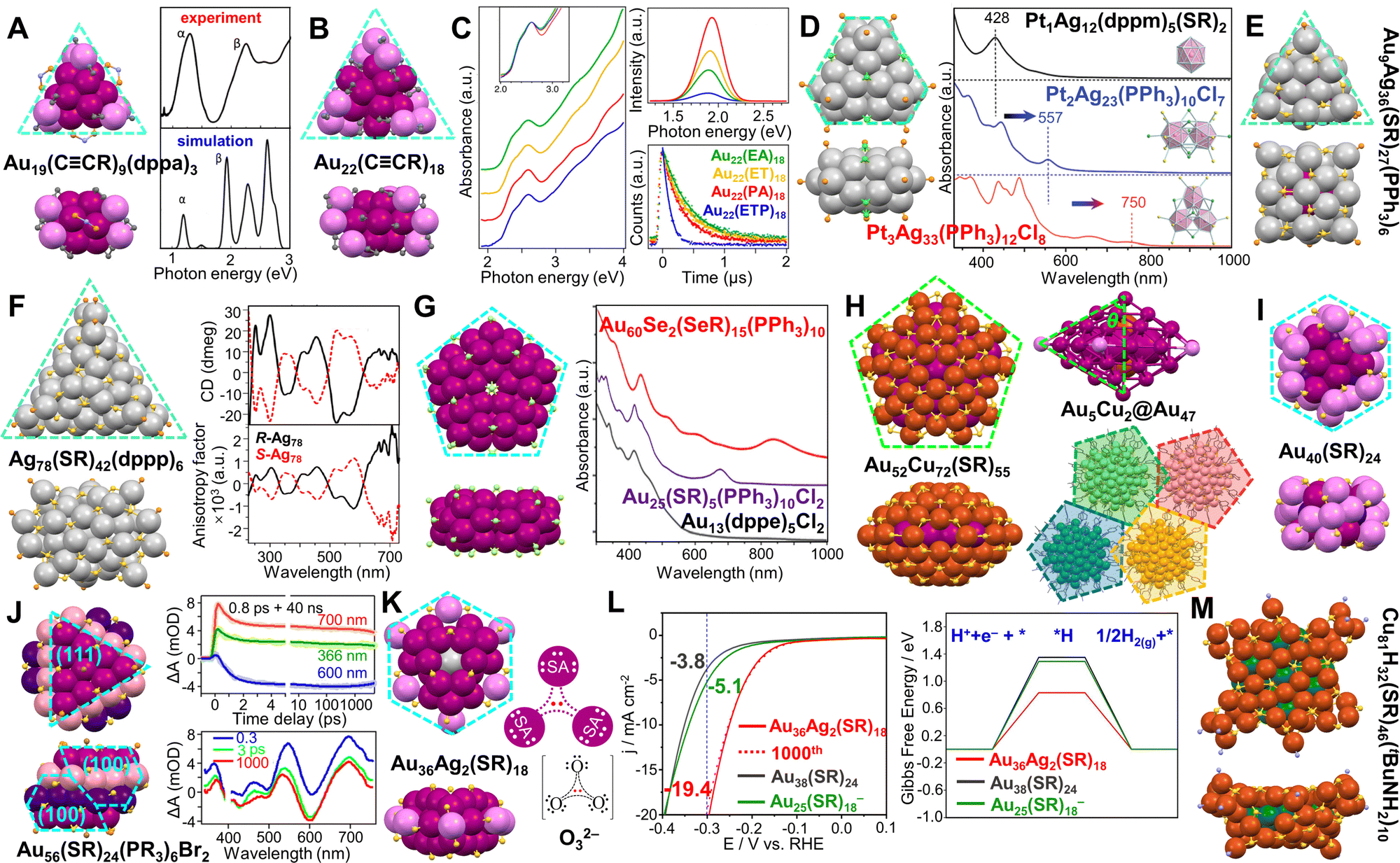 | ||
| Fig. 8 (A) Top and side views of Au19(CCR)9(dppa)3, and its experimental and simulated optical absorption spectra. Reproduced with permission.232 Copyright 2014, American Chemical Society. (B) Top and side views of Au22(CCR)18. (C) UV-vis spectra, PL spectra and lifetimes of Au22(CCR′)18 with different ligands. Reproduced with permission.131 Copyright 2019, American Chemical Society. (D) Top and side views of Pt3Ag33(PPh3)12Cl8, and UV-vis spectra of Pt1Ag12(dppm)5(SR)2, Pt2Ag23(PPh3)10Cl7, and Pt3Ag33(PPh3)12Cl8. Reproduced with permission.235 Copyright 2018, The Royal Society of Chemistry. (E) Top and side views of Au9Ag36(SR)27(PPh3)6. (F) Top and side views of Ag78(SR)42(dppp)6, and the CD spectra and corresponding anisotropy factors of R- and S-Ag78 enantiomers. Reproduced with permission.237 Copyright 2017, American Chemical Society. (G) Top and side views of Au60Se2(SeR)10(PPh3)15, and its UV-Vis spectrum compared to Au25(SR)5(PPh3)10Cl2 and Au13(dppe)5Cl2. Reproduced with permission.238 Copyright 2015, Wiley-VCH GmbH. (H) Top and side views of Au52Cu72(SR)55, its Au5Cu2@Au47 decahedral kernel, and the interactions among four nearest pentagonal NCs. Reproduced with permission.239 Copyright 2020, The author(s). (I) Top and side views of Au40(SR)24. (J) Top and side views of Au56(SR)24(PR3)6Br2, the kinetic traces of transient absorption and corresponding fits at selected wavelengths, and spectra at selected time-delays. Reproduced with permission.133 Copyright 2022, The author(s). (K) Top and side views of Au36Ag2(SR)18, and the Lewis structure of the [Au30Ag2]12+ kernel compared to O32−, SA = superatom. (L) HER voltammograms of Au36Ag2(SR)18, Au38(SR)24 and Au25(SR)18− catalysts, and the calculated Gibbs free energy of H2(g) formation on the catalysts. Reproduced with permission.132 Copyright 2021, American Chemical Society. (M) Top and side views of Cu81H32(SR)46(tBuNH2)10. Color codes: magenta/violet/purple/pink = Au, light grey = Ag, orange/green = Cu, yellow = S, light orange = P, light green = Se, light purple = N. R groups are omitted or simplified for clarity. | ||
Not only the rod-shaped structure can be achieved by connecting three Ih units via vertex sharing,188 a cyclic structure (i.e., an oblate shape) is also realized in bimetallic Pt3Ag33(PPh3)12Cl8 (Fig. 8D, left) and trimetallic Pt3Au12Ag21(PPh3)12Cl8 NCs with three Pt atoms doped at the centers of three Ih M13 (M = Ag/Au) units.235 Since the cyclic structure is composed of three Ih units, the whole shape can be regarded as a truncated triangular disk. A clear red-shift of the lowest energy peak was observed from 428 nm for mono-Ih Pt1Ag12(dppm)5(SR)2 (dppm = Ph2PCH2PPh2, SR = SPh(CH3)2) to 557 nm for bi-Ih Pt2Ag23(PPh3)10Cl7, then to 750 nm for cyclic tri-Ih Pt3Ag33(PPh3)12Cl8 (Fig. 8D, right).235
Zheng et al. reported that Au9Ag36(SR)27(PPh3)6 (SR = SPhCl2) has an Au9 kernel encaged in a trigonal prismatic Ag36(SR)27(PPh3)6 shell.236 This bimetallic NC can be viewed to have two dented (111) facets and three distorted (100) facets of fcc arrangement (Fig. 8E). The same group also revealed an Ag78(SR)42(dppp)6 NC (SR = SPhCF3, dppp = Ph2P(CH2)3PPh2) with a triangular prismatic structure conformed to ideal D3 symmetry (Fig. 8F, left). When replacing the achiral dppp with chiral diphosphine, i.e., (2s,4s) or (2r,4r)-2,4-bis(diphenylphosphino) pentane, the flexible C–C–C of diphoshpine restricts the relative orientation of the four Ag–thiolate–phosphine moieties at the three side edges, resulting in enantioselectivity of the metal core (R- and S-Ag78), with the maximum anisotropy factor up to 2 × 10−3 at 678 nm (Fig. 8F, right).237
Five Ih Au13 building blocks can even form a closed ring by vertex sharing in the Au60Se2(SePh)15(PPh3)10 structure, which shows a pentagonal oblate shape.238 The two neighboring Au13 units are connected by selenolate linkages, and two Se atoms are capped at the top/bottom centers of the NC. The Au60 NC not only maintains the optical property of the Au13 unit, the interactions between neighboring Au13 units also lead to further red-shift of the low-energy absorption peak to 835 nm compared to the 670 nm peak of bi-Ih Au25(SR)5(PPh3)10Cl2 (Fig. 8G).238
Oblate decahedral NCs have been reported when the kernel diameter reaches 2 nm. The thiolate (SR = SPhtBu)-stabilized NC containing 374 Ag atoms was reported to become metallic with the emergence of surface plasmon resonance.240 From the structural perspective, both Ag136 and Ag374 NCs are regarded as miniatures of five-fold twinned NPs. Ag136 has a pentagonal bipyramidal Ag54 kernel with ten (111) facets, whereas Ag374 has an elongated pentagonal bipyramidal Ag207 kernel with five (100) faces at the five sides and five (111) facets at each end.240 A decahedral Ag51 kernel with D5h symmetry was also found in Ag146(SR′)80Br2 (SR′ = SPhCH(CH3)2) with an overall pentagonal oblate shape.241 Interestingly, the Ag146(SR′)80Br2 NC shows an optical bandgap and power-independent electron dynamics, indicating its molecular-like nature, as opposed to the plasmonic Ag136 NC with a smaller size. The bimetallic Au130−xAgx(SR)55 (avg. x = 98) also has a decahedral M54 kernel (M = Au/Ag) inside the Ag-SR shell.242 In the M54 kernel, all ~32 Au atoms are localized in the truncated M49 Marks decahedron, whereas the five corners of the regular M54 decahedron are all Ag.242 An interesting question is why homogold NCs of similar size can only have the truncated Au49 kernel,20,85 while Ag or Au/Ag alloy NCs show M54 full decahedrons inside the NCs. The case of Au52Cu72(SR′′)55 (SR′′ = SPhCH3) gives insights into the Marks decahedron truncation mechanism (Fig. 8H). When comparing the Au5Cu2@Au47 kernel (Fig. 8H, top right) in the Au-Cu bimetallic NC and the Au7@Au42 kernel in Au102(SR)44 or Au103S2(SR)41, it was found that the axial Au–Cu–Cu–Au length is contracted by 7% due to Cu doping, and the angle (θ) also expends, giving enough space for an Au atom to fit in (total five corners), completing a decahedral M54 core. The same phenomenon was also observed in Ag or Ag–Au bimetallic NCs, so that M54 full decahedrons are formed. Moreover, the Cu70(SR′′)55 exterior cage of Au52Cu72(SR′′)55 resembles 3D Penrose tiling, and interparticle interactions in the supercrystal give rise to a “quadruple-gear-like” interlocking pattern (Fig. 8H, bottom right).239
Tetrahedral Au4 units coil up into a Kekulé-like ring along two vertex-sharing Au4 units to constitute the fcc kernel in Au40(SR)24 (SR = SPhCH3).87 Since the surface motifs are distributed along the C3 axis, the whole NC exhibits a hexagonal oblate structure (Fig. 8I).87 In the seed-sized Au56(SR′)24(PR3)6Br2 (SR′ = SPhtBu, R = PhCF3, PhCl, or PhF) nanoprism, the side Au{100} facets are covered by bridging thiolates, whereas the top or bottom {111} facet is capped by phosphine ligands at three corners and Br− at the center (Fig. 8I, left).133 The bromide is key to effectively stabilize the Au{111} to fulfill a complete fcc core, while the halide-free gold precursor results in Au55(SR′′)24(PPh3)6 (SR′′ = SPhCH3) with one missing gold on one of the two (111) facets.243 Moreover, the femtosecond transient absorption spectrum of Au56 (Fig. 8J, right) is similar to that of larger-sized gold NCs with n > 100, which is ascribed to the completeness of the prismatic fcc core.133
Beyond the Au3 face-fusion of two Ih Au13 units to form the dimeric kernel of homogold and Ih-center-doped Au38(SR)24 NCs (see Fig. 3A, C, D, G and H), the Jin group recently reported a Au36Ag2(SR)18 (SR = SC5H10) NC in which three Ih units face-fused together in a cyclic way.132 The series of NCs, i.e., mono-Ih Au25(SR′)18−, di-Ih Au38(SR′)24 (SR′ = SC2H4Ph) and tri-Ih Au36Ag2(SR)18 (Fig. 8K) is achieved with free valence electron number increasing from 8e− to 14e− to 20e−, in contrast to the vertex-sharing Ih NC series with 8e− to 16e− to 24e− electron configurations (Fig. 8D). The two Ag atoms at the centers of the NC are found to be critical in achieving the trimeric face-fused structure, since the Ih central Au would take more electron density than the shell atoms244 and the much less electronegative Ag atoms (χAg = 1.93 vs. χAu = 2.54) would be preferred at the two positions that are shared by all three Ih units. Since the [Au30Ag2]12+ kernel can be viewed as a union of three face-fused superatoms, Cheng et al. performed chemical bonding analysis suggesting a three-superatom-center two-electron bond for the octet rule of each superatom, which mimics the bonding framework of the D3h O32− molecular anion (Fig. 8K, right).245 Moreover, the new series of NCs was used in a comparative study in hydrogen evolution reaction (HER).245 The tri-Ih NC exhibited high catalytic activity for HER due to its low ligand-to-metal ratio, low-coordinated Au atoms and unfilled superatomic orbitals, providing a new strategy for constructing highly active catalysts from HER-inert metals (e.g. Au) via atomically precise nanochemistry. The current density of Au36Ag2(SR)18 at −0.3 V vs. RHE was 3.8 and 5.1 times that of Au25(SR′)18− and Au38(SR′)24, respectively. DFT calculations also revealed lower hydrogen binding energy and higher electron affinity of Au36Ag2(SR)18 (Fig. 8L).132
Bakr et al. reported a Cu81H32(SPh)46(tBuNH2)10 NC with an unprecedented planar Cu17 kernel inside a hemispherical shell comprised of a curved surface layer and a planar surface layer (Fig. 8M). This copper NC was synthesized with a mild reducing agent (borane tert-butylamine complex).246 However, this NC does not have any free valence electron.
Challenges and perspectives
The optical and chemical properties of NPs are strongly dependent on the particle size and shape. As the size of NPs shrinks to the ultrasmall regime (<3 nm in diameter), distinct optical properties with successive multiple absorption bands appear because these ultrasmall NPs start to show quantized or molecular states.One of the challenges is to design anisotropic NCs of atomic precision in hope that their optical properties can be tailored via anistropic growth. Indeed, as NCs grow from isotropic or spherical structures to the rod shapes, one can see that the lowest-energy absorption band red-shifts to longer wavelengths (from visible to NIR region). Note that only NCs with free valence electrons show tunable absorption bands, but not metal complexes. The NIR absorption of atomically precise NCs shows great importance due to the potential applications including photothermal conversion and NIR emmision which are very useful in medical therapy and bioimaging, especially because the NCs are smaller than the 5.5 nm—the renal clearance limit—and thus can undergo rapid excretion by kidney.247–249 But before we can effectively apply these atomically precise anisotropic NCs to real applications, some challenges should be addressed.
The first challenge pertains to the methodology for the synthesis of anisotropic NCs, which is still not well developed. However, some clues for future work are provided herein. Dimeric homogold and Pt/Pd doped Au38(SR)24 can be prepared by either direct reduction,75 or the recently developed method of fusion-mediated synthesis in which monomeric units are used as the precursor.158 Both rod-shaped Au25(SR)5(PPh3)10Cl2 and Au37(SR)10(PPh3)10Cl2 NCs are synthesized by reducing AuI(PPh3)Cl with NaBH4 to cluster-sized seeds, which further react with excess thiol upon moderate heating.187,188 Whether the growth mechanism of rod-shaped NCs is similar to that of plasmonic nanorods250 or not remains to be found out. Moreover, for rod-shaped Au NCs with fcc or hcp kernels, slow reduction is required to introduce anisotropic growth. Ultrathin Au nanorods with a constant diameter of ∼2 nm (within the NC size regime) and tunable length (5–20 nm) were obtained by slow reducing AuI in the presence of oleylamine, then ligand exchanging with thiolates.251 The ultrathin Au-SR nanorods show an intense band in the NIR and are not plamonic.251 The phenomenon is later proved in the resolved Au42(SR)32 nanorods of atomic precision.127 We expect that even longer close-packed kernels can be achieved by delicate kinetic control, however, the crystal growth of NCs with larger aspect ratios would be very challenging. In such cases, high-angle annular dark field scanning TEM with ultra-low dose would be necessary to characterize the shape of the NCs.252
No matter how exciting the atomcially precise structure is, it is required to convert the organic soluble NCs into aqua phase for broader biological applications. In the case of rod-shaped Au25−xAgx(SR)5(PPh3)10Cl2 (x = 1–13), the organic soluble thiolates are replaced by SC2H4OH so that the NCs can be used in fluorescence confocal imaging for living cancer cells.179 Au22(SG)18 (SG = glutathione) NC can be used as biocompatible light absorber to overcome the slow kinetics of electron transfer, enabling photosynthesis of acetic acid from CO2 when translocating into non-photosynthetic bacteria.253 The Zhang group investigated the peptide ligands stablized Au25 NCs with NIR-II emission (1100–1350 nm).254 The bright luminescence enhanced by Cu or Zn doping could penetrate deeper into the tissue, and be applied in in vivo brain vessel imaging and tumor metastasis.254 So far, very few biocompatable Au NCs of atomic precision have been synthesized using water-soluble ligands since it is very difficult for crystallization.18,255,256 Therefore, looking for an effective way to transfer the organo-soluble NCs into aqua solution is highly desirable in future work.
The second challenge is to design NCs with regioselective surface functionalization. Anisotropic NPs are usually associated with distinctive self-assembly behavior,257,258 making them quite attractive in fundamental research and various applications. In the NC regime, we have indeed observed the outstanding self-assemblies achieved by heterodimeric Au29(SR)19 NCs.38 But many of the properties of this NC have not been fully understood, e.g., its much longer excited-state lifetime compared to that of the homodimeric counterpart, and its NIR emission, either. When coupling a pair of nonequivalent NPs to form a heterodimer, the out-of-phase plasmon mode is no longer silent, whereas the in-phase mode is only allowed for homodimers.259 Thus, more experimental and theoretical works are needed on heterodimeric NCs to reveal the mechanisms in a more profound way. Moreover, noble metal (e.g., Ag) NPs can combine with magnetic (e.g., Fe3O4) NPs to form a bifunctional heterodimer for both fluorescence imaging and magnetic manipulation.260 It has been shown that a single (SR)3CdBr motif can be attached to the Au NC in atomic precision,122 which can be regarded as a simplified heterodimeric combination. It is worth trying to attach heteroatoms, e.g., Zn, Ni, Rh, on the surface of group-10 metal NCs to achieve a regioselective surface with specific functionalization.
The third challenge is how to grow oblate NCs into large sizes. The anisotropy of triangular metal nanoprisms show strong quadrupole plasmon excitation.261–263 Introducing iodide ion leads to triangular Au nanoprisms,264 and gold nanoprism growth could be controlled by slow reduction of Au ions onto the surface of seeding nanoprisms with the quadrupole plasmon resonance λmax shifting with the nanoprism edge length.265 Since the longitudinal transition has been confirmed in atomically precise NCs of rod-shape, we expect that quadrupole transitions might also be present in triangular oblate NCs. A delicate control over the aspect ratio with different ligands is still necessary.
In conclusion, atomically precise metal NCs with anisotropic structures reported so far have been summarized in this review, and their advantages over isotropic counterparts have been highlighted through comparison. While the synthesis of anisotropic NCs still remains a challenge, generally speaking, the presence of isotropic NC seeds in the precursor could be a way to prepare dimeric and rod-shaped NCs, and a very mild reducing environment is often required to prepare anisotropic nanostructures. With the longitudinal transition appeared in rod-shaped NCs and its tunability with the aspect ratio of the anisotropic structure, new physicochemical properties and applications have been observed and worth further studies in future research. The 2D or disk-shaped NCs also remain to be explored in terms of their unique properties and applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
R. J. acknowledges the financial support from the NSF (DMR-1808675).Notes and references
- C. Burda, X. Chen, R. Narayanan and M. A. El-Sayed, Chem. Rev., 2005, 105, 1025–1102 CrossRef CAS PubMed.
- M. Grzelczak, J. Pérez-Juste, P. Mulvaney and L. M. Liz-Marzán, Chem. Soc. Rev., 2008, 37, 1783–1791 RSC.
- R. Jin, Y. Charles Cao, E. Hao, G. S. Métraux, G. C. Schatz and C. A. Mirkin, Nature, 2003, 425, 487–490 CrossRef CAS PubMed.
- R. Narayanan and M. A. El-Sayed, Nano Lett., 2004, 4, 1343–1348 CrossRef CAS.
- C. J. Orendorff, T. K. Sau and C. J. Murphy, Small, 2006, 2, 636–639 CrossRef CAS PubMed.
- J. N. Anker, W. P. Hall, O. Lyandres, N. C. Shah, J. Zhao and R. P. Van Duyne, Nat. Mater., 2008, 7, 442–453 CrossRef CAS PubMed.
- S. Cao, F. (Feng) Tao, Y. Tang, Y. Li and J. Yu, Chem. Soc. Rev., 2016, 45, 4747–4765 RSC.
- J. Turkevich, P. C. Stevenson and J. Hillier, Discuss. Faraday Soc., 1951, 55–75 RSC.
- G. Ferns, Nat. Phys. Sci., 1973, 241, 20–22 CrossRef.
- G. Schmid, Chem. Rev., 1992, 92, 1709–1727 CrossRef CAS.
- M.-C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS PubMed.
- M. Giersig and P. Mulvaney, Langmuir, 1993, 9, 3408–3413 CrossRef CAS.
- R. Elghanian, J. J. Storhoff, R. C. Mucic, R. L. Letsinger and C. A. Mirkin, Science, 1997, 277, 1078–1081 CrossRef CAS PubMed.
- M. Brust, J. Fink, D. Bethell, D. J. Schiffrin and C. Kiely, J. Chem. Soc., Chem. Commun., 1995, 1655–1656 RSC.
- M. J. Hostetler, J. E. Wingate, C.-J. Zhong, J. E. Harris, R. W. Vachet, M. R. Clark, J. D. Londono, S. J. Green, J. J. Stokes, G. D. Wignall, G. L. Glish, M. D. Porter, N. D. Evans and R. W. Murray, Langmuir, 1998, 14, 17–30 CrossRef CAS.
- R. L. Whetten, J. T. Khoury, M. M. Alvarez, S. Murthy, I. Vezmar, Z. L. Wang, P. W. Stephens, C. L. Cleveland, W. D. Luedtke and U. Landman, Adv. Mater., 1996, 8, 428–433 CrossRef CAS.
- S. Chen, R. S. Ingram, M. J. Hostetler, J. J. Pietron, R. W. Murray, T. G. Schaaff, J. T. Khoury, M. M. Alvarez and R. L. Whetten, Science, 1998, 280, 2098–2101 CrossRef CAS PubMed.
- Y. Negishi, Y. Takasugi, S. Sato, H. Yao, K. Kimura and T. Tsukuda, J. Am. Chem. Soc., 2004, 126, 6518–6519 CrossRef CAS PubMed.
- Y. Negishi, K. Nobusada and T. Tsukuda, J. Am. Chem. Soc., 2005, 127, 5261–5270 CrossRef CAS PubMed.
- P. D. Jadzinsky, G. Calero, C. J. Ackerson, D. A. Bushnell and R. D. Kornberg, Science, 2007, 318, 430–433 CrossRef CAS PubMed.
- M. Zhu, C. M. Aikens, F. J. Hollander, G. C. Schatz and R. Jin, J. Am. Chem. Soc., 2008, 130, 5883–5885 CrossRef CAS PubMed.
- M. W. Heaven, A. Dass, P. S. White, K. M. Holt and R. W. Murray, J. Am. Chem. Soc., 2008, 130, 3754–3755 CrossRef CAS PubMed.
- R. Jin, Y. Zhu and H. Qian, Chem. – Eur. J., 2011, 17, 6584–6593 CrossRef CAS PubMed.
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed.
- I. Chakraborty and T. Pradeep, Chem. Rev., 2017, 117, 8208–8271 CrossRef CAS PubMed.
- M. Zhou, T. Higaki, G. Hu, M. Y. Sfeir, Y. Chen, D. Jiang and R. Jin, Science, 2019, 364, 279–282 CrossRef CAS PubMed.
- Q. Yao, T. Chen, X. Yuan and J. Xie, Acc. Chem. Res., 2018, 51, 1338–1348 CrossRef CAS PubMed.
- N. Xia and Z. Wu, Chem. Sci., 2021, 12, 2368–2380 RSC.
- T. Omoda, S. Takano and T. Tsukuda, Small, 2021, 17, 2001439 CrossRef CAS PubMed.
- R. Jin, G. Li, S. Sharma, Y. Li and X. Du, Chem. Rev., 2021, 121, 567–648 CrossRef CAS PubMed.
- Y. Jiang, S. Alvarez and R. Hoffmann, Inorg. Chem., 1985, 24, 749–757 CrossRef CAS.
- Y. Li, M. Zhou and R. Jin, Adv. Mater., 2021, 2006591 CrossRef CAS PubMed.
- R. Jin and T. Higaki, Commun. Chem., 2021, 4, 28 CrossRef CAS PubMed.
- H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2012, 41, 370–412 RSC.
- C. N. R. Rao, G. U. Kulkarni, P. J. Thomas and P. P. Edwards, Chem. Soc. Rev., 2000, 29, 27–35 RSC.
- M. Walter, J. Akola, O. Lopez-Acevedo, P. D. Jadzinsky, G. Calero, C. J. Ackerson, R. L. Whetten, H. Grönbeck and H. Häkkinen, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9157–9162 CrossRef CAS PubMed.
- T. Higaki, C. Liu, C. Zeng, R. Jin, Y. Chen, N. L. Rosi and R. Jin, Angew. Chem., Int. Ed., 2016, 55, 6694–6697 CrossRef CAS PubMed.
- Y. Li, M. Zhou, Y. Song, T. Higaki, H. Wang and R. Jin, Nature, 2021, 594, 380–384 CrossRef CAS PubMed.
- C. E. Anson, A. Eichhöfer, I. Issac, D. Fenske, O. Fuhr, P. Sevillano, C. Persau, D. Stalke and J. Zhang, Angew. Chem., Int. Ed., 2008, 47, 1326–1331 CrossRef CAS PubMed.
- M.-L. Fu, I. Issac, D. Fenske and O. Fuhr, Angew. Chem., Int. Ed., 2010, 49, 6899–6903 CrossRef CAS PubMed.
- S. C. Riha, B. A. Parkinson and A. L. Prieto, J. Am. Chem. Soc., 2011, 133, 15272–15275 CrossRef CAS PubMed.
- P. K. Santra and P. V. Kamat, J. Am. Chem. Soc., 2013, 135, 877–885 CrossRef CAS PubMed.
- M. Zhou, T. Higaki, Y. Li, C. Zeng, Q. Li, M. Y. Sfeir and R. Jin, J. Am. Chem. Soc., 2019, 141, 19754–19764 CrossRef CAS PubMed.
- T. Higaki, M. Zhou, K. J. Lambright, K. Kirschbaum, M. Y. Sfeir and R. Jin, J. Am. Chem. Soc., 2018, 140, 5691–5695 CrossRef CAS PubMed.
- B. Yoon, W. D. Luedtke, R. N. Barnett, J. Gao, A. Desireddy, B. E. Conn, T. P. Bigioni and U. Landman, Nat. Mater., 2014, 13, 807–811 CrossRef CAS PubMed.
- C. Zeng, Y. Chen, K. Kirschbaum, K. J. Lambright and R. Jin, Science, 2016, 354, 1580–1584 CrossRef CAS PubMed.
- J. Yan, S. Malola, C. Hu, J. Peng, B. Dittrich, B. K. Teo, H. Häkkinen, L. Zheng and N. Zheng, Nat. Commun., 2018, 9, 3357 CrossRef PubMed.
- Y. Li and R. Jin, J. Am. Chem. Soc., 2020, 142, 13627–13644 CrossRef CAS PubMed.
- G. Li and R. Jin, Acc. Chem. Res., 2013, 46, 1749–1758 CrossRef CAS PubMed.
- M. M. Montemore, M. A. van Spronsen, R. J. Madix and C. M. Friend, Chem. Rev., 2018, 118, 2816–2862 CrossRef CAS PubMed.
- T. Higaki, Y. Li, S. Zhao, Q. Li, S. Li, X.-S. Du, S. Yang, J. Chai and R. Jin, Angew. Chem., Int. Ed., 2019, 58, 8291–8302 CrossRef CAS PubMed.
- Y. Du, H. Sheng, D. Astruc and M. Zhu, Chem. Rev., 2020, 120, 526–622 CrossRef CAS PubMed.
- X.-K. Wan, S.-F. Yuan, Z.-W. Lin and Q.-M. Wang, Angew. Chem., Int. Ed., 2014, 53, 2923–2926 CrossRef CAS PubMed.
- R. L. Whetten, H.-C. Weissker, J. J. Pelayo, S. M. Mullins, X. López-Lozano and I. L. Garzón, Acc. Chem. Res., 2019, 52, 34–43 CrossRef CAS PubMed.
- Y. Li, T. Higaki, X. Du and R. Jin, Adv. Mater., 2020, 1905488 CrossRef CAS PubMed.
- S. Chen, W. Du, C. Qin, D. Liu, L. Tang, Y. Liu, S. Wang and M. Zhu, Angew. Chem., Int. Ed., 2020, 59, 7542–7547 CrossRef CAS PubMed.
- J. Pérez-Juste, I. Pastoriza-Santos, L. M. Liz-Marzán and P. Mulvaney, Coord. Chem. Rev., 2005, 249, 1870–1901 CrossRef.
- A. R. Tao, S. Habas and P. Yang, Small, 2008, 4, 310–325 CrossRef CAS.
- M. Rycenga, C. M. Cobley, J. Zeng, W. Li, C. H. Moran, Q. Zhang, D. Qin and Y. Xia, Chem. Rev., 2011, 111, 3669–3712 CrossRef CAS PubMed.
- M. L. Personick and C. A. Mirkin, J. Am. Chem. Soc., 2013, 135, 18238–18247 CrossRef CAS PubMed.
- E. C. Dreaden, A. M. Alkilany, X. Huang, C. J. Murphy and M. A. El-Sayed, Chem. Soc. Rev., 2012, 41, 2740–2779 RSC.
- H. Duan, D. Wang and Y. Li, Chem. Soc. Rev., 2015, 44, 5778–5792 RSC.
- X. M. Lin, H. M. Jaeger, C. M. Sorensen and K. J. Klabunde, J. Phys. Chem. B, 2001, 105, 3353–3357 CrossRef CAS.
- J. Th and G. Overbeek, Adv. Colloid Interface Sci., 1982, 15, 251–277 CrossRef.
- K. R. Brown, D. G. Walter and M. J. Natan, Chem. Mater., 2000, 12, 306–313 CrossRef CAS.
- N. R. Jana, L. Gearheart and C. J. Murphy, Langmuir, 2001, 17, 6782–6786 CrossRef CAS.
- Y. Zheng, X. Zhong, Z. Li and Y. Xia, Part. Part. Syst. Charact., 2014, 31, 266–273 CrossRef CAS.
- Z. Wu, J. Suhan and R. Jin, J. Mater. Chem., 2009, 19, 622–626 RSC.
- W. Zhang, J. Kong, Y. Li, Z. Kuang, H. Wang and M. Zhou, Chem. Sci., 2022, 13, 8124–8130 RSC.
- H. Qian, Y. Zhu and R. Jin, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 696–700 CrossRef CAS PubMed.
- T. Higaki, M. Zhou, G. He, S. D. House, M. Y. Sfeir, J. C. Yang and R. Jin, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 13215–13220 CrossRef CAS PubMed.
- R. Jin, Nanoscale, 2010, 2, 343–362 RSC.
- H. Qian, M. Zhu, Z. Wu and R. Jin, Acc. Chem. Res., 2012, 45, 1470–1479 CrossRef CAS PubMed.
- R. Jin, H. Qian, Z. Wu, Y. Zhu, M. Zhu, A. Mohanty and N. Garg, J. Phys. Chem. Lett., 2010, 1, 2903–2910 CrossRef CAS.
- H. Qian, Y. Zhu and R. Jin, ACS Nano, 2009, 3, 3795–3803 CrossRef CAS PubMed.
- H. Qian, W. T. Eckenhoff, Y. Zhu, T. Pintauer and R. Jin, J. Am. Chem. Soc., 2010, 132, 8280–8281 CrossRef CAS PubMed.
- H. Qian and R. Jin, Nano Lett., 2009, 9, 4083–4087 CrossRef CAS PubMed.
- N. Yan, N. Xia, L. Liao, M. Zhu, F. Jin, R. Jin and Z. Wu, Sci. Adv., 2018, 4, eaat7259 CrossRef CAS PubMed.
- C. Zeng, H. Qian, T. Li, G. Li, N. L. Rosi, B. Yoon, R. N. Barnett, R. L. Whetten, U. Landman and R. Jin, Angew. Chem., Int. Ed., 2012, 51, 13114–13118 CrossRef CAS PubMed.
- C. Zeng, T. Li, A. Das, N. L. Rosi and R. Jin, J. Am. Chem. Soc., 2013, 135, 10011–10013 CrossRef CAS PubMed.
- C. Zeng, C. Liu, Y. Chen, N. L. Rosi and R. Jin, J. Am. Chem. Soc., 2014, 136, 11922–11925 CrossRef CAS PubMed.
- A. Das, T. Li, G. Li, K. Nobusada, C. Zeng, N. L. Rosi and R. Jin, Nanoscale, 2014, 6, 6458–6462 RSC.
- C. Zeng, Y. Chen, K. Kirschbaum, K. Appavoo, M. Y. Sfeir and R. Jin, Sci. Adv., 2015, 1, e1500045 CrossRef PubMed.
- H. Dong, L. Liao and Z. Wu, J. Phys. Chem. Lett., 2017, 8, 5338–5343 CrossRef CAS PubMed.
- T. Higaki, C. Liu, M. Zhou, T.-Y. Luo, N. L. Rosi and R. Jin, J. Am. Chem. Soc., 2017, 139, 9994–10001 CrossRef CAS PubMed.
- Y. Chen, C. Zeng, D. R. Kauffman and R. Jin, Nano Lett., 2015, 15, 3603–3609 CrossRef CAS PubMed.
- C. Zeng, Y. Chen, C. Liu, K. Nobusada, N. L. Rosi and R. Jin, Sci. Adv., 2015, 1, e1500425 CrossRef PubMed.
- Y. Chen, C. Zeng, C. Liu, K. Kirschbaum, C. Gayathri, R. R. Gil, N. L. Rosi and R. Jin, J. Am. Chem. Soc., 2015, 137, 10076–10079 CrossRef CAS PubMed.
- Z. Gan, N. Xia and Z. Wu, Acc. Chem. Res., 2018, 51, 2774–2783 CrossRef CAS PubMed.
- S. Wang, Q. Li, X. Kang and M. Zhu, Acc. Chem. Res., 2018, 51, 2784–2792 CrossRef CAS PubMed.
- S. Hossain, Y. Niihori, L. V. Nair, B. Kumar, W. Kurashige and Y. Negishi, Acc. Chem. Res., 2018, 51, 3114–3124 CrossRef CAS PubMed.
- A. Ghosh, O. F. Mohammed and O. M. Bakr, Acc. Chem. Res., 2018, 51, 3094–3103 CrossRef CAS PubMed.
- S. Eustis and M. A. El-Sayed, Chem. Soc. Rev., 2006, 35, 209–217 RSC.
- J. E. Millstone, S. J. Hurst, G. S. Métraux, J. I. Cutler and C. A. Mirkin, Small, 2009, 5, 646–664 CrossRef CAS PubMed.
- T. S. Ahmadi, Z. L. Wang, T. C. Green, A. Henglein and M. A. El-Sayed, Science, 1996, 272, 1924–1925 CrossRef CAS PubMed.
- Y. Sun and Y. Xia, Science, 2002, 298, 2176–2179 CrossRef CAS PubMed.
- M. R. Langille, M. L. Personick, J. Zhang and C. A. Mirkin, J. Am. Chem. Soc., 2012, 134, 14542–14554 CrossRef CAS PubMed.
- C. J. Murphy, A. M. Gole, S. E. Hunyadi and C. J. Orendorff, Inorg. Chem., 2006, 45, 7544–7554 CrossRef CAS PubMed.
- S. E. Lohse and C. J. Murphy, Chem. Mater., 2013, 25, 1250–1261 CrossRef CAS.
- A. Gole and C. J. Murphy, Chem. Mater., 2004, 16, 3633–3640 CrossRef CAS.
- F. Kim, J. H. Song and P. Yang, J. Am. Chem. Soc., 2002, 124, 14316–14317 CrossRef CAS PubMed.
- Y.-Y. Yu, S.-S. Chang, C.-L. Lee and C. R. C. Wang, J. Phys. Chem. B, 1997, 101, 6661–6664 CrossRef CAS.
- C. Sönnichsen and A. P. Alivisatos, Nano Lett., 2005, 5, 301–304 CrossRef PubMed.
- J. Becker, A. Trügler, A. Jakab, U. Hohenester and C. Sönnichsen, Plasmonics, 2010, 5, 161–167 CrossRef CAS.
- L. Vigderman, B. P. Khanal and E. R. Zubarev, Adv. Mater., 2012, 24, 4811–4841 CrossRef CAS PubMed.
- Y. W. C. Cao, R. Jin and C. A. Mirkin, Science, 2002, 297, 1536–1540 CrossRef CAS PubMed.
- H. Chen, L. Shao, Q. Li and J. Wang, Chem. Soc. Rev., 2013, 42, 2679–2724 RSC.
- P. Zijlstra, P. M. R. Paulo and M. Orrit, Nat. Nanotechnol., 2012, 7, 379–382 CrossRef CAS PubMed.
- V. Amendola, R. Pilot, M. Frasconi, O. M. Maragò and M. A. Iatì, J. Phys., 2017, 29, 203002 Search PubMed.
- M. Yu and J. Zheng, ACS Nano, 2015, 9, 6655–6674 CrossRef CAS PubMed.
- L. Tong, Q. Wei, A. Wei and J.-X. Cheng, Photochem. Photobiol., 2009, 85, 21–32 CrossRef CAS PubMed.
- J. Chen, C. Glaus, R. Laforest, Q. Zhang, M. Yang, M. Gidding, M. J. Welch and Y. Xia, Small, 2010, 6, 811–817 CrossRef CAS PubMed.
- S. M. Moghimi, A. C. Hunter and J. C. Murray, Pharmacol. Rev., 2001, 53, 283 CAS.
- M. Ferrari, Nat. Rev. Cancer, 2005, 5, 161–171 CrossRef CAS PubMed.
- N. L. Rosi and C. A. Mirkin, Chem. Rev., 2005, 105, 1547–1562 CrossRef CAS PubMed.
- X. Huang, P. K. Jain, I. H. El-Sayed and M. A. El-Sayed, Nanomedicine, 2007, 2, 681–693 CrossRef CAS PubMed.
- A. Albanese, P. S. Tang and W. C. W. Chan, Annu. Rev. Biomed. Eng., 2012, 14, 1–16 CrossRef CAS PubMed.
- A. Desireddy, B. E. Conn, J. Guo, B. Yoon, R. N. Barnett, B. M. Monahan, K. Kirschbaum, W. P. Griffith, R. L. Whetten, U. Landman and T. P. Bigioni, Nature, 2013, 501, 399–402 CrossRef CAS PubMed.
- Y. Wang, X.-K. Wan, L. Ren, H. Su, G. Li, S. Malola, S. Lin, Z. Tang, H. Häkkinen, B. K. Teo, Q.-M. Wang and N. Zheng, J. Am. Chem. Soc., 2016, 138, 3278–3281 CrossRef CAS PubMed.
- F. Hu, Z.-J. Guan, G. Yang, J.-Q. Wang, J.-J. Li, S.-F. Yuan, G.-J. Liang and Q.-M. Wang, J. Am. Chem. Soc., 2021, 143, 17059–17067 CrossRef CAS PubMed.
- I. Dolamic, S. Knoppe, A. Dass and T. Bürgi, Nat. Commun., 2012, 3, 798 CrossRef PubMed.
- Y. Li, M. J. Cowan, M. Zhou, M. G. Taylor, H. Wang, Y. Song, G. Mpourmpakis and R. Jin, ACS Nano, 2020, 14, 6599–6606 CrossRef CAS PubMed.
- Z. Wu, G. Hu, D. Jiang, D. R. Mullins, Q.-F. Zhang, L. F. Allard, L.-S. Wang and S. H. Overbury, Nano Lett., 2016, 16, 6560–6567 CrossRef CAS PubMed.
- V. K. Kulkarni, B. N. Khiarak, S. Takano, S. Malola, E. L. Albright, T. I. Levchenko, M. D. Aloisio, C.-T. Dinh, T. Tsukuda, H. Häkkinen and C. M. Crudden, J. Am. Chem. Soc., 2022, 144, 9000–9006 CrossRef CAS PubMed.
- L. Liao, C. Wang, S. Zhuang, N. Yan, Y. Zhao, Y. Yang, J. Li, H. Deng and Z. Wu, Angew. Chem., Int. Ed., 2020, 59, 731–734 CrossRef CAS PubMed.
- Z. Gan, Y. Lin, L. Luo, G. Han, W. Liu, Z. Liu, C. Yao, L. Weng, L. Liao, J. Chen, X. Liu, Y. Luo, C. Wang, S. Wei and Z. Wu, Angew. Chem., Int. Ed., 2016, 55, 11567–11571 CrossRef CAS PubMed.
- Y. Li, Y. Song, X. Zhang, T. Liu, T. Xu, H. Wang, D. Jiang and R. Jin, J. Am. Chem. Soc., 2022, 144, 12381–12389 CrossRef CAS PubMed.
- L. Luo, Z. Liu, X. Du and R. Jin, J. Am. Chem. Soc., 2022, 144, 19243–19247 CrossRef CAS PubMed.
- Y. Shichibu and K. Konishi, Inorg. Chem., 2013, 52, 6570–6575 CrossRef CAS PubMed.
- M. Zhou, R. Jin, M. Y. Sfeir, Y. Chen, Y. Song and R. Jin, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E4697–E4705 CrossRef CAS PubMed.
- S. Ito, S. Takano and T. Tsukuda, J. Phys. Chem. Lett., 2019, 10, 6892–6896 CrossRef CAS PubMed.
- Y. Li, S. Li, A. V. Nagarajan, Z. Liu, S. Nevins, Y. Song, G. Mpourmpakis and R. Jin, J. Am. Chem. Soc., 2021, 143, 11102–11108 CrossRef CAS PubMed.
- Y. Song, Y. Li, M. Zhou, H. Li, T. Xu, C. Zhou, F. Ke, D. Huo, Y. Wan, J. Jie, W. W. Xu, M. Zhu and R. Jin, Nat. Commun., 2022, 13, 1235 CrossRef CAS PubMed.
- G. Chen, Y. Wang, L. H. Tan, M. Yang, L. S. Tan, Y. Chen and H. Chen, J. Am. Chem. Soc., 2009, 131, 4218–4219 CrossRef CAS PubMed.
- K. L. Wustholz, A.-I. Henry, J. M. McMahon, R. G. Freeman, N. Valley, M. E. Piotti, M. J. Natan, G. C. Schatz and R. P. Van Duyne, J. Am. Chem. Soc., 2010, 132, 10903–10910 CrossRef CAS PubMed.
- X. Liu, G. Saranya, X. Huang, X. Cheng, R. Wang, M. Chen, C. Zhang, T. Li and Y. Zhu, Angew. Chem., Int. Ed., 2020, 59, 13941–13946 CrossRef CAS PubMed.
- X. Cai, W. Hu, S. Xu, D. Yang, M. Chen, M. Shu, R. Si, W. Ding and Y. Zhu, J. Am. Chem. Soc., 2020, 142, 4141–4153 CrossRef CAS PubMed.
- Z.-H. Gao, K. Wei, T. Wu, J. Dong, D. Jiang, S. Sun and L.-S. Wang, J. Am. Chem. Soc., 2022, 144, 5258–5262 CrossRef CAS PubMed.
- S.-F. Yuan, J.-J. Li, Z.-J. Guan, Z. Lei and Q.-M. Wang, Chem. Commun., 2020, 56, 7037–7040 RSC.
- J. Xu, L. Xiong, X. Cai, S. Tang, A. Tang, X. Liu, Y. Pei and Y. Zhu, Chem. Sci., 2022, 13, 2778–2782 RSC.
- A. Das, T. Li, K. Nobusada, Q. Zeng, N. L. Rosi and R. Jin, J. Am. Chem. Soc., 2012, 134, 20286–20289 CrossRef CAS PubMed.
- J. Chen, Q.-F. Zhang, T. A. Bonaccorso, P. G. Williard and L.-S. Wang, J. Am. Chem. Soc., 2014, 2014, 92–95 CrossRef PubMed.
- J. Dong, Z.-H. Gao, Q.-F. Zhang and L.-S. Wang, Angew. Chem., Int. Ed., 2021, 60, 2424–2430 CrossRef CAS PubMed.
- S. Jin, X. Zou, L. Xiong, W. Du, S. Wang, Y. Pei and M. Zhu, Angew. Chem., Int. Ed., 2018, 57, 16768–16772 CrossRef CAS PubMed.
- O. Lopez-Acevedo, H. Tsunoyama, T. Tsukuda, H. Häkkinen and C. M. Aikens, J. Am. Chem. Soc., 2010, 132, 8210–8218 CrossRef CAS PubMed.
- L. Cheng, C. Ren, X. Zhang and J. Yang, Nanoscale, 2013, 5, 1475–1478 RSC.
- L. Cheng and J. Yang, J. Chem. Phys., 2013, 138, 141101 CrossRef PubMed.
- Y. Li, R. Juarez-Mosqueda, Y. Song, Y. Zhang, J. Chai, G. Mpourmpakis and R. Jin, Nanoscale, 2020, 12, 9423–9429 RSC.
- M. S. Devadas, S. Bairu, H. Qian, E. Sinn, R. Jin and G. Ramakrishna, J. Phys. Chem. Lett., 2011, 2, 2752–2758 CrossRef CAS.
- C. Kumara and A. Dass, Nanoscale, 2012, 4, 4084–4086 RSC.
- C. Kumara, K. J. Gagnon and A. Dass, J. Phys. Chem. Lett., 2015, 6, 1223–1228 CrossRef CAS PubMed.
- J. Chai, Y. Lv, S. Yang, Y. Song, X. Zan, Q. Li, H. Yu, M. Wu and M. Zhu, J. Phys. Chem. C, 2017, 121, 21665–21669 CrossRef CAS.
- Y. Negishi, K. Igarashi, K. Munakata, W. Ohgake and K. Nobusada, Chem. Commun., 2012, 48, 660–662 RSC.
- M. Kim, Q. Tang, A. V. Narendra Kumar, K. Kwak, W. Choi, D. Jiang and D. Lee, J. Phys. Chem. Lett., 2018, 9, 982–989 CrossRef CAS PubMed.
- B. Zhang, S. Kaziz, H. Li, D. Wodka, S. Malola, O. Safonova, M. Nachtegaal, C. Mazet, I. Dolamic, J. Llorca, E. Kalenius, L. M. L. Daku, H. Häkkinen, T. Bürgi and N. Barrabés, Nanoscale, 2015, 7, 17012–17019 RSC.
- N. Barrabés, B. Zhang and T. Bürgi, J. Am. Chem. Soc., 2014, 136, 14361–14364 CrossRef PubMed.
- E. Ito, S. Takano, T. Nakamura and T. Tsukuda, Angew. Chem., Int. Ed., 2021, 60, 645–649 CrossRef CAS PubMed.
- E. Ito, S. Ito, S. Takano, T. Nakamura and T. Tsukuda, JACS Au, 2022, 2, 2627–2634 CrossRef CAS PubMed.
- X. Liu, E. Wang, M. Zhou, Y. Wan, Y. Zhang, H. Liu, Y. Zhao, J. Li, Y. Gao and Y. Zhu, Angew. Chem., Int. Ed., 2022, 61, e202207685 CAS.
- S. Lee, M. S. Bootharaju, G. Deng, S. Malola, H. Häkkinen, N. Zheng and T. Hyeon, J. Am. Chem. Soc., 2021, 143, 12100–12107 CrossRef CAS PubMed.
- T. Dainese, S. Antonello, S. Bogialli, W. Fei, A. Venzo and F. Maran, ACS Nano, 2018, 12, 7057–7066 CrossRef CAS PubMed.
- S. Tian, Y.-Z. Li, M.-B. Li, J. Yuan, J. Yang, Z. Wu and R. Jin, Nat. Commun., 2015, 6, 8667 CrossRef CAS PubMed.
- Z. Wang, R. Senanayake, C. M. Aikens, W.-M. Chen, C.-H. Tung and D. Sun, Nanoscale, 2016, 8, 18905–18911 RSC.
- K. Nunokawa, M. Ito, T. Sunahara, S. Onaka, T. Ozeki, H. Chiba, Y. Funahashi, H. Masuda, T. Yonezawa, H. Nishihara, M. Nakamoto and M. Yamamoto, Dalton Trans., 2005, 2726–2730 RSC.
- X.-K. Wan, Z.-W. Lin and Q.-M. Wang, J. Am. Chem. Soc., 2012, 134, 14750–14752 CrossRef CAS PubMed.
- Y. Li, M. G. Taylor, T.-Y. Luo, Y. Song, N. L. Rosi, G. Mpourmpakis and R. Jin, J. Phys. Chem. Lett., 2020, 11, 7307–7312 CrossRef CAS PubMed.
- L. Xiong, B. Peng, Z. Ma, P. Wang and Y. Pei, Nanoscale, 2017, 9, 2895–2902 RSC.
- Y. Chen, C. Liu, Q. Tang, C. Zeng, T. Higaki, A. Das, D.-E. Jiang, N. L. Rosi and R. Jin, J. Am. Chem. Soc., 2016, 138, 1482–1485 CrossRef CAS PubMed.
- W. Du, S. Deng, S. Chen, S. Jin, Y. Zhen, Y. Pei and M. Zhu, J. Phys. Chem. Lett., 2021, 12, 6654–6660 CrossRef CAS PubMed.
- X. Wei, H. Shen, C. Xu, H. Li, S. Jin, X. Kang and M. Zhu, Inorg. Chem., 2021, 60, 5931–5936 CrossRef CAS PubMed.
- Y. Song, S. Weng, H. Li, H. Yu and M. Zhu, Inorg. Chem., 2019, 58, 7136–7140 CrossRef CAS PubMed.
- Q. Li, K. J. Lambright, M. G. Taylor, K. Kirschbaum, T.-Y. Luo, J. Zhao, G. Mpourmpakis, S. Mokashi-Punekar, N. L. Rosi and R. Jin, J. Am. Chem. Soc., 2017, 139, 17779–17782 CrossRef CAS PubMed.
- S. Li, A. V. Nagarajan, D. R. Alfonso, M. Sun, D. R. Kauffman, G. Mpourmpakis and R. Jin, Angew. Chem., Int. Ed., 2021, 60, 6351–6356 CrossRef CAS PubMed.
- Y. Kamei, Y. Shichibu and K. Konishi, Angew. Chem., Int. Ed., 2011, 50, 7442–7445 CrossRef CAS PubMed.
- Y. Shichibu, Y. Kamei and K. Konishi, Chem. Commun., 2012, 48, 7559–7561 RSC.
- N. Kobayashi, Y. Kamei, Y. Shichibu and K. Konishi, J. Am. Chem. Soc., 2013, 135, 16078–16081 CrossRef CAS PubMed.
- K. Konishi, M. Iwasaki and Y. Shichibu, Acc. Chem. Res., 2018, 51, 3125–3133 CrossRef CAS PubMed.
- M. A. Bakar, M. Sugiuchi, M. Iwasaki, Y. Shichibu and K. Konishi, Nat. Commun., 2017, 8, 576 CrossRef PubMed.
- S. Wang, X. Meng, A. Das, T. Li, Y. Song, T. Cao, X. Zhu, M. Zhu and R. Jin, Angew. Chem., Int. Ed., 2014, 53, 2376–2380 CrossRef CAS PubMed.
- H. Shen, G. Deng, S. Kaappa, T. Tan, Y.-Z. Han, S. Malola, S.-C. Lin, B. K. Teo, H. Häkkinen and N. Zheng, Angew. Chem., Int. Ed., 2019, 58, 17731–17735 CrossRef CAS PubMed.
- S.-F. Yuan, C.-Q. Xu, W.-D. Liu, J.-X. Zhang, J. Li and Q.-M. Wang, J. Am. Chem. Soc., 2021, 143, 12261–12267 CrossRef CAS PubMed.
- T.-H. Chiu, J.-H. Liao, F. Gam, I. Chantrenne, S. Kahlal, J.-Y. Saillard and C. W. Liu, J. Am. Chem. Soc., 2019, 141, 12957–12961 CrossRef CAS PubMed.
- B. K. Teo and H. Zhang, Inorg. Chem., 1991, 30, 3115–3116 CrossRef CAS.
- B. K. Teo, H. Zhang and X. Shi, J. Am. Chem. Soc., 1990, 112, 8552–8562 CrossRef CAS.
- B. K. Teo and H. Zhang, Coord. Chem. Rev., 1995, 143, 611–636 CrossRef CAS.
- Y. Shichibu, Y. Negishi, T. Tsukuda and T. Teranishi, J. Am. Chem. Soc., 2005, 127, 13464–13465 CrossRef CAS PubMed.
- Y. Shichibu, Y. Negishi, T. Watanabe, N. K. Chaki, H. Kawaguchi and T. Tsukuda, J. Phys. Chem. C, 2007, 111, 7845–7847 CrossRef CAS.
- R. Jin, C. Liu, S. Zhao, A. Das, H. Xing, C. Gayathri, Y. Xing, N. L. Rosi, R. R. Gil and R. Jin, ACS Nano, 2015, 9, 8530–8536 CrossRef CAS PubMed.
- Y. Shichibu and K. Konishi, Small, 2010, 6, 1216–1220 CrossRef CAS PubMed.
- D. M. P. Mingos, J. Chem. Soc., Chem. Commun., 1985, 1352–1354 RSC.
- K. Nobusada and T. Iwasa, J. Phys. Chem. C, 2007, 111, 14279–14282 CrossRef CAS.
- M. Y. Sfeir, H. Qian, K. Nobusada and R. Jin, J. Phys. Chem. C, 2011, 115, 6200–6207 CrossRef CAS.
- S. Park and D. Lee, Langmuir, 2012, 28, 7049–7054 CrossRef CAS PubMed.
- S. Chen, H. Ma, J. W. Padelford, W. Qinchen, W. Yu, S. Wang, M. Zhu and G. Wang, J. Am. Chem. Soc., 2019, 141, 9603–9609 CrossRef CAS PubMed.
- X. Kang, J. Xiang, Y. Lv, W. Du, H. Yu, S. Wang and M. Zhu, Chem. Mater., 2017, 29, 6856–6862 CrossRef CAS.
- L. V. Nair, S. Hossain, S. Takagi, Y. Imai, G. Hu, S. Wakayama, B. Kumar, W. Kurashige, D. Jiang and Y. Negishi, Nanoscale, 2018, 18969–18979 RSC.
- M. S. Bootharaju, S. M. Kozlov, Z. Cao, M. Harb, N. Maity, A. Shkurenko, M. R. Parida, M. N. Hedhili, M. Eddaoudi, O. F. Mohammed, O. M. Bakr, L. Cavallo and J.-M. Basset, J. Am. Chem. Soc., 2017, 139, 1053–1056 CrossRef CAS PubMed.
- S. Hossain, S. Miyajima, T. Iwasa, R. Kaneko, T. Sekine, A. Ikeda, T. Kawawaki, T. Taketsugu and Y. Negishi, J. Chem. Phys., 2021, 155, 024302 CrossRef CAS PubMed.
- F. Gam, C. W. Liu, S. Kahlal and J.-Y. Saillard, Nanoscale, 2020, 12, 20308–20316 RSC.
- S.-F. Yuan, Z.-J. Guan, W.-D. Liu and Q.-M. Wang, Nat. Commun., 2019, 10, 4032 CrossRef PubMed.
- C. Zeng, Y. Chen, K. Iida, K. Nobusada, K. Kirschbaum, K. J. Lambright and R. Jin, J. Am. Chem. Soc., 2016, 138, 3950–3953 CrossRef CAS PubMed.
- W. W. Xu, X. Duan and X. C. Zeng, J. Phys. Chem. Lett., 2020, 11, 536–540 CrossRef CAS PubMed.
- S. Zhuang, L. Liao, Y. Zhao, J. Yuan, C. Yao, X. Liu, J. Li, H. Deng, J. Yang and Z. Wu, Chem. Sci., 2018, 9, 2437–2442 RSC.
- S. Takano, S. Yamazoe, K. Koyasu and T. Tsukuda, J. Am. Chem. Soc., 2015, 137, 7027–7030 CrossRef CAS PubMed.
- W. W. Xu, Y. Li, Y. Gao and X.-C. Zeng, Nanoscale, 2016, 8, 7396–7401 RSC.
- Z. Ma, P. Wang, G. Zhou, J. Tang, H. Li and Y. Pei, J. Phys. Chem. C, 2016, 120, 13739–13748 CrossRef CAS.
- S. Yang, J. Chai, Y. Song, X. Kang, H. Sheng, H. Chong and M. Zhu, J. Am. Chem. Soc., 2015, 137, 10033–10035 CrossRef CAS PubMed.
- H. Yang, J. Lei, B. Wu, Y. Wang, M. Zhou, A. Xia, L. Zheng and N. Zheng, Chem. Commun., 2013, 49, 300–302 RSC.
- C. Liu, T. Li, H. Abroshan, Z. Li, C. Zhang, H. J. Kim, G. Li and R. Jin, Nat. Commun., 2018, 9, 744 CrossRef PubMed.
- H. Yang, J. Yan, Y. Wang, H. Su, L. Gell, X. Zhao, C. Xu, B. K. Teo, H. Häkkinen and N. Zheng, J. Am. Chem. Soc., 2017, 139, 31–34 CrossRef CAS PubMed.
- M. J. Alhilaly, M. S. Bootharaju, C. P. Joshi, T. M. Besong, A.-H. Emwas, R. Juarez-Mosqueda, S. Kaappa, S. Malola, K. Adil, A. Shkurenko, H. Häkkinen, M. Eddaoudi and O. M. Bakr, J. Am. Chem. Soc., 2016, 138, 14727–14732 CrossRef CAS PubMed.
- C. Zeng, C. Liu, Y. Chen, N. L. Rosi and R. Jin, J. Am. Chem. Soc., 2016, 138, 8710–8713 CrossRef CAS PubMed.
- S. Zhuang, L. Liao, M.-B. Li, C. Yao, Y. Zhao, H. Dong, J. Li, H. Deng, L. Li and Z. Wu, Nanoscale, 2017, 9, 14809–14813 RSC.
- J.-Q. Wang, S. Shi, R.-L. He, S.-F. Yuan, G.-Y. Yang, G.-J. Liang and Q.-M. Wang, J. Am. Chem. Soc., 2020, 142, 18086–18092 CrossRef CAS PubMed.
- M. Zhu, H. Qian and R. Jin, J. Phys. Chem. Lett., 2010, 1, 1003–1007 CrossRef CAS.
- Y. Pei, R. Pal, C. Liu, Y. Gao, Z. Zhang and X. C. Zeng, J. Am. Chem. Soc., 2012, 134, 3015–3024 CrossRef CAS PubMed.
- Y. Song, S. Wang, J. Zhang, X. Kang, S. Chen, P. Li, H. Sheng and M. Zhu, J. Am. Chem. Soc., 2014, 136, 2963–2965 CrossRef CAS PubMed.
- A. Das, C. Liu, H. Y. Byun, K. Nobusada, S. Zhao, N. L. Rosi and R. Jin, Angew. Chem., Int. Ed., 2015, 54, 3140–3144 CrossRef CAS PubMed.
- S. Chen, S. Wang, J. Zhong, Y. Song, J. Zhang, H. Sheng, Y. Pei and M. Zhu, Angew. Chem., Int. Ed., 2015, 54, 3145–3149 CrossRef CAS PubMed.
- J. Xiang, P. Li, Y. Song, X. Liu, H. Chong, S. Jin, Y. Pei, X. Yuan and M. Zhu, Nanoscale, 2015, 7, 18278–18283 RSC.
- T. Higaki, C. Liu, C. Zeng, R. Jin, Y. Chen, N. L. Rosi and R. Jin, Angew. Chem., Int. Ed., 2016, 55, 6694–6697 CrossRef CAS PubMed.
- M. R. K. Ali, M. A. Rahman, Y. Wu, T. Han, X. Peng, M. A. Mackey, D. Wang, H. J. Shin, Z. G. Chen, H. Xiao, R. Wu, Y. Tang, D. M. Shin and M. A. El-Sayed, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E3110 CAS.
- X. Jiang, B. Du, S. Tang, J.-T. Hsieh and J. Zheng, Angew. Chem., Int. Ed., 2019, 58, 5994–6000 CrossRef CAS PubMed.
- W. Fan, Y. Yang, Q. You, J. Li, H. Deng, N. Yan and Z. Wu, J. Phys. Chem. C, 2023, 127, 816–823 CrossRef CAS.
- Z. Lei, Z.-J. Guan, X.-L. Pei, S.-F. Yuan, X.-K. Wan, J.-Y. Zhang and Q.-M. Wang, Chem. – Eur. J., 2016, 22, 11156–11160 CrossRef CAS PubMed.
- L. He, J. Yuan, N. Xia, L. Liao, X. Liu, Z. Gan, C. Wang, J. Yang and Z. Wu, J. Am. Chem. Soc., 2018, 140, 3487–3490 CrossRef CAS PubMed.
- L. He, Z. Gan, N. Xia, L. Liao and Z. Wu, Angew. Chem., Int. Ed., 2019, 58, 9897–9901 CrossRef CAS PubMed.
- S.-S. Zhang, H.-F. Su, Z. Wang, X.-P. Wang, W.-X. Chen, Q.-Q. Zhao, C.-H. Tung, D. Sun and L.-S. Zheng, Chem. – Eur. J., 2018, 24, 1998–2003 CrossRef CAS PubMed.
- J.-W. Liu, L. Feng, H.-F. Su, Z. Wang, Q.-Q. Zhao, X.-P. Wang, C.-H. Tung, D. Sun and L.-S. Zheng, J. Am. Chem. Soc., 2018, 140, 1600–1603 CrossRef CAS PubMed.
- Y.-M. Su, Z. Wang, G.-L. Zhuang, Q.-Q. Zhao, X.-P. Wang, C.-H. Tung and D. Sun, Chem. Sci., 2019, 10, 564–568 RSC.
- J.-W. Liu, H.-F. Su, Z. Wang, Y.-A. Li, Q.-Q. Zhao, X.-P. Wang, C.-H. Tung, D. Sun and L.-S. Zheng, Chem. Commun., 2018, 54, 4461–4464 RSC.
- X.-K. Wan, Q. Tang, S.-F. Yuan, D.-E. Jiang and Q.-M. Wang, J. Am. Chem. Soc., 2015, 137, 652–655 CrossRef CAS PubMed.
- Z. Lei, X.-K. Wan, S.-F. Yuan, Z.-J. Guan and Q.-M. Wang, Acc. Chem. Res., 2018, 51, 2465–2474 CrossRef CAS PubMed.
- X.-S. Han, X. Luan, H.-F. Su, J.-J. Li, S.-F. Yuan, Z. Lei, Y. Pei and Q.-M. Wang, Angew. Chem., Int. Ed., 2020, 59, 2309–2312 CrossRef CAS PubMed.
- S. Yang, J. Chai, Y. Lv, T. Chen, S. Wang, H. Yu and M. Zhu, Chem. Commun., 2018, 54, 12077–12080 RSC.
- L. Huang, J. Yan, L. Ren, B. K. Teo and N. Zheng, Dalton Trans., 2017, 46, 1757–1760 RSC.
- H. Yang, J. Yan, Y. Wang, G. Deng, H. Su, X. Zhao, C. Xu, B. K. Teo and N. Zheng, J. Am. Chem. Soc., 2017, 139, 16113–16116 CrossRef CAS PubMed.
- Y. Song, F. Fu, J. Zhang, J. Chai, X. Kang, P. Li, S. Li, H. Zhou and M. Zhu, Angew. Chem., Int. Ed., 2015, 54, 8430–8434 CrossRef CAS PubMed.
- Y. Song, Y. Li, H. Li, F. Ke, J. Xiang, C. Zhou, P. Li, M. Zhu and R. Jin, Nat. Commun., 2020, 11, 478 CrossRef CAS PubMed.
- H. Yang, Y. Wang, X. Chen, X. Zhao, L. Gu, H. Huang, J. Yan, C. Xu, G. Li, J. Wu, A. J. Edwards, B. Dittrich, Z. Tang, D. Wang, L. Lehtovaara, H. Häkkinen and N. Zheng, Nat. Commun., 2016, 7, 12809 CrossRef CAS PubMed.
- Y. Song, K. J. Lambright, M. Zhou, K. Kirschbaum, J. Xiang, A. Xia, M. Zhu and R. Jin, ACS Nano, 2018, 12, 9318–9325 CrossRef CAS PubMed.
- T. Higaki, C. Liu, D. J. Morris, G. He, T.-Y. Luo, M. Y. Sfeir, P. Zhang, N. L. Rosi and R. Jin, Angew. Chem., Int. Ed., 2019, 58, 18798–18802 CrossRef CAS PubMed.
- X.-K. Wan, J.-Q. Wang and Q.-M. Wang, Angew. Chem., Int. Ed., 2021, 60, 20748–20753 CrossRef CAS PubMed.
- Y. Li, T.-Y. Luo, M. Zhou, Y. Song, N. L. Rosi and R. Jin, J. Am. Chem. Soc., 2018, 140, 14235–14243 CrossRef CAS PubMed.
- C. Xu, Y. Zhou, L. Shi and L. Cheng, J. Phys. Chem. Lett., 2022, 13, 10147–10152 CrossRef CAS PubMed.
- R.-W. Huang, J. Yin, C. Dong, A. Ghosh, M. J. Alhilaly, X. Dong, M. N. Hedhili, E. Abou-Hamad, B. Alamer, S. Nematulloev, Y. Han, O. F. Mohammed and O. M. Bakr, J. Am. Chem. Soc., 2020, 142, 8696–8705 CrossRef PubMed.
- H. Soo Choi, W. Liu, P. Misra, E. Tanaka, J. P. Zimmer, B. Itty Ipe, M. G. Bawendi and J. V. Frangioni, Nat. Biotechnol., 2007, 25, 1165–1170 CrossRef PubMed.
- B. Du, X. Jiang, A. Das, Q. Zhou, M. Yu, R. Jin and J. Zheng, Nat. Nanotechnol., 2017, 12, 1096–1102 CrossRef CAS PubMed.
- C. N. Loynachan, A. P. Soleimany, J. S. Dudani, Y. Lin, A. Najer, A. Bekdemir, Q. Chen, S. N. Bhatia and M. M. Stevens, Nat. Nanotechnol., 2019, 14, 883–890 CrossRef CAS PubMed.
- C. J. Murphy and N. R. Jana, Adv. Mater., 2002, 14, 80–82 CrossRef CAS.
- R. Takahata, S. Yamazoe, K. Koyasu, K. Imura and T. Tsukuda, J. Am. Chem. Soc., 2018, 140, 6640–6647 CrossRef CAS PubMed.
- Q. Yao, L. Liu, S. Malola, M. Ge, H. Xu, Z. Wu, T. Chen, Y. Cao, M. F. Matus, A. Pihlajamäki, Y. Han, H. Häkkinen and J. Xie, Nat. Chem., 2023, 15, 230–239 CrossRef CAS PubMed.
- H. Zhang, H. Liu, Z. Tian, D. Lu, Y. Yu, S. Cestellos-Blanco, K. K. Sakimoto and P. Yang, Nat. Nanotechnol., 2018, 13, 900–905 CrossRef CAS PubMed.
- H. Liu, G. Hong, Z. Luo, J. Chen, J. Chang, M. Gong, H. He, J. Yang, X. Yuan, L. Li, X. Mu, J. Wang, W. Mi, J. Luo, J. Xie and X.-D. Zhang, Adv. Mater., 2019, 31, 1901015 CrossRef CAS PubMed.
- S. Kumar and R. Jin, Nanoscale, 2012, 4, 4222–4227 RSC.
- T. Chen, V. Fung, Q. Yao, Z. Luo, D. Jiang and J. Xie, J. Am. Chem. Soc., 2018, 140, 11370–11377 CrossRef CAS PubMed.
- L. H. Tan, H. Xing, H. Chen and Y. Lu, J. Am. Chem. Soc., 2013, 135, 17675–17678 CrossRef CAS PubMed.
- G. Chen, K. J. Gibson, D. Liu, H. C. Rees, J.-H. Lee, W. Xia, R. Lin, H. L. Xin, O. Gang and Y. Weizmann, Nat. Mater., 2019, 18, 169–174 CrossRef CAS PubMed.
- S. Sheikholeslami, Y. Jun, P. K. Jain and A. P. Alivisatos, Nano Lett., 2010, 10, 2655–2660 CrossRef CAS PubMed.
- J. Jiang, H. Gu, H. Shao, E. Devlin, G. C. Papaefthymiou and J. Y. Ying, Adv. Mater., 2008, 20, 4403–4407 CrossRef CAS.
- R. Jin, Y. W. Cao, C. A. Mirkin, K. L. Kelly, G. C. Schatz and J. G. Zheng, Science, 2001, 294, 1901–1903 CrossRef CAS PubMed.
- S. S. Shankar, A. Rai, B. Ankamwar, A. Singh, A. Ahmad and M. Sastry, Nat. Mater., 2004, 3, 482–488 CrossRef CAS PubMed.
- L. J. Sherry, R. Jin, C. A. Mirkin, G. C. Schatz and R. P. Van Duyne, Nano Lett., 2006, 6, 2060–2065 CrossRef CAS PubMed.
- T. H. Ha, H.-J. Koo and B. H. Chung, J. Phys. Chem. C, 2007, 111, 1123–1130 CrossRef CAS.
- J. E. Millstone, G. S. Métraux and C. A. Mirkin, Adv. Funct. Mater., 2006, 16, 1209–1214 CrossRef CAS.
Footnote |
| † Present address: Department of Chemistry and Chemical Biology, Harvard University, Cambridge, MA 02138, USA. |
| This journal is © The Royal Society of Chemistry 2023 |