DOI:
10.1039/D3NA00413A
(Paper)
Nanoscale Adv., 2023,
5, 5036-5044
Nanobiocatalysts with inbuilt cofactor recycling for oxidoreductase catalysis in organic solvents†
Received
14th June 2023
, Accepted 15th August 2023
First published on 1st September 2023
Abstract
The major stumbling block in the implementation of oxidoreductase enzymes in continuous processes is their stark dependence on costly cofactors that are insoluble in organic solvents. We describe a chemical strategy that allows producing nanobiocatalysts, based on an oxidoreductase enzyme, that performs biocatalytic reactions in hydrophobic organic solvents without external cofactors. The chemical design relies on the use of a silica-based carrier nanoparticle, of which the porosity can be exploited to create an aqueous reservoir containing the cofactor. The nanoparticle core, possessing radial-centred pore channels, serves as a cofactor reservoir. It is further covered with a layer of reduced porosity. This layer serves as a support for the immobilisation of the selected enzyme yet allowing the diffusion of the cofactor from the nanoparticle core. The immobilised enzyme is, in turn, shielded by an organosilica layer of controlled thickness fully covering the enzyme. Such produced nanobiocatalysts are shown to catalyse the reduction of a series of relevant ketones into the corresponding secondary alcohols, also in a continuous flow fashion.
Introduction
In a world facing major environmental and resource scarcity issues, the quest for sustainable manufacturing is gaining undeniable significance.1,2 The ambition to transform the current throwaway economy to a circular one calls, among others, for energy-efficient chemical processes avoiding catalysts based on once-ubiquitous yet scarce transition metals.3,4 In this context, enzymes appear to be of pivotal importance.5–7 Besides exquisite catalytic properties both in terms of efficiency and selectivity, they combine several crucial advantages. These advantages include low energy demands and the opportunity to be produced from renewable feedstocks.8 Moreover, the native enantioselectivity of a large majority of enzymes places biocatalysis in a pivotal position when it comes to the production of chiral active pharmaceutical ingredients.9–12 Consequently, the contribution of biocatalysis to industrial catalysis has been significantly increasing over the past two decades.8,13,14 The expected paradigm shift from metal-to bio-catalysis is, however, hampered by several constraints inherent to the use of native enzymes in industrial processes. This holds particularly true when it comes to the processing of molecules displaying limited water solubility. This necessitates the reaction to be carried out in organic solvents (OSs). Indeed, besides esterases that can withstand high proportions of organic solvents, a large majority of enzymes display very limited stability in these conditions.15 This is explained by the importance of structural water molecules that support the macromolecular structural integrity and hence biocatalytic activity.16,17 Scientists have devoted considerable effort to develop effective strategies to enhance enzyme stability in OSs. Strategies that have been proven successful are mainly based on either the isolation of novel enzymes from extreme environments,18 protein immobilisation and cross-linking, or the use of modern enzyme engineering methods.19 A rational strategy of enzyme engineering for improving compatibility in OSs has been recently published;20 it is however limited to lipases and the general challenge of increasing enzyme stability in organic solvents remains mainly unmet. Additionally, a significant number of industrially relevant processes involve both solvophilic and solvophobic reaction partners.21,22 This is particularly the case when it comes to the use of oxidoreductases, as they are dependent on strictly water-soluble cofactors (mainly based on nicotinamide dinucleotides), to process hydrophobic substrates. Oxidoreductases represent certainly one of the most relevant classes of enzymes for industrial biocatalysis, especially for the production of chiral compounds. Oxidoreductases, however, necessitate cofactors, and cofactor recycling is the key to industrial cofactor-dependent enzyme use. There have been studies and successful implementations in aqueous systems where the cofactor is recycled by the addition of either an additional recycling oxidoreductase enzyme, a cosubstrate or a combination thereof.23,24 For example, Contente and Paradisi recently developed a system in which both the oxidoreductase used for the targeted reaction (i.e., production of tyrosol and 2-arylpropanols) and the recycling one were immobilised on a resin packed in a column.25 Another study approached cofactor recycling by applying a biomimetic enzyme engineering strategy. Scott et al. developed a strategy where a flexible arm, to which the cofactor is conjugated, swings between two catalytic domains of a fusion protein.26 Other studies focused on batch mode synthesis in neat organic solvent using various immobilisation supports for the selected oxidoreductase enzymes.27,28 It has also been demonstrated that few alcohol dehydrogenases from Geotrichum candidum retain the cofactor in the catalytic site.29 Herein, we report the synthesis of oxidoreductase nanobiocatalysts capable of self-sustaining cofactor recycling in neat organic solvents. As carrier for the nanobiocatalyst, we chose a mesoporous silica nanoparticle (MSN) displaying a dual porosity. Nanostructured carriers provide crucial advantages for the preparation of biocatalytic material as highlighted for biofuel cells design,30 single-atom enzyme-metal hybrids31 and metal–organic frameworks-based nanobiocatalysts preparation.32,33 The inner core of the selected MSNs was designed so to display large center-radial conical pores, which served as cofactor reservoir (Fig. 1). It was further coated by a layer of reduced mesoporosity which allowed to restrict the immobilisation of the selected cofactor-dependent oxidoreductase enzyme to the surface of the nanoparticle and to avoid its diffusion within the nanoparticle core. At the surface of the MSN where the enzyme was immobilised, we grew a protective organosilica layer, protecting the enzyme against the deleterious effect of the solvent; Fig. 1. The overall silica architecture, when loaded with water containing the cofactor and transferred into a water-immiscible hydrophobic solvent, is expected to act as an aqueous reservoir. This reservoir is expected to retain the cofactor owing to its stark preference for the aqueous phase compared to hydrophobic OSs. Indeed, the cofactor displays a markedly stronger affinity for the aqueous phase (predicted log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P value of −7 for octanol/water)34 than for a hydrophobic organic solvent. This is expected to strongly limit cofactor leaching when the loaded MSNs will be transferred in OSs. The catalytic potential of such prepared nanobiocatalyst was tested in a series of organic solvents of increasing hydrophobicity; this allowed demonstrating the independence of the nanobiocatalyst to external cofactor molecules.
P value of −7 for octanol/water)34 than for a hydrophobic organic solvent. This is expected to strongly limit cofactor leaching when the loaded MSNs will be transferred in OSs. The catalytic potential of such prepared nanobiocatalyst was tested in a series of organic solvents of increasing hydrophobicity; this allowed demonstrating the independence of the nanobiocatalyst to external cofactor molecules.
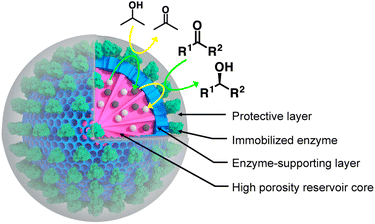 |
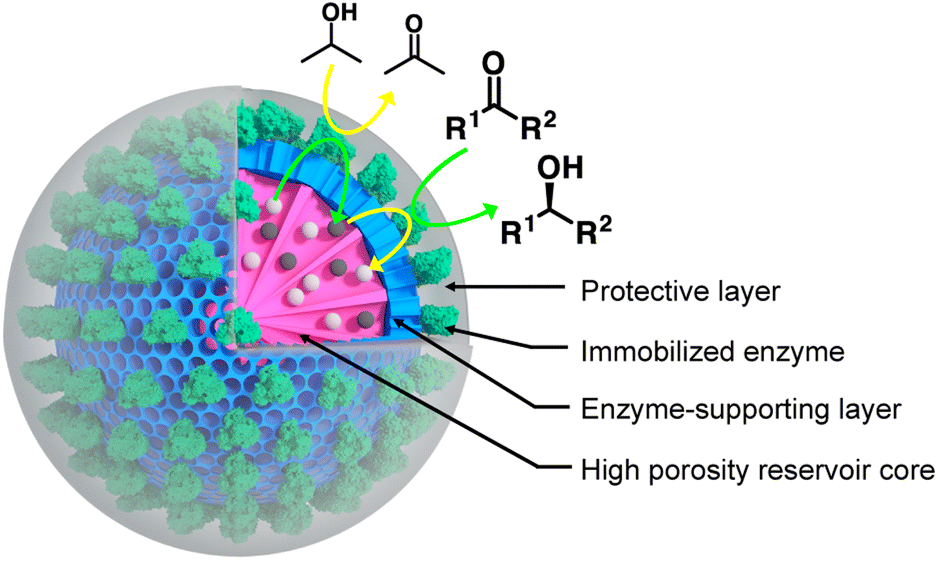
| | Fig. 1 Nanobiocatalyst design. It is based on the use of mesoporous silica nanoparticles (MSNs) with a radial pore architecture. While the inner core (pink) is acting as cofactor reservoir, the outer layer (blue) is exploited to immobilise the target enzyme, namely a secondary alcohol dehydrogenase from Thermoanaerobacter ethanolicus. The immobilised enzyme is shielded in an organosilica protective layer of controlled thickness, providing the enzyme with higher stability when submitted to organic solvents. The studied ketone substrates are expected to migrate from the organic solvent to the enzyme biocatalytic site to be transformed to the corresponding alcohol, at the cost of the oxidation of a cofactor molecule NADPH (grey spheres), to NADP+ (white spheres). The latter is, in turn, recycled by being reduced by the same enzyme concomitantly to the oxidation of an isopropanol molecule provided in the organic phase. | |
Results and discussion
Nanobiocatalyst synthesis and characterisation
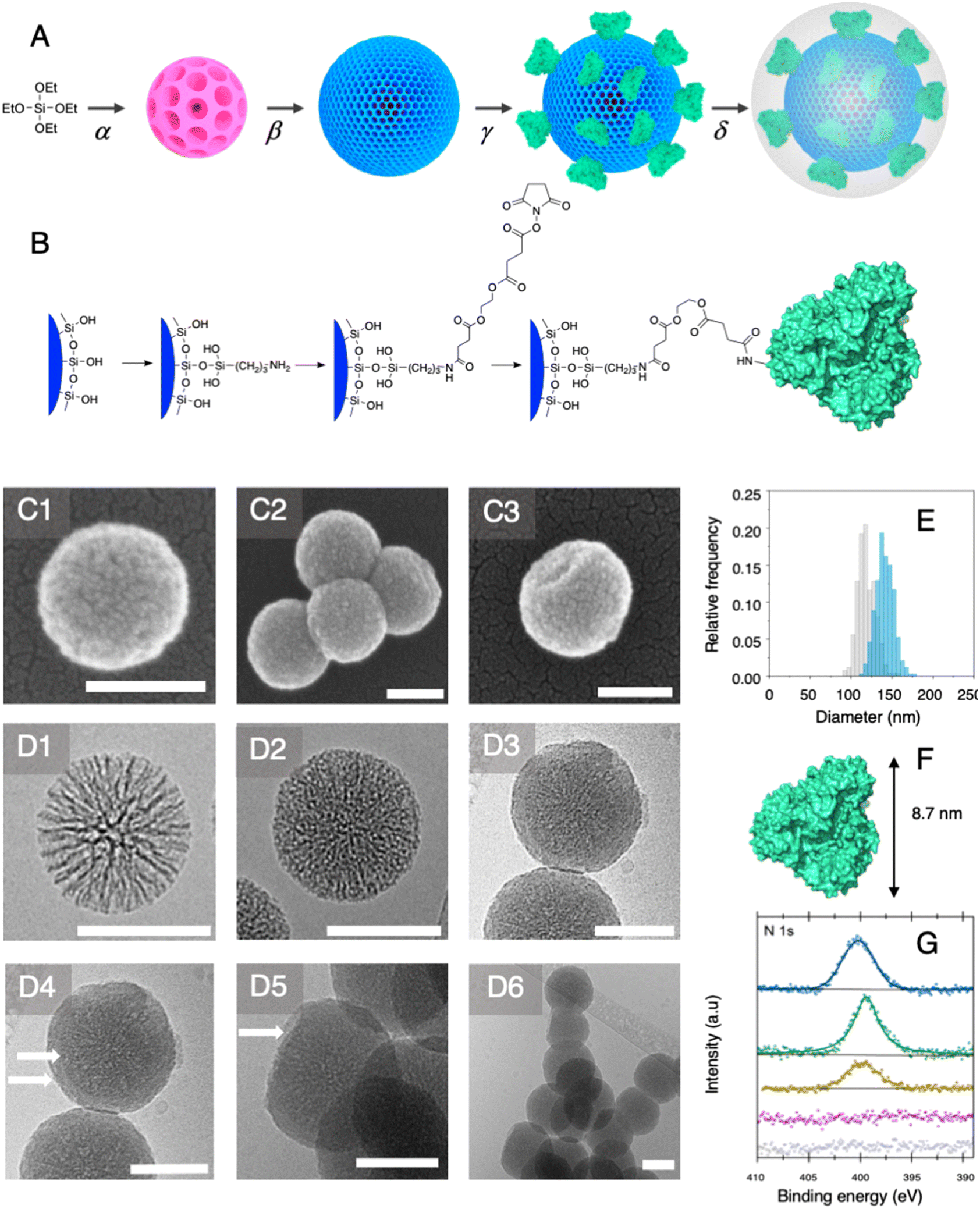
Among established porous solid supports, we chose hierarchical mesoporous silica nanoparticles possessing a radial pore architecture (MSNs).35 Those systems display the advantage of being monodisperse in size; they provide the opportunity to design multiple concentric layers with varying yet controlled porosity. This design can be used to construct a core displaying a large pore volume serving as a reservoir and a second shell of lower porosity. The latter is expected to restrict prospective enzyme immobilisation at the surface of the MSNs and prevent protein mass transfer within the core of the particles. To produce these MSNs, we applied a bi-phase stratification approach using tetraethyl orthosilicate (TEOS) as silica precursor and cetyltrimethylammonium chloride (CTAC) as a template-forming surfactant. The reaction conditions were adapted from the work published by Shen et al. to produce a core with large radial pores (MSN1G) and a second layer with smaller pores (MSN2G).35 The MSNs produced were characterised by means of field-emission scanning electron microscopy (SEM) and cryogenic transmission electron microscopy (cryoEM); representative micrographs are provided in Fig. 2. The SEM micrographs show spherical nanoparticles with an uneven surface and an average size of 119 ± 11 nm (Fig. 2C1). CryoEM experiments confirmed the concentric radial pore architecture of MSN1G (Fig. 2D1); they also confirmed that the growth of the second layer, using the established bi-phase stratification approach, yields an outer shell of limited porosity (Fig. 2D2). Additional nitrogen adsorption experiments (Brunauer–Emmett–Teller method) confirmed the dual porosity of the nanoparticles as previously demonstrated by Shen et al.35 Characterisation of the MSNs by thermogravimetric analysis and X-ray photoelectron spectroscopy (XPS) confirmed the absence of surfactant in the MSNs after the work-up procedure (Fig. 2G and ESI, Fig. S1–S3†). To introduce chemical moieties that will allow the further anchoring of the enzyme at the surface of the MSNs, the surface of the MSNs were amino-modified by reaction with (3-aminopropyl)triethoxysilane (APTES) to yield MSN-NH2. The presence of amine functions at the surface of MSN-NH2 was confirmed by XPS (Fig. 2G and ESI, Fig. S3†). As a model enzyme, we selected a variant of the secondary alcohol dehydrogenase from Thermoanaerobacter ethanolicus (TeSADH W110A).36,37 TeSADH W110A is a homotetrameric enzyme with an estimated diameter of 8.7 nm (PDB code: 7JNQ, Fig. 2F). When compared to the wild type enzyme, this mutant was shown to possess a larger catalytic pocket, which allows accommodating large substrates such as phenol-containing ketones.27,36 Our initial attempts for the immobilisation of TeSADH W110A on the surface of MSN-NH2 to yield MSNENZ were made using glutaraldehyde as a bifunctional cross-linker, as previously described for the immobilisation on silica of a series of enzymes (β-galactosidase, laccase, acid phosphatase, alcohol dehydrogenase).38 This resulted in excellent enzyme immobilisation (91%), as measured by analysing the soluble fraction of the immobilisation reaction mixture with a protein quantification assay. The activity of the immobilised enzyme was tested with isopropanol (i-PrOH, 2 M) as substrate and nicotinamide adenine dinucleotide phosphate (NADP+) as cofactor; the results showed an unsatisfactory activity retention of only 1.8% when compared to the amount of enzymatic activity added to the reaction mixture. We then selected a different homobifunctional cross-linker displaying a longer spacer arm, namely ethylene glycol bis(succinimidyl succinate).
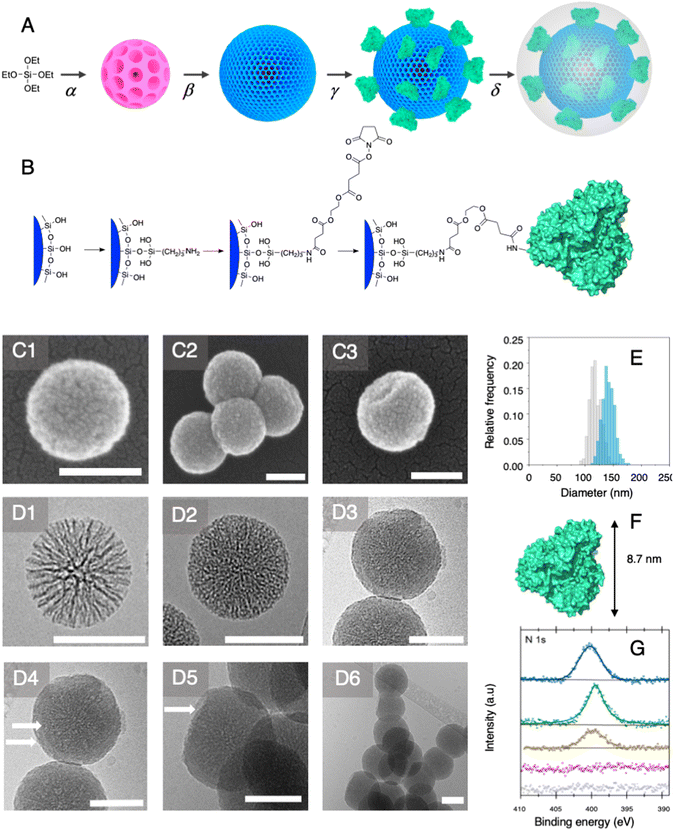 |
| | Fig. 2 Nanoparticle synthesis and characterisation. (A) General synthetic route: a: TEOS polycondensation via an interfacial bi-phase stratification yielding the core SiO2 nanoparticles (α) and the second layer with reduced pore size (β); enzyme immobilisation (γ) and protective layer growth (δ) towards the formation of MSNENZ-S. (B) Enzyme immobilisation route. The silica surface of hierarchical mesoporous silica nanoparticles with a radial pore architecture is partially amino-modified using APTES. After reaction with ethylene glycol bis(succinimidyl succinate), the nanoparticles were reacted with TeSADH W110A in order to yield MSNENZ. (C) SEM characterisation of a bare MSN (C1) and organosilica shielded MSNs (C2 and C3). Cryo-EM micrographs of MSN1G particles revealing the radial pore structure (D1) and MSN2G with the second layer of lower porosity (D2) and shielded MSN (D3–D6). White arrows (D4) point to the phase boundary between the MSN core and the organosilica layer. The white arrow in D5 points to a region where the layer was broken revealing the inner core of the MSNs. (E) Distribution histograms of MSN before (grey) and after 20 hours (blue) of organosilica layer growth reaction measured on at least 250 MSNs measured on SEM micrographs; average diameters are 119 nm and 141 nm, respectively. Scale bars represent 100 nm in all micrographs. (F) Crystal structure of TeSADH W110A, the longest dimension of the enzyme (8.7 nm) is indicated. (G) Nitrogen sensitive XPS analysis (N 1s) of core MSN (MSN1G, grey), MSN2G (purple) evidencing the absence of surfactant (bottom two lines) and of amino-modified MSN2G (yellow), MSNENZ (green) and MSNENZ-S (blue) evidencing the successful functionalisation. | |
The amount of immobilised TeSADH W110A was determined to be 30 μg per mg of MSN – 76.9 U mg−1, which represents satisfactory conservation of 70% of the enzymatic activity. Further, to load the mesoporous reservoir by diffusion, MSNENZ were dispersed in a solution containing NADP+ (1.5 mM). It is expected that this cofactor can freely diffuse withing the MSNs' porosity until concentration equilibrium is reached. The so-treated MSNENZ were further reacted with TEOS and APTES at 20 °C for 20 h to produce an organosilica layer shielding the immobilised enzyme and ensuring higher structural stability against detrimental effects owing to solvent exposure. The targeted layer thickness, to fully cover the immobilised enzyme, was of at least 9 nm. We previously developed a method to grow organosilica on non-porous silica nanoparticles for producing virus recognition materials39 and nanobiocatalysts.38 However, this method resulted for MSNs in a layer of insufficient thickness (5 nm) and prompted us to adapt the precursor concentration. The optimised method required concentrations of TEOS and APTES of 49.9 and 9.4 mM, respectively. This method allowed producing a shielding layer of 11 nm after 20 h of reaction (MSNENZ-S, Fig. 2E). It is noteworthy that, to avoid cofactor leaching from the MSN, the layer growth reaction was carried out in the presence of NADP+ at a concentration of 1.5 mM. Additionally, the standard deviation for the size distribution remained constant (10 nm) confirming that the shielding layer was grown evenly on the particles. The MSNENZ-S were characterised by cryo-EM (Fig. 2D3–D6). At the edge of the particles was the organosilica layer visible (indicated with white arrows). On some particles, a crack in the outer layer is visible while the core MSN is still intact. This highlighted the morphological difference between these two different materials and rigidity differences. The pore structure of the core MSN is still visible. This suggests that the reaction mainly occurred at the surface of MSNENZ and maintained the porosity of the core reservoir. MSNENZ and MSNENZ-S were stored at 4 °C in buffer; after 30 days of storage, MSNENZ-S preserved as much as 57% of activity while no relevant activity could be measured for MSNENZ or for the soluble TeSADH W110A. This result represents the first evidence of the stabilisation effect of the shielding organosilica layer on the studied enzyme.
Biotransformations
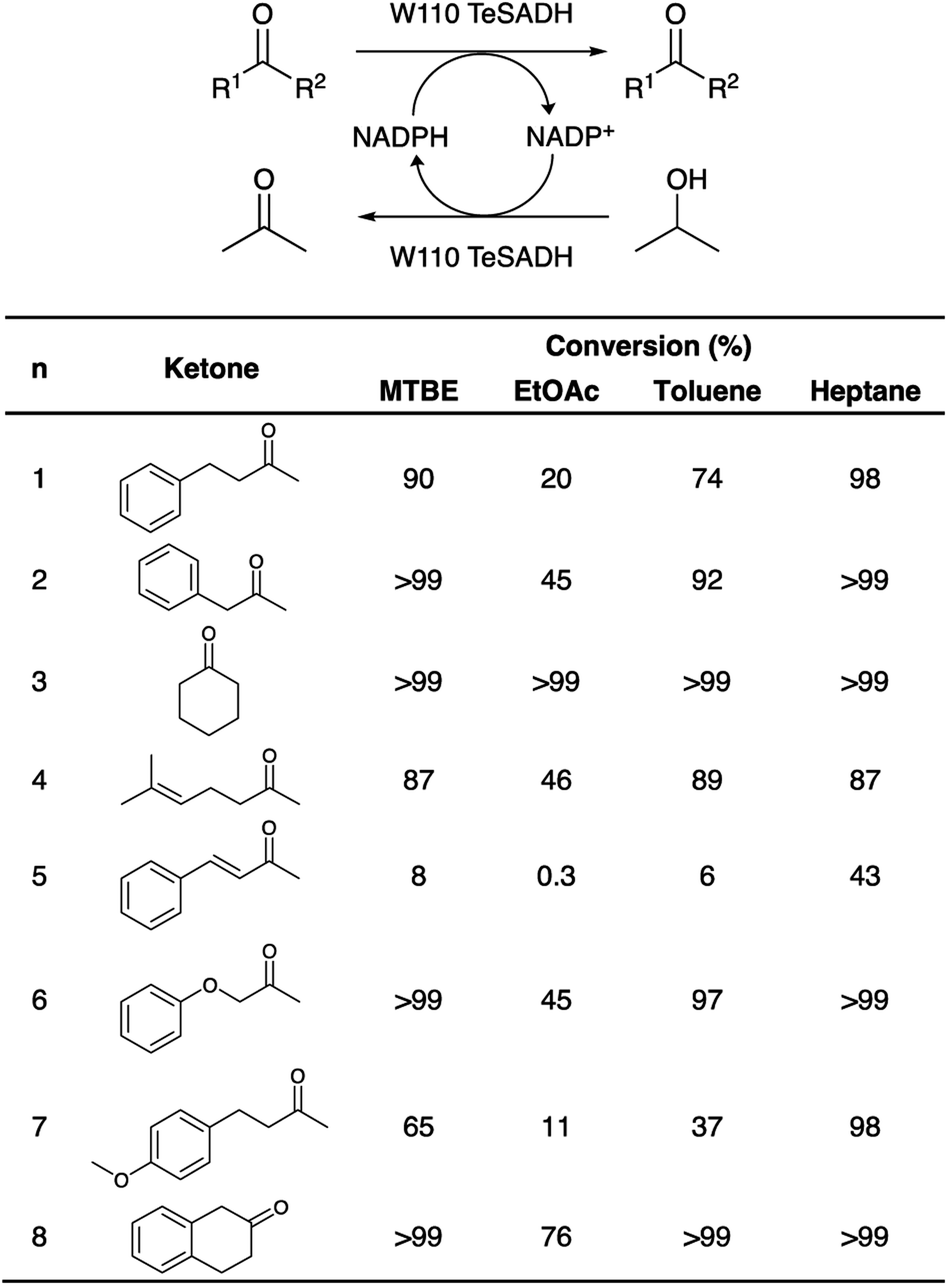
In order to assess the biocatalytic and cofactor recycling properties of the produced MSNENZ-S in OSs, we selected a series of relevant TeSADH W110A substrates, namely benzylacetone (1), phenylacetone (2), cyclohexanone (3), 6-methyl-5-hepten-2-one (4), 4-phenyl-3-buten-2-one (5), phenoxy-2-propanone (6), 4-(4-methoxyphenyl)-2-butanone (7), β-tetralone (8).36 As solvents, we tested 4 solvents of increasing hydrophobicity [i.e., ethyl-acetate (EtOAc), methyl-tert-butyl-ether (MTBE), toluene and heptane]. Biotransformations were carried out by removing the aqueous buffer from the MSNENZ-S and then dispersing them in a reaction solution consisting of 30 mM of the ketones and 2 M of the co-substrate i-PrOH as a hydride source dissolved in the OS. A large excess of i-PrOH with regard to the ketone was used to favour the ketone reduction reaction. It is noteworthy that all reactions were carried out without adding NADP+/NADPH to the reaction. The mixtures were maintained for 18 hours at 45 °C and the conversion of the ketone to the corresponding alcohol was determined by means of gas chromatography (GC); Fig. 3. Of note is that the tested cofactors being fully insoluble in the tested solvents, no control using free cofactor or soluble enzymes was feasible. It can be seen in Fig. 3 that, under the experimental conditions applied, all substrates were transformed by MSNENZ-S, with, as the only source of the cofactor, NADP+ contained in the core of MSNENZ-S. Of note is that MSNENZ showed high aggregation in the organic solvent studied; no relevant activity could be measured after such aggregation. In order to evaluate the amount of NADP+ encapsulated, we considered a full equilibration of the cofactor by passive diffusion within the mesopores and a pore volume of 0.527 cm3 per gram of MSN. This represented a NADP+ loading of 0.7 μmol g−1 MSN; the amount of NADP+ present in the reaction mixture (in 11.92 mg of MSN) was consequently of ca. 1.4 nmol. This is drastically less than the amount of substrate that is 6 μmol. Additionally, the reduction reaction required the reduced form of the cofactor while MSNENZ-S were loaded with the oxidised counterpart. Therefore, the successful reduction reactions confirmed that, besides stabilising the enzyme to endure detrimental OS effects, the mesoporous reservoir system allowed the cofactor to reach the enzyme to be oxidised to NADPH and the cofactor recycling took place within the nanoparticles. When considering reaction efficiency, conversion levels as high as 98% were reached for 6 out of 8 substrates tested (1, 2, 3, 6, 7 and 8). For all tested solvents, 3 was converted with more than 99% efficiency. In the case of 8, 99% efficiency was measured in heptane. In contrast, such high efficiency was reached for 2 and 6 in MTBE and heptane. In the case of 4, the conversion level is satisfactory, reaching 89% in toluene. Oppositely, the conversion of 5 reached only 43% in heptane; this value remained very low for the other solvents, not exceeding 8%. This low conversion level was in agreement with the results published for the same enzyme/substrate in water: i-PrOH mixtures.36 Overall, this set of results showed that the catalytic competence of the protected enzyme has been preserved. This suggested that the enzyme's conformation was maintained within the protective organosilica layer; the affinity of the enzyme's active site for both the cofactor and the substrate was not drastically altered. In a previous study, we demonstrated that an organosilica shielding layer can alter the substrate promiscuity of an esterase enzyme.40 This phenomenon was not observed in the present work since the promiscuity of the enzyme appeared unchanged, with the tested substrate, before and after shielding.36 The results shown in Fig. 3 also suggested that the higher the hydrophobicity of the solvent, the higher the catalytic efficiency of the nanobiocatalyst. This trend can be explained by the limited ability of solvents with high hydrophobicity to strip water molecules required to support enzyme conformation and thus activity16 (Fig. 4). There was, however, an exception with MTBE, solvent for which the conversion for most substrates studied was higher than with toluene. This can be explained by toluene molecules acting as competitive inhibitors when accommodated in the hydrophobic pocket (Ala85, Ala110 and Tyr267) of the enzyme, known to accommodate aromatic moieties.27,41 Our results also pointed to the importance of the solvent for the reaction efficiency. Initially, it was expected that the enzyme could catalyse the reaction in a highly hydrophobic solvent, namely heptane, as shown previously with hexane.27 However, high conversions in solvents with lower hydrophobicity, which were expected to be more detrimental to the enzyme by removing more efficiently structural water molecules, was measured. One may assign the preserved catalytic activity of the enzyme to a combined effect of the presence of the water reservoir, in the vicinity of the enzyme, and the ability of the organosilica layer to establish a set of interactions with the surface of the enzyme which preserved its three-dimensional structure.38
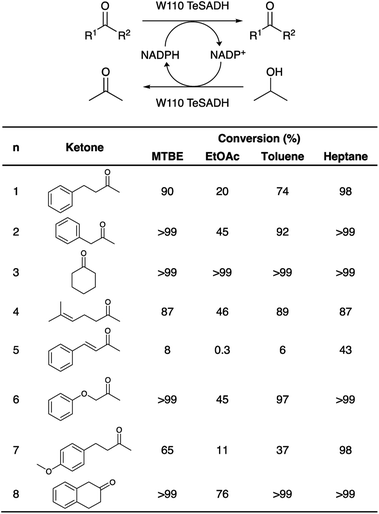 |
| | Fig. 3 Biotransformation reactions of ketone reduction to the corresponding alcohol, and cofactor recycling in OSs. (Top): the ketone is reduced to the corresponding chiral alcohol concomitantly to the oxidation of a NADPH molecule. The oxidised cofactor NADP+ is, in turn, reduced upon the oxidation of isopropanol to acetone. (Bottom): table listing the conversion efficiencies depending on the ketone used in the process. Reactions were carried out at 45 °C under stirring in MTBE, EtOAc, toluene and heptane in the absence of additional cofactor. The soluble phase was collected, and conversions rates were measured by gas chromatography and rationalised with the starting substrate quantity (values are provided in % of the initial amount of ketone). | |
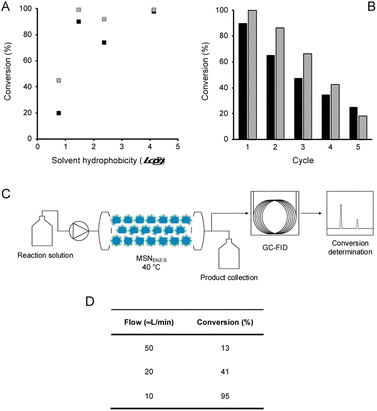 |
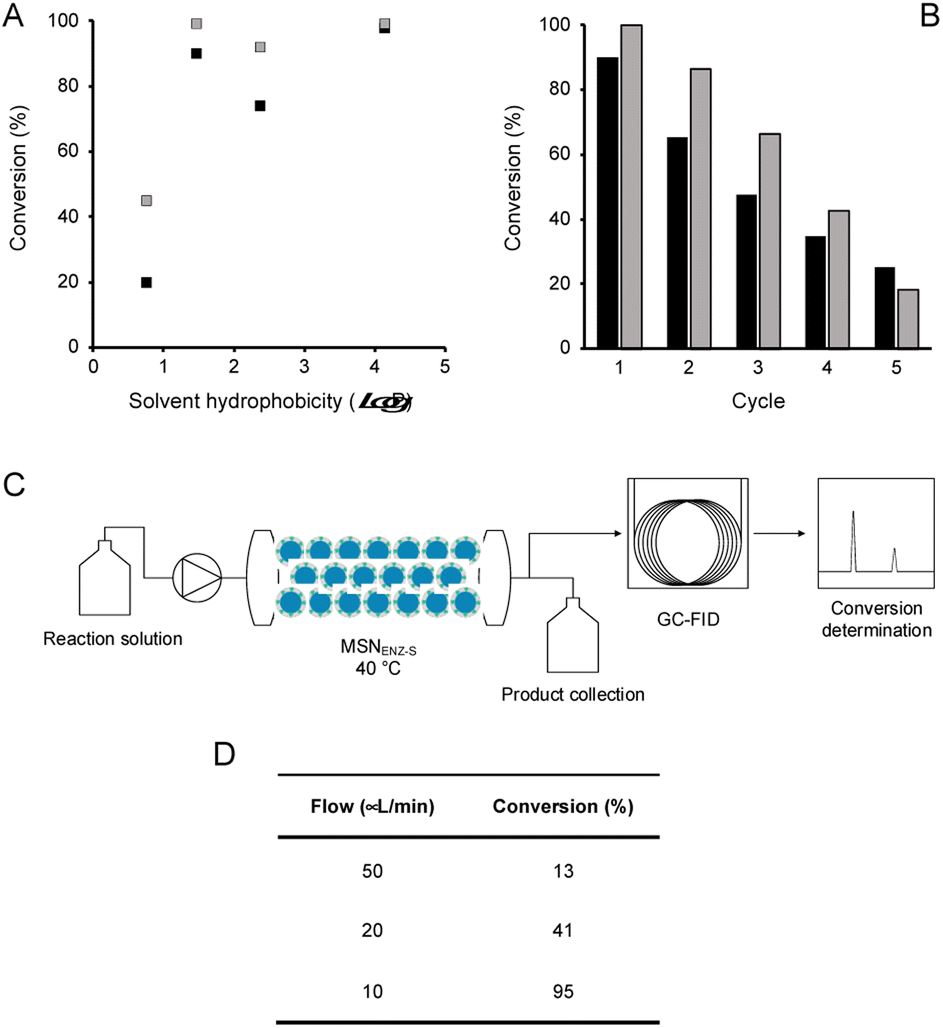
| | Fig. 4 Effect of solvent hydrophobicity, recyclability, and continuous flow reaction. (A) Conversion levels of benzylacetone (1, black squares) and phenylacetone (2, grey square) reached after 24 hours of reaction in OSs at 45 °C in function of the hydrophobicity of the OS (logP values for EtOAc, MTBE, toluene and heptane are 0.76, 1.48, 2.39 and 4.16, respectively). (B) Recyclability of MSNENZ-S carried out using MTBE as OS, conversion levels are presented as the fraction (in %) of substrate 1 (black bars) or 2 (grey bars) transformed. (C) Schematic representation of the continuous flow reactor used to measure the conversion levels shown in (D) for substrate 1. | |
Next, in order to assess the recyclability of MSNENZ-S and the long-term stability of the enzyme in OSs, the biotransformation experiment was repeated 5 times with substrates 1 and 2, after regenerating MSNENZ-S in buffer supplemented with NADP+ between each cycle; Fig. 4. Our results showed that, while a decrease in activity was measured for consecutive cycles, as much as 65% and 86% of conversion were measured for the second cycle for 1 and 2, respectively. After 5 cycles implying that the enzyme was maintained in MTBE for 120 hours, relevant conversions were determined at 25% and 18%. When omitting the regeneration step, much steeper reductions in activity were measured with only 7 and 13% of activity measured for the second conversion cycle for 1 and 2, respectively. No consistent activity was measured under these conditions for the third cycle. Considering the lack of solubility of the cofactor in the solvents studied, one can safely rule out cofactor leaching. The observed loss in activity without regeneration steps might be attributed to the loss of enzymes' structural water molecules, which are recovered after the regeneration step. Limited cofactor leaching during column regeneration or cofactor instability may also explain the need of cofactor reloading after every cycle.
Continuous flow bioconversion
In order to take advantage of the in-built recycling system and stability in OSs, the experimental work was focused on the initial demonstration of the possibility of using the nanobiocatalysts developed in a continuous flow system. To that end, MSNENZ-S were packed into a packed bed reactor and used for continuous bioconversion (Fig. 4C). The column used had a volume of 831 μL and a quantity of 140 mg of MSNENZ-S was applied to fill the column. The column was subsequently flushed with a buffer solution containing NADP+ to ensure that the core of the particles was saturated with the cofactor. Next, the column was flushed with a solution of MTBE containing benzylacetone (30 mM) and i-PrOH (3 M). Initially, we applied a relatively high flow rate of 200 μL min−1 to eliminate water possibly remaining in the column. The flow was then decreased to 50, 20 or 10 μL min−1 and the flow through was collected at different flow rates to determine the conversion of 1 to its corresponding alcohol. Our results show that, while at high flow rates of 50 and 20 μL min−1, conversion levels were low with values of 13 and 41%, it reached an excellent level of 98% at a flow rate of 10 μL min−1. This can be explained by the longer residence time at lower flow rates, which allowed for a longer reaction time. Overall, these results provide additional evidence that the cofactor is retained and recycled within the core of the particle. Indeed, if this were not be the case, the cofactor would have been flushed out with the excess water and no conversion would occur. Additionally, the reaction occurred with an enantiomeric excess of 98%; this is consistent with that measured for soluble TeSADH W110A.27,36
Conclusion
We constructed a nanobiocatalyst competent for oxidoreductase biocatalytic ketone reduction in organic solvents in the absence of an externally added cofactor. In order to build such nanobiocatalysts, we used hierarchical mesoporous silica nanoparticles with a radial pore architecture. This provided the possibility to create an aqueous reservoir within the core displaying large pores and to immobilise the selected enzyme on the outer layer possessing smaller pores. The organosilica layer further grown on these systems, besides providing protection to the enzyme, represents an environment where oxidation and biocatalytic reactions can take place. In the conditions used in the present work, the selected enzyme, a mutated version of a secondary alcohol dehydrogenase from Thermoanaerobacter ethanolicus did catalyse the reduction of a series of hydrophobic ketones into the corresponding alcohol. Concomitantly, the same enzyme catalysed the oxidation of isopropanol to acetone. Out of the 8 substrates tested, conversion yields greater than or equal to 98% were reached for 6 substrates. The nanobiocatalysts produced were also shown to display consistent biocatalytic activity for at least 5 cycles; this was made possible by the stabilisation of the enzyme that was maintained in the organic solvent for 120 hours. We expect that the reported method can be applied to a number of oxidoreductases necessitating a water-soluble cofactor.
Experimental
Materials
Unless otherwise stated, were all chemicals and solvents purchased from commercial suppliers and used without further purification (Sigma Aldrich, Thermo Fischer Scientific, Roth, TCI, Toronto, ThermoFischer Scientific – Acros).
MSN synthesis
The MSNs were prepared as previously described.35 In brief, 60 mL of cetyltrimethylammonium chloride (CTAC, 25 wt%), 620 mL of triethylamine and 90 mL of water were equilibrated at 60 °C, under magnetic stirring for 1 hour. A volume of 50 mL of 10% (v/v) TEOS in cyclohexane was added slowly to the solution and the reaction was left to react for 14 hours. Consequently, the aqueous phase was transferred to another flask and 50 mL of 20% (v/v) TEOS in octadecene was added to the solution. The reaction was left to react for 6 hours under magnetic stirring. The aqueous phase was submitted to centrifugation at 20000g for 20 min and the resulting pellet was resuspended in ethanol. This procedure, called the washing step, was repeated thrice. For the last resuspension, the pellet was resuspended in 0.6 wt% NH4NO3 in ethanol. The MSNs produced were subsequently incubated at 60 °C for 6 h to allow for template extraction. Subsequently, the suspension was centrifuged at 20000g for 20 min and the extraction was then repeated two more times. After the last extraction was the washing step repeated four more times, twice with ethanol and twice with water. The final particles were stored at 4 °C.
Protein production and purification
The gene encoding TeSADH W110A was purchases as subcloned into pET28c(+) vector from Eurofins Genomics. The restriction sites NdeI and XhoI were used for insertion into the plasmid and competent cells of E. coli BL21(DE3) were used for the expression. Protein expression was carried out with ZYM-5052 medium for autoinduction supplemented with 100 μg per mL kanamycin at 37 °C for 24 hours incubation. The bacteria were harvested by centrifugation at 4 °C and 5000g and then frozen. The pellet was thawed on ice and was resuspended in wash buffer (50 mM NaPi, 300 mM NaCl and 10 mM imidazole pH 8) and then lysed by sonification (5 min of 4 °C, 30 s pulses at 50%, 30 s off). The cell lysate was centrifuged (5000g for 20 min at 4 °C) and the supernatant was then heat-treated by incubation at 70 °C for 15 minutes. The heat-treated cell lysate was once more centrifuged (20000g for 10 min at 4 °C) and then filtered through a 0.22 μm filter. The enzyme was expressed with an N-terminal 6xHis-tag and was purified using a His trap column (GE Life sciences) and was eluted using a gradient of elution buffer (wash buffer but with 300 mM imidazole). The fractions were analysed with SDS-PAGE to determine purity. The fractions with the protein of interest were pooled together and were then desalted on a HiTrap desalting column (GE Life sciences). Enzyme concentration was determined with BCA assay. The enzyme stocks were aliquoted, snap-frozen and stored at −20 °C.
Nanobiocatalyst synthesis
The MSNs were functionalised with 5.6 mM aminopropyltriethoxysilane (APTES) for 90 min at 20 °C. The washing step to remove the unreacted APTES was then carried out four times by centrifugation (20000g, 15 min) and resuspension of the particles in water. The MSN-NH2 were stored at 4 °C. MSN-NH2 (3.2 mg mL−1 MSN-NH2 in 10 mM NaPi, pH 7.5) were then further functionalised with 0.16 mM EGS for 20 min at 20 °C and 400 rpm stirring to yield MSN-EGS. The particles were washed by centrifugation (20000g, 5 min) and then resuspended in immobilisation buffer 50 mM NaPi, pH 6. The washing was repeated four times. 200 μg mL−1 of the enzyme was used for immobilisation on MSN-EGS for 30 min at 20 °C and 400 rpm stirring to yield MSNENZ. The MSNENZ were washed by centrifugation (1500g, 5 min) and then resuspended in layer growth buffer (50 mM NaPi, pH 8). The washing was repeated three times. The immobilisation yield was determined by BCA assay (Pierce™ BCA Protein assay Kit, Thermo Fischer Scientific) by analysing the protein content in the first supernatant after immobilisation. 1.5 mM of NADP+ was added to the MSNENZ and they were incubated for 20 minutes. Then 49.9 μM of TEOS was added to the MSNENZ and they were incubated for 1 h at 20 °C with 400 rpm stirring. 9.4 μM APTES was added and the MSNENZ was incubated at 20 °C with 400 rpm stirring for 20 h. After incubation, the particles were collected and washed by centrifugation (1500g, 5 min) 3 times. MSNENZ-S were stored at 20 °C for 14 hours to allow curing before being stored at 4 °C.
Microscopic characterisation
Scanning electron microscopy was carried out using a Zeiss SUPRA 40VP system. Samples were prepared by spreading 2 μL of the nanoparticle suspension in water on a silicon substrate. The sample was dried under ambient conditions and then sputter-coated with a gold-platinum alloy (15 s, 20 mA). Micrographs were acquired using the InLens mode with an accelerating voltage of 10kV. Particle sizes were measured on micrographs acquired at a magnification of 200000× using the Olympus Analysis software package. To ensure size measurement accuracy, at least 250 measurements were made per sample. For transmission electron microscopy measurements, a 4 μL aliquot of sample was adsorbed onto a holey carbon-coated grid (Lacey, Tedpella), blotted with Whatman 1 filter paper and plunge-frozen into liquid ethane at −180 °C using a vitrobot (FEI). Frozen grids were transferred onto a Talos electron microscope (FEI) using a Gatan 626 cryo-holder. Electron micrographs were recorded at an accelerating voltage of 200 kV using a low-dose system (50 e− Å−2) and keeping the sample at −175 °C. Defocus values were −3 μm. Micrographs were recorded on a 4 × 4 K Ceta CMOS camera.
Synthesis of phenylacetone and 4(4-methoxyphenyl)-2-butanol
Phenylpropane-2-one was synthesised according to a described procedure42 but with graphite electrodes and EtOAc as solvent. 1H- and 13C-NMR spectra are consistent with those reported.43 4-(4-methoxyphenyl)-2-butanol was synthesised as previously described.441H- and 13C-NMR spectra were consistent with those reported.44,45
Activity measurement in buffer
The typical reaction mixture for measuring TeSADH activity contained 500 mM isopropanol and 1 mM of NADP+ in 50 mM NaPi pH 8. The assay was run at 60 °C with 1 μg mL−1 of soluble TeSADH W110A, MSNENZ or MSNENZ-S. The assay was run for 5 minutes and was then stopped with thiourea (0.9 M) and the mixture was thereafter centrifuged (20000g, 2 min) to pellet the particles. A volume of 250 μL from the supernatant was then transferred to a 96 UV well plate and absorbance was measured at 340 nm in a plate reader.
Activity measurement in neat organic solvents
The tested substrates [30 mM, benzylacetone (1), phenylacetone (2), cyclohexanone (3), 6-methyl-5-hepten-2-one (4), 4-phenyl-3-buten-2-one (5), phenoxy-2-propanone (6), 4-(4-methoxyphenyl)-2-butanone (7), β-tetralone (8)] and 2 M i-PrOH were respectively dissolved in the organic solvents tested (heptane, EA, toluene or MTBE). A quantity of 1.92 μg of MSNENZ-S was re-suspended in 200 μL of the reaction mixture and incubated with stirring at 400 rpm for 18 hours at 45 °C. The reaction mixture was centrifuged to pellet the MSNENZ-S and the supernatant was collected and analysed with GC-FID (Agilent 7820A) on an Agilent HP-5 column (30 m, 0.32 mm i.d., df 0.25 μm) to determine the conversion from the substrate to product (via calibration curves of substrate and product).
Recycling experiments
The recycling experiment was performed in the same way as the biotransformations with the addition of the recycling step and 24 hour incubation. The substrates were either benzylacetone or phenylacetone. The MSNENZ-S were, after centrifugation after the first biotransformation, washed once with 200 μL of MTBE by centrifugation (3200g, 5 min) before being incubated in 200 μL of 1.5 mM NADP+ in 50 mM NaPi, pH 8 for 15 minutes to re-equilibrate the mesopores within MSNENZ-S. The MSNENZ-S were then collected by centrifugation (3200g, 5 min) and resuspended in 200 μL fresh and then incubated for the next biotransformation cycle. The supernatant after each recycling step was collected and analysed with GC-FID as previously stated.
Continuous flow biotransformation
A quantity of 140 mg of MSNENZ-S was packed into a metal column (4.6 mm i.d. × 50 mm length) by vacuum suction resulting in the drying of the MSNENZ-S material. To compensate for the loss in cofactor during the column drying by vacuum suction, it was then rinsed with 1.5 mM NADP+ in 50 mM NaPi, pH 8 for 20 min (flow 0.3 mL min−1). A reaction solution was prepared containing 30 mM benzylacetone and 3 M i-PrOH in MTBE. The reaction solution was pumped through the column for 20 min (flow 200 μL min−1) to remove all remaining water. The flow was reduced to 10 μL min−1, the column was heated to 40 °C and fractions were collected and analysed with GC-FID to determine the conversion from the substrate to product (8 fractions collected for 330 min). The flow was increased to 20 μL min−1 and 50 μL min−1, and the procedure was repeated for these flow rates as well. The absolute configuration of the product was determined by derivatization with N-methyl-bis(trifluoroacetamide) followed by analysis with GC-FID on a Chiraldex B-DM (50 m, 0.25 mm i.d., df 0.12 μm).
X-ray photoelectron spectroscopy
As a complementary measure to monitor the hierarchical synthesis of MSNs, X-ray photoelectron spectroscopy experiments have been performed. It is important to note that the information depth of this technique at the photoelectron energies of the Si 2p and N 1s lines is in the order of ca. 6 nm at normal emission, thus mostly sensitive to the outermost layer composition of the MSNs. A Specs FOCUS 500 monochromator equipped with an XR 50 M Al Kα (hν = 1486.7 eV) excitation source was used, and the emitted photoelectron energies were analysed with a PHOIBOS 150 electron analyser. For all measurements, a shift towards higher binding energies (>10 eV) was observed in the Si 2p spectra. This effect was attributed to the insulating nature of the core–shell particles and their size in the order of 100 nm. For this reason, the photoelectron current has been compensated by using a flood gun; this allowed translating the Si 2p peak to the standard position before acquiring N 1s signals. Samples for XPS analysis were prepared as follows. Suspensions of MSN1G, MSN2G, MSN-NH2 and MSNENZ were snap-frozen in liquid nitrogen and freeze-dried for 12 hours under reduced pressure. The resulting powders were compressed onto indium foils to produce flat pellets. XPS spectra were acquired on the MSN pellets, at 20 °C, in normal emission mode.
Thermogravimetric analysis (TGA)
TGA experiments were carried out using a TGA analyser (TGA 4000, PerkinElmer). Reference samples (MSN + CTAC) were prepared by mixing 20 μL of CTAC (25 wt% in H2O) with 20 mg of MSN2G. Samples were snap-frozen and freeze-dried prior to analysis. The system was maintained under a nitrogen atmosphere and heated from 30 °C to 900 °C applying a temperature ramp of 10 °C min−1 and a purge rate of 19.8 mL min−1.
Analysis of nitrogen adsorption–desorption isotherms
Nitrogen adsorption experiments were carried out by means of a Micromeritics Gemini VII surface area analyser. MSN powders obtained by freeze-drying were further degassed under a nitrogen flow for 12 h. All measurements were carried out at a temperature of 77 K. The Brunauer–Emmett–Teller (BET) specific surface area was calculated from nitrogen adsorption data. The pore size distribution was calculated from the adsorption branch of the isotherms using the density functional theory (DFT).
Author contributions
PS designed the general concept and supervised the work. JS carried out the protein production and purification, nanomaterial synthesis and biocatalytic transformation studies. CS carried out the synthesis of phenylacetone and 4(4-methoxyphenyl)-2-butanol. TAJ, RSK, NHR and AB carried out the XPS measurement. CW, SAN and AB carried out the physicochemical characterization of the MSNs. JS and PS wrote the manuscript with the help of TAJ.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
This research was funded by Swissuniversities through the program Innovation in Catalysis: A toolbox for sustainable bio-based production and European Union's Horizon 2020 Research and Innovation Programme, grant agreement no. 101000327 (Project FuturEnzyme).
Notes and references
- J. B. Zimmerman, P. T. Anastas, H. C. Erythropel and W. Leitner, Science, 2020, 367, 397–400 CrossRef CAS PubMed.
- R. A. Sheldon and J. M. Woodley, Chem. Rev., 2018, 118, 801–838 CrossRef CAS PubMed.
- J. R. Ludwig and C. S. Schindler, Chem, 2017, 2, 313–316 CAS.
- Price, pressures on metals, Nat. Catal., 2019, 2, 735, DOI:10.1038/s41929-019-0359-7.
- P. N. Devine, R. M. Howard, R. Kumar, M. P. Thompson, M. D. Truppo and N. J. Turner, Nat. Rev. Chem., 2018, 2, 409–421 CrossRef.
- R. A. Sheldon, D. Brady and M. L. Bode, Chem. Sci., 2020, 11, 2587–2605 RSC.
- E. L. Bell, W. Finnigan, S. P. France, A. P. Green, M. A. Hayes, L. J. Hepworth, S. L. Lovelock, H. Niikura, S. Osuna, E. Romero, K. S. Ryan, N. J. Turner and S. L. Flitsch, Nat. Rev. Methods Primers, 2021, 1, 46 CrossRef CAS.
- R. A. Sheldon and D. Brady, ChemSusChem, 2019, 12, 2859–2881 CrossRef CAS PubMed.
- V. Steck, D. M. Carminati, N. R. Johnson and R. Fasan, ACS Catal., 2020, 10, 10967–10977 CrossRef CAS PubMed.
- S. Simić, E. Zukić, L. Schmermund, K. Faber, C. K. Winkler and W. Kroutil, Chem. Rev., 2022, 122, 1052–1126 CrossRef PubMed.
- N. J. Turner and E. O'Reilly, Nat. Chem. Biol., 2013, 9, 285–288 CrossRef CAS PubMed.
- U. Hanefeld, F. Hollmann and C. E. Paul, Chem. Soc. Rev., 2022, 51, 594–627 RSC.
- U. T. Bornscheuer, G. W. Huisman, R. J. Kazlauskas, S. Lutz, J. C. Moore and K. Robins, Nature, 2012, 485, 185–194 CrossRef CAS PubMed.
-
O. May, in Industrial Enzyme Applications, ed. A. Vogel and O. May, Wiley-VCH, 2019, pp. 1–24 Search PubMed.
- E. Busto, V. Gotor-Fernández and V. Gotor, Chem. Soc. Rev., 2010, 39, 4504–4523 RSC.
-
S. Kara and A. Liese, in Industrial Enzyme Applications, Wiley-VCH, 2019, pp. 71–94 Search PubMed.
- H. Cui, L. Zhang, L. Eltoukhy, Q. Jiang, S. K. Korkunç, K.-E. Jaeger, U. Schwaneberg and M. D. Davari, ACS Catal., 2020, 10, 14847–14856 CrossRef CAS.
- D. Zarafeta, D. Moschidi, E. Ladoukakis, S. Gavrilov, E. D. Chrysina, A. Chatziioannou, I. Kublanov, G. Skretas and F. N. Kolisis, Sci. Rep., 2016, 6, 38886 CrossRef CAS PubMed.
- V. Stepankova, S. Bidmanova, T. Koudelakova, Z. Prokop, R. Chaloupkova and J. Damborsky, ACS Catal., 2013, 3, 2823–2836 CrossRef CAS.
- H. Cui, T. H. J. Stadtmüller, Q. Jiang, K.-E. Jaeger, U. Schwaneberg and M. D. Davari, ChemCatChem, 2020, 12, 4073–4083 CrossRef CAS.
- F. H. Arnold, Angew. Chem., Int. Ed., 2019, 58, 14420–14426 CrossRef CAS PubMed.
- M. M. C. H. van Schie, J.-D. Spöring, M. Bocola, P. Domínguez de María and D. Rother, Green Chem., 2021, 23, 3191–3206 RSC.
- F. G. Mutti, T. Knaus, N. S. Scrutton, M. Breuer and N. J. Turner, Science, 2015, 349, 1525–1529 CrossRef CAS PubMed.
- S. L. Montgomery, J. Mangas-Sanchez, M. P. Thompson, G. A. Aleku, B. Dominguez and N. J. Turner, Angew. Chem., Int. Ed., 2017, 56, 10491–10494 CrossRef CAS PubMed.
- M. L. Contente and F. Paradisi, Nat. Catal., 2018, 1, 452–459 CrossRef CAS.
- C. J. Hartley, C. C. Williams, J. A. Scoble, Q. I. Churches, A. North, N. G. French, T. Nebl, G. Coia, A. C. Warden, G. Simpson, A. R. Frazer, C. N. Jensen, N. J. Turner and C. Scott, Nat. Catal., 2019, 2, 1006–1015 CrossRef CAS.
- M. M. Musa, K. I. Ziegelmann-Fjeld, C. Vieille, J. G. Zeikus and R. S. Phillips, Angew. Chem., Int. Ed., 2007, 46, 3091–3094 CrossRef CAS PubMed.
- M. Heidlindemann, G. Rulli, A. Berkessel, W. Hummel and H. Gröger, ACS Catal., 2014, 4, 1099–1103 CrossRef CAS.
- T. Matsuda, T. Harada, N. Nakajima and K. Nakamura, Tetrahedron Lett., 2000, 41, 4135–4138 CrossRef CAS.
- P. G. Le, Q. Wu, D. Y. Kong, J. Ge and M. Il Kim, ACS Appl. Energy Mater., 2022, 5, 13113–13127 CrossRef CAS.
- X. Li, Y. Cao, K. Luo, L. Zhang, Y. Bai, J. Xiong, R. N. Zare and J. Ge, Nat. Commun., 2022, 13, 2189 CrossRef CAS PubMed.
- F. Lyu, Y. Zhang, R. N. Zare, J. Ge and Z. Liu, Nano Lett., 2014, 14, 5761–5765 CrossRef CAS PubMed.
- Y. Chen, P. Li, H. Noh, C. W. Kung, C. T. Buru, X. Wang, X. Zhang and O. K. Farha, Angew. Chem., Int. Ed., 2019, 58, 7682–7686 CrossRef CAS PubMed.
- T. Cheng, Y. Zhao, X. Li, F. Lin, Y. Xu, X. Zhang, Y. Li, R. Wang and L. Lai, J. Chem. Inf. Model., 2007, 47, 2140–2148 CrossRef CAS PubMed.
- D. Shen, J. Yang, X. Li, L. Zhou, R. Zhang, W. Li, L. Chen, R. Wang, F. Zhang and D. Zhao, Nano Lett., 2014, 14, 923–932 CrossRef CAS PubMed.
- M. M. Musa, K. I. Ziegelmann-Fjeld, C. Vieille, J. G. Zeikus and R. S. Phillips, J. Org. Chem., 2007, 72, 30–34 CrossRef CAS PubMed.
- M. M. Musa, R. S. Phillips, M. Laivenieks, C. Vieille, M. Takahashi and S. M. Hamdan, Org. Biomol. Chem., 2013, 11, 2911–2915 RSC.
- M. R. Correro, N. Moridi, H. Schützinger, S. Sykora, E. M. Ammann, E. H. Peters, Y. Dudal, P. F.-X. Corvini and P. Shahgaldian, Angew. Chem., Int. Ed., 2016, 55, 6285–6289 CrossRef CAS PubMed.
- A. Cumbo, B. Lorber, P. F. Corvini, W. Meier and P. Shahgaldian, Nat. Commun., 2013, 4, 1503 CrossRef PubMed.
- C. I. Giunta, I. Cea-Rama, S. Alonso, M. L. Briand, R. Bargiela, C. Coscolín, P. F. X. Corvini, M. Ferrer, J. Sanz-Aparicio and P. Shahgaldian, ACS Nano, 2020, 14, 17652–17664 CrossRef CAS PubMed.
- M. M. Musa, N. Lott, M. Laivenieks, L. Watanabe, C. Vieille and R. S. Phillips, ChemCatChem, 2009, 1, 89–93 CrossRef CAS.
- T. Inokuchi, S. Matsumoto and S. Torii, J. Org. Chem., 1991, 56, 2416–2421 CrossRef CAS.
- G. Zhang, X. Hu, C.-W. Chiang, H. Yi, P. Pei, A. K. Singh and A. Lei, J. Am. Chem. Soc., 2016, 138, 12037–12040 CrossRef CAS PubMed.
- T. C. Atack and S. P. Cook, J. Am. Chem. Soc., 2016, 138, 6139–6142 CrossRef CAS PubMed.
- S. Rezazadeh, V. Devannah and D. A. Watson, J. Am. Chem. Soc., 2017, 139, 8110–8113 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence bc and
Patrick
Shahgaldian
bc and
Patrick
Shahgaldian