
cRGD-modified nanoparticles of multi-bioactive agent conjugate with pH-sensitive linkers and PD-L1 antagonist for integrative collaborative treatment of breast cancer†
Chenming
Zou‡
 a,
Yuepeng
Tang‡
a,
Ping
Zeng‡
a,
Derong
Cui‡
b,
Majdi Al
Amili
a,
Ya
Chang
a,
Zhu
Jin
a,
Yuanyuan
Shen
*a,
Songwei
Tan
*c and
Shengrong
Guo
*a
a,
Yuepeng
Tang‡
a,
Ping
Zeng‡
a,
Derong
Cui‡
b,
Majdi Al
Amili
a,
Ya
Chang
a,
Zhu
Jin
a,
Yuanyuan
Shen
*a,
Songwei
Tan
*c and
Shengrong
Guo
*a
aSchool of Pharmacy, Shanghai Jiao Tong University, 800 Dongchuan Road, Shanghai 200240, China. E-mail: shenyuanyuan@sjtu.edu.cn; srguo@sjtu.edu.cn
bDepartment of Anesthesiology, Shanghai Jiao Tong University Affiliated Sixth People's Hospital, 600 Yishan Road, Shanghai 200233, China
cSchool of Pharmacy, Tongji Medical College, Huazhong University of Science and Technology, 13 Hangkong Road, Wuhan 430030, China. E-mail: Tansw@hust.edu.cn
First published on 13th March 2023
Abstract
Targeted co-delivery and co-release of multi-drugs is essential to have an integrative collaborative effect on treating cancer. It is valuable to use few drug carriers for multi-drug delivery. Herein, we develop cRGD-modified nanoparticles (cRGD-TDA) of a conjugate of doxorubicin as cytotoxic agent, adjudin as an anti-metastasis agent and D-α-tocopherol polyethylene glycol 1000 succinate (TPGS) as a reactive oxygen species inducer linked with pH-sensitive bonds, and then combine the nanoparticles with PD-L1 antagonist to treat 4T1 triple-negative breast cancer. cRGD-TDA NPs present tumor-targeted co-delivery and pH-sensitive co-release of triple agents. cRGD-TDA NPs combined with PD-L1 antagonist much more significantly inhibit tumor growth and metastasis than single-drug treatment, which is due to their integrative collaborative effect. It is found that TPGS elicits a powerful immunogenic cell death effect. Meanwhile, PD-L1 antagonist mitigates the immunosuppressive environment and has a synergistic effect with the cRGD-TDA NPs. The study provides a new strategy to treat refractory cancer integratively and collaboratively.
New conceptsCancer cannot be adequately treated only using cytotoxic agents or anti-metastasis agents or immunotherapeutic agents, owing to its malignant cell proliferation, metastasis and low immunogenicity or immunosuppressive microenvironment. Inspired by the effectiveness of multi-drug combination therapy and chemoimmunotherapy, we proposed a simple and effective method to construct a novel integrative collaborative nanoparticle: cRGD-modified nanoparticle (denoted as cRGD-TDA NPs) of amphiphilic compounds of doxorubicin (DOX), adjudin (ADD) and D-α-tocopherol polyethylene glycol 1000 succinate (TPGS) conjugated with pH-sensitive Schiff base and hydrazone bonds with high drug loading efficiency and tumor-targeted accumulation. Besides the cytotoxic and metastasis-preventing effects of DOX and ADD, respectively, for the first time we found that TPGS could elicit a powerful immunogenic cell death (ICD) effect in tumor cells and promote maturation of dendritic cells in vivo. This special finding might broaden the application range of TPGS in chemoimmunotherapy. We verified the promising anti-tumor and anti-metastasis outcomes of cRGD-TDA NPs benefitting from the integrative collaborative effect of these three therapeutic agents. Furthermore, combination therapy using the cRGD-TDA NPs plus PD-L1 antagonist significantly enhanced the anti-tumor and anti-metastasis effects. We believe that this work provides a novel facile integrative collaborative strategy for refractory cancer treatment in nanomedicine and chemoimmunotherapy. |
1. Introduction
Cancer is a refractory disease. The complex and heterogeneous tumor microenvironment (TME), such as insufficient tumor immunogenicity and the immunosuppression, frequently compromises the therapeutic efficacy of chemotherapeutic drugs and preferentially leads to metastasis and recurrence.1 Metastasis accounts for approximately 90% of cancer mortality and is much more challenging compared to primary tumors. For instance, the low immunogenicity and immunosuppressive TME in triple-negative breast cancer (TBNC), one of the most aggressive and metastatic breast cancers, can mediate extravasation and metastatic outgrowth of tumor cells in the lung, and the effect of current chemotherapeutic regimens is unsatisfactory.2,3 Despite frustrating outcomes, chemotherapy is still the first-line treatment for various cancers. Nevertheless, the routine non-targeted systemic administration can lead to lymphodepletion and immunosuppression of the whole system,4 and can cultivate the potential metastatic capacity through the epithelial–mesenchymal transition (EMT) effect as well as damaging the barrier of the tumor extracellular matrix.5–7 There is an urgent need to solve the deficiencies of conventional chemotherapy and apply the multi-agent approach to address heterogeneous factors for synergistic effects.8,9 Multi-drug combination strategy based on nanotechnology is a promising way to fight against advanced malignancy, because it can deliver several agents to the target site simultaneously.10–12In the past decade, immunotherapy has emerged as a powerful strategy for cancer therapy.13 Many types of cancer immunotherapy have been explored, including monoclonal antibodies, adoptive T-cell therapy, cancer vaccines, and immune checkpoint blockade therapy.14,15 Immunosurveillance is compromised under the immunosuppressive microenvironment and exhaustion of tumor-infiltrating lymphocytes. Immunity can be aroused by immune checkpoint blockers (ICB) like antibodies against cytotoxic T-lymphocyte associated protein 4 (CTLA-4), antibodies against PD-L1 and antibodies against PD-1.16,17 Besides removing the suppressive tumor microenvironment, how to elicit a potent antitumor immune response is also of vital influence, because the loss of immunogenicity in different types of cancer cells can lead to the failure to recognize tumor cells or the quiescence of DCs, and result in a poor level of immune response.18,19
The high prevalence of programmed cell death ligand-1 (PD-L1) expression on the surface of various tumor cells, such as melanoma, renal cell carcinoma, and TNBC, hints at the benefits from programmed cell death protein-1 (PD-1)/PD-L1 pathway blockade and further supports its therapeutic application clinically.20,21 The expression of PD-L1 can be increased by the secretion of interferon-γ (IFN-γ) in the tumor microenvironment from persistent exposure to chemodrugs, which has been referred to as a mechanism of adaptive immune resistance and exhaustion.22,23 These lethal effects imply the high possibility of a combination between multi-chemotherapy and immunotherapy, and such a combination has already demonstrated prominent efficacy in breast cancer. Recently, clinical trials for combination treatment of chemotherapy plus immuno-checkpoint blockade therapy showed encouraging outcomes.10,24,25 The immunogenic cell death (ICD) effect plays a crucial role in cancer immunotherapy by stimulating antitumor immunity and awakening immunosurveillance.26 The ICD effect can trigger the release of the molecules of damage associated molecular patterns (DAMP), including the translocation of calreticulin (CRT), the excretion of adenosine triphosphate (ATP), and the release of high mobility group box 1 protein (HMGB-1), stimulating immature dendritic cells (DCs) to proliferate into mature DCs, which could help present tumor-associated antigens to T cells so as to activate a subsequent immune response.27 This effect can revert “cold” tumors into “hot” tumors, and amplify the immunotherapeutic efficacy with ICB antagonists.28–31 Of note, oxaliplatin, cyclophosphamide and anthracyclines such as doxorubicin (DOX) and mitoxantrone have been verified to trigger immunogenic cell death at a certain dosage.32,33 Although it has been found that DOX could induce immunogenic cell death of breast cancer cells to elicit a certain level of adaptive immune response, DOX is relatively inefficient in robustly promoting ICD due to a variety of restrictive circumstances and a complicated microenvironment.34–36 Therefore, more effective ICD inducers are urgently needed to boost the release and recognition of tumor-associated antigens (TAA) so as to strengthen DCs’ maturation and improve the tumor-specific immune response. Recently, reactive oxygen species (ROS) have been found to be positively correlated with ICD and this inspired the search for ICD inducers or amplifiers. For instance, Fenton and Fenton-like reaction-induced ROS generation can amplify the DOX-based ICD effect.37,38 Moreover, Chen designed and synthesized a mitochondrial-targeting AIE photosensitizer TPE-DPA-TCyP that caused mitochondrial oxidative stress under light excitation and greatly amplified ICD induction. It is the first report showing that mitochondrial oxidative stress can massively evoke ICD.39 Inspired by this research, D-α-tocopherol polyethylene glycol 1000 succinate (TPGS) comes into our sight. TPGS, a type of drug adjuvant approved by the FDA, is a powerful ROS inducer through its interaction with the ubiquinone-binding sites of mitochondrial respiratory complex II.40–43 ROS induced by TPGS in cytoplasm may amplify the ICD effect of DOX in tumor cells and achieve maturation of DCs to a higher extent than conventional chemotherapy.
Metastasis is a big challenge for cancer therapy. Although conventional clinical chemotherapy suppresses cancer progression, it also cultivates the metastatic potential by inducing CSC (cancer stem-like cells) traits, triggering EMT effects and causing an inflammatory response in the tumor microenvironment, which leads to unfavourable prognosis for patients.44,45 Therefore, novel therapeutic strategies against cancer metastasis are urgently needed. Adjudin (ADD), an analog of lonidamine, was reported to enhance the cytotoxic sensitivity of breast cancer cells to chemodrugs in previous studies.46 It also plays an anti-metastasis role by inhibiting the PI3K/Akt/mTOR or Wnt signaling pathways to reverse the EMT-inducing metastasis, as recently reported.47–51 Meanwhile, the expression of immune checkpoints is associated with tumor metastasis. On the one hand, upregulation of surface-expressed PD-L1 on tumor cells or their exosomes causes immune escape and promotes metastasis. On the other hand, the state of tumor metastasis, such as EMT or tumor stemness, is closely associated with PD-L1 upregulation by complex signaling pathways.52,53 This feedback loop between immune checkpoint expression and metastasis promoting tumor progression urgently needs to be broken for satisfactory prognosis.
In this work, we synthesized a conjugate composed of doxorubicin (DOX), adjudin (ADD) and TPGS through dual pH-sensitive Schiff base and hydrazone bond, then constructed it into targeting cocktail nanoparticles (denoted as cRGD-TDA NPs) with cyclic oligopeptide Arg-Gly-Asp (cRGD) modification. cRGD-TDA NPs could achieve actively targeted drug delivery, ratiometric stability, and acidic pH environment-triggered drug release (Scheme 1(A)). The cRGD-TDA NPs demonstrated predominant antitumor and anti-metastasis effects in vitro and in vivo. It is worth noting that TPGS produced high levels of ROS in tumor cells and elicited a powerful ICD effect in the tumor microenvironment, and this superior ICD effect elicited53 by cRGD-TDA NPs was also elucidated in vitro and in vivo. In addition, of PD-L1 ligand, a D-peptide (NYSKPTDRQYHF) antagonist widely used as a blockade of PD-1/PD-L1 pathway to rouse antitumor immunity,54,55 was injected subcutaneously around the tumors simultaneously with the intravenous injection of the cRGD-TDA NPs to further reduce the immunosuppressive and exhausted states (Scheme 1(B)). Remarkably, the antitumor and anti-metastasis therapeutic effects of cRGD-TDA NPs combined with anti-PD-L1 therapy were verified on an orthotopic murine 4T1 breast tumor model. TPGS can be used as an ICD inducer, which will be endowed with more functional possibilities in the pharmaceutical and immunotherapeutic fields. Last but not least, multifunctional cRGD-TDA NPs provide a promising strategy for cancer treatment and a robust nanoplatform for chemoimmunotherapy.
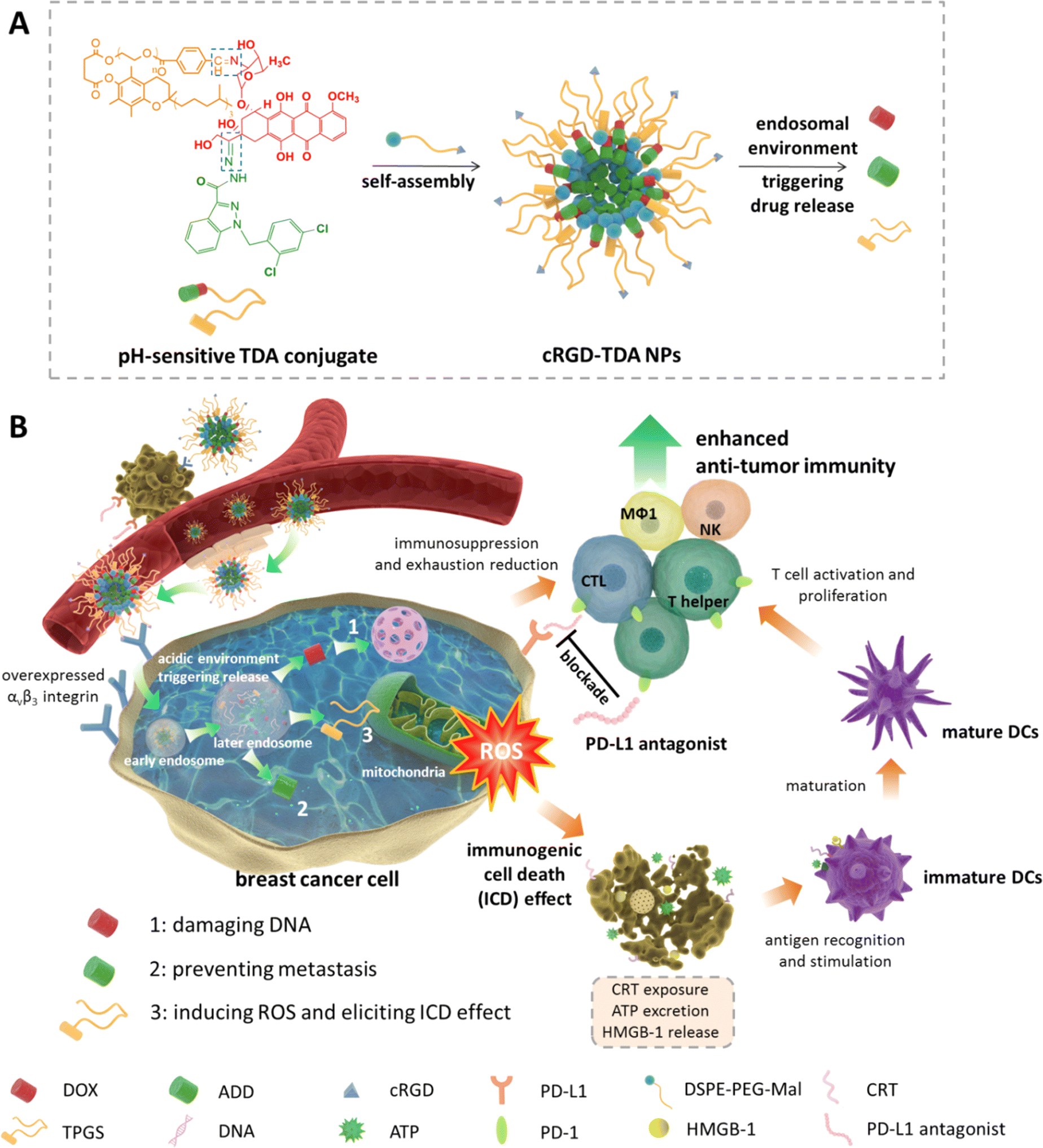 | ||
| Scheme 1 Schematic illustration of construction and antitumor mechanisms of cRGD-TDA NPs. (A) Self-assembly and pH-sensitive property of cRGD-TDA NPs. (B) cRGD-TDA NPs target 4T1 breast cancer cells through overexpressed αvβ3 integrin receptors. After endocytosis into the cytoplasm, the Schiff base and hydrazone bond were cleaved under the acidic endosomal environment, leading to disruption of the cRGD-TDA NPs and the release of DOX, ADD and TPGS. Free DOX and ADD can damage the DNA and prevent metastasis of aggressive 4T1 cells, respectively. Meanwhile, TPGS can induce high levels of ROS through interrupting normal functions of mitochondria and elicit a strong ICD effect to promote the maturation of DCs. Subsequently, matured DCs recognize and present tumor-associated antigen to activate and proliferate neoantigen-specific T cells. In addition, combination therapy with PD-L1 antagonist is introduced to reverse the immunosuppressive environment and attenuate exhausted states of tumor-infiltrating lymphocytes. The cocktail cRGD-TDA NPs are a novel approach for chemoimmunotherapy and a promising strategy to treat malignant cancer. | ||
2. Results and discussion
2.1. Synthesis of TDA conjugate
The stability of the Schiff base and hydrazone bond was different during the conjugation reaction, so the synthesis route was first investigated. The synthetic procedure shown in Scheme 2(A) was used to synthesize TPGS-TPA first. The second step involved the conjugation of TPGS-TPA and DOX in an acidic environment with glacial acetic acid to obtain the intermediate product, TPGS-DOX (TD). Then, the final step involved the conjugation between TD and ADD. ADD was added to a TD solution containing TD and 1% TFA to react for 24 h. The 1H-NMR spectra of TD and TDA are shown in Fig. S1 (ESI†). The conjugation degree of DOX in TD was 80%, while the conjugation degrees of DOX and ADD were only 20% and 10%, respectively, calculated by peak areas in 1H-NMR spectrum. Because of its low conjugation degree, another method was conducted whereby DOX and ADD were first combined through hydrazone bonding and subsequently conjugated with TPGS-TPA by Schiff base (Scheme 2(B)). The structure of the DOX–ADD intermediate was characterized by 1H-NMR, and the yield was 65%. After conjugation of DOX–ADD to TPGS-TPA was completed, the structure of the TDA product was characterized by 1H-NMR, FTIR and UV-vis spectrophotometry. As the spectrum in Fig. S2 (ESI†) shows, the emerging peaks at 4.4 ppm (a) and 10.1 ppm (h) belong to protons of –CH2– and –NH– adjacent to the hydrazone bond between DOX and ADD, and the peak at 8.4 ppm (i) belongs to the –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N– of the Schiff base adjacent to TPGS-TPA. The signals at 3.9 ppm (e), 4.6 ppm (b), and 5.2 ppm (c) were attributed to the DOX part of TDA. Meanwhile, the signals at 5.8 ppm (f) belong to protons of –CH2– between the indole ring and the benzene ring in ADD. In addition, the signal at 4.6 ppm of –NH2– in ADD vanished, which confirmed the successful formation of the new hydrazone bond.
N– of the Schiff base adjacent to TPGS-TPA. The signals at 3.9 ppm (e), 4.6 ppm (b), and 5.2 ppm (c) were attributed to the DOX part of TDA. Meanwhile, the signals at 5.8 ppm (f) belong to protons of –CH2– between the indole ring and the benzene ring in ADD. In addition, the signal at 4.6 ppm of –NH2– in ADD vanished, which confirmed the successful formation of the new hydrazone bond.
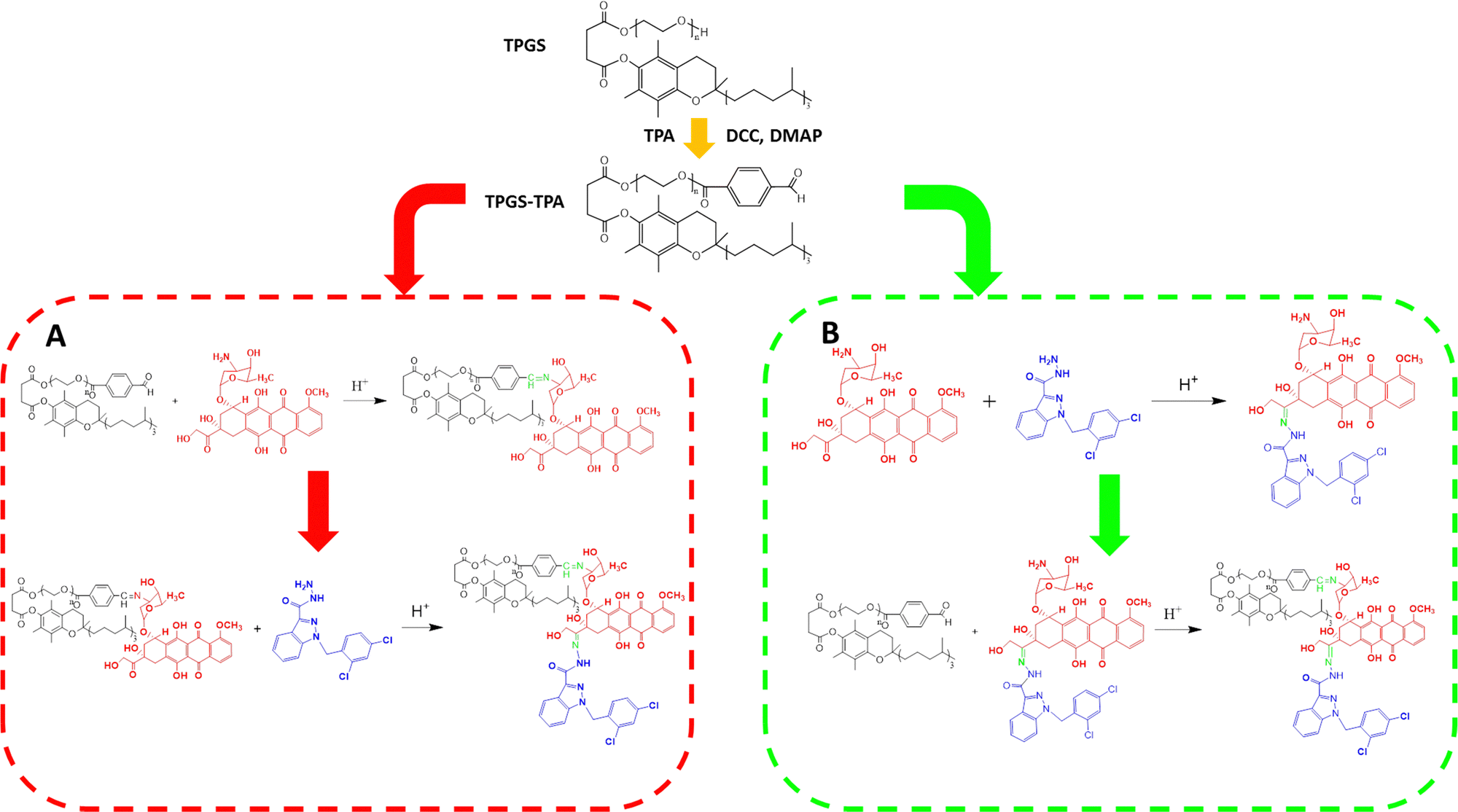 | ||
| Scheme 2 Two different synthesis method for TDA. (A) Conjugate DOX with TPGS first (TD), then conjugate TD with ADD, resulting in low grafting rates of DOX and ADD. (B) Conjugate DOX and ADD first (DOX–ADD), then conjugate TPGS with DOX–ADD, resulting in high grafting rates of DOX and ADD. | ||
In the Fourier-transform infrared spectrum shown in Fig. S3 (ESI†), the peaks between 3300 and 3600 cm−1 of TDA belong to –OH groups in DOX and amino groups in ADD. Also, the peaks overlapped between 2800 and 3000 cm−1 in the spectrum of TDA were attributed to the stretching vibrations of –CH3 (νC–H), –CH2– (νC–H) and –CH– (νC–H). The strong peaks at 1120 cm−1 were attributed to the stretching vibration of polyethylene glycol (νC–O) in TPGS. The peaks at 1746 and 1725 cm−1 belong to the stretching vibration of the ester in TPGS and carbonyl groups of the anthracene ring of DOX, respectively. Meanwhile, the peaks at 1665 and 1635 cm−1 typically belong to hydrazide groups of ADD. Moreover, the peaks appearing at 1675 cm−1 represent the stretching vibration of the hydrazone bond (νCN) between DOX and ADD as well as TPGS and DOX.
In the UV-vis spectrum shown in Fig. S4 (ESI†), the peaks around 475 nm and 295 nm in TDA belong to the anthracene group of DOX and the indole group of ADD, respectively, which confirmed the successful synthesis of the TDA conjugate. From the matrix-assisted laser desorption/ionization time of flight mass spectrum (MALDI-TOF-MS) (Fig. S6, ESI†), the peak at m/z of about 2500 was in accordance with the calculated molecular weight. All of the spectra discussed above indicate that TDA was successfully synthesized through Schiff base and hydrazone bond, and the grafting rates of DOX and ADD in TDA were calculated to be 85.0% from the 1H-NMR results.
2.2. Preparation and characterization of the TDA NPs
After formation of the TDA NPs through nano-precipitation method, the diameter was measured by dynamic light scattering (DLS). As shown in Fig. 1(A), TDA could self-assemble into nanoparticles with size of 133.7 ± 1.7 nm and narrow polydispersity index of 0.182. The zeta potential was −6.27 ± 0.66 mV (Fig. S7, ESI†). Transmission electron microscopy (TEM) images showed that the TDA NPs had a uniform spherical shape with diameters around 100 nm (Fig. 1(B)). These results proved that the TDA conjugate was suitable to be constructed into nanoparticles.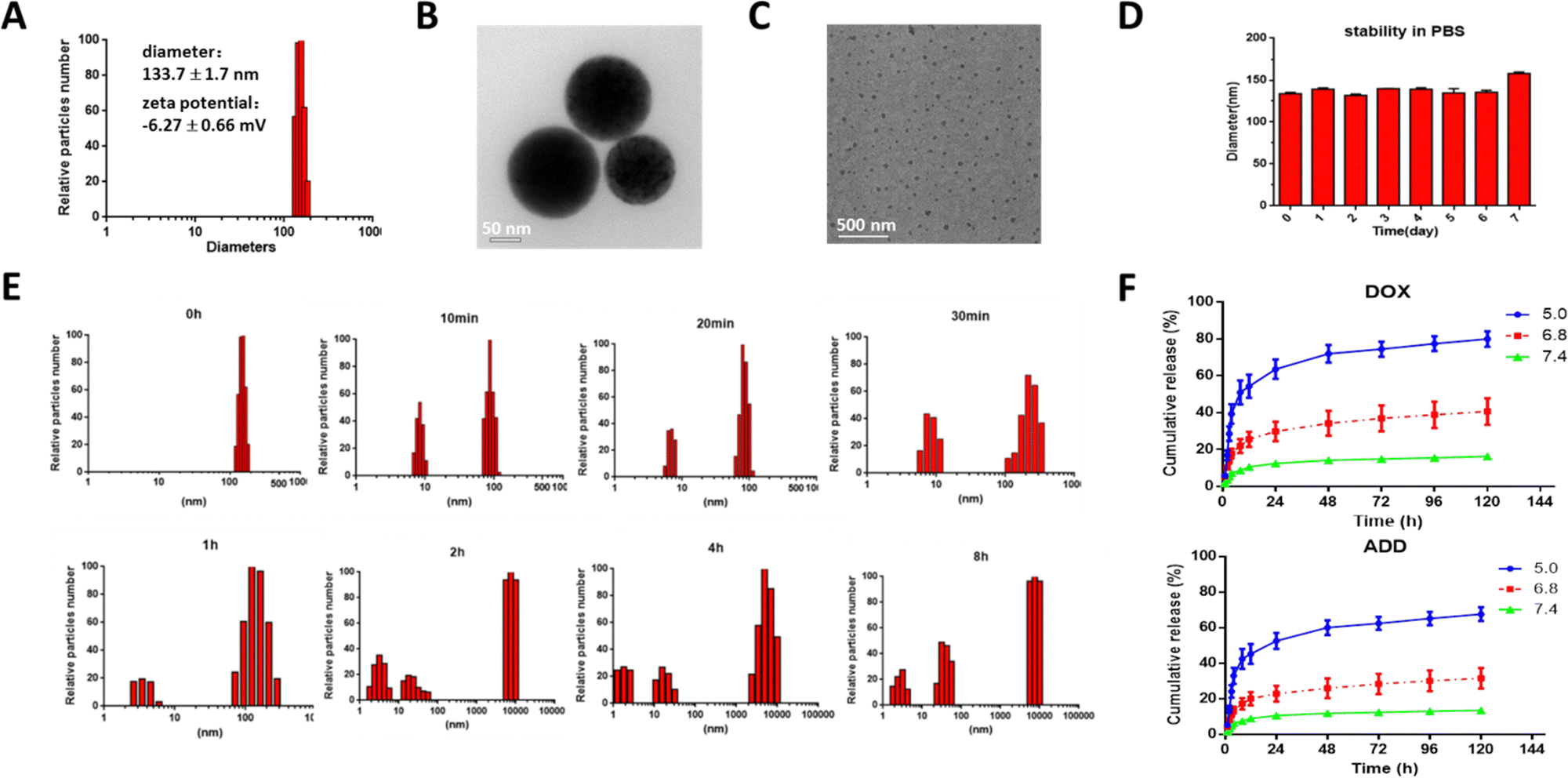 | ||
| Fig. 1 Characterization of TDA NPs. (A) Diameter and zeta potential of TDA NPs measured by DLS. (B) TEM image of TDA NPs in phosphate buffer (pH = 7.4) (scale bar = 1.0 μm). (C) TEM image of TDA NPs incubated in acetate buffer (pH = 5.0) for 2 h (scale bar = 500 nm). (D) Stability of TDA NPs in PBS (pH = 7.4) at 25 °C. (E) Particle size changes of TDA NPs in acetate buffer (pH = 5.0) at different time points (0 min, 10 min, 20 min, 30 min, 1 h, 2 h, 4 h and 8 h). (F) Cumulative drug release from TDA NPs in different pH buffers. | ||
Nanoparticles composed of conjugates can maintain the ratiometric stability of drugs after trafficking from the injection site to the targeted site in the body compared with drug-loaded nano-delivery systems. The sensitivity of a conjugate is one of the most important properties in the design, because it is critical that the conjugate can reduce unexpected drug release in the blood and ensure precise drug delivery or adequate release at the targeted sites. Dual acid-labile bonds endowed the TDA NPs with fast acidic environment-responsive properties when the TDA NPs were endocytosed into cellular endo/lysosomes with pH of around 5. Therefore, we incubated the TDA NPs in acetate buffer (pH = 5.0) and detected the diameter changes at designated time points (0, 10 min, 20 min, 30 min, 1 h, 2 h, 4 h, 8 h). As shown in Fig. 1(E), the TDA NPs were almost all distributed in a narrow range with low polydispersity index at 0 min. With increasing time, a cluster of nanoparticles with diameter of around 10 nm emerged, resulting from the cleavage of linkers and self-assembly of dissociated TPGS. When time exceeded 2 h, the majority of the particles were about 10 nm in diameter, as observed by TEM (Fig. 1(C)). Another cluster of nanoparticles around 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 nm arose on account of cleavage of the hydrazone bond between DOX and ADD, and aggregation of released hydrophobic ADD. Concurrently, the drug release study (Fig. 1(F)) showed that DOX and ADD were released dramatically faster under pH = 5.0 condition than pH = 6.8 or pH 7.4 (80.00% vs. 40.68% or 16.38% of DOX; 67.75% vs. 31.71% or 13.75% of ADD at 120 h), indicating that the drug release rates increased with decreasing pH due to the higher dissociation speed of the Schiff base and hydrazone bond under the acidic pH environment. In summary, we inferred that the TDA NPs could dissociate rapidly and selectively release DOX and ADD under an acidic endo/lysosome environment inside tumor cells, while remaining stable under neutral physiological conditions (Fig. 1(D)). The TDA NPs are therefore suitable for precise ratiometric delivery of different drugs.
000 nm arose on account of cleavage of the hydrazone bond between DOX and ADD, and aggregation of released hydrophobic ADD. Concurrently, the drug release study (Fig. 1(F)) showed that DOX and ADD were released dramatically faster under pH = 5.0 condition than pH = 6.8 or pH 7.4 (80.00% vs. 40.68% or 16.38% of DOX; 67.75% vs. 31.71% or 13.75% of ADD at 120 h), indicating that the drug release rates increased with decreasing pH due to the higher dissociation speed of the Schiff base and hydrazone bond under the acidic pH environment. In summary, we inferred that the TDA NPs could dissociate rapidly and selectively release DOX and ADD under an acidic endo/lysosome environment inside tumor cells, while remaining stable under neutral physiological conditions (Fig. 1(D)). The TDA NPs are therefore suitable for precise ratiometric delivery of different drugs.
2.3. Cellular uptake of the cRGD-TDA NPs in vitro
Nano-drug delivery systems depend on the enhanced permeability and retention (EPR) effect to passively target tumor sites. However, there is still unsatisfactory accumulation efficiency because of complicated trafficking conditions and thus a need for improvement.26 Because 4T1 cells overexpress αvβ3 integrin receptors on the surface, we utilized its ligand, cRGD peptides (containing cysteine residue), to modify TDA NPs through thiol-maleimide click chemistry with DSPE-PEG2000-Mal (Fig. S5, ESI†). In order to explore the increased targeting ability of the cRGD-TDA NPs, cellular uptake of free DOX, TDA NPs and cRGD-TDA NPs were investigated after incubation with 4T1 cells for 1, 3, and 6 h. As shown in Fig. 2(B), the red fluorescence intensity of free DOX exceeded that of the counterparts of TDA NPs and cRGD-TDA NPs in 1 and 3 h, owing to the faster diffusion rate of free DOX than the endocytosis rate of the NPs. Moreover, the fluorescence intensities of the TDA NPs and cRGD-TDA NPs were stronger than that of free DOX at 6 h (Fig. 2(A)), as a consequence of the diminished concentration gradient of DOX between the extracellular medium and the intracellular fluid. More importantly, the cRGD-TDA NPs displayed 1.24-fold, 1.27-fold and 1.37-fold higher mean fluorescence intensities than the TDA NPs at 1, 3, and 6 h by flow cytometry analysis, respectively (Fig. 2(B) and (C)), which confirmed that cRGD modification could facilitate the active targeting ability of TDA NPs towards 4T1 cells. | ||
| Fig. 2 Cellular uptake, cytotoxicity, anti-migration and anti-invasion effects of TDA NPs. (A) LSCM images of cellular uptake of free DOX, TDA NPs and cRGD-TDA NPs incubated for 6 h (scale bar = 50 μm). (B) Histogram and (C) mean fluorescence intensity of 4T1 cells incubated with free DOX, TDA NPs and cRGD-TDA NPs for 1 h, 3 h, 6 h measured by flow cytometry (n = 3). (D) The cell viability after treatment with free DOX, free ADD, free DOX + ADD, free DOX–ADD, TD NPs and TDA NPs for 24 h and 48 h (n = 5). (E) Microscopy images of scratched lines at 0 and 24 h in wound healing study (scale bar = 500 μm, the scratched areas were circled with yellow lines). (F) Migration rates calculated by the degree of change in wound area (n = 3). (G) Images of cells invading into the lower chamber in the transwell invasion assay (scale bar = 100 μm). (***p < 0.001; **p < 0.01; *p < 0.05; NS, no significant difference). | ||
2.4. Cytotoxicity, anti-migration and anti-invasion effects of the TDA NPs
To investigate the cytotoxicity of the TDA NPs, MTT colorimetric assay was conducted on 4T1 cells. As shown in Fig. 2(D) and Table 1, the IC50 at 24 h for free DOX + ADD was 1.27 × 10−2 μg mL−1, smaller than those for free DOX (5.65 × 10−2 μg mL−1) and free ADD (3.60 × 10−1 μg mL−1). The combination index (CI) of DOX and ADD was 0.48, <1, calculated using the Chou–Talalay method,56 indicating there is a synergistic effect of DOX and ADD. TDA NPs exhibited the strongest cytotoxicity (IC50 = 5.25 × 10−3 μg mL−1) among all the groups. The IC50 values at 48 h were lower than those at 24 h for all the groups.| IC50 (μg mL−1) | Free DOX | Free ADD | Free DOX + ADD | Free DOX–ADD | TD NPs | TDA NPs |
|---|---|---|---|---|---|---|
| 24 h | 5.65 × 10−2 | 3.60 × 10−1 | 1.27 × 10−2 | 2.90 | 2.65 × 10−2 | 5.25 × 10−3 |
| 48 h | 3.74 × 10−3 | 5.93 × 10−3 | 1.41 × 10−3 | 7.56 × 10−2 | 3.59 × 10−3 | 9.67 × 10−4 |
Wound healing assay was conducted to evaluate the anti-migration efficacy in vitro. As shown in Fig. 2(E) and (F), free ADD (migration rate: 17.00 ± 3.12%) and free DOX + ADD (−14.52 ± 3.61%) inhibited tumor migration much more than free DOX (32.84 ± 4.10%), especially when free DOX possessed higher cytotoxicity than free ADD, which indicated the anti-migration ability of ADD. Except for free DOX + ADD, the TDA NPs (−3.93 ± 2.61%) exhibited a stronger migration inhibitory effect than the other groups, proving its potential for anti-metastasis. In addition, we observed that only a small number of 4T1 cells invaded into the lower transwell chamber after being treated with free DOX + ADD and ADD, which indicated that ADD has the ability to prevent cell invasion. Moreover, the quantity of invaded cells was further reduced in the TDA NPs group, demonstrating a good inhibitory effect of TDA NPs on 4T1 cell invasion (Fig. 2(G)). The above results demonstrated that the cocktail TDA NPs have potent antitumor and anti-metastasis effects, and are promising to be employed in malignant and metastatic breast cancers.
2.5. Enhanced ROS production and ICD effects by the cRGD-TDA NPs in vitro
According to reports, intracellular ROS are mainly generated in mitochondria and a high dose of ROS can induce ICD, enhancing the specific antitumor immunity. Traditional photodynamic therapy (PDT) is widely utilized as a ROS generator in cancer therapy, but faces a variety of disadvantages, such as the penetration limitation of exogenous stimuli and restricted light exposure time and area. Chemodynamic therapy can circumvent these drawbacks by generating a high ROS dose in situ.57 Here, we ingeniously introduced TPGS, reported as a powerful ROS inducer, into the TDA NPs to generate high levels of ROS in the cytoplasm.2′,7′-Dichlorofluorescein diacetate (DCFH-DA) can be rapidly oxidized into dichlorofluorescein (DCF) by ROS to emit green fluorescence. For quantitative analysis (Fig. 3(B)), free DOX increased ROS production approximately the same as free ADD, but free DOX + ADD elevated levels of ROS higher than the individual components (vs. free DOX, p < 0.001; vs. free ADD, p < 0.01), revealing the synergistic effects. In addition, significantly stronger green fluorescence intensity was measured in the TDA NPs, 2.3-fold, 2.1-fold, 1.6-fold and 1.2-fold higher than free DOX, free ADD, free DOX + ADD and TD NPs, respectively, which demonstrated that the TDA NPs could accelerate ROS production to a great extent. More importantly, we wondered whether these enhanced ROS-producing properties arose from TPGS; thus, the same equivalent dosage of free DOX + TPGS was compared with TDA NPs (Fig. S5, ESI†). As we expected, free DOX + TPGS displayed a slightly higher intensity than TDA NPs, which confirmed that the powerful ROS-producing ability of TDA NPs came from TPGS. Furthermore, we checked whether the ROS promotion was induced in a dose-dependent manner. Along with the higher TPGS concentration supplemented, the stronger fluorescence was detected by flow cytometry (Fig. S8, ESI†). This dose-dependent ROS-producing profile endowed TPGS with the capacity to be employed as a promising chemodynamic agent to enhance the intracellular ROS level for cancer therapy.
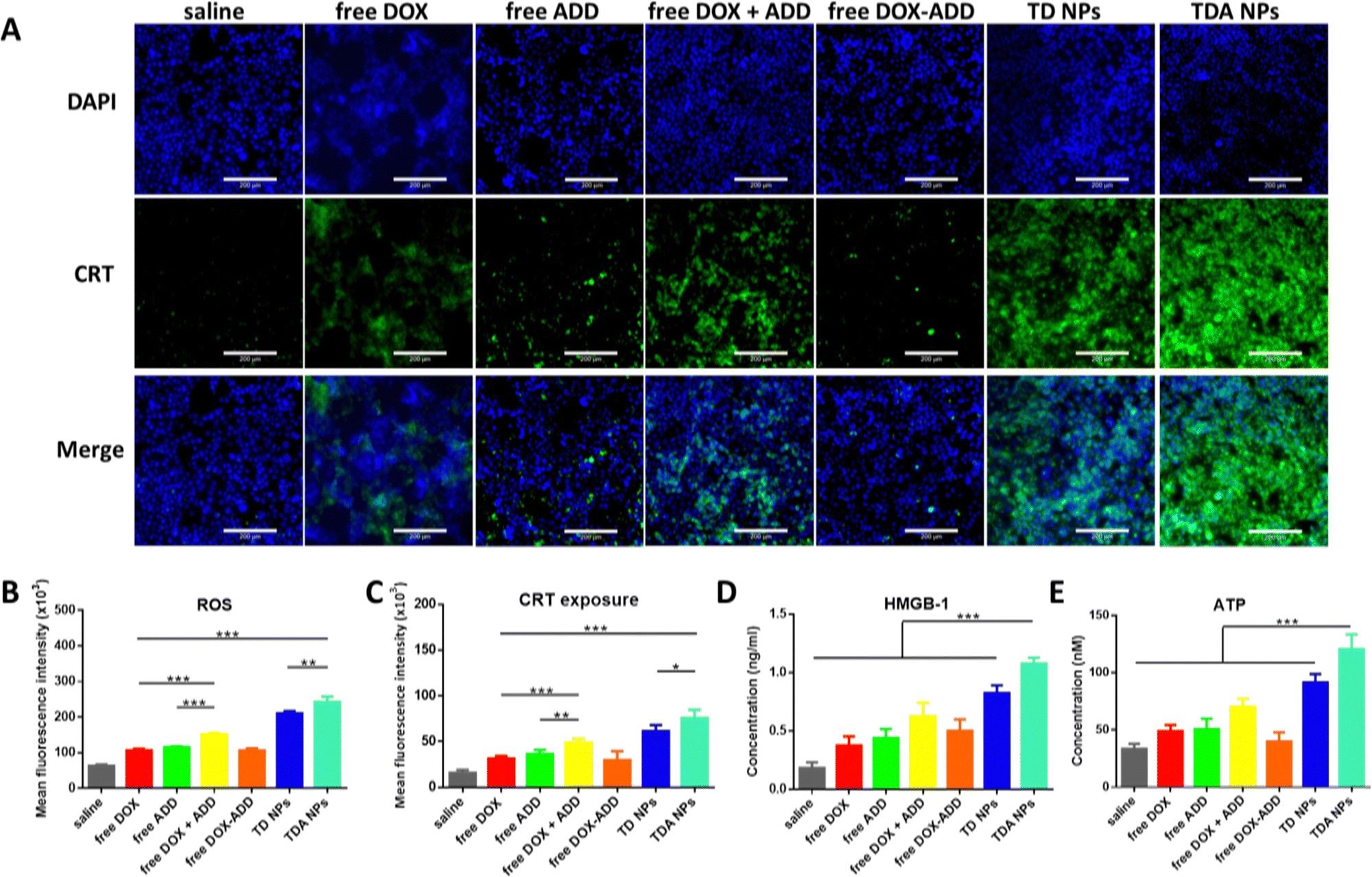 | ||
| Fig. 3 In vitro enhanced ROS level and ICD effect in 4T1 cells. (A) Microscopy images of CRT exposure (green signals) after incubated with saline, free DOX, free ADD, free DOX + ADD, free DOX–ADD, TD NPs and TDA NPs at same dosage for 24 h (scale bar = 200 μm). (B) ROS levels of different treatments for 24 h measured by flow cytometry (n = 3). (C) Quantitative measurement of CRT exposure by flow cytometry (n = 3). Concentrations of HMGB-1 (D) and ATP (E) in medium after different treatments for 24 h measured by HMGB-1 ELISA kit and ATP assay kit (n = 3). (***p < 0.001; **p < 0.01; *p < 0.05; NS, no significant difference). | ||
Considering the high levels of ROS produced by TPGS and TDA NPs in 4T1 cells, we further verified the hypothesis that TPGS or TDA NPs have great potential to induce the ICD effect. The identical ICD markers including translocation of CRT, release of HMGB-1 and excretion of ATP were investigated in 4T1 cells. CRT was labelled with green fluorescence, and the expression remarkably increased on the cell surface in the TDA NPs group, as displayed by microscopy images shown in Fig. 3(A). The free DOX and free DOX + ADD showed moderate enhancement (2.0-fold and 3.1-fold, respectively) compared to saline (Fig. 3(C)), indicating that DOX could only induce the ICD effect to some extent. Notably, TDA NPs demonstrated 2.4-fold and 1.6-fold enhancement of fluorescence intensity compared to free DOX and free DOX + ADD, while the highest TPGS concentration (8 μM) elicited more CRT exposure than the lower concentration counterparts (1.3-fold vs. 4 μM TPGS; 1.8-fold vs. 2 μM TPGS), as depicted in Fig. S9A and B (ESI†). These results were basically consistent with the earlier in vitro ROS assays. Concurrently, higher levels of HMGB-1 and ATP were measured in the medium after treatment with TDA NPs (Fig. 3(D) and (E)), and an increasing trend was observed as the TPGS concentration increased (Fig. S9C and D, ESI†).
These inspiring discoveries not only confirmed the potent ROS-generating function of TPGS, but also indicated that TPGS or TDA NPs were able to induce a strong ICD effect in cancer cells and this effect urgently needs to be confirmed in vivo.
2.6. Biodistribution of cRGD-TDA NPs
To evaluate the 4T1 breast cancer targeting properties of cRGD-TDA NPs in vivo, near infrared fluorescence probe Dir was encapsulated in TDA NPs and cRGD-TDA NPs. As shown in Fig. 4(A), compared with the negligible fluorescence in the free Dir group, both TDA and cRGD-TDA NPs exhibited much higher fluorescence intensity at tumor sites, confirming the superior accumulating ability of TDA and the cRGD-TDA NPs due to the EPR effect. The modification of the cRGD peptide upon the cRGD-TDA NPs was supposed to enhance the active targeting properties of the TDA NPs towards 4T1 cells, because of the overexpressed αvβ3 integrin on the surface. From the results in Fig. 4(A), the cRGD-TDA NP group maintained higher fluorescence intensity than the TDA NPs group after intravenous injection. For quantitative comparison (Fig. 4(B) and (C)), we further calculated the AUC of free Dir, TDA NPs and cRGD-TDA NPs based on the fluorescence intensity–time curve at tumor sites (Table 2). The AUC(0-120h) of the cRGD-TDA NPs improved by 1.30-fold compared with the non-decorated TDA NPs, showing the active targeting benefit of cRGD decoration. In addition, both the TDA NPs and cRGD-TDA NPs accumulated at tumor sites much more than other main organs (Fig. 4(C) and Fig. S7C, ESI†). In line with this result, free Dir displayed extremely low fluorescence distribution in the blood, while considerably high blood fluorescence intensity was observed in both the TDA NPs and cRGD-TDA NPs groups, demonstrating the long-circulating effect of the nanoparticle system (Fig. 4(D) and (E)). Conclusively, these results confirmed the improved targeting ability of cRGD-modified NPs and it was helpful for enhancing the capacity of our nanosystem in immunity elicitation and antitumor therapy.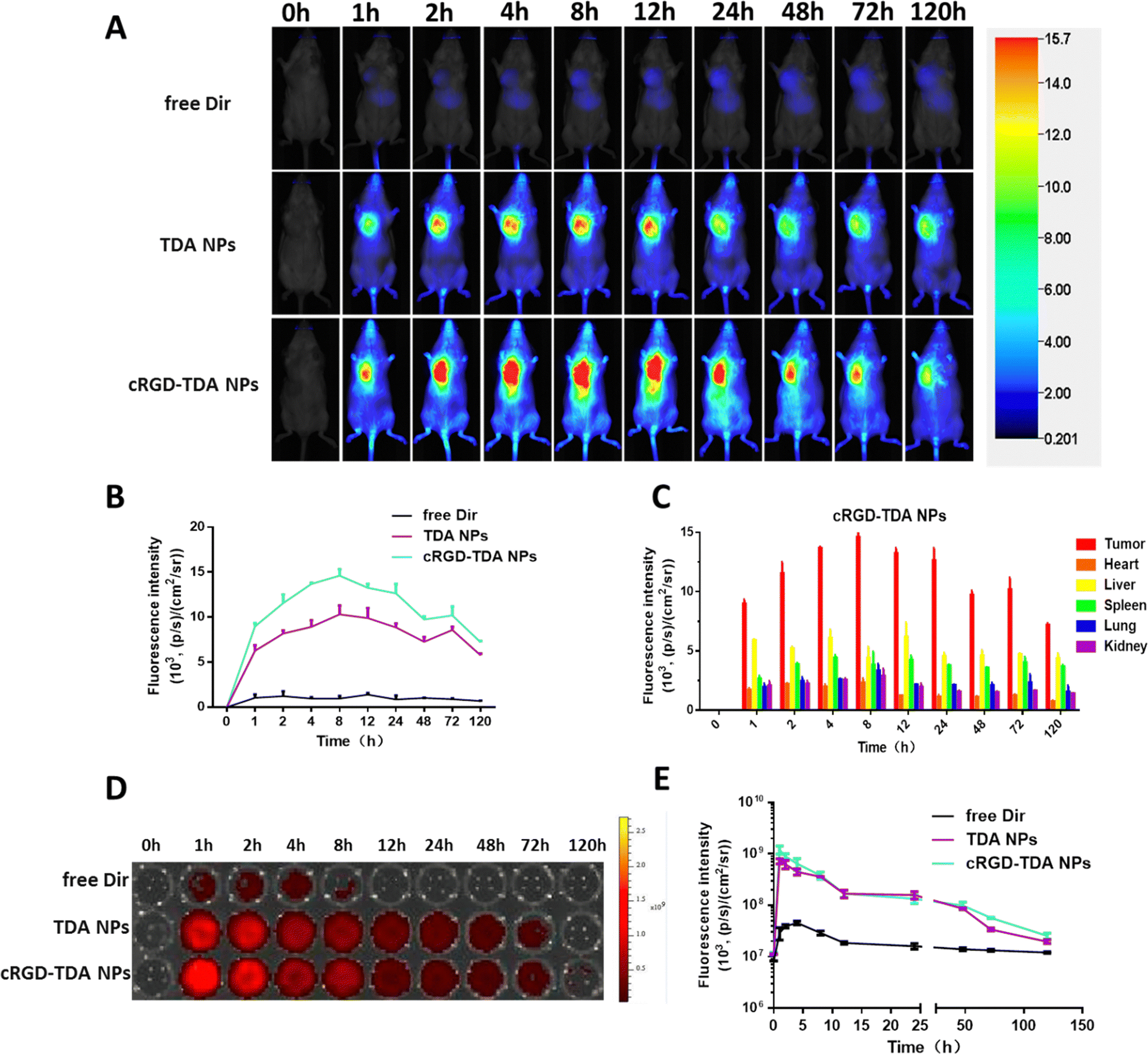 | ||
| Fig. 4 In vivo tumor targeting properties and biodistribution of cRGD-TDA NPs. (A) The in vivo fluorescent images of 4T1 tumor-bearing mice at different time points (0 h, 1 h, 2 h, 4 h, 8 h, 12 h, 24 h, 48 h, 72 h, 120 h) after i.v. administration with free Dir, Dir@TDA NPs, and Dir@cRGD-TDA NPs. (B) The quantitative mean fluorescent intensity of dissected tumor sections at different time points (0 h, 1 h, 2 h, 4 h, 8 h, 12 h, 24 h, 48 h, 72 h, 120 h) after i.v. administration with free Dir, Dir@TDA NPs, and Dir@cRGD-TDA NPs. (C) The mean fluorescence intensity of different dissected organs at different time points (0 h, 1 h, 2 h, 4 h, 8 h, 12 h, 24 h, 48 h, 72 h, 120 h) in the cRGD-TDA NPs group (n = 3). (D) Fluorescence images and (E) quantitative analysis of blood samples from mice treated with free Dir, Dir@TDA NPs, and Dir@cRGD-TDA NPs at different time points (0 h, 1 h, 2 h, 4 h, 8 h, 12 h, 24 h, 48 h, 72 h, 120 h). (***p < 0.001; **p < 0.01; *p < 0.05; NS, no significant difference). | ||
| Area under curvea (AUC) | Tumor | Heart | Liver | Spleen | Lung | Kidney | AUCtumor/AUCliver |
|---|---|---|---|---|---|---|---|
| a Unit of AUC of fluorescence intensity: [(p s−1)/(cm2 sr−1)] h. | |||||||
| Free Dir | 110.02 | 52.34 | 594.19 | 383.34 | 191.50 | 44.63 | 0.19 |
| TDA NPs | 948.88 | 156.25 | 594.73 | 471.56 | 228.10 | 194.34 | 1.60 |
| cRGD-TDA NPs | 1237.02 | 141.70 | 569.82 | 462.68 | 255.60 | 199.65 | 2.17 |
2.7. Enhanced ROS production and ICD effects of the cRGD-TDA NPs in vivo
After elucidating the upregulated ROS production and enhanced ICD effect in vitro, the 4T1 homogeneous tumor model was established to investigate the in vivo efficacy. Three days after administering either saline, free DOX, free DOX + ADD and cRGD-TDA NPs, ROS production and CRT exposure were observed by microscopy (Fig. 5(A) and (B)). The free DOX and free DOX + ADD groups produced slightly stronger ROS green signals than the saline group, while negligibly different ROS intensity was observed in both the free DOX and free DOX + ADD groups, which showed that ADD was not the dominant source of ROS production. Of note, the strongest red signal of DOX and the highest green signal of ROS were observed in the tumor section from the cRGD-TDA NPs group, indicating that the cRGD-TDA NPs delivered significantly more DOX into the tumor sites and induced significantly higher ROS production than free DOX or free DOX +ADD. The yellow regions in the overlapped images were observed considerably more in the cRGD-TDA NPs than the saline and other groups, implying that ROS were generated where TDA NPs were delivered.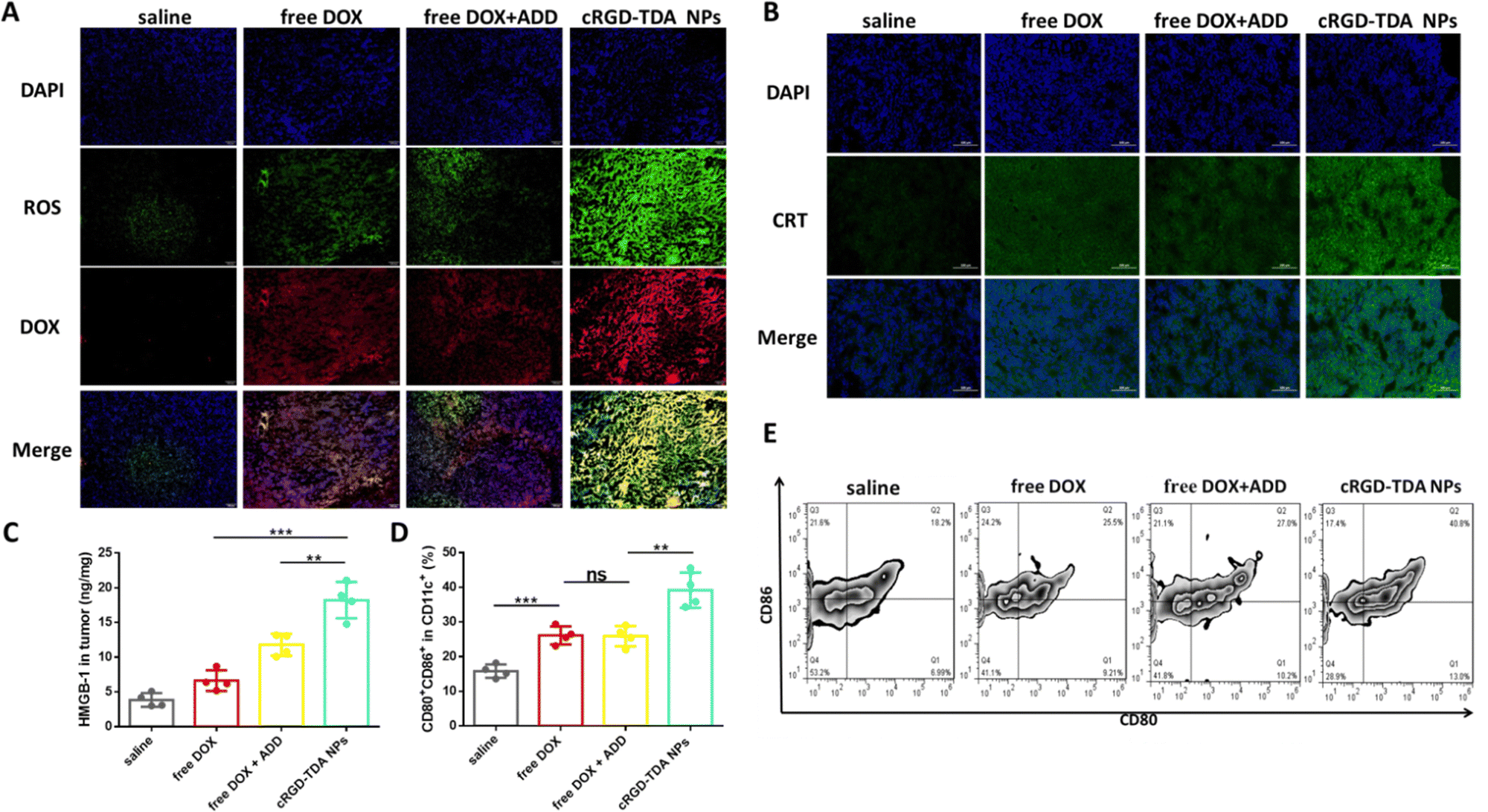 | ||
| Fig. 5 In vivo enhanced ROS level, ICD effect and DCs maturation. (A) Microscopy images of ROS production (green signals) after i.v. administration of saline, free DOX, free DOX + ADD or cRGD-TDA NPs (scale bar = 200 μm). (B) Microscopy images of CRT exposure after i.v. administration three times of saline, free DOX, free DOX + ADD or cRGD-TDA NPs every three days (scale bar = 200 μm). (C) The amount of released HMGB-1 in tumor interstitium measured by ELISA kit after i.v. administration three times with different groups every three days (n = 4). (D) The proportion of CD80+CD86+ cells on CD11c+ gates measured by flow cytometry and the scatter plots (E) of CD80+CD86+ cells on CD11c+ gates after treatment three times with saline, free DOX, free DOX + ADD or cRGD-TDA NPs every three days (n = 4) (***p < 0.001; **p < 0.01; *p < 0.05; NS, no significant difference). | ||
Concurrently, the in vivo ICD effect was investigated by the detection of CRT translocation on cell membrane and HMGB-1 release in the tumor tissue. As shown in Fig. 5(B), extensive CRT signals were observed for the cRGD-TDA NP group that exceeded other groups, and the concentration of HMGB-1 in tumors exhibited 2.8-fold and 1.5-fold enhancement compared to the counterparts of free DOX and free DOX + ADD (Fig. 5(C)). These data effectively confirmed the hypothesis that nanoparticles with TPGS had a powerful capacity to produce ROS and induce the ICD effect, thereby holding great potential to enhance tumor immunogenicity and subsequently elicit the antitumor immune response.
2.8. Antitumor and anti-metastasis efficacy in a 4T1 tumor-bearing model
We first established the H22 tumor model to evaluate the antitumor effect of the cRGD-TDA NPs. After being treated with different formulations, the cRGD-TDA NPs showed the highest tumor inhibition rate (89.49%) among other groups (free DOX: 54.84%, free DOX + ADD: 65.65%, free DOX–ADD: 67.18%, TD NPs: 74.95%, TDA NPs: 84.30%) (Fig. S11, ESI†). The antitumor inhibition was well correlated with the cytotoxicity results. Meanwhile, significantly more cRGD-TDA NPs accumulated in the tumors and caused more apoptotic nuclei and abnormal cellular morphology in the tumor sections than other groups (Fig. S12, ESI†). The immunohistology also showed the fewest vessels and the largest positive areas of TUNEL in the cRGD-TDA NPs group (Fig. S13, ESI†). Considering this superior antitumor efficacy of cRGD-TDA NPs, cRGD-TDA NPs rather than TD NPs or TDA NPs were selected for combination therapy with PD-L1 antagonists in the 4T1 tumor model.The antitumor and anti-metastasis efficacy of the cRGD-TDA NPs and combination therapy were further evaluated in the 4T1 orthotopic model. PD-L1 is overexpressed on the surface of tumor cells, resulting from the immunosuppressive microenvironment or persistent antigen exposure from the lysates of apoptotic tumor cells, and contributing to the exhaustion and anergy of effector immune cells. Anti-PD-L1 was introduced into combination therapy in order to reprogram the antitumor immunity and reinforce the therapeutic effect of the cRGD-TDA NPs.
As shown in Fig. 6(B), (C) and (H), anti-PD-L1 therapy slightly inhibited tumor progression by 38.05%, similar to the free DOX group (33.71%), which may be due to the loss of tumor immunogenicity and immature states of DCs. Moreover, the free DOX + ADD group showed minor improvement in remission by 9.94% compared to free DOX, which implied the relatively weaker antitumor efficacy of ADD. Furthermore, the cRGD-TDA NPs showed a significantly aggressive regression effect with a 67.41% inhibition rate, revealing the superior antitumor efficacy compared to the free drug groups. Notably, combination therapy of cRGD-TDA NPs plus anti-PD-L1 achieved the highest synergistic inhibition rate (84.08%), extremely boosting the efficacy of single anti-PD-L1 therapy. These results correlated well with the tumor weights in Fig. 6(D). As shown in the H&E and ki67 assays (Fig. 6(I)), the largest area of necrosis in the tumor sections was observed for the combination therapy. Conversely, the minimum positive area of expressed ki67 was found for the combination therapy. These histology results were well consistent with the tumor inhibition study. Moreover, negligible fluctuation was observed in the cRGD-TDA NPs and combination groups, while free DOX and free DOX + ADD showed heavy fluctuation (Fig. 6(G)), reflecting the lower systemic toxicity of the nano-formulation than the free chemodrugs.
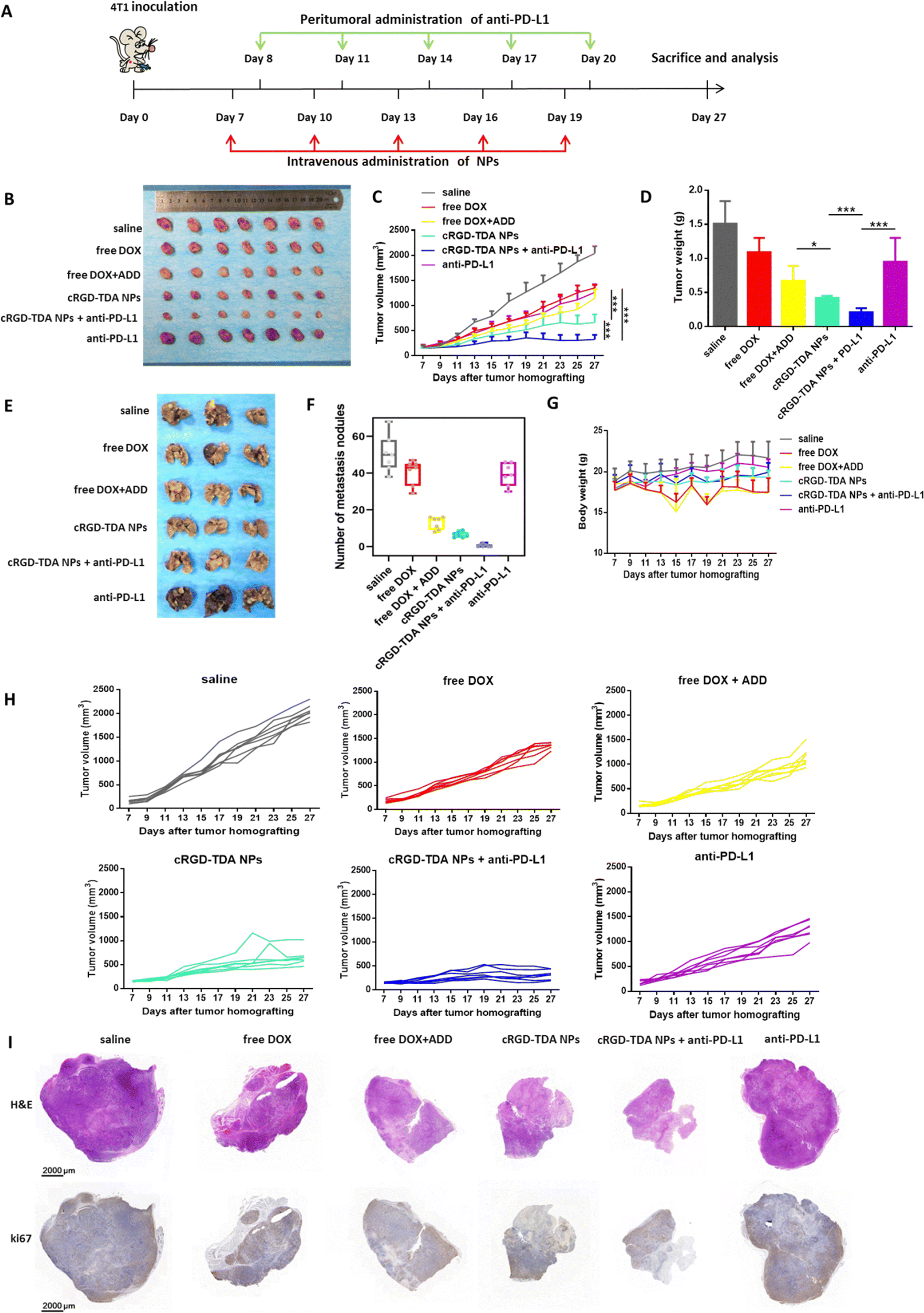 | ||
| Fig. 6 In vivo antitumor and anti-metastasis study in 4T1 tumor-bearing model. (A) Schematic illustration of procedures in inhibition study. (B) Images of dissected tumors, (C) tumor growth curves and (D) the weight of dissected tumor tissues in antitumor study after treatment with saline, free DOX, free DOX + ADD, cRGD-TDA NPs, cRGD-TDA NPs + anti-PD-L1 or anti-PD-L1 (n = 7). (E) Images of metastasis nodules in lungs stained with Bousin's fixative buffer and (F) the number of metastasis nodules in anti-metastasis study after treatment with saline, free DOX, free DOX + ADD, cRGD-TDA NPs, cRGD-TDA NPs + anti-PD-L1 or anti-PD-L1 (n = 7). (G) The body weight changes in the inhibition study (n = 7). (I) Scanning images of H&E and ki67 staining assays. | ||
In addition, luciferase-expressing 4T1 cells were utilized as a metastatic model because of their aggressive and metastatic property. During the treatment, luciferin was injected to monitor the metastasis states. As shown in Fig. S14 (ESI†), negligible luminescence area appeared in cRGD-TDA NPs + anti-PD-L1 group and cRGD-TDA NPs group also showed fewer metastatic spots compared with saline and free drug groups. At the end of the study, it was easily found that the number of pulmonary metastatic nodules obviously decreased after treatment with free DOX + ADD, being evidently lower than the free DOX group or the saline group in Fig. 6(E) and (F) (number of lung nodules: 12.86 ± 2.93 vs. 40.57 ± 4.82 or 50.57 ± 7.73), showing the powerful anti-metastasis ability of ADD, consistent with the in vitro results. Furthermore, the cRGD-TDA NPs achieved better attenuated metastasis (6.57 ± 1.41), and the nodules could barely be observed in the combination therapy group (0.57 ± 0.80). These results demonstrated that the cRGD-TDA NPs prominently possess potent antitumor and anti-metastasis effects, and both of them could be enhanced to a great extent when combined with anti-PD-L1 therapy.
2.9. Antitumor immune response of cRGD-TDA NPs and combination therapy
To further elucidate the mechanism of the cRGD-TDA NPs and the synergistic therapy, tumor-infiltrating lymphocytes were isolated and analyzed by flow cytometry. It is well known that eliciting the antigen presentation and maturation of DCs will further activate CD4+ T cells (helper T lymphocytes) and enhance CD8+ T-cell (cytotoxic T lymphocytes) infiltration in tumor sites. As shown in Fig. 7(A), infiltrating CD8+ T cells in the cRGD-TDA NPs + anti-PD-L1 group accounted for the maximum percentage of 10.05 ± 0.92% and cRGD-TDA NPs elevated CD8+ T cells to 7.50 ± 0.72%. Both of them were obviously higher than the counterparts of saline, free DOX, free DOX + ADD, and single anti-PD-L1 therapy. Meanwhile, combination therapy upregulated the proportion of infiltrating CD4+ T cells to 21.93 ± 2.37%, higher than for the cRGD-TDA NPs (14.35 ± 1.51%) or anti-PD-L1 groups (13.70 ± 0.52%), prominently demonstrating elevated infiltrating T cells in the tumor microenvironment. CD69 is a vital marker representing the activated state of functional lymphocytes. The expression of CD69 declines when T cells are in exhausted condition due to persistent antigen exposure or immunosuppressive microenvironment.58 As we expected, increased CD69 expression on CD4+ and CD8+ T cells was detected after anti-PD-L1 therapy, especially in the CD4+ subtype, more than the saline and other free drug groups (Fig. 7(B) and (C)). This intriguing phenomenon revealed that anti-PD-L1 therapy had a powerful capacity of reversing the exhausted or anergic states under consecutive chemo-inducing apoptosis condition or immunosuppressive tumor environment. More importantly, IFN-γ secreting T cells (IFN-γ+T) and natural killing T (NKT) cells were strong killing immune cells and represented the level of specific adaptive antitumor immune response.59–62 The proportions of these armed immune cells were investigated at the same time. Fig. 7(D) and (E) showed combination therapy elevated the amount of IFN-γ+T and NKT cells to 16.63 ± 1.40% and 6.24 ± 0.78%, respectively, significantly higher than the other groups. Moreover, we found that free DOX (1.24%) and free DOX + ADD (1.54%) reduced the proportion of NKT cells in comparison with saline (3.36%), reflecting the fact that the cRGD-TDA NPs could reduce the negative impact on NKT cells, mainly due to their tumor targeting ability and superior immunostimulatory functions. In addition, the amount of immunosuppressive cells such as regulatory T cells (Tregs, CD25+Foxp3+ cells) in the combination group drastically declined to 0.42 ± 0.06% compared with the other groups. Furthermore, the cRGD-TDA NPs achieved greater reduction of Tregs than free DOX and free DOX + ADD, which could be attributed to the enhanced immunogenic response by the cRGD-TDA NPs (Fig. 7(F)). Furthermore, type I macrophages (MΦ1) were upregulated to 20.53 ± 1.86% after combination treatment (Fig. 7(G)) compared with the negligible enhanced percentage in the anti-PD-L1 group (8.00%), which verified the immunostimulation and immune-environment reprogramming effects of the cRGD-TDA NPs. In summary, these promising findings confirmed that the cRGD-TDA NPs tremendously activated the specific immune response and have potential to reverse the immunosuppressive microenvironment in tumor regions. Furthermore, these capacities could be amplified through combination therapy with anti-PD-L1 to achieve a prominent tumor growth inhibition effect.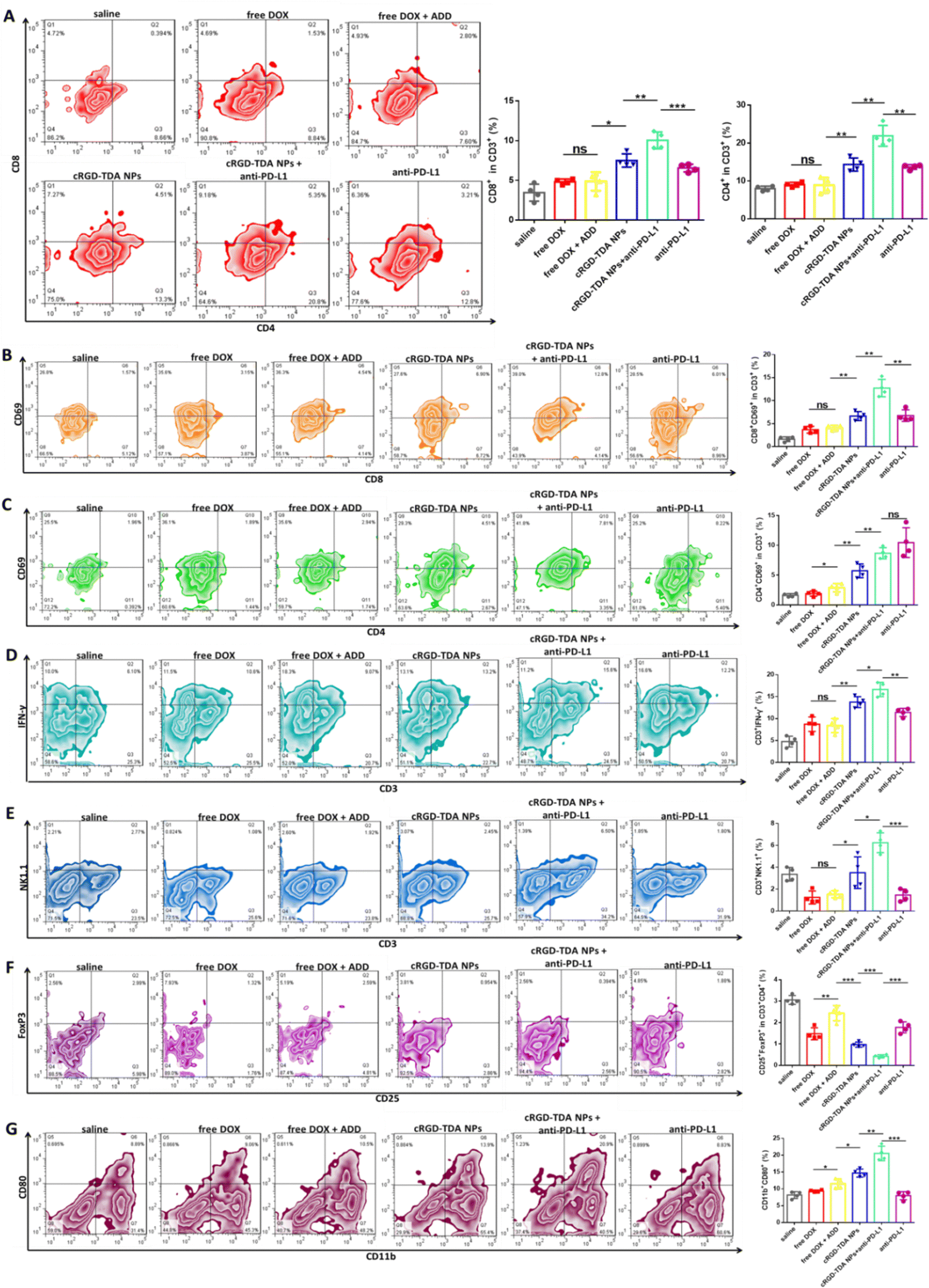 | ||
| Fig. 7 In vivo antitumor immune response of cRGD-TDA NPs and combination therapy. The scatter plots and column graphs of the proportion of (A) CD4+ and CD8+ lymphocytes in CD3+ gates, (B) CD8+CD69+ in CD3+ gates, (C) CD4+CD69+ in CD3+ gates, (D) IFN-γ+ T cells, (E) NKT cells, (F) CD25+FoxP3+ (Tregs) in CD3+CD4+ gates and (G) CD11b+CD80+ (type I macrophages, MΦ1) after treatment with saline, free DOX, free DOX + ADD, cRGD-TDA NPs, cRGD-TDA NPs + anti-PD-L1 or anti-PD-L1 (n = 4). (***p < 0.001; **p < 0.01; *p < 0.05; NS, no significant difference). | ||
2.10. Biosafety evaluation
To evaluate the biosafety of the cRGD-TDA NPs in vivo, BALB/c mice were administrated with saline, free DOX, free DOX + ADD or cRGD-TDA NPs by the same procedures used in the earlier inhibition study. Blood biochemical and histological analysis were conducted. As depicted in Fig. S15 and S16 (ESI†), compared with the saline group, all the parameters (total protein, TP; albumin, ALB; globulin, GLO; albumin/globulin, A/G; alanine transaminase, ALT; aspartate transaminase, AST; creatinine, CRE and total bilirubin, TBIL) in the cRGD-TDA NPs group showed no statistically significant difference. In contrast, free DOX and free DOX + ADD slightly reduced the level of GLO, demonstrating that the free chemodrugs caused more toxicity than the cRGD-TDA NPs. Furthermore, the histological analysis of the main organs showed no obvious damage in the cRGD-TDA NPs group, demonstrating that the cRGD-TDA NPs had no noticeable acute systemic toxicity and had good biocompatibility to be utilized as candidates for cancer therapy.It is still a big challenge to overcome the hurdles of cancer metastasis. Conventional clinical chemotherapy not only suppresses cancer cell development, but also induces CSC traits, triggers EMT effects and causes an inflammatory response in the tumor microenvironment.44 As a result of these phenomena, the metastatic potential is elevated, leading to fatal elements for patients. Thus, therapeutic strategies against metastasis are urgently needed.
Recent studies have focused on reinforcing the anti-metastasis effect of chemotherapy. The existence of CSCs, referred to as tumor-initiating cells, is evidence of one of the vital elements of metastasis in multiple malignancies.45 All-trans retinoic acid (ATRA) has the ability to reduce the proportion of CSCs in various tumors. Mo et al. proposed a convergent therapeutic strategy using a cell-differentiation-regulator, ATRA, to form a polymer–drug conjugate (nitroimidazole-modified hyaluronic acid–oxalate–CPT conjugate: n-HA–oxa–CPT) for overcoming the relapse and metastasis of the CSC-enriched heterogeneous breast tumors in the mouse models.63 As recently reported, the inflammatory TME can trigger the process of metastasis through activating the NF-κB pathway and upregulating pro-metastasis molecules downstream.64 Anti-inflammation agents can reduce inflammation in the TME to prevent the occurrence of metastasis. For example, berberine can contribute to the inhibition of chemotherapy-inducing metastasis by blocking the arachidonic acid (AA) metabolic pathway.65 Based on this theory, Yuan Huang et al. formulated celecoxib (COX-2 inhibitor) encapsulated in Lip-Gel to inhibit inflammation in the TME through blocking the COX-2/PGE2 pathway. This strategy downregulated inflammation and alleviated the metastasis potential of TNBC.66 Sun et al. introduced Digoxin (DIG) into DOX-loaded 4T1 membrane-coated PLGA nanoparticles to inhibit the migration and metastasis of breast cancer. Due to its Na+/K+-ATPase-inhibiting function, DIG elevated the intracellular Ca2+ concentration, restrained cell–cell junctions between breast cancer cells, inhibited the EMT effect, and thereby suppressed metastasis.67 Chen et al. designed systemic metastasis-targeting nanotherapeutic H@CaPP for co-delivering an anti-inflammatory agent, piceatannol (PIC), and an anti-thrombotic agent, low molecular weight heparin (LMWH), to combat tumor metastasis by hindering the multiple steps of tumor metastasis. Because PIC effectively inhibited metastasis by suppressing signal transducers and activators of STAT-3 and NF-κB, H@CaPP successfully inhibited the EMT process in the invasion phase. Combined with chemotherapy or surgical resection, H@CaPP significantly reduced lung tumor metastasis and prolonged the overall survival of 4T1 tumor-bearing mice.68
Inspired by these promising results, we planned to combine DOX and ADD into one nanosystem through chemical conjugating method to detect whether the anti-metastasis agent, ADD, can achieve attenuated metastasis together with DOX treatment. ADD is an analog of lonidamine, interfering with mitochondrial function to impact energy metabolism in cancer cells and is normally applied for enhancing the chemo-sensitivity of breast cancer cells in chemotherapy.46 Recent research demonstrated that ADD has an anti-inflammation effect through inhibiting the NF-κB pathway for neuroprotection,69 and also plays an anti-metastasis role by inhibiting the PI3K/Akt/mTOR or Wnt signaling pathways to reverse the EMT-inducing metastasis.47–51 These interesting findings remind us of the possibility of the combination of ADD plus chemotherapy for reducing the occurrence of metastasis. As a proof of concept, our study finds that DOX + ADD indeed shows prominent anti-migration ability in vitro (Fig. 2) and a potent anti-metastasis effect to a certain extent in vivo (Fig. 6 and Fig. S14, ESI†).
In order to ensure the ratiometric stability and adequate drug release in the acidic endosome environment, in this study, we precisely conjugated DOX and ADD by hydrazone bond. Due to the hydrophobic profile of the DOX–ADD conjugate, we introduced amphiphilic TPGS as a carrier to conjugate TPGS with DOX–ADD together so as to form self-assembling nanoparticles to increase the active targeting ability and circulation time in body. TPGS is an adjuvant approved by the FDA, meanwhile recent works find it is also a powerful ROS inducer because of its interaction with ubiquinone-binding sites of mitochondrial respiratory complex II.40–42 According to recent research, a high dose of intracellular ROS can induce the ICD effect, lately enhancing the maturation of DCs. Based on this idea, we want to confirm whether TPGS is capable of inducing the ICD effect. As our results show (Fig. 3, 5 and Fig. S5, S6, ESI†), upregulated ROS and enhanced ICD effect in vitro or in vivo are observed and the strength of the ICD effect is proportionally related to the concentration of TPGS. This novel finding makes us believe that TPGS can widely act as an endogenous chemodynamic therapy agent to supplement the inadequate ICD effect of chemodrugs in the field of chemoimmunotherapy. More importantly, we elucidate that our three-in-one cocktail cRGD-TDA NPs possess anti-proliferation, anti-metastasis and ICD effects in vitro (Fig. 2 and 3), and also in the orthotopic 4T1 murine breast cancer model (Fig. 5–7).
Thanks to the positive feedback of cancer immunotherapy in malignancy, the combination therapy between nanomedicine and immunotherapy is blooming into one of the hottest research orientations. Obviously, a large amount of relevant researches are being conducted.70 As far as we know, there are some articles about combining chemodrugs, anti-metastasis agents and immunotherapies together to investigate the synergistic anti-proliferation and anti-metastasis effects.71–73 Considering these promising results and the ICD effect of the cRGD-TDA NPs, it is reasonable to infer that the ability of reducing the immunosuppressive condition by ICB can improve the antitumor and anti-metastasis outcomes. To verify the combination effect of immunotherapy and the cRGD-TDA NPs, we administered the cRGD-TDA NPs combined with PD-L1 antagonist in a 4T1 breast cancer model to evaluate the antitumor and anti-metastasis effects. Excitingly, the combination therapy inhibits tumor progression better than cRGD-TDA NPs and anti-PD-L1 alone. Meanwhile, the metastatic nodules in lungs can be barely observed after combination treatment (Fig. 6). The immune response analysis (Fig. 7) shows that the combination therapy elevates the proportion of infiltrating CD4+ and CD8+ T cells in the tumor environment, also enhances the activated states of CD4+ and CD8+ T cells, which means the immunosuppressive environment and anergic state are reversed by combination therapy. In addition, the proportions of positive immune cells, like IFN-γ+T, NKT cells and MΦ1, are upregulated after combination therapy; the proportions of negative immune cells, like Tregs, are downregulated after combination therapy. These phenomena verify the antitumor and anti-metastasis capacities of the cRGD-TDA NPs and the beneficial outcomes when combined with anti-PD-L1 therapy. In summary, the cRGD-TDA NPs prove the powerful antitumor and anti-metastasis effects of the combination of chemodrug, anti-metastasis agent and ICD effect inducer. At the same time, the combination of multifunctional nanoparticles and immunotherapy provides more possibilities for advanced and metastatic cancer therapies.
3. Conclusions
Overall, we successfully synthesized a dual pH-sensitive conjugate composed of three functional agents (DOX, ADD and TPGS) and constructed cocktail TDA NPs with good morphology and stability. The TDA NPs exhibited a quick disassembling response and efficient drug release under an acidic environment through cleavage of pH-sensitive linkers. Because of the cocktail effects of the different agents, the TDA NPs showed superior cytotoxicity and anti-metastasis effects in 4T1 cells. Significantly, we found that the FDA-approved pharmaceutic excipient, TPGS, possessed a powerful ICD-inducing capacity, and this special profile might broaden the range of TPGS in scientific and clinical applications. Meanwhile, the introduction of TPGS empowered the TDA NPs to elicit a robust ICD effect and promote maturation and antigen presentation of DCs in the 4T1 breast tumor model. After being modified with targeting peptides cRGD, cRGD-TDA NPs demonstrated preferential tumor accumulation, and also displayed robust tumor recession and anti-metastasis effects. Additionally, the combination with PD-L1 antagonists significantly improved recruitment of infiltrating lymphocytes and reduced immunosuppressive and exhausted states in the tumor microenvironment, which resulted in stronger tumor inhibition and metastasis prevention. We believe cocktail cRGD-TDA NPs offer a promising strategy for malignant cancer therapy and have great potential for broad applications in chemoimmunotherapy.Author contributions
Chenming Zou: methodology, software, investigation, visualization, writing – original draft. Yuepeng Tang: methodology, software, investigation, visualization. Ping Zeng: software, validation, formal analysis, visualization. Derong Cui: investigation, writing – review & editing. Majdi Al Amili: methodology, validation. Ya Chang: methodology, validation. Zhu Jin: resources, data curation. Yuanyuan Shen: conceptualization, resources, supervision. Songwei Tan: conceptualization, supervision, writing – review & editing. Shengrong Guo: conceptualization, resources, supervision, writing – original draft, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Natural Science Foundation of China (grant number 82073776 and 82104075), the Shanghai Sailing Program (grant number 21YF1420400), the Shanghai Jiao Tong University Foundation (YG2021QN33, YG2021GD04, YG2021GD03), and the Startup Fund for Young Faculty at SJTU (21X010500792). The authors thank LOGAN INSTRUMENT CORP. and the Analytical and Testing Center of Shanghai Jiao Tong University for sample testing. We also thank Dr. Liu Tian, Dr. Zhaoyang Chen and Dr. Ning Yang for valuable experimental assistance.References
- D. F. Quail and J. A. Joyce, Nat. Med., 2013, 19, 1423–1437 CrossRef CAS PubMed.
- S. Loibl, P. Poortmans, M. Morrow, C. Denkert and G. Curigliano, Lancet, 2021, 397, 1750–1769 CrossRef CAS PubMed.
- P. Schmid, H. S. Rugo, S. Adams, A. Schneeweiss, C. H. Barrios, H. Iwata, V. Dieras, V. Henschel, L. Molinero, S. Y. Chui, V. Maiya, A. Husain, E. P. Winer, S. Loi, L. A. Emens and I. M. Investigators, Lancet Oncol., 2020, 21, 44–59 CrossRef CAS PubMed.
- D. Mathios, J. E. Kim, A. Mangraviti, J. Phallen, C.-K. Park, C. M. Jackson, T. Garzon-Muvdi, E. Kim, D. Theodros, M. Polanczyk, A. M. Martin, I. Suk, X. Ye, B. Tyler, C. Bettegowda, H. Brem, D. M. Pardoll and M. Lim, Sci. Transl. Med., 2016, 8, 370ra180 Search PubMed.
- F. Marcucci, G. Stassi and R. De Maria, Nat. Rev. Drug Discovery, 2016, 15, 311–325 CrossRef CAS PubMed.
- D. R. Welch and D. R. Hurst, Cancer Res., 2019, 79, 3011–3027 CrossRef CAS PubMed.
- D. M. Gilkes, G. L. Semenza and D. Wirtz, Nat. Rev. Cancer, 2014, 14, 430–439 CrossRef CAS PubMed.
- W. Yu, C. Hu and H. Gao, Adv. Drug Delivery Rev., 2021, 178, 113909 CrossRef CAS PubMed.
- C. Liang, L. Xu, G. Song and Z. Liu, Chem. Soc. Rev., 2016, 45, 6250–6269 RSC.
- J. Nam, S. Son, K. S. Park, W. Zou, L. D. Shea and J. J. Moon, Nat. Rev. Mater., 2019, 4, 398–414 CrossRef.
- M. J. Mitchell, M. M. Billingsley, R. M. Haley, M. E. Wechsler, N. A. Peppas and R. Langer, Nat. Rev. Drug Discovery, 2021, 20, 101–124 CrossRef CAS PubMed.
- Q. Hu, W. Sun, C. Wang and Z. Gu, Adv. Drug Delivery Rev., 2016, 98, 19–34 CrossRef CAS PubMed.
- P. Dobosz and T. Dzieciatkowski, Front. Immunol., 2019, 10, 2965 CrossRef CAS PubMed.
- A. D. Waldman, J. M. Fritz and M. J. Lenardo, Nat. Rev. Immunol., 2020, 20, 651–668 CrossRef CAS PubMed.
- S. K. Wculek, F. J. Cueto, A. M. Mujal, I. Melero, M. F. Krummel and D. Sancho, Nat. Rev. Immunol., 2020, 20, 7–24 CrossRef CAS PubMed.
- S. P. Kubli, T. Berger, D. V. Araujo, L. L. Siu and T. W. Mak, Nat. Rev. Drug Discovery, 2021, 20, 899–919 CrossRef CAS PubMed.
- W. Zou, J. D. Wolchok and L. Chen, Sci. Transl. Med., 2016, 8, 328rv4 Search PubMed.
- P. S. Hegde and D. S. Chen, Immunity, 2020, 52, 17–35 CrossRef CAS PubMed.
- J. Galon and D. Bruni, Nat. Rev. Drug Discovery, 2019, 18, 197–218 CrossRef CAS PubMed.
- A. Bassez, H. Vos, L. Van Dyck, G. Floris, I. Arijs, C. Desmedt, B. Boeckx, M. Vanden Bempt, I. Nevelsteen, K. Lambein, K. Punie, P. Neven, A. D. Garg, H. Wildiers, J. Qian, A. Smeets and D. Lambrechts, Nat. Med., 2021, 27, 820–832 CrossRef CAS PubMed.
- S. L. Topalian, J. M. Taube, R. A. Anders and D. M. Pardoll, Nat. Rev. Cancer, 2016, 16, 275–287 CrossRef CAS PubMed.
- P. Sharma, S. Hu-Lieskovan, J. A. Wargo and A. Ribas, Cell, 2017, 168, 707–723 CrossRef CAS PubMed.
- J. L. Benci, L. R. Johnson, R. Choa, Y. Xu, J. Qiu, Z. Zhou, B. Xu, D. Ye, K. L. Nathanson, C. H. June, E. J. Wherry, N. R. Zhang, H. Ishwaran, M. D. Hellmann, J. D. Wolchok, T. Kambayashi and A. J. Minn, Cell, 2019, 178, 933–948 CrossRef CAS PubMed.
- P. Schmid, S. Y. Chui and L. A. Emens, N. Engl. J. Med., 2019, 380, 987–988 Search PubMed.
- J. Cortes, D. W. Cescon, H. S. Rugo, Z. Nowecki, S.-A. Im, M. M. Yusof, C. Gallardo, O. Lipatov, C. H. Barrios, E. Holgado, H. Iwata, N. Masuda, M. Torregroza Otero, E. Gokmen, S. Loi, Z. Guo, J. Zhao, G. Aktan, V. Karantza, P. Schmid and K. Investigators, Lancet, 2020, 396, 1817–1828 CrossRef PubMed.
- D. V. Krysko, A. D. Garg, A. Kaczmarek, O. Krysko, P. Agostinis and P. Vandenabeele, Nat. Rev. Cancer, 2012, 12, 860–875 CrossRef CAS PubMed.
- L. Galluzzi, A. Buque, O. Kepp, L. Zitvogel and G. Kroemer, Nat. Rev. Immunol., 2017, 17, 97–111 CrossRef CAS PubMed.
- X. Duan, C. Chan and W. Lin, Angew. Chem., Int. Ed., 2019, 58, 670–680 CrossRef CAS PubMed.
- M. Kyu Shim, S. Yang, I.-C. Sun and K. Kim, Adv. Drug Delivery Rev., 2022, 183, 114177 CrossRef CAS PubMed.
- L. Galluzzi, L. Senovilla, L. Zitvogel and G. Kroemer, Nat. Rev. Drug Discovery, 2012, 11, 215–233 CrossRef CAS PubMed.
- J. Li, H. Wang, Y. Wang, X. Gong, X. Xu, X. Sha, A. Zhang, Z. Zhang and Y. Li, Adv. Mater., 2020, 32, 2002380 CrossRef CAS PubMed.
- G. Schiavoni, A. Sistigu, M. Valentini, F. Mattei, P. Sestili, F. Spadaro, M. Sanchez, S. Lorenzi, M. T. D'Urso, F. Belardelli, L. Gabriele, E. Proietti and L. Bracci, Cancer Res., 2011, 71, 768–778 CrossRef CAS PubMed.
- M. Obeid, A. Tesniere, F. Ghiringhelli, G. M. Fimia, L. Apetoh, J. L. Perfettini, M. Castedo, G. Mignot, T. Panaretakis, N. Casares, D. Metivier, N. Larochette, P. van Endert, F. Ciccosanti, M. Piacentini, L. Zitvogel and G. Kroemer, Nat. Med., 2007, 13, 54–61 CrossRef CAS PubMed.
- L. Apetoh, F. Ghiringhelli, A. Tesniere, M. Obeid, C. Ortiz, A. Criollo, G. Mignot, M. C. Maiuri, E. Ullrich, P. Saulnier, H. Yang, S. Amigorena, B. Ryffel, F. J. Barrat, P. Saftig, F. Levi, R. Lidereau, C. Nogues, J.-P. Mira, A. Chompret, V. Joulin, F. Clavel-Chapelon, J. Bourhis, F. Andre, S. Delaloge, T. Tursz, G. Kroemer and L. Zitvogel, Nat. Med., 2007, 13, 1050–1059 CrossRef CAS PubMed.
- Z. Yu, J. F. Guo, M. Y. Hu, Y. Q. Gao and L. A. Huang, ACS Nano, 2020, 14, 4816–4828 CrossRef CAS PubMed.
- Z. C. Wang, C. Sun, H. J. Wu, J. Z. Xie, T. Zhang, Y. M. Li, X. L. Xu, P. L. Wang and C. Wang, Mater. Today Bio, 2022, 13, 100189 CrossRef CAS PubMed.
- L. S. Xie, G. H. Wang, W. Sang, J. Li, Z. Zhang, W. X. Li, J. Yan, Q. Zhao and Y. L. Dai, Biomater., 2021, 269, 120638 CrossRef CAS PubMed.
- J. Zhang, X. Sun, X. Zhao, C. Yang, M. Shi, B. Zhang, H. Hu, M. Qiao, D. Chen and X. Zhao, Acta Pharm. Sin. B, 2022, 12, 3694–3709 CrossRef CAS PubMed.
- C. Chen, X. Ni, S. R. Jia, Y. Liang, X. L. Wu, D. L. Kong and D. Ding, Adv. Mater., 2019, 31, 1904914 CrossRef CAS PubMed.
- Z. Su, M. Chen, Y. Xiao, M. Sun, L. Zong, S. Asghar, M. Dong, H. Li, Q. Ping and C. Zhang, J. Controlled Release, 2014, 196, 370–383 CrossRef CAS PubMed.
- S. Tan, C. Zou, W. Zhang, M. Yin, X. Gao and Q. Tang, Drug Delivery, 2017, 24, 1831–1842 CrossRef CAS PubMed.
- S. Fulda, L. Galluzzi and G. Kroemer, Nat. Rev. Drug Discovery, 2010, 9, 447–464 CrossRef CAS PubMed.
- Y. L. Bao, M. X. Yin, X. M. Hu, X. T. Zhuang, Y. Sun, Y. Y. Guo, S. W. Tan and Z. P. Zhang, J. Controlled Release, 2016, 235, 182–194 CrossRef CAS PubMed.
- Y. W. Zhao, M. H. He, L. Z. Cui, M. H. Gao, M. Zhang, F. L. Yue, T. F. Shi, X. H. Yang, Y. Pan, X. Zheng, Y. Jia, D. Shao, J. Li, K. He and L. Chen, Br. J. Cancer, 2020, 122, 1638–1648 CrossRef CAS PubMed.
- X. Ye, W. L. Tam, T. Shibue, Y. Kaygusuz, F. Reinhardt, E. N. Eaton and R. A. Weinberg, Nature, 2015, 525, 256–260 CrossRef CAS PubMed.
- Q. Wang, C. Zou, L. Wang, X. Gao, J. Wu, S. Tan and G. Wu, Acta Biomater., 2019, 94, 469–481 CrossRef CAS PubMed.
- Y. Huang, G. Sun, X. Sun, F. Li, L. Zhao, R. Zhong and Y. Peng, Cancers, 2020, 12, 3332 CrossRef CAS PubMed.
- G. Cheng, Q. Zhang, J. Pan, Y. Lee, O. Ouari, M. Hardy, M. Zielonka, C. R. Myers, J. Zielonka, K. Weh, A. C. Chang, G. Chen, L. Kresty, B. Kalyanaraman and M. You, Nat. Commun., 2019, 10, 2205 CrossRef PubMed.
- X. Wang, Q. Zeng, Z. Li, X. Yang, W. Xia and Z. Chen, Thorac. Cancer, 2019, 10, 642–658 CrossRef CAS PubMed.
- A. W. Lambert and R. A. Weinberg, Nat. Rev. Cancer, 2021, 21, 325–338 CrossRef CAS PubMed.
- J. Yang, P. Antin, G. Berx, C. Blanpain, T. Brabletz, M. Bronner, K. Campbell, A. Cano, J. Casanova, G. Christofori, S. Dedhar, R. Derynck, H. L. Ford, J. Fuxe, A. Garcia de Herreros, G. J. Goodall, A.-K. Hadjantonakis, R. Y. J. Huang, C. Kalcheim, R. Kalluri, Y. Kang, Y. Khew-Goodall, H. Levine, J. Liu, G. D. Longmore, S. A. Mani, J. Massague, R. Mayor, D. McClay, K. E. Mostov, D. F. Newgreen, M. A. Nieto, A. Puisieux, R. Runyan, P. Savagner, B. Stanger, M. P. Stemmler, Y. Takahashi, M. Takeichi, E. Theveneau, J. P. Thiery, E. W. Thompson, R. A. Weinberg, E. D. Williams, J. Xing, B. P. Zhou, G. Sheng and E. M. T. I. A. Temtia, Nat. Rev. Mol. Cell Biol., 2021, 22, 834 CrossRef CAS PubMed.
- G. Chen, A. C. Huang, W. Zhang, G. Zhang, M. Wu, W. Xu, Z. Yu, J. Yang, B. Wang, H. Sun, H. Xia, Q. Man, W. Zhong, L. F. Antelo, B. Wu, X. Xiong, X. Liu, L. Guan, T. Li, S. Liu, R. Yang, Y. Lu, L. Dong, S. McGettigan, R. Somasundaram, R. Radhakrishnan, G. Mills, Y. Lu, J. Kim, Y. H. Chen, H. Dong, Y. Zhao, G. C. Karakousis, T. C. Mitchell, L. M. Schuchter, M. Herlyn, E. J. Wherry, X. Xu and W. Guo, Nature, 2018, 560, 382–386 CrossRef CAS PubMed.
- M. Taki, K. Abiko, M. Ukita, R. Murakami, K. Yamanoi, K. Yamaguchi, J. Hamanishi, T. Baba, N. Matsumura and M. Mandai, Clin. Cancer Res., 2021, 27, 4669–4679 CrossRef CAS PubMed.
- H.-N. Chang, B.-Y. Liu, Y.-K. Qi, Y. Zhou, Y.-P. Chen, K.-M. Pan, W.-W. Li, X.-M. Zhou, W.-W. Ma, C.-Y. Fu, Y.-M. Qi, L. Liu and Y.-F. Gao, Angew. Chem., Int. Ed., 2015, 54, 11760–11764 CrossRef CAS PubMed.
- C. Zou, G. Jiang, X. Gao, W. Zhang, H. Deng, C. Zhang, J. Ding, R. Wei, X. Wang, L. Xi and S. Tan, Nanomedicine, 2019, 22, 102092 CrossRef CAS PubMed.
- T.-C. Chou, Cancer Res., 2010, 70, 440–446 CrossRef CAS PubMed.
- W. Zhang, X. Hu, Q. Shen and D. Xing, Nat. Commun., 2019, 10, 1704 CrossRef PubMed.
- D. Cibrian and F. Sanchez-Madrid, Eur. J. Immunol., 2017, 47, 946–953 CrossRef CAS PubMed.
- E. Vivier, S. Ugolini, D. Blaise, C. Chabannon and L. Brossay, Nat. Rev. Immunol., 2012, 12, 239–252 CrossRef CAS PubMed.
- D. Lindau, P. Gielen, M. Kroesen, P. Wesseling and G. J. Adema, Immunology, 2013, 138, 105–115 CrossRef CAS PubMed.
- A. M. Gocher, C. J. Workman and D. A. A. Vignali, Nat. Rev. Immunol., 2022, 22, 158–172 CrossRef CAS PubMed.
- J. Fourcade, Z. Sun, O. Pagliano, P. Guillaume, I. F. Luescher, C. Sander, J. M. Kirkwood, D. Olive, V. Kuchroo and H. M. Zarour, Cancer Res., 2012, 72, 887–896 CrossRef CAS PubMed.
- S. Shen, X. Xu, S. Lin, Y. Zhang, H. Liu, C. Zhang and R. Mo, Nat. Nanotechnol., 2021, 16, 104–113 CrossRef CAS PubMed.
- H. Z. Huang, Y. X. Huang, Y. Chen, Z. Y. Luo, Z. Q. Zhang, R. Z. Sun, Z. Y. Wan, J. J. Sun, B. F. Lu, L. Zhang, J. Hu and S. Li, Biomater., 2021, 270, 120708 CrossRef CAS PubMed.
- Y. W. Zhao, X. H. Yang, J. T. Zhao, M. H. Gao, M. Zhang, T. F. Shi, F. Zhang, X. Zheng, Y. Pan, D. Shao, J. Li, K. He and L. Chen, Eur. J. Pharmacol., 2021, 895, 173887 CrossRef CAS PubMed.
- X. L. Yi, Y. Yan, L. Li, R. Zhou, X. R. Shen and Y. Huang, J. Controlled Release, 2022, 341, 753–768 CrossRef CAS PubMed.
- D. Li, Y. L. Wang, C. Li, Q. Wang, B. J. Sun, H. T. Zhang, Z. G. He and J. Sun, Acta Pharm. Sin. B, 2021, 11, 3262–3271 CrossRef CAS PubMed.
- M. J. Xu, K. L. Hu, Y. P. Liu, Y. K. Huang, S. S. Liu, Y. Chen, D. Y. Wang, S. L. Zhou, Q. Zhang, N. Mei, H. P. Lu, F. A. Li, X. L. Gao and J. Chen, Nat. Commun., 2021, 12, 3187 CrossRef CAS PubMed.
- T. T. Zhang, X. Yang, T. Y. Liu, J. X. Shao, N. Z. Fu, A. J. Yan, K. Y. Geng and W. L. Xia, Stem Cell Res. Ther., 2017, 8, 248 CrossRef PubMed.
- J. Nam, S. Son, K. S. Park, W. P. Zou, L. D. Shea and J. J. Moon, Nat. Rev. Mater., 2019, 4, 398–414 CrossRef.
- C. Hu, X. He, Y. Chen, X. Yang, L. Qin, T. Lei, Y. Zhou, T. Gong, Y. Huang and H. Gao, Adv. Funct. Mater., 2021, 31, 2007149 CrossRef CAS.
- Y. Xu, J. Xiong, X. Sun and H. Gao, Acta Pharm. Sin. B, 2022, 12, 4327–4347 CrossRef CAS PubMed.
- Y. Chu, T. Sun and C. Jiang, Chin. Chem. Lett., 2022, 33, 4157–4168 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nh00590e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |