
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAssessing the impact of ultra-thin diamond nanothreads on the glass transition temperature of a bituminous binder†
Yingying
Pang
a,
Liangfeng
Sun
bc,
Haifei
Zhan
 *abd,
Xianglong
Zheng
be,
Jiandong
Zhang
a,
Chengyou
Bian
f and
Chaofeng
Lü
*ga
*abd,
Xianglong
Zheng
be,
Jiandong
Zhang
a,
Chengyou
Bian
f and
Chaofeng
Lü
*ga
aCollege of Civil Engineering and Architecture, Zhejiang University, Hangzhou 310058, P. R. China. E-mail: zhan_haifei@zju.edu.cn; Tel: +86 571 8898 1940
bCenter for Balance Architecture, Zhejiang University, Hangzhou 310028, P. R. China
cArchitectural Design and Research Institute of Zhejiang University Co., Ltd, Hangzhou 310028, China
dSchool of Mechanical, Medical and Process Engineering, Queensland University of Technology (QUT), Brisbane, QLD 4001, Australia
eFaculty of Engineering, Hangzhou City University, Hangzhou 310015, P. R. China
fZCCC Hongtu Transportation Construction Co., Ltd, Hangzhou 310051, P. R. China
gFaculty of Mechanical Engineering & Mechanics, Ningbo University, Ningbo 315211, P. R. China. E-mail: lucf@nbu.edu.cn; Tel: +86 571 8820 8473
First published on 31st October 2023
Abstract
Low-temperature cracking and rutting are the most destructive problems of bitumen that hinder the application of high-performance bitumen engineering, which is dependent on its glass transition temperature (Tg). Through in silico studies, this work has systematically investigated the Tg of a bituminous binder with the addition of diamond nanothread (DNT) fillers with varying filler content, alignment, distribution, and functional groups. In general, the glass transition phenomenon of the bitumen is determined by the mobility of its constituent molecules. Tg is found to increase gradually with the increase in the weight percentage of DNT and then decreases when the weight percentage exceeds 5.05 wt%. The enhancement effect on Tg is weakened when DNTs are distributed vertically or functionalized with functional groups. Specifically, DNT fillers induce inhomogeneity, which promotes the motion of small molecules while hindering the motion of large molecules. The aggregation of DNTs and the molecular environment in the vicinity of DNTs directly affect Tg. In summary, aggregation and adhesion are the dominant mechanisms affecting the mobility of the constituent molecules in the DNT/bitumen system and thus its glass transition temperature. This work provides in-depth insights into the underlying mechanisms for the glass transition of a bituminous binder, which could serve as theoretical guidance for tuning the low-temperature performance of the bituminous binder.
1. Introduction
Bituminous binder is an essential thermal material in engineering applications, with an estimated global yearly consumption of approximately 74 million ton in pavement projects.1 However, it usually suffers from temperature-dependent problems, such as cracking, rutting, and low durability.2–4 At higher temperatures, the bituminous binder becomes flexible and susceptible to rutting, while at low temperatures, it becomes susceptible to cracking that can cause secondary damage, which exacerbates in the glassy state at the glass transition temperature.5–7 Therefore, bituminous binder requires good low-temperature performance and relaxation capacity to prevent damage, especially in winters and cold regions.8In this regard, the glass transition temperature (Tg) of bitumen can serve as an indicator to evaluate its low-temperature performance.9Tg is a critical parameter indicating the transition status from the rubbery state to the glassy state.10 Understanding Tg can provide valuable insights into the material design and control measures for preventing low-temperature damage in bitumen. Various additive modification technologies have been applied to decrease Tg and extend the properties of pavements in colder climates. Among the suggested additives, nanomaterials have attracted significant attention for improving bitumen pavements due to their higher specific surface area and strong binding capability with bitumen.11 They account for more than 10% of the entire modified bitumen market.12 Carbon-based nanomaterials, such as carbon black, carbon nanotubes (CNT), fullerenes, and graphene sheets, are particularly popular additives for a bituminous binder.7,13–19 Literature reports have highlighted the enhancements in the performance of bituminous binders achieved by incorporating these nano-additives using various techniques.
Through the solid-state reaction of benzene under high pressure, researchers have reported the successful synthesis of an ultra-thin one-dimensional carbon nanostructure, the diamond nanothread (DNT),20 which is an sp3-bonded carbon structure. Previous work has demonstrated its excellent stiffness (850 GPa), bending rigidity (5.35 × 10−28 N m2), and enhancement effects on the mechanical strength of polymers.21 Compared to sp2 carbon nanotubes, DNTs exhibit resonance quality factors that are twice as high.22 Their hydrogenated surface provides the functionalization opportunity without introducing defects to their backbone structure,23 making them the most suitable for applications in nanoscale synthesized materials.20,24 Further studies have reported their unique thermal transport properties that are promising for thermal management applications.25 In particular, DNT is regarded as a promising reinforcer for nanocomposites due to its good interfacial interactions and mechanical interlocking effect with the polymer matrix.20,26–28 An earlier report showed that DNT can significantly increase the Tg of poly(methyl methacrylate) composites.29 To this end, in this study, we have assessed the Tg of bituminous binder with the DNT as a new type of nano-additive.
There are different experimental methods for exploring the Tg of materials, such as differential scanning calorimetry (DSC)30,31 and dilatometry.32 Although these methods can provide a good measurement of Tg, they are unable to provide underlying insights.33,34 Bitumen binder is a complex matter that contains millions of different molecules and thus it is a significant challenge to determine the role of different molecules from experiments.35 Hindered by the complexities and challenges, the understanding of bitumen's glass transition process is ambiguous at the molecular level,36–38 and the detailed mechanism for the aggregation and adhesion behaviors between bitumen mixtures and nanofillers can hardly be assessed through experiments. In this regard, molecular dynamics (MD) simulation is an effective and feasible approach to exploring the microscopic mechanisms of molecules at varying temperatures.
With the aid of MD simulations, this work has adopted a four-component bituminous binder model to investigate in-depth the microscopic behaviors during the glass transition process. By monitoring the movements of different molecules, the glass transition process can be identified. Various models were constructed by varying the dispersion pattern and the weight percentage of DNTs and functional groups to systematically examine the influence of the DNT fillers. It was found that aggregation and adhesion are the dominant mechanisms affecting the mobility of the constituent molecules in the DNT/bitumen system, and thus its glass transition temperature. The findings in this work could provide theoretical guidelines for selecting nano-additives for high-performance bituminous binders.
2. Methods
2.1 Modelling approach
Bituminous binder is produced during the petroleum refining process, known for its highly complex molecular structure, which makes it challenging to construct an atomistic model containing all types of constituent molecules. Thus, only representative molecules are usually taken into consideration, with appropriate proportions that can accurately reproduce the physical and chemical properties of bitumen binder. There are currently different secondary molecules being adopted in literature.39–41 In our work, the 4-component bitumen molecular structure proposed by Li and Greenfield42 was applied. The structures of each molecule are shown in Fig. 1a. Following previous work,43 bitumen molecules were assembled to meet the elemental mass fraction and the atom ratio of real bitumen from experiments. The overall compositions of the atomistic bitumen model are shown in Table 1. The elemental mass fractions of the proposed bitumen model are in good agreement with experimental measurements,42 as compared in Table 2. The amorphous molecular model of bitumen was created using the random walking algorithm with the Amorphous Cell module implemented in Material Studio. The initial density was set as the experimentally measured value of 1.15 g cm−3 and the corresponding temperature was 300 K.43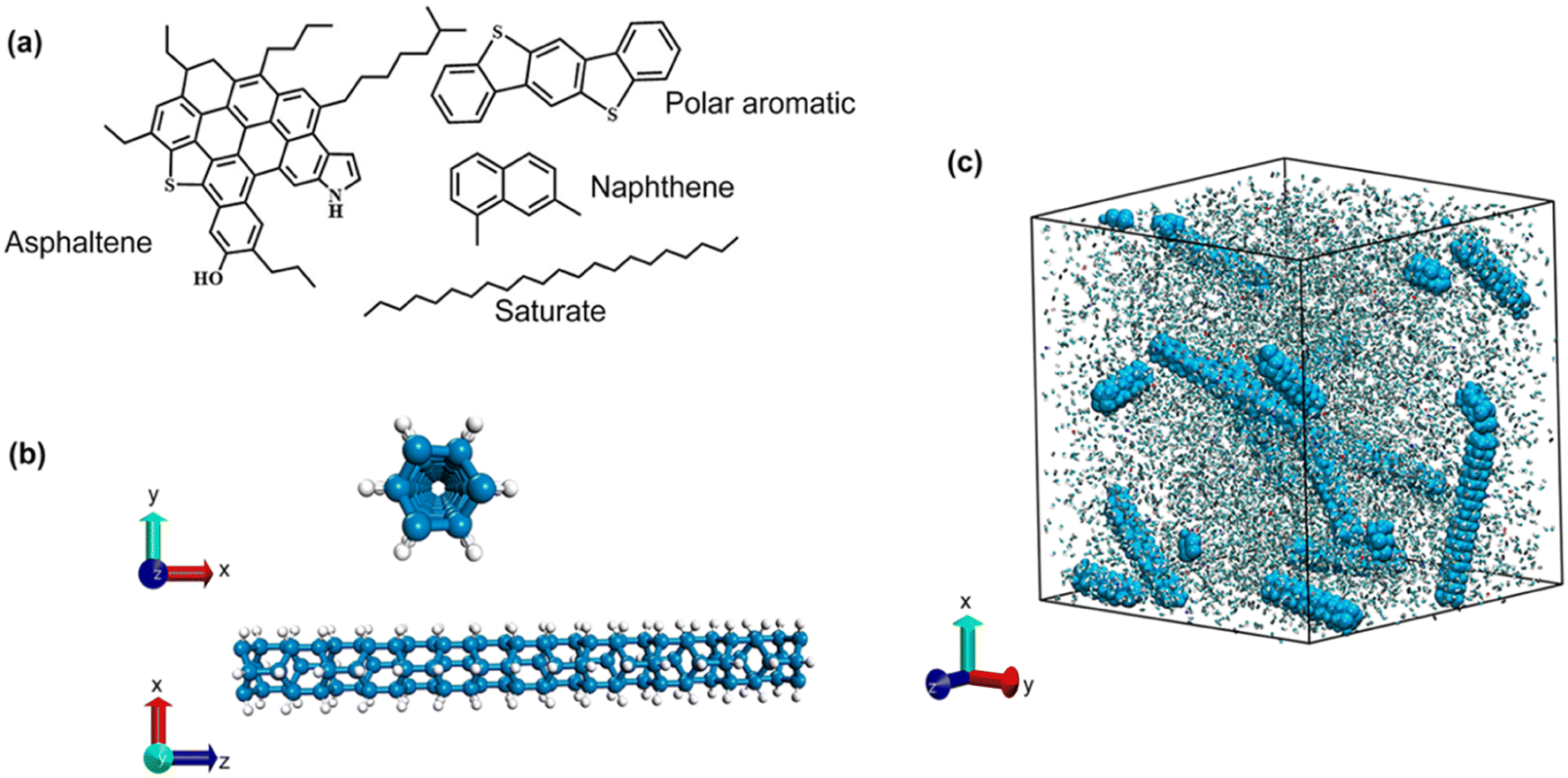 | ||
| Fig. 1 A schematic view of the simulation model. (a) Four representative molecules of the bituminous binder. (b) The atomic structure of the selected diamond nanothread. (c) The molecular structure of bituminous binder with 5.05 wt% randomly dispersed DNTs. | ||
| Bitumen model | Mass (g mol−1) | Chemical formula | Number of molecules | Mass fraction (%) |
|---|---|---|---|---|
| Asphaltene | 754.04 | C53H55NOS | 28 | 25.56 |
| Naphthene | 156.22 | C12H12 | 42 | 7.94 |
| Polar aromatic | 290.38 | C18H10S2 | 48 | 16.87 |
| Saturate | 310.59 | C22H46 | 132 | 49.63 |
| Bitumen binder | 250 |
| System | Elemental mass fraction (%) | Atom ratio (%) | ||||
|---|---|---|---|---|---|---|
| C | H | N | O | S | C/H | |
| Bitumen model | 83.68 | 10.49 | 0.47 | 0.54 | 4.81 | 1.49 |
| Experiment43 | 83.9 | 10.00 | 0.50 | 0.60 | 5.50 | 1.49 |
To assess the influences from 1D nanofillers, the ultra-thin diamond nanothread (DNT) was adopted. Previous reports have shown that chiral DNTs can effectively enhance the mechanical performance of nanocomposites due to their good interfacial interactions and mechanical interlocking effect.28 Thus, this work adopted one type of chiral DNT as the target nanofiller (Fig. 1b), which is analogous to the fully hydrogenated (3,0) carbon nanotubes. Currently, the sample box adopted in most literature reports is smaller than 60 Å.13,43 Considering the potential influence on the aggregation state from the model size, we used a larger box, i.e., 75 × 75 × 75 Å3. Periodic boundary conditions were employed in our simulations to reduce the potential size impact. In general, an initial box with a size of 75 × 75 × 75 Å3 was created, and DNT was randomly placed in the box. Afterward, the representative molecules were filled in the box to reach the preset density (Fig. 1c). To systematically explore the influence of the DNT filler, different models were constructed by varying the dispersion pattern and the weight percentage of DNTs and functional groups. Details of the atomistic models examined in this work are listed in ESI S1.†
2.2 Glass transition temperature calculation
The glass transition temperature (Tg) is a critical indicator for the engineering properties of the bitumen system. For molecular dynamics simulation, the glass transition temperature refers to the temperature when noticeable variations in the volume of the system and the molecular diffusion rate are observed while changing the temperature of the system.44 In this work, the model was first optimized through energy minimization and relaxation. The Consistent Valence Force Field (CVFF) was adopted to describe the atomic interactions in the bitumen systems. The long-range van der Waals interactions were represented by the Lenard-Jones potentials with a cutoff distance of 12.5 Å. The particle–particle particle–mesh (PPPM) algorithm was adopted to calculate the long-range electrostatic interactions with an accuracy of 10−4. The system was equilibrated for 0.5 ns, 1 ns, and 0.5 ns at a constant pressure of 1 atm and constant temperature of 300 K under canonical (NVT), isothermal-isobaric (NPT), and micro-canonical (NVE) ensembles, respectively, to obtain a reasonable and stable bituminous model. To confirm the rationality of the bitumen models, the density, and elastic modulus of the system were selected as the verification parameters for the bitumen system. The equilibrium density of bitumen was approximately 0.96 g cm−3, which is close to the laboratory data of 0.95–1.05 g cm−3,45 and the density of actual bitumen varies from the components and source of the bitumen. The elastic modulus was calculated from E = σ/ε, where σ refers to the stress and ε refers to the strain. We simulated the system under tensile stress in the x-direction and the estimated elastic modulus was about 1313 MPa, which aligns well with the experimental value of 1000–1400 MPa18,46 and simulation results.32 Details of the elastic modulus calculated by tensile simulation in this work are shown in ESI S2.† A cooling cycle was performed for the Tg calculation at a pressure of 1 atm. That is, the model was first warmed up to 400 K after 1 ns in the NPT ensemble and kept at a constant temperature for 0.5 ns, then it was cooled down to 100 K. A total of 25 cooling cycles were carried out with a temperature interval of 20 K. Due to the difference in time scales, the cooling rates in molecular dynamics simulations differ by orders of magnitude (1012) from experimental cooling rates. At such a high cooling rate, the cooling rate effect on the Tg is not substantial for MD simulations. The chosen cooling cycle can reproduce the kinetics of molecules during glass transition states according to previous research.47,48 As the temperature was progressively reduced, the volume and density of the sample were usually tracked to determine the glass transition temperature by previous researchers.49,50 Since the volume and density share the same changing profile, the volume parameter was chosen to fit the glass transition temperature in this work.3. Results and discussion
3.1 Glass transition temperature of the pristine bituminous binder
Fig. 2a shows the change in the volume of the pristine bituminous binder during the cooling process. Tg was determined by the intersection of two linear fitting lines that represent the glassy and rubbery states, respectively. To reduce numerical errors, each model was simulated three times with different relaxation steps, and Tg was averaged from these three calculations. The cooling rate in the simulation was 1012 faster than that in the experiments. Since the Tg decreases with the cooling rate in experiments,51–53 the Tg acquired from simulation can be treated as the estimation of the lower boundary value for the Tg in experiments.50,54,55 Extrapolating the Tg value to the experimental cooling rate is quite complex because the changing rate varies for different molecular structures and relaxation processes,34,56 and some of them are beyond the computation capability of MD simulation for large systems with macromolecules. Thus, in this research, only the Tg with the same cooling rate are compared, i.e., 20 K per cycle, to show the effects from DNT addition. The estimated Tg in this work is about 259 ± 6.3 K, which is comparable with that reported in literature. For instance, the 3-component neat bitumen model was reported with a Tg of 261.8 K.28,57 We should note that there were some reports of higher Tg, which were attributed to several factors. For example, the AAA-1 and AAM-1 bitumen systems58 were reported with Tg of 350.0 ± 6.9 K and 348.3 ± 7.7 K, respectively.48 In the molecular rheology field, flow behavior is usually categorized into three stages that are connected by two transition states based on the manner of molecular motion, i.e., the glassy state, glass transition, rubbery (high elasticity) state, flow transition, and flow state.59 Some researchers considered the temperature where the high elastic state remains as the glass transition temperature,51 while others regarded the initial temperature entering the glassy state as the glass transition temperature.36,60 Many studies selected the middle temperature within the glass transition region as the glass transition temperature.40,44,48,61 Meanwhile, different temperature ranges have been adopted for the Tg calculation. For example, some researchers heated bitumen to 400 K before cooling it down,47 while others heated it to 600 K.62 Adopting different temperature ranges will affect the estimated Tg the of bitumen binder. Moreover, due to the different molecular species, proportions, relaxation processes, force field selections, and ensemble selections adopted in the simulation, the calculation results may also differ from each other. A comprehensive discussion of the influences of these factors will be carried out in our following work. | ||
| Fig. 2 Simulation results for the pristine bituminous binder. (a)The volume as a function of temperature for the bituminous binder between 100 and 400 K. The mean square displacement (MSD) of bituminous binder molecules at (b) 300 K and (c) 240 K. | ||
The glass transition phenomenon of the bitumen is closely related to the mobility of atoms/molecules within the model. The mean square displacement (MSD) is commonly used to describe the mobility of atoms for a given duration, which has been widely used to investigate the special distribution, self-healing process, and bitumen-aggregate interactions.63 Specifically, MSD was calculated according to MSD(t) = 〈Δri(t)2〉 = 〈(ri(t) − ri(0))2〉, where ri(t) and ri(0) are the position vectors of the ith bitumen molecule at time t and the initial stage, respectively. The volume–temperature curve indicates that there is a significant change in the volume of the bituminous sample while changing from the glass state to the rubber state (as the temperature increases). To illustrate the influence of temperature on the trend of molecular activity, the models (at the equilibrium status) at the glass state (under 240 K) and the rubber state (under 300 K) were selected. The MSD of each molecule was then calculated by performing MD simulations under the NPT ensemble over 1 ns. As shown in Fig. 2b, the MSD magnitude of each molecule follows the order of saturate > naphthene > polar aromatic > asphaltene at the rubber state (300 K). Asphaltenes with the slowest migration rate are attributed to the relatively large molecular weight. Although polar aromatics also have a small molecular weight, their adsorption effects on asphaltene hinder their mobility and thus reduce their migration rate.64 At 240 K, except for saturated molecules, the MSD curves for other molecules exhibited a saturated line (Fig. 2c), indicating insignificant molecular movement. Such results suggest the transition from a free state to a “frozen state”. However, the MSD curve of the saturated molecules still exhibited an increasing trend in the “frozen state” at 240 K, perhaps because they are more likely to undergo stretching and entangling, ensuring that they remain active at low temperatures. Additionally, it was found that the movement of the polar aromatics decreased remarkably in the frozen state, which even fell below that of the asphaltene. This result implies that the mobility of polar molecules is particularly sensitive to temperature changes, which is consistent with previous studies.65
3.2 Influences from the weight percentage and distribution of DNT fillers
With the above understanding, we investigated the effects of DNT on the glass transition temperature of the bituminous binder. Firstly, we considered the models with different weight percentages of DNTs. For discussion convenience, we named DNTs with the randomly dispersed scenario as DNTR. The boxplot in Fig. 3a represents the range of Tg fluctuations in the glass transition state observed in three independent simulations for the same model. The relatively large fluctuations of Tg indicated the strong impact of the microstructural arrangement of molecules. As compared in Fig. 3a, the addition of DNT fillers resulted in insignificant changes to Tg as compared with that of the pristine bituminous binder. The sample with 5.05 wt% DNTs exhibited the highest glass transition temperature, while the sample with 1.29 wt% DNTs showed a slightly decreased Tg. Overall, Tg generally increased with the increase in the weight percentage of DNT and then decreased when the weight percentage exceeded 5.05 wt%. Here, three different models were established for each case to account for the potential influences from the random distribution of the bituminous molecules.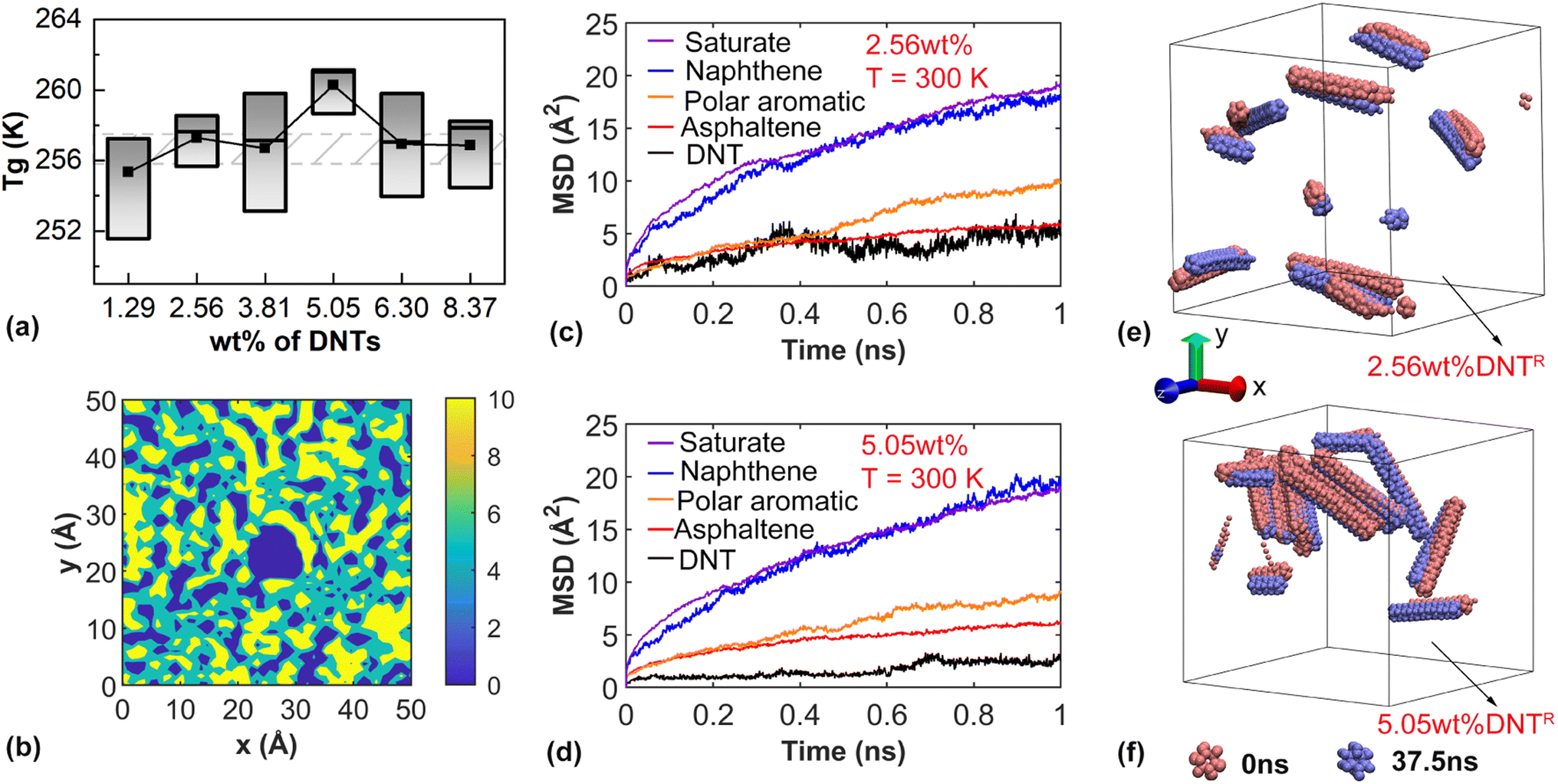 | ||
| Fig. 3 Simulation results for the bituminous binder with randomly dispersed DNTs. (a) The glass transition temperature of the DNT-bitumen system with various weight percentages of DNTs. The grey region represents the fluctuation range of the Tg of the pristine bituminous binder. (b) The projected C atom density contour in the xy-plane of the sample model. The mean square displacement (MSD) of different molecules at 300 K for the sample with (c) 2.56 wt% DNT and (d) 5.05 wt% DNT. The orientation of DNTs at the initial state and the simulation time of 37.5 ns during the cooling process for the sample with (e) 2.56 wt% DNT and (f) 5.05 wt% DNT. DNTs are colored according to the simulation time. | ||
To illustrate the effects of DNT on the microstructural arrangement of bitumen, a sample model with an individual DNT in a smaller supercell was constructed. Fig. 3b shows the distribution of C atoms at the vicinity of the DNT by projecting the number of C atoms onto the xy-plane (with a thickness of 20 Å at the center of the model along the axis of the DNT). Only C atoms in the bituminous molecules were counted. With the existence of relatively large asphaltene molecules, there were both coarse and dense regimes. Specifically, a higher density zone was observed surrounding DNT, suggesting the adsorption effect from DNT.
Earlier reports on DNT/polymer systems indicated that the attractive force between the polymer and DNT reduced the sample volume (passivation effect), while the repulsive force between DNTs increased the sample volume (activation effect).66 With the increase of DNT, the passivation effect between DNTs became stronger than the activation effect between DNTs and polymer, leading to a parabolic relationship. In our work, there is localized aggregation of DNT, which leads to the marginal volume change of the bituminous binder, resulting in a weak relationship. To reveal the underlying mechanisms behind the increased Tg observed in the sample containing 5.05 wt% DNT, a comparison of the MSD curves was conducted with its counterpart containing 2.56 wt% DNT at 300 K. According to Fig. 3c and d, both samples share similar MSD profiles with certain slight differences. The gap between the polar aromatic and asphaltene molecules became smaller for higher DNT contents, suggesting the suppressed mobility of the polar molecules. With increasing DNT content, the mobility of DNTs themselves decreased, which is understandable due to the repulsive interactions between each DNT. Specifically, a remarkable increase in MSD for the polar aromatic molecules was observed between 0.4 and 0.6 ns for the sample with 2.56 wt% DNTs. In comparison, the sample with 5.05 wt% DNT did not exhibit such a sudden increase phenomenon. This observation suggests that the addition of DNTs promotes the motion of small molecules but passivates the motion of large molecules of the bitumen. Since the high proportion of small molecules in the bitumen system dominates the mobility of the whole sample, it eventually leads to an increase in Tg with a suitable content of DNTs.
According to the MSD curves, no significant displacements of DNTs occurred during the cooling process due to their comparatively high molecular weight. Compared with the initial configurations, aggregation between DNTs was observed during the cooling process. Fig. 3e and f show that there were different aggregation patterns within the sample. Since the relative positions of the DNTs are randomly assembled during modeling, different assembly situations yield different molecular environments around the DNTs.67 With 5.05 wt% DNTs, the aggregation of DNT became obvious, leading to a large degree of inhomogeneity within the sample and thus, increased Tg Overall, the aggregation of DNT fillers and the enhanced mobility of small molecules can explain the increased Tg in Fig. 3a (with 5.05 wt% DNTs). Since these effects are highly correlated with the microstructure of the sample, a separate study is desired to clarify which effect plays the dominant role.
The above results signify that the dispersion of DNTs will affect the aggregation of different molecules, and thus the mobility of the constituent molecules. To further demonstrate the potential impacts, we established bituminous binder models with controlled dispersion of DNTs. Besides the sample with randomly distributed DNTs (named model I), five samples (denoted as model II to model VI) with different distribution patterns were constructed. All samples had an identical weight percentage of DNTs, i.e., 5.05 wt%. Specifically, model II initially contained evenly distributed DNTs along the xy plane, which were placed in the middle of the sample and vertical to the xy plane; model III contained vertically aligned DNTs but their positions were not constrained in the x, y, and z directions; model IV contained four vertically aligned DNT bundles (each bundle contained three DNTs); model V contained two vertically aligned DNT bundles; model VI contained only on vertically aligned DNT bundles. In models IV to VI, the DNT bundles were placed in the middle of the sample along the thickness direction (z-axis) and were evenly distributed in the xy plane. As shown in Fig. 4a, the preset orientations of DNTs or DNT bundles changed during the relaxation stage due to the movement of bituminous molecules. Fig. 4b compares the Tg of these models, from which both reduced and increased Tg were observed. Overall, model II (with controlled distribution of individual DNTs) and model V exhibited a much smaller Tg as compared with model I, which is similar to that of the neat bitumen. A large variation range was observed for models III, IV, and VI.
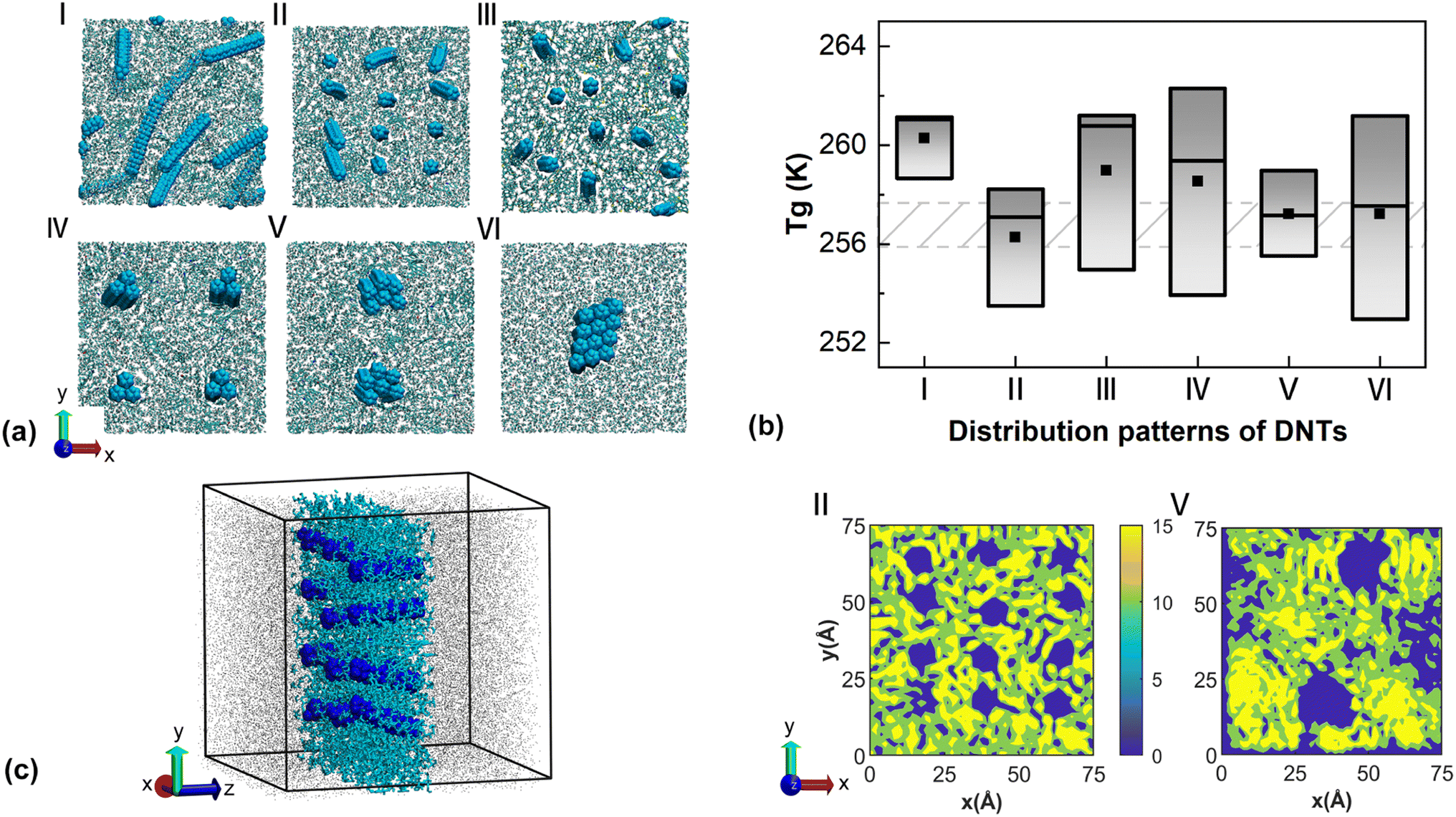 | ||
| Fig. 4 Simulation results for the bituminous binder with vertically aligned DNTs. (a) Cross-sectional view of the model with different DNT construction methods. (b) The glass transition temperature for the sample with different distribution patterns. (c) Atomistic view of the sample (left panel, the highlighted region is the density calculation regime) and the corresponding C atom density distribution (right panel). The DNT fillers were excluded while calculating the C atom density profile. | ||
Following the previous discussion, there are two coexisting mechanisms affecting Tg, i.e., the uniformly distributed system forms a dense network that restricts the mobility of molecules;68 the movement of molecules is reduced due to the strong adsorption effect from DNTs.69 For illustration, we calculated the distribution of C atoms of model II for a given slab (left panel in Fig. 4c) with a thickness of 20 Å. As shown in the right panel of Fig. 4c, the C distribution near the DNTs region is quite uniform without a clear formation of networks. This observation aligns with previous work, as the networks formed within asphalt systems are temporary.9 Due to the orientation change, the left corner DNT is not clearly depicted (by the blue region). Different from model II, clear clusters of C atoms were observed near the DNT bundles in model V together with relatively large voids. In other words, the DNT bundles with larger effective diameters have a stronger adsorption effect on the bituminous molecules. Based on the colloid theory of bitumen, there is also mutual adsorption and bonding between bitumen molecules, with asphaltene being the dispersed phase while saturates and aromatics act as dispersants. In neat bitumen, the asphaltene exhibits a tendency to aggregate.70,71 According to the distribution of C atoms, it is anticipated that the uniform distribution of DNTs in model II hinders the aggregation of asphaltene to a certain extent, and thus improves its low-temperature properties with a decreased Tg. In comparison, the decreased Tg in models V and VI are attributed to the strong adsorption of DNT bundles on bitumen molecules, which enhances its ability to resist cooling.
3.3 Bituminous binder with functionalized DNT
Given the influence of aggregated DNTs (DNT bundles), we further examined the Tg of bituminous binder with functionalized DNTs, where functional groups were frequently grafted onto nanofillers to avoid their aggregation. Here, the sample with 5.05 wt% randomly dispersed DNTs was considered. Common functional groups, including –CH3, –COOH, –C2H5, and –C6H5, were adopted, which randomly replaced the H atoms on the DNT surface (Fig. 5a). For an even comparison, the functionalization percentage was kept the same at 15%, and the functionalization sites were kept the same for all models. From Fig. 5b, the introduction of –CH3 and –C6H5 resulted in a slight decrease in Tg, while the existence of –C2H5 and –COOH exerted a marginal impact on Tg. Interestingly, Tg of the sample with –C6H5 groups exhibited a large variation. One potential explanation for such an observation is the benzene rings in the –C6H5 groups, which result in strong π-bonds with the asphaltene molecules. Thus, a minor change in the orientation of –C6H5 groups can lead to a greater influence on the structure.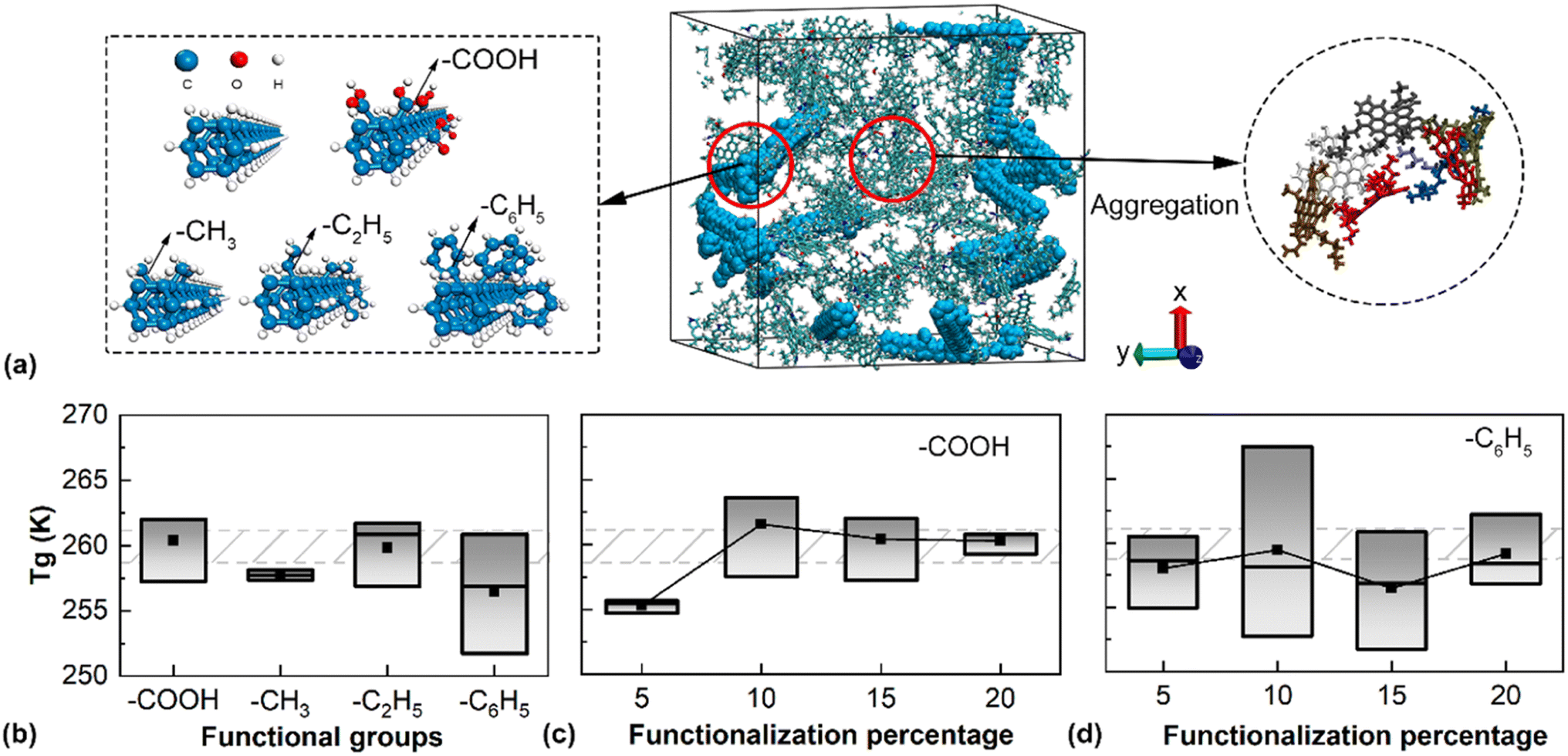 | ||
| Fig. 5 Simulation results for the bituminous binder with functionalized DNTs. (a) The atomistic structure (left panel) and the bituminous binder model (right panel). (b) The glass transition temperature for models containing different functional groups. (c) The functionalization percentage of –COOH and (d) –C6H5. The dashed region represents the Tg of the sample without functionalization. | ||
Considering the existence of π-bonds in –C6H5 and H-bonds in the –COOH-functionalized samples, we also examined the Tg of the samples with varying functionalization percentages. As displayed in Fig. 5c, the functionalization percentage of –COOH did not influence the Tg except in the case with a 5% functionalization ratio, where a reduced Tg was observed. Additional calculations were conducted for three different models with 5% –COOH (by varying the dispersion of the DNTs), from which a uniformly decreased Tg was observed. For the sample with –C6H5 groups, a relatively large variation of Tg was detected, while the mean value of Tg appeared irrelevant to the functionalization ratio. Such large variations in Tg could be because the –C6H5 groups are more flexible on the surface of DNTs. With the formation and breaking of π-bonds, such flexibility induces stronger microstructural rearrangements during the simulation. Here, the formation and breakage of π-bonds can be easily identified by tracking the movements of adjacent molecules that contain benzene rings, which are controlled by the vdW interactions in the applied force field. Based on the atomic configurations, we monitored the distance between the axes of two DNTs. It was found that the most dispersed DNT distribution and the lowest degree of aggregation of asphaltene always resulted in the lowest Tg (lower boundary of the box plot in Fig. 5c). Calculation details are given in ESI S3.†
The aggregation of DNTs and the molecular environment in the vicinity of DNTs directly affect Tg.72 To quantify the effects of functional groups, we counted the number of atoms for different molecules surrounding the DNTs. Given that the long-distance van der Waals cutoff distance was set as 12.5 Å and the diameter of DNT was approximately 5 Å, a cylindrical domain with a radius of 15 Å was selected to investigate the adsorption capacity of the DNT fillers, which shared the axis of the corresponding DNT filler. The error bars in Fig. 6a represent the calculation data from ten frames (with a time interval of 5 ps) at the equilibrium state for each model to ensure accuracy. The introduction of functional groups increased the attractive force of DNT toward neighboring molecules, especially at low temperatures. The sample containing –C6H5 groups had the fewest surrounding atoms at the rubber state (300 K, Fig. 6a), while the models with –CH3 and –COOH groups had the highest number of surrounding atoms. Since the –CH3 group has a shorter length as compared with other groups, more molecules can approach the surface of the DNT fillers. The –COOH group enhanced the adsorption of bituminous molecules onto DNTs due to the presence of hydrogen bonds. In comparison, the sample with –C2H5 groups exhibited the highest temperature sensitivity with a relatively large variation in the number of surrounding atoms. It is estimated that the longer –C2H5 group provides many binding sites at low temperatures, which is beneficial for attracting bituminous molecules to the vicinity of DNTs. However, at higher temperatures, there are fewer nearby molecules with increasing molecular mobility, and the branching chain instead hinders the adsorption of molecules. The introduction of functional groups leads to a similar number of unsaturated molecules around DNTs at higher temperatures. However, significant differences were observed under low-temperature conditions, with a noticeable increase in non-saturated molecules near DNTs with –COOH and –C6H5 groups. In comparison, the sample with –C2H5 groups exhibited a slightly decreased number of non-saturated molecules. It is worth noting that the number of asphaltene molecules around the –C6H5 group showed a significant increase as the temperature decreased, once again demonstrating the strong adsorption effect of π-bonds on asphaltene. From the energy point of view, the introduction of polar functional groups can increase the intermolecular non-bond interactions, such as the van der Waals and electrostatic interactions. Thus, polar functional groups are expected to strengthen intermolecular bonding and increase the resistance to deformation (caused by external forces or temperature rise).73
 | ||
| Fig. 6 Simulation results for bituminous binders with functionalized DNTs. The atomic number of each molecule surrounding the axis of the DNT fillers for the sample: (a) with different types of functional groups; (b) with different functionalization percentages of –COOH and (c) –C6H5. (d) The pairwise relationship between the number of atoms in the bitumen (left panel) and naphthene molecules (right panel) around DNTs and Tg at different temperatures. Here, PCC refers to the Pearson correlation coefficient; the red marker represents the DNT/bitumen system without functional groups, while the green, pink, yellow, and black markers represent those modified with –CH3, –C2H5, –COOH, and –C6H5 groups, respectively; and the light-colored region represents the 95% confidence interval. | ||
Fig. 6b and c further compare the number of surrounding atoms between the samples with different functionalization percentages. When the functionalization ratio is below 20%, the introduction of –COOH or –C6H5 groups will rearrange the overall molecular distributions as the temperature changes. At 10% functionalization of –C6H5 groups, there are notable changes in the number and arrangement of atoms around DNTs, suggesting that the formation and breaking of π-bonds is most prominent at this point. This aligns with the earlier mention of significant fluctuations in Fig. 5d. To identify which molecule around DNT has the highest correlation to Tg, we systematically analyzed the Pearson correlation coefficients (PCCs)74 between each molecule and Tg in ESI S4.† For data accuracy, ten frames (with a time interval of 5 ps) in the equilibrium state for each model were selected for calculation, which were averaged for fitting purposes. The strongest correlation was found between the number of naphthenes clustered around the DNTs under 300 K with a PCC value of about −0.75, and the second strongest was for bitumen molecules with a PCC value of about −0.62 (Fig. 6d). It was observed that DNT with –C6H5 groups is more likely to deviate from the fitted line, which further suggests the impacts from the π-bonds. Above all, to further quantify the impact of functionalized DNTs on the Tg of bitumen, additional systematic work is desirable, considering a wider range of factors such as the size of the filler, the location of the functional groups, and the composition molecules of bitumen.
4. Conclusions
Based on large-scale molecular dynamics simulation, we systematically analyzed the impact of DNT on the glass transition temperature (Tg) of bitumen for the first time. The glass transition phenomenon of the bitumen is closely related to the mobility of its constituent atoms/molecules, and the mobility of polar molecules is particularly sensitive to temperature changes. By varying the content of randomly distributed DNT, Tg was found to generally increase with the increase of the weight percentage of DNT and then decrease when the weight percentage exceeds 5.05 wt%. The enhancement effect was weakened when DNT was distributed vertically or functionalized with functional groups, especially when –C6H5 groups were grafted. From a microscopic molecular viewpoint, the presence of DNTs leads to an inhomogeneous bituminous matrix, thereby enhancing the mobility of small molecules while inhibiting the mobility of large molecules in the bitumen. The dispersion of DNTs was found to alter the aggregation of different molecules, and thus affect the mobility of the constituent molecules within the bituminous binder. For instance, the sample with vertically and uniformly aligned DNTs exhibited an obviously reduced Tg due to the formation of temporary networks that obstruct molecular motion. In comparison, the sample with two bundles of DNTs also exhibited an obviously reduced Tg, which was caused by the strong adsorption effect on the bituminous molecules. The introduction of –CH3 and –C6H5 functional groups on DNTs resulted in a slight decrease in Tg, while the existence of –C2H5 and –COOH exerted a marginal impact on Tg. Specifically, the sample with –C6H5 groups showed a relatively large variation in Tg due to the formation and breakage of π bonds. Overall, the sample with the dispersed DNTs and the lower degree of aggregation of asphaltene always exhibited a lower Tg.In summary, this work provides an in-depth analysis of the glass transition of bituminous binder, focusing on the microstructural changes induced by the presence of DNT nano-additives. Aggregation and adhesion are the dominant mechanisms observed in the DNT/bitumen system that affect the mobility of its constituent molecules. These findings provide theoretical guidance for selecting nano-additives to achieve the desired Tg. It is important to note that this work only considers a bituminous binder model with four types of molecules. Therefore, further investigation is warranted to explore the interactions between different bituminous molecules and DNTs. Given the vast number of molecules present in real bituminous binders, high-throughput computation techniques should be employed for such purposes. Meanwhile, with the development of the machine-learning interatomic potentials, the simulation accuracy can be improved,75,76 compared to the universal potential currently used, which also requires building a customized dataset for various bituminous molecules. The establishment of the bitumen-customized dataset and high-throughput algorithm is scheduled for future studies.
Data availability
The data that support the findings of this study are available from the corresponding authors on reasonable request.Author contributions
Yingying Pang: conceptualization, methodology, investigation, data curation, characterization, formal analysis, visualization, writing – original draft preparation. Liangfeng Sun: investigation and formal analysis. Haifei Zhan: conceptualization, methodology, formal analysis, supervision, funding acquisition, project administration, writing – reviewing and editing. Xianglong Zheng: investigation and formal analysis. Jiandong Zhang: methodology, investigation and formal analysis. Chengyou Bian: investigation and formal analysis. Chaofeng Lü: supervision, resources, funding acquisition, project administration, discussion and writing – reviewing.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
H. Z. would like to thank the financial support from the National Natural Science Foundation of China (12172325) and the Zhejiang Provincial Natural Science Foundation (LR22A020006); C. L. would like to thank the financial support National Natural Science Foundation of China (11925206). This work was also funded by the Center for Balance Architecture, Zhejiang University; the ZCCC Hongtu Transportation Construction Co., Ltd, and the ZJU-ZCCC Institute of Collaborative Innovation (No. ZDJG2021003).References
- S. Pyshyev, V. Gunka, Y. Grytsenko and M. Bratychak, Polymer Modified Bitumen: Review, Chem. Chem. Technol., 2016, 10(4s), 631–636 CrossRef CAS.
- A. Cannone Falchetto and K. H. Moon, Comparison of Thermal Stress Calculation: Hopkins and Hamming's Algorithm and Laplace Transformation Approach, J. Mater. Civ. Eng., 2016, 28(9), 04016076 CrossRef.
- K. Zhang and J. Kevern, Review of porous asphalt pavements in cold regions: the state of practice and case study repository in design, construction, and maintenance, Journal of Infrastructure Preservation and Resilience, 2021, 2(1), 4 CrossRef.
- Z. Suo and W. G. Wong, Nonlinear properties analysis on rutting behaviour of bituminous materials with different air void contents, Constr. Build. Mater., 2009, 23(12), 3492–3498 CrossRef.
- P. Lu, Y. Ma, K. Ye and S. Huang, Analysis of high-temperature performance of polymer-modified asphalts through molecular dynamics simulations and experiments, Constr. Build. Mater., 2022, 350, 128903 CrossRef CAS.
- X. Lu and U. Isacsson, Effect of Binder Rheology on the Low-Temperature Cracking of Asphalt Mixtures, Road Mater. Pavement Des., 2011, 2(1), 29–47 CrossRef.
- T. Xu and X. Huang, Investigation into causes of in-place rutting in asphalt pavement, Constr. Build. Mater., 2012, 28(1), 525–530 CrossRef.
- R. I. H. D. T. Amoussou, H. Tanoue, M. Sasaki and M. Shigeishi, Hydrothermal recovery of asphalt from asphalt concrete, Constr. Build. Mater., 2016, 125, 1196–1204 CrossRef CAS.
- Z. Lei, T. Yi-qiu and H. Bahia, Relationship between glass transition temperature and low temperature properties of oil modified binders, Constr. Build. Mater., 2016, 104, 92–98 CrossRef CAS.
- P. Kriz, J. Stastna and L. Zanzotto, Glass Transition and Phase Stability in Asphalt Binders, Road Mater. Pavement Des., 2011, 9(sup1), 37–65 CrossRef.
- Y. Li, X. Huang, L. Zeng, R. Li, H. Tian, X. Fu, Y. Wang and W.-H. Zhong, A review of the electrical and mechanical properties of carbon nanofiller-reinforced polymer composites, J. Mater. Sci., 2018, 54(2), 1036–1076 CrossRef.
- D. Lesueur, The colloidal structure of bitumen: consequences on the rheology and on the mechanisms of bitumen modification, Adv. Colloid Interface Sci., 2009, 145(1–2), 42–82 CrossRef CAS.
- Z. Long, S. Zhou, S. Jiang, W. Ma, Y. Ding, L. You, X. Tang and F. Xu, Revealing compatibility mechanism of nanosilica in asphalt through molecular dynamics simulation, J. Mol. Model., 2021, 27(3), 81 CrossRef CAS PubMed.
- W. Cui, W. Huang, H. M. Z. Hassan, X. Cai and K. Wu, Study on the interfacial contact behavior of carbon nanotubes and asphalt binders and adhesion energy of modified asphalt on aggregate surface by using molecular dynamics simulation, Constr. Build. Mater., 2022, 316, 125849 CrossRef CAS.
- C. Li, Y. Nie, H. Zhan, J. Bai, T. Liu and Y. Gu, Mechanical properties of polymer nanocomposites with randomly dispersed and cross-linked two-dimensional diamond, Compos. Sci. Technol., 2022, 230, 109722 CrossRef CAS.
- T. Ramanathan, A. A. Abdala, S. Stankovich, D. A. Dikin, M. Herrera-Alonso, R. D. Piner, D. H. Adamson, H. C. Schniepp, X. Chen, R. S. Ruoff, S. T. Nguyen, I. A. Aksay, R. K. Prud'Homme and L. C. Brinson, Functionalized graphene sheets for polymer nanocomposites, Nat. Nanotechnol., 2008, 3(6), 327–331 CrossRef CAS.
- M. K. Alam, M. T. Islam, M. F. Mina and M. A. Gafur, Structural, mechanical, thermal, and electrical properties of carbon black reinforced polyester resin composites, J. Appl. Polym. Sci., 2014, 131(13), 40421 CrossRef.
- U. Tayfun, Y. Kanbur, U. Abaci, H. Y. Guney and E. Bayramli, Mechanical, flow and electrical properties of thermoplastic polyurethane/fullerene composites: Effect of surface modification of fullerene, Composites, Part B, 2015, 80, 101–107 CrossRef CAS.
- Q. Yang and C. Yu, Multiscale Enhancement Mechanism of Carbon Nanotube-Modified Asphalt at High Temperature by Oxidative Aging: A Molecular Dynamics Simulation Investigation, Energy Fuels, 2022, 36(24), 15279–15296 CrossRef CAS.
- H. Zhan, G. Zhang, V. B. C. Tan and Y. Gu, The best features of diamond nanothread for nanofibre applications, Nat. Commun., 2017, 8(1), 14863 CrossRef CAS PubMed.
- R. E. Roman, K. Kwan and S. W. Cranford, Mechanical Properties and Defect Sensitivity of Diamond Nanothreads, Nano Lett., 2015, 15(3), 1585–1590 CrossRef CAS PubMed.
- K. Duan, Y. Li, L. Li, Y. Hu and X. Wang, Diamond nanothread based resonators: ultrahigh sensitivity and low dissipation, Nanoscale, 2018, 10(17), 8058–8065 RSC.
- J. Chen, L. Yan, W. Song and D. Xu, Interfacial characteristics of carbon nanotube-polymer composites: A review, Composites, Part A, 2018, 114, 149–169 CrossRef CAS.
- J. Gao, G. Zhang, B. I. Yakobson and Y. W. Zhang, Kinetic theory for the formation of diamond nanothreads with desired configurations: a strain-temperature controlled phase diagram, Nanoscale, 2018, 10(20), 9664–9672 RSC.
- T. Zhu and E. Ertekin, Phonons, Localization, and Thermal Conductivity of Diamond Nanothreads and Amorphous Graphene, Nano Lett., 2016, 16(8), 4763–4772 CrossRef CAS PubMed.
- H. Zhan, G. Zhang, V. B. C. Tan, Y. Cheng, J. M. Bell, Y.-W. Zhang and Y. Gu, Diamond Nanothread as a New Reinforcement for Nanocomposites, Adv. Funct. Mater., 2016, 26(29), 5279–5283 CrossRef CAS.
- H. Zhan, G. Zhang, V. B. C. Tan, Y. Cheng, J. M. Bell, Y.-W. Zhang and Y. Gu, From brittle to ductile: a structure dependent ductility of diamond nanothread, Nanoscale, 2016, 8(21), 11177–11184 RSC.
- L. W. Zhang, W. M. Ji and K. M. Liew, Mechanical properties of diamond nanothread reinforced polymer composites, Carbon, 2018, 132, 232–240 CrossRef CAS.
- W. M. Ji and L. W. Zhang, Diamond nanothread reinforced polymer composites: Ultra-high glass transition temperature and low density, Compos. Sci. Technol., 2019, 183, 107789 CrossRef CAS.
- R. Zhang, F. Du, K. Jariyavidyanont, E. Zhuravlev, C. Schick and R. Androsch, Glass transition temperature of poly(d,l-lactic acid) of different molar mass, Thermochim. Acta, 2022, 718, 179387 CrossRef CAS.
- H. Soenen, J. Besamusca, H. R. Fischer, L. D. Poulikakos, J.-P. Planche, P. K. Das, N. Kringos, J. R. A. Grenfell, X. Lu and E. Chailleux, Laboratory investigation of bitumen based on round robin DSC and AFM tests, Mater. Struct., 2013, 47(7), 1205–1220 CrossRef.
- L. You, T. Spyriouni, Q. Dai, Z. You and A. Khanal, Experimental and molecular dynamics simulation study on thermal, transport, and rheological properties of asphalt, Constr. Build. Mater., 2020, 265, 120358 CrossRef CAS.
- Y. Sun, B. Huang, J. Chen, X. Jia and Y. Ding, Characterizing rheological behavior of asphalt binder over a complete range of pavement service loading frequency and temperature, Constr. Build. Mater., 2016, 123, 661–672 CrossRef CAS.
- D. Hu, X. Gu, G. Wang, Z. Zhou, L. Sun and J. Pei, Performance and mechanism of lignin and quercetin as bio-based anti-aging agents for asphalt binder: A combined experimental and ab initio study, J. Mol. Liq., 2022, 359, 119310 CrossRef CAS.
- G. Li, Y. Tan, Y. Fu, P. Liu, C. Fu and M. Oeser, Density, zero shear viscosity and microstructure analysis of asphalt binder using molecular dynamics simulation, Constr. Build. Mater., 2022, 345, 128332 CrossRef CAS.
- Y. Kang, D. Zhou, Q. Wu, R. Liang, S. Shangguan, Z. Liao and N. Wei, Molecular dynamics study on the glass forming process of asphalt, Constr. Build. Mater., 2019, 214, 430–440 CrossRef.
- K. H. Choi, S.-Y. Lee, I. Hong, S. Son, J.-C. An and S. Kim, Molecular-level investigation of coal-tar pitch treated by air blowing: Revealing the restructure of aromatic compounds via radical reactions, Carbon, 2023, 203, 377–385 CrossRef CAS.
- S. Liu, X. Qi and L. Shan, Effect of molecular structure on low-temperature properties of bitumen based on molecular dynamics, Constr. Build. Mater., 2022, 319, 126029 CrossRef CAS.
- M. Mousavi, F. Pahlavan, D. Oldham, S. Hosseinnezhad and E. H. Fini, Multiscale Investigation of Oxidative Aging in Biomodified Asphalt Binder, J. Phys. Chem. C, 2016, 120(31), 17224–17233 CrossRef CAS.
- H. Yao, Q. Dai and Z. You, Molecular dynamics simulation of physicochemical properties of the asphalt model, Fuel, 2016, 164, 83–93 CrossRef CAS.
- G. Sun, J. Zhang, Z. Chen, Z. Niu and Y. Li, Interfacial Performance of Asphalt-Aggregate System under Different Conditions Based on Molecular Dynamics Simulation, J. Mater. Civ. Eng., 2023, 35(6), 04023116 CrossRef CAS.
- D. D. Li and M. L. Greenfield, Chemical compositions of improved model asphalt systems for molecular simulations, Fuel, 2014, 115, 347–356 CrossRef CAS.
- F. Nie, W. Jian and D. Lau, An atomistic study on the thermomechanical properties of graphene and functionalized graphene sheets modified asphalt, Carbon, 2021, 182, 615–627 CrossRef CAS.
- H. A. Tabatabaee, R. Velasquez and H. U. Bahia, Predicting low temperature physical hardening in asphalt binders, Constr. Build. Mater., 2012, 34, 162–169 CrossRef.
- L. Zhang and M. L. Greenfield, Analyzing Properties of Model Asphalts Using Molecular Simulation, Energy Fuels, 2007, 21(3), 1712–1716 CrossRef CAS.
- J. Ouyang, J. Zhao and Y. Tan, Modeling Mechanical Properties of Cement Asphalt Emulsion Mortar with Different Asphalt to Cement Ratios and Temperatures, J. Mater. Civ. Eng., 2018, 30(10), 04018263 CrossRef.
- L. Luo, L. Chu and T. F. Fwa, Molecular dynamics analysis of oxidative aging effects on thermodynamic and interfacial bonding properties of asphalt mixtures, Constr. Build. Mater., 2021, 269, 121299 CrossRef CAS.
- F. Khabaz and R. Khare, Glass Transition and Molecular Mobility in Styrene-Butadiene Rubber Modified Asphalt, J. Phys. Chem. B, 2015, 119(44), 14261–14269 CrossRef CAS PubMed.
- H. Yao, J. Liu, M. Xu, J. Ji, Q. Dai and Z. You, Discussion on molecular dynamics (MD) simulations of the asphalt materials, Adv. Colloid Interface Sci., 2022, 299, 102565 CrossRef CAS PubMed.
- J. E. K. Schawe, The influence of hydrogen bonds on the glass transition in amorphous binary systems, J. Mol. Liq., 2022, 368, 120598 CrossRef CAS.
- J. Liu, Y. Sun, W. Wang and J. Chen, Using the viscoelastic parameters to estimate the glass transition temperature of asphalt binders, Constr. Build. Mater., 2017, 153, 908–917 CrossRef.
- P. Apostolidis, M. Elwardany, L. Porot, S. Vansteenkiste and E. Chailleux, Glass transitions in bituminous binders, Mater. Struct., 2021, 54(3), 01726–6 CrossRef.
- X. Xia, J. Li, J. Zhang and G. J. Weng, Uncovering the glass-transition temperature and temperature-dependent storage modulus of graphene-polymer nanocomposites through irreversible thermodynamic processes, Int. J. Eng. Sci., 2021, 158, 103411 CrossRef CAS.
- F. Godey, A. Fleury, A. Ghoufi and A. Soldera, The extent of the glass transition from molecular simulation revealing an overcrank effect, J. Comput. Chem., 2018, 39(5), 255–261 CrossRef CAS.
- F. Godey, A. Fleury and A. Soldera, Local dynamics within the glass transition domain, Sci. Rep., 2019, 9(1), 9638 CrossRef PubMed.
- J. W. P. Schmelzer, T. V. Tropin, V. M. Fokin, R. Zhang, A. Abdelaziz, Y. Z. Chua, V. Madhavi, T. D. Shaffer and C. Schick, Correlation between glass transition temperature and the width of the glass transition interval, Int. J. Appl. Glass Sci., 2019, 10(4), 502–513 CrossRef CAS.
- D. Sun, T. Lin, X. Zhu, Y. Tian and F. Liu, Indices for self-healing performance assessments based on molecular dynamics simulation of asphalt binders, Comput. Mater. Sci., 2016, 114, 86–93 CrossRef CAS.
- J. C. Petersen and R. Glaser, Asphalt Oxidation Mechanisms and the Role of Oxidation Products on Age Hardening Revisited, Road Mater. Pavement Des., 2011, 12(4), 795–819 CrossRef.
- Q. Qin, J. F. Schabron, R. B. Boysen and M. J. Farrar, Field aging effect on chemistry and rheology of asphalt binders and rheological predictions for field aging, Fuel, 2014, 121, 86–94 CrossRef CAS.
- L. Yang, D. Zhou and Y. Kang, Rheological Properties of Graphene Modified Asphalt Binders, Nanomater., 2020, 10(11), 2197 CrossRef CAS PubMed.
- J. Liu, Q. Liu, S. Wang, X. Zhang, C. Xiao and B. Yu, Molecular dynamics evaluation of activation mechanism of rejuvenator in reclaimed asphalt pavement (RAP) binder, Constr. Build. Mater., 2021, 298, 123898 CrossRef CAS.
- W. M. Ji and L. W. Zhang, Diamond nanothread reinforced polymer composites: Ultra-high glass transition temperature and low density, Compos. Sci. Technol., 2019, 183, 107789 CrossRef CAS.
- Z. Long, L. You, X. Tang, W. Ma, Y. Ding and F. Xu, Analysis of interfacial adhesion properties of nano-silica modified asphalt mixtures using molecular dynamics simulation, Constr. Build. Mater., 2020, 255, 119354 CrossRef CAS.
- L. He, G. Li, S. Lv, J. Gao, K. J. Kowalski, J. Valentin and A. Alexiadis, Self-healing behavior of asphalt system based on molecular dynamics simulation, Constr. Build. Mater., 2020, 254, 119225 CrossRef CAS.
- X. Zheng, W. Xu, K. Cao and K. Li, Self-healing behavior of recycled asphalt prepared by residue oil of straw liquefaction based on molecular dynamics simulation, Sci. Rep., 2022, 12(1), 2718 CrossRef CAS PubMed.
- R. A. A. Khan, X. Chen, H.-K. Qi, J.-H. Huang and M.-B. Luo, A novel shift in the glass transition temperature of polymer nanocomposites: a molecular dynamics simulation study, Phys. Chem. Chem. Phys., 2021, 23(21), 12216–12225 RSC.
- L. Zhang and M. L. Greenfield, Effects of Polymer Modification on Properties and Microstructure of Model Asphalt Systems, Energy Fuels, 2008, 22(5), 3363–3375 CrossRef CAS.
- G. Polacco, J. Stastna, Z. Vlachovicova, D. Biondi and L. Zanzotto, Temporary networks in polymer-modified asphalts, Polym. Eng. Sci., 2004, 44(12), 2185–2193 CrossRef CAS.
- L. Q. Zhang and M. L. Greenfield, Effects of polymer modification on properties and microstructure of model asphalt systems, Energy Fuels, 2008, 22(5), 3363–3375 CrossRef CAS.
- M. Nikookar, M. R. Omidkhah, G. R. Pazuki and A. H. Mohammadi, An insight into molecular weight distributions of asphaltene and asphalt using Gel Permeation Chromatography, J. Mol. Liq., 2022, 362, 119736 CrossRef CAS.
- X. Liu, H. Zhu, S. Li, Y. Gao, H. Wang, Y. Zhou, H. Peng, Y. Lei and P. Yu, Effect of Temperature on the Aggregation Kinetic and Interaction Mode of Asphaltene in Toluene-Heptane System at Molecular Level Using Molecular Dynamics (MD) Simulation, J. Mol. Liq., 2023, 122167 CrossRef CAS.
- K. Debbarma, B. Debnath and P. P. Sarkar, A comprehensive review on the usage of nanomaterials in asphalt mixes, Constr. Build. Mater., 2022, 361, 129634 CrossRef CAS.
- H. Hashemzadeh and H. Raissi, The functionalization of carbon nanotubes to enhance the efficacy of the anticancer drug paclitaxel: a molecular dynamics simulation study, J. Mol. Model., 2017, 23(8), 222 CrossRef PubMed.
- P. Wang, H. Zhan and Y. Gu, Molecular Dynamics Simulation of Chiral Carbon Nanothread Bundles for Nanofiber Applications, ACS Appl. Nano Mater., 2020, 3(10), 10218–10225 CrossRef CAS.
- B. Mortazavi, B. Javvaji, F. Shojaei, T. Rabczuk, A. V. Shapeev and X. Zhuang, Exceptional piezoelectricity, high thermal conductivity and stiffness and promising photocatalysis in two-dimensional MoSi2N4 family confirmed by first-principles, Nano Energy, 2021, 82, 105716 CrossRef CAS.
- B. Mortazavi, M. Silani, E. V. Podryabinkin, T. Rabczuk, X. Zhuang and A. V. Shapeev, First-Principles Multiscale Modeling of Mechanical Properties in Graphene/Borophene Heterostructures Empowered by Machine-Learning Interatomic Potentials, Adv. Mater., 2021, 33(35), 2102807 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Including the model information for the DNT-modified bituminous binder; the distance between the axes of two adjacent DNTs; and the Pearson correlation coefficient for different molecules. See DOI: https://doi.org/10.1039/d3na00622k |
| This journal is © The Royal Society of Chemistry 2023 |