
Advances in nanoparticle-based mRNA delivery for liver cancer and liver-associated infectious diseases
Seokhwan
Chung
,
Chan Mi
Lee
and
Miqin
Zhang
 *
*
Department of Materials Science and Engineering, University of Washington, Seattle, Washington 98195, USA. E-mail: mzhang@uw.edu
First published on 12th October 2022
Abstract
The liver is a vital organ that functions to detoxify the body. Liver cancer and infectious diseases such as influenza and malaria can fatally compromise liver function. mRNA delivery is a relatively new means of therapeutic treatment which enables expression of tumor or pathogenic antigens, and elicits immune responses for therapeutic or prophylactic effect. Novel nanoparticles with unique biological properties serving as mRNA carriers have allowed mRNA-based therapeutics to become more clinically viable and relevant. In this review, we highlight recent progress in development of nanoparticle-based mRNA delivery systems for treatment of various liver diseases. First, we present developments in nanoparticle systems used to deliver mRNAs, with specific focus on enhanced cellular uptake and endosomal escape achieved through the use of these nanoparticles. To provide context for diseases that target the liver, we provide an overview of the function and structure of the liver, as well as the role of the immune system in the liver. Then, mRNA-based therapeutic approaches for addressing HCC are highlighted. We also discuss nanoparticle-based mRNA vaccines for treating hepatotropic infectious diseases. Finally, we present current challenges in the clinical translation of nanoparticle-based mRNA delivery systems and provide outlooks for their utilization in treating liver-related diseases.
 Seokhwan Chung | Seokhwan Chung earned his BS in materials and science engineering from the University of Illinois at Urbana-Champaign. He is currently pursuing his PhD degree in materials science and engineering at the University of Washington. His current research interest is in synthesis and application of iron oxide and carbon-based nanomaterials for cancer therapy and diagnostics. |
 Chan Mi Lee | Chan Mi Lee earned her BS in chemistry from University of Illinois at Urbana-Champaign. She is currently pursuing her Master's degree in materials science and engineering at the University of Washington. Her current research interests are in polyplex and lipoplex-mediated drug delivery and gene delivery. |
 Miqin Zhang | Miqin Zhang is Kyocera Chair Professor of Materials Science & Engineering and Neurological Surgery, and adjunct professor in the Departments of Bioengineering, Radiology, and Orthopaedics & Sports Medicine at the University of Washington. She received her PhD from University of California at Berkley. Her research focuses on nanomedicine for cancer diagnosis and treatment, biomaterials for tissue engineering, and biosensors for detection of chemical and biological agents. |
1. Introduction
The liver, a vital organ in the body, performs key functions including removal of toxins, regulation of blood sugar levels, and synthesis of proteins. Hence, impaired liver function from various diseases can lead to fatal outcomes. Liver cancer has had the greatest increase in incidence in recent years, and exhibited the second-lowest 5 year relative survival rate compared to all other cancer types from 2008 to 2014.1,2 The most common case of liver cancer is hepatocellular carcinoma (HCC). Current treatments for HCC are surgical removal for early-stage patients and chemotherapy for more advanced stages of HCC. Surgical ablation is a viable treatment option for early-stage tumors but not for advanced stages of HCC due to impaired regenerative capability of the liver at later stages; however, patients are often diagnosed with advanced stages of HCC, making them ineligible for this method.2 Administrative routes of chemotherapy include trans-arterial chemoembolization and oral dosage with sorafenib, a kinase inhibitor, for late-stage patients.3 Improvement in clinical outcome has been limited, however, as the tumor develops resistance to the chemotherapeutic agent within six months into the treatment regimen. These data indicate a need for a different approach to improve the outcome for liver cancer patients. In addition to HCC, several infectious diseases are known to be hepatotropic. As the primary organ responsible for removal of foreign material from the blood, the liver is prone to infection from viruses and parasites.4–6 The immune cells in the liver offer protection and memory against the antigens over time, but these infections can be fatal with insufficient levels of immune response. While vaccines exist for some of these infectious diseases, challenges remain in development of delivery vehicles that are that do not elicit immune response themselves. For development of vaccines against novel pathogens, rapid turnaround is required between identification of target antigen and large-scale production, especially for pathogens capable of antigenic drift and shift.7 Conventional vaccines consisting of antigens or engineered viruses require months to reach viable mass production. These issues highlight a need for a novel platform for vaccine development.Gene therapy utilizes nucleic acids to alter the errant genetic expression of target cells and correct a disease. In case of cancer, which is directly caused by genetic mutation, genetic material is delivered to upregulate the expression specific genes such as tumor suppressor genes, or silence the expression of oncogenes.8 As it utilizes the molecular machinery to alter gene expression, gene therapy exhibits sustained therapeutic effects upon successful stable transfection; in contrast, traditional chemotherapy regimen consists of repeated administration of the therapeutic agent for long-term effects.9 However, several risks and barriers are present for gene therapy. In addition to altering the genes in the target cells, altering the genes of off-target cells may cause genotoxicity in otherwise healthy cells.10 Furthermore, in order to access the host genome, materials used for gene therapy need to be transported across the cell membrane, and into the nucleus, without degradation of the cargo by nucleases and other proteins present in the cytoplasm.11 Current methods for gene therapy utilize retrovirus and other engineered viruses as a transfection vector which could elicit unwanted immune responses.
Messenger RNA (mRNA) coding for specific tumor antigens or parts of foreign pathogens can prime the immune system to recognize tumor or infected cells, which are then subject to apoptosis induced by the innate cytotoxic immune cells in the body. Compared to delivering DNA, mRNA delivery does not require crossing the nuclear membrane. The delivery of mRNA implies that mRNA only needs to be translated into target protein, eliminating the chance of erroneous transcription.12 There is no risk of insertional mutagenesis associated with DNA delivery as mRNA is not integrated into the host genome.13 Compared to peptide vaccines, mRNA vaccines can encode full length tumor antigens, and allows delivery of multiple antigens.14,15 Furthermore, mRNA can be synthesized in large scale without the use of cells.16 Despite these advantages, mRNA-based therapeutics had limited development due to their poor stability and excessive immunogenicity. A single naked strand of mRNA is relatively unstable and subject to degradation in vivo, and this was the case when the first mRNA-based therapeutics were explored in 1990.17 The relatively large size (300–5000 kDa) and highly negative charge of mRNA also present obstacles for efficient transfection in vivo. In order to overcome these barriers, novel nanoparticles, largely based on cationic polymers and lipids, have been developed to protect the mRNA from degradation and stabilize the charge through electrostatic interactions. In addition to protection of the cargo, nanoparticles allow targeted delivery of mRNA to the intended cells and tissues through targeting ligands and moieties.18,19 Advances in nanotechnology has also shown that properties of nanoparticles can be tailored so that nanoparticle-mediated mRNA delivery would enhance cellular uptake and endosomal escape, improving the transfection efficiency.20–22 Through the use of nanoparticles, stable and safe mRNA-based delivery could lead the way in developing innovative and effective cancer treatments, as well as vaccines for infectious diseases. mRNA-based cancer vaccines in clinical trials are highlighted in Table 1.
| Cancer type | Target antigen | Combination therapy | Nanoparticle type | Route of administration | Trial Phase | Ref. |
|---|---|---|---|---|---|---|
| Breast | Shared tumor antigens and patient-specific mutated neoantigens | Surgery and adjuvant chemotherapy | Size & charge based RNA-lipoplex | IV | I | NCT0231645723 |
| Alphaviral vector encoding portion of HER2 (VRP-HER2) | Pembrolizumab (anti PD-1) | IV | II | NCT0363294124 | ||
| Melanoma | Melan-A | N/A | Protamine complexed mRNA | ID | II | NCT0020460725 |
| Mage A1 | ||||||
| Mage A3 | ||||||
| Survivin | ||||||
| GP100 | ||||||
| Tyrosinase | ||||||
| 4 TAAs | ||||||
| – RBL001.1, RBL002.2, RBL003.1, and RBL004 (Lipo-MERIT) | N/A | Liposome complexed mRNA | IV | I | NCT0241073326 | |
| Personalized vaccine targeting 20 TAAs (mRNA-4157) | N/A | Lipid encapsulated mRNA | IM | II | NCT0389788127 | |
| Prostate | TAAs (CV9103) | N/A | Protamine complexed mRNA | ID | 2008-003967-3728 | |
| – PSA | ||||||
| – Prostate stem cell antigen | ||||||
| – PSMA | ||||||
| – 6-Transmembrane epithelial antigen of prostate 1 (STEAP1) | ||||||
| Non-small cell lung cancer | TAAs (CV9201) | N/A | RNActive® technology | ID | II | NCT0092331229 |
| – 5 formulated mRNAs | ||||||
| TAAs (CV9202) | Anti-PDL1 (durvalumab), or with anti-PDL1 and anti-CTLA-4 (durvalumab + tremelimumab) | RNActive® technology | ID, IV | II | NCT0316477230 | |
| – 6 formulated mRNAs | ||||||
| Digestive tract adeno-carcinoma | Personalized tumor neoantigen | N/A | N/A | SC | N/A | NCT0346824431 |
| Colorectal | Tumor antigen (BNT 122) | N/A | Size & charged based RNA lipoplex | IV | II | NCT0448637832 |
| Advanced malignancies | OX40 ligand (mRNA-2416) | Alone, or with anti-PDL1 (durvalumab) | LNP | IT | II | NCT0332339833 |
| OX40L, IL-23, IL36γ (mRNA-2752) | Alone, or with anti-PDL1 (durvalumab) | LNP | IT, IV | I | NCT0373993134 |
In this review, we present an overview of advances in mRNA delivery and application in treating liver cancer and liver-associated infectious diseases. We first provide a brief summary of recent developments on methods for mRNA delivery, including viral vectors and non-viral vectors including various classes of nanoparticles. Then, a brief overview of the function of the liver, as well as its response to diseases and potential targets for mRNA therapy are presented. Next, we highlight mRNA delivery for treatment of liver cancer by discussing the role of liver resident T-cells and targets for mRNA-mediated gene regulation, cancer vaccines, and concurrent therapy. We also feature mRNA delivery for treatment and vaccine for other infectious diseases targeting the liver. Finally, we present the current challenges in developing nanoparticle-based mRNA delivery systems and provide outlooks for their utilization in treating liver-related diseases.
2. Methods for mRNA delivery
One of the most important challenges for effective mRNA delivery is its stability in the physiological environment. The molecular design of therapeutic mRNA plays a key role in overcoming this hurdle. Factors such as the length of the poly(A) tail and structure of the 5′ cap can improve the stability of the mRNA by controlling the type and amount of protein binding.35,36 In addition, codon optimization and nucleoside modification of the mRNA sequence has shown to improve the translation efficiency and reduce immunogenicity.37 Recent efforts at designing modified therapeutic mRNA have been covered extensively in otherworks.12,38,39While modification of mRNA is important in addressing the in vivo stability and translation efficiency, the mRNA still requires a delivery vesicle that can carry the mRNA from the site of administration to inside the cytoplasm of the target cells. Several physiological barriers must be taken into consideration in designing delivery systems for mRNA. First, the delivery system needs to protect the mRNA from degradation by nucleases that are present throughout the skin and blood which are the most common routes of therapeutic administration.40,41 Then, the mRNA needs to be transported through the permeable, yet selective cell membrane in order to reach the cytoplasm. Because the cell membrane is composed of negatively charged phospholipids and contains ion pumps and channels that maintain a highly negative potential in the membrane, it is not an ideal environment for transfer of a large and negatively charged molecule such as mRNA.8 Finally, the mRNA must be released from the delivery vesicle in order to interact with the intracellular machinery for protein translation. Various systems, including viral vectors and nanoparticle-based delivery systems, have been developed to overcome these barriers and achieve effective mRNA delivery. Fig. 1 schematically represents these vectors.
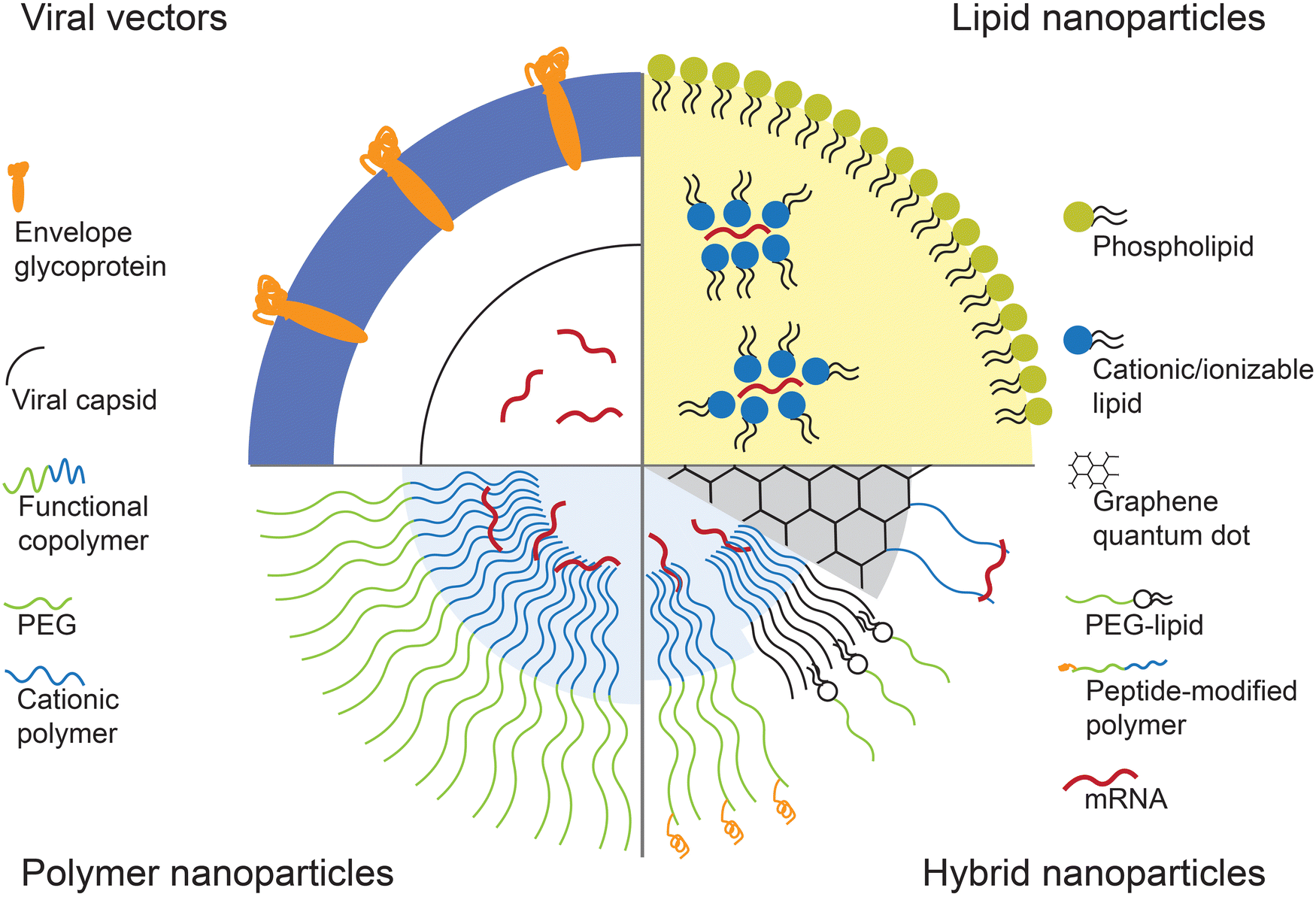 | ||
| Fig. 1 Types of mRNA carriers. Viral vectors utilize modified forms of viruses such as retrovirus to encapsulate and deliver mRNA. Lipid-based vectors use amphiphilic lipids with positive charge which can bind and stabilize mRNA. Block-copolymer consisting of PEG and cationic polymer is used to form structures that can stabilize the mRNA in the core of the vector. Hybrid vector systems consist of different classes of materials to take the advantageous aspects of each material. | ||
2.1 Viral vectors
Viruses used for gene delivery include retrovirus, adenovirus, adeno-associated viruses, and herpes simplex viruses. These viruses are genetically modified so that they are partially or fully substituted with the therapeutic genes. Viral vectors allow high specific delivery of genes to target cells, and have highly efficient and long-term gene expression compared to other transfection vectors. However, they can also elicit severe immune response and cytotoxic effects. Furthermore, integration of viral genetic material into the host genome could be fatal. Hence, viral vectors utilize viruses that have minimal pathogenicity, and with defective replication mechanism through modification of the viral genome.42 Retrovirus have been of interest for RNA delivery as their machinery are specifically designed to inject the viral RNA, rather than DNA, and utilize reverse transcription to alter the host genome. Non-integrating retroviruses contain mutations that specifically prevent the reverse transcription of the viral mRNA. Non-integrating gamma-retroviral vectors were used to deliver mRNA for expression of zinc-finger nuclease, a restriction enzyme that is prominent in the field of genome editing.43Instead of utilizing the entire viruses, recent works have resorted to use of “virus-like” particles or viral proteins to overcome limitations associated with viral vectors. Virus-like particles (VLPs) consisting of Vesicular stomatitis virus (VSV-G) and RNA-binding ribosomal protein L7Ae were developed for transfection of EGFP (enhanced green fluorescent protein) mRNA. While the exact mechanism of mRNA incorporation remains to be elucidated, the VLPs were able to efficiently deliver EGFP mRNA in multiple cell lines. The L7Ae RNA-binding domain was crucial as the presence of the L7Ae led to significant increase in fluorescence observed in the cells.44
Adenoviral vectors have been shown to induce acute hepatotoxicity by activating immune cells in the liver, which would release excessive levels of cytokine and lead to inflammation.45 This effect is not limited to the liver, and once the viral vector enters blood circulation, it can trigger release of pro-inflammatory cytokines.46 While blockage of specific cytokines could complement the immunogenic nature of viral vectors, more effective methods of reducing the immune response are required. Furthermore, adenovirus is so common to humans that most humans have developed preexisting immunity to adenoviral vectors, reducing their efficacy in gene expression.47,48 Furthermore, the scalability of viral vectors at a commercial scale remains as another challenge. The most common method of producing viral vectors is transfection of human embryonic kidney (HEK)293 cells with plasmids for viral components. Efforts made to improve the yield of vectors include adaptation of HEK293 cells to suspension cells,49 altering host protein expression to enhance vector replication,50 or use of insect cell-based expression system.51 These barriers have stagnated the clinical translation of viral vectors.
2.2 Nanoparticle vectors
Due to the aforementioned challenges associated with viral vector-mediated gene delivery, much research has been focused on development of non-viral vectors for mRNA delivery. Most commonly utilized types of non-viral vectors include lipid nanoparticles, polymeric nanoparticles, and protein-based nanoformulations. These materials are often positively charged for electrostatic interaction with both the mRNA and the cell membrane, and have to be biocompatible with minimal induction of cytotoxicity.In addition to amphiphilic lipid molecules, phospholipids, cholesterol, and polyethylene glycol (PEG) are often used as components in lipid nanoparticles. Phospholipids help improve the stability of the nanoparticle, as well as neutralizing some of the positive charged in cationic and ionizable lipids.56,57 Furthermore, phospholipids have shown to aid endosomal escape by disrupting the endosomal membrane.58 Cholesterol also plays a role in stabilizing the lipid structure by filling in the gaps between hydrophobic chains of the lipid molecules.59 PEG, which is often incorporated into the nanoparticle in the form of lipid-anchored-PEG, provides steric stability to the lipid nanoparticle by preventing aggregation and non-specific protein binding.60,61 A library consisting of various factors of each component, i.e., different ionizable lipid molecules, phospholipids, PEG-lipids, was screened to optimize the formulation for delivery of ovalbumin (OVA)-coding mRNA. In vivo CD8-T cell levels in mice as a response to expressed OVA were measured to find the optimal formulation. It was found that formulations without phospholipids did not induce T cell response at all. Longer PEG chains led to smaller hydrodynamic size of the liposomes, which also yielded the greatest T cell levels.62 In another study, a library of ionizable lipids synthesized from alkyl chains and polyamine cores was screened by delivering luciferase mRNA to Jurkat cells. The formulation consisting of the best performing ionizable lipid, lipid-anchored PEG, cholesterol, phospholipid, and mRNA, was synthesized via a microfluidics device. The resultant nanoparticle demonstrated efficient delivery of mRNA to T cells compared to electroporation and induced lower toxicity.63
Development of optimal conditions for efficient delivery of mRNA often starts with screening of a library of different molecules and various compositions, as demonstrated by these studies.64 While these studies can find the best performing combination, the link between the optimized condition and the subsequent efficient transfection results is still obscured. While efforts have been made to elucidate the mechanism behind efficient mRNA delivery by lipid nanoparticles, such as modulation of the mTOR pathway,65 further investigation of the interaction between cellular components and various aspects of lipid nanoparticles would streamline the development of optimized lipid nanoparticles for mRNA delivery. In addition, it has been reported that components in lipid-based mRNA vaccines cause significant inflammatory responses in humans. Certain formulations of lipid-based mRNA vaccines can increase the release of IL-1 cytokines, which induces the release of pro-inflammatory cytokines. Even some empty lipid nanoparticles were sufficient for release of IL-1, highlighting the need to further investigate the interaction of lipid components with immune receptors.66,67
Dimethylaminoethyl methacrylate (DMAEMA) and Diethylaminoethyl methacrylate (DEAEMA) have been explored as vectors for mRNA delivery, as these cationic polymers have shown to change into structures that destabilize the endosomal membrane at endosomal pH. Copolymers synthesized from DMAEMA or DEAEMA, PEG, and hydrophobic alkyl methacrylate monomers were used to encapsulate mRNA. In this study, increasing the cationic density of the polymer and shortening the alkyl chains on the methacrylate monomers led to the most efficient mRNA encapsulation. The formulation with smallest molecular weight led to reduced cytotoxic effects and exhibited increased transfection efficiency, outperforming PEI.68
Polymers containing hydrolysable ester bonds have also been investigated for mRNA delivery due to their good biodegradability. Poly(amino-co-ester) (PACE), synthesized from diesters, polyamines, and lactones, has been used for delivery of various nucleic acid therapeutics. The effect of the polymer molecular weight was explored by formation of lower molecular weight PACE by exposing high molecular weight PACE in air to allow hydrolysis. The use of the shortened PACE resulted in greater transfection efficiency of mRNA and a lower cytotoxicity profile.69
Endosomal escape is an important mechanism in any intracellular therapeutic delivery, as high encapsulation by a transfection agent and high cellular uptake is rendered irrelevant if the cargo is unable to be released from the endosome. PACE was modified with various end groups to investigate the relationship between mRNA cellular uptake, endosomal escape, and transfection efficiency. A novel luciferase-based probe that is fluorescently active only in the cytosol was used to assess endosomal escape. In total, a library of 31 end groups on PACE was tested, and a linear regression analysis showed no strong correlation between uptake and transfection efficiency, while a strong correlation was found between endosomal escape and transfection efficiency.70 This study shows that ensuring endosomal escape is more important than improving cellular uptake of transfection agents. Tuning the hydrophobicity of the polymer has shown to improve endosomal escape due to the interaction between the endosomal membrane and the hydrophobic moieties. In addition, hydrophobic interaction between the polymer molecules improves the stability of the polymeric complex. Nanoparticles encapsulating mRNA were synthesized from hydrophobic cationic polymers with pH-dependent aqueous solubility. These nanoparticles showed enhanced transfection efficiency compared to commercially available transfection agents.71
Altering the molecular interaction between the polymer and the mRNA has shown to affect the stability of the complexes, which is important when considering the routes of administration of the mRNA therapeutics. The effect of replacing primary amines on a cationic polymer with a guanidine group on the stability and transfection efficiency was explored. While both polymers formed micelles with mRNA, the polymer with guanidine group displayed greater stability against polyanion exchange, urea addition, and nuclease attack. The protection against these degradative elements increased the bioavailability of the mRNA, and led to greater transfection efficiency.72 While each of these studies shows each type of nanoparticles can be optimized for most effective mRNA delivery, the manner in which they can be optimized is not universal across the types of polymer and nanoparticle. For example, changing the molecular weight of PEI will likely not have the same effect as changing the molecular weight of PACE on transfection efficiency. Due to the diverse and customizable nature of polymers, advantages from various systems can be harnessed into a multifunctional nanocarrier, but the unique formulation will have to be individually assessed. Polymeric systems present a great opportunity in improving mRNA delivery through optimization of their structure and composition.
Proteins and peptides have also been used in combination with polymeric systems as they can perform specific tasks via their affinity with intracellular components. Fusogenic peptides can disrupt the endosomal membrane, and addition of fusogenic peptide on the surface of transfection vectors can enhance their endosomal escape. Polymeric micelles assembled from copolymer of polylactic acid and poly(N-acroloxy succinimide-co-N-vinyl pyrrolidone) (P(NAS-co-NVP)) were modified with cationic fusogenic peptide RALA and mRNA. These structures provided protection for the mRNA in serum, and showed high transfection efficiency.75 In another approach, proteins involved in translation of mRNA were complexed with polyamines to form ribonucleoprotein complexes. The affinity between mRNA and initiation factor eIF4E protein was used to form the initial ribonucleoprotein structure, which was then complexed with polyamine carriers. In addition to using the natural affinity between translational protein and mRNA, this approach mimics the translational steps necessary to express the gene encoded by the mRNA (Fig. 2a and b). Luciferase mRNA was used to assess the role of the eIF4E protein. Mice injected with preassembled mRNA-eIF4E showed much greater levels of luciferase expression than mice injected with mRNA only (Fig. 2c).76 The authors devised a similar structure consisting of poly(A) tail binding protein and mRNA containing a poly(A) tail, which showed high levels of mRNA transfection efficiency.77
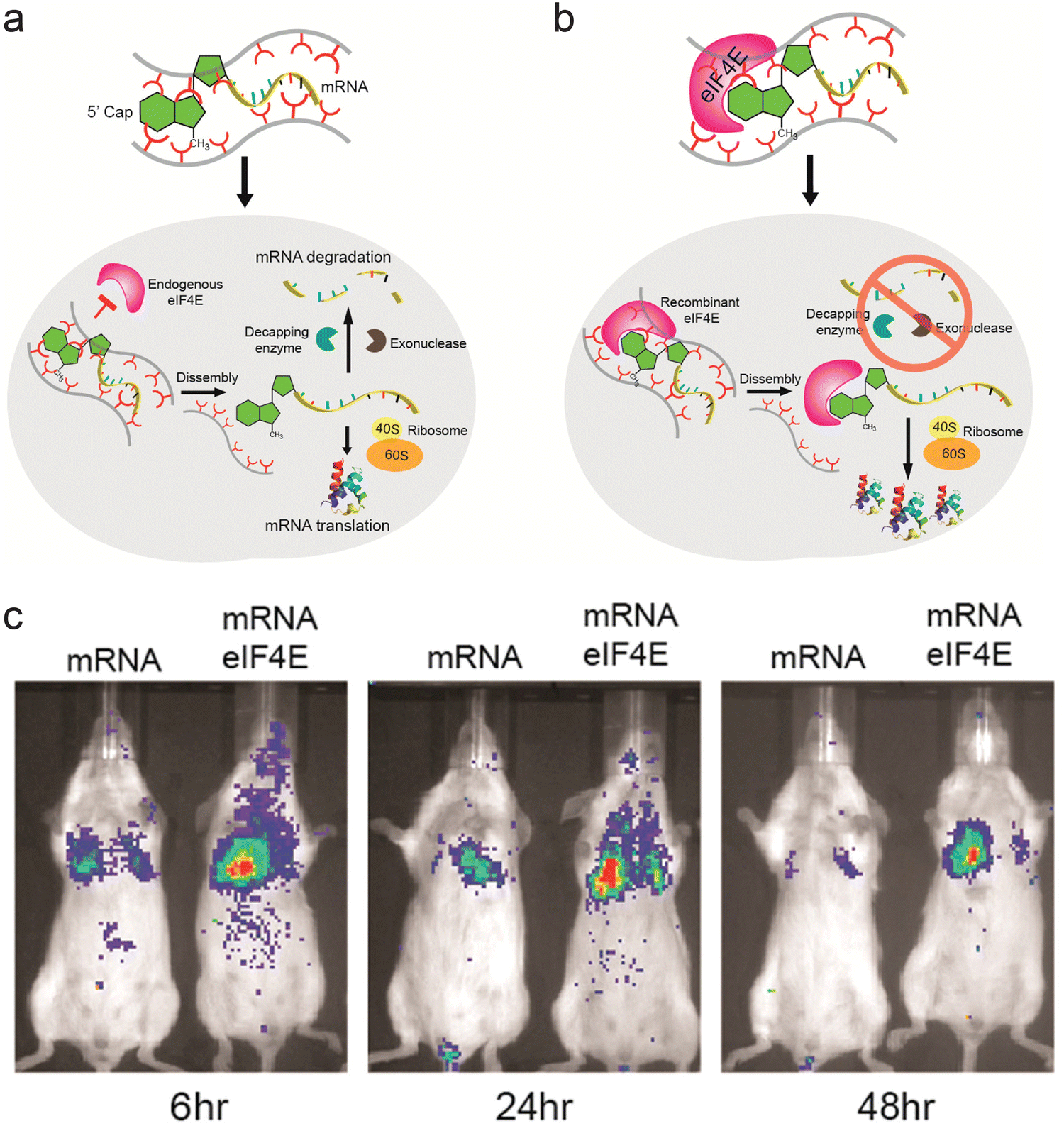 | ||
| Fig. 2 Ribonucleoprotein consisting of mRNA and eIF4E as a delivery vector. (a) Schematic representation of pathway for mRNA delivered into cells without preformation of ribonucleoprotein complex. (b) Schematic representation of suggested pathway for mRNA-eIF4E delivered into cells. (c) Fluorescence image of Balb/c mice injected with luciferase mRNA and mRNA-eIF4E over 48 h. Adapted with permission from ref. 74. Copyright 2017, American Chemical Society. | ||
In addition to lipid, polymers, and proteins, other molecular platforms have been explored to construct novel mRNA delivery systems. Aminoglycosides, primarily used as antibiotics in clinical and research settings, penetrate the cell membrane of pathogens. This interaction with pathogenic membranes was used to form lipid-based nanoparticles with aminoglycoside coatings to enhance endosomal escape. The aminoglycosides were incorporated in the form of cationic lipid-modified aminoglycosides, which enhanced the stability of the structure.78 Graphene quantum dots (GQDs) have recently garnered interest as imaging probes and therapeutic delivery systems.79,80 GQDs synthesized from citric acid were modified with PEI to impart positive charge onto the GQDs, which were then used to deliver mRNA. This design exhibited greater toxicity and lower transfection efficiency than lipid-based mRNA systems; as this was reported as the first application of GQD for mRNA delivery, fine-tuning of the synthesis parameters still remains to be solved. However, the GQD-based system displayed greater shear tolerance than lipid nanoparticles, and present a new avenue of developing multifunctional delivery platform for mRNA therapeutics.81 The field of nanoparticles for mRNA delivery presents an exciting opportunity to develop novel nanocarriers that could combine advantageous properties of different classes of materials. With each combination, however, careful evaluation of properties such as surface chemistry, shape, charge, size, and protein adsorption is required to create a potent and safe mRNA delivery vector.
3. Liver function and immunology
Prior to the discussion and presentation of recent works on mRNA-based therapeutics for liver-related diseases, it is important to understand the function of the liver and the immune response to pathogens in the liver. The structure and function of the liver, as well as the response of resident immune cells to HCC and infectious diseases will be briefly discussed in this section in order to shed light on potential targets for liver-bound mRNA therapy.3.1 Function and structure of the liver
The liver is the largest internal solid organ of the human body, and performs an array of functions including metabolism, blood volume regulation, and protein synthesis. Also, it plays a role in supporting the immune system and removing pathogens and exogenous antigens from the body.82–84 The versatility of the liver can be attributed to the presence of various cell types present in the organ. The main cell type that occupies around 80% of the total liver tissue is the parenchymal cells, or hepatocytes. Non-parenchymal cells, such as liver sinusoidal endothelial cells (LSECs), Kupffer cells, hepatic stellate cells, and resident lymphocytes, are responsible for 5–6% of the total liver tissue, and the rest is composed of extracellular space.85 The hepatocytes form a hexagonal pattern around the central vein into a structure called the hepatic lobule. At the vertices of these hexagonal arrangements are the portal triads, consisting of portal vein, hepatic artery, and bile duct. LSECs can be found in the lining of liver sinusoids which are capillaries through which antigen-containing blood from the portal vein is passed. Kupffer cells are macrophages that exist in the liver sinusoids and eliminate antigens and endotoxins through phagocytosis; in fact, Kupffer cells account for around 80% of the total macrophage population in the body.86 The combination of the resident cell types, vascular structure, and location of the liver enables it to perform the important detoxification of the systemic blood circulation.Understanding the structure and function of the liver can provide strategies for developing methods for mRNA delivery into the liver. Nanoparticles for mRNA delivery will face clearance by the Kupffer cells and the LSECs. As negatively charge nanoparticles and particles larger than 200 nm are removed from the system, this puts a constraint on the physicochemical characteristics of the nanocarrier for mRNA.87 Various methods have been developed to enhance the delivery of mRNA into hepatocytes. By utilizing the mannose receptors on LSECs, mannose-modified lipid nanoparticles have shown to accumulate in the liver in an in vivo study.88 The method of administration of mRNA can also affect the uptake of mRNA in the liver. Hydrodynamic delivery is a rapid injection of genetic material to alter the hydrodynamic pressure in capillaries to increase cell permeability temporarily. Reports of hydrodynamic delivery of pDNA have shown increased gene expression in the liver, presenting an avenue for improved mRNA delivery into the liver.89,90 The use of ultrasound targeted microbubbles techniques was employed in a study to further enhance the cellular permeability for gene delivery.91 In contrast to intravenous or intraportal administration, intrabiliary injection allows the delivery of therapeutics and evade phagocytosis by direct access to hepatocytes. Several studies have shown interbiliary infusion and biliary hydrodynamic delivery lead to greater transfection efficiency when compared to methods that utilize other vasculature.92,93 However, in order for biliary mRNA delivery to be more feasible, the nanocarriers must be able to protect the mRNA from degradation, as well as aggregation prior to reaching the hepatocytes.
3.2 Immune response to infections in the liver
The immune system in the liver consists of various cell types that have distinct functions in response to infections. Lymphocytes in the liver include T-cells, B cells, natural killer (NK) cells, and NK T-cells, which can identify specific antigens and play a role in directly or indirectly breaking down the recognized molecules.94,95 T-cells are identified by the presence of T-cell receptors (TCR) on the cell surface, and depending on the expression of TCR can function as cytotoxic cells or regulate the immune response through cytokine expression. Intrahepatic T cells are mostly comprised of CD8+ T-cells compared to CD4+ T cells, and are observed to have an activated phenotype. T-cells require priming by antigen presenting cells (APCs), during which APCs uptake antigens and present them on the cell surface via major histocompatibility complex I and II. T-cells are then activated by recognition of these antigens through TCR as well as interactions between co-stimulatory ligands and receptors.96,97 Different types of APCs have been found in the liver, including hepatocytes, LSECs, and dendritic cells trafficked through the liver.98–101 NK cells and NKT cells (T cells that express the NK marker CD56), are cytotoxic lymphocytes that are found more frequently in the liver compared to any other organ with resident lymphocytes. Liver-resident T-cells, NK cells, and NKT cells express the liver homing chemokine receptors CXCR3 and CXCR6, responsible for the accumulation of the lymphocytes, as well as regulation of NKT cell activity.102–104Tissue-resident memory T-cells (TRM) are also found in the liver, where they can provide rapid and potent responses to reinfection through cytolytic activity and regulation of proinflammatory cytokines. CD8+ TRM in the liver have been shown to patrol the intrahepatic vasculature and act as a first line of defense in pathogen infection. In response to a viral infection, hepatic CD8+ TRM cells exhibit a stronger immune response compared to non-resident memory T cells. Memory T cells specific to hepatitis C virus were shown to last for up to a few years after primary viral infection, and a secondary infection was able to be subdued quickly by the cytolytic activity of liver resident TRM cells.105 Expression of perforin is also elevated in TRM cells, which may aid the cytolytic activity against infected hepatocytes. In cases of hepatitis B virus-related HCC, CD8+ TRM cells were enriched in comparison to non-viral-related HCC, and corresponded to good prognosis.106 However, there have been cases where an over-stimulated antiviral response resulted in over-production of cytokines, leading to further liver injury in addition to direct toxicity on the liver by the virus.107,108 While NK cells display elevated levels of receptor expression such as NKG2D and NKp44 which can recognize viral-associated antigens, other evidence has shown that elevation in immunosuppressive molecules such as NKG2A in response to viral infection can prevent recruitment of peripheral NK cells into the liver, suppressing the immune response.109,110 NKT cells exhibit increased interferon-γ secretion which stimulates adaptive immune response and also inhibits viral replication in the early stages of viral infection.111
Research on hepatic immune response to parasitic infection has largely been focused on understanding the role of resident memory T-cells in combatting Malaria. CD8+ TRM cells survey the liver sinusoids, acting as the first line of defense of malaria liver infection. Anti-parasitic activity can be enhanced by priming CD8+ TRM cells with malaria antigens delivered by adeno-associated virus or presented by dendritic cells. Intravenous injection of malaria vaccine has also resulted in expansion of malaria-specific CD8+ cells to provide prophylactic effects.112,113 The roles of other lymphocytes in response to parasitic liver infection has not been elucidated.
3.3 Immune response to cancer in the liver
Understanding anti-tumor immune response in the liver is important for development of therapeutic approaches to HCC, as well as metastatic tumors common to the liver. Liver resident NK cells play an important role in controlling the tumor. Much of the NK cells found in HCC tumors display liver-resident phenotype. However, the tumor microenvironment is not conducive to strong anti-tumor activity, as evidenced by down-regulation of NKG2D and cytokine secretion and diminished cytotoxic activity of the NK cells. The anti-tumor activity was observed to be recovered with administration of IL-15.114 NKT cells have been associated with anti-tumor immunity. Stimulation of NKT cells with HCC-derived antigens showed suppression of tumor growth and elimination of hepatoma cells in murine liver. Other studies showed a relationship between the bile acid metabolism controlled by the gut microbiome and immunosurveillance activity of the NKT cells. Depletion of gut commensal bacteria led to accumulation of CXCR6+ NKT cells into the liver. The accumulated NKT cells produced more IFN-γ, which in turn, led to tumor growth inhibition.115The liver is a complex organ capable of many tasks vital to the body. The immune system of the liver in particular is important in combating infections and diseases as the liver is able to break down and eliminate antigens and foreign substances. Further understanding of the role of the structure and components of the liver in the scope of immune response to infections would accelerate advances in development of novel and sophisticated therapeutic approaches to circumvent current clinical obstacles.
4. Nanoparticle-mediated mRNA delivery for treatment of hepatocarcinoma
Prior to the development and release of SARS-Cov-2 vaccine, much of the effort in mRNA vaccine technology aimed at development of cancer vaccines. In fact, there are several mRNA-based cancer vaccines undergoing clinical trials. mRNA can encode specific antigens, and provide a safer and cost-effective alternative to other methods of vaccination for cancer vaccines. In many cases, lipid nanoparticles or protein-based polyplexes were used to enhance the therapeutic effects of these mRNA vaccines. In addition to vaccines, mRNA can be delivered using nanoparticles to upregulate cancer-associated genes that would lead to tumor cell death and inhibition of tumor growth. These approaches include delivery of mRNA coding for tumor suppressor genes or apoptotic proteins to cancer cells, immunostimulatory cytokines to macrophages, and tumor antigens to dendritic cells (Fig. 3).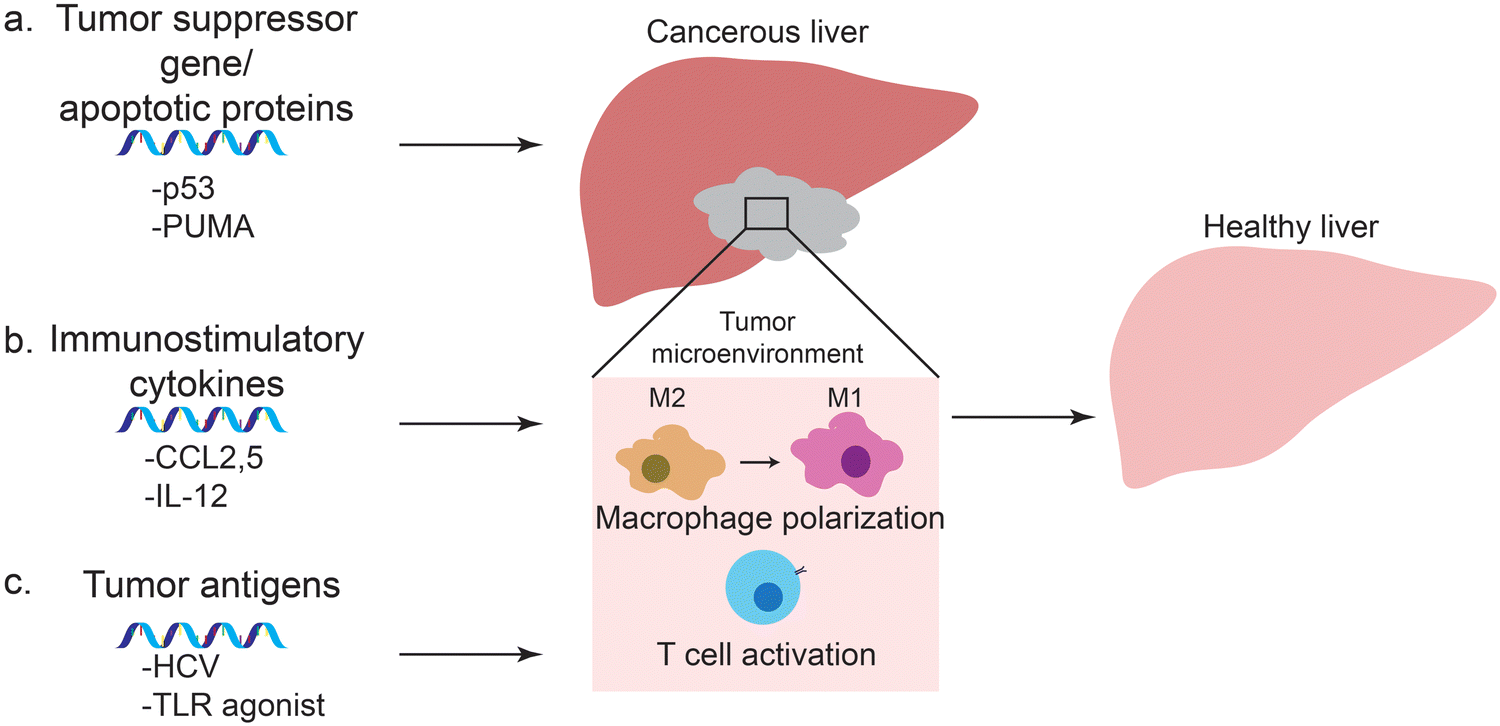 | ||
| Fig. 3 Approaches using mRNA-based therapeutics and vaccines for treatment of HCC. (a) Tumor suppressor gene and apoptotic protein encoding mRNA leads to apoptosis of tumor cells and inhibition of tumor growth. (b) Immunostimulatory cytokines coded by mRNA polarize macrophages to tumor-suppressive M1 state. (c) Cancer vaccines use mRNA encoding tumor antigens to elicit immune response and prime T cells against these antigens. | ||
4.1 Tumor suppressor genes and apoptotic proteins
Tumor suppressor genes encode for proteins that regulate the cell cycle and prevent the replication of problematic cells (Fig. 3a). Loss of function of these genes leads to deregulation in intracellular signaling pathways and contributes to initiation and development of tumors. The TP 53 gene encodes the transcription factor p53 that is involved in cell cycle regulation, apoptosis, DNA repair, angiogenesis and its inactivation has been reported in 30–60% of HCC tumors. Restoration of p53 expression was observed to elicit significant tumor regression116 and recently, many groups investigated the antitumor effects of nanoparticle-meditated delivery of mRNA targeting p53.117,118 The synergistic effects of mRNA delivery was demonstrated in combination with everolimus, a mammalian target of rapamycin (mTOR) inhibitor. mTOR inhibitors have poor drug efficacies – notably in advanced HCC – due to autophagy activation that acts as a resistance mechanism.119 Delivery of p53-mRNA using redox-responsive lipid-polymer nanoparticles sensitized p53 deficient HCC tumors to everolimus because restoration of gene expression inhibited autophagy and activated the apoptosis pathway; subsequent therapeutic efficacies were observable in vivo.117Another target for liver cancers are apoptotic proteins such as PUMA or Caspase. PUMA (p53 upregulated modulator of apoptosis), a member of the Bcl-2 protein family, is a downstream protein of the p53 pathway that plays a critical role in caspase activation during p53-dependent and -independent apoptosis. Co-delivery of apoptotic mRNA with miRNA (miR-122) allowed selective expression of target proteins, leading to apoptosis only in diseased cells.120 The noncoding miRNA targeted complementary sequences at the 3′ UTR of the mRNA, leading to mRNA destabilization, cleavage and repression of translation.121 In HCC cells, this innate silencing mechanism was hampered, and display markedly lower levels of miRNA than healthy hepatic counterparts. Hence co-delivery of mRNA-miRNA by evaded PUMA expression in miR122-high healthy hepatocytes and triggered apoptosis only in miR-122 low HCC cells, significantly reducing liver toxicity. The miRNA strategy enabled the therapy at fatally high doses of mRNA, compatible with intratumoral and systemic administration.
The studies mentioned here demonstrated the efficacies of mRNA therapeutics in reactivating mutant TP53 genes and downstream proteins. It is important to note that the cargo did not consist solely of mRNA; strongest anti-tumor effects occurred when delivered in conjunction with other inhibitory/regulatory agents. Despite such promising reports, studies of p53 restoration via mRNA delivery for liver cancers are limited and p53 gene therapy is more popular in the community.
4.2 Immunostimulatory cytokines
mRNA therapeutics are also effective for delivery of immunostimulatory cytokines (Fig. 3b). HCCs are highly resistant to chemotherapy due to their unique immunotolerant microenvironment and have limited treatment modalities. Macrophages in the tumor environment can be polarized to a cancer-promoting or cancer-inhibitory state. Polarization of the macrophages can be achieved by expression of stimulatory cytokines through delivery of cytokine-coding mRNA. An mRNA/LNP system was used to transfect HCC cells with mRNA encoding a bispecific single-antibody (BisCCL2/5i). Dlin-MC-DMA, a two-tailed lipid with a single dimethylamine headgroup was used as it is able to encapsulate mRNA through its ionizable headgroup (Fig. 4a). Subsequent blockade of CCL2 and CCL5 pathways polarized macrophage from cancer-promoting M2 phenotype to cancer-inhibitory M1 phenotype (Fig. 4b and c). Reversal of immunosuppression, confirmed via increased levels of CD8+ T and NK cells, facilitated synergistic antitumor effects when combined with PD-1 inhibition therapy (Fig. 4d and e).122 Combination of macrophage polarization through mRNA delivery with checkpoint blockade therapy activates T cells, elicits significant tumor destruction and increases long-term survival rates in syngeneic mouse models of primary and metastatic HCC.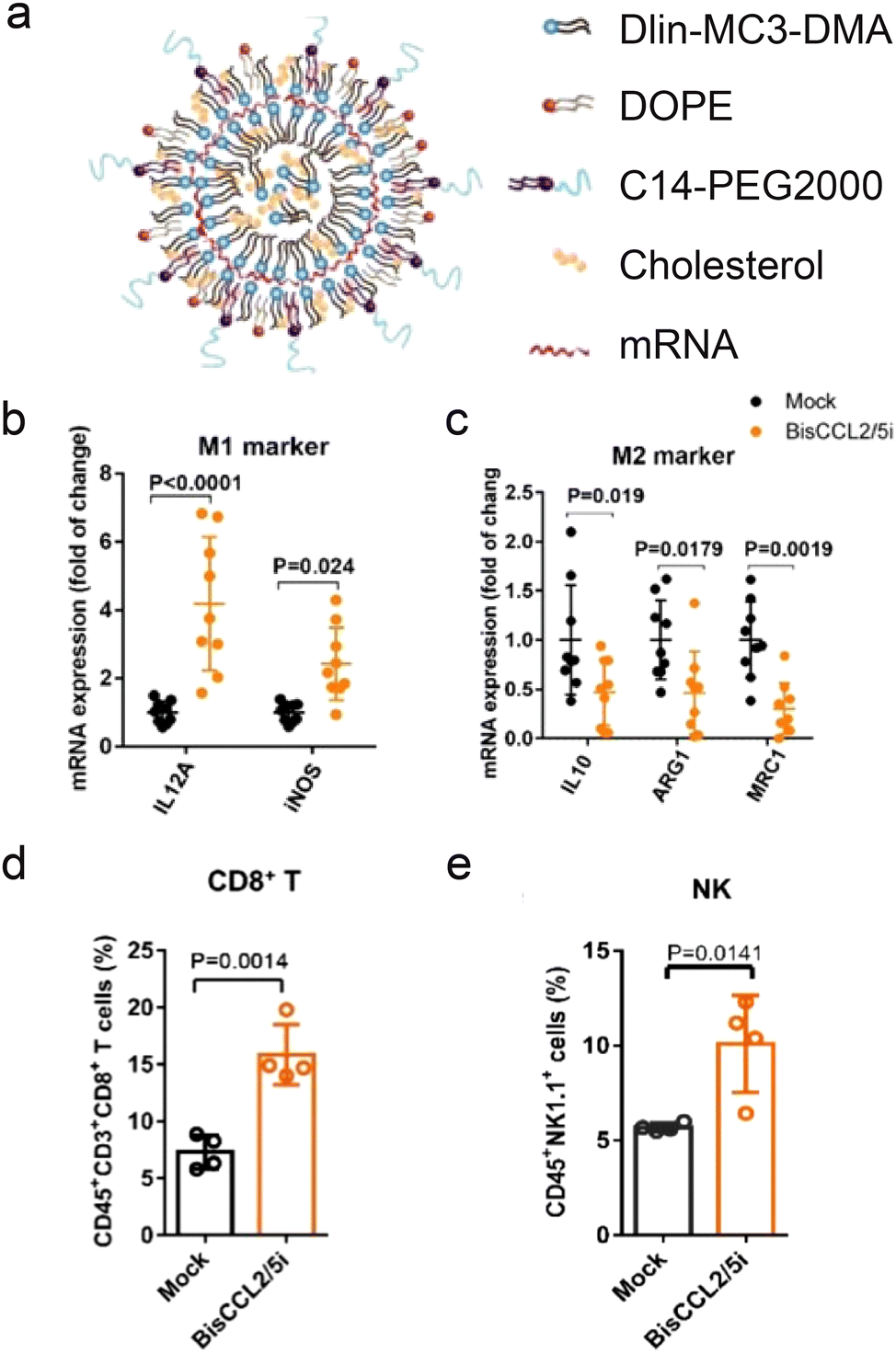 | ||
| Fig. 4 Delivery of mRNA for macrophage polarization and synergistic immunotherapy. (a) Schematic representation of mRNA/LNP. (b and c) mRNA expression of classic M1 (b) and M2 (c) markers in HCC tumor tissues 48 h after systemic administration of mRNA/LNP. (d) Percentage of CD8+ T cells in CD3+ T cells. (e) Percentage of nk cells in CD45+CD3− cells. Immune cells derived from mice 48 h after injection with mRNA/LNP. | ||
In another study, lipid nanoparticle delivery of mRNA encoding Interleukin-12 (IL-12-LNP), a strategic signaling mediator in T cell activation and M1 macrophage polarization, reduced tumor burden and increased survival in transgenic mice model of MYC-driven HCC. IL-12-LNP induced HCC regression by eliciting anti-tumor immunological response rather than suppressing MYC oncogene levels; recruitment of CD44 + CD + 3 CD + 4 T helper cells to tumor sites and neighboring tissues was confirmed along with upregulated IFNγ levels.123 Additionally, overexpression of IL-12 polarized macrophage to M1 phenotype via downregulation of Stat-3 and its downstream transcription factor c-myc, a key activator of M2 polarization.124 Such relation with c-myc may explain why MYC-driven HCC is sensitive to immune therapies like IL-12-LNP. Although direct molecular targeting of MYC is an ideal solution to treating aggressive HCCs, the toxic effects of MYC inhibitors on healthy tissues are unknown. Thus the study provided an alternative, non-toxic approach that targeted the immuno-vulnerability of HCC tumors rather than direct suppression of MYC proto-oncogene.
The studies discussed here involved direct transfection of HCC with mRNA coding appropriate cytokines and relied on the host immuno-repertoire to induce anti-tumor effects in vivo. They utilized the immunosuppressed nature of HCCs to minimize effects to neighboring, immunologically active healthy cells. The use of nanoparticles as medium for mRNA delivery showed that mRNA was stabilized and efficiently delivered to the target cells. For further development of the therapy, drug biodistribution within the liver, efficacy on metastatic tumors and accuracy of murine models to simulate immune responses in humans (including immunotherapy-related adverse events) must be addressed.
4.3 Tumor antigens
mRNA-based cancer vaccines are another promising modality in cancer immunotherapy (Fig. 3c). Delivery of tumor antigen-encoding mRNA teaches the immune system to activate cytotoxic responses upon recognition of antigen presenting tumor cells. Thus mRNA cancer vaccines involve the transfection of immune cells rather than tumor cells. In contrast to delivery of cytokine mRNA, tumor antigen mRNA is to be delivered to APCs in order to activate T cells to the specific tumor antigens to utilize the anti-tumor activity of T cells. Various types of antigens, including epitopes from immunogenic neoantigens, predicted neoantigens, and mutations in tumor suppressor genes or oncogenes in HCC have been explored.125–127 Studies showed that compared to other cancer types, HCC cells seldom express neoantigens and targeting tumor-associated antigens may be more feasible for the purpose.128,129 Several studies used NP-assisted mRNA delivery to express tumor antigens for activation of cytotoxic immunity in various cancer models.62,130,131 The transfection of dendritic cells with modified mRNA encoding for Hepatitis C virus (HCV) and the successful expression of target neoantigen on effector cells were presented. This study presented possibilities of mRNA vaccines as a preventive modality of liver cirrhosis and HCC.132 Nevertheless, identification of HCC neoantigens remains a challenge, but with the advent of next generation sequencing and algorithms, neoantigen selection for personalized mRNA-vaccines is achievable. Phase I/II clinical trial (NCT03480152) to evaluate safety and immunogenicity of a multi-epitope mRNA vaccine for HCC and metastatic liver tumors is currently underway.133Therapeutic effects of cancer vaccines have been investigated in murine models of aggressive melanoma, lymphoma, and prostate cancers. Pre-vaccination of mice with antigen encoding mRNA elicited endogenous T cell response once injected with cancer cells and stalled disease progression and suppression than their control counterparts.62,134–136 Recently, studies have geared towards co-delivery of mRNA antigen and adjuvant127,135–137 following the discovery that type 1 IFNs induced from unmodified mRNA may hamper with T cell response and vaccine efficacy.136,138 Subsequently, co-delivery of TLR agonist with nucleoside-modified mRNA was explored to recover for the immunogenic loss, and with considerable success. Nucleoside modified mRNA retained translational capacity, enhanced activity of antigen presenting cells and prevented type 1 IFN over secretion. Adjuvant co-delivery using lipid nanoparticles and lipid-PEG nanoparticles enabled dendritic cell maturation and subsequent cytotoxic T cell activation, effectively harboring both innate and adaptive immune response.135,136 However, mRNA – adjuvant combinations as preventive therapies have not yet been explored for liver cancers. The challenges lie in identifying an appropriate antigen for liver targets, optimizing the dynamics and kinetics of mRNA expression to achieve dendritic cell activation at minimal type 1 IFN release, and discovering an adjuvant that pairs with the mRNA to produce synergistic anti-tumor response.
5. Nanoparticle-mediated mRNA delivery for treatment of hepatotropic infectious diseases
Following the success of mRNA vaccines against SARS-Cov-2 virus, clinical application of mRNA vaccines for other infectious diseases have gathered great interest from academic research to clinical development. Compared to other types of vaccines such as live-attenuated vaccines or subunit vaccines, mRNA vaccines are much easier to manufacture, and induce a strong and potent T cell and humoral immune responses. Progress and development of vaccines for SARS-Cov-2 have been thoroughly reviewed elsewhere.38,139–142 In addition to identification of effective target antigens in the infectious pathogen, advances in nanoparticle technology have enhanced the effectiveness of the vaccines by improving the stability and transfection efficiency of mRNA in vivo. Developments in nanoparticle-mediated mRNA vaccines against other viral and parasitic diseases targeting the liver are presented in this section.5.1 Nanoparticle-mRNA vaccines for viral infections
The liver is exposed to various infectious pathogens, ranging from viruses to parasites. These pathogens can directly and indirectly affect the liver, and priming the immune system of the liver against such infections is important in eliminating the pathogens and progression of diseases.Influenza infection has been reported to induce liver involvement, especially in patients with underlying liver conditions.6,143–145 Influenza A virus genomes change their sequences coding for the hemagglutinin (HA) and neuraminidase (NA) antigens over time. This complicates the development of effective influenza vaccine as the antigenic variability could render a vaccine ineffective if a new strain of the virus not covered by the vaccine arises. Long production times and variable yields of antigens encumber a timely response to a new strain of influenza A. mRNA vaccines present a method for developing more effective influenza vaccine owing to the ease of mRNA synthesis. HA-expressing mRNA complexed with protamine was tested in animal models as a prophylactic vaccine against influenza. By eliciting B and T-cell responses, the vaccine led to increased survival in mice, ferret, and pigs. Protection against heterologous viral infections was also observed.146 Self-replicating mRNA encoding HA was able to induce the same level of protection as non-replicating mRNA at 64-fold less concentration. The study also showed that trivalent mRNA vaccination offered protection against multiple strains.147
Nucleoprotein (NP) in influenza viruses is a relatively conserved antigen between various strains, and presents a target for a more universal influenza vaccine. Administration of NP mRNA induced stronger T-cell response in mice compared to DNA vaccination, and also offered modest protection against heterologous strains.148 In a study, segments of antigens that are known to be conserved were identified, and mRNA encoding these antigens were loaded in a lipid nanoparticle. Along with HA and NP, NA and M2 protein, a viral transmembrane protein, encoding mRNA were administered in mice models (Fig. 5a and b). The mRNA strands were encapsulated in a lipid nanoparticle that mimicked the size of an influenza virion (∼80 nm). Compared to administration of mRNA encoding each of these antigens, the combination approach led to significantly increased antibody response. The combined vaccine also induced potent protection from virus challenge (Fig. 5c–f).149 Clinical trials of lipid nanoparticle-mRNA vaccine for potentially pandemic avian H10N8 and H7N9 influenza viruses showed robust immune response, with minimal side effects.150 Efforts are being made to further elucidate the mechanism of mRNA vaccines against influenza, such as modification of nucleosides in the mRNA and the modification of the antigen structure and function.151,152
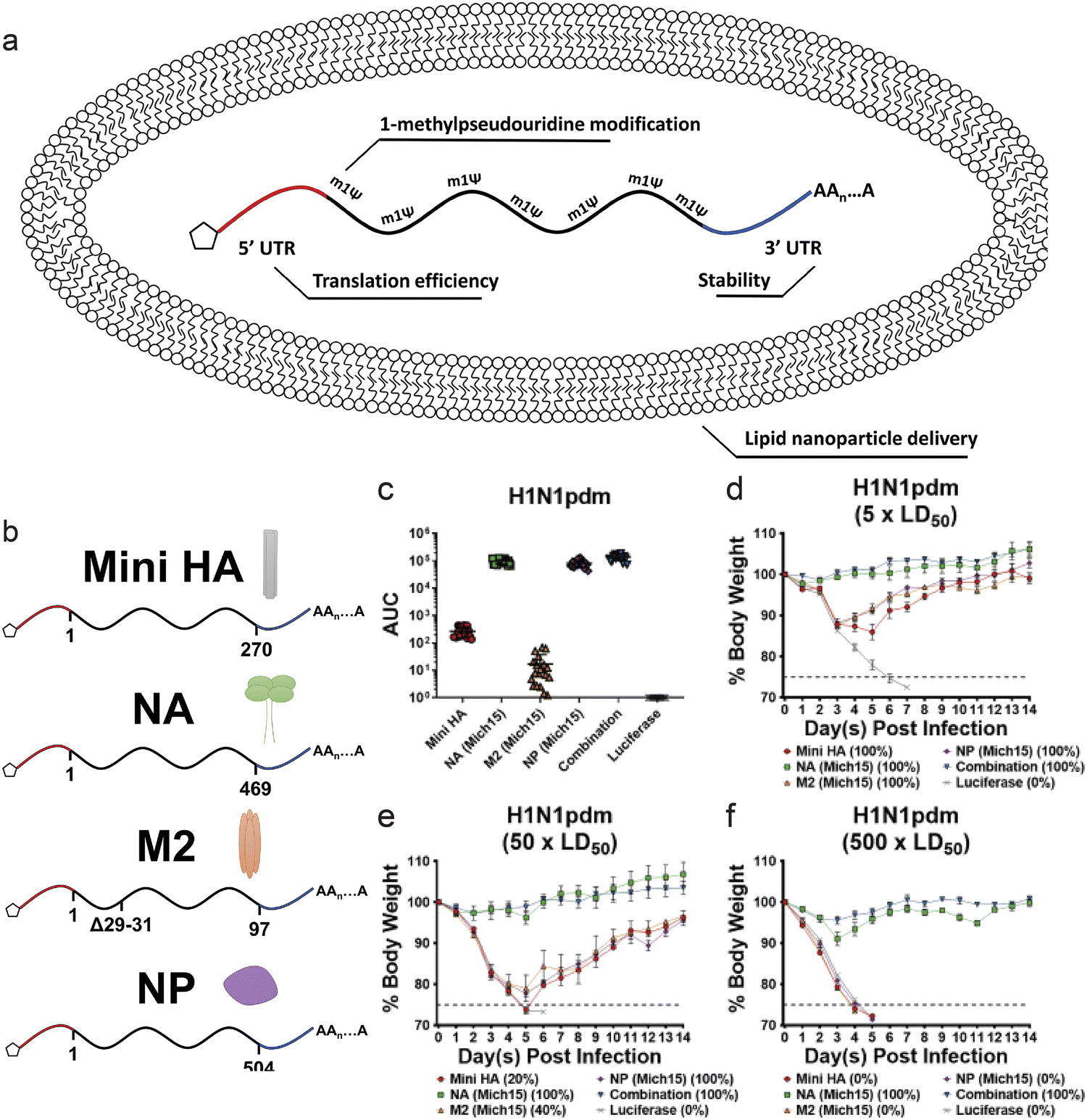 | ||
| Fig. 5 (a and b) Schematic of lipid nanoparticle and nucleoside modified mRNA encoding influenza antigens. (c) Sera were collected from mice 28 days after mRNA-LNP vaccination. Antibodies against corresponding antigens were measured via ELISA. (d–f) Body weight loss over time of mice challenged with (d) 5 × LD50, (e) 50 × LD50 or (f) 500 × LD50 of H1N1pdm influenza. (n = 5 per group.) Adapted with permission from ref. 138. Copyright 2020, ASGCT. | ||
In addition to influenza, several other viral infections can target the liver. Dengue is caused by dengue virus (DENV) and presents a varying range of feverish symptoms. While not limited to the liver, liver dysfunction and clinical manifestations such as jaundice and acute liver failure is associated with dengue infection.153,154 DENV1-NS, composed of regions of non-structural proteins, has been reported as a candidate for dengue vaccine that can induce a strong T cell response. Strong CD8 T cell responses were observed in mice administered with DENV1-NS encoding mRNA encapsulated by lipid nanoparticles, and significantly reduced viremia was detected after viral challenge.155 mRNA vaccines encoding prME and E80 as DENV antigens have also induced antibody and T cell responses, and protective effects against DENV1 and DENV2, distinct serotypes of DENV.156,157
Human cytomegalovirus (HCMV) can infect various cells within the host and cause significant hepatitis, especially in immunocompromised hosts.158 Glycoprotein B (gB) on HCMV is a viral fusogen that is responsible for entry into cells and has been identified as a target for efficacious vaccine against HCMV. Nucleoside modified gB mRNA encapsulated with lipid nanoparticles were administered to rabbits. The mRNA vaccine induced improved durability and peptide binding compared to administration of full length gB.159 In another study, mRNA expressing pentameric complex (PC), another vaccine candidate, and gB were administered in mice and nonhuman primates. The vaccine induced a potent and durable antibody response which lasted even after several months post-vaccination.160
Hepatitis is often caused by hepatitis B virus (HBV) and hepatitis C virus (HCV), and can lead to carcinoma, including HCC, liver failure, and chronic inflammation. Adequate immune response induced by vaccination is crucial in prevention of viral hepatitis. While HBV vaccines are clinically available, no vaccines exist for HCV. A strategy for immunization against HCV was adoptive transfer of dendritic cells transfected with mRNA. NS3 protein in HCV is considered as a vaccine candidate due to its limited genetic variability, and CD4+ T helper epitope and several cytotoxic T lymphocyte epitopes. NS4A is a cofactor of the NS3 protease and is able to target the NS3/4A complex to intracellular membranes, as well as increase the half-life of NS3 inside the cell. Hence, dendritic cells were transfected with mRNA expressing NS3/4A. Mice were vaccinated with dendritic cells transfected with NS3/4A mRNA or green fluorescent protein mRNA, non-transfected dendritic cells, and PBS. Mice injected with dendritic cells transfected with mRNA showed significantly higher NS3-specific lymphocyte proliferation, increased number of IFN-γ secreting cells, and greater cytotoxic T lymphocyte response. Vaccinated mice were challenged with recombinant virus 10 days after vaccination, and the virus level was determined five days after the challenge. The virus level was decreased by at least five orders of magnitude in mice vaccinated with NS3/4A-transfected dendritic cells.161
Nanoparticle-based mRNA vaccines for viral infections can address the shortcomings of current viral vaccine regimen. Efforts are aimed at identifying candidate antigens or subunits of antigens and utilizing mRNA encoding multiple targets to enhance the immune response. Some formulations have already entered clinical trials, and present opportunities for development of vaccines for unaddressed viral diseases.
5.2 Nanoparticle-mRNA vaccines for parasitic infections
While much effort in development of mRNA vaccines is geared towards cancer and viral infections, vaccines for parasitic infections are also being developed. Plasmodium falciparum malaria is an infectious and life-threatening disease, and presents a major global health problem. Often transmitted through mosquito bites, the parasite rapidly infects hepatocytes in sporozoite form and develop into schizonts. These schizonts can rupture, killing the hepatocyte, releasing merozoites into the bloodstream, and further progressing the infection. Hence, controlling and preventing the infection while it is contained in the liver is critical in treating malaria.4,5,162,163 Plasmodium macrophage inhibitory factor (PMIF) was identified as a potential target for malaria vaccines. PMIF interferes with development of T cells into long-lived memory T cells, and PMIF-deficient Plasmodium yoelii was associated with delayed parasitic growth in the liver. Self-amplifying mRNA expressing PMIF complexed, as a nano-emulsion consisting of cationic lipids and surfactants, was administered in mice as a vaccine against plasmodium. Potent cellular and humoral immune responses were induced and the vaccine was able to confer protection to re-infection.164 In another study, plasmodium falciparum circumsporozoite protein (PfCSP) was identified as the target antigen for malaria vaccine. PfCSP is the dominant coat antigen on sporozoite-stage Plasmodium falciparum. PfCSP mRNA was encapsulated with lipid nanoparticles, and the immune response in vaccinated mice was observed. A secondary boost administration was necessary to induce potent immune response, as a single-dose regimen resulted in significantly lower antibody titer (Fig. 6a). Against a parasitic challenge in mice, a prime:boost regimen of mRNA-LNP vaccine was shown to confer protection, with greater dosage leading to improved survival (Fig. 6b). The vaccine administration schedule was also shown to significantly affect the survival, as 3 week immunization schedule was less effective than a 6 week immunization schedule in mice. The effect of mRNA modification was more pronounced in the groups with 6 week immunization schedule, as mRNA with nucleoside modification, labelled UPenn in Fig. 6c and d, resulted in greater survival compared to commercially available, non-modified mRNA (TriLink in Fig. 6c and d).165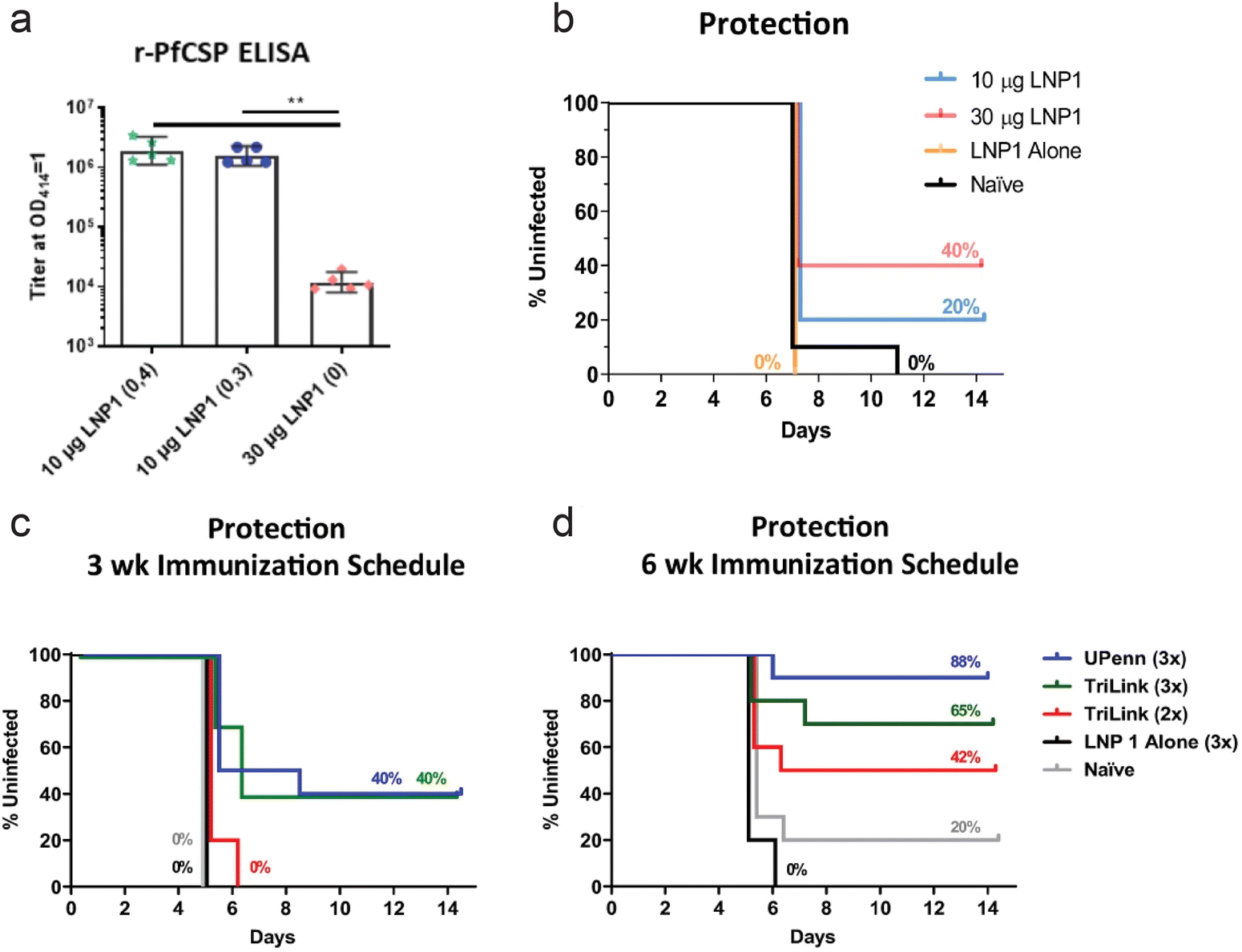 | ||
| Fig. 6 PfCSP mRNA for immunization against malaria. (a) PfCSP-specific antibody assessed by r-PfCSP titration ELISA for mice immunized twice at 4 week interval (10 μg LNP1 (0,4)), 3 week interval (10 μg LNP1 (0,3)) and once (30 μg LNP1 (0)). (b) Kaplan–Meier survival curves for infected mice after administration of different doses of mRNA-LNP, LNP only, and naïve control. (c and d) Kaplan–Meier curves of parasitic challenged mice with 3 week (c) and 6 week (d) immunization schedule. mRNA from university of pennsylvania with nucleoside modification (UPenn) and commercially available mRNA (TriLink) were administered with prime:boost (2×) or prime:boost:boost (3×) regimen. Adapted with permission from ref. 154. Copyright 2021, Springer Nature. | ||
In contrast, few studies have shown successful development of mRNA vaccines for other parasitic liver infections, such as schistosomiasis. However, recent studies have revealed potential candidate antigens for these infections.166,167 Plans for clinical trials for mRNA vaccines against malaria have been announced by BioNTech, building on its success in developing the SARS-Cov-2 vaccine. These efforts could advance the understanding of vaccine design for other parasitic infections.
6. Conclusions and outlook
mRNA technology has become a subject generating significant amount of coverage and discussion not only in academic and clinical research, but also the political sphere. The ease of manufacturing, elimination of insertional mutagenesis risk, and the demonstrated effectiveness in the success of the COVID19 vaccines has highlighted mRNA-based vaccines to be a viable alternative to traditional vaccine types. Furthermore, barriers to effective mRNA delivery in vivo can be potentially overcome with novel nanoparticle systems that are able to stabilize mRNA and improve their cellular uptake and expression. In this review, we present recent efforts in development of nanoparticle-mediated delivery of mRNA-based vaccines and therapeutics against liver diseases. Modifications to the mRNA can modulate the immunogenicity and stability of the mRNA molecules in biological settings. Transfection agents, including viral vectors and nanoparticles, consisting of lipids and polymers, protect the mRNA from degradative environment in vivo, and improve the transfection efficiency of the mRNA. The unique function, structure, and composition of the liver allows it to perform vital tasks, and the resident immune system plays an important role in combatting infections and tumorigenesis. mRNA-based therapeutics and vaccines have been developed for HCC, and identification of other genetic targets for gene therapy present further opportunities for mRNA technology to be applied in addressing liver cancer. Further, immune response to hepatotropic pathogens such as viruses and parasites can be boosted with mRNA vaccines, and many studies have shown improved immune response as well as conferred protection.Despite the advances made in mRNA-based therapeutics and vaccines, challenges still remain. The mRNA vaccine formulations had to be kept at ultra-cold temperatures during transport and storage.168 Due to the poor thermal stability of mRNA, a cold chain is required in order to ensure that the mRNA formulation is at optimal stability prior to clinical use, which can be costly when considering transport of millions of doses. It should also be considered that infectious diseases are prevalent in developing nations in the tropics, amplifying the need for mRNA vaccine formulations that do not need an ultra-low temperature storage.38 Currently, the effect of interaction between mRNA and lipid molecules is not well understood, and data from empirical studies on long term storage of free and encapsulated mRNA rarely show stability after more than a month. Various approaches such as addition of excipients and lyophilization have shown increased stability at temperatures above 0 °C.169–172 Further exploration of the effect of these processes, as well as a more fundamental understanding of how lipid–mRNA interaction retards the hydrolysis of mRNA would ameliorate the prospects of cold chain-free distribution of mRNA vaccines.
The apparent success of the COVID19 vaccines has spurred the industry to pursue development of mRNA vaccines for other diseases. Prior to that, clinical trials for mRNA therapeutic cancer vaccines had already been underway. These results have propelled a multitude of efforts aimed at improving mRNA delivery ranging from modification of the mRNA sequence to fine-tune immunogenicity and stability to design of novel transfection vectors to enhance antigen expression in the target cells. The use of nanoparticles, mostly based on lipids, were important in implementing mRNA delivery in vivo to prevent degradation in physiological environment and to improve the uptake of the mRNA-nanoparticle structure and subsequent antigen expression. While translation of these therapeutic effects to human clinical trials remains to be addressed, mRNA vaccines present a new avenue for treatment of cancer and prevention of infectious diseases.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by National Institutes of Health Grant (NIH/NIBIB R01EB026890). M. Z. acknowledges the support of Kyocera Chair Professor Endowment.Notes and references
- R. L. Siegel, K. D. Miller and A. Jemal, Ca-Cancer J. Clin., 2019, 69, 7–34 CrossRef PubMed.
- D. Anwanwan, S. K. Singh, S. Singh, V. Saikam and R. Singh, Biochim. Biophys. Acta, Rev. Cancer, 2020, 1873, 188314 CrossRef CAS PubMed.
- G. M. Keating and A. Santoro, Drugs, 2009, 69, 223–240 CrossRef CAS PubMed.
- M. R. Hollingdale, Hepatology, 1985, 5, 327–335 CrossRef CAS PubMed.
- I. J. Reuling, G. M. de Jong, X. Z. Yap, M. Asghar, J. Walk, L. A. van de Schans, R. Koelewijn, A. Färnert, Q. de Mast, A. J. van der Ven, T. Bousema, J. J. van Hellemond, P. J. J. van Genderen and R. W. Sauerwein, EBioMedicine, 2018, 36, 131–139 CrossRef PubMed.
- R. Carrillo Esper, E. Pérez Bustos, S. Ornelas Arroyo, J. Albores Saavedra and M. Uribe, Ann. Hepatol., 2010, 9, 107–111 CrossRef PubMed.
- N. Pardi, M. J. Hogan, F. W. Porter and D. Weissman, Nat. Rev. Drug Discovery, 2018, 17, 261–279 CrossRef CAS PubMed.
- G. Lin, R. A. Revia and M. Zhang, Adv. Funct. Mater., 2021, 31, 2007096 CrossRef CAS PubMed.
- C. E. Dunbar, K. A. High, J. K. Joung, D. B. Kohn, K. Ozawa and M. Sadelain, Science, 2018, 359(6372), eaan4672 CrossRef PubMed.
- F. D. Bushman, Mol. Ther., 2020, 28, 352–356 CrossRef CAS PubMed.
- H. Bai, G. M. S. Lester, L. C. Petishnok and D. A. Dean, Biosci. Rep., 2017, 37, BSR20160616 CrossRef CAS PubMed.
- Y. Liang, L. Huang and T. Liu, Front. Bioeng. Biotechnol., 2021, 9, 658 Search PubMed.
- F. Kowalzik, D. Schreiner, C. Jensen, D. Teschner, S. Gehring and F. Zepp, Vaccines, 2021, 9, 390 CrossRef CAS PubMed.
- D. Magini, C. Giovani, S. Mangiavacchi, S. Maccari, R. Cecchi, J. B. Ulmer, E. D. Gregorio, A. J. Geall, M. Brazzoli and S. Bertholet, PLoS One, 2016, 11, e0161193 CrossRef PubMed.
- K. Lee, M. Kim, Y. Seo and H. Lee, Nano Res., 2018, 11, 5173–5192 CrossRef CAS.
- H. Niederholtmeyer, L. Xu and S. J. Maerkl, ACS Synth. Biol., 2013, 2, 411–417 CrossRef CAS PubMed.
- J. A. Wolff, R. W. Malone, P. Williams, W. Chong, G. Acsadi, A. Jani and P. L. Felgner, Science, 1990, 247, 1465–1468 CrossRef CAS PubMed.
- Z. R. Stephen, F. M. Kievit, O. Veiseh, P. A. Chiarelli, C. Fang, K. Wang, S. J. Hatzinger, R. G. Ellenbogen, J. R. Silber and M. Zhang, ACS Nano, 2014, 8, 10383–10395 CrossRef CAS PubMed.
- C. Sun, C. Fang, Z. Stephen, O. Veiseh, S. Hansen, D. Lee, R. G. Ellenbogen, J. Olson and M. Zhang, Nanomedicine, 2008, 3, 495–505 CrossRef CAS PubMed.
- P. Lönn, A. D. Kacsinta, X.-S. Cui, A. S. Hamil, M. Kaulich, K. Gogoi and S. F. Dowdy, Sci. Rep., 2016, 6, 32301 CrossRef PubMed.
- S. A. Smith, L. I. Selby, A. P. R. Johnston and G. K. Such, Bioconjugate Chem., 2019, 30, 263–272 CrossRef CAS PubMed.
- C. Sun, R. Sze and M. Zhang, J. Biomed. Mater. Res., Part A, 2006, 78A, 550–557 CrossRef CAS PubMed.
- M. Schmidt and A.-S. Heimes, Cancers, 2021, 13, 4883 CrossRef CAS PubMed.
- E. J. Crosby, C. R. Acharya, A.-F. Haddad, C. A. Rabiola, G. Lei, J.-P. Wei, X.-Y. Yang, T. Wang, C.-X. Liu, K. U. Wagner, W. J. Muller, L. A. Chodosh, G. Broadwater, T. Hyslop, J. H. Shepherd, D. P. Hollern, X. He, C. M. Perou, S. Chai, B. K. Ashby, B. G. Vincent, J. C. Snyder, J. Force, M. A. Morse, H. K. Lyerly and Z. C. Hartman, Clin. Cancer Res., 2020, 26, 4670–4681 CrossRef CAS PubMed.
- B. Weide, S. Pascolo, B. Scheel, E. Derhovanessian, A. Pflugfelder, T. K. Eigentler, G. Pawelec, I. Hoerr, H.-G. Rammensee and C. Garbe, J. Immunotherapy, 2009, 32, 498–507 CrossRef CAS PubMed.
- U. Sahin, P. Oehm, E. Derhovanessian, R. A. Jabulowsky, M. Vormehr, M. Gold, D. Maurus, D. Schwarck-Kokarakis, A. N. Kuhn, T. Omokoko, L. M. Kranz, M. Diken, S. Kreiter, H. Haas, S. Attig, R. Rae, K. Cuk, A. Kemmer-Brück, A. Breitkreuz, C. Tolliver, J. Caspar, J. Quinkhardt, L. Hebich, M. Stein, A. Hohberger, I. Vogler, I. Liebig, S. Renken, J. Sikorski, M. Leierer, V. Müller, H. Mitzel-Rink, M. Miederer, C. Huber, S. Grabbe, J. Utikal, A. Pinter, R. Kaufmann, J. C. Hassel, C. Loquai and Ö. Türeci, Nature, 2020, 585, 107–112 CrossRef CAS PubMed.
- ModernaTX, Inc., A Phase 2 Randomized Study of Adjuvant Immunotherapy With the Personalized Cancer Vaccine mRNA-4157 and Pembrolizumab Versus Pembrolizumab Alone After Complete Resection of High-Risk Melanoma (KEYNOTE-942), 2021, https://www.clinicaltrials.gov.
- H. Kübler, B. Scheel, U. Gnad-Vogt, K. Miller, W. Schultze-Seemann, F. vom Dorp, G. Parmiani, C. Hampel, S. Wedel, L. Trojan, D. Jocham, T. Maurer, G. Rippin, M. Fotin-Mleczek, F. von der Mülbe, J. Probst, I. Hoerr, K.-J. Kallen, T. Lander and A. Stenzl, J. Immunother. Cancer, 2015, 3, 26 CrossRef PubMed.
- M. Fotin-Mleczek, K. M. Duchardt, C. Lorenz, R. Pfeiffer, S. Ojkic-Zrna, J. Probst and K.-J. Kallen, J. Immunotherapy, 2011, 34, 1–15 CrossRef CAS PubMed.
- L. Gandhi, K. Aufiero Ramirez, P. Schwarzenberger, T. Ricciardi, M. J. Macri, A. Ryan and R. R. Venhaus, JCO, 2018, 36, TPS9107 Search PubMed.
- X. Zhan, B. Wang, Y. Wang, L. Chen, X. Peng, J. Li, M. Wu, L. Zhang and S. Tang, JCO, 2020, 38, e15269 Search PubMed.
- BioNTech SE, A Multi-site, Open-label, Phase II, Randomized, Controlled Trial to Compare the Efficacy of RO7198457 Versus Watchful Waiting in Resected, Stage II (High Risk) and Stage III Colorectal Cancer Patients Who Are ctDNA Positive Following Resection, 2021, https://www.clinicaltrials.gov.
- ModernaTX, Inc., A Phase 1/2, Open-Label, Multicenter, Dose Escalation and Efficacy Study of mRNA-2416, a Lipid Nanoparticle Encapsulated mRNA Encoding Human OX40L, for Intratumoral Injection Alone or in Combination With Durvalumab for Patients With Advanced Malignancies, 2021, https://www.clinicaltrials.gov.
- D. Srikrishna and K. Sachsenmeier, Genome Med., 2021, 13, 120 CrossRef PubMed.
- K. K. W. To and W. C. S. Cho, Expert Opin. Drug Discovery, 2021, 16, 1307–1317 CrossRef CAS PubMed.
- N. Z. Rouf, S. Biswas, N. Tarannum, L. M. Oishee and M. M. Muna, RNA Biol., 2022, 19, 386–410 CrossRef CAS PubMed.
- M. K. Franco and K. S. Koutmou, Biophys. Chem., 2022, 285, 106780 CrossRef CAS PubMed.
- L. Schoenmaker, D. Witzigmann, J. A. Kulkarni, R. Verbeke, G. Kersten, W. Jiskoot and D. J. A. Crommelin, Int. J. Pharm., 2021, 601, 120586 CrossRef CAS PubMed.
- S. C. Kim, S. S. Sekhon, W.-R. Shin, G. Ahn, B.-K. Cho, J.-Y. Ahn and Y.-H. Kim, Mol. Cell. Toxicol., 2022, 18, 1–8 CrossRef CAS PubMed.
- J. Houseley and D. Tollervey, Cell, 2009, 136, 763–776 CrossRef CAS PubMed.
- N. B. Tsui, E. K. Ng and Y. D. Lo, Clin. Chem., 2002, 48, 1647–1653 CrossRef CAS.
- T. Ura, K. Okuda and M. Shimada, Vaccines, 2014, 2, 624–641 CrossRef PubMed.
- S. Bobis-Wozowicz, M. Galla, J. Alzubi, J. Kuehle, C. Baum, A. Schambach and T. Cathomen, Sci. Rep., 2014, 4, 4656 CrossRef PubMed.
- Y. Zhitnyuk, P. Gee, M. S. Y. Lung, N. Sasakawa, H. Xu, H. Saito and A. Hotta, Biochem. Biophys. Res. Commun., 2018, 505, 1097–1102 CrossRef CAS PubMed.
- Z. Wang and X. Zhang, Cytotherapy, 2021, 23, 1045–1052 CrossRef CAS PubMed.
- J. L. Shirley, Y. P. de Jong, C. Terhorst and R. W. Herzog, Mol. Ther., 2020, 28, 709–722 CrossRef CAS PubMed.
- S. Deng, H. Liang, P. Chen, Y. Li, Z. Li, S. Fan, K. Wu, X. Li, W. Chen, Y. Qin, L. Yi and J. Chen, Microorganisms, 2022, 10, 1450 CrossRef CAS PubMed.
- M. Alhashimi, A. Elkashif, E. E. Sayedahmed and S. K. Mittal, Viruses, 2021, 13, 1493 CrossRef CAS PubMed.
- D. Blessing, G. Vachey, C. Pythoud, M. Rey, V. Padrun, F. M. Wurm, B. L. Schneider and N. Déglon, Mol. Ther. Methods Clin. Dev., 2018, 13, 14–26 CrossRef PubMed.
- J. J. Aponte-Ubillus, D. Barajas, J. Peltier, C. Bardliving, P. Shamlou and D. Gold, Biotechnol. Prog., 2019, 35, e2725 CrossRef PubMed.
- J. H. Kurasawa, A. Park, C. R. Sowers, R. A. Halpin, A. Tovchigrechko, C. L. Dobson, A. E. Schmelzer, C. Gao, S. D. Wilson and Y. Ikeda, Mol. Ther.--Methods Clin. Dev., 2020, 19, 330–340 CrossRef CAS PubMed.
- P. S. Kowalski, A. Rudra, L. Miao and D. G. Anderson, Mol. Ther., 2019, 27, 710–728 CrossRef CAS PubMed.
- G. Basha, T. I. Novobrantseva, N. Rosin, Y. Y. C. Tam, I. M. Hafez, M. K. Wong, T. Sugo, V. M. Ruda, J. Qin, B. Klebanov, M. Ciufolini, A. Akinc, Y. K. Tam, M. J. Hope and P. R. Cullis, Mol. Ther., 2011, 19, 2186–2200 CrossRef CAS PubMed.
- G. Sahay, W. Querbes, C. Alabi, A. Eltoukhy, S. Sarkar, C. Zurenko, E. Karagiannis, K. Love, D. Chen, R. Zoncu, Y. Buganim, A. Schroeder, R. Langer and D. G. Anderson, Nat. Biotechnol., 2013, 31, 653–658 CrossRef CAS PubMed.
- T. Yang, C. Li, X. Wang, D. Zhao, M. Zhang, H. Cao, Z. Liang, H. Xiao, X.-J. Liang, Y. Weng and Y. Huang, Bioactive Mater., 2020, 5, 1053–1061 CrossRef PubMed.
- D. Hirsch-Lerner, M. Zhang, H. Eliyahu, M. E. Ferrari, C. J. Wheeler and Y. Barenholz, Biochim. Biophys. Acta, Biomembr., 2005, 1714, 71–84 CrossRef CAS PubMed.
- J.
![[S with combining breve]](https://www.rsc.org/images/entities/char_0053_0306.gif) misterová, A. Wagenaar, M. C. A. Stuart, E. Polushkin, G. ten Brinke, R. Hulst, J. B. F. N. Engberts and D. Hoekstra, J. Biol. Chem., 2001, 276, 47615–47622 CrossRef PubMed.
misterová, A. Wagenaar, M. C. A. Stuart, E. Polushkin, G. ten Brinke, R. Hulst, J. B. F. N. Engberts and D. Hoekstra, J. Biol. Chem., 2001, 276, 47615–47622 CrossRef PubMed. - V. Kumar, J. Qin, Y. Jiang, R. G. Duncan, B. Brigham, S. Fishman, J. K. Nair, A. Akinc, S. A. Barros and P. V. Kasperkovitz, Mol. Ther.--Nucleic Acids, 2014, 3, e210 CrossRef PubMed.
- D. E. Large, R. G. Abdelmessih, E. A. Fink and D. T. Auguste, Adv. Drug Delivery Rev., 2021, 176, 113851 CrossRef CAS PubMed.
- M. C. Woodle, Adv. Drug Delivery Rev., 1995, 16, 249–265 CrossRef CAS.
- B. L. Mui, Y. K. Tam, M. Jayaraman, S. M. Ansell, X. Du, Y. Y. C. Tam, P. J. Lin, S. Chen, J. K. Narayanannair, K. G. Rajeev, M. Manoharan, A. Akinc, M. A. Maier, P. Cullis, T. D. Madden and M. J. Hope, Mol. Ther.--Nucleic Acids, 2013, 2, e139 CrossRef CAS PubMed.
- M. A. Oberli, A. M. Reichmuth, J. R. Dorkin, M. J. Mitchell, O. S. Fenton, A. Jaklenec, D. G. Anderson, R. Langer and D. Blankschtein, Nano Lett., 2017, 17, 1326–1335 CrossRef CAS PubMed.
- M. M. Billingsley, N. Singh, P. Ravikumar, R. Zhang, C. H. June and M. J. Mitchell, Nano Lett., 2020, 20, 1578–1589 CrossRef CAS PubMed.
- H. Zhang, J. Leal, M. R. Soto, H. D. C. Smyth and D. Ghosh, Pharmaceutics, 2020, 12, 1042 CrossRef CAS PubMed.
- S. Patel, N. Ashwanikumar, E. Robinson, A. DuRoss, C. Sun, K. E. Murphy-Benenato, C. Mihai, Ö. Almarsson and G. Sahay, Nano Lett., 2017, 17, 5711–5718 CrossRef CAS PubMed.
- S. Tahtinen, A.-J. Tong, P. Himmels, J. Oh, A. Paler-Martinez, L. Kim, S. Wichner, Y. Oei, M. J. McCarron, E. C. Freund, Z. A. Amir, C. C. de la Cruz, B. Haley, C. Blanchette, J. M. Schartner, W. Ye, M. Yadav, U. Sahin, L. Delamarre and I. Mellman, Nat. Immunol., 2022, 23, 532–542 CrossRef CAS PubMed.
- H. Parhiz, J. S. Brenner, P. N. Patel, T. E. Papp, H. Shahnawaz, Q. Li, R. Shi, M. E. Zamora, A. Yadegari, O. A. Marcos-Contreras, A. Natesan, N. Pardi, V. V. Shuvaev, R. Kiseleva, J. W. Myerson, T. Uhler, R. S. Riley, X. Han, M. J. Mitchell, K. Lam, J. Heyes, D. Weissman and V. R. Muzykantov, J. Controlled Release, 2022, 344, 50–61 CrossRef CAS PubMed.
- D. Ulkoski, M. J. Munson, M. E. Jacobson, C. R. Palmer, C. S. Carson, A. Sabirsh, J. T. Wilson and V. R. Krishnamurthy, ACS Appl. Bio Mater., 2021, 4, 1640–1654 CrossRef CAS PubMed.
- Y. Jiang, A. Gaudin, J. Zhang, T. Agarwal, E. Song, A. C. Kauffman, G. T. Tietjen, Y. Wang, Z. Jiang, C. J. Cheng and W. M. Saltzman, Biomaterials, 2018, 176, 122–130 CrossRef CAS PubMed.
- Y. Jiang, Q. Lu, Y. Wang, E. Xu, A. Ho, P. Singh, Y. Wang, Z. Jiang, F. Yang, G. T. Tietjen, P. Cresswell and W. M. Saltzman, Nano Lett., 2020, 20, 1117–1123 CrossRef CAS PubMed.
- J. I. Solomun, G. Cinar, P. Mapfumo, F. Richter, E. Moek, F. Hausig, L. Martin, S. Hoeppener, I. Nischang and A. Traeger, Int. J. Pharm., 2021, 593, 120080 CrossRef CAS PubMed.
- T. Miyazaki, S. Uchida, H. Hatano, Y. Miyahara, A. Matsumoto and H. Cabral, Eur. Polym. J., 2020, 140, 110028 CrossRef CAS.
- J. C. Kaczmarek, A. K. Patel, L. H. Rhym, U. C. Palmiero, B. Bhat, M. W. Heartlein, F. DeRosa and D. G. Anderson, Biomaterials, 2021, 275, 120966 CrossRef CAS PubMed.
- H. Yasar, A. Biehl, C. De Rossi, M. Koch, X. Murgia, B. Loretz and C.-M. Lehr, J. Nanobiotechnol., 2018, 16, 72 CrossRef PubMed.
- C. Lacroix, A. Humanes, C. Coiffier, D. Gigmes, B. Verrier and T. Trimaille, Pharm. Res., 2020, 37, 30 CrossRef CAS PubMed.
- J. Li, W. Wang, Y. He, Y. Li, E. Z. Yan, K. Zhang, D. J. Irvine and P. T. Hammond, ACS Nano, 2017, 11, 2531–2544 CrossRef CAS PubMed.
- J. Li, Y. He, W. Wang, C. Wu, C. Hong and P. T. Hammond, Angew. Chem., Int. Ed., 2017, 56, 13709–13712 CrossRef CAS PubMed.
- X. Yu, S. Liu, Q. Cheng, T. Wei, S. Lee, D. Zhang and D. J. Siegwart, Adv. Healthcare Mater., 2020, 9, 1901487 CrossRef CAS PubMed.
- H. Wang, Q. Mu, K. Wang, R. A. Revia, C. Yen, X. Gu, B. Tian, J. Liu and M. Zhang, Appl. Mater. Today, 2019, 14, 108–117 CrossRef PubMed.
- H. Wang, R. Revia, K. Wang, R. J. Kant, Q. Mu, Z. Gai, K. Hong and M. Zhang, Adv. Mater., 2017, 29, 1605416 CrossRef PubMed.
- Y. Liu, C. Zhao, A. Sabirsh, L. Ye, X. Wu, H. Lu and J. Liu, ChemistryOpen, 2021, 10, 666–671 CrossRef CAS PubMed.
- S. R. Z. Abdel-Misih and M. Bloomston, Surg. Clin. North Am., 2010, 90, 643–653 CrossRef PubMed.
- E. Trefts, M. Gannon and D. H. Wasserman, Curr. Biol., 2017, 27, R1147–R1151 CrossRef CAS PubMed.
- A. Kalra, E. Yetiskul, C. J. Wehrle and F. Tuma, StatPearls, StatPearls Publishing, Treasure Island (FL), 2021 Search PubMed.
- D. P. Bogdanos, B. Gao and M. E. Gershwin, Compr. Physiol., 2013, 3, 567–598 Search PubMed.
- I. N. Crispe, Annu. Rev. Immunol., 2009, 27, 147–163 CrossRef CAS PubMed.
- L. Gu, F. Zhang, J. Wu and Y. Zhuge, Front. Mol. Biosci, 2022, 8, 804396 CrossRef PubMed.
- Q. Zheng, F. Qin, R. Luo, C. Jin, H. Huang, H. Xi, W. Xiao, M. Guo, S. Yang, S. He, L. Cheng, N. Fan, S. Yao and X. Song, Adv. Funct. Mater., 2021, 31, 2011068 CrossRef CAS.
- M. Gao and D. Liu, Sci. Rep., 2019, 9, 13427 CrossRef PubMed.
- K. Xu, J. Sun, S. Chen, Y. Li, X. Peng, M. Li and Y. Li, Biochem. Biophys. Res. Commun., 2019, 508, 198–202 CrossRef CAS PubMed.
- X.-F. Yang, H.-Y. Wang, W.-L. Lu, W. Ma, H. Zhang and F.-R. Li, Am. J. Transl. Res., 2020, 12, 7275–7286 CAS.
- H. Dai, X. Jiang, G. C. Tan, Y. Chen, M. Torbenson, K. W. Leong and H.-Q. Mao, Int. J. Nanomed., 2006, 1, 507–522 CrossRef CAS PubMed.
- R. L. Kruse, Y. Huang, T. Shum, L. Bai, H. Ding, Z. Z. Wang, F. M. Selaru and V. Kumbhari, Gastrointest. Endosc., 2021, 94, 1119–1130.e4 CrossRef PubMed.
- B. Gao, Cell Mol. Immunol., 2016, 13, 265–266 CrossRef CAS PubMed.
- C. Selmi, I. R. Mackay and M. E. Gershwin, Semin. Liver Dis., 2007, 27, 129–139 CrossRef CAS PubMed.
- H. Kita, J. Van De Water, M. E. Gershwin and I. R. Mackay, Gastroenterology, 2001, 120, 1485–1501 CrossRef CAS PubMed.
- Y. Wang and C. Zhang, Front. Immunol., 2019, 10, 1582 CrossRef CAS PubMed.
- Y. C. Lee, L. Lu, F. Fu, W. Li, A. W. Thomson, J. J. Fung and S. Qian, Transplant. Proc., 1999, 31, 784 CrossRef CAS PubMed.
- P. Bertolino, M.-C. Trescol-Biémont and C. Rabourdin-Combe, Eur. J. Immunol., 1998, 28, 221–236 CrossRef CAS PubMed.
- S. Qian, L. Lu, F. Fu, Y. Li, W. Li, T. E. Starzl, J. J. Fung and A. W. Thomson, J. Immunol., 1997, 158, 4654–4661 CAS.
- P. Bertolino, D. G. Bowen, G. W. McCaughan and B. F. de S. Groth, J. Immunol., 2001, 166, 5430–5438 CrossRef CAS PubMed.
- F. Geissmann, T. O. Cameron, S. Sidobre, N. Manlongat, M. Kronenberg, M. J. Briskin, M. L. Dustin and D. R. Littman, PLoS Biol., 2005, 3, e113 CrossRef PubMed.
- E. Germanov, L. Veinotte, R. Cullen, E. Chamberlain, E. C. Butcher and B. Johnston, J. Immunol., 2008, 181, 81–91 CrossRef CAS PubMed.
- A. Wehr, C. Baeck, F. Heymann, P. M. Niemietz, L. Hammerich, C. Martin, H. W. Zimmermann, O. Pack, N. Gassler, K. Hittatiya, A. Ludwig, T. Luedde, C. Trautwein and F. Tacke, J. Immunol., 2013, 190, 5226–5236 CrossRef CAS PubMed.
- N. H. Shoukry, A. Grakoui, M. Houghton, D. Y. Chien, J. Ghrayeb, K. A. Reimann and C. M. Walker, J. Exp. Med., 2003, 197, 1645–1655 CrossRef CAS PubMed.
- C. J. Lim, Y. H. Lee, L. Pan, L. Lai, C. Chua, M. Wasser, T. K. H. Lim, J. Yeong, H. C. Toh, S. Y. Lee, C. Y. Chan, B. K. Goh, A. Chung, M. Heikenwälder, I. O. Ng, P. Chow, S. Albani and V. Chew, Gut, 2019, 68, 916–927 CrossRef CAS PubMed.
- M. Luo, M. P. Ballester, U. Soffientini, R. Jalan and G. Mehta, Hepatol. Int., 2022, 16, 755–774 CrossRef PubMed.
- L. Xu, J. Liu, M. Lu, D. Yang and X. Zheng, Liver Int., 2020, 40, 998–1004 CrossRef CAS PubMed.
- P. D. Krueger, S. Narayanan, F. A. Surette, M. G. Brown, S.-S. J. Sung and Y. S. Hahn, J. Leukocyte Biol., 2017, 101, 329–338 CrossRef CAS PubMed.
- C. Harmon, M. W. Robinson, R. Fahey, S. Whelan, D. D. Houlihan, J. Geoghegan and C. O’Farrelly, Eur. J. Immunol., 2016, 46, 2111–2120 CrossRef CAS PubMed.
- K. Kakimi, L. G. Guidotti, Y. Koezuka and F. V. Chisari, J. Exp. Med., 2000, 192, 921–930 CrossRef CAS PubMed.
- D. Fernandez-Ruiz, W. Y. Ng, L. E. Holz, J. Z. Ma, A. Zaid, Y. C. Wong, L. S. Lau, V. Mollard, A. Cozijnsen, N. Collins, J. Li, G. M. Davey, Y. Kato, S. Devi, R. Skandari, M. Pauley, J. H. Manton, D. I. Godfrey, A. Braun, S. S. Tay, P. S. Tan, D. G. Bowen, F. Koch-Nolte, B. Rissiek, F. R. Carbone, B. S. Crabb, M. Lahoud, I. A. Cockburn, S. N. Mueller, P. Bertolino, G. I. McFadden, I. Caminschi and W. R. Heath, Immunity, 2016, 45, 889–902 CrossRef CAS PubMed.
- A. S. Ishizuka, K. E. Lyke, A. DeZure, A. A. Berry, T. L. Richie, F. H. Mendoza, M. E. Enama, I. J. Gordon, L.-J. Chang, U. N. Sarwar, K. L. Zephir, L. A. Holman, E. R. James, P. F. Billingsley, A. Gunasekera, S. Chakravarty, A. Manoj, M. Li, A. J. Ruben, T. Li, A. G. Eappen, R. E. Stafford, N. K. C. T. Murshedkar, H. DeCederfelt, S. H. Plummer, C. S. Hendel, L. Novik, P. J. M. Costner, J. G. Saunders, M. B. Laurens, C. V. Plowe, B. Flynn, W. R. Whalen, J. P. Todd, J. Noor, S. Rao, K. Sierra-Davidson, G. M. Lynn, J. E. Epstein, M. A. Kemp, G. A. Fahle, S. A. Mikolajczak, M. Fishbaugher, B. K. Sack, S. H. I. Kappe, S. A. Davidson, L. S. Garver, N. K. Björkström, M. C. Nason, B. S. Graham, M. Roederer, B. K. L. Sim, S. L. Hoffman, J. E. Ledgerwood and R. A. Seder, Nat. Med., 2016, 22, 614–623 CrossRef CAS PubMed.
- N. J. W. Easom, K. A. Stegmann, L. Swadling, L. J. Pallett, A. R. Burton, D. Odera, N. Schmidt, W.-C. Huang, G. Fusai, B. Davidson and M. K. Maini, Front. Immunol., 2018, 9, 1009 CrossRef PubMed.
- C. Ma, M. Han, B. Heinrich, Q. Fu, Q. Zhang, M. Sandhu, D. Agdashian, M. Terabe, J. A. Berzofsky, V. Fako, T. Ritz, T. Longerich, C. M. Theriot, J. A. McCulloch, S. Roy, W. Yuan, V. Thovarai, S. K. Sen, M. Ruchirawat, F. Korangy, X. W. Wang, G. Trinchieri and T. F. Greten, Science, 2018, 360, eaan5931 CrossRef PubMed.
- A. Ventura, D. G. Kirsch, M. E. McLaughlin, D. A. Tuveson, J. Grimm, L. Lintault, J. Newman, E. E. Reczek, R. Weissleder and T. Jacks, Nature, 2007, 445(7128), 661–665 CrossRef CAS PubMed.
- N. Kong, W. Tao, X. Ling, J. Wang, Y. Xiao, S. Shi, X. Ji, A. Shajii, S. T. Gan, N. Y. Kim, D. G. Duda, T. Xie, O. C. Farokhzad and J. Shi, Sci. Transl. Med., 2019, 11(523), eaaw1565 CrossRef CAS PubMed.
- C. Zhang, X. Zhang, W. Zhao, C. Zeng, W. Li, B. Li, X. Luo, J. Li, J. Jiang, B. Deng, D. W. McComb and Y. Dong, Nano Res., 2019, 12(4), 855–861 CrossRef CAS PubMed.
- L. Rosich, S. Xargay-Torrent, M. López-Guerra, E. Campo, D. Colomer and G. Roué, Clin. Cancer Res., 2012, 18, 5278–5289 CrossRef CAS PubMed.
- R. Jain, J. P. Frederick, E. Y. Huang, K. E. Burke, D. M. Mauger, E. A. Andrianova, S. J. Farlow, S. Siddiqui, J. Pimentel, K. Cheung-Ong, K. M. McKinney, C. Köhrer, M. J. Moore and T. Chakraborty, Nucleic Acid Ther., 2018, 28, 285–296 CrossRef CAS PubMed.
- E. J. Sontheimer, Nat. Rev. Mol. Cell Biol., 2005, 6(2), 127–138 CrossRef CAS PubMed.
- Y. Wang, K. Tiruthani, S. Li, M. Hu, G. Zhong, Y. Tang, S. Roy, L. Zhang, J. Tan, C. Liao and R. Liu, Adv. Mater., 2021, 33, 2007603 CrossRef CAS PubMed.
- I. Lai, S. Swaminathan, V. Baylot, A. Mosley, R. Dhanasekaran, M. Gabay and D. W. Felsher, J. Immunother. Cancer, 2018, 6, 1–11 Search PubMed.
- Q. Wang, F. Cheng, T. Ma, H.-Y. Xiong, Z.-W. Li, C.-L. Xie, C.-Y. Liu and Z.-G. Tu, Mol. Cell. Biochem., 2016, 415(1), 157–168 CrossRef CAS PubMed.
- F. Tada, M. Abe, M. Hirooka, Y. Ikeda, Y. Hiasa, Y. Lee, N.-C. Jung, W.-B. Lee, H.-S. Lee, Y.-S. Bae and M. Onji, Int. J. Oncol., 2012, 41, 1601–1609 CrossRef CAS PubMed.
- L. Buonaguro and M. Tagliamonte, Vaccines, 2020, 8, 615 CrossRef CAS PubMed.
- H. Yang, L. Sun, A. Guan, H. Yin, M. Liu, X. Mao, H. Xu, H. Zhao, X. Lu, X. Sang, S. Zhong, Q. Chen and Y. Mao, Cancer Immunol. Immunother., 2020, 70(3), 667–677 CrossRef PubMed.
- P. Chen, Q.-X. Fang, D.-B. Chen and H.-S. Chen, World J. Gastrointestinal Oncology, 2021, 13, 673 CrossRef PubMed.
- L. Lu, J. Jiang, M. Zhan, H. Zhang, Q.-T. Wang, S.-N. Sun, X.-K. Guo, H. Yin, Y. Wei, S.-Y. Li, J. O. Liu, Y. Li and Y.-W. He, Hepatology, 2021, 73, 821–832 CrossRef PubMed.
- E. Gilboa and J. Vieweg, Immunol. Rev., 2004, 199, 251–263 CrossRef CAS PubMed.
- M. Sebastian, A. Schröder, B. Scheel, H. S. Hong, A. Muth, L. von Boehmer, A. Zippelius, F. Mayer, M. Reck, D. Atanackovic, M. Thomas, F. Schneller, J. Stöhlmacher, H. Bernhard, A. Gröschel, T. Lander, J. Probst, T. Strack, V. Wiegand, U. Gnad-Vogt, K.-J. Kallen, I. Hoerr, F. von der Muelbe, M. Fotin-Mleczek, A. Knuth and S. D. Koch, Cancer Immunol. Immunother., 2019, 68(5), 799–812 CrossRef CAS PubMed.
- Z. Sharifnia, M. Bandehpour, B. Kazemi and N. Zarghami, Iran Biomed. J., 2019, 23, 57–67 CrossRef PubMed.
- G. Cafri, J. J. Gartner, T. Zaks, K. Hopson, N. Levin, B. C. Paria, M. R. Parkhurst, R. Yossef, F. J. Lowery, M. S. Jafferji, T. D. Prickett, S. L. Goff, C. T. McGowan, S. Seitter, M. L. Shindorf, A. Parikh, P. D. Chatani, P. F. Robbins and S. A. Rosenberg, J. Clin. Invest., 2020, 130, 5976–5988 CrossRef CAS PubMed.
- Y.-N. Fan, M. Li, Y.-L. Luo, Q. Chen, L. Wang, H.-B. Zhang, S. Shen, Z. Gu and J. Wang, Biomater. Sci., 2018, 6, 3009–3018 RSC.
- M. A. Islam, J. Rice, E. Reesor, H. Zope, W. Tao, M. Lim, J. Ding, Y. Chen, D. Aduluso, B. R. Zetter, O. C. Farokhzad and J. Shi, Biomaterials, 2021, 266, 120431 CrossRef CAS PubMed.
- R. Verbeke, I. Lentacker, L. Wayteck, K. Breckpot, M. Van Bockstal, B. Descamps, C. Vanhove, S. C. De Smedt and H. Dewitte, J. Controlled Release, 2017, 266, 287–300 CrossRef CAS PubMed.
- K. Lee, S. Young Kim, Y. Seo, M. Hee Kim, J. Chang and H. Lee, Biomater. Sci., 2020, 8, 1101–1105 RSC.
- A. De Beuckelaer, C. Pollard, S. Van Lint, K. Roose, L. Van Hoecke, T. Naessens, V. K. Udhayakumar, M. Smet, N. Sanders, S. Lienenklaus, X. Saelens, S. Weiss, G. Vanham, J. Grooten and S. De Koker, Mol. Ther., 2016, 24, 2012–2020 CrossRef CAS PubMed.
- A. Vitiello and F. Ferrara, Inflammopharmacology, 2021, 29, 645–649 CrossRef CAS PubMed.
- J. W. Park, P. N. P. Lagniton, Y. Liu and R.-H. Xu, Int. J. Biol. Sci., 2021, 17, 1446–1460 CrossRef CAS PubMed.
- P. Anand and V. P. Stahel, Patient Saf. Surg., 2021, 15, 1–9 CrossRef PubMed.
- S. P. Teo, J. Pharmacy Practice, 2021, 08971900211009650 Search PubMed.
- N. Papic, A. Pangercic, M. Vargovic, B. Barsic, A. Vince and I. Kuzman, Influenza Other Respir. Viruses, 2012, 6, e2–e5 CrossRef PubMed.
- A. Schütte, S. Ciesek, H. Wedemeyer and C. M. Lange, J. Hepatol., 2019, 70, 797–799 CrossRef PubMed.
- J. R. Whitworth, C. L. Mack, J. A. O’Connor, M. R. Narkewicz, S. Mengshol and R. J. Sokol, J. Pediatr. Gastroenterol. Nutr., 2006, 43, 536–538 CrossRef PubMed.
- B. Petsch, M. Schnee, A. B. Vogel, E. Lange, B. Hoffmann, D. Voss, T. Schlake, A. Thess, K.-J. Kallen, L. Stitz and T. Kramps, Nat. Biotechnol., 2012, 30, 1210–1216 CrossRef CAS PubMed.
- A. B. Vogel, L. Lambert, E. Kinnear, D. Busse, S. Erbar, K. C. Reuter, L. Wicke, M. Perkovic, T. Beissert, H. Haas, S. T. Reece, U. Sahin and J. S. Tregoning, Mol. Ther., 2018, 26, 446–455 CrossRef CAS PubMed.
- P. T. Joe, I. Christopoulou, L. van Hoecke, B. Schepens, T. Ysenbaert, C. Heirman, K. Thielemans, X. Saelens and J. L. Aerts, J. Transl. Med., 2019, 17, 242 CrossRef PubMed.
- A. W. Freyn, J. Ramos da Silva, V. C. Rosado, C. M. Bliss, M. Pine, B. L. Mui, Y. K. Tam, T. D. Madden, L. C. de Souza Ferreira, D. Weissman, F. Krammer, L. Coughlan, P. Palese, N. Pardi and R. Nachbagauer, Mol. Ther., 2020, 28, 1569–1584 CrossRef CAS PubMed.
- R. A. Feldman, R. Fuhr, I. Smolenov, A. (Mick) Ribeiro, L. Panther, M. Watson, J. J. Senn, M. Smith, Ö. Almarsson, H. S. Pujar, M. E. Laska, J. Thompson, T. Zaks and G. Ciaramella, Vaccine, 2019, 37, 3326–3334 CrossRef CAS PubMed.
- E. V. Starostina, S. V. Sharabrin, D. N. Antropov, G. A. Stepanov, G. Y. Shevelev, A. E. Lemza, A. P. Rudometov, M. B. Borgoyakova, N. B. Rudometova, V. Y. Marchenko, N. V. Danilchenko, A. N. Chikaev, S. I. Bazhan, A. A. Ilyichev and L. I. Karpenko, Vaccines, 2021, 9, 452 CrossRef CAS PubMed.
- A. W. Freyn, M. Pine, V. C. Rosado, M. Benz, H. Muramatsu, M. Beattie, Y. K. Tam, F. Krammer, P. Palese, R. Nachbagauer, M. McMahon and N. Pardi, Mol. Ther.--Methods Clin. Dev., 2021, 22, 84–95 CrossRef CAS PubMed.
- S. Fernando, A. Wijewickrama, L. Gomes, C. T. Punchihewa, S. D. P. Madusanka, H. Dissanayake, C. Jeewandara, H. Peiris, G. S. Ogg and G. N. Malavige, BMC Infect. Dis., 2016, 16, 319 CrossRef PubMed.
- J. Samanta and V. Sharma, World J. Clin. Cases, 2015, 3, 125–131 CrossRef PubMed.
- C. Roth, T. Cantaert, C. Colas, M. Prot, I. Casadémont, L. Levillayer, J. Thalmensi, P. Langlade-Demoyen, C. Gerke, K. Bahl, G. Ciaramella, E. Simon-Loriere and A. Sakuntabhai, Front. Immunol., 2019, 10, 1424 CrossRef CAS PubMed.
- M. Zhang, J. Sun, M. Li and X. Jin, Mol. Ther.--Methods Clin. Dev., 2020, 18, 702–712 CrossRef CAS PubMed.
- C. J. Wollner, M. Richner, M. A. Hassert, A. K. Pinto, J. D. Brien and J. M. Richner, J. Virology, 2021, 95, e02482–20 CrossRef CAS PubMed.
- T. Da Cunha and G. Y. Wu, J. Clin. Transl. Hepatol., 2021, 9, 106–115 Search PubMed.
- C. S. Nelson, J. A. Jenks, N. Pardi, M. Goodwin, H. Roark, W. Edwards, J. S. McLellan, J. Pollara, D. Weissman and S. R. Permar, J. Virology, 2020, 94, e00186–20 CAS.
- S. John, O. Yuzhakov, A. Woods, J. Deterling, K. Hassett, C. A. Shaw and G. Ciaramella, Vaccine, 2018, 36, 1689–1699 CrossRef CAS PubMed.
- H. Yu, L. A. Babiuk and S. van Drunen Littel-van den Hurk, Vaccine, 2007, 25, 1701–1711 CrossRef CAS PubMed.
- P. Viriyavejakul, V. Khachonsaksumet and C. Punsawad, Malar. J., 2014, 13, 106 CrossRef PubMed.
- Y. Sato, S. Ries, W. Stenzel, S. Fillatreau and K. Matuschewski, Front. Immunol., 2019, 10, 2554 CrossRef CAS PubMed.
- A. Baeza Garcia, E. Siu, T. Sun, V. Exler, L. Brito, A. Hekele, G. Otten, K. Augustijn, C. J. Janse, J. B. Ulmer, J. Bernhagen, E. Fikrig, A. Geall and R. Bucala, Nat. Commun., 2018, 9, 2714 CrossRef PubMed.
- K. L. Mallory, J. A. Taylor, X. Zou, I. N. Waghela, C. G. Schneider, M. Q. Sibilo, N. M. Punde, L. C. Perazzo, T. Savransky, M. Sedegah, S. Dutta, C. J. Janse, N. Pardi, P. J. C. Lin, Y. K. Tam, D. Weissman and E. Angov, npj Vaccines, 2021, 6, 1–12 CrossRef PubMed.
- S. Dias, M. Boroni, E. Rocha, T. Lüscher-Dias, D. Laet-Souza, F. Oliveira, M. Bitar, A. Macedo, C. Machado, M. Caliari and G. Franco, Front. Genetics, 2014, 5, 174 Search PubMed.
- R. E. Ridi and H. Tallima, Vaccine, 2009, 27, 666–673 CrossRef PubMed.
- D. J. A. Crommelin, T. J. Anchordoquy, D. B. Volkin, W. Jiskoot and E. Mastrobattista, J. Pharm. Sci., 2021, 110, 997–1001 CrossRef CAS PubMed.
- M. N. Uddin and M. A. Roni, Vaccines, 2021, 9, 1033 CrossRef CAS PubMed.
- P. Zhao, X. Hou, J. Yan, S. Du, Y. Xue, W. Li, G. Xiang and Y. Dong, Bioactive Mater., 2020, 5, 358–363 CrossRef PubMed.
- W. Abdelwahed, G. Degobert, S. Stainmesse and H. Fessi, Adv. Drug Delivery Rev., 2006, 58, 1688–1713 CrossRef CAS PubMed.
- A.-L. Fabre, M. Colotte, A. Luis, S. Tuffet and J. Bonnet, Eur. J. Hum. Genet., 2014, 22, 379–385 CAS.
| This journal is © The Royal Society of Chemistry 2023 |