
Transition metal single atom-optimized g-C3N4 for the highly selective electrosynthesis of H2O2 under neutral electrolytes†
Hongcen
Yang‡
,
Fei
Ma‡
,
Niandi
Lu
,
Shuhao
Tian
,
Guo
Liu
,
Ying
Wang
,
Zhixia
Wang
,
Di
Wang
,
Kun
Tao
 ,
Hong
Zhang
* and
Shanglong
Peng
*
,
Hong
Zhang
* and
Shanglong
Peng
*
School of Physical Science and Technology, School of Materials and Energy, Electron Microscopy Centre of Lanzhou University, Lanzhou University, Lanzhou 730000, China. E-mail: pengshl@lzu.edu.cn
First published on 17th March 2023
Abstract
Neutral electrosynthesis of H2O2via the 2e− ORR is attractive for numerous applications, but the low activity and high cost of electrocatalysts have become important constraints. Therefore, the development of cheap and efficient electrocatalysts for the 2e− ORR is necessary. Herein, we report the embedding of transition metal single atoms (TM SAs) in g-C3N4 nanosheets (CNNS). The introduction of TM SAs increases the N–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N content and reduces the C–C/CC content in CNNS, which contributes to the increased selectivity of TM SA/CNNS for the 2e− ORR. TM SA is the main reason for the enhanced activity of the 2e− ORR. Based on the results obtained by replacing a series of TM SA, the Ni0.10 SA/CNNS with optimal N–CN content exhibited the best selectivity (∼98%) and highest yield of H2O2 (∼503 mmol gcat−1 h−1), which is ∼14.6 times higher than that of CNNS (∼34.4 mmol gcat−1 h−1). Other TM SA/CNNS also exhibited high activity and selectivity. This study demonstrates the ability of TM SA to modulate the selectivity and activity of CNNS, making it a promising candidate for the 2e− ORR and providing more reference ideas for the preparation of H2O2.
N content and reduces the C–C/CC content in CNNS, which contributes to the increased selectivity of TM SA/CNNS for the 2e− ORR. TM SA is the main reason for the enhanced activity of the 2e− ORR. Based on the results obtained by replacing a series of TM SA, the Ni0.10 SA/CNNS with optimal N–CN content exhibited the best selectivity (∼98%) and highest yield of H2O2 (∼503 mmol gcat−1 h−1), which is ∼14.6 times higher than that of CNNS (∼34.4 mmol gcat−1 h−1). Other TM SA/CNNS also exhibited high activity and selectivity. This study demonstrates the ability of TM SA to modulate the selectivity and activity of CNNS, making it a promising candidate for the 2e− ORR and providing more reference ideas for the preparation of H2O2.
New conceptsTransition metal single atom-optimized g-C3N4 have been successfully synthesized via a two-step thermal oxidation etching method for the efficient neutral electrosynthesis of H2O2. Compared with CNNS, the H2O2 selectivity of Ni0.10 SA/CNNS increased from 61% to 98% in 0.1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) M PBS. The H2O2 production rate of Ni0.10 SA/CNNS is 503 mmol gcat−1 h−1, which is 14.6 times that of CNNS (34.4 mmol gcat−1 h−1). The excellent electrochemical activity is due to the introduction of TM SA, optimising the structure of CNNS and promoting the increase in N–CN sites and the decrease in C–C/CC sites in the CNNS, and improving the selectivity of the electrocatalysts for the 2e− ORR. The high electrochemical activities exhibited by TM SA/CNNS are due to the isolated TM SA on CNNS, which enhanced the 2e− ORR activity of electrocatalysts. M PBS. The H2O2 production rate of Ni0.10 SA/CNNS is 503 mmol gcat−1 h−1, which is 14.6 times that of CNNS (34.4 mmol gcat−1 h−1). The excellent electrochemical activity is due to the introduction of TM SA, optimising the structure of CNNS and promoting the increase in N–CN sites and the decrease in C–C/CC sites in the CNNS, and improving the selectivity of the electrocatalysts for the 2e− ORR. The high electrochemical activities exhibited by TM SA/CNNS are due to the isolated TM SA on CNNS, which enhanced the 2e− ORR activity of electrocatalysts.
|
1. Introduction
As a high-value and environmentally friendly oxidant, hydrogen peroxide (H2O2) has a wide range of applications in various industrial environments such as chemical synthesis, environmental remediation, and paper/pulp bleaching.1–4 Currently, the synthesis of H2O2 relies on anthraquinone processes.5 However, anthraquinone processes are limited by complex infrastructure, large energy input, and the generation of large quantities of waste chemicals.6 In addition, the high concentration of H2O2 (up to 70 wt%) obtained by the anthraquinone method has huge safety hazards in transportation and does not match the end-user demand that requires low concentrations of H2O2.7 Based on this, green and sustainable electrochemical synthesis (two-electron oxygen reduction reaction, 2e− ORR) that is powered by renewable electricity is highly sought-after for the direct on-site production of H2O2.8 However, the electrochemical synthesis of H2O2 is constrained by the four-electron oxygen reduction reaction (4e− ORR),9 so electrocatalysts with high activity and selectivity are essential. At present, noble metal catalysts (e.g., Pt, Pd, Ag, Au, Pd–Hg, and Au–Pd) exhibit excellent selectivity and activity for the synthesis of H2O210–16 but these catalysts are expensive, scarce, and have inferior stability, which restrict their large-scale production.13,17 Therefore, it is of great interest to develop highly selective, efficient, and durable electrocatalysts that can replace noble metals for electrochemical H2O2 synthesis.Single-atom electrocatalysts (SACs) benefit from the desirable properties of isolated metal sites to enable the end-on type of adsorption of O2, thereby reducing the probability of the O–O bond cleavage and improving their selectivity for electrochemical H2O2 synthesis.18,19 At the same time, the realization of atomical dispersion, the utilization of the unique geometry, and the regulation of the electronic structure of SACs are all conducive to the further improvement of the electrocatalytic activity and selectivity.7,15 Therefore, the study of carbon-supported non-noble metal single-atom electrocatalysts with tailored coordination environments for electrocatalytic H2O2 synthesis is of great interest. To date, non-noble metal SACs (e.g., Fe, Mn, Cu, Zn, Mo, Co, and Ni) based on carbon-based supports have emerged as promising schemes for the electrocatalytic synthesis of H2O2 in alkaline solutions.20–25 However, H2O2 has the problem of easy decomposition in alkaline solution, which is detrimental to the preservation of H2O2.10 Therefore, the electrosynthesis of H2O2 under neutral conditions is very attractive for many applications where neutral pH operation is required, such as H2O2 fuel cells, disinfection, environmental purification, and other on-site H2O2 applications.26 However, the low activity for the electrochemical synthesis of H2O2 at neutral pH has become an important factor restricting its development.27,28 This is due to the relatively low concentration of hydroxide ions in the neutral electrolyte hindering the efficient incorporation of OOH*, a key intermediate of the 2e− ORR, resulting in the inadequate performance of non-noble metal single-atom electrocatalysts with high overpotentials and low selectivity.29,30 Although the selectivity of carbon-supported non-noble metal SACs for the 2e− ORR can be enhanced by the selection of appropriate precursors such as metal–organic frameworks, the preparation of such electrocatalysts requires expensive precursors (such as terephthalic acid) and harsh synthesis conditions (such as strong acids).30,31 Furthermore, the resulting carbon-supported non-noble metal SACs still suffer from inefficiencies.31,32 Therefore, the selection, optimization, and modification of carbon-based supports are important for enhancing the 2e− ORR selectivity of non-noble metal SACs under neutral conditions because carbon-based materials are not only excellent supports but also excellent 2e− ORR catalysts.
It is well known that nitrogen doping is a way to increase the 2e− ORR capability of carbon-based catalysts.31,33 There is a linear relationship between the activity of the 2e− ORR and the nitrogen content of nitrogen-doped carbon materials.34 Various carbon–nitrogen coordination structures exist in nitrogen-doped carbon materials, such as pyridine-N, pyrrole-N, and N-oxide.11,35,36 The carbon–nitrogen coordination can effectively improve the adsorption energy of OOH* and generate more active sites, which can improve the selectivity for both H2O2 and the 2e− ORR activity.36 Some studies have shown that pyridine N is suitable for the 2e− ORR, and it has been reported that carbon atoms near pyridine N are the active centers of the 2e− ORR.1 Recently, the formation of nitrogen-doped graphene oxide on a carbon paper was reported by Kim et al. The authors identified the active center of the catalyst through various characterization techniques and observed that the sp2 carbon sites near the region of sp3 carbon after nitrogen doping were the catalytic active centers of the 2e− ORR.1,3 In addition, it was found that partially reduced graphene oxide achieved 99% H2O2 selectivity in acidic and basic solutions;2,3 however, its activity for H2O2 electrosynthesis at neutral pH is still limited. Based on this, graphitic carbon nitride (g-C3N4) has infinite potential in electrocatalytic H2O2 synthesis via the 2e− ORR because g-C3N4 nanosheets (CNNS) have abundant carbon–nitrogen coordination, including sp2 carbon bond (N–CN), sp3 carbon bond (C–NHX) and the graphite-like carbon (C–C/CC) and so on, which can create more active conditions for the 2e− ORR and achieve the stable anchoring of isolated non-noble metal single atoms through the formation of metal–nitrogen bonds.37,38 CNNS also has the advantages of being low-cost, non-toxic and harmless, with a large surface area and good stability of the carbon–nitrogen structure.38,39
Herein, relying on a simple and universal two-step thermal oxidation etching method, transition metal single atoms (TM SA: Ni, Fe, Co, Mn, Cu, Zn) have been successfully anchored on CNNS (TM SA/CNNS) for efficient H2O2 synthesis via the 2e− ORR in neutral electrolytes. Compared with CNNS, all the TM SA/CNNS exhibited high selectivity for the 2e− ORR in the following order: Ni0.10 SA/CNNS (98%) > Mn0.10 SA/CNNS (95%) > Zn0.10 SA/CNNS (93%) > Cu0.10 SA/CNNS (90%) > Fe0.10 SA/CNNS (86%) > Co0.10 SA/CNNS (76%) > CNNS (61%). The high selectivity of TM SA/CNNS may be ascribed to the introduction of TM SA, which optimized the N–CN (increased) and C–C/CC (decreased) contents in CNNS, which can improve the selectivity of TM SA/CNNS for the 2e− ORR. This demonstrates that the N–CN site favors the 2e− ORR, while the C–C/CC site hinders the electrochemical synthesis of H2O2. The linear relationship between the content of the N–CN bond in TM SA/CNNS and its 2e−ORR selectivity further supports these conclusions. In addition, TM SA further promotes the enhancement of 2e− ORR activity. Secondly, the introduction of TM SA lowers the reaction potential of the 2e− ORR and improves the electrocatalytic activity of TM SA/CNNS, which is one of the important reasons why H2O2 yields could reach industrial levels. All of these are verified by characterization methods including high-angle annular dark field-scanning transmission electron microscopy (HAADF-STEM), X-ray photoelectron spectroscopy (XPS), density functional theory (DFT), and so on.
2. Results and discussion
2.1 Synthesis and characterization of electrocatalysts
The synthetic scheme for TM SA/CNNS (TM: Ni, Mn, Fe, Co, Cu, Zn) is as follows: three precursors (urea, dicyandiamide (DCDA), NiCl2·6H2O and other transition metal chlorides) were used. In the first step, urea, DCDA and transition metal chlorides were thoroughly and uniformly mixed by mechanical grinding. The obtained mixture was treated at 550 °C for 4 h in the atmosphere to obtain the initial TM SA/g-C3N4 nanocomposites. Finally, the initial TM SA/g-C3N4 nanocomposites were further heated to 500 °C for 2 h with 10% Ar/H2 flow to achieve TM SA/g-C3N4 nanosheets (TM SA/CNNS). Based on the excellent activity and selectivity of Ni SA/CNNS electrocatalysts, a series of samples with different Ni contents (0.05, 0.10, and 0.20 mmol) were also synthesized to explore the optimal loading of Ni single atoms. They were denoted as CNNS, Ni0.05 SA/CNNS, Ni0.10 SA/CNNS, Ni0.20 SA/CNNS, respectively. Among them, Ni0.10 SA/CNNS exhibited the best 2e− ORR activity and selectivity. Similar to the method of Ni0.10 SA/CNNS, 0.10 mmol of transition metal chlorides (TM: Mn, Zn, Cu, Fe, Co) were used as the metal source to prepare Mn0.10 SA/CNNS, Fe0.10 SA/CNNS, Co0.10 SA/CNNS, Cu0.10 SA/CNNS, Zn0.10 SA/CNNS; all of these were named TM SA/CNNS. It can be seen that the synthesis method is simple, safe, universal and suitable for the industrial production of TM SA on CNNS.The XRD results of CNNS and TM SA/CNNS are displayed in Fig. 1(a) and Fig. S1 (ESI†). The characteristic peaks exhibited at 13.2° and 27.6°, respectively, correspond to the in-plane structural packing motif of the continuous heptazine network and the interlayer stacking of aromatic segments in g-C3N4, which belongs to the (100) and (002) diffraction plane of g-C3N4 (JCPDS no. 87-1526).37,40 There were no obvious diffraction peaks of Ni, Mn, Zn, Cu, Fe, and Co among the XRD, which might be due to the lower transition metal content or the formation of transition metal single atoms or clusters on CNNS. Based on XRD patterns, the Debye–Scherrer equation was used to calculate the crystallite size of CNNS, Ni0.10 SA/CNNS and other TM SA/CNNS. As shown in Table S1 (ESI†), the crystallite sizes of CNNS, Ni0.10 SA/CNNS, Mn0.10 SA/CNNS, Zn0.10 SA/CNNS, Cu0.10 SA/CNNS, Fe0.10 SA/CNNS and Co0.10 SA/CNNS were 3.45 nm, 4.10 nm, 5.88 nm, 5.27 nm, 5.91 nm, 5.44 nm, 4.75 nm, respectively. The results show that TM SA enhanced the crystallite size of CNNS, but the change in the crystallite size of TM SA/CNNS is inconsistent with its corresponding electrochemical properties. It can be seen that with the increase in crystallite size of CNNS via TM SA, it is possible to achieve improved electrocatalytic performance but the crystallite size is not the main cause of the electrocatalytic performance.
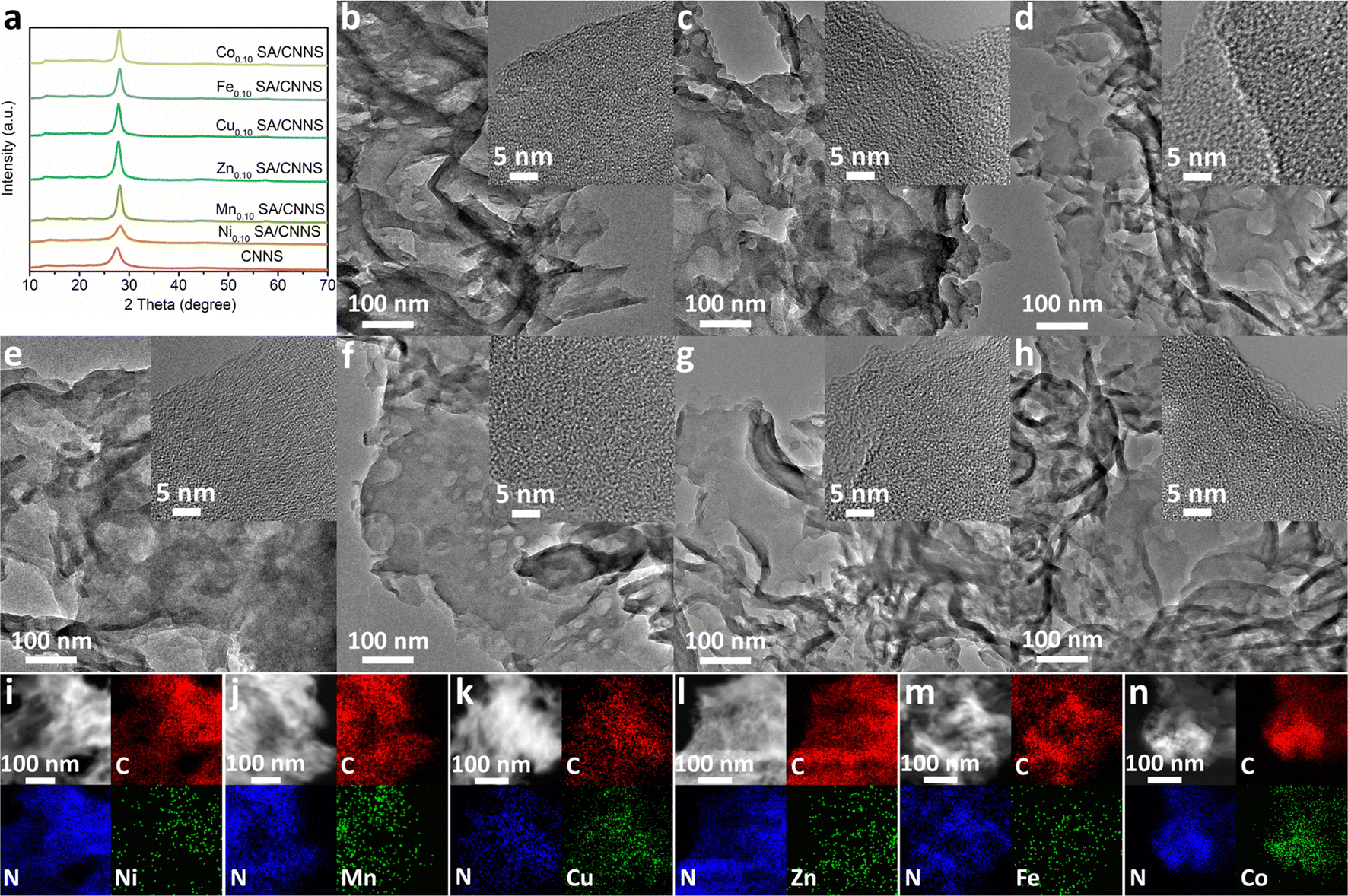 | ||
| Fig. 1 (a) XRD patterns of CNNS, Ni0.10 SA/CNNS and other TM SA/CNNS. TEM images (insets show the HRTEM images) of (b) CNNS, (c) Ni0.10 SA/CNNS, (d) Mn0.10 SA/CNNS, (e) Zn0.10 SA/CNNS, (f) Cu0.10 SA/CNNS, (g) Fe0.10 SA/CNNS and (h) Co0.10 SA/CNNS. HAADF-STEM images and EDS mapping of (i) Ni0.10 SA/CNNS, (j) Mn0.10 SA/CNNS, (k) Zn0.10 SA/CNNS, (l) Cu0.10 SA/CNNS, (m) Fe0.10 SA/CNNS and (n) Co0.10 SA/CNNS. | ||
As shown in Fig. S2a (ESI†), the Raman spectroscopy of CNNS, Ni0.10 SA/CNNS and other TM SA/CNNS was conducted using a 785 nm laser. CNNS, Ni0.10 SA/CNNS, Mn0.10 SA/CNNS, Zn0.10 SA/CNNS, Cu0.10 SA/CNNS, Fe0.10 SA/CNNS and Co0.10 SA/CNNS exhibited similar Raman vibration modes at 900–1700 cm−1. The Raman spectra at 900–1400 cm−1 were related to the typical heptazine units. The peaks located at 987 cm−1, 1168 cm−1, 1294 cm−1, 1429 cm−1, 1520 cm−1 and 1646 cm−1 originated from the vibration modes of CN heterocycles in CNNS. In detail, the diffraction peak at 1294 cm−1 is caused by the C (sp2) bending vibration. The two peaks at 1429 cm−1 and 1520 cm−1 are assigned to the D and G bands of a typical graphitic structure, which are related to the sp2 carbon materials of CNNS. This means that the TM SA does not make other significant changes to the CNNS.
Further research on materials by Fourier Transform infrared (FT-IR) spectroscopy revealed the functional groups present in CNNS, Ni0.10 SA/CNNS and other TM SA/CNNS (Fig. S2b, ESI†). Several major bands centered at about 700–1000 cm−1, 1100–1800 cm−1 and 2800–3700 cm−1 were observed. In comparison with CNNS, Ni0.10 SA/CNNS and other TM SA/CNNS showed similar spectra. The absorption peak at 812 cm−1 is caused by the out-of-plane bending vibration of the triazine/heptazine ring in CNNS. The band at 888 cm−1 is due to the deformation vibrations of the N–H bonds. The absorption peaks at 1237 cm−1, 1323 cm−1 1402 cm−1, 1461 cm−1, 1569 cm−1 and 1643 cm−1 arise from the stretching vibrations of the C–N bonds. Several broad bands near 3182 cm−1 are caused by stretching vibrations of the N–H or O–H bonds of the residual amino groups or adsorbed H2O molecules. These show that all materials maintain the classical structure of g-C3N4.
The morphologies and structures of CNNS and TM SA/CNNS were characterized via SEM, TEM, and HRTEM. As shown in Fig. 1(b)–(h) and Fig. S3, S4 (ESI†), CNNS and TM SA/CNNS have similar structures. The SEM images (Fig. S3 and S4, ESI†) show that CNNS and TM SA/CNNS are composed of ultrathin flexible nanosheets with wrinkles, and a large number of pore structures are observed in the TEM images (Fig. 1(b)–(h)). All these indicate the formation of ultrathin flexible nanosheets with porous structures in CNNS and TM SA/CNNS. The insets of Fig. 1(b)–(h) show the HRTEM of these electrocatalysts, no obvious transition metal nanoparticles were observed on the surface of CNNS. However, the elemental mapping of TM SA/CNNS (Fig. 1(i)–(n)) shows that C, N, and transition metals (Ni, Mn, Zn, Cu, Fe, and Co) are present and uniformly distributed. These prove that Ni, Mn, Zn, Cu, Fe, and Co may also exist in TM SA/CNNS in the form of single atoms or clusters. From HAADF-STEM images, the isolated bright dots are observed in TM SA/CNNS (Fig. 2(a)–(f)), which are identified as transition metal single atoms considering the atomic number of transition metals is larger than that of C and N.7,19,41 In addition, XAFS (X-ray absorption fine structure) was used to further demonstrate the presence of Ni single atoms in Ni0.10 SA/CNNS, which has the best 2e− ORR performance in all TM SA catalysts. The detailed electronic structures and coordination information of Ni single atoms in Ni0.10 SA/CNNS were also obtained. The Ni k-edge XANES (X-ray absorption near-edge structure) spectra in Fig. 2(g) show that the white-line intensity of Ni0.10 SA/CNNS is between Ni foil and NiO, indicating that Ni single atoms are positively charged. The EXAFS (extended X-ray absorption fine structure) data in Fig. 2(h) show that the peak at 2.18 Å in Ni foil is a metallic Ni–Ni bond. This is not found in Ni0.10 SA/CNNS, indicating that Ni does not exist as clusters or nanoparticles. Meanwhile, the main peak of Ni0.10 SA/CNNS at 1.44 Å is assigned to the Ni–N bond. As shown in Fig. S5 and Table S2 (ESI†), the fitting results of Ni0.10 SA/CNNS show Ni–N3 coordination. Combined with HAADF-STEM, all these results indicate that Ni in Ni0.10 SA/CNNS does exist in the form of single atoms.
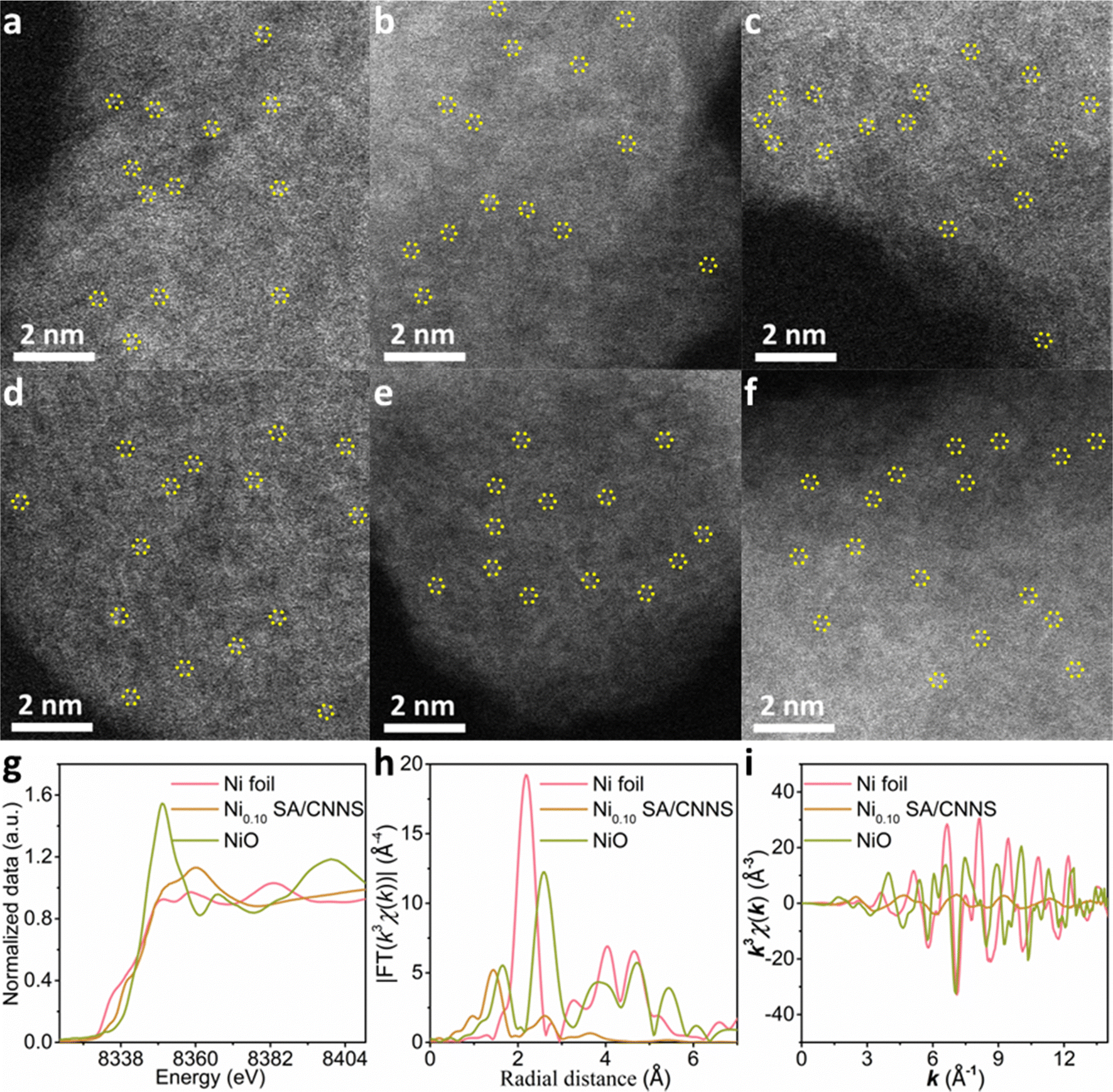 | ||
| Fig. 2 HAADF-STEM images of (a) Ni0.10 SA/CNNS, (b) Mn0.10 SA/CNNS, (c) Zn0.10 SA/CNNS, (d) Cu0.10 SA/CNNS, (e) Fe0.10 SA/CNNS and (f) Co0.10 SA/CNNS. (g) The normalized XANES spectra at the Ni K-edge of Ni foil, Ni0.10 SA/CNNS and NiO. (h) Ni R-space of the EXAFS for Ni foil, Ni0.10 SA/CNNS and NiO. (i) Ni k-space of the EXAFS for Ni foil, Ni0.10 SA/CNNS and NiO. | ||
To further clarify the interactions between the TM SA and CNNS, the chemical states of CNNS and TM SA/CNNS were investigated by XPS. Fig. S6 and S7 (RDI), show that CNNS and TM SA/CNNS are composed of C and N. At the same time, Ni, Mn, Zn, Cu, Fe, and Co were found in TM SA/CNNS, respectively. The spectra of Ni 2p (Fig. S7a, ESI†) can be decomposed into four peaks at 853.40 and 870.90 eV, 860.10, and 878.80eV, which are ascribed to Ni 2p3/2 and Ni 2p1/2, and the satellite peaks of Ni 2p3/2 and Ni 2p1/2, which are similar to the binding energy of NiO.39,40 Similarly, the chemical states of atomically dispersed Mn, Zn, Cu, Fe and Co species in TM SA/CNNS are close to (II and III), (II), (II), (II and III), and (II) (Fig. S7b–f, ESI†), respectively.39,42–50 All of these prove the existence of the isolated transition metal single atoms. The high-resolution C 1s spectra (Fig. 3 and Fig. S8, ESI†) show three component peaks at 284.7 eV, 284.40 eV, and 288.15 eV for CNNS and TM SA/CNNS, which correspond to C–C/CC, C–NHX and N–CN, respectively.37,49 The typical components around 284.7 eV, 284.40 eV, and 288.15 eV in CNNS and TM SA/CNNS are almost the same. These results indicate that the chemical state of the carbon remains almost unchanged, meaning that carbon atoms hardly interact with the metal ions. Fig. S9 and S10 (ESI†) display the N 1s spectra of CNNS and TM SA/CNNS; four peaks were observed at 398.60, 399.80, 400.94, and 404.30 eV. The peaks at 398.60 eV, 399.80, 400.94, and 404.30 eV correspond to the N sp2 bond, the tertiary nitrogen group (N–C3), the quaternary N three carbon atom amino functional group (N–HX) in the aromatic ring and π excitation, respectively.38,40 It is notable that nitrogen peaks appeared at around 398.20, 397.82, 398.10, 297.90, 397.90, and 398.00 eV in the N 1s spectra of TM SA/CNNS, which could be attributed to N–TM.40,44 Besides, as shown in Table S3 (ESI†), the contents of N–CN in CNNS and TM SA/CNNS follow the order of Ni0.10 SA/CNNS (92%) > Mn0.10 SA/CNNS (90%) > Zn0.10 SA/CNNS (86%) > Cu0.10 SA/CNNS (85%) > Fe0.10 SA/CNNS (80%) > CNNS (73%) > Co0.10 SA/CNNS (67%). The C–C/CC content has the exact opposite pattern. These indicate that the introduction of TM SA increases the content of N–CN sites and reduces the C–C/CC content. This may be beneficial to improve the selectivity of TM SA/CNNS, which will be further verified by electrochemical properties, as well as DFT calculations.
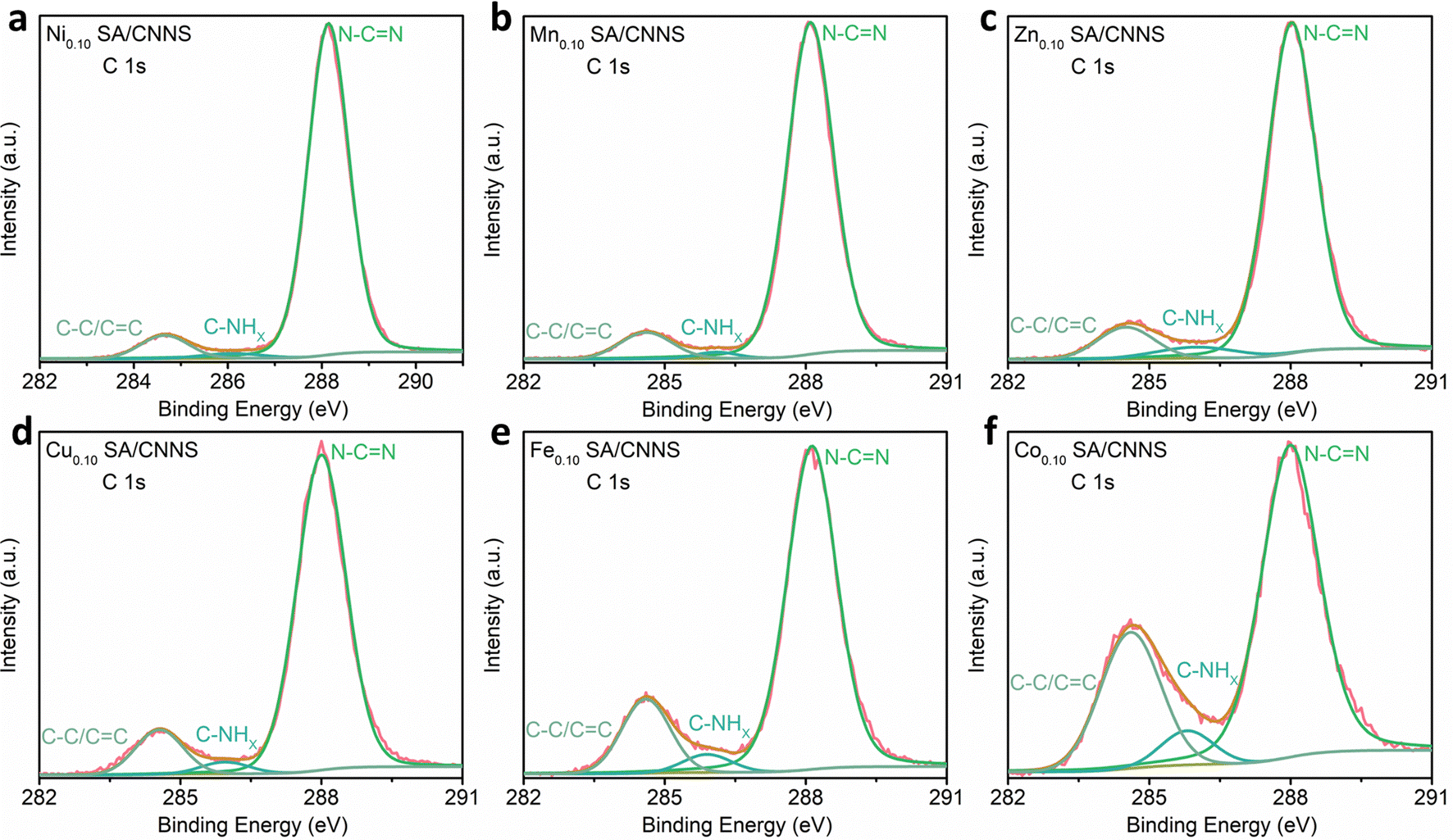 | ||
| Fig. 3 High-resolution XPS of the C 1s spectra of (a) Ni0.10 SA/CNNS, (b) Mn0.10 SA/CNNS, (c) Zn0.10 SA/CNNS, (d) Cu0.10 SA/CNNS, (e) Fe0.10 SA/CNNS and (f) Co0.10 SA/CNNS. | ||
2.2 The electrochemical properties of catalysts
To verify the above conclusions, the electrochemical properties of CNNS and TM SA/CNNS were analyzed. The 2e− ORR performances of CNNS and TM SA/CNNS were determined in 0.1M PBS. As shown in Fig. S11 (ESI†), the linear scan voltammogram (LSV) profiles were obtained by subtracting the capacitive current in the N2-saturated electrolyte from the O2-saturated ORR current. The most promising Ni SA/CNNS electrocatalysts were optimized by adjusting the Ni content (Fig. S12, ESI†). Among them, Ni0.10 SA/CNNS exhibited the highest 2e− ORR activity and selectivity (the selectivity was ∼98% at 0.3 V vs. RHE; the faradaic efficiency (FE%) was ∼97%). Based on this, other TM SA/CNNS (Mn0.10 SA/CNNS, Zn0.10 SA/CNNS, Cu0.10 SA/CNNS, Fe0.10 SA/CNNS, and Co0.10 SA/CNNS) were also synthesized. Fig. 4(a) shows the LSV curves of CNNS and TM SA/CNNS, which contain the disk and ring currents. Compared with CNNS (∼0.54 V vs. RHE) and Ni0.10 SA/CNNS (∼0.55 V vs. RHE), other TM SA/CNNS have similar initial potentials. These are below the equilibrium potential of the 2e− ORR (0.70 V vs. RHE), indicating that CNNS and TM SA/CNNS are close to the thermodynamic equilibrium potential of the 2e− ORR, which was also confirmed by the ORR selectivity. The selectivity, n, and FE% of CNNS and TM SA/CNNS are presented in Fig. 4(b) and Fig. S13 (ESI†). The selectivities of CNNS, Ni0.10 SA/CNNS, Mn0.10 SA/CNNS, Zn0.10 SA/CNNS, Cu0.10 SA/CNNS, Fe0.10 SA/CNNS, and Co0.10 SA/CNNS are ∼61%, ∼98%, ∼95%, ∼93%, ∼90%, ∼86%, and ∼76% at 0.3 V vs. RHE (Fig. 4(b)), where n is ∼2.73, ∼2.02, ∼2.11, ∼2.14, ∼2.19, ∼2.27, and ∼2.48, (Fig. S13a, ESI†) respectively. FE% is ∼44%, ∼97%, ∼89%, ∼86%, ∼82%, ∼76%, and ∼61%, respectively (Fig. S13b, ESI†). These reflect that the electrocatalytic activities of CNNS and TM SA/CNNS follow the order Ni0.10 SA/CNNS > Mn0.10 SA/CNNS > Zn0.10 SA/CNNS > Cu0.10 SA/CNNS > Fe0.10 SA/CNNS > Co0.10 SA/CNNS > CNNS, which is consistent with the changes in the N–CN and C–C/CC contents in the high-resolution C 1s spectra, except for Co0.10 SA/CNNS. Tafel plots of CNNS and TM SA/CNNS (Fig. 4(c)) were calculated from the RRDE data. TM SA/CNNS exhibited significantly faster kinetics for the 2e− ORR than CNNS under neutral conditions in the order of Ni0.10 SA/CNNS > Mn0.10 SA/CNNS > Zn0.10 SA/CNNS > Cu0.10 SA/CNNS > Fe0.10 SA/CNNS > CNNS > Co0.10 SA/CNNS. The electrochemically active surface areas (ECSA) of CNNS and TM SA/CNNS were measured via the electrochemical double-layer capacitance (Fig. 4(d) and Fig. S14, ESI†). The ECSA of Ni0.10 SA/CNNS, Mn0.10 SA/CNNS, Zn0.10 SA/CNNS, Cu0.10 SA/CNNS, Fe0.10 SA/CNNS and Co0.10 SA/CNNS are 21.9 mF cm−2, 21.1 mF cm−2, 19.0 mF cm−2, 18.0 mF cm−2, 16.4 mF cm−2 and 15.4 mF cm−2, respectively. These results also clearly display the higher electrochemical activity of TM SA/CNNS for the 2e− ORR as compared with CNNS (7.80 mF cm−2); Ni0.10 SA/CNNS showed the highest electrochemical activity. This means that the isolated TM SA could improve the kinetics of the 2e− ORR and the electrochemically active area of TM SA/CNNS. The activities of the electrocatalytic H2O2 reduction reaction (H2O2RR) of CNNS and TM SA/CNNS were measured in N2-saturated 0.1 M PBS containing 10 mM H2O2. For Fig. 4(e), when the potential of Ni0.10 SA/CNNS is 0.3 V vs. RHE, the H2O2RR current density of Ni0.10 SA/CNNS is about −0.03 mAcm−2. Meanwhile, the H2O2RR current densities of Mn0.10 SA/CNNS, Zn0.10 SA/CNNS, Cu0.10 SA/CNNS, Fe0.10 SA/CNNS, and Co0.10 SA/CNNS were about −0.042 mAcm−2, −0.050 mAcm−2, −0.087 mAcm−2, −0.125 mAcm−2, and −0.143 mAcm−2, respectively. This means that TM SA/CNNS have poorer activity towards the H2O2RR as compared to CNNS (−0.18 mAcm−2 at 0.3 V vs. RHE), which indicates that TM SA/CNNS are more conducive to the retention of H2O2 during the 2e− ORR. Therefore, the introduction of the isolated TM SA can improve the yield of H2O2 and is also beneficial to the synthesis of high-concentration H2O2 solutions. From the Tafel slopes, ECSA and H2O2RR, it was found that the introduction of the isolated TM SA effectively enhanced the activity and selectivity for the 2e− ORR. Combined with the relationship between the change in carbon species and H2O2 selectivity in electrocatalysts (Fig. 4(f)), it was observed that the isolated TM SA could optimize the structure of CNNS, adjust the content of N–CN and C–C/CC in CNNS and improve the selectivity for the 2e− ORR. Although the N–CN content of Co0.10 SA/CNNS is significantly lower than that of CNNS, it still possesses better activity and selectivity for the 2e− ORR. It might be due to the introduction of Co SA optimising the charge distribution, the lowering of the reaction potential of the 2e− ORR and the improvement of the electrochemically active area, which can compensate for the loss of N–CN content. This indicates that N–CN plays an important role in improving the 2e− ORR selectivity, but it is not the only factor affecting the activity and selectivity of electrocatalysts.
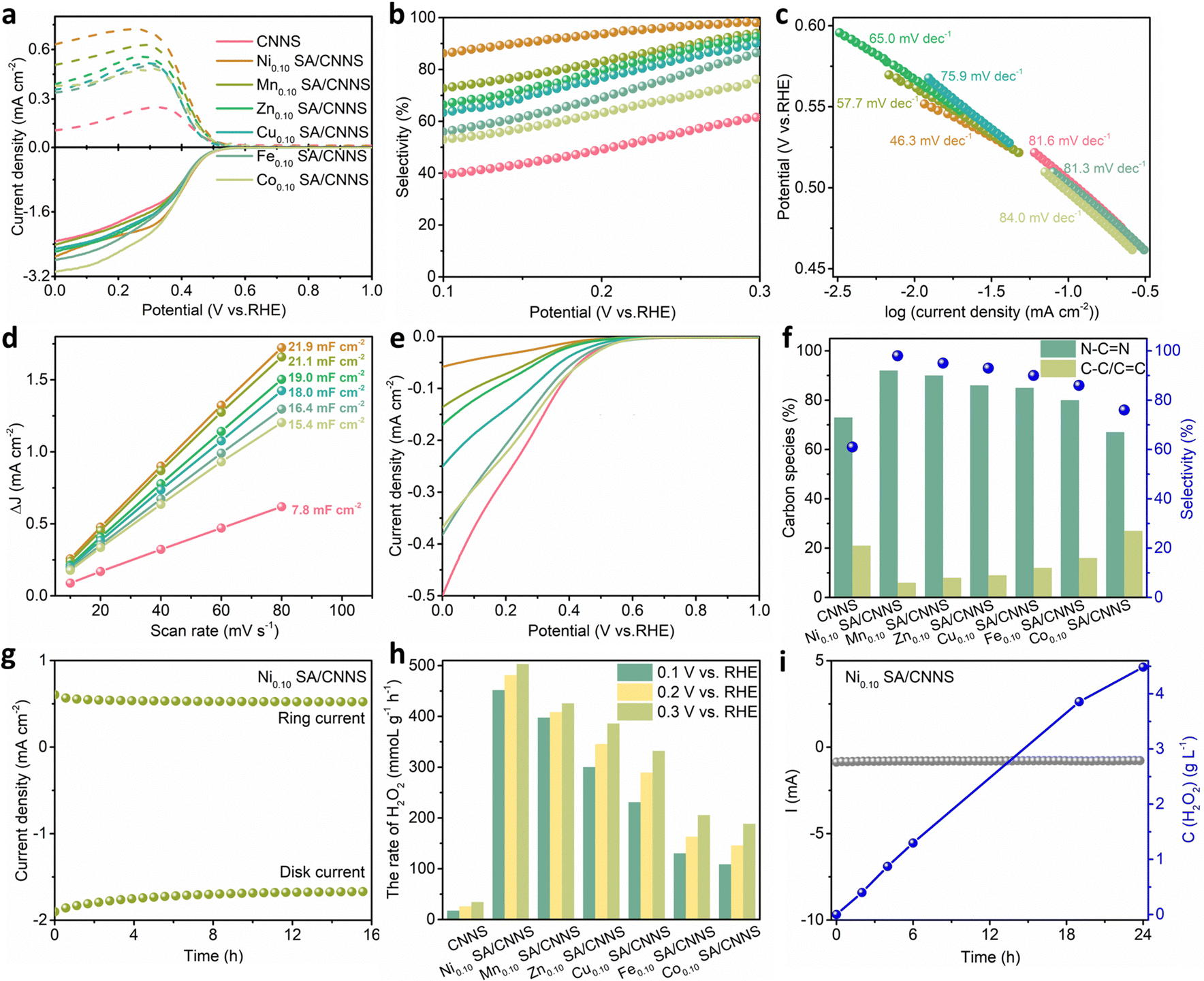 | ||
| Fig. 4 Electrocatalytic performances of CNNS, Ni0.10 SA/CNNS and other TM SA/CNNS in 0.1 M PBS: (a) ORR disk current density together with the ring currents at a fixed potential of 1.20 V vs. RHE. (b) H2O2 selectivity (H2O2%). (c) Tafel plots. (d) Current density differences at 0.75 V vs. the scan rates. (e) LSV curves of H2O2 reduction reaction recorded in N2-saturated 0.1M PBS containing 10 mM H2O2. (f) The atomic percentages of carbon species in high-resolution C 1s spectra of CNNS, Ni0.10 SA/CNNS and other TM SA/CNNS and their corresponding H2O2 selectivities at 0.3 V vs. RHE. (g) Stability measurement of Ni0.10 SA/CNNS at a fixed disk potential of 0.3 V vs. RHE. (h) The H2O2 production rate of CNNS, Ni0.10 SA/CNNS and other TM SA/CNNS. (i) Stability measurement and H2O2 concentration of Ni0.10 SA/CNNS at 0.3 V vs. RHE. | ||
The stability of Ni0.10 SA/CNNS was also further assessed. The chronoamperometric currents of the ring and disk electrodes were recorded in O2-saturated 0.1 M PBS, when they were biased at 1.2 and 0.3 V vs. RHE, respectively. Fig. 4(g) shows that both idisk and iring remained stable through the 16 h test with negligible decay. The selectivity for H2O2 was maintained in the range of 96.6–98.8% during the entire test (Fig. S15, ESI†), indicating that the Ni0.10 SA/CNNS has excellent electrochemical stability. The physical stability of Ni0.10 SA/CNNS was also tested by comparing the 2e− ORR activity and selectivity of the Ni0.10 SA/CNNS ink before and after 68 days at room temperature. Fig. S16 (ESI†) shows that the H2O2 selectivity of Ni0.10 SA/CNNS after standing for 68 days was 94%, which is 4% lower than the original Ni0.10 SA/CNNS (the selectivity for H2O2 was 98%), indicating that the Ni0.10 SA/CNNS has high physical stability. Lastly, to demonstrate the feasibility of CNNS and TM SA/CNNS for the 2e− ORR in the fuel cell, we deposited CNNS and TM SA/CNNS electrocatalysts (1 mg cm−2) on carbon paper as the working electrode and constructed an H-cell for the direct synthesis of H2O2. The titration method was used to obtain the amount of H2O2 produced in an H-cell (Fig. S17–S24, ESI†). As shown in Fig. 4(h), the H2O2 production rate of Ni0.10 SA/CNNS is also highly promising with up to ∼503.0 mmol gcat−1 h−1 at 0.3 V vs. RHE, which is 14.6 times that of CNNS (∼34.4 mmol gcat−1 h−1). Therefore, the H2O2 production rates of Mn0.10 SA/CNNS, Zn0.10 SA/CNNS, Cu0.10 SA/CNNS, Fe0.10 SA/CNNS, and Co0.10 SA/CNNS (Fig. 4(h)) were also measured, which were ∼425.5 mmol gcat−1 h−1, ∼385.9 mmol gcat−1 h−1, ∼332.0 mmol gcat−1 h−1, ∼205.6 mmol gcat−1 h−1, and ∼188.4 mmol gcat−1 h−1 at 0.3 V vs. RHE, respectively. Compared with CNNS, TM SA/CNNS exhibited a significantly improved yield of H2O2, which is consistent with the test results of the three-electrode system. Fig. 4(i) shows the stability measurements and H2O2 concentration of Ni0.10 SA/CNNS at 0.3 V vs. RHE for 24 h. In the i–t test, the change in the current density was negligible, indicating that the Ni0.10 SA/CNNS has very good stability. The accumulated H2O2 concentration increased with the operation time and approached 4.5 g L−1 for 24 h (the titration method was also used, as shown in Fig. S25 (ESI†)). The rate of H2O2 was also maintained in the range of 0.43–0.51 mol g−1 h−1 (Fig. S26, ESI†) in the i–t test, indicating that the production of H2O2 increased linearly with time, which further demonstrated that Ni0.10 SA/CNNS has high activity, selectivity, and stability in the electrochemical synthesis of H2O2. Consequently, the TM SA/CNNS are considered the candidates with the greatest potential for electrocatalytic H2O2 preparation due to their high electrochemical activity and selectivity, low H2O2RR activity, and excellent stability, which surpass most of the reported work (Table S4, ESI†).
2.3 The DFT calculations of catalysts
The above results indicate that N–CN is the main active site for the 2e− ORR, while C–C/CC sites can hinder the electrochemical synthesis of H2O2. The mechanisms of C–C/CC and N–CN sites in CNNS were investigated by DFT calculations (Fig. 5(a)–(d)). Fig. 5(e) and (f) show the Gibbs free energies (ΔG) of C–C/CC and N–CN at different potentials (U). The ΔG of OOH* on C–C/CC is about −3.70 eV. The high free energy is more favorable for the adsorption of O2 and the breakage of the O–O bond, which inhibits the formation of the OOH* intermediates in the 2e− ORR process. The ΔG of OOH* on N–CN is ∼0.46 eV, and the lower free energy is favorable for the formation of OOH* in the 2e− ORR process. These verify that N–CN is an important active site for the electrochemical preparation of H2O2, while C–C/CC hinders the formation of H2O2. Therefore, it is clear that N–CN in CNNS is more inclined to the 2e− ORR but it needs to overcome a potential barrier of 0.46 eV during the formation of OOH*, which affects the reaction rate of the 2e− ORR. To further understand the role of TM SA, a model of Ni SA/CNNS was constructed. As shown in Fig. 6(a) and (b), Ni SA in CNNS can improve the adsorption energy of N–CN sites for OOH* (where the yellow part shows the increase of charge density and the blue part shows the decrease of charge density). It is known that the rate-determining step (RDS) of the ORR depends on the thermodynamic step with the smallest change in free energy. From the Gibbs free energy of Ni SA/CNNS (Fig. 6(c)), the ΔG of OOH* on Ni SA/CNNS is −0.65 eV in the 2e− ORR and all reaction steps are thermodynamically decreasing, so the process of OOH* is the RDS of the 2e− ORR. ΔG of OOH* is closer to the equilibrium potential of the 2e− ORR (Uequilibrium = 1.4/2 = 0.7), indicating that Ni SA/CNNS has excellent capability for the 2e− ORR. During the 4e− ORR, the overpotential of Ni SA/CNNS is ∼0.58 eV (Fig. 6(d)), and the high overpotential leads to its slow reaction kinetics. The introduction of Ni single atoms improves the activity and selectivity for the 2e− ORR and hinders the 4e− ORR. Fig. 6(e) and (f) show the Gibbs free energy of Ni SA/CNNS and CNNS in the 2e− ORR pathway, respectively. On comparing the Gibbs free energy of Ni SA/CNNS and CNNS, it was found that Ni SA/CNNS has a negligible barrier potential in the RDS of the 2e− ORR. This indicates that Ni SA can improve the adsorption energy of N–CN sites for OOH*, and reduce the overpotential of OOH* intermediates, thus enhancing the reaction rate of the 2e− ORR.
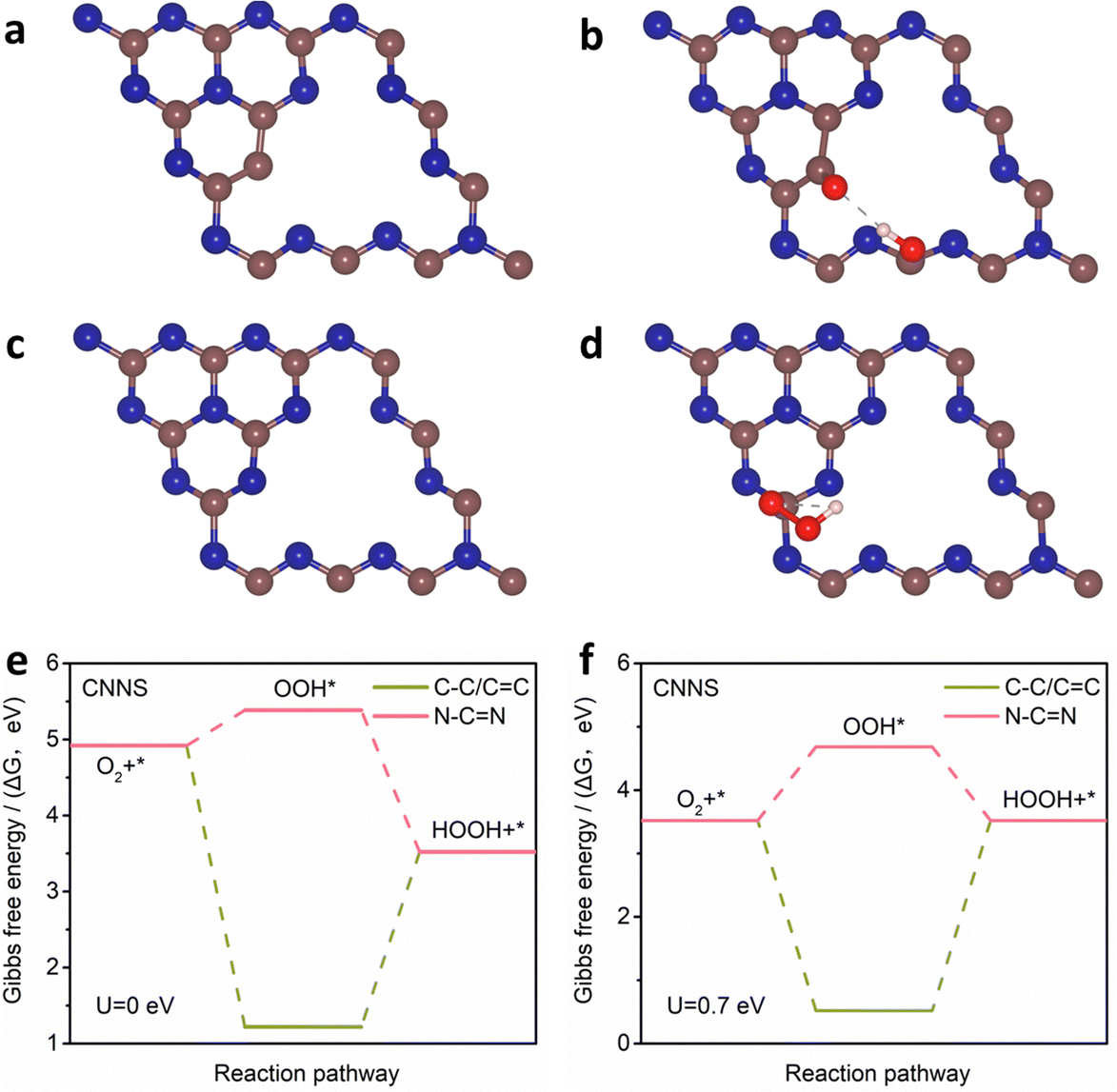 | ||
| Fig. 5 (a)–(d) The optimized configuration and (e), (f) calculated free energy (U = 0 and 0.7 eV) of C–C/CC and N–CN species in CNNS for the synthesis of H2O2. | ||
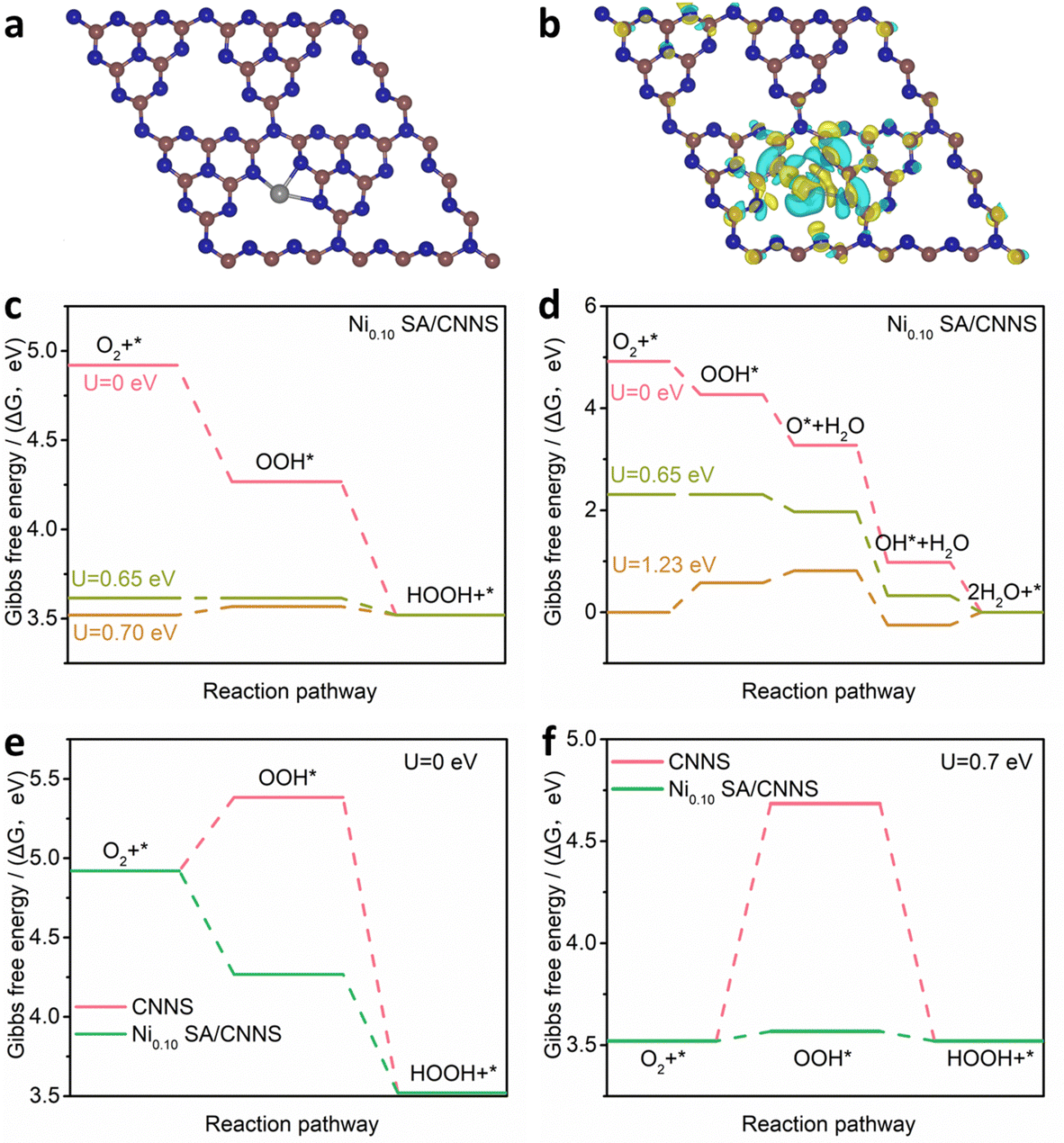 | ||
| Fig. 6 (a) and (b) The optimized configuration of Ni0.10 SA/CNNS. (c) and (d) The calculated free energy of Ni0.10 SA/CNNS for the synthesis of H2O and H2O2. (e) and (f) The calculated free energy (U = 0 and 0.7 eV) of N–CN species in CNNS and Ni0.10 SA/CNNS for the synthesis of H2O2. | ||
3. Conclusions
TM SA/CNNS have been successfully synthesized via a simple and universal two-step thermal oxidation etching method for electrochemical H2O2 synthesis in neutral electrolytes. Among them, Ni0.10 SA/CNNS showed the highest selectivity, which is up to ∼98%. The H2O2 yield of Ni0.10 SA/CNNS is ∼503 mmol gcat−1 h−1, which is ∼14.6 times higher than that of CNNS (∼34.4 mmol gcat−1 h−1). Notably, compared with CNNS, TM SA/CNNS showed efficient electrochemical H2O2 production. Combined with the characterization of XPS and HAADF-STEM, electrochemical tests and theoretical calculations, it was found that the N–CN and C–C/CC sites in CNNS belong to the 2e− ORR and 4e− ORR pathways, respectively. Therefore, the highly efficient electrochemical activities exhibited by TM SA/CNNS are due to the isolated TM SA on CNNS, which effectively enhances the activity of the 2e− ORR of electrocatalysts. On the other hand, the introduction of TM SA optimizes the structure of CNNS and promotes the increase of N–CN sites and the decrease of C–C/CC sites in CNNS, thereby further improving the 2e− ORR selectivity. The results indicate that TM SA/CNNS are promising candidates for the 2e− ORR, and provide more reference ideas for the preparation of H2O2 by 2e− ORR.
Author contributions
Hongcen Yang and Fei Ma: conceptualization, methodology, data curation, writing – original draft, formal analysis, investigation, and visualization. Niandi Lu and Kun Tao are in the analysis of data fitting. Hong Zhang performed HAADF-STEM analysis. Shuhao Tian and Guo Liu: investigation, visualization, and so ware analysis. Ying Wang, Zhixia Wang and Di Wang: validation, data curation, and investigation. All authors discussed the results and participated in writing the manuscript. Shanglong Peng directed the research.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
All the DFT calculation works were supported by the Supercomputing Center of Lanzhou University. All SEM, TEM and HAADF-STEM were supported by the Electron Microscopy Centre of Lanzhou University. This work was financially supported by the Gansu Provincial Natural Science Foundation of China (No. 21JR7RA493, 17JR5RA198, 2020HZ-2, 21JR7RA470), the Cooperation project of Gansu Academy of Sciences (2020HZ-2), the Fundamental Research Funds for the Central Universities (No. lzujbky-2018-119, lzujbky-2018-ct08, lzujbky-2019-it23), and Key Areas Scientific and Technological Research Projects in Xinjiang Production and Construction Corps (No. 2018AB004). All the DFT calculation works were supported by the Supercomputing Center of Lanzhou University. All SEM, TEM and HAADF-STEM were supported by the Electron Microscopy Center of Lanzhou University.References
- Y. Bu, Y. Wang, G. F. Han, Y. Zhao, X. Ge, F. Li, Z. Zhang, Q. Zhong and J.-B. Baek, Adv. Mater., 2021, 33, 2103266 CrossRef CAS PubMed.
- K. Dong, Y. Lei, H. Zhao, J. Liang, P. Ding, Q. Liu, Z. Xu, S. Lu, Q. Li and X. Sun, J. Mater. Chem. A, 2020, 8, 23123–23141 RSC.
- N. Wang, S. Ma, P. Zuo, J. Duan and B. Hou, Adv. Sci., 2021, 8, 2100076 CrossRef CAS PubMed.
- F. Zhang, Y. Zhu, C. Tang, Y. Chen, B. Qian, Z. Hu, Y. C. Chang, C. W. Pao, Q. Lin, S. A. Kazemi, Y. Wang, L. Zhang, X. Zhang and H. Wang, Adv. Funct. Mater., 2022, 32, 2110224 CrossRef CAS.
- W. Zhou, L. Xie, J. Gao, R. Nazari, H. Zhao, X. Meng, F. Sun, G. Zhao and J. Ma, Chem. Eng. J., 2021, 410, 128368 CrossRef CAS.
- X. Yang, Y. Zeng, W. Alnoush, Y. Hou, D. Higgins and G. Wu, Adv. Mater., 2022, 34, 2107954 CrossRef CAS PubMed.
- E. Zhang, L. Tao, J. An, J. Zhang, L. Meng, X. Zheng, Y. Wang, N. Li, S. Du, J. Zhang, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2022, 61, e202117347 CAS.
- C. Zhang, R. Lu, C. Liu, J. Lu, Y. Zou, L. Yuan, J. Wang, G. Wang, Y. Zhao and C. Yu, Adv. Sci., 2022, 9, 2104768 CrossRef CAS PubMed.
- C. Yan, Q. Ma, F. Wang, L. Zhou, X. Lv, J. Du, B. Zheng and Y. Guo, Chem. Eng. J., 2022, 433, 133651 CrossRef CAS.
- H. Li, P. Wen, D. S. Itanze, Z. D. Hood, S. Adhikari, C. Lu, X. Ma, C. Dun, L. Jiang, D. L. Carroll, Y. Qiu and S. M. Geyer, Nat. Commun., 2020, 11, 3928 CrossRef CAS PubMed.
- S. Yang, A. Verdaguer-Casadevall, L. Arnarson, L. Silvioli, V. Čolić, R. Frydendal, J. Rossmeisl, I. Chorkendorff and I. E. L. Stephens, ACS Catal., 2018, 8, 4064–4081 CrossRef CAS.
- A. Verdaguer-Casadevall, D. Deiana, M. Karamad, S. Siahrostami, P. Malacrida, T. W. Hansen, J. Rossmeisl, I. Chorkendorff and I. E. L. Stephens, Nano Lett., 2014, 14, 1603–1608 CrossRef CAS PubMed.
- J. S. Jirkovský, I. Panas, E. Ahlberg, M. Halasa, S. Romani and D. J. Schiffrin, J. Am. Chem. Soc., 2011, 133, 19432–19441 CrossRef PubMed.
- C. Yang, S. Bai, Z. Yu, Y. Feng, B. Huang, Q. Lu, T. Wu, M. Sun, T. Zhu, C. Cheng, L. Zhang, Q. Shao and X. Huang, Nano Energy, 2021, 89, 106480 CrossRef CAS.
- N. Wang, X. Zhao, R. Zhang, S. Yu, Z. H. Levell, C. Wang, S. Ma, P. Zou, L. Han, J. Qin, L. Ma, Y. Liu and H. L. Xin, ACS Catal., 2022, 12, 4156–4164 CrossRef CAS.
- R. Shen, W. Chen, Q. Peng, S. Lu, L. Zheng, X. Cao, Y. Wang, W. Zhu, J. Zhang, Z. Zhuang, C. Chen, D. Wang and Y. Li, Chem, 2019, 5, 2099–2110 CAS.
- E. Jung, H. Shin, B. H. Lee, V. Efremov, S. Lee, H. S. Lee, J. Kim, W. Hooch Antink, S. Park, K. S. Lee, S. P. Cho, J. S. Yoo, Y. E. Sung and T. Hyeon, Nat. Mater., 2020, 19, 436–442 CrossRef CAS PubMed.
- X. Song, N. Li, H. Zhang, L. Wang, Y. Yan, H. Wang, L. Wang and Z. Bian, ACS Appl. Mater. Interfaces, 2020, 12, 17519–17527 CrossRef CAS PubMed.
- X. Li, S. Tang, S. Dou, H. J. Fan, T. S. Choksi and X. Wang, Adv. Mater., 2022, 34, 2104891 CrossRef CAS PubMed.
- C. Liu, H. Li, F. Liu, J. Chen, Z. Yu, Z. Yuan, C. Wang, H. Zheng, G. Henkelman, L. Wei and Y. Chen, J. Am. Chem. Soc., 2020, 142, 21861–21871 CrossRef CAS PubMed.
- C. Tang, Y. Jiao, B. Shi, J. N. Liu, Z. Xie, X. Chen, Q. Zhang and S. Z. Qiao, Angew. Chem., Int. Ed., 2020, 59, 9171–9176 CrossRef CAS PubMed.
- Z. Zhou, Y. Kong, H. Tan, Q. Huang, C. Wang, Z. Pei, H. Wang, Y. Liu, Y. Wang, S. Li, X. Liao, W. Yan and S. Zhao, Adv. Mater., 2022, 34, 2106541 CrossRef CAS PubMed.
- H. Wu, T. He, M. Dan, L. Du, N. Li and Z. Q. Liu, Chem. Eng. J., 2022, 435, 134863 CrossRef CAS.
- C. Liu, H. Li, J. Chen, Z. Yu, Q. Ru, S. Li, G. Henkelman, L. Wei and Y. Chen, Small, 2021, 17, 2007249 CrossRef CAS PubMed.
- J. Zhang, W. Liu, F. He, M. Song, X. Huang, T. Shen, J. Li, C. Zhang, J. Zhang and D. Wang, Chem. Eng. J., 2022, 438, 135619 CrossRef CAS.
- J. Lee, S. W. Choi, S. Back, H. Jang and Y. J. Sa, Appl. Catal., B, 2022, 309, 121265 CrossRef CAS.
- H. Shen, N. Qiu, L. Yang, X. Guo, K. Zhang, T. Thomas, S. Du, Q. Zheng, J. P. Attfield, Y. Zhu and M. Yang, Small, 2022, 18, 2200730 CrossRef CAS PubMed.
- M. Gao, Z. Y. Wang, Y. R. Yuan, W. W. Li, H. Q. Liu and T. Y. Huang, Chem. Eng. J., 2022, 434, 134788 CrossRef CAS.
- Q. Zhao, Y. Wang, W. H. Lai, F. Xiao, Y. Lyu, C. Liao and M. Shao, Energy Environ. Sci., 2021, 14, 5444–5456 RSC.
- Y. Pang, K. Wang, H. Xie, Y. Sun, M. M. Titirici and G. L. Chai, ACS Catal., 2020, 10, 7434–7442 CrossRef CAS.
- Y. Sun, I. Sinev, W. Ju, A. Bergmann, S. Dresp, S. Kühl, C. Spöri, H. Schmies, H. Wang, D. Bernsmeier, B. Paul, R. Schmack, R. Kraehnert, B. Roldan Cuenya and P. Strasser, ACS Catal., 2018, 8, 2844–2856 CrossRef CAS.
- Q. Yang, W. Xu, S. Gong, G. Zheng, Z. Tian, Y. Wen, L. Peng, L. Zhang, Z. Lu and L. Chen, Nat. Commun., 2020, 11, 5478 CrossRef CAS PubMed.
- J. Zhang, G. Zhang, S. Jin, Y. Zhou, Q. Ji, H. Lan, H. Liu and J. Qu, Carbon, 2020, 163, 154–161 CrossRef CAS.
- Z. Lu, G. Chen, S. Siahrostami, Z. Chen, K. Liu, J. Xie, L. Liao, T. Wu, D. Lin, Y. Liu, T. F. Jaramillo, J. K. Nørskov and Y. Cui, Nat. Catal., 2018, 1, 156–162 CrossRef CAS.
- K. Jiang, S. Back, A. J. Akey, C. Xia, Y. Hu, W. Liang, D. Schaak, E. Stavitski, J. K. Nørskov, S. Siahrostami and H. Wang, Nat. Commun., 2019, 10, 3997 CrossRef PubMed.
- Y. Wang, Y. Zhou, Y. Feng and X. Y. Yu, Adv. Funct. Mater., 2022, 32, 2110734 CrossRef CAS.
- H. Yang, J. Yin, R. Cao, P. Sun, S. Zhang and X. Xu, Sci. Bull., 2019, 64, 1510–1517 CrossRef CAS PubMed.
- H. Yang, R. Cao, P. Sun, J. Yin, S. Zhang and X. Xu, Appl. Catal., B, 2019, 256, 117862 CrossRef CAS.
- R. Du, K. Xiao, B. Li, X. Han, C. Zhang, X. Wang, Y. Zuo, P. Guardia, J. Li, J. Chen, J. Arbiol and A. Cabot, Chem. Eng. J., 2022, 441, 135999 CrossRef CAS.
- Z. Teng, W. Cai, W. Sim, Q. Zhang, C. Wang, C. Su and T. Ohno, Appl. Catal., B, 2021, 282, 119589 CrossRef CAS.
- W. Liu, J. Feng, R. Yin, Y. Ni, D. Zheng, W. Que, X. Niu, X. Dai, W. Shi, F. Wu, J. Yang and X. Cao, Chem. Eng. J., 2022, 430, 132990 CrossRef CAS.
- L. Wang, Y. Zhu, D. Yang, L. Zhao, H. Ding and Z. Wang, Appl. Surf. Sci., 2019, 488, 728–738 CrossRef CAS.
- J. Ma, N. Jia, C. Shen, W. Liu and Y. Wen, J. Hazard. Mater., 2019, 378, 120782 CrossRef CAS PubMed.
- S. Hu, X. Qu, P. Li, F. Wang, Q. Li, L. Song, Y. Zhao and X. Kang, Chem. Eng. J., 2018, 334, 410–418 CrossRef CAS.
- A. Malik, P. K. Prajapati, B. M. Abraham, S. Bhatt, P. Basyach and S. L. Jain, Catal. Sci. Technol., 2022, 12, 2688–2702 RSC.
- C. Lai, H. Yan, D. Wang, S. Liu, X. Zhou, X. Li, M. Zhang, L. Li, Y. Fu, F. Xu, X. Yang and X. Huo, Chemosphere, 2022, 293, 133472 CrossRef CAS PubMed.
- Z. Huang, M. Shen, J. Liu, J. Ye and T. Asefa, J. Mater. Chem. A, 2021, 9, 14841–14850 RSC.
- W. Niu, K. Marcus, L. Zhou, Z. Li, L. Shi, K. Liang and Y. Yang, ACS Catal., 2018, 8, 1926–1931 CrossRef CAS.
- X. Yan, Z. Jia, H. Che, S. Chen, P. Hu, J. Wang and L. Wang, Appl. Catal., B, 2018, 234, 19–25 CrossRef CAS.
- C. Wang, S. Zhao, X. Song, N. Wang, H. Peng, J. Su, S. Zeng, X. Xu and J. Yang, Adv. Energy Mater., 2022, 12, 2200157 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nh00564f |
| ‡ Hongcen Yang and Fei Ma contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |