
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNovel two-dimensional Janus β-Ge2XY (X/Y = S, Se, Te) structures: first-principles examinations
Nguyen Dinh
Hien
 ab,
D. V.
Lu
*c and
Le C.
Nhan
d
ab,
D. V.
Lu
*c and
Le C.
Nhan
d
aInstitute of Research and Development, Duy Tan University, Da Nang 550000, Vietnam
bSchool of Engineering & Technology, Duy Tan University, Da Nang 550000, Vietnam
cFaculty of Physics, The University of Danang – University of Science and Education, Da Nang, 550000, Vietnam. E-mail: dvlu@ued.udn.vn
dFaculty of Environmental Science, Saigon University, 273 An Duong Vuong Street, Ward 3, District 5, Ho Chi Minh City, Vietnam
First published on 1st August 2023
Abstract
Two-dimensional (2D) structures can stably exist in different allotropes. In this manuscript, we propose a new series of Janus structures based on the β-phase of germanium monochalcogenides, namely, β-Ge2XY (X/Y = S, Se, and Te) monolayers. Our calculations indicate that Janus β-Ge2XY monolayers have a stable crystal structure and possess anisotropic mechanical properties. At the ground state, β-Ge2XY monolayers are semiconductors with a large bandgap and their electronic properties depend strongly on a biaxial strain. Strains not only change the bandgap but can also lead to a change in the bandgap characteristic, namely transitions from indirect to direct bandgap. Our findings not only introduce a new structure of germanium chalcogenide compounds but also show that they have superior physical properties suitable for applications in nanoelectronics.
1 Introduction
Two-dimensional (2D) nanomaterials have become a major issue of interest in the materials research community since the discovery of graphene.1 With outstanding physical properties, graphene is expected to greatly contribute to the creation of a new generation of high-performance electronic and optoelectronic devices.2,3 In parallel with the study of the mechanical and physical properties of graphene, the search for 2D graphene-like structures has been intensely promoted. As a result, the 2D family is constantly expanding with a series of new materials that have been successfully synthesized in recent times, namely silicene,4 transition metal dichalcogenides,5 and post-transition metal monochalcogenide compounds.6 Among them, group IV monochalcogenides are of particular interest due to their extraordinary physical features, environmental friendliness, and potential applications in new technologies.7The physical features of 2D layered materials depend greatly on the number of atomic sublayers, stacking configuration, and especially their symmetric group. Along with the discovery of new materials, the search for different allotropes of existing materials is also of interest to scientists. Xu et al. have shown that group IV monochalcogenide compounds can stably exist in different structural phases, including α and β phases.8 Moreover, the γ-phase of group IV monochalcogenides (γ-GeSe) has been experimentally released recently.9 It has been indicated that, with different symmetry structures, the physical properties of phases of group IV monochalcogenides are significantly different.8,10,11 Particularly, β-GeSe exhibits extremely high electron mobility, up to 2.93 × 104 cm2 V−1 s−1,8 which is suitable for nanoelectronic applications.
The Janus vertical asymmetric structures of 2D materials have been successfully synthesized experimentally.12,13 The successful fabrication of Janus materials by atomic layer substitution has opened up great opportunities for creating many new materials with fascinating physical properties.14,15 With the absence of mirror symmetry, Janus structures exhibit many new and outstanding physical properties that are not available to symmetric structures, such as the appearance of in-built electric fields16 or high out-of-plane piezoelectricity.17 With their outstanding physical properties, Janus asymmetrical structures have attracted special attention in recent times. Huang et al. indicated that Janus group-III monochalcogenide monolayers are perfect candidates for photocatalytic applications.18 In particular, Seixas showed that Janus structures created from the α-phase of group IV monochalcogenide materials have many physical properties that are superior to those of their original symmetric structures.19
Stimulated by the development trend of 2D Janus materials as above, in this study, we propose a series of Janus structures β-Ge2XY (X/Y = S, Se, Te) and thoroughly investigate their crystal structures, stability, mechanical features, and electronic characteristics based on the density functional theory (DFT) method. We first focus on structural stabilities through the calculations for the phonon spectrum and other stability tests. The electronic properties and the effects of mechanical strains on the electronic characteristics have been investigated thoroughly and systematically in this paper.
2 Methodology
The DFT calculations in this paper were carried out using the Vienna ab initio simulation package,20,21 using a plane-wave basis set with an energy cut-off of 500 eV to extend the Kohn–Sham orbitals. The projector augmented wave (PAW) approximation22 parameterized by Perdew, Burke, and Ernzerhof (PBE)23 was selected to describe the exchange–correlation potential. Besides, the energy gaps of the studied structures were also corrected by using the Heyd–Scuseria–Ernzerhof (HSE06) functional.24 We used the DFT-D3 method25 to describe the van der Waals in the studied layered-structures. The first Brillouin zone (BZ) was sampled by a 15 × 15 × 1 k-point grid. The threshold for forces and energy in the structural relaxation is selected to be 1 × 10−3 eV Å−1 and 1 × 10−6 eV, respectively. The phonon dispersions were carried out by using finite displacement approximation through the PHONOPY package.26 We used ab initio molecular dynamics (AIMD) calculations with the canonical ensemble to test the thermodynamical stability of the studied structures.273 Results and discussion
3.1 Crystal lattice and structural stabilities
The crystal structures of Janus β-Ge2XY (X/Y = S, Se, and Te) monolayers are shown in Fig. 1. The optimized lattice parameters of β-Ge2XY are tabulated in Table 1. The shortest lattice constants a and b (along the x and y axes, respectively) are found for β-Ge2SSe being 3.55 Å and 5.79 Å, respectively, while those of β-Ge2STe and β-Ge2SeTe are found to be higher. This results from the increase of ionic radii in the order of S, Se, and Te. The crystal structure of Janus β-Ge2XY is highly anisotropic. However, the anisotropic ratio b/a for all three structures is almost the same despite the large difference between their lattice constants. It is calculated that the anisotropic ratio of β-Ge2XY is about 1.6 as listed in Table 1. The anisotropic ratio of β-Ge2XY is much higher than that of α-Ge2XY.28 The cation–cation bond lengths d are also scrutinized and listed in Table 1. The Ge–X bond length d1 and Ge–Y bond length d3 follow a similar trend of lattice constant when the atomic size of the chalcogen elements increases.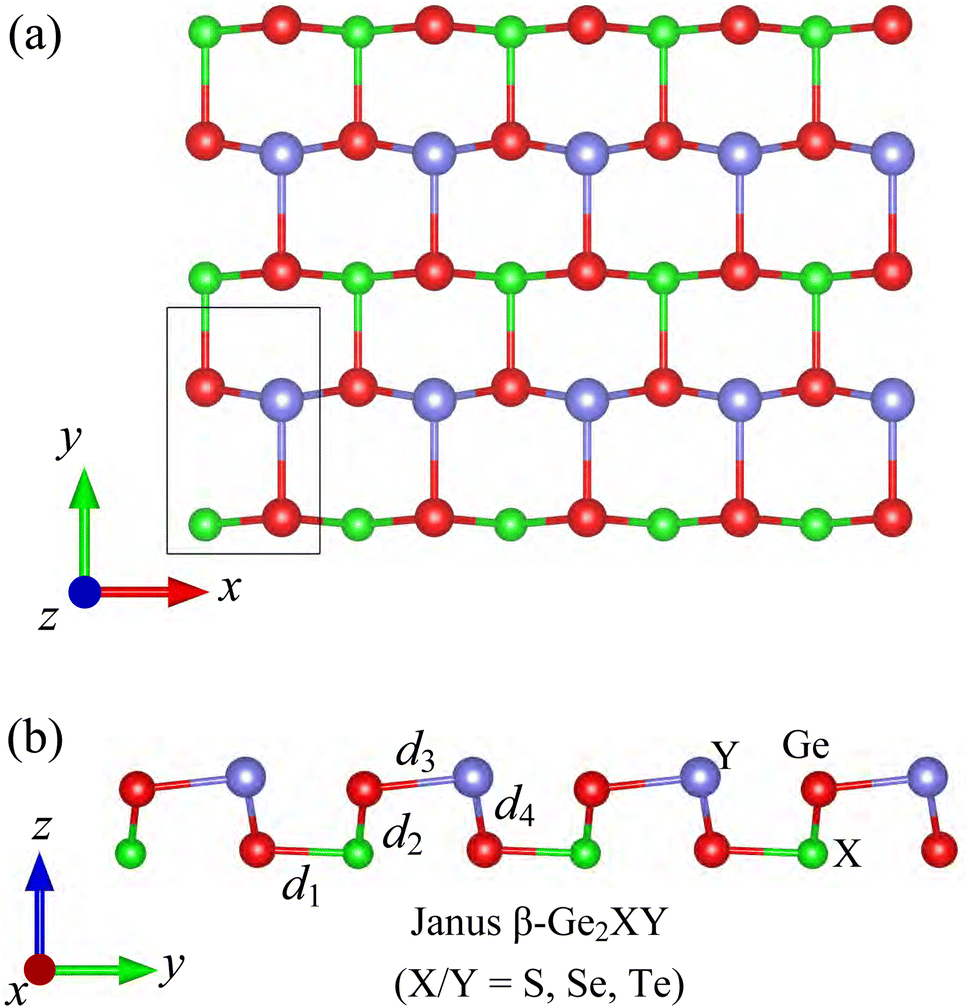 | ||
| Fig. 1 (a) Top and (b) side views of crystal structures of β-Ge2XY (X = S, Se, Te) monolayers. | ||
| a | b | d 1 | d 2 | d 3 | d 4 | Δh | b/a | E coh | C 11 | C 12 | C 22 | C 66 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| β-Ge2SSe | 3.55 | 5.79 | 2.42 | 2.54 | 2.53 | 2.69 | 1.79 | 1.63 | −4.45 | 40.34 | 5.83 | 41.55 | 7.47 |
| β-Ge2STe | 3.71 | 6.03 | 2.44 | 2.52 | 2.73 | 2.91 | 1.95 | 1.63 | −4.25 | 33.97 | 5.68 | 41.95 | 4.25 |
| β-Ge2SeTe | 3.77 | 6.08 | 2.69 | 2.56 | 2.92 | 2.72 | 2.04 | 1.61 | −4.10 | 35.42 | 3.82 | 28.20 | 4.24 |
We next calculate the cohesive energy Ecoh to evaluate the energetic stability and also the chemical bond strength in the studied monolayers. The cohesive energy can be written as
 | (1) |
To check the structural stabilities, we first evaluate the phonon spectra of the investigated materials as presented in Fig. 2(a). The phonon spectra of β-Ge2XY have 12 vibrational modes because their primitive cell contains four atoms as depicted in Fig. 1(a). The patterns of the phonon dispersion curves of all three structures are found to be similar. No negative frequencies exist throughout the Brillouin zone, implying that the studied materials are dynamically stable. In addition, we also carry out the AIMD simulations to evaluate the thermal stability. The AIMD calculations are performed within 8 ps (a time step of 1 fs) at 500 K. We present the total energy fluctuation to simulation time of the β-Ge2XY at 500 K in Fig. 2(b). It is indicated that the total energies of β-Ge2XY monolayers fluctuate slightly during the 8 ps AIMD simulation. No structural transitions nor breaking of chemical bonds in the studied materials after the test reveal that they have good thermal structural integrity at 500 K.
 | ||
| Fig. 2 Phonon spectra (a) and fluctuation of the total energies to simulation time at 500 K by AIMD calculations (b) of β-Ge2XY monolayers. | ||
We also evaluate the mechanical stability of the studied systems based on considering the elastic constants Cij. Here, the standard Voigt notation for Cij is utilized. For the 2D structure, C11, C12, C22, and C66 are four independent coefficients that should be calculated. The calculations for Cij in the present study are based on the method suggested by Duerloo and co-workers.29 The in-plane stiffness coefficients can be calculated by parabolic fitting the uniaxial strain-dependent elastic energies of the examined materials.29 We here apply uniaxial strains from −2% to +2% in 0.5% increments along the two x- and y-directions. This method has been successfully used to evaluate the elastic constants of the similar 2D structures.29–31
The obtained results for Cij of β-Ge2XY are also presented in Table 1. It is found that all four independent stiffness coefficients Cij are positive and also satisfy the condition of C11C22 > C122.32,33 It implied that they meet Born–Huang's elastic stability conditions, demonstrating that the investigated materials are mechanically stable. From the obtained elastic constants Cij, the directional dependent Young's modulus Y2D and Poisson's ratio ν (indicated by angle α) can be given by:34,35
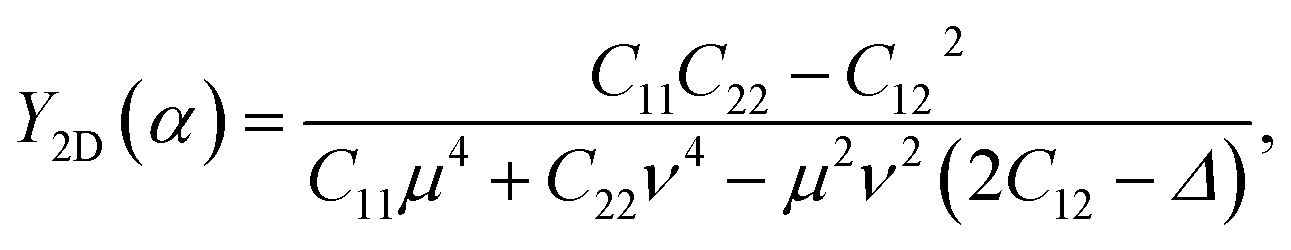 | (2) |
 | (3) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) α, ν = cosα, and Δ = (C11C22 − C122)/C66. α denotes the angle formed by the armchair axis and the considered direction.
α, ν = cosα, and Δ = (C11C22 − C122)/C66. α denotes the angle formed by the armchair axis and the considered direction.
The calculated Y2D(α) and P(α) of β-Ge2XY are presented in Fig. 3. It is demonstrated that Y2D(α) and P(α) of the examined materials are strongly directional anisotropic due to their lattice anisotropy. From Fig. 3(a), we can see that β-Ge2SSe and β-Ge2STe exhibit the highest Y2D along the direction corresponding to α = 90°. Meanwhile, the maximum Young's modulus of β-Ge2SeTe is found at α = 0°. This is due to the large difference in the chemical bond length between Ge–Te and Ge–S. The calculated values for the highest Young's modulus of β-Ge2SSe and β-Ge2STe are found to be 40.71 and 40.90 N m−1, respectively. However, the difference between Y2D(90°) and Y2D(0°) for β-Ge2SSe is very small. The obtained value for Y2D(0°) of β-Ge2SSe is 39.52 N m−1. Young's modulus of β-Ge2XY is minimum at α = 45° and α = 135°. The Y2D of β-Ge2XY is comparable with that of α group IV monochalcogenides36 and their Janus structures.37,38 The directional anisotropy of Poisson's ratio is also shown in Fig. 3(b).
 | ||
| Fig. 3 Polar diagrams of Young's modulus Y2D(α) (a) and Poisson's ratio P(α) (b) for β-Ge2XY monolayers. | ||
3.2 Electronic properties
We here use the band structures and weighted bands to investigate the electronic features of the studied Janus materials. The calculated band diagrams of β-Ge2XY monolayers are presented in Fig. 4, at both PBE and HSE06 levels. Our calculated results indicate that β-Ge2XY monolayers exhibit semiconducting characteristics with an indirect bandgap. At the PBE level, the bandgaps of β-Ge2SSe, β-Ge2STe, and β-Ge2SeTe are found to be 1.72, 1.04, and 1.15 eV, respectively. For comparison, the bandgap of β-Ge2XY is wider than that of α-Ge2XY, including α-Ge2SSe (1.32 eV), α-Ge2SSe (0.97 eV), and α-Ge2SSe (0.88 eV).19 To correct the bandgaps of the studied materials, we also plot the band structures by using the HSE06 functional as also shown in Fig. 4. Our calculated results indicate that, near the Fermi level, the HSE06 band diagrams are similar profiles to the PBE band structures. However, there is an important difference that needs to be mentioned, which is that β-Ge2STe is a direct semiconductor with both the CBM and VBM located at the S point at the HSE06 level as presented in Fig. 4(b). It should also be emphasized that, at the HSE06 level, the difference in energy between the CBM and minimum energy of the conduction band on the Y–Γ line of β-Ge2STe is not very small. The obtained results demonstrate that, at the HSE06 level, meanwhile β-Ge2STe exhibits direct semiconducting characteristics with a bandgap of 1.53 eV, β-Ge2SSe and β-Ge2SeTe monolayers are indirect semiconductors with bandgaps of 2.44 and 1.67 eV, respectively. The HSE06 band gap is much wider than those evaluated by the PBE approach. | ||
| Fig. 4 Band structures of β-Ge2SSe (a), β-Ge2Ste (b), and β-Ge2SeTe (c) calculated using the PBE/HSE06 functional. | ||
To get inside the nature of the formation of electronic structures, we studied the weighted band structures of β-Ge2XY monolayers as shown in Fig. 5. The obtained results show that the weighted bands of the studied monolayers are quite similar. The conduction band minimum (CBM) is formed from the contribution mainly from the p-orbitals of Ge atoms while the p-orbitals of Y atoms make a major contribution to the valence band maximum (VBM). The contribution of the p-orbitals of the chalcogen atoms X and Y to the valence band is significant. Meanwhile, the contributions of the p-orbitals of Ge atoms to the conduction band are more prominent in comparison with other orbitals of all compound constituents.
 | ||
| Fig. 5 The PBE weighted bands of β-Ge2Sse (a), β-Ge2Ste (b), and β-Ge2SeTe (c). | ||
The physical features of nanostructured materials are susceptible to change under the influence of external conditions such as mechanical strains, pressure, or doping. Pereira and co-workers showed that mechanical strain changed the hopping energy and consequently changed the electronic characteristics of the material significantly.39 The change in the crystal structure can give rise to many new physical features in 2D structures. Here, we evaluate the effects of biaxial strains εb on the band structure characteristics of the studied monolayers based on the DFT calculations. Quantitatively, we define the biaxial strain as εb = (δ – δ0)/δ0, where δ0 and δ are the lengths of the computed cell before and after strain. In our calculations, the biaxial strain ranging from 0 to ±6% is applied to evaluate the effect of the biaxial strain on the energy band structure of the material. To confirm the stability of the crystal structures under strain, the crystal structures of strained β-Ge2XY monolayers at εb = −6% and εb = +6% are studied by the AIMD simulations within 5 ps at 500 K as shown in Fig. 6. It is found that no structural transitions nor chemical bonds are breaking in the examined strained structures. In Fig. 7, we show the HSE06 band diagrams of β-Ge2XY monolayers under various values of εb. It is shown that the band structures of β-Ge2XY monolayers depend highly on the applied biaxial strain εb. While the strain does not change the indirect semiconductor characteristics of β-Ge2Sse, the CBM of β-Ge2Ste and β-Ge2SeTe is shifted towards the Γ–X path when the compressive strain is introduced. As a result, the β-Ge2STe monolayer becomes the direct bandgap semiconductor with both the CBM and VBM located at the S-point when the tensile strain (εb > 0) is applied as shown in Fig. 7(b). Meanwhile, the tensile strain is the cause of the indirect–direct bandgap transition in β-Ge2SeTe as shown in Fig. 7(c). The strain not only drastically moves the CBM position, which leads to the indirect-to-direct bandgap transition, but also significantly changes the bandgap of all the examined materials. The dependence of the bandgap of β-Ge2XY monolayers on εb is depicted in Fig. 8. It is found that the graph showing the strain-dependence of the bandgap of the examined structures is of the same form. While the bandgap of monolayers changes slightly in the presence of tensile strain, compressive strain rapidly reduces their bandgap as depicted in Fig. 8.
 | ||
| Fig. 6 Crystal structures of strained β-Ge2XY at εb = −6% and εb = +6% after 5 ps at 500 K by AIMD simulations. | ||
 | ||
| Fig. 7 Band structures of (a) β-Ge2SSe, (b) β-Ge2STe, and (c) β-Ge2SeTe monolayers at various values of εb. | ||
 | ||
| Fig. 8 Dependence of bandgaps of β-Ge2XY on biaxial strain εb. The filled and empty symbols indicate the indirect and direct bandgaps, respectively. | ||
4 Conclusive remarks
We have constructed and examined the structural, mechanical, and electronic characteristics of a series of novel Janus structures β-Ge2XY based on the DFT calculations. β-Ge2XY materials have been predicted to be stable and they are promising to be experimentally synthesized. Our calculated results have indicated that β-Ge2XY materials possess highly anisotropic mechanical characteristics due to their anisotropic crystal structures. All of these proposed 2D Janus structures are semiconductors with an indirect energy bandgap. The electronic characteristics of β-Ge2XY monolayers can be easily modulated by mechanical strains and in particular phase transitions can be found at some suitable values of strain. The obtained results not only introduce novel Janus materials but also show that they have attractive physical properties with potential applications in nanoelectronics.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This research is funded by the Vietnam Ministry of Education and Training under Grant No. B2023.DNA.23.References
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666 CrossRef CAS PubMed.
- K. S. Novoselov, D. Jiang, F. Schedin, T. J. Booth, V. V. Khotkevich, S. V. Morozov and A. K. Geim, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10451–10453 CrossRef CAS PubMed.
- N. A. Poklonski, S. A. Vyrko, A. I. Siahlo, O. N. Poklonskaya, S. V. Ratkevich, N. N. Hieu and A. A. Kocherzhenko, Mater. Res. Express, 2019, 6, 042002 CrossRef.
- B. Lalmi, H. Oughaddou, H. Enriquez, A. Kara, S. Vizzini, B. Ealet and B. Aufray, Appl. Phys. Lett., 2010, 97, 223109 CrossRef.
- J. N. Coleman, M. Lotya, A. O'Neill, S. D. Bergin, P. J. King, U. Khan, K. Young, A. Gaucher, S. De, R. J. Smith, I. V. Shvets, S. K. Arora, G. Stanton, H.-Y. Kim, K. Lee, G. T. Kim, G. S. Duesberg, T. Hallam, J. J. Boland, J. J. Wang, J. F. Donegan, J. C. Grunlan, G. Moriarty, A. Shmeliov, R. J. Nicholls, J. M. Perkins, E. M. Grieveson, K. Theuwissen, D. W. McComb, P. D. Nellist and V. Nicolosi, Science, 2011, 331, 568 CrossRef CAS PubMed.
- P. Hu, Z. Wen, L. Wang, P. Tan and K. Xiao, ACS Nano, 2012, 6, 5988 CrossRef CAS PubMed.
- C. Chowdhury, S. Karmakar and A. Datta, J. Phys. Chem. C, 2017, 121, 7615–7624 CrossRef CAS.
- Y. Xu, H. Zhang, H. Shao, G. Ni, J. Li, H. Lu, R. Zhang, B. Peng, Y. Zhu, H. Zhu and C. M. Soukoulis, Phys. Rev. B, 2017, 96, 245421 CrossRef.
- S. Lee, J.-E. Jung, H. g. Kim, Y. Lee, J. M. Park, J. Jang, S. Yoon, A. Ghosh, M. Kim, J. Kim, W. Na, J. Kim, H. J. Choi, H. Cheong and K. Kim, Nano Lett., 2021, 21, 4305–4313 CrossRef CAS PubMed.
- L. Xu, M. Yang, S. J. Wang and Y. P. Feng, Phys. Rev. B, 2017, 95, 235434 CrossRef.
- V. V. Tuan, A. A. Lavrentyev, O. Y. Khyzhun, N. T. T. Binh, N. V. Hieu, A. I. Kartamyshev and N. N. Hieu, Phys. Chem. Chem. Phys., 2022, 24, 29064–29073 RSC.
- A.-Y. Lu, H. Zhu, J. Xiao, C.-P. Chuu, Y. Han, M.-H. Chiu, C.-C. Cheng, C.-W. Yang, K.-H. Wei, Y. Yang, Y. Wang, D. Sokaras, D. Nordlund, P. Yang, D. A. Muller, M.-Y. Chou, X. Zhang and L.-J. Li, Nat. Nanotechnol., 2017, 12, 744 CrossRef CAS PubMed.
- J. Zhang, S. Jia, I. Kholmanov, L. Dong, D. Er, W. Chen, H. Guo, Z. Jin, V. B. Shenoy, L. Shi and J. Lou, ACS Nano, 2017, 11, 8192–8198 CrossRef CAS PubMed.
- Z. Guan and S. Ni, Nanoscale, 2020, 12, 22735–22742 RSC.
- Z. Guan, S. Ni and S. Hu, J. Phys. Chem. C, 2018, 122, 6209–6216 CrossRef CAS.
- C.-F. Fu, J. Sun, Q. Luo, X. Li, W. Hu and J. Yang, Nano Lett., 2018, 18, 6312–6317 CrossRef CAS PubMed.
- T. V. Vu, H. V. Phuc, A. I. Kartamyshev and N. N. Hieu, Appl. Phys. Lett., 2023, 122, 061601 CrossRef CAS.
- A. Huang, W. Shi and Z. Wang, J. Phys. Chem. C, 2019, 123, 11388 CrossRef CAS.
- L. Seixas, J. Appl. Phys., 2020, 128, 045115 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- A. Togo, L. Chaput and I. Tanaka, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 094306 CrossRef.
- S. Nosé, J. Chem. Phys., 1984, 81, 511 CrossRef.
- Y. Zhu, X. Wang and W. Mi, Phys. E, 2020, 117, 113802 CrossRef CAS.
- K.-A. N. Duerloo, M. T. Ong and E. J. Reed, J. Phys. Chem. Lett., 2012, 3, 2871–2876 CrossRef CAS.
- W.-Z. Xiao, G. Xiao and L.-L. Wang, J. Chem. Phys., 2016, 145, 174702 CrossRef PubMed.
- Y. Guo, S. Zhou, Y. Bai and J. Zhao, Appl. Phys. Lett., 2017, 110, 163102 CrossRef.
- F. Mouhat and F.-X. Coudert, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90, 224104 CrossRef.
- Z. j. Wu, E. j. Zhao, H. p. Xiang, X. f. Hao, X. j. Liu and J. Meng, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 054115 CrossRef.
- N. T. Hung, A. R. T. Nugraha and R. Saito, J. Phys. D: Appl. Phys., 2018, 51, 075306 CrossRef.
- P. Xiang, S. Sharma, Z. M. Wang, J. Wu and U. Schwingenschlögl, ACS Appl. Mater. Interfaces, 2020, 12, 30731 CrossRef CAS PubMed.
- L. C. Gomes, A. Carvalho and A. H. Castro Neto, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92, 214103 CrossRef.
- K. Cheng, W. Hu, X. Guo, L. Wu, S. Guo and Y. Su, Phys. Chem. Chem. Phys., 2023, 25, 5663–5672 RSC.
- K. D. Pham, RSC Adv., 2021, 11, 36682–36688 RSC.
- V. M. Pereira, A. H. Castro Neto and N. M. R. Peres, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 045401 CrossRef.
| This journal is © The Royal Society of Chemistry 2023 |