
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFabrication of membrane proteins in the form of native cell membrane nanoparticles using novel membrane active polymers†
Thi Kim
Hoang Trinh
 ab,
Claudio
Catalano
ab and
Youzhong
Guo
*ab
ab,
Claudio
Catalano
ab and
Youzhong
Guo
*ab
aDepartment of Medicinal Chemistry, School of Pharmacy, Virginia Commonwealth University, Richmond, VA 23298, USA. E-mail: yguo4@vcu.edu
bInstitute for Structural Biology, Drug Discovery and Development, School of Pharmacy, Virginia Commonwealth University, Richmond, VA 23219, USA
First published on 9th October 2023
Abstract
Membrane proteins are a widespread class of bio-macromolecules responsible for numerous vital biological processes and serve as therapeutic targets for a vast array of contemporary medications. For membrane protein isolation and purification, detergents have historically been used. Despite this, detergents frequently result in protein instability. Consequently, their application was limited. Recent detergent-free approaches have been invented. Among these, styrene–maleic acid lipid particle (SMALP), diisobutylene–maleic acid lipid particle (DIBMALP), and native cell membrane nanoparticle (NCMN) systems are the most prevalent. The NCMN system intends to create a library of membrane-active polymers suitable for high-resolution structure determination of membrane protein. Design, synthesis, characterization, and comparative application evaluations of three novel classes of NCMN polymers, NCMNP13-x, NCMNP21-x, and NCMNP21b-x, are presented in this article. Although each NCMN polymer can solubilize distinct model membrane proteins and retain native lipids in NCMN particles, only the NCMNP21b-x family produces lipid–protein particles with ideal buffer compatibility and high homogeneity suitable for single-particle cryo-EM analysis. NCMNP21b-x polymers that generate high-quality NCMN particles are particularly desirable for membrane protein structural biology.
Introduction
Membrane proteins are a collection of bio-macromolecules with diverse structures and activities that are involved in a wide variety of biological processes. To comprehend the active mechanism of a given protein, precise structural knowledge is required, especially for membrane proteins implicated in protein–lipid interactions.1 As prominent pharmacological targets, structural and mechanistic understanding of membrane proteins offers tremendous prospects for drug development.2,3 For structural investigation, it is typically necessary to extract membrane proteins from the cell membrane milieu using detergents. Protein–lipid interactions are crucial for maintaining a membrane protein's native structure and function. During membrane protein solubilization, however, detergents frequently cause excessive delipidation. Synthetic membrane-active polymers can wrap around the transmembrane domain and retain the native cell membrane lipids associated with the membranous protein without affecting lipid–protein interactions. Therefore, they may provide superior alternatives for membrane protein structural research.4 In polymer–lipid–protein particles, hydrophilic side groups of polymeric belts face the aqueous phase, which suspends and stabilizes the particles for subsequent purification and analysis.5–8Styrene–maleic acid (SMA) co-polymer, diisobutylene–maleic acid (DIBMA) co-polymers, and various NCMN polymers within the native cell membrane nanoparticles system can retain and stabilize membrane protein–lipid complexes endogenous to the cell membrane.9–13 In the case of SMA, the Escherichia coli (E. coli) membrane exhibited significant increases in solubilization efficiency at a small shift toward an alkaline pH. As a result of the comparatively large negative charge density coming from the basic condition, polymeric chains can get into the lipid bilayer and ultimately induce membrane solubilization.9,14 Nevertheless, changes in chemical structure and hydrophobicity of polymeric hydrophobic domains are significant drivers of yield, size, purity, and long-term durability of resultant particles. The hydrophobic effect is typically the driving force behind membrane solubilization.5 Thus, it is unsurprising that the SMA (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) copolymer with prominent hydrophobicity of phenyl groups has a far greater solubilizing effect on specific proteins, such as ZipA and ABC transporter BmrA, than DIBMA, which contains significantly fewer hydrophobic sections.10,15 In most instances, DIBMA treatment resulted in protein particles that were bigger in diameter and less homogeneous.10,15 Perhaps the larger-sized particles accommodated more impurities and were, as a result, incompatible with structural characterization using single-particle cryo-electron microscopy (cryo-EM). In addition, SMA particles were more stable during long-term storage due to the close packing of lipid acyl chains due to the insertion and bending of phenyl rings around the original lipid bilayer.10,15
:1) copolymer with prominent hydrophobicity of phenyl groups has a far greater solubilizing effect on specific proteins, such as ZipA and ABC transporter BmrA, than DIBMA, which contains significantly fewer hydrophobic sections.10,15 In most instances, DIBMA treatment resulted in protein particles that were bigger in diameter and less homogeneous.10,15 Perhaps the larger-sized particles accommodated more impurities and were, as a result, incompatible with structural characterization using single-particle cryo-electron microscopy (cryo-EM). In addition, SMA particles were more stable during long-term storage due to the close packing of lipid acyl chains due to the insertion and bending of phenyl rings around the original lipid bilayer.10,15
With the above superiority, SMA co-polymers have gained much attention in structural studies of membrane proteins.16,17 In practice, SMA co-polymers have limits. The carboxylate function group could contribute to non-covalent aggregation in acidic pH and divalent cation buffers, essential for solubilizing and purifying certain membrane proteins.14,18 To solve present disadvantages, the SMALP community has investigated SMA side chains by modifying carboxylates into novel functional groups, such as alcohol (SMA–EA),19,20 amino (SMA–ED),21 thiol (SMA–SH)22 and phosphobetaine (zSMA).23 The stability of polymer–lipid–protein particles formed from various SMA variations is either partially or entirely altered. Nevertheless, none of them has been successful in the high-resolution structure determination of membrane proteins. Notably, Marconnet et al. have recently exposed a novel cryo-EM compatible polymer, cycloalkane-modified amphipols (CyclAPols), which can extract native membrane proteins but cannot preserve the well-organized natural lipids plug within transmembrane (TM) domains.24,25
Among membrane-active polymers, the commercial SMA2000 with a specified ratio of 2:1 hydrophobic: hydrophilic domains has proven to be the most efficient solubilizer for various membrane protein model systems.9,11,14,26 To overcome the limitations of SMA2000 and maintain a good extraction efficiency, three classes novel polymers were produced by grafting isopropylamine (IPA), N,N-dimethylethylenediamine (DMEDA), and 3-((2-aminoethyl)-dimethylammonio)propane-1-sulfonate (DMEDA-PS) onto the side chains of SMA2000 at various controlled levels (Scheme 1). The polymers, including IPA, DMEDA, and DMEDA-PS units, were designated NCMNP13-x, NCMNP21-x, and NCMNP21b-x, with “x” representing the desired amine grafting level. While previous studies only employed the grafting level of 50 or 100%, herein, variable “x” values were used to investigate how the amines' chemical compositions and structures affect membrane solubilization, protein purification, and stability.
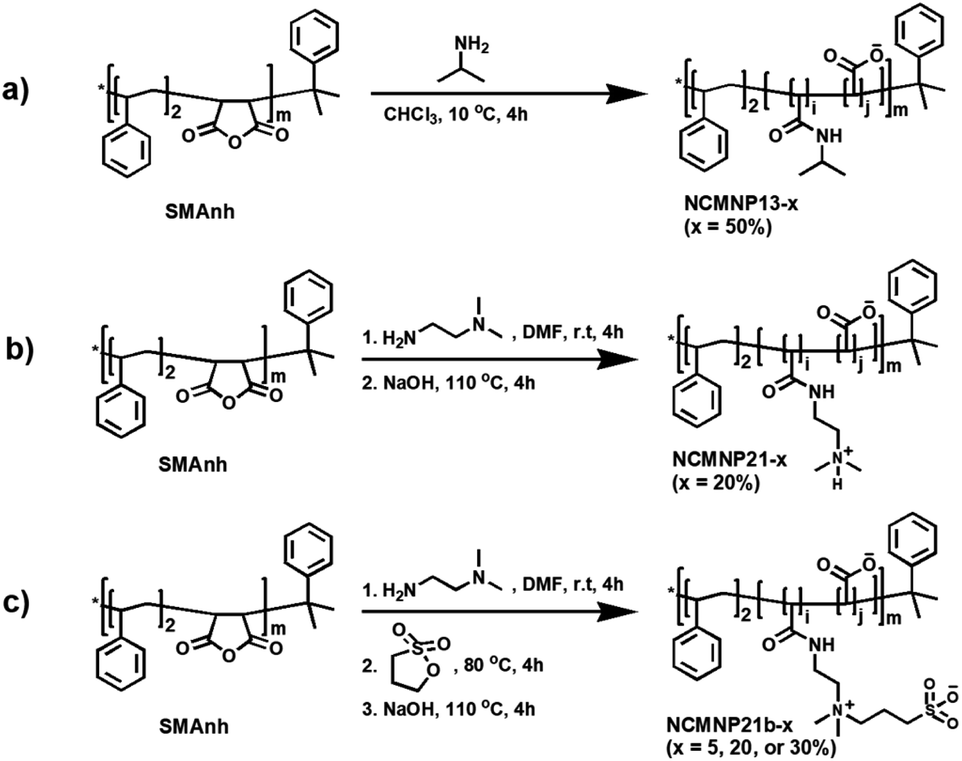 | ||
| Scheme 1 The general synthetic strategy. (a) NCMNP13-x. Isopropylamine (IPA) was used with a slight excess amount of maleic anhydride to obtain a complete ring-opening process. (b) NCMNP21-x. (c) NCMNP21b-x. | ||
Here, we describe the design, synthesis, characterization, and comparative application evaluation of NCMNP13-x, NCMNP21-x, and NCMNP21b-x. The comparative application analyses include four components: (a) solubilization efficiency and long-term stability of NCMN particles. The solubilization effectiveness of two distinct proteins, E. coli Acriflavine resistance channel protein B (AcrB) and small-conductance mechanosensitive channel protein, was evaluated (MscS). At pH 7.8, AcrB (ExPASy—ProtParam tool calculated pI 5.35) has a pI 5.35.27 has an overall negative charge, and MscS (ExPASy—ProtParam tool calculated pI ∼7.9)27 has an overall near-neutral charge that allowed us to elucidate the role of electrostatic interaction for NCMN particle stability. (b) Stability of NCMN towards pH conditions and Ca2+. (c) Lipidomic analysis of NCMN particles. Lipidomic analysis was done on AcrB-NCMN particles to see if native lipids were encapsulated and to identify any compositional alterations. TM domains are prevented from collapsing by the lipid bilayer patch. Consequently, confirming the presence of the lipid bilayer patch is one factor used to justify the structural study suitability of NCMN particles. (d) Single-particle electron microscopic analysis of NCMN particles. The comparative results indicate that NCMNP21b-x has great promise as a membrane-active polymer for high-resolution single-particle cryo-EM analysis of membrane proteins
Results and discussions
Design, synthesis, and characterization of NCMNP13-x, NCMNP21-x, and NCMNP21b-x
The NCMN polymers were made by opening the styrene–maleic anhydride (SMAnh) ring with various nucleophiles, followed by hydrolysis of unreacted maleic anhydrides (MAnhs) with NaOH (Scheme 1). Initial preparation of NCMNP13-x and NCMNP21-x utilized a set molar ratio of amine to MAnh (50 percent for IPA, 20 percent for DMEDA) to produce NCMNP13-x and NCMNP21-x. This ratio was changed to 5, 20, and 30 percent for NCMNP21b-x to determine their potential effect on the ability to extract membrane proteins. Following hydrolysis, all polymers became soluble and were precipitated out of the solution to separate from unreacted residues. The freeze-dried polymers were obtained with high purity and excellent yields (>90%). Using 1H NMR and FTIR, the structures and molecular weight of the NCMN polymers were validated (Fig. S1–S3†).Comparative application analyses of NCMNP13-x, NCMNP21-x, and NCMNP21b-x
Effect of grafted amine moieties. Using AcrB as a model protein, the solubilization efficiency of NCMNP13-50, NCMNP21-20, and NCMNP21b-20 at a final concentration of 2.5% (w/v) was evaluated. After adding NCMN polymers, all membrane solutions eventually became transparent, showing that these polymers could solubilize the membrane. A nickel affinity column was used to purify AcrB-NCMN particles in a single step. On SDS-PAGE gels, apparent bands of almost equivalent strength at 110 kDa confirm the purification of the AcrB monomer. NCMNP21b-20 prepared samples contain some contaminated protein (Fig. 1a).
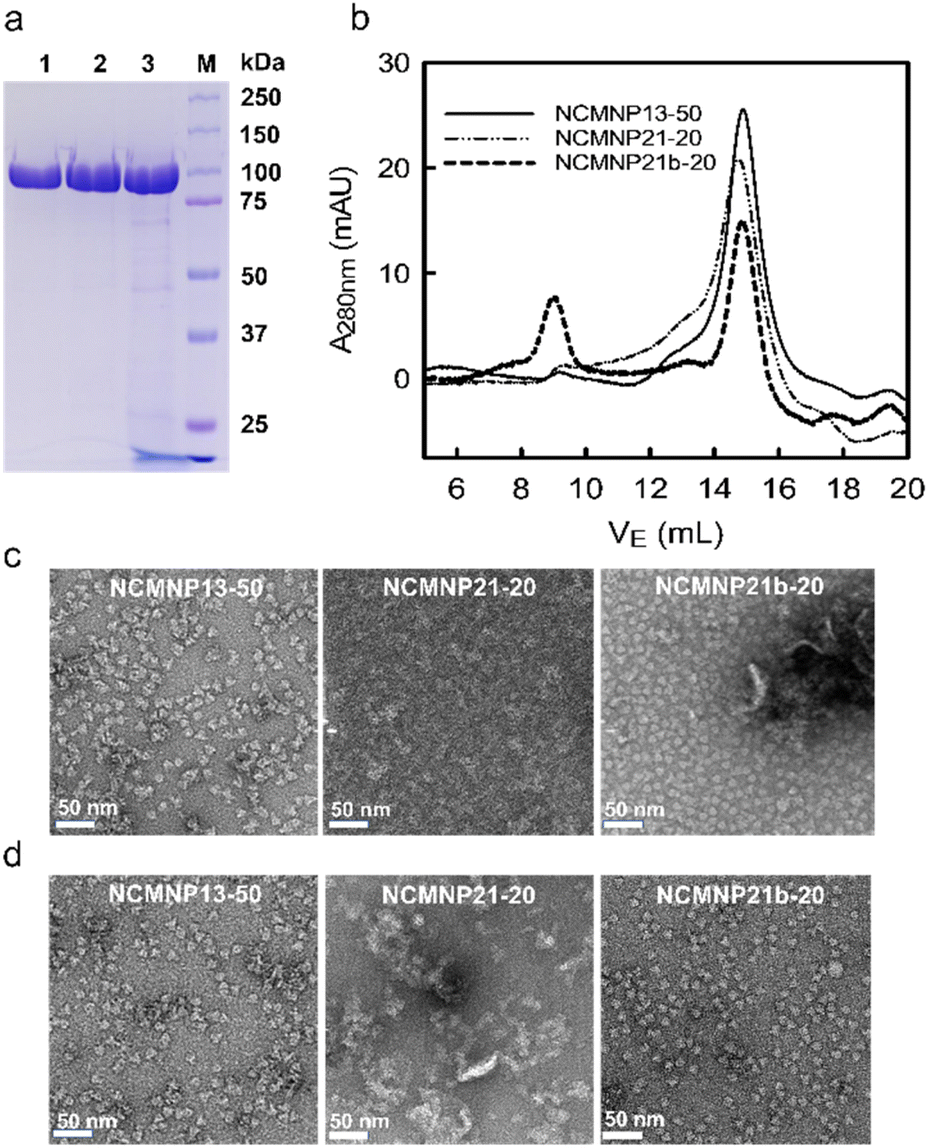 | ||
| Fig. 1 Effect of alkyl amine units of NCMN polymers on the solubilization of AcrB. (a) SDS-PAGE of AcrB particles after Ni-NTA purification visualized by Blue Coomassie (lane 1. AcrB in NCMNP13-50, lane 2. AcrB in NCMNP21-20, lane 3. AcrB in NCMNP21b-20 and M. protein ladder). (b) SEC elution profiles of AcrB particles treated with different NCMN polymers. (c) Negative stain images of one-day-old AcrB particles after Ni-NTA purification (White scale bar represents 50 nm). (d) Negative stain images of one-day-old AcrB particles after SEC purification. | ||
Moreover, the size-exclusion chromatography (SEC) purification profiles show that AcrB particles in NCMNP13-50 and NCMNP21-20 were predominantly eluted at 15 mL. However, a peak in the void volume (9 mL) was identified in NCMNP21b-20, indicating a bigger particle size (Fig. 1b). Large particles may be responsible for the decreased purity of the NCMNP21b-20 sample, as the larger proportion of formed lipids can encapsulate additional proteins. Nevertheless, the contaminants were perfectly removed via SEC as shown in Fig. S4.† These results demonstrate that our functionalized polymers suit membrane solubilization and purification.
The morphology of pure AcrB-NMCN particles was analyzed using negative staining EM (Fig. 1c and d). These polymers produce AcrB-NCMN particles with unique morphologies. Even after SEC purification, AcrB-NCMNP21-20 particles exist primarily as aggregates. Both AcrB-NCMNP13-50 and AcrB-NCMNP21b-20 particles exist as single particles with a typical triangular shape and sizes of 10 nm, consistent with prior SMA2000 studies.11 As seen, NCMNP21b-20 produced a higher monodisperse population of particles than NCMNP13-50, demonstrating the beneficial effects of the changed side chains.
The morphological divergence may be the result of unfavorable interactions mediated by the chemical nature of the novel head groups, which are arbitrarily distributed along polymeric belts, particularly in regions unrelated to TM domains. Notably, at temperatures lower than the lower critical solution temperature (LCST) of NCMNP13-50 (32 °C),28,29 the nonpolar isopropyl anchors might engage hydrophobically with the bound lipids of neighboring particles, which triggered the non-covalent aggregation of the NCMN particles.30 Similarly, the amino groups of DEADA units incorporated on NCMNP21-20 with a pKa value of 8.51 (calculated based on ChemAxon) are probably protonated in the elution buffer pH 7.8, and hence electrostatically interact with carboxylic units, anionic lipids, and cytoplasmic domains,31 of which are negative-charged species, consequently destabilizing the NCMN particles. Lastly, in the case of NCMNP21b-20, the less aggregation probably relates to the zwitterionic nature of propane-1-sulfonate (PS),32 where its oppositely charged ions on the same unit associate with each other in contrast to other ionic species. Thus, the total charge on the AcrB-NCMNP21b-20 particle surface stays negative, maintaining the repulsion between the particles and reducing their potential for aggregation.
Effect of membrane proteins. Except for AcrB, we expanded our analysis to include MscS, a member of the mechanosensitive channel family that is structurally distinct from AcrB. MscS is a homoheptamer, with each monomer containing a cytoplasmic and TM domain and a molar mass of 31 kDa.26 For these three NCMN polymers, there were no significant changes in the yields and purity of MscS-NCMN particles following elution from the Ni-NTA column, as determined by SDS-PAGE (Fig. S5a†). Nevertheless, their size exclusion chromatography profiles varied. As depicted in Fig. S5b,† AcrB-NCMNP21b-20 elutes as a single peak at 14 mL, which corresponds to the typical retention volume of MscS-SMA2000.26 In contrast, both AcrB-NCMNP21 and AcrB-NCMNP13-50 exhibit two large peaks at 9 mL and 15 mL. The change to a reduced elution volume implies particle aggregation, whereas the increased elocution volume is attributable to the fragmentation of the particle complex. These were confirmed by additional characterization using negative-stain TEM. As demonstrated in Fig. S5c,† the NCMNP21b-20 formed homogenous particles with a high mono-distribution, whereas variable amounts of aggregation were seen for the MscS-NCMNP13-50 and MscS-NCMNP21-20 samples. More aggregation was seen for NCMNP21-20, which, as stated for AcrB-NCMN particles, is a result of electrostatic interactions between positively charged side groups of DMEDA units and negatively charged clusters in neighboring MscS-NCMNP21-20 particles. These electrostatic connections are far stronger than those between MscS-NCMNP13-50 particles, with only non-specific hydrophobic interactions taking place at IPA segments. Surprisingly, the MscS complexes purified with NCMNP13-50 and NCMNP21-20 may have been fractured during the SEC purification, as shown by the presence of smaller, undefined-shaped particles on the TEM image (Fig. S5c and d†). The flexibility of MscS-TM domains may reduce their propensity for binding to polymers. When the NCMN particles pass through the SEC column, the mechanical shearing forces may further deteriorate the loose association between the protein particles and polymers. In contrast, the well-defined form of MscS can be observed in the SEC elution fraction of NCMNP21-20 independent of aggregation (Fig. S5d†). We hypothesize that at pH 7.8, the negative charge on MscS with an isoelectric point (pI) below 7.9 is less than that on AcrB (pI below 5.35). Obviously, this could also be a result of the weak electrostatic contact between amino groups and NCMNP21-20, which reduces particle aggregation. These data imply that the solubilizing activity of NCMN polymers may be protein-independent; nevertheless, the remarkable long-term stability of the resultant NCMN particles depends on the composition of the NCMN polymers and membrane proteins.31
The effect of grafting percentage. NCMNP21b-20 generated superior NCMN particles compared to NCMNP13-50 and NCMNP20-20. Therefore, NCMNP21b-5 and NCMNP21b-30 were utilized to investigate the effect of grafting degree on protein solubilization. The solubilization results indicate that the percentage of grafting drastically alters the purification yield, purity, and particle morphology (Fig. 2).
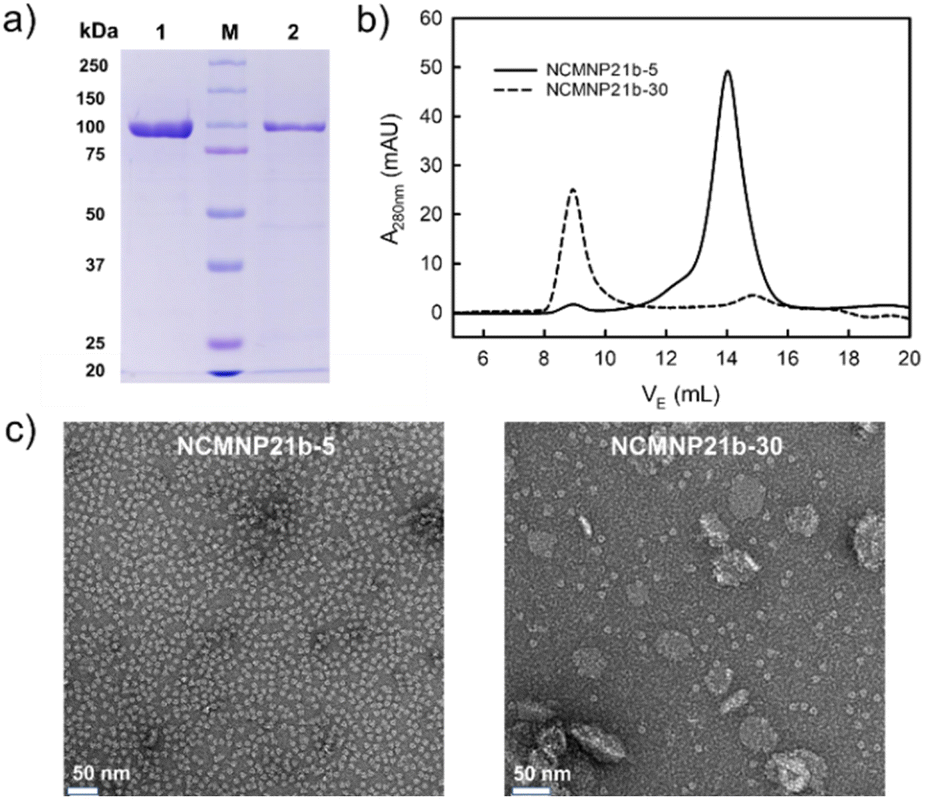 | ||
| Fig. 2 Effect of the grafting degree of NCMN polymers on the solubilization of AcrB. (a) SDS-PAGE of AcrB particles after Ni-NTA purification visualized by Blue Coomassie (lane 1. AcrB in NCMNP21b-5, lane 2. AcrB in NCMNP21b-30 and M. protein ladder). (b) SEC elution profiles of AcrB particles treated with different NCMN polymers. (c) Negative stain images of one-day-old AcrB particles after Ni-NTA purification (White scale bar represents 50 nm). | ||
Fascinatingly, NCMNP21b-5 produced the highest yield and purity of AcrB particles, which decreased in the following order: NCMNP21b-5 is superior to NCMNP21b-20 and NCMNP21b-30 (Fig. 2a and b). Similar to SMA2000,11 NCMNP21b-5 produced only single particles with diameters of approximately 10 nm (Fig. 2c). small single particles dominated AcrB-NCMNP21b-20, while AcrB-NCMNP21b-30 was dominated by larger patches. It appears that the ability to self-assemble into single particles diminishes as PS grafting levels rise. However, this raises the question of why the NCMNP13-50 containing 50% IPA units produced only single particles. Previous work of Scheidelaar et al. depicting the SMA-induced solubilization mechanism,5 where the efficiency of SMA is mainly driven by hydrophobic interactions and satisfied with a relatively small cross-sectional area given by its –COOH side groups. Fundamentally, the hydrophobic insertion of the phenyl ring into the lipid bilayer, recognized as a crucial prerequisite for membrane assembly, is favorable with –COOH as a neighboring unit because it can reduce steric hindrance. Substituting these typical units with larger alkyl groups likely generates a relatively large cross-sectional area that can hinder phenyl–lipid interactions, resulting in a larger particle size and lower particle yield.33–36 As described here, despite having a large number of –COOH replaced, NCMNP13-50 is still capable of producing single particles due to the fact that its IPA units are too small to have significant adverse effects. In contrast, the high grafting degree of PS, a much larger group than –COOH and IPA, made it difficult to insert NCMNP21b-x into the membrane to initiate solubilization, resulting in a decrease in solubilization efficiency. These findings allow us to comprehend the relationship between modification levels and membrane solubilization action, paving the way for future research to tailor NCMN polymers optimally.
| Polymer | Amine/MAnh feeding ratioa | Grafting degree of amineb (%) | M n (g mol−1) | Optimum pHd | Tolerance to [Ca2+]d (mM) |
|---|---|---|---|---|---|
| a Molar ratio. b Experimental grafting percentage of amine calculated based on 1H NMR characterization. c Calculated based on 1H-NMR (see ESI on Section 3). d Stability of AcrB particles in various buffer environments determined based on turbidity experiments (Fig. S6 and S7). | |||||
| SMA2000 | 0 | 0 | 3000 | >5 | <1.25 |
| NCMNP13-50 | 1.05 | 47.4 | 3300 | >3 | 7.5 |
| NCMNP21-20 | 0.40 | 17.7 | 3200 | >3 | 2.5 |
| NCMNP21b-5 | 0.10 | 4.4 | 3100 | >4 | 5.0 |
| NCMNP21b-20 | 0.40 | 18.0 | 3600 | >3 | 15.0 |
| NCMNP21b-30 | 0.60 | 27.5 | 3900 | >2 | 25.0 |
AcrB-NCMNP13-50 particles begin to aggregate at pH values below 3 and in the presence of [Ca2+] concentrations greater than 7.5 mM, as shown by the increased absorption bands (Fig. S6a and S7a†). The compatibility of wider pH ranges and divalent cations with the NCMNP13-50 polymer as compared to SMA2000 is most likely due to the reduction of approximately 50 percent of the polymer's negative charge.6 In contrast, NCMNP21-20 particles, which have a lower Ca2+-tolerance capacity than NCMNP13-50, can withstand a pH as low as 3 with only 20 percent supplants (Fig. S6b and S7b†). This enhanced acidic pH stability may be attributable to the presence of positively charged groups, which inhibit non-covalent aggregation caused by protonation of –COOH groups.21,37,38 In-depth examination of the NCMNP21b-x series revealed that, with the same grafting levels as NCMNP21-20, the NCMNP21b-20 particles are more stable in the presence of Ca2+ at concentrations up to 15 mM. For the NCMNP21b-x, the tolerance to Ca2+ follows the order of NCMNP21b-5 < NCMNP21b20 < NCMNP21b-30 (Fig. S7c–e†). A similar trend is observed in the pH tolerance, which increases as the grafting degree increases (Fig. S6c–e†). The presence of pH-insensitive PS units results in the formation of negatively charged holes on the surface of the belt, which allows Ca2+ to bind while remaining highly soluble.39–41 Therefore, it may account for the improvement of NCMNP21b-x's notable Ca2+ resistance. In conclusion, the stability of NCMN particles is dependent upon the nature of the side groups and the percentage of grafting.
 | ||
| Fig. 3 Lipid analysis and structural determination of AcrB by cryo-EM. (a) Abundance comparison of three significant phospholipids in membrane fractions and in AcrB particles displayed as the sum for each. (b) Representative 2D class averages of AcrB-NCMNP21b-20. (c) The cryo-EM map of AcrB-NCMNP21b- 20 is coloured by local resolution. (d) Endogenous lipid located with the TM of the pseudo-symmetric AcrB trimer. | ||
Conclusions
In this work, we describe novel SMA variants known as NCMN polymers and investigate the effects of their side chains on membrane solubilization, morphology, and stability of NCMN particles. Various alkyl amide units were substituted for the –COOH groups of commercial SMA2000 to produce these polymers. A systematic comparison of the application of these polymers to two distinct membrane proteins revealed:• All NCMN polymers can directly induce the solubilization of AcrB and MscS proteins into NCMNs with various sizes and morphologies.
• AcrB-NCMN particle size and homogeneity are primarily determined by the coupling levels of amines. Many large alkyl side groups can interfere with the interaction between styrene and lipids, decreasing the likelihood of obtaining a high yield of small single particles.
• The long-term stability of NCMN particles is contingent upon the chemical nature of polymers and protein structure. MscS-NCMN particles in the same polymer were theoretically more stable than AcrB-NCMN particles at pH 7.8 due to the positive charge on the sidechain of NCMNP21-20.
• When more non-chelating units are added to NCMN polymers, their compatibility with a broad pH range and divalent cations is enhanced.
• Side groups do not interact with lipids, allowing NCMN polymers to retain lipids in NCMN particles close to their native state.
• NCMNP21b-x polymers with vastly improved divalent ion and pH range compatibility are highly desirable for membrane protein structural biology.
These findings provide valuable insights into critical factors that will enable the future engineering of SMA with desirable properties for membrane protein research.
Experimental
Materials
Styrene–maleic anhydride copolymer (SMAnh, St:MAnh = 2:1, Mn = 3000 g mol−1, Mw = 7500 g mol−1, acid number: 358 g KOH per mol) was purchased from Cray Valley USA, LLC (Exton, PA). N,N-Dimethylformamide (DMF, 99.8%, extra dry, anhydrous, AcroSeal), chloroform (CHCl3, 99.9%, extra dry, stabilized, AcroSeal), and deuterium oxide (D2O, 99.8 atom % D) was brought from Acros. Isopropylamine (IPA, >99%, reagent grade), 1,3-propanesultone (PS, 98%), N,N-dimethylethylenediamine (DMEDA) and tris(2-carboxyethyl) phosphine hydrochloride (TCEP) were obtained from TCI, Sigma, and GoldBio, respectively. Sodium hydroxide (NaOH, pellets, certified ACS), calcium chloride dihydrate (CaCl2·2H2O, certified ACS), sodium chloride (NaCl, crystalline, certified ACS), diethyl ether (laboratory), hydrochloric acid (HCl, 36.5 to 38.0%, certified ACS plus), sodium acetate anhydrous (white crystals), glycine (white crystals), HEPES (white crystals, molecular biology) and imidazole (molecular biology), were bought from Fisher. All chemicals were used as received.
Polymer synthesis
000g for 20 min. Subsequently, the HCl was decanted, and the polymer was rinsed again with water (twice). Finally, the polymer was re-dissolved in NaOH (0.6 M), and the pH of the polymeric solution was adjusted to 7.8 before lyophilization. Finally, 5.876 g product was obtained (yield: 98.1%).
000g for 20 min. Afterward, the HCl was decanted, and the polymer was rinsed again with water (twice). Finally, the polymer was re-dissolved in NaOH (0.6 M), and the pH of the polymer solution was adjusted to 7.8 before lyophilization. Finally, 5.327 g product was obtained (yield: 95.8%).
000g for 20 min. Afterward, the HCl was decanted, and the polymer was rinsed again with water (twice). Finally, the polymer was re-dissolved in NaOH (0.6 M), and the pH of the polymer solution was adjusted to 7.8 before lyophilization. Finally, 5.831 g product was obtained (yield: 91.5%).
- NCMNP21b-5: DMEDA (172 μL, 1.58 mmol, 0.1 equiv.) and PS (306 μL, 3.49 mmol, 0.22 equiv.).
- NCMNP21b-30: DMEDA (1038 μL, 9.52 mmol, 0.6 equiv.) and PS (862 μL, 9.84 mmol, 0.62 equiv.).
Finally, 5.214 g (yield: 93.7%) and 6.483 g (yield: 92.1%) were obtained for NCMNP21b-5 and NCMNP21b-30.
Polymer characterization
Expression and purification of AcrB and MscS
Both 8 His-tagged AcrB and 10 His-tagged MscS were over-expressed in the E. coli BL21DE3PlysS strain, and membrane fractions were prepared as described previously.26,46,47 1 g of membrane fraction was suspended and homogenized in 10 mL NCMN Buffer A using a Dounce homogenizer, according to a standard solubilization protocol. The membrane was then transferred to a 50 mL polypropylene tube and blended with NCMN polymer at a final concentration of 2.5% w/v. The incubation was carried out for 2 h at 20 °C, and the insoluble species were then spun down at 200000g for 1 h at 20 °C. The supernatant was collected and loaded onto a 5 mL Ni-NTA column (GE Healthcare) pre-equilibrated with NCMN Buffer A at a flow rate of 0.5 mL min−1. The column was washed with 25 mL of each NCMN Buffer B and NCMN Buffer C prior to the elution of the protein with 20 mL of a mixture buffer of the NCMN Buffer C and NCMN Buffer D (1:1 v/v). Collected membrane protein fractions were loaded onto a Superpose 6 increase 10/300 GL column (GE Healthcare) and eluted with 30 mL of NCMN Buffer E. The NCMN polymer was added to all washing and elution buffers with a final concentration of 0.05% (w/v).
All the buffers were filtered with 0.22 μm MCE Membrane (MF-Millipore™) before use, and their compositions are listed below:
- NCMN Buffer A: 50 mM HEPES, pH 7.8, 500 mM NaCl, 5% glycerol, 20 mM imidazole, 0.1 mM TCEP.
- NCMN Buffer B: 25 mM HEPES, pH 7.8, 500 mM NaCl, 40 mM imidazole, 0.1 mM TCEP.
- NCMN Buffer C: 25 mM HEPES, pH 7.8, 500 mM NaCl, 75 mM imidazole, 0.1 mM TCEP.
- NCMN Buffer D: 25 mM HEPES, pH 7.8, 500 mM NaCl, 500 mM imidazole, 0.1 mM TCEP.
- NCMN Buffer E: 40 mM HEPES, pH 7.8, 500 mM NaCl, 0.1 mM TCEP.
NCMN particles characterization
- Ca2+-based buffers (pH 7.8): 40 mM HEPES, 100 mM NaCl, 0.1 mM TCEP and a given CaCl2 concentration (0–100 mM).
- pH buffers:
• Tris buffer (pH 10): 40 mM Tris, 100 mM NaCl, 0.1 mM TCEP.
• HEPES buffer (pH 7.8): 40 mM HEPES, 100 mM NaCl, 0.1 mM TCEP.
• Sodium acetate buffer (pH 4 and 5): 40 mM sodium acetate, 100 mM NaCl, 0.1 mM TCEP.
• Glycine buffer (pH 2 and 3): 40 mM glycine, 100 mM NaCl, 0.1 mM TCEP.
Ca2+ sensitivity. To 100 μL of AcrB-NCMN particles (OD280 ∼ 1 mg mL−1) in elution buffer (pH 7.8) was added dropwise 100 μL of Ca2+-based buffer to achieve the final Ca2+ concentration (0–50 mM). After 1 h incubation, the transparent changes were monitored by a NanoDrop spectrophotometer.
Every experiment was conducted using a NanoDropTM2000 spectrophotometer (Thermo Scientific). All spectra with a normalized 10 mm pathlength absorbance were collected using the preconfigured Protein A280 mode with a bandwidth of 1 nm and a scan speed of 800 nm min−1.
000 magnification at specimen level.
:1 v/v) and allowed to shake for 1 h at 4 °C. Afterward, the mixture was centrifuged at 12000g and 4 °C for 10 min to give phase separation. The aqueous layer (upper phase) was removed, while the organic layer (lower phase) was washed twice with 1 volume of cold water and evaporated to complete dryness. The total lipid extract was analyzed by the Thermo Scientific™ Vanquish™ UHPLC system utilizing a C18+ 2.1 (i.d.) × 150 mm reverse-phase column with 1.5 μm particles. The mass spectrometer was operated at 55 °C with a binary solvents system, mobile phase A1 (CH3CN/H2O, 50/50, v/v, with 5 mM ammonium formate and 0.1% formic acid) and mobile phase B1 (CH3CHOHCH3/CH3CN/H2O, 88/10/2, v/v/v, with 5 mM ammonium formate and 0.1% formic acid) at a flow rate of 0.26 mL min−1. The lipid sample was characterized in both positive and negative ionization modes. Full mass spectra were recorded in a range of 300 to 2000 m/z with the resolution set to 30000. All data were analyzed via Thermo Scientific's Lipid Search 4.2 software.
000 nominal magnification, corresponding to a calibrated pixel size of 0.8256 Å per pixel. An initial dataset of 8591 micrographs for AcrB was obtained by automated data collection using Leginon, with nominal defocus values ranging between 0.4 and 2.9 μm at a dose rate of 44.18 e− Å−2 s−1 with a total exposure of 1.36 s, for an accumulated dose of 60.22 e− Å−2.49 The full images dataset was processed using CryoSparc v3.3.1: beam-induced motion and CTF were corrected using patch motion and CTF correction function modules.50,51 The exposures were manually curated, removing all the micrographs with more than 1000 Å astigmatism and more than 5 Å CTF fit resolution. ∼1.5 million particles were automatically picked with Topaz and extracted with a box size of 360.52 Several rounds of 2D classifications were performed to remove junk particles and duplicates. The best-looking 2D classes and their respective particles were subjected to ab initio reconstruction. Non-uniform reconstruction was then performed on 85980 particles with C1 symmetry, and the resulting sharpened model had a resolution of 3.6 Å according to the gold-standard Fourier shell correlation (Fig. S9†). The cryo-EM data collection and processing parameters are summarized in Table S1.†
Author contributions
Y. G. conceived the project. Y. G. and T. K. H. T. designed the experiments. T. K. H. T. performed the polymer synthesis, characterization, NCMN sample preparation, analysis, and cryo-EM data processing. CCparticipated in growing cell culture and preparation of the cryo-EM map. Y. G. oversaw and supervised the project. T. K. H. T. drafted the manuscript. Y. G., T. K. H. T. and C. C. revised the manuscript.Conflicts of interest
Y. G. and T. K. H. T. are inventors of NCMNP13-x, NCMNP21-x and NCMNP21b-x polymers (Patent pending). Y. G. is the founder of NCMNtech LLC.Acknowledgements
Y. G. was supported by the VCU School of Pharmacy and the Department of Medicinal Chemistry through startup funds, the Institute for Structural Biology, Drug Discovery and Development through laboratory space and facilities, and NIH Grant R01-GM132329 (to YG). The funders had no role in study design, data collection, analysis, publication decision, or manuscript preparation. The content is solely the authors' responsibility and does not necessarily represent the National Institutes of Health or other funding organizations' official views. The lipidomic analyses were performed at the VCU Lipidomics/Metabolomics Core with the NIH-NCI Cancer Center Support Grant P30 CA016059 to the VCU Massey Cancer Center and shared resource grant (S10RR031535) from the National Institutes of Health. We also thank ChemAxon for providing us with the academic license to access the ChemAxon 20.11.0 version for pKa calculation. We are grateful to Edward T. Eng, Charlie Dubbeldam, Carolina Hernandez, Kashyap Maruthi for their great help for cryo-EM data collection at the National Center for CryoEM Access and Training (NCCAT) and the Simons Electron Microscopy Center located at the New York Structural Biology Center, supported by the NIH Common Fund Transformative High-Resolution Cryo-Electron Microscopy program (U24 GM129539), and by grants from the Simons Foundation (SF349247) and NY State Assembly. Last, we also thank Abu Bakkar Siddique for his constructive comments and suggestions.References
- M. Ernst and J. L. Robertson, J. Mol. Biol., 2021, 433, 167103 CrossRef CAS PubMed.
- H. Yin and A. D. Flynn, Annu. Rev. Biomed. Eng., 2016, 18, 51–76 CrossRef CAS PubMed.
- K. R. Vinothkumar and R. Henderson, Q. Rev. Biophys., 2010, 43, 65–158 CrossRef CAS PubMed.
- Y. Guo, Crystals, 2020, 10, 86 CrossRef CAS PubMed.
- S. Scheidelaar, M. C. Koorengevel, J. D. Pardo, J. D. Meeldijk, E. Breukink and J. A. Killian, Biophys. J., 2015, 108, 279–290 CrossRef CAS PubMed.
- Z. Stroud, S. C. L. Hall and T. R. Dafforn, Methods, 2018, 147, 106–117 CrossRef CAS PubMed.
- B. Krishnarjuna, S.-C. Im, T. Ravula, J. Marte, R. J. Auchus and A. Ramamoorthy, Anal. Chem., 2022, 94, 11908–11915 CrossRef CAS PubMed.
- T. Ravula and A. Ramamoorthy, Angew. Chem., Int. Ed., 2021, 60, 16885–16888 CrossRef CAS PubMed.
- A. H. Kopf, J. M. Dorr, M. C. Koorengevel, F. Antoniciello, H. Jahn and J. A. Killian, Biochim. Biophys. Acta, Biomembr., 2019, 1862, 183125 CrossRef PubMed.
- A. O. Oluwole, J. Klingler, B. Danielczak, J. O. Babalola, C. Vargas, G. Pabst and S. Keller, Langmuir, 2017, 33, 14378–14388 CrossRef CAS PubMed.
- W. Qiu, Z. Fu, G. G. Xu, R. A. Grassucci, Y. Zhang, J. Frank, W. A. Hendrickson and Y. Guo, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 12985–12990 CrossRef CAS PubMed.
- Y. Guo, Biochem. Soc. Trans., 2021, 49, 1361–1374 CrossRef CAS PubMed.
- C. Sun, S. Benlekbir, P. Venkatakrishnan, Y. Wang, S. Hong, J. Hosler, E. Tajkhorshid, J. L. Rubinstein and R. B. Gennis, Nature, 2018, 557, 123–126 CrossRef CAS PubMed.
- S. Scheidelaar, M. C. Koorengevel, C. A. van Walree, J. J. Dominguez, J. M. Dörr and J. A. Killian, Biophys. J., 2016, 111, 1974–1986 CrossRef CAS PubMed.
- A. A. Gulamhussein, R. Uddin, B. J. Tighe, D. R. Poyner and A. J. Rothnie, Biochim. Biophys. Acta, Biomembr., 2020, 1862, 183281 CrossRef CAS PubMed.
- F. Li, P. F. Egea, A. J. Vecchio, I. Asial, M. Gupta, J. Paulino, R. Bajaj, M. S. Dickinson, S. Ferguson-Miller, B. C. Monk and R. M. Stroud, J. Biol. Chem., 2021, 296, 100557 CrossRef CAS PubMed.
- C. L. Bon, B. Michon, J.-L. Popot and M. Zoonens, Q. Rev. Biophys., 2021, 54, E6 CrossRef PubMed.
- J. M. Dörr, S. Scheidelaar, M. C. Koorengevel, J. J. Dominguez, M. Schäfer, C. A. van Walree and J. A. Killian, Eur. Biophys. J., 2016, 45, 3–21 CrossRef PubMed.
- T. Ravula, S. K. Ramadugu, G. Di Mauro and A. Ramamoorthy, Angew. Chem., Int. Ed., 2017, 129, 11624–11628 CrossRef.
- T. Ravula, N. Z. Hardin, G. M. Di Mauro and A. Ramamoorthy, Eur. Polym. J., 2018, 108, 597–602 CrossRef CAS PubMed.
- T. Ravula, N. Z. Hardin, S. K. Ramadugu and A. Ramamoorthy, Langmuir, 2017, 33, 10655–10662 CrossRef CAS PubMed.
- S. Lindhoud, V. Carvalho, J. W. Pronk and M.-E. Aubin-Tam, Biomacromolecules, 2016, 17, 1516–1522 CrossRef CAS PubMed.
- M. C. Fiori, Y. Jiang, G. A. Altenberg and H. Liang, Sci. Rep., 2017, 7, 7432 CrossRef PubMed.
- A. Marconnet, B. Michon, C. Le Bon, F. Giusti, C. Tribet and M. Zoonens, Biomacromolecules, 2020, 21, 3459–3467 CrossRef CAS PubMed.
- A. J. Higgins, A. J. Flynn, A. Marconnet, L. J. Musgrove, V. L. G. Postis, J. D. Lippiat, C. Chung, T. Ceska, M. Zoonens, F. Sobott and S. P. Muench, Commun. Biol., 2021, 4, 1–9 CrossRef PubMed.
- C. Catalano, D. Ben-Hail, W. Qiu, P. Blount, A. des Georges and Y. Guo, Membranes, 2021, 11, 849 CrossRef CAS PubMed.
- ProtParam References, https://web.expasy.org/protparam/protpar-ref.html, accessed 19 December 2021.
- K. Tauer, D. Gau, S. Schulze, A. Völkel and R. Dimova, Colloid Polym. Sci., 2009, 287, 299 CrossRef CAS.
- M. Bahram and P. Najafi-Moghaddam, Chiang Mai J. Sci., 2017, 44, 224–236 CAS.
- G. Bokias, D. Hourdet, I. Iliopoulos, G. Staikos and R. Audebert, Macromolecules, 1997, 30, 8293–8297 CrossRef CAS.
- T. Ravula, N. Z. Hardin, J. Bai, S.-C. Im, L. Waskell and A. Ramamoorthy, Chem. Commun., 2018, 54, 9615–9618 RSC.
- M.-C. Sin, S.-H. Chen and Y. Chang, Polym. J., 2014, 46, 436–443 CrossRef CAS.
- M. Esmaili, C. Acevedo-Morantes, H. Wille and M. Overduin, Biochim. Biophys. Acta, Biomembr., 2020, 1862, 183360 CrossRef CAS PubMed.
- N. Z. Hardin, T. Ravula, G. D. Mauro and A. Ramamoorthy, Small, 2019, 15, 1804813 CrossRef PubMed.
- J. L. Thomas, B. P. Devlin and D. A. Tirrell, Biochim. Biophys. Acta, 1996, 1278, 73–78 CrossRef PubMed.
- J. C. Chung, D. J. Gross, J. L. Thomas, D. A. Tirrell and L. R. Opsahl-Ong, Macromolecules, 1996, 29, 4636–4641 CrossRef CAS.
- T. Ravula, N. Z. Hardin, S. K. Ramadugu, S. J. Cox and A. Ramamoorthy, Angew. Chem., Int. Ed., 2018, 57, 1342–1345 CrossRef CAS PubMed.
- S. C. L. Hall, C. Tognoloni, J. Charlton, É. C. Bragginton, A. J. Rothnie, P. Sridhar, M. Wheatley, T. J. Knowles, T. Arnold, K. J. Edler and T. R. Dafforn, Nanoscale, 2018, 10, 10609–10619 RSC.
- J. Xu, W. Sun, G. Li, J. Xu, L. Zheng, H. Wang and G. Zhang, Chem. J. Chin., 2007, 28, 496 CAS.
- Y. Cui, S. Gao, R. Zhang, L. Cheng and J. Yu, Polymers, 2020, 12, 98 CrossRef CAS PubMed.
- X. Ji, M. Tian, D. Ma, Y. Zhu, Z.-H. Zhou, Q. Zhang and Y. Wang, Langmuir, 2018, 34, 291–301 CrossRef CAS PubMed.
- B. Krishnarjuna and A. Ramamoorthy, Biomolecules, 2022, 12, 1076 CrossRef CAS PubMed.
- B. Krishnarjuna, J. Marte, T. Ravula and A. Ramamoorthy, J. Colloid Interface Sci., 2023, 634, 887–896 CrossRef CAS PubMed.
- I. Prabudiansyah, I. Kusters, A. Caforio and A. J. M. Driessen, Biochim. Biophys. Acta, Biomembr., 2015, 1848, 2050–2056 CrossRef CAS PubMed.
- C. R. Raetz, Microbiol. Rev., 1978, 42, 614–659 CrossRef CAS PubMed.
- Y. Guo, R. C. Kalathur, Q. Liu, B. Kloss, R. Bruni, C. Ginter, E. Kloppmann, B. Rost and W. A. Hendrickson, Science, 2015, 347, 551–555 CrossRef CAS PubMed.
- K. G. Kroeck, W. Qiu, C. Catalano, T. K. H. Trinh and Y. Guo, J. Visualized Exp., 2020, e61298 Search PubMed.
- S. J. Hesketh, D. P. Klebl, A. J. Higgins, M. Thomsen, I. B. Pickles, F. Sobott, A. Sivaprasadarao, V. L. G. Postis and S. P. Muench, Biochim. Biophys. Acta, Biomembr., 2020, 1862, 183192 CrossRef CAS PubMed.
- C. Suloway, J. Pulokas, D. Fellmann, A. Cheng, F. Guerra, J. Quispe, S. Stagg, C. S. Potter and B. Carragher, J. Struct. Biol., 2005, 151, 41–60 CrossRef CAS PubMed.
- A. Punjani, J. L. Rubinstein, D. J. Fleet and M. A. Brubaker, Nat. Methods, 2017, 14, 290–296 CrossRef CAS PubMed.
- A. Punjani, H. Zhang and D. J. Fleet, Nat. Methods, 2020, 17, 1214–1221 CrossRef CAS PubMed.
- T. Bepler, A. Morin, M. Rapp, J. Brasch, L. Shapiro, A. J. Noble and B. Berger, Nat. Methods, 2019, 16, 1153–1160 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3na00381g |
| This journal is © The Royal Society of Chemistry 2023 |