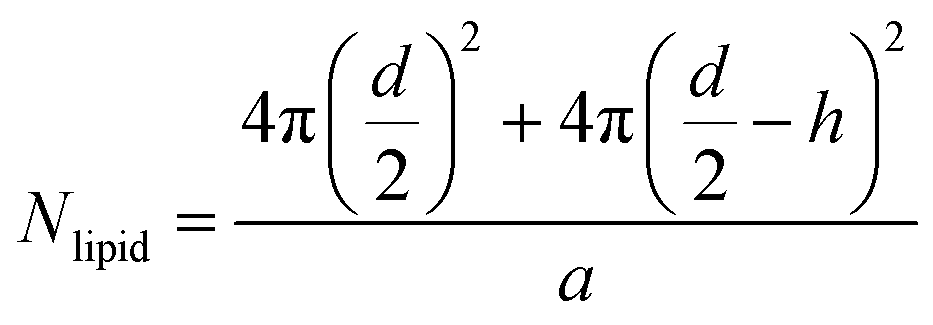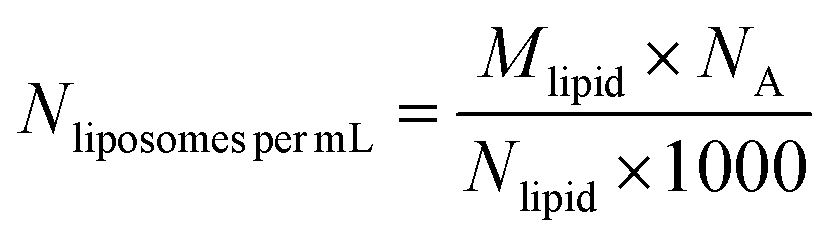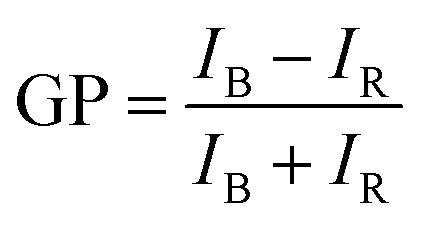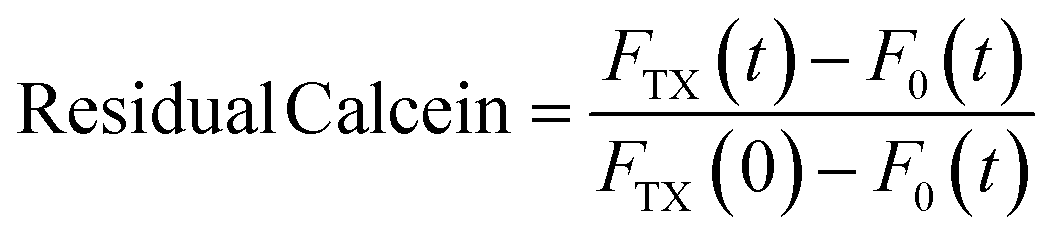DOI:
10.1039/D3NA00608E
(Paper)
Nanoscale Adv., 2023,
5, 6533-6541
Analytical probing of membranotropic effects of antimicrobial copper nanoparticles on lipid vesicles as membrane models†
Received
7th August 2023
, Accepted 13th October 2023
First published on 16th October 2023
Abstract
Copper nanoparticles (CuNPs) are antimicrobial agents that are increasingly being used in several real-life goods. However, concerns are arising about their potential toxicity and thus, appropriate legislation is being issued in various countries. In vitro exploration of the permeability and the distribution of nanoparticles in cell membranes should be explored as the first step towards the investigation of the toxicity mechanisms of metal nanoantimicrobials. In this work, phosphatidylcholine-based large unilamellar vesicles have been explored as mimics of cellular membranes to investigate the effect of ultra-small CuNPs on the physicochemical features of phospholipid membranes. 4 nm-sized CuNPs were synthesized by a wet-chemical route that involves glutathione as a stabilizer, with further characterization by UV-vis absorption spectroscopy, fluorescence spectroscopy, transmission electron microscopy, X-ray photoelectron spectroscopy (XPS), and Fourier transform infrared (FTIR) spectroscopy. Two fluorescent membrane probes bearing naphthalene moieties (laurdan and prodan) were used to monitor the bilayer structure and dynamics, as well as to demonstrate the strong membranotropic effects of CuNPs. The fluorescence spectroscopic studies were supported by dynamic light scattering (DLS) measurements and the calcein leakage assay. Additionally, the degree of perturbation of the phospholipid bilayer by CuNPs was compared against that of Cu2+ ions, the latter resulting in negligible effects. The findings suggested that CuNPs are able to damage the phospholipid membranes, leading to their agglomeration or disruption.
Introduction
Metals, such as copper and silver, have been utilized for thousands of years in antimicrobial applications such as water disinfection, food preservation, surgical bandages and sutures. In particular, with the growing interest in nanomaterials and the rising emergence of antibiotic resistance, there has been reinvigorated interest in the use of metal nanomaterials as antimicrobial agents. Among them, the use of copper nanoantimicrobial agents is widespread.1 Copper is less expensive than other metals (such as gold and silver) and it is a trace element well tolerated by humans. Many aerobic organisms require copper for their metabolism as well as for the electron transport chain. However, if its concentration exceeds the biologically required, it becomes toxic and inhibits microbial growth.1 Several applications of CuNP- antimicrobials are based on their use as nano-reservoirs, providing a controlled release of Cu2+ bioactive ions. Specifically, solid-state surfaces/films can embed biocidal NPs, allowing only the release of metal ions from their surface rather than the release of the entire nanoparticle. In this sense, CuNPs have been used as a source of cupric ions in polymeric matrices to develop bioactive coatings for food packaging applications.2 The bioactivity of such coatings correlates with the Cu2+ release from the nanocomposite surface. In general, the effect is long-lasting, until the nanoparticle corrosion completely depletes the metal contained in the packaging outer layers.
In addition, in the last few years, CuNPs have been successfully involved in various antiviral applications, also exhibiting a significant efficacy against the recent SARS-CoV-2.3–5
Considering the current growth of use of CuNPs as antimicrobials, and also their recent use as bioselective reagents for cellular imaging applications,6 the investigation of the ecotoxicological effect of inorganic nanomaterials is arising, aiming to assess their influence on both human health and ecosystem processes. In addition, the legislation related to CuNP uses needs deeper toxicological evaluation.7 To this aim, it is essential to understand how CuNPs and Cu2+ ions interact with cell membranes, which are the first barriers to be crossed to enter cells.8 The investigation of toxicokinetics in the organism – including internal transport, metabolism, and excretion – is just the last step of human health risk assessments. Before that, elucidating the mechanisms at the molecular level in vitro, and inferring permeability and distribution of contaminants in cell membranes is a critical onset process that supports the prediction and assessment of toxicity pathways,9–11 in line with the EU REACH (Registration, Evaluation, Authorisation and restriction of Chemicals) regulations to promote in vitro testing against ecotoxicity assays.
In this sense, liposomes have been consolidated as biomimetic artificial models of eukaryotic cell membranes. They are composed mainly of phospholipids, the same major component in biological membranes, and thus they are useful for the in vitro exploration of supramolecular interactions of xenobiotics with the lipid bilayer.12–14 Therefore, their interaction with inorganic nanoparticles, as potentially toxic materials, should also be explored. Several studies have been published on the effect of gold nanoparticles on the membranes.15–17 Lipid bilayers are highly flexible and can be deformed due to NP adhesion on its surface followed by NP engulfment that might end with the full NP uptake. The morphology, size and composition of the shell stabilizing the inorganic NP core have a great impact on its interaction with biological systems.18 Indeed, the NP–membrane interactions can lead to different scenarios: NP internalisation within the membrane bilayer, full NP engulfment (similar to non-specific cellular endocytosis) or NP adsorption on the external membrane surface.8,16
Here, the study is focused on ultra-small copper nanoparticles (CuNPs). To the best of our knowledge, few studies have been carried out on the investigation of the effect of CuNPs on the phospholipid membranes, focusing mainly on the cytotoxicity studies of Cu- or CuO-NPs on cell lines.19–21 No papers have yet been published on the investigation of the membranotropic effects of CuNPs, especially with respect to ultra-small CuNPs. The tiny NP size allows for obtaining colourless colloidal solutions,22 which is sought for the development of transparent bioactive nanocomposites. Such CuNP feature makes them the best candidates for the preparation of antimicrobial coatings, useful to cover common-touched surfaces, textiles or other industrial goods. However, potential health risks related to the small size of CuNPs should be considered.23 For this study, 4 nm-sized CuNPs were synthesized by means of a wet-chemical route that involves glutathione (GSH) as a stabilizer. The resulting CuNPs were characterized by UV-vis absorption spectroscopy, fluorescence spectroscopy, transmission electron microscopy (TEM), X-ray photoelectron spectroscopy (XPS), and Fourier transform infrared (FTIR) spectroscopy. Phosphatidylcholine (PC) is the most abundant phospholipid in eukaryotic membrane cells. Therefore, in situ synthesized large unilamellar vesicles (LUVs)/liposomes from natural sources of PC containing both saturated and unsaturated chains have been herein selected as models of biological membranes.10,11 The incorporation of environment-sensitive fluorescent membrane dyes was used to monitor the structure, dynamics and interactions of CuNPs with biomembranes. In particular, two fluorescent membrane probes bearing naphthalene moieties (viz., 6-propionyl-2-dimethylaminonaphthalene (prodan) and 6-dodecanoyl-2-dimethylaminonaphthalene (laurdan)) have been chosen to ascertain low-resolution changes in bilayer organization by generalized polarization (GP), that serves to indicate alterations in lipid ordering, packing and hydration.11,24,25 The physicochemical studies were supported by dynamic light scattering (DLS) measurements and calcein leakage assay. Additionally, the comparison of the membranotropic effects of CuNPs versus Cu2+ ions was investigated in detail, so as to decouple the role of the whole particle from that associated with ions released from its surface.
Materials and methods
Materials
Ascorbic acid (AA) and PBS (phosphate buffered saline) tablets were purchased from Sigma-Aldrich/Merck KGaA. Copper(II) chloride dihydrate (CuCl2·2H2O, reagent grade ACS) was received from Scharlau. L-Glutathione (GSH, reduced, 98+%) was obtained from AlfaAesar. Natural soybean L-α-phosphatidylcholine, LIPOID S100, was purchased from LIPOID GmbH (Ludwigshafen, Germany) with a concentration of L-α-phosphatidylcholine not less than 94% and a lipid tail distribution of linoleic acid (C18:2, (9Z,12Z)-octadeca-9,12-dienoic acid) as the main fatty acid, followed by palmitic acid (C16:0, n-hexadecanoic acid) and oleic acid (C18:1, cis-9-octadecenoic acid) with percentages of ca. 63%, 15% and 11%, respectively. The fluorescent membrane probes 6-dodecanoyl-N,N-dimethyl-2-naphthylamine (laurdan) and N,N-dimethyl-6-propionyl-2-naphthylamine (prodan) were also obtained from Sigma-Aldrich/Merck KGaA.
Synthesis of ultra-small CuNPs@GSH
CuNPs were prepared following the wet-chemical synthesis proposed by Huang et al.,22 using CuCl2 and ascorbic acid as precursor and reducing agent, respectively. The GSH was used as stabilizer. Typically, a solution of 0.15 mM CuCl2 in 0.2 mM GSH was prepared. Then, 112.5 μL of freshly prepared 100 mM AA was added to 5 mL of the CuCl2 – GSH solution and the reaction was kept under stirring for 4 h at 65 °C. Afterwards, the colloid was allowed to cool to room temperature, and stored at 4 °C for further use.
Theoretical estimation of CuNP concentration
The common practice for the theoretical estimation of the metal nanoparticle concentration involves calculations based on their optical properties, such as extinction coefficients relevant to surface plasmon resonance (SPR) features.26 Nevertheless, it could not be used in the present work because of the lack of suitable SPR signals. For this reason, a theoretical calculation based on the copper density and assuming that the Cu(II) precursor was quantitatively converted into elemental nanosized copper phases was carried out. The resulting estimation returns the highest available CuNP concentration, which in turn leads to the worst case scenario. In particular, assuming that all the amount of Cu2+ from the metal precursor used in the synthesis (0.15 mM) was quantitatively turned into elemental Cu, the mass of copper in 1 mL of colloid was calculated (9.8 μg). Then, using the CuNP average diameter estimated from TEM images (3.7 nm) to determine the average volume of a single spherical CuNP, the mass of one CuNP was calculated from the copper density (8940 kg m−3). Hence, the total mass to single NP mass ratio gives an estimation of the number of CuNPs, in our case, 4 × 1013 particles per mL of colloid solution.
Synthesis of large unilamellar vesicles (LUVs)
Liposomes were prepared by the lipid film hydration method27 followed by extrusion28 with some changes to improve the quality of the final product. To this end, a solution of 50 mM of Soy PC was prepared in a mixture of chloroform : methanol at a ratio 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Then 200 μL of this solution was added to a 25 mL round-bottom flask covered by aluminium foil to protect lipids from photooxidation. For laurdan-containing liposomes, 100 μL of 1 mM laurdan solution prepared in the same organic solvent mixture was added. Then, the solution of PC alone or with laurdan was diluted up to 3 mL with the same organic mixture. The increase of the volume was intended to improve the formation of a thin lipid film by increasing the surface area of deposition after evaporation. To this end, the solution of PC was dried using a rotary evaporator at 30 °C and reduced pressure (290 mbar) for 2 hours to obtain a well-dispersed homogeneous lipid film on the walls of the flask. Subsequently, the flask was kept under vacuum for 2 more hours to ensure the complete removal of any organic solvent traces that might have remained tightly attached to the lipid film. The hydration of the dry lipid film was achieved by adding 1 mL of filtered aqueous phosphate buffered saline (PBS) solution to obtain a final concentration of 10 mM PC and 100 μM fluorescent probe (ratio 100:1), and then vortexing for 1 minute every 5 minutes over a period of 1 hour to aid in the swelling and complete resuspension of the PC layers. The obtained milky suspension of multilamellar vesicles (MLVs) was refrigerated overnight to stabilize the emulsion. Finally, the size, lamellarity and homogeneity of MLV suspension were controlled by extrusion using a mini extruder (Avanti Lipids) at room temperature through a 100 nm polycarbonate membrane. The procedure was repeated 29 times, to enable obtaining large unilamellar vesicles (LUVs).
:1. Then 200 μL of this solution was added to a 25 mL round-bottom flask covered by aluminium foil to protect lipids from photooxidation. For laurdan-containing liposomes, 100 μL of 1 mM laurdan solution prepared in the same organic solvent mixture was added. Then, the solution of PC alone or with laurdan was diluted up to 3 mL with the same organic mixture. The increase of the volume was intended to improve the formation of a thin lipid film by increasing the surface area of deposition after evaporation. To this end, the solution of PC was dried using a rotary evaporator at 30 °C and reduced pressure (290 mbar) for 2 hours to obtain a well-dispersed homogeneous lipid film on the walls of the flask. Subsequently, the flask was kept under vacuum for 2 more hours to ensure the complete removal of any organic solvent traces that might have remained tightly attached to the lipid film. The hydration of the dry lipid film was achieved by adding 1 mL of filtered aqueous phosphate buffered saline (PBS) solution to obtain a final concentration of 10 mM PC and 100 μM fluorescent probe (ratio 100:1), and then vortexing for 1 minute every 5 minutes over a period of 1 hour to aid in the swelling and complete resuspension of the PC layers. The obtained milky suspension of multilamellar vesicles (MLVs) was refrigerated overnight to stabilize the emulsion. Finally, the size, lamellarity and homogeneity of MLV suspension were controlled by extrusion using a mini extruder (Avanti Lipids) at room temperature through a 100 nm polycarbonate membrane. The procedure was repeated 29 times, to enable obtaining large unilamellar vesicles (LUVs).
Theoretical estimation of liposome concentration
To determine the number of liposomes, the average number of PC molecules in a liposome is first calculated using eqn (1) and considering that the liposomes are unilamellar.| | 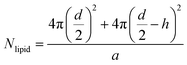 | (1) |
where “d” is the diameter of the LUV, in our case 100 nm, “h” is the average thickness of the lipid bilayer (c.a. 5 nm) and “a” is the lipid head group area that for PC corresponds to 0.71 nm2. The number of liposomes per mL (eqn (2)) is obtained by dividing the initial concentration of lipids (Mlipid) multiplied by the Avogrado number over the number of lipids that were previously calculated, giving a final concentration of 1012 liposome per mL.| | 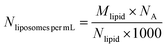 | (2) |
Spectroscopic and morphological CuNPs@GSH characterization
UV-vis spectra were acquired with a double beam spectrophotometer (Shimadzu UV-1601) in the 250–800 nm wavelength range. Excitation and emission fluorescence spectra were acquired using a Varian Cary Eclipse fluorescence spectrometer (Agilent Technologies). Quartz cuvettes (optical path 1 cm, Optech, München, Germany) were used. Transmission electron microscopy (TEM) was performed with a FEI Tecnai 12 microscope (Eindhoven, Netherlands), equipped with a LaB6 filament operating at 120 kV. Size distribution histograms were obtained after TEM image processing using ImageJ software,29 manually highlighting individual NPs on each micrograph. Histograms were produced on three replicates, counting more than 500 nanoparticles. X-ray photoelectron spectroscopy measurements were performed on CuNP colloids deposited on silicon substrates using a PHI Versaprobe II (Chanhassen, MN, USA) spectrometer equipped with monochromatized Al-Kα radiation (1486.6 eV). The binding energy (BE) scale was corrected using the C 1s component at 284.8 eV. The Cu 2p3/2 region was fitted using MultiPak® version 9.9.0.8, which selects an asymmetric function for the Cu(0)/Cu(I) component and a Gaussian–Lorentz function (with a Gauss% = 90%) for the satellite peaks. Infrared spectra were recorded using a PerkinElmer Spectrum-Two. Dried samples were analysed in transmission with attenuated total reflection (ATR) mode, using an ATR accessory with a diamond crystal at a fixed 45° incidence angle. Each spectrum was averaged over 32 scans in the range 400–4000 cm−1, at a spectral resolution of 2 cm−1.
Identification of CuNP permeation across lipids by fluorescence measurements
The potential alteration of phospholipid packing and ordering at distinct depths of the membrane of LUVs in the presence of bioavailable contaminants was investigated using fluorescent membrane probes (laurdan and prodan) at 37 °C. A stock solution of liposomes was prepared and diluted to 100 μM PC in PBS and stored in a brown glass vial. As mentioned before, the laurdan probe was added to the phospholipid solution during the lipid film formation, to ensure appropriate molecule stabilization across the bilayer. On the contrary, a metered volume of prodan in 3:1 chloroform/methanol was added to the liposomal suspension after their preparation and kept in darkness at room temperature for 15 min, to allow its partition into the membrane. In both cases, a final probe concentration of 1 μM was used. Finally, an appropriate volume of copper colloid was added, to have a final concentration of 1011 CuNPs per mL. Steady-state fluorescence assays were performed using a Varian Cary Eclipse fluorescence spectrometer (Agilent Technologies). The samples were incubated at 37 °C in the Peltier multicell holder for 5 min prior to starting the measurements. Fluorescence emission spectra were recorded from 400 to 600 nm by setting the excitation wavelength to 360 nm, and the photomultiplier detector voltage to 540 V with slit widths of 10 nm. The potential modification of lipid packing and ordering, thus hydration of membrane upon interaction with CuNPs into the bilayer was measured using the generalized polarization (GP) equation:| | 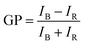 | (3) |
where IB and IR stand for the fluorescence emission intensities at 440 nm and 490 nm, respectively, that correspond to the maximum emission of the membrane probes in nonpolar media and polar environment, respectively. Theoretically, GP values range from −1 to +1. Negative values refer to a liquid-crystalline phase, while positive values are obtained with the gel phase, which is characterized by a more ordered and dehydrated membrane. The change in the GP values was monitored for 24 hours by recording an emission spectrum each hour. Biological membranes are usually characterized by having negative GP values, due to the presence of highly unsaturated acyl chains that allow the permeation of metabolites and other molecules for their proper functioning. The same instrumentation was used to measure the fluorescence emission of calcein from vesicles. The excitation and the emission wavelengths were 495 nm and 516 nm, respectively. Excitation and emission slits were changed, depending on the concentration of total calcein in the solution analyzed. The measured calcein fluorescence was used to quantify the calcein leakage from the vesicles. However, the strong quenching by the CuNP does not allow the measurement of customary leakage kinetics. To overcome this aspect, the ad hoc strategy described in the Results and discussion section has been used.
Results and discussion
Spectroscopic and morphological CuNPs@GSH characterization
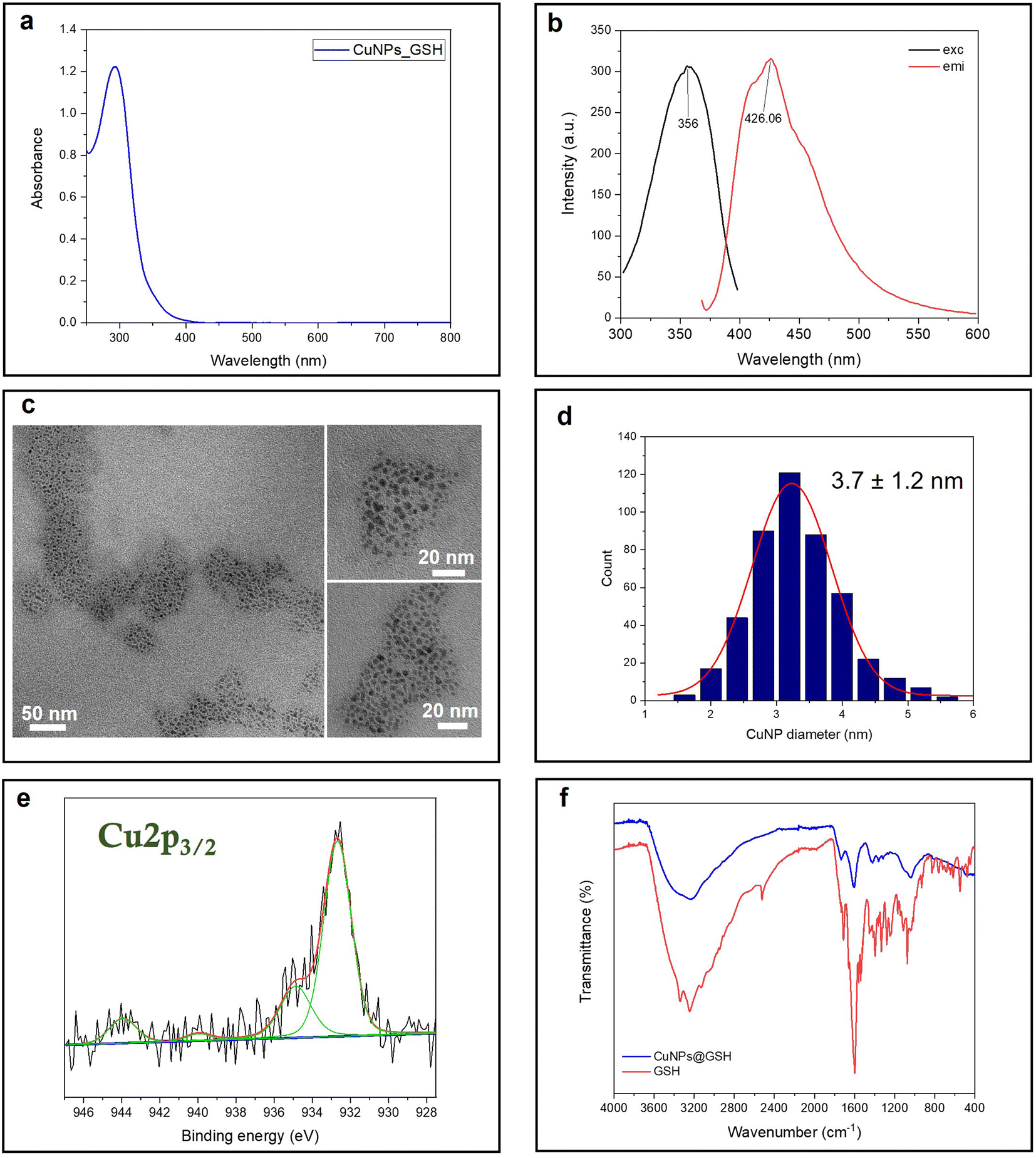
Glutathione is a natural tripeptide widely used to prepare ultra-fine metal nanoparticles.22,30,31 Herein, it is utilized as a stabilizing agent for the synthesis of small CuNPs. The as-prepared copper colloid is almost colorless in visible light. In fact, the UV-vis absorption spectrum shows a strong absorption in the UV region, ascribed to the molecule-like properties of tiny CuNPs. The typical plasmonic peak of CuNPs, falling at around 490 nm, is indeed absent for these fine particles (Fig. 1a). The fluorescent CuNPs showed maximum excitation and emission peaks at 356 nm and 426 nm, respectively, which are in agreement with the findings of Huang and coworkers22 (Fig. 1b). No UV absorption and fluorescence emission was observed for the reactants at room temperature (Fig. S1†). This demonstrated that the formation of CuNPs only occurred when the synthesis was carried out at the activation temperature of 65 °C. Although the CuNP excitation wavelength is very close to the laurdan and prodan's excitation wavelength, we tested that the emission fluorescence intensity of CuNPs was neglectable at the concentrations used for GP studies. The size of CuNPs was assessed by TEM analysis (Fig. 1c and d). According to the size analysis of more than 500 individual particles, the mean diameter of CuNPs was 3.7 ± 1.2 nm. Fig. 1e shows the Cu 2p3/2 XP high-resolution spectrum relevant to the CuNPs@GSH sample. Two photoelectron components were detected: the component centered at 932.7 ± 0.2 eV, attributed to Cu species in a lower oxidation state, Cu0 and Cu1+; and the second one, at 934.8 ± 0.2 eV, attributed to Cu2+.32 The presence of cupric species was confirmed by the shake-up features, fitted by two peaks falling at 941.2 ± 0.2 eV and 943.7 ± 0.2 eV. These results suggest that CuNPs undergo a partial surface oxidation, reasonably due to the unavoidable exposure of colloidal solution to air, both during the synthesis and the sample preparation for the XPS characterization. In addition, the ultra-small size scale of NPs implies an extremely high surface/volume ratio, thus increasing the availability of Cu atoms on the NP surface for potential oxidation. Fig. 1f shows the ATR-FTIR spectra of CuNPs@GSH and pure GSH. As expected, the –SH stretching band (2519 cm−1) of GSH disappears from the surface of CuNPs, indicating the formation of the covalent bonds (Cu–S-R) between GSH and CuNPs.22 The IR spectra also showed several typical glutathione IR peaks, which are consistent with the glutathione spectrum in SpectraBase.33 Among them, the ν(–COOH) stretching vibration at 1730 cm−1, the asymmetric carboxylate vibration νas(COO–) at 1610 cm−1, the COO– symmetric stretching vibration at 1410 cm−1, and ν(C–N) around 1050–1000 cm−1 are the most significant.34
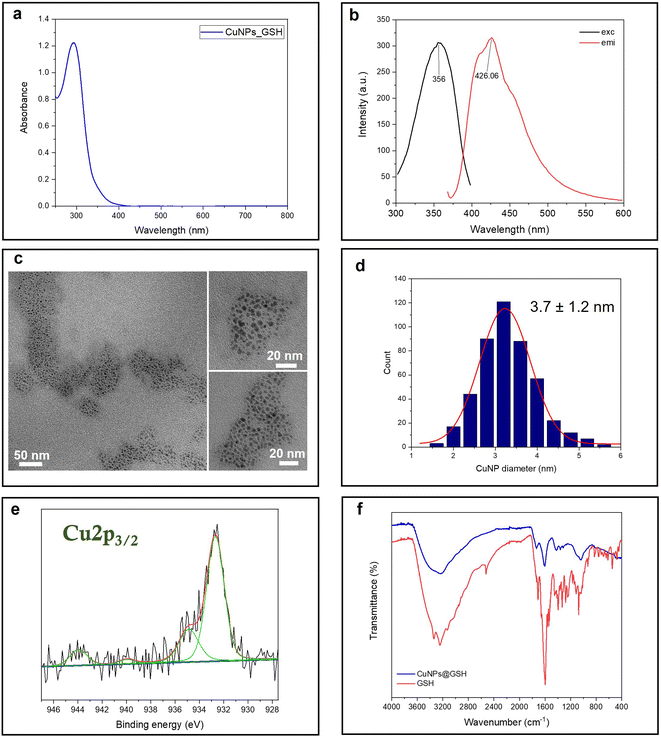 |
| | Fig. 1 Spectroscopic and morphological characterization of CuNPs: (a) UV-vis spectrum; (b) fluorescence spectra: (c) TEM images; (d) size histogram; (e) Cu 2p3/2 XPS spectrum; and (f) ATR-FTIR spectra. | |
Fluorescence generalized polarization spectroscopy
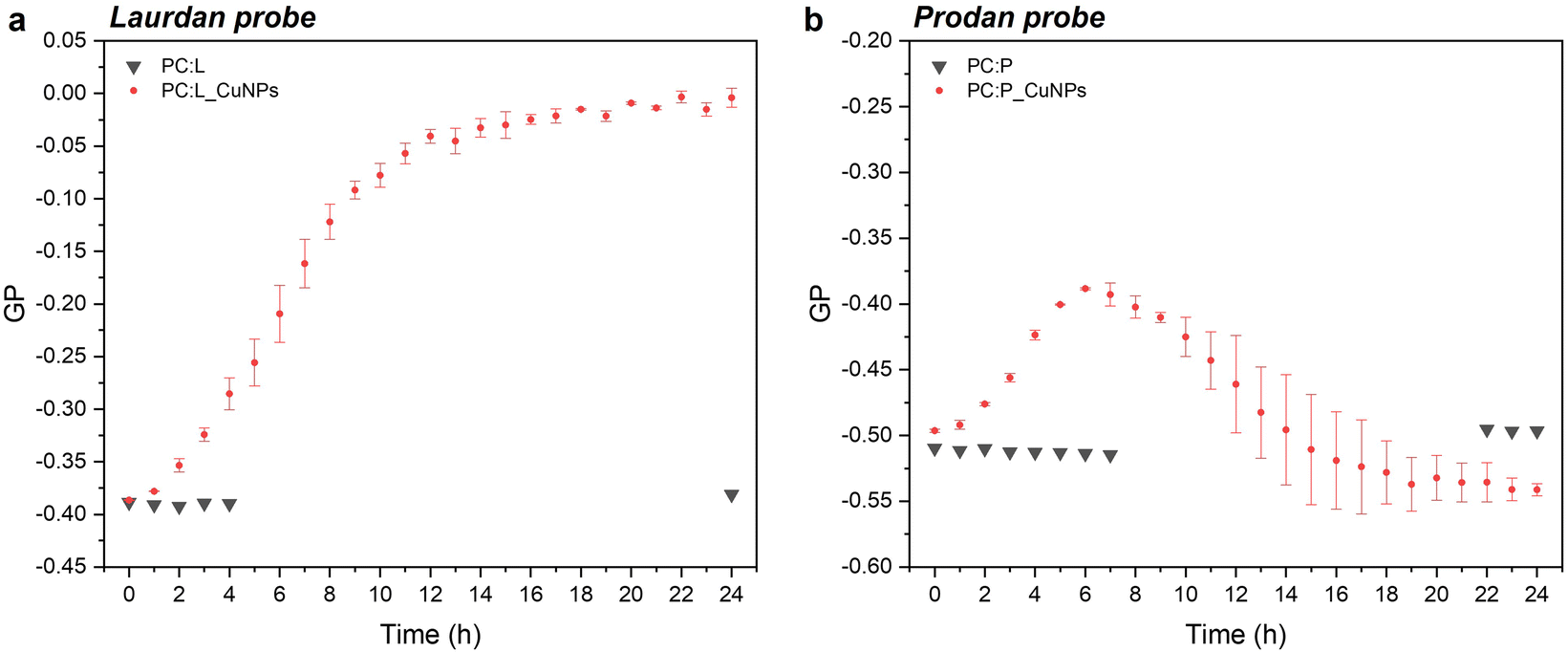
Steady-state fluorescence emission spectra and Stokes shifts of the fluorophore-bearing laurdan and prodan probes are good indicators of structural and dynamic alterations of liposome lipid bilayers. The studies were carried out in 24 hours, to examine the GP trend over time as a function of the CuNP interaction with the liposomes. The emission spectra showed a decrease in the peak maximum throughout time for both probes, as shown in Fig. S2.† In particular, the emission spectra of laurdan showed a remarkable blue shift of the emission peak up to 470 nm (Fig. S2a†), with the subsequent significant variation of the GP (calculated according to eqn (1)), which increased towards positive values from −0.39 to 0 (see Fig. 2a). Moreover, it reached a plateau level around 20 hours. On the other side, the GP values relative to prodan fluorescent emission, characterized by a short acyl chain composed of a propyl group, showed a smaller variation over time, from −0.50 to −0.40. This observation can be attributed to the ability of prodan to interact with both the external water molecules and the polar head group of the phospholipids as corroborated by the more negative GP values compared to those of laurdan.10 Consequently, the higher polarity of the medium around prodan is much less affected by the presence of CuNPs. Interestingly, the increment of GP-prodan values did not keep constant throughout time. As shown in Fig. 2b, it reached a maximum GP value around 6 hours and then decreased again towards more negative GP values. Additionally, the mobility of the prodan probe is identified by the higher variability of GP values after an incubation time of 10 h.
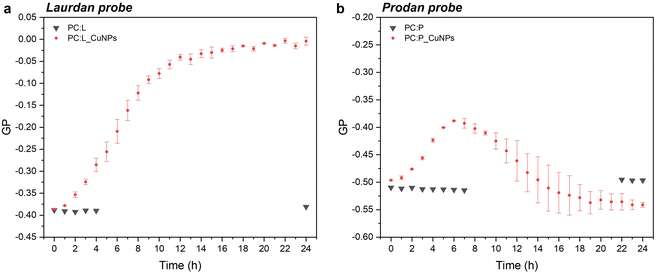 |
| | Fig. 2 . Time-dependent GP variation for laurdan (a) and prodan (b) in the presence of 1011 CuNPs per mL. | |
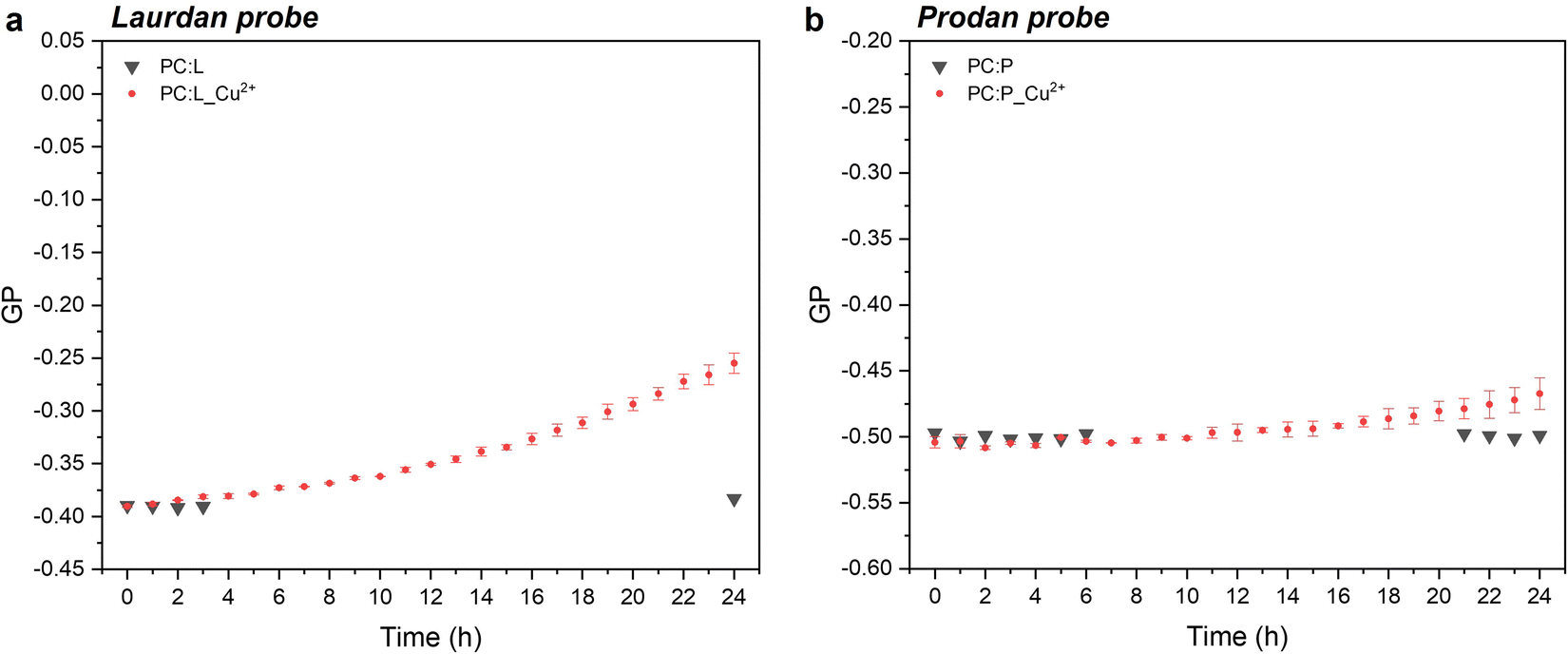
In the case of laurdan, the increment (hypsochromic shift) of GP values is indicative of the packed structure of the lipid bilayer in the presence of CuNPs, thereby suggesting enhanced lipid packing and ordering at the glycerol level with concomitant membrane dehydration.10 Our hypothesis is that CuNPs interact firstly with the polar head group of the external leaflet of the membrane, and then penetrate through the outermost part of the liposome membrane during the first incubation hours (prodan-responsive zone). During this period, CuNPs accumulate on the membrane/water interface. With longer assay times, CuNPs are able to reach the wider bilayer zone and interact with the phospholipid acyl chains (laurdan-responsive zone) with potential accumulation in the bilayer zone or penetration into the inner liposome part (vide infraFig. 5). A similar trend was reported by Luchini and colleagues17 on lipid-functionalized AuNPs, although they observed a more significant GP variation in a time-frame of about one hour. Such discrepancies are probably due to the different metals, as well as to the different liposome/NP concentrations ([18-lysophosphocholine]AuNPs = 250 μM). Indeed, to the best of our knowledge, no papers have been published about the study of membranotropic effects of CuNPs to date. However, it was demonstrated by means of density functional theory and molecular dynamics simulations that Cu2+ ions can cause the decrease of phospholipid bilayer fluidity, due to the phospholipid coupling by a Cu–Cu bond.35 It has been reported that upon Cu2+ binding to the negatively charged phosphate group of the polar head of the phospholipid, Cu2+ is reduced to Cu+. As a consequence of the anomalous attraction between Cu+ cations, the binding between two close lipids is triggered, resulting in the formation of the PL–Cu–Cu–PL structure. Thus, in order to investigate the membranotropic effects of the Cu2+ ions, which could be potentially released from the surface of the CuNPs, steady-state fluorescence studies involving the fluorophore probes were also carried out on Cu2+ rather than CuNPs. To this aim, Cu2+ ions were added to the liposome solution using CuCl2 at the same concentration as the CuNP synthesis. Fig. 3a and b show the GP temporal profiles of laurdan and prodan, respectively, as a function of the Cu2+ interaction with the liposome. It is evident that the effect of the Cu2+ ions is less significant than that of CuNPs, especially for the outer part of the lipidic membrane (prodan-responsive probe). In fact, the GP increase for the laurdan was just moderate, compared to that obtained for the CuNPs, and the GP increment for the prodan was almost negligible. These findings indicate that the main reason for the change of the membranotropic properties of the membrane is occasioned by CuNPs; however, the Cu2+ ions potentially released from the CuNP surface may still play an additional or synergistic role in deeper lipidic layer alterations. The potential effect caused by the stabilizing agent GSH was also evaluated, showing no significant changes in the laurdan GP values over the course of 24 h (data not shown).
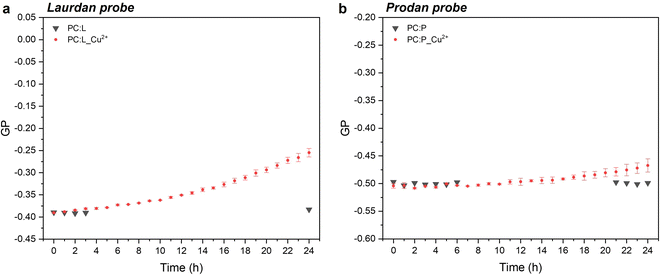 |
| | Fig. 3 Temporal profiles of the GP value for laurdan (a) and prodan (b) without and with Cu2+ ions at the same concentration as that of the synthesis of CuNPs, diluted 100 times as in the case of CuNPs (0.0015 mM). | |
Fluorescence leakage assay
The effect of CuNPs on membrane permeability can be investigated by measuring the fluorescence of liposomes entrapping a high concentration of calcein. Calcein is an aqueous soluble fluorophore that self-quenches at high concentrations, so in the customary leakage assay, one probes the kinetics of fluorescence emission associated with the vesicle breakdown and subsequent calcein release and dilution and detection in the outer medium.14 Such an approach cannot be regrettably used in our case because both the CuNPs or the Cu2+ ions are very efficient quenchers of calcein emission. Indeed, the addition of CuNPs or Cu2+ to a diluted solution of highly fluorescent calcein (without any vesicle) induces in a few minutes the quenching of more than 90% of the signal. Accordingly, the fluorescence emission of LUV-containing calcein challenged by CuNPs does not change in time because even in the presence of slow leakage events the emission of calcein molecules in the matrix solution is efficiently quenched by the CuNPs. For this reason, we modified the leakage assay as follows: various samples having the same concentration of LUV and CuNPs have been prepared simultaneously and let to incubate at different times. After a given incubation time, the vesicles were disrupted by adding the detergent Triton X100 (TX), which induced the prompt release of all the calcein molecules entrapped in the LUVs, and the fluorescence emission was collected immediately. Operating in such a way, the emission of calcein that permeated during the incubation time (before TX addition) is lost because of the CuNP quenching and the fluorescence emission recorded is due only to the remaining calcein that is released from the liposomes after the TX addition. The fraction of calcein that remained in the intact vesicles during the incubation time can be evaluated from eqn (4):| | 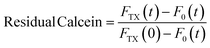 | (4) |
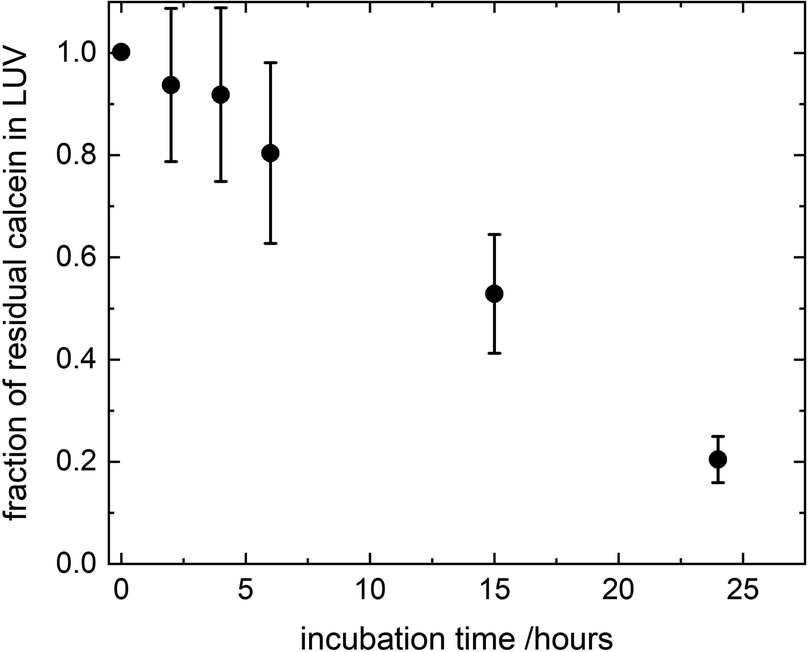
where FTX(t) and F0(t) are the fluorescence intensity values measured immediately after and before, respectively, the addition of TX to a solution of LUV incubated with CuNPs for a time t. FTX(0) is the fluorescence emission taken by adding simultaneously TX and CuNPs. The dependence of the residual calcein in intact liposomes on the incubation time with the CuNPs is shown in Fig. 4. The fraction of calcein entrapped in intact liposomes decreased linearly with the time they were in contact with the CuNPs. After 24 hours almost all the calcein (80%) was quenched by the CuNPs. This indicates that in the presence of CuNPs the phospholipid membrane becomes gradually more permeable and the calcein is released in a controlled manner.
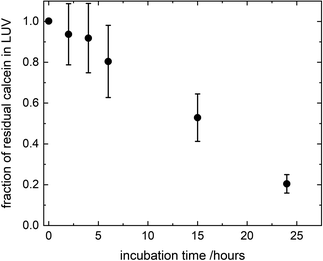 |
| | Fig. 4 Fraction of calcein molecules remaining in liposomes after mixing with CuNPs at different incubation times, as obtained by liposome solubilization with TritonX100. | |
Liposome hydrodynamic diameter over time
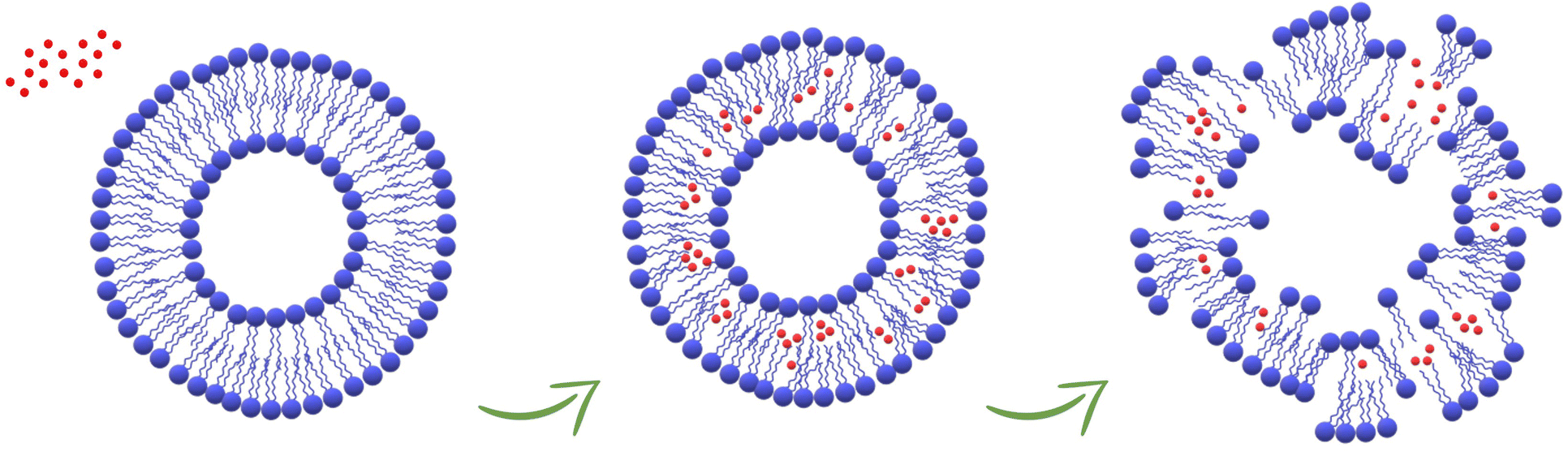
Our findings in this work indicate that CuNPs might trigger changes in liposome size over time. The hydrodynamic diameter was then monitored for 24 hours by DLS measurements. As shown by the trend in Fig. S4,† the liposome size significantly increased approximately 11 hours after mixing with CuNPs, suggesting agglomeration of vesicles or their disruption and transformation into worm-like micelles (which are expected to have a larger hydrodynamic size). Table 1 compiles the temporal size profiles of the liposomes before and after mixing with CuNPs (1011 CuNPs per mL) or Cu2+ (0.0015 mM). Interestingly, the vesicle size does not undergo any change over time when Cu2+ ions are added to the liposome solution. These results definitively are consistent with the fluorescence studies, providing evidence that ultra-small CuNPs are able to alter the physicochemical features of the phospholipid membranes and jeopardise the stability of liposomes.
Table 1 Hydrodynamic diameter of liposomes over time measured by DLS after interaction with CuNPs (1011 CuNPs per mL) or Cu2+ (0.15 mM)
| Time (h) |
Liposome (nm) |
CuNPs-liposome (nm) |
Cu2+-liposome (nm) |
| 0 |
110 ± 4 |
105.3 ± 1.1 |
103.5 ± 1.1 |
| 2 |
104.5 ± 1.1 |
108.3 ± 2.0 |
107.5 ± 2.1 |
| 4 |
106.1 ± 0.5 |
108 ± 3 |
107.4 ± 1.2 |
| 20 |
110 ± 3 |
700 ± 160 |
107.2 ± 0.8 |
| 24 |
116.7 ± 0.1 |
550 ± 350 |
111.0 ± 0.8 |
Conclusions
In this work, the in vitro supramolecular interactions of 4 nm-CuNPs with phospholipids integrating biological membranes were studied. For this purpose, PC-liposomal nanoparticles tailored with two distinct polarity-sensitive membrane probes, prodan and laurdan, were harnessed for discrimination of membranotropic effects at the polar head group and glycerol level/acyl chains of PC, respectively. The use of the two fluorescence probes is suitable for ascertaining low-resolution changes in bilayer organization by GP measurements that indicate alterations in lipid ordering and membrane hydration due to the presence of CuNPs. In particular, the increase of laurdan GP towards more positive values suggested the enhancement of lipid packing and the order at the glycerol level, with concomitant membrane dehydration. In addition, the investigation over time of calcein leakage and liposome hydrodynamic diameter confirms that ultra-small CuNPs are able to damage the phospholipid membranes, with the subsequent vesicle agglomeration or disruption and transformation into worm-like micelles (Fig. 5). Interestingly, the comparison of CuNPs against Cu2+ demonstrated that the potential deleterious effects are mainly attributed to the presence of whole nanoparticles themselves, rather than to Cu2+ released from their surface. This is an interesting finding for the antimicrobial application of CuNPs, corroborating once more the advantages of their use as nano-reservoirs of bioactive Cu2+ ions, entrapped in solid state films/coatings because in this way the direct release of potentially toxic CuNPs is avoided. Fluorescence spectroscopy proved to be a suitable tool for the investigation of membranotropic effects of nanoantimicrobial particles on the phospholipid membranes, paving the way for further insights about the potential toxicity of nanoantimicrobials. Work is in progress to extend the study to antimicrobial nanoparticles of different size and composition.
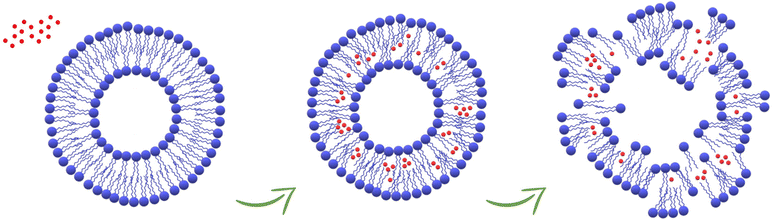 |
| | Fig. 5 Schematic representation of the damage of the liposomes caused by the accumulation of CuNPs in the phospholipid membranes. | |
Author contributions
Conceptualization: M. M., N.·C., M. I., and M. O.; methodology: M. O, M. I., M. M., G. P., and N.·C.; investigation: M. I., M. O., and H. M.; writing – original draft: M. I.; writing – review & editing: M. O., H. M., G. P., N.·C., and M. M.; supervision: M. M. and N.·C.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was partially carried out during the PhD secondment of M. I. at the University of the Balearic Islands. Prof. Rosaria Anna Picca is kindly acknowledged for her contribution to XPS analyses. Dr Maria Chiara Sportelli is kindly acknowledged for suggestions on the CuNP synthesis. Partial financial support is acknowledged from HORIZON EUROPE SEEDS – Codice identificativo progetto S12 (N. C.), HORIZON EUROPE SEEDS - SCOOP – Codice identificativo progetto S52 (G. P.) and from the MUR PNRR Extended Partnership initiative on Emerging Infectious Diseases – project no. PE00000007, INF-ACT (G. P.). H. M. was funded by POR PUGLIA FESR-FSE 2014/2020 Research for Innovation (REFIN)-Codice Pratica: 7BDC8679. M.M. acknowledges financial support from the Spanish State Research Agency (Agencia Estatal de Investigación, AEI/10.13039/501100011033), and the Spanish Ministry of Science and Innovation (Ministerio de Ciencia e Innovación, MCIN) through project PID2020-117686RB-C33 (AEI/MCIN). M. O. is grateful to the Spanish Ministry of Universities and the European Union (NextGenerationEU) for granting a “Margarita Salas” post-doctoral position in the framework of the Recovery, Transformation and Resilience Plan.
References
- M. L. Ermini and V. Voliani, ACS Nano, 2021, 15, 6008–6029 CrossRef CAS PubMed.
- M. C. Sportelli, M. Izzi, A. Volpe, V. Lacivita, M. Clemente, C. Di Franco, A. Conte, M. A. Del Nobile, A. Ancona and N. Cioffi, Food Packag. Shelf Life, 2019, 22, 100422 CrossRef.
- A. Purniawan, M. I. Lusida, R. W. Pujiyanto, A. M. Nastri, A. A. Permanasari, A. A. H. Harsono, N. H. Oktavia, S. T. Wicaksono, J. R. Dewantari, R. R. Prasetya, K. Rahardjo, M. Nishimura, Y. Mori and K. Shimizu, Sci. Rep., 2022, 12, 4835 CrossRef CAS PubMed.
- A. A. Cortes and J. M. Zuñiga, Diagn. Microbiol. Infect. Dis., 2020, 98, 115176 CrossRef CAS PubMed.
- V. Govind, S. Bharadwaj, M. R. Sai Ganesh, J. Vishnu, K. V. Shankar, B. Shankar and R. Rajesh, BioMetals, 2021, 34, 1217–1235 CrossRef CAS PubMed.
- R. Rajamanikandan, B. Aazaad, S. Lakshmipathi and M. Ilanchelian, Microchem. J., 2020, 158, 105253 CrossRef CAS.
-
Directorate-General for Health and Food Safety, SCCS – Final Opinion on Copper (Nano) and Colloidal Copper (Nano), https://health.ec.europa.eu/latest-updates/sccs-final-opinion-copper-nano-and-colloidal-copper-nano-2021-03-05_en, (accessed July 25, 2023) Search PubMed.
- K. L. Chen and G. D. Bothun, Environ. Sci. Technol., 2014, 48, 873–880 CrossRef CAS PubMed.
- J.-J. Ortega-Calvo, J. Harmsen, J. R. Parsons, K. T. Semple, M. D. Aitken, C. Ajao, C. Eadsforth, M. Galay-Burgos, R. Naidu, R. Oliver, W. J. G. M. Peijnenburg, J. Römbke, G. Streck and B. Versonnen, Environ. Sci. Technol., 2015, 49, 10255–10264 CrossRef CAS PubMed.
- M. Oliver, A. Bauzá, A. Frontera and M. Miró, Environ. Sci. Technol., 2016, 50, 7135–7143 CrossRef CAS PubMed.
- M. Oliver, M. Adrover, A. Frontera, J. Ortega-Castro and M. Miró, Sci. Total Environ., 2020, 738, 140096 CrossRef CAS PubMed.
- S. Andrade, M. J. Ramalho, J. A. Loureiro and M. C. Pereira, J. Mol. Liq., 2021, 334, 116141 CrossRef CAS.
- L. Šturm and N. Poklar Ulrih, Int. J. Mol. Sci., 2021, 22, 6547 CrossRef PubMed.
- H. Mateos, A. Mallardi, F. Blasi and G. Palazzo, Colloids Surf., B, 2022, 220, 112885 CrossRef CAS PubMed.
- A. Bhat, L. W. Edwards, X. Fu, D. L. Badman, S. Huo, A. J. Jin and Q. Lu, Appl. Phys. Lett., 2016, 109, 263106 CrossRef PubMed.
- C. Contini, J. W. Hindley, T. J. Macdonald, J. D. Barritt, O. Ces and N. Quirke, Commun. Chem., 2020, 3, 1–12 CrossRef PubMed.
- A. Luchini, G. D'Errico, S. Leone, Z. Vaezi, A. Bortolotti, L. Stella, G. Vitiello and L. Paduano, Colloids Surf., B, 2018, 168, 2–9 CrossRef CAS PubMed.
- C. Contini, M. Schneemilch, S. Gaisford and N. Quirke, J. Exp. Nanosci., 2018, 13, 62–81 CrossRef CAS.
- H. L. Karlsson, P. Cronholm, Y. Hedberg, M. Tornberg, L. De Battice, S. Svedhem and I. O. Wallinder, Toxicology, 2013, 313, 59–69 CrossRef CAS PubMed.
- J. Hedberg, H. L. Karlsson, Y. Hedberg, E. Blomberg and I. Odnevall Wallinder, Colloids Surf., B, 2016, 141, 291–300 CrossRef CAS PubMed.
- N. Li, H. J. Sui and D. M. Gao, Adv. Mater. Res., 2012, 356–360, 2274–2277 CAS.
- H. Huang, H. Li, J.-J. Feng, H. Feng, A.-J. Wang and Z. Qian, Sens. Actuators, B, 2017, 241, 292–297 CrossRef CAS.
-
I. Pacheco and C. Buzea, in Metal Nanoparticles, John Wiley & Sons, Ltd, 2018, pp. 237–293 Search PubMed.
- T. Parasassi, E. K. Krasnowska, L. Bagatolli and E. Gratton, J. Fluoresc., 1998, 8, 365–373 CrossRef CAS.
- G. Gunther, L. Malacrida, D. M. Jameson, E. Gratton and S. A. Sánchez, Acc. Chem. Res., 2021, 54, 976–987 CrossRef CAS PubMed.
- W. Haiss, N. T. K. Thanh, J. Aveyard and D. G. Fernig, Anal. Chem., 2007, 79, 4215–4221 CrossRef CAS PubMed.
- A. D. Bangham, J. De Gier and G. D. Greville, Chem. Phys. Lipids, 1967, 1, 225–246 CrossRef CAS.
- M. J. Hope, M. B. Bally, G. Webb and P. R. Cullis, Biochim. Biophys. Acta, Biomembr., 1985, 812, 55–65 CrossRef CAS PubMed.
-
ImageJ, https://imagej.nih.gov/ij/, (accessed May 31, 2019) Search PubMed.
- X. Yuan, Y. Tay, X. Dou, Z. Luo, D. T. Leong and J. Xie, Anal. Chem., 2013, 85, 1913–1919 CrossRef CAS PubMed.
- G. Zhang, Y. Li, J. Xu, C. Zhang, S. Shuang, C. Dong and M. M. F. Choi, Sens. Actuators, B, 2013, 183, 583–588 CrossRef CAS.
- N. Cioffi, L. Torsi, N. Ditaranto, G. Tantillo, L. Ghibelli, L. Sabbatini, T. Bleve-Zacheo, M. D'Alessio, P. G. Zambonin and E. Traversa, Chem. Mater., 2005, 17, 5255–5262 CrossRef CAS.
-
Glutathione (Reduced) – SpectraBase, https://spectrabase.com/compound/6507B5KJYT1, (accessed September 9, 2023) Search PubMed.
- J. P. Monrás, V. Díaz, D. Bravo, R. A. Montes, T. G. Chasteen, I. O. Osorio-Román, C. C. Vásquez and J. M. Pérez-Donoso, PLoS One, 2012, 7, e48657 CrossRef PubMed.
- X. Jiang, J. Zhang, B. Zhou, P. Li, X. Hu, Z. Zhu, Y. Tan, C. Chang, J. Lü and B. Song, Sci. Rep., 2018, 8, 14093 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Miquel
Oliver‡
b,
Helena
Mateos
a,
Gerardo
Palazzo
a,
Miquel
Oliver‡
b,
Helena
Mateos
a,
Gerardo
Palazzo