
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGold nanorods derivatized with CTAB and hydroquinone or ascorbic acid: spectroscopic investigation of anisotropic nanoparticles of different shapes and sizes†
Simone
Amatori
 a,
Alberto
Lopez
a,
Carlo
Meneghini
a,
Annarica
Calcabrini
b,
Marisa
Colone
b,
Annarita
Stringaro
b,
Sofia
Migani
b,
Ivan
Khalakhan
c,
Giovanna
Iucci
a,
Iole
Venditti
a and
Chiara
Battocchio
*a
a,
Alberto
Lopez
a,
Carlo
Meneghini
a,
Annarica
Calcabrini
b,
Marisa
Colone
b,
Annarita
Stringaro
b,
Sofia
Migani
b,
Ivan
Khalakhan
c,
Giovanna
Iucci
a,
Iole
Venditti
a and
Chiara
Battocchio
*a
aRoma Tre University, Dept of Sciences, Via della Vasca Navale 79, Rome 00146, Italy. E-mail: chiara.battocchio@uniroma3.it
bNational Center for Drug Research and Evaluation, Istituto Superiore di Sanità, Rome 00161, Italy
cDepartment of Surface and Plasma Science, Faculty of Mathematics and Physics, Charles University, V Holešovičkách 2, Prague 18000, Czech Republic
First published on 21st June 2023
Abstract
Gold nanorods stabilized by binary ligand mixtures of cetyltrimethylammonium bromide (CTAB, primary ligand) and ascorbic acid or hydroquinone were investigated by complementary synchrotron radiation-induced spectroscopies and microscopies, with the aim to find evidence of the influence of the secondary ligand molecular and chemical structure on the nanorod shapes and size ratios. Indeed, as it is well known that the CTAB interaction with Ag(I) ions at the NR surface plays a key role in directing the anisotropic growth of nanorods, the possibility to finely control the NR shape and dimension by opportunely selecting the secondary ligands opens new perspectives in the design and synthesis of anisotropic nanoparticles.
1. Introduction
Noble metal nanoparticles (MNPs) have been attracting a huge amount of attention from the scientific community over the last two decades thanks to their peculiar physicochemical properties arising from the maximized surface-to-volume ratio, such as the well-known surface plasmon resonance (SPR).1 The most-exploited bottom-up approach for MNP synthesis is a chemical method involving the reduction of metal salts (AuHCl4, AgNO3) in a medium containing capping ligands, usually organic molecules with thiol or amine end-groups, able to graft the metal surface competing with the metal particle growth. By opportunely modulating the stoichiometric ratio between metal salts, reducing agents and capping ligands, as well as the specific synthesis conditions (time of reaction, temperature) it is nowadays possible to obtain almost monodispersed nanoparticles with desired sizes and shapes.2 It is noteworthy that the opportune selection of capping ligands is a crucial step in the MNP synthesis since on the one hand they prevent disordered growth and avoid agglomeration,3 and on the other hand they can confer to the stabilized MNPs desired properties arising from the specific organic moiety (optical properties, biocompatibility, hydrophilic behaviour, etc.).4,5Among others, anisotropic (i.e., non-spherical) particles have attracted much attention due to their size- and shape-dependent optical properties. Rod-shaped nanoparticles are particularly interesting since they display two plasmon bands: a transverse plasmon band corresponding to an electron oscillation along the short axis of the rod, at around 510–550 nm for gold nanorods (AuNRs), and a longitudinal plasmon band, in the range 600–1200 nm.6 Indeed, the nanorods' aspect ratio can be modified by the strict control of the experimental parameters during their chemical synthesis, allowing the opportune tuning of the position of the plasmon bands, for example obtaining nanomaterials active in the NIR, which is of major interest for applications such as therapeutic/imaging (“theragnostic”) agents. Another characteristic of anisotropic nanoparticles in general, and specifically nanorods, that attracts great attention from the research community, is their size-related SPR tunability; for example, AuNRs can experience a 50 nm red shift of the longitudinal plasmon mode for a change in aspect ratio (i.e., length/diameter) from about 2.5 to nearly 3; this is a considerable shift, if we recall that for spherical AuNPs a circa 50 nm shift in the plasmon resonance requires a diameter size modification from 10 to 100 nm (λmax increases with mean diameter, from about 520 nm for smaller NPs to about 570 nm for the larger ones).7 Moreover, it is noteworthy that the optical properties (optical absorption or scattering wavelengths) of the NRs strictly depend on the rods' length and are insensitive to their diameter.8
All the above reported considerations concur to substantiate the appealing of gold nanorods, but they also introduce a central issue in the design and synthesis of such anisotropic nanoparticles, that is the ability to finely control AuNR aspect ratio, homogeneity and dispersion, since the plasmonic optical properties are much sensitive to these parameters.9 To attain this goal, a mandatory step is the comprehension as accurately as possible of the mechanism driving the anisotropic growth that is at the base of NR formation. Since “there is strong experimental evidence that every ingredient/parameter in the synthesis procedure plays a critical role in determining the shape and resulting surface chemistry”,10 the comprehension of the role played by each participant in the synthetic path has been and still is subject to much experimental and theoretical research.
The synthetic procedure most used to prepare AuNRs is a two-step protocol named “seed mediated-method” developed by Murphy and co-workers.11 In the first step, tetra chloroauric acid (HAuCl4) is reduced with a strong reducing agent as for example sodium borohydride (NaBH4) in the presence of a stabilizing agent, citrate or cetyltrimethylammonium bromide (CTAB), producing 2–4 nm quasi-spherical Au seeds. Then, in the second step, the seeds are added to a growth solution containing AgNO3, a weak reducing agent as for example ascorbic acid, and CTAB, producing AuNRs. Nowadays, the role played by some parameters has been fully elucidated by several research reports. There is general accordance about the formation of a CTAB bilayer on the AuNR surface.10 The roles of the seeds, silver ions and CTAB concentration have been investigated systematically,12 the importance of bromine ion as the counterion of CTA+ as a shape-directing agent, through inhibition of the side growth during the elongation of nanorods, has been largely discussed13 as well as the role played in favouring the anisotropic growth by the complex formed in situ between CTAB and Ag(I) ions.14,15
As for the silver ions and atoms, there is still debate because while several authors observe metallic silver near the AuNR surface, for example by applying X-ray photoelectron spectroscopy (XPS) and X-ray absorption spectroscopy (XAS),14 suggesting the role of silver in directing the anisotropic growth by forming a sub-monolayer on selected AuNR side-facets (namely, under potential deposition of silver on AuNRs14), other authors point out the absence of preferential absorption of Ag(I) ions on certain facets, in contrast with this hypothesis.16 However, it is well established that the presence of silver ions is necessary to improve the AuNR synthesis yield and to control the aspect ratio, a fact that justifies further investigation of the local structure of both silver atoms and silver ions, since, to the best of our knowledge, a unique picture of Ag species distribution in the AuNR bulk and surface has still not been attained.17 Another aspect that deserves further investigation is the role played by the secondary ligands, when AuNR synthesis involves the use of a binary ligand mixture. This aspect is still poorly investigated, lacking a systematic investigation leading to correlate the secondary ligand molecular structure with AuNR dispersion, size-ratio and self-organization.
In this work, we apply complementary microscopic (FE-SEM, TEM) and spectroscopic techniques, in particular state-of-the-art synchrotron radiation-induced X-ray photoelectron spectroscopy (SR-XPS) and X-ray absorption spectroscopy (XAS) to study the molecular, electronic and chemical structure, as well as local structure, particularly around silver and gold atoms, and the morphology of gold nanorods prepared with two different secondary ligands, the weak reducing agents ascorbic acid (AA) and hydroquinone (HQ). The two series of AuNRs, namely AuNRs-AA and AuNRs-HQ, differ in the plasmon band position, morphology and aspect ratio in a reproducible way, due to the influence of the different secondary ligands on the anisotropic growth process. The accurate investigation reported in this work will shed light on the possibility of designing AuNRs of desired aspect ratios and shapes by opportunely selecting the secondary ligand, as well as on the destiny of silver atoms and ions in the AuNRs-AA and AuNRs-HQ nanosystems.
2. Materials and methods
2.1. Materials
Freshly bidistilled water was used as a solvent in all the procedures. All reactants required were purchased and used as received, without further purification: cetyltrimethylammonium bromide (C19H42BrN, CTAB, ≥97% Millipore Corporation), tetrachloroauric(III) acid trihydrate (HAuCl4·3H2O, ≥99.9%, Sigma-Aldrich), sodium borohydride (NaBH4, 99.99%, Sigma-Aldrich), silver nitrate (AgNO3, 99.9%, Carlo Erba), L-ascorbic acid (C6H8O6, 99%, Sigma-Aldrich) and hydroquinone (C6H602, ≥99%, Sigma-Aldrich). A mini spin Eppendorf centrifuge was used for the purification of AuNR samples. Uv-visible spectra were acquired with a Shimadzu 2401 PC spectrophotometer (200–800 nm), while NIR spectra were measured with a Nicolet IS50 FT-IR Thermo scientific spectrophotometer (400–1100 nm) using quartz cells with a 1 cm optical path.2.2. Synthesis
CTAB stabilized AuNRs were prepared by a seed-growth approach as reported in the literature:18 a seed solution of quasi-spherical nanoparticles is dissolved in a growth solution containing its precursor species (HAuCl4·3H2O and CTAB), AgNO3 and either ascorbic acid or hydroquinone as a reducing agent. This synthesis relies on a complex and delicate equilibrium of reactants to achieve anisotropic growth conditions: silver ions and a ligand agent show preferential absorption onto specific facets of gold and hinder reduction in these directions in the presence of a weak-enough reducing agent. The seed solution is prepared with 5 mL of HAuCl4·3H2O (5 × 10−4 M) and 5 mL of CTAB (0.2 M) solutions in deionized water in an inert atmosphere (Ar(g)), to which 0.6 mL of a NaBH4 solution (10−2 M) is added as a reducing agent. After 5 minutes, the solution becomes yellow-brownish and is then stored for 1 hour before use. Various experiments were carried out under different conditions both with AA and with HQ and then the best samples were compared. The syntheses were repeated in duplicate at least three times, always giving consistent results. Table S1 (see the ESI†) shows the most summarized experimental data: the bests results, as indicated in Table S1,† are the subject of this study. It is noteworthy that only anisotropic rod-shaped nanoparticles were considered for this study (neither spherical nor other non-rod-like nanoparticles were considered).2.3. Microscopic and spectroscopic methods
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O 1s → π* transition, and the resonance at 400.00 eV, assigned to the 1s → σ* transition of the positively charged nitrogen, respectively. The raw C and N K-edge NEXAFS spectra were normalized to the incident photon flux by dividing the sample spectrum by the spectrum collected on a freshly sputtered gold surface. Spectra were then normalized subtracting a straight line that fits the part of the spectrum below the edge and assessing to 1 the value at 330.00 and 420.00 eV for C and N, respectively.
O 1s → π* transition, and the resonance at 400.00 eV, assigned to the 1s → σ* transition of the positively charged nitrogen, respectively. The raw C and N K-edge NEXAFS spectra were normalized to the incident photon flux by dividing the sample spectrum by the spectrum collected on a freshly sputtered gold surface. Spectra were then normalized subtracting a straight line that fits the part of the spectrum below the edge and assessing to 1 the value at 330.00 and 420.00 eV for C and N, respectively.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 919 eV) and at the Ag K-edge (25514 eV) respectively, at the 11.1R-XAFS beamline at ELETTRA synchrotron radiation facility20 and at the LISA-BM08 beamline21 at ESRF (European Synchrotron Radiation Facility). The AuNR samples suspended in water were mixed with cellulose, dried in vacuum, and pressed to obtain homogeneous pellets suitable for handling. Both beamlines were equipped with double crystal, fixed exit monochromators with harmonic rejection mirrors. The Au LIII-edge XAS spectra were acquired in transmission geometry keeping the samples in vacuum at liquid nitrogen temperature, with two gas-filled ionizing chambers measuring the incident (I0) and transmitted (I1) X-ray beam intensities. Pure Au foil was placed in vacuum after the I1 chamber, and the transmitted intensity (I2) was measured with a third ionization chamber placed right after the reference foil. The absorption signals of samples 2 (AuNRs-AA) and 4 (ANRs-HQ), calculated as
919 eV) and at the Ag K-edge (25514 eV) respectively, at the 11.1R-XAFS beamline at ELETTRA synchrotron radiation facility20 and at the LISA-BM08 beamline21 at ESRF (European Synchrotron Radiation Facility). The AuNR samples suspended in water were mixed with cellulose, dried in vacuum, and pressed to obtain homogeneous pellets suitable for handling. Both beamlines were equipped with double crystal, fixed exit monochromators with harmonic rejection mirrors. The Au LIII-edge XAS spectra were acquired in transmission geometry keeping the samples in vacuum at liquid nitrogen temperature, with two gas-filled ionizing chambers measuring the incident (I0) and transmitted (I1) X-ray beam intensities. Pure Au foil was placed in vacuum after the I1 chamber, and the transmitted intensity (I2) was measured with a third ionization chamber placed right after the reference foil. The absorption signals of samples 2 (AuNRs-AA) and 4 (ANRs-HQ), calculated as 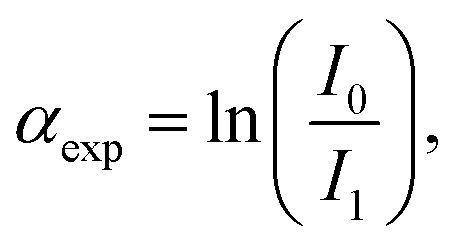 and the one of the reference foil, equal to
and the one of the reference foil, equal to 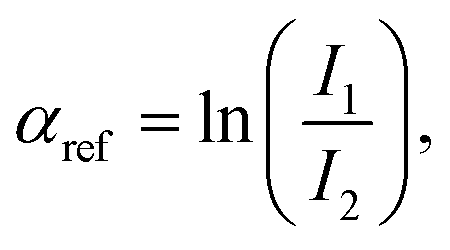 were acquired, aligned and averaged to improve the data statistics. The Ag K-edge XAS spectra were measured in fluorescence geometry, using a high purity germanium (HP-Ge) multidetector (13 elements, (ORTEC)). The Ag Kα fluorescence signal for each detector Ifi was electronically selected from the total fluorescence yield using the analyser's multichannel electronics. The absorption spectrum of pure Ag foil placed after the sample was measured and used to check the energy calibration during data collection and eventually align the energy scale of the spectra. The spectra of all detector channels (except those with a lower signal-to-noise ratio) were summed, and the absorption signal was calculated as
were acquired, aligned and averaged to improve the data statistics. The Ag K-edge XAS spectra were measured in fluorescence geometry, using a high purity germanium (HP-Ge) multidetector (13 elements, (ORTEC)). The Ag Kα fluorescence signal for each detector Ifi was electronically selected from the total fluorescence yield using the analyser's multichannel electronics. The absorption spectrum of pure Ag foil placed after the sample was measured and used to check the energy calibration during data collection and eventually align the energy scale of the spectra. The spectra of all detector channels (except those with a lower signal-to-noise ratio) were summed, and the absorption signal was calculated as 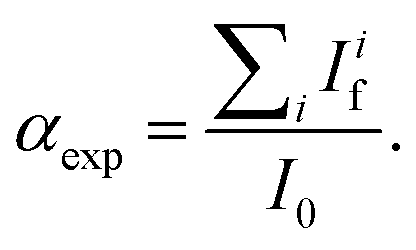 Due to the weak Ag content, multiple spectra (19 for AuNRs-AA and 8 for AuNRs-HQ samples) were measured, checked for energy calibration, and averaged to obtain data statistics suitable for quantitative analysis.
Due to the weak Ag content, multiple spectra (19 for AuNRs-AA and 8 for AuNRs-HQ samples) were measured, checked for energy calibration, and averaged to obtain data statistics suitable for quantitative analysis.
To extract EXAFS structural signal χexp, the experimental spectra, αexp, were treated according to standard procedures22 including the linear pre-edge subtraction (α′ = αexp − αpre), bare atomic background (αb) subtraction, and normalization, to extract EXAFS structural signals: 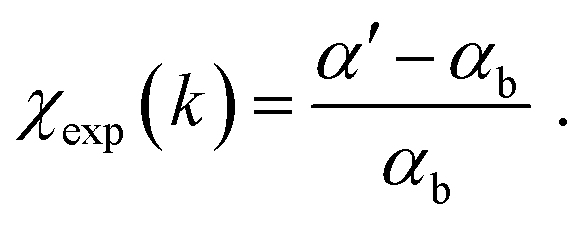 The edge energy E0, origin of the photoelectron wavenumber
The edge energy E0, origin of the photoelectron wavenumber 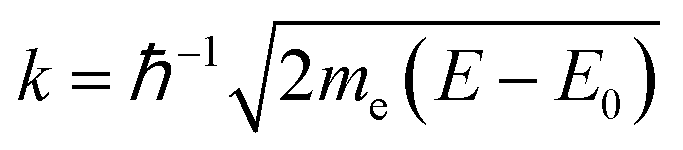 (me being the electron mass), was defined at the first inflection point (maximum of the first derivative) of the pre-edge subtracted spectra α′.
(me being the electron mass), was defined at the first inflection point (maximum of the first derivative) of the pre-edge subtracted spectra α′.
The quantitative analysis of the EXAFS signals was carried out fitting the kw-weighted theoretical curves kwχth to the raw experimental data kwχexp, applying a non-linear least-square procedure implemented in the program FiteEXA.22 The Au-LIII data analysis has been carried out in the 3–12 (AuNRs-AA) and 3–15 Å−1 (AuNRs-HQ) k range with w = 2. The Ag–K edge spectra of AuNRs exhibit a weaker signal-to-noise ratio, which is attributed to either the lower Ag content (that affects the count statistics due to the reduction of the fluorescence signal) or the more disordered/lighter local coordination chemistry (decreasing the amplitude of the EXAFS structure signal). The Ag EXAFS spectra were analyzed in the 3–8 (AuNRs-AA) and 3–12 Å−1 (AuNRs-HQ) k range with w = 1. The theoretical curves χth(k) were calculated as a sum of partial contributions χi, calculated using a Gaussian pair distribution function model and the standard EXAFS formula23,24 with the Gaussian disorder model. The theoretical photoelectron scattering amplitude and phase functions were calculated using the FEFF8 program.25
3. Results and discussion
3.1. AuNR morphology: TEM and FE-SEM images
Transmission electron microscopy analysis revealed different AuNR dimensions depending on the secondary ligand used during the synthesis. AuNR-AA (Fig. 1a) was shorter (mean length = 40 ± 6, mean width = 10 ± 2 nm) than AuNR-HQ (Fig. 1b, mean length = 100 ± 8, mean width = 14 ± 4 nm).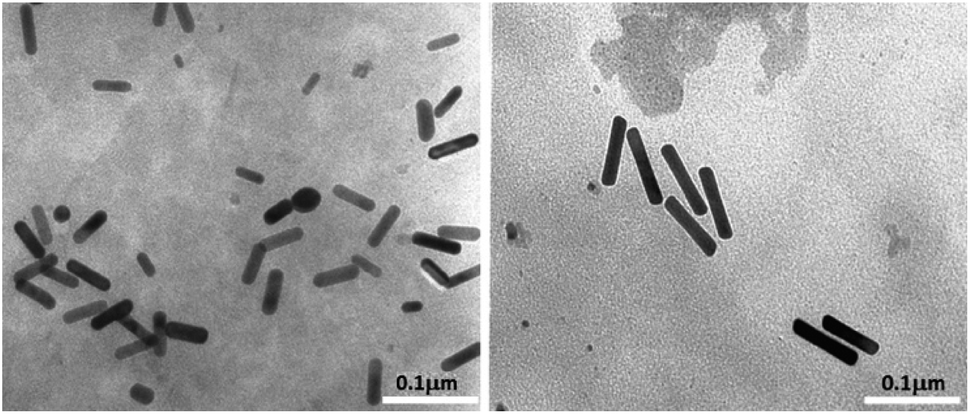 | ||
| Fig. 1 TEM images of AuNRs: (a) AuNR-AA; (b) AuNR-HQ. | ||
The variation in the dimensions of AuNRs resulting from the use of different secondary ligands during synthesis is further confirmed by scanning electron microscopy, as illustrated in Fig. 2. Specifically, the AuNRs-AA sample at different magnifications in Fig. 2a and c exhibited smaller nanorods, whereas the AuNRs-HQ sample in Fig. 2b and d is composed of significantly larger nanorods. To highlight the comparison, Fig. 2e summarizes the corresponding histograms of the nanorod size distribution, yielding a mean width of 10 ± 4 nm and length of 41 ± 5 nm for the AuNRs-AA sample (average aspect ratio 4.1) and a mean width of 22 ± 4 nm and mean length of 106 ± 14 nm for the AuNRs-HQ sample (average aspect ratio 4.8).
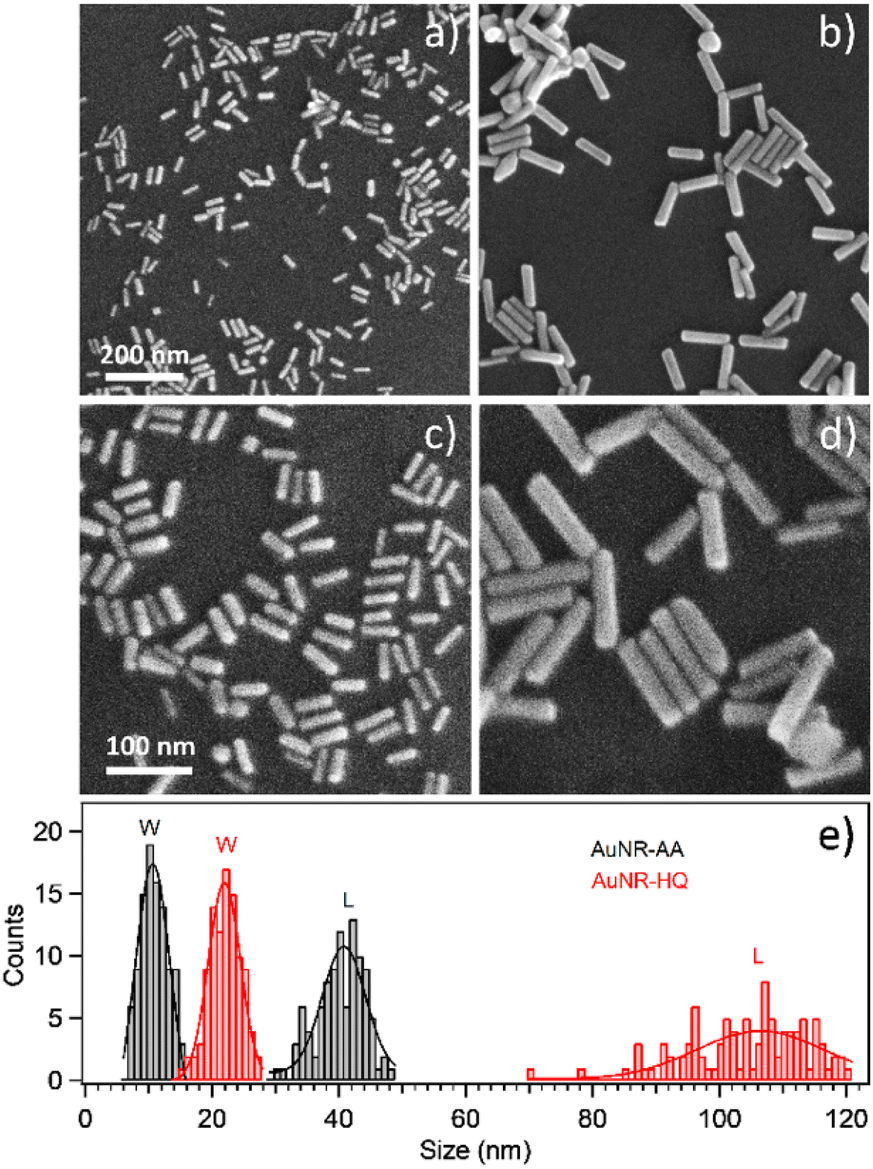 | ||
| Fig. 2 SEM images of: (a) and (c) AuNR-AA; (b) and (d) AuNR-HQ; (e) corresponding histograms for their size distribution (W stands for width, L stands for length), obtained by analyzing 100 nanorods for each sample. | ||
3.2. Molecular and electronic structure: SR-XPS and NEXAFS results
SR-XPS measurements were carried out on the two AuNRs-AA and AuNRs-HQ in the solid state, as described in the “Materials and methods” section. All SR-XPS data were analyzed by following a peak-fitting procedure, allowing deconvolution of the components arising from chemical elements with different atomic environments; all peak position BEs (binding energies), FWHM values, atomic ratios (relative intensities) and assignments are reported in ESI Table S2.†C 1s, N 1s, Br 3d, Ag 3d and Au 4f core level signals of AuNRs-AA and AuNRs-HQ were collected and analyzed. C 1s spectra are reported in Fig. 3a (AuNRs-AA) and Fig. 3d (AuNRs-HQ). Both spectra appear composite and consist of at least four components corresponding respectively to aliphatic C–C groups of CTAB and AA or aromatic carbons of HQ (BE = 285.0 eV), to C–N (CTAB) and C–O (AA, HQ) signals (286.5 eV), which can't be deconvoluted with the used resolution, to the CO carbons of AA, with a contribution from adventitious carbon (287.8 eV)17 and finally to carboxylic groups of contaminants (289.2 eV).26 (The in-depth description of each spectral component of C 1s spectra is reported in the caption of Fig. S2†).
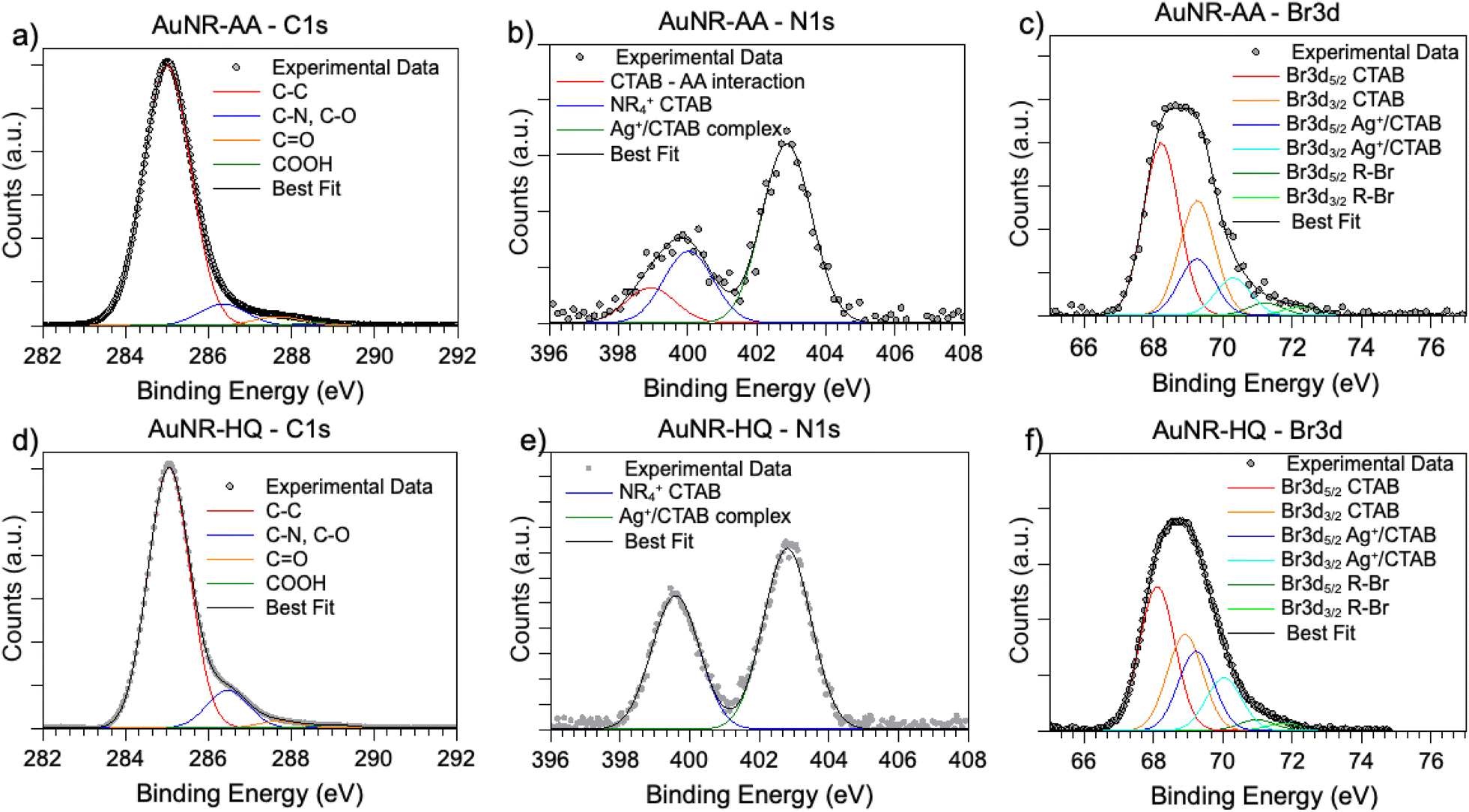 | ||
| Fig. 3 SR-XPS C 1s, N 1s and Br 3d spectra of AuNRs-AA (a–c) and AuNRs-HQ (d–f). | ||
N 1s spectra (Fig. 3b and e) are made of two groups of components: the peaks at lower BE (400 eV in AuNRs-HQ, 399 eV + 400 eV in AuNRs-AA) are associated with amine-like N atoms of CTAB (400 eV);27 the low-intensity peak at 399 eV in AuNRs-AA indicates amine groups of CTAB shifted at lower BE by ion–ion and/or ion-dipole CTAB–AA electrostatic interactions, as suggested in;28 the reproducible N 1s signal observed in both spectra at around 402.8 eV can be attributed to neatly protonated nitrogens of the Ag+/Br−/CTA+ coordination compound. As observed by some authors, the positive shift in N 1s binding energy can also be interpreted as due to the gold surface vicinity.15 Correspondingly, Br3d spectra (Fig. 3c and f) show a couple of spin–orbit pairs at lower BE (Br3d5/2 BE = 68.2 eV) attributed to unperturbed CTAB molecules15,29 pointing out of the surface in the CTAB bilayer (see Scheme 1), while the Br 3d signal at higher BE (Br3d5/2 BE = 69.1 eV) is assigned to less negatively charged bromine anions in the “sandwich” structure of the Ag+/Br−/CTA+ at the interface between CTAB and AuNR surface.30 This structure is, at least from the SR-XPS point of view, independent of the identity of the secondary ligand, since HQ and AA-containing nanorods display very similar N 1s and Br3d photoelectron spectra, indicative of analogous electronic structures of nitrogen and bromine atoms. This finding suggests that the presence of HQ or AA as a secondary ligand does not affect the CTAB double layer arrangement, as well as the nitrogen and bromide local structure. The very low-intensity signal observed at high BE (70 eV Br3d5/2) in the Br3d spectra (4–6% of Br species, almost at the detection limit of the technique also considering the very complex sample matrix31) is attributed to bromine atoms covalently bonded to C,32,33 and its presence is probably due to some degradation arising under the X-ray beam, or to contaminants.
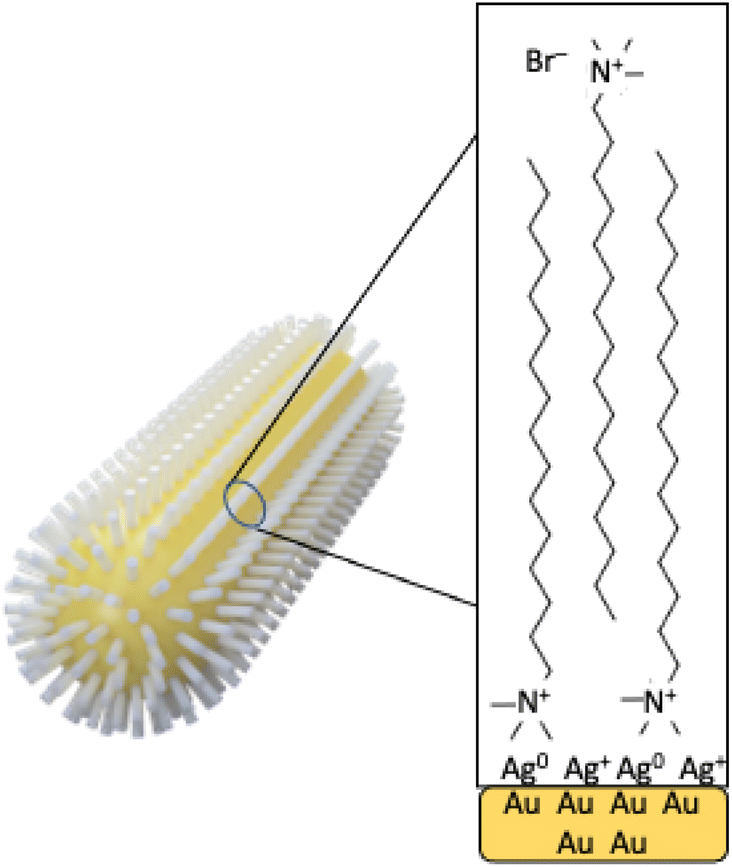 | ||
| Scheme 1 Schematic representation of the AuNR surface with the evidenced CTAB bilayer. | ||
As for the gold and silver electronic structure, Au4f and Ag3d core level spectra are reported in Fig. 4. Au4f signals (Fig. 4a and c) are the composite of two spin–orbit pairs (Au4f7/2, Au4f5/2), coherent with literature findings on noble metal nanoparticles of several shapes, either spherical or anisotropic.34 The most intense Au4f7/2 signal at about 83.7 eV is assigned to metallic Au(0) atoms in the NR bulk, while the less intense doublet (Au4f7/2 BE = 84.2 eV) is attributed to a low percentage of gold atoms at the NR surface and interacting with the ligands. The Ag3d spectra (Fig. 4b and d) are also very similar for AuNRs-AA and AuNRs-HQ and still composite allowing individuation of two spin orbit doublets. The most intense signal at lower BE (Ag3d5/2 BE = 367.3 eV) is assigned to Ag+ ions at the NR surface leading to the Ag+/Br−/CTA+, according to the literature on analogous systems.15,17 It is noteworthy that this signal percentage is about 90% of all the silver sampled by XPS, i.e., in the first nm of the AuNR surface (XPS sampling depths of a few nanometers35). The Ag3d5/2 signal at higher BE (nearly 368 eV) is indicative of the presence of metallic Ag(0) atoms,17 also located at the NR surface. In the literature there is still an ongoing debate about the presence of metallic silver atoms in AuNRs and their location. The finding presented here locates a low amount of Ag(0) on the nanoparticle surface, and the XAS data discussed in the following will help to shed light on the local structure of silver atoms and ions also in the NR bulk.
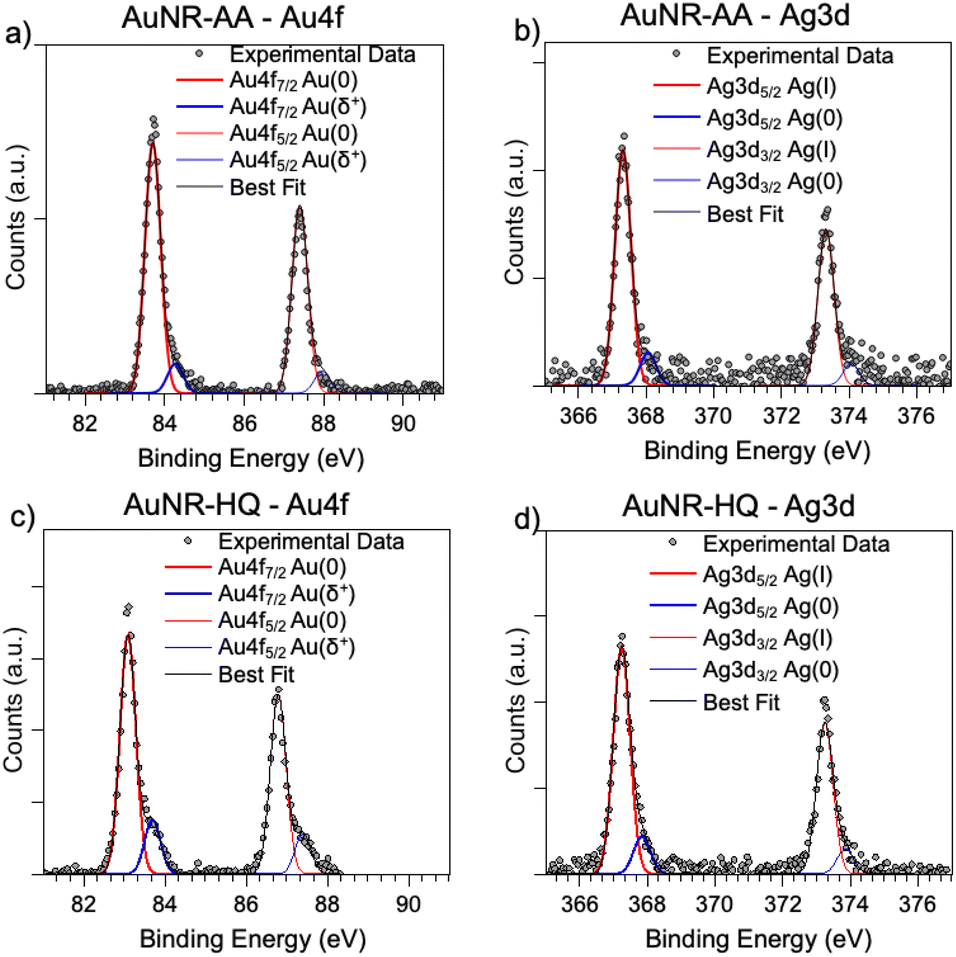 | ||
| Fig. 4 SR-XPS Au4f and Ag3d spectra of AuNRs-AA (a, b) and AuNRs-HQ (c, d). | ||
NEXAFS spectra collected on AuNRs-AA and AuNRs-HQ in the solid state, prepared in the same manner as the samples for SR-XPS measurements, are reported in Fig. 5. The spectra of the two samples are very similar, therefore they will be discussed together. In the C K-edge spectra of AuNRs-AA and AuNRs-HQ, reported in Fig. 4a and c, respectively, several resonances due to electronic transitions from the C 1s core level to antibonding molecular orbitals can be observed, and assigned by comparison with literature data as follows: the sharp feature at about 288.7 eV is associated with the C 1s → π* transition of CO molecular orbital, the shoulder at about 288 eV to a σ* resonance by the C–H groups and Rydberg features, additional features around 293 and 303 eV can be assigned to 1s → σ* transitions by C–C and CO molecular groups, respectively.20 The pre-edge peak at 285.1 eV in the spectrum of AuNRs-HQ and appearing as a shoulder in the spectrum of AuNRs-AA is probably an artifact related to carbon contamination of the beamline mirrors. In N K-edge spectra (Fig. 5b for AuNRs-AA, Fig. 5d for AuNRs-HQ) only one sharp peak appears at 402 eV, assigned to the N 1s → σ* transition of positively charged nitrogen as expected from the CTAB molecular structure, and coherently with SR-XPS findings. The low intensity peaks around 395 eV in Fig. 5d are spurious peaks related to problems with noise and background subtraction for the low intensity N K edge signal.
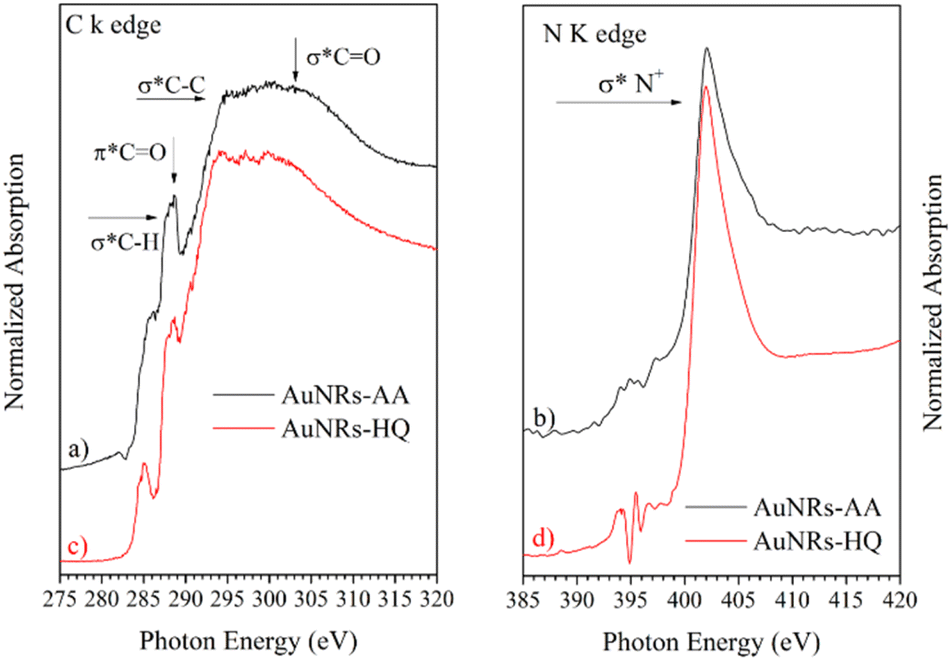 | ||
| Fig. 5 Carbon K-edge and nitrogen K-edge spectra of AuNRs-AA (a, b) and of AuNRs-HQ (c, d). | ||
3.3. Local structure around Au and Ag: XAS analysis
The XAS spectra of gold and silver were analysed in both XANES and EXAFS regions, which provide complementary information on the local ordering around the average absorber. The XANES features provide information on the average valence state of the absorber, the density of empty states near the Fermi level, and the symmetry of the coordination environment.36 In the near edge region, the effect of structural disorder is smaller than in the extended region; therefore, the analysis of the XANES region is particularly suitable for revealing weaker electronic and/or structural differences between samples. Analysis of the EXAFS data provides further details on the average atomic coordination around the absorber in terms of the average distance, number, and mean squared relative displacement (MSRD) of the neighbouring shells.23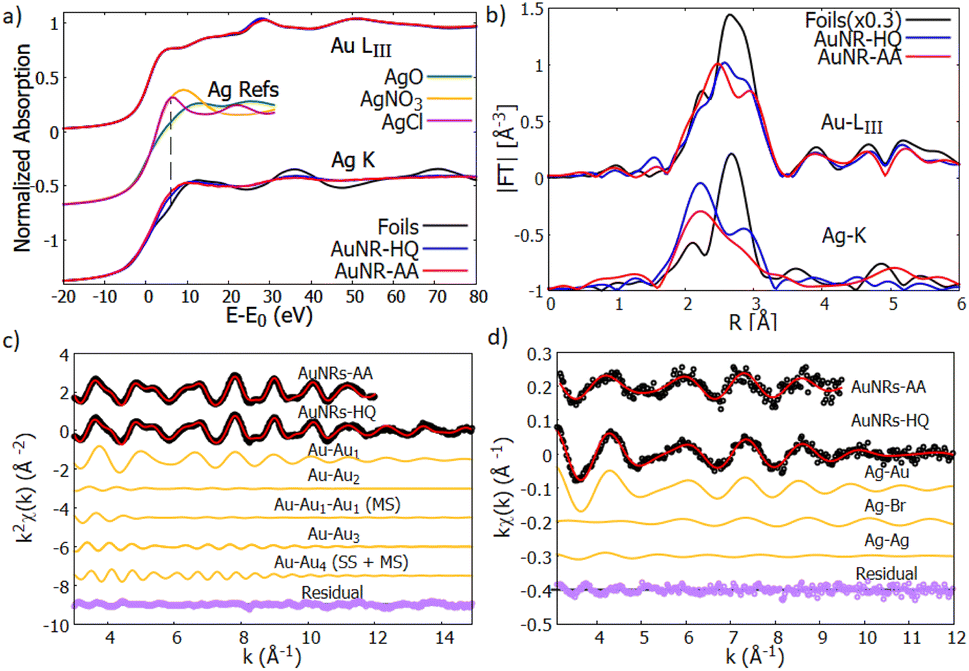 | ||
| Fig. 6 (a) Normalized XANES spectra of Au-LIII (top curves) and Ag–K edge (bottom curves) measured on reference foils (Au and Ag, black lines) and on AuNR samples (AuNR—HQ: blue and AuNR—AA: red). The reference spectra of Ag+ (AgNO3 and AgCl) and Ag2+ (AgO) are shown for the sake of comparison (from ref. 39–41). (b) Moduli of the Fourier transforms |FT| of kw weighted EXAFS spectra of Au-LIII (top curves) and Ag–K (bottom curves) edges for the reference foils (black lines) and AuNRs (AuNR-HQ: blue, AuNR-AA: red). (c) Results of the Au EXAFS data fitting for AuNR-HQ and AuNR-AA samples. The experimental (black dots), best fit (red lines) and the partial contributions used for the analysis (orange lines) are shown (vertically shifted for clarity). The bottom curves (purple dots) represent the residues (experimental data minus best fit). (d) Results of the Ag–K edge EXAFS fitting for AuNR-HQ and AuNR-AA samples. The black dots represent the experimental data, the red line the best fit, the orange ones represent the contribution of every path to the final fit and the purple dots represent the residual. All plots are shifted vertically for the sake of clarity. The larger noise on AuNR-AA forces restriction of the range for the analysis (see text). | ||
The first shoulder in the Au-LIII XANES at about 11920 eV is commonly referred to as the “white line” of gold and is related to the photoelectron transitions from the 2p3/2 state to the 5d state, the intensity of the white line being proportional to the density of free states in the 5d valence state.37 The very similar shape of the XANES measured on AuNRs and on Au reference foil suggests that the larger fraction of Au consists of zero-valent Au0 atoms located in the inner volume of the NR, with negligible fraction of Au atoms located at the NR surface. This is consistent with relatively large nanostructures having a sharp surface.
Looking at the Ag–K edge (Fig. 6a) differences between the XANES measured on AuNR samples and on silver reference foil become apparent indicating that the Ag coordination chemistry in these samples is definitively different from metallic silver. In AuNPs, the rise of the Ag K edge XANES signal around 25520 eV is similar to what was observed in Ag+ composites (i.e., AgNO3 or AgCl) so it can be attributed to the rise of the Ag valence state38 in our samples. Notice that the AgO XANES features are definitively different so the presence of Ag2+ can be excluded.
The quantitative analysis of Au LIII-edge EXAFS spectra has been carried out selecting the relevant contribution from the Au-fcc bulk structure (crystallographic information file from the crystallography open database, COD #9008463 (ref. 42)), the k2χ(k) experimental data and best fit curves for AuNR samples are shown in Fig. 6c, the single (SS) and multiple (MS) shell contributions used for the analysis and the residues (experimental data minus best fit curves) are shown for the sake of comparison.
To select the relevant contribution and reduce the correlations among the fitting parameters we applied constraints based on the Au crystallographic structure.43 After an initial trial and error procedure 4 single scattering (SS) and 3 multiple scattering (MS) were found as a suitable set to refine the Au EXAFS experimental data. The paths used in the EXAFS fit describe the coordination shells of metallic gold up to the 4th neighboring atoms (around 5.8 Å). Considering the absorber at the unit cell cube, the 4 single scattering paths correspond, considering the absorber at the unit cell cube edge, to 12 Au1 nearest neighbours at the center of the fcc face (SS1), 6 second neighbours Au2 along the fcc cube edge (SS2), 24 third neighbours Au3 on the opposite fcc face center (SS3), and 12 fourth neighbours Au4 along the fcc face diagonal (SS4). The MS contributions correspond to the triple scattering (MS1: Au–Au1–Au1) between two first neighbors as well as the triple (MS3: Au–Au1–Au4) and quadruple (MS4: Au–Au1–Au4–Au1) scattering paths along the fcc face diagonal. We fixed the multiplicity numbers of the SS and MS contributions to crystallographic values, the coordination lengths of the Au–Au bond (RSS1) were left free, the coordination lengths of the next neighbour shells were constrained to a single free parameter a = RSS2 (the edge of the fcc unit cell):  The MSRD (σi2) were refined independently, only we constrained σMS32 = (σSS42 + σMS42)/2 The same energy shift (ΔE = 6.8 eV) and the same S02 = 0.9 (representing the passive electron reduction factor) were applied to all the paths and dataset. Table 1 resumes the best fit parameters.
The MSRD (σi2) were refined independently, only we constrained σMS32 = (σSS42 + σMS42)/2 The same energy shift (ΔE = 6.8 eV) and the same S02 = 0.9 (representing the passive electron reduction factor) were applied to all the paths and dataset. Table 1 resumes the best fit parameters.
| Paths | AuNR-HQ | AuNR-AA | ||||
|---|---|---|---|---|---|---|
| N | R (Å) | σ 2 (×10−2 Å−2) | N | R (Å) | σ 2 (×10−2 Å−2) | |
| SS1 | 12* | 2.85(1) | 0.93(1) | 12* | 2.85(1) | 0.83(1) |
| SS2 | 6* | 4.01(2) | 1.87(3) | 6* | 3.97(1) | 2.35(2) |
| MS1 | 48* | 4.32(2) | 0.36(5) | 48* | 4.32(1) | 0.11(5) |
| SS3 | 24* | 4.98* | 1.34(4) | 24* | 4.99(2) | 1.35(2) |
| SS4 | 12* | 5.76* | 1.25(2) | 12* | 5.73(4) | 2.96(3) |
| MS2 | 24* | 5.76* | 1.33* | 24* | 5.73(4) | 2.34(2) |
| MS3 | 12* | 5.76* | 1.40(6) | 12* | 5.73(4) | 1.71(5) |
The values obtained from the Au EXAFS analysis agree well with the Au bulk structure, with a weak (1%) compression of the lattice parameter; we found a = Rss2 = 4.01(2) Å with respect to the gold lattice parameter aAu = 4.065 (Å), which can be attributed to thermal compression, since the measurements were performed at LN temperature.
The Ag EXAFS (Fig. 6d) spectra have larger noise with respect to the Au ones, this forces reduction of the k-range of the analysis. To individuate the suitable contributions we proceeded by trial and error selecting contributions from a shortlist of possible Ag local coordination structures, based on the sample synthesis and composition: Ag–metal, Ag–Au amalgams, and Ag–bromine (of the ionic head of CTAB). The preliminary attempts allow exclusion of a sizable amount of Ag–O contribution. Model structures used to calculate the photoelectron amplitude and phase functions were taken from structures in the Crystallography Open Database.44,45 After some testing S02 was fixed at 0.9 for both spectra and the energy scale shift ΔE0, which was calculated in the analysis with the less noisy spectra (AuNP-HQ), was fixed for the analysis of the sample AuNP-AA.
The AuNR-HQ spectrum has a better signal-to-noise ratio. We found that a satisfactory best fit is obtained using a first shell Ag–Au, around 2.83(2) Å with multiplicity close to 5, which is much less than 12, expected for Ag dispersed in an Au matrix. We found two other contributions relevant for the analysis, namely Ag–Br and Ag–Ag. Due to the weakness of these contributions, the antiphase effect between Ag–Au and Ag–Br contributions and the known correlation between multiplicity number and MSRD, the obtained values are affected by quite large uncertainty. However, they demonstrate Ag bonded either to Au or Br. Assuming crisp cylindrical surfaces, we may calculate the NR's surface-to-volume ratio (S/V) at roughly 5% for AuNR-HQ and slightly less, around 4% for AuNR-AA samples; however, surface flaws and roughness may result in higher actual S/V values for the samples. Given the total Ag fraction in the samples (around 3% from EDX analysis) the EXAFS results indicate that Ag atoms likely form an Ag–Au layer on the NR surface (Ag–Au and Ag–Ag neighbours), some of the Ag atoms connecting Br from one side and the NR inner Au atoms from the other side. The hypothesis that a fraction of unreacted Ag binds only to the amine group in the CTAB head can be excluded because the washing procedure would have removed that fraction, leaving therefore only Ag bound to the gold nanorods. The EXAFS data on the AuNR-AA sample are much noisier and the results are surely less accurate, but they generally confirm the trend observed on AuNR-AA with a slightly higher fraction of Ag–Ag neighbours, which is consistent with smaller S/V in this sample. The results of the Ag–K edge EXAFS data analysis are shown in Table 2.
| Paths | AuNR-HQ | AuNR-AA | ||||
|---|---|---|---|---|---|---|
| N | R (Å) | σ 2 (×10−2 Å−2) | N | R (Å) | σ 2 (×10−2 Å−2) | |
| Ag–Au | 5.0(5) | 2.83(1) | 1.2(2) | 4.1(6) | 2.82(1) | 1.8(2) |
| Ag–Br | 0.5(1) | 2.63(2) | 0.5* | 0.5(2) | 2.69(3) | 0.29(3) |
| Ag–Ag | 0.7(1) | 2.81(2) | 1.5* | 1.3(3) | 3.03(4) | 1.5* |
4. Conclusions
The investigation of the morphology, molecular, electronic structure and local structure around silver and gold atoms reported in this work sheds light on the correlation between the secondary ligand used in the synthesis path, i.e., HQ or AA, and the optical properties displayed by two sets of AuNRs. The here investigated AuNRs-AA and AuNRs-HQ differ in their plasmon band position, morphology and aspect ratio in a reproducible way, and our multi-technique investigation evidenced the influence of the different secondary ligands on the anisotropic growth process, leading to the conclusion that the moderate reducing agent AA induces the formation of AuNRs of lower size ratio, while HQ allows obtaining longer rods. The destiny of silver atoms and ions was also deeply investigated, and the analogous presence of Ag(I) and Ag(0) species on the two sets of AuNR surfaces revealed by SR-XPS and XAS suggested that the observed different size ratio of AuNRs-AA and AuNRs-HQ nanosystems is induced by the secondary ligand only.Author contributions
C. B.: conceptualization; C. B., C. M., S. A., A. L.: data curation, formal analysis; C. B., C. M., I. V., G. I.: funding acquisition; C. B., C. M., A. L., S. A., G. I., I. K., A. S., A. C., M. C., S. M.: investigation, methodology, validation, visualization; C. B., A. L., S. A.: writing – original draft; C. B., G. I., C. M., I. V.: writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the CERIC-ERIC Consortium for the access to experimental facilities LISA (ESRF), XAFS (ELETTRA), SuperESCA (ELETTRA) and FE-SEM Charles University, and financial support. They also acknowledge ELETTRA for the access to BEAR beamline and partial financial support. Authors from Roma Tre also acknowledge Ministry of Education, Universities and Research: FINANZIAMENTO DIPARTIMENTI DI ECCELLENZA 2023–2027 (Art. 1, commi 314–337 Legge 11/12/2016, n. 232). Finally, the authors gratefully thank Prof. Luca Tortora and Dr Eleonora Marconi of Roma Tre University, Dept. of Sciences, for NIR measurements (data reported in the ESI, Table S1†).Notes and references
- M. A. El-Sayed, Some interesting properties of metals confined in time and nanometer space of different shapes, Acc. Chem. Res., 2001, 34(4), 257e64 CrossRef PubMed.
- D. Maccora, V. Dini, C. Battocchio, I. Fratoddi, A. Cartoni, D. Rotili, M. Castagnola, R. Faccini, I. Bruno, T. Scotognella, A. Giordano and I. Venditti, Gold nanoparticles and nanorods in nuclear medicine: a mini review, Appl. Sci., 2019, 9(16), 3232 CrossRef CAS.
- M. Grzelczak, J. Vermant, E. M. Furst and L. M. Liz-Marzan, Directed self-assembly of nanoparticles, ACS Nano, 2010, 4(7), 3591e605 CrossRef PubMed.
- G. Pasparakis, Recent developments in the use of gold and silver nanoparticles in biomedicine, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2022, 14(5), e1817 CAS.
- L. M. Rossi, J. L. Fiorio, M. A. S. Garcia and C. P. Ferraz, The role and fate of capping ligands in colloidally prepared metal nanoparticle catalysts, Dalton Trans., 2018, 47(17), 5889e915 RSC.
- H. Yin, N. Li, Y. Si, H. Zhang, B. Yang and J. Wang, Gold nanonails for surface-enhanced infrared absorption, Nanoscale Horiz., 2020, 5, 1200–1212 RSC.
- N. Garg, C. Scholl, A. Mohanty and R. Jin, The role of bromide ions in seeding growth of Au nanorods, Langmuir, 2010, 26(12), 10271–10276 CrossRef CAS PubMed.
- S. Eustis and M. El-Sayed, Aspect ratio dependence of the enhanced fluorescence intensity of gold nanorods: experimental and simulation study, J. Phys. Chem. B, 2005, 109(34), 16350–16356 CrossRef CAS PubMed.
- P. K. Jain, X. H. Huang, I. H. El-Sayed and M. A. El-Sayed, Noble metals on the nanoscale: optical and photothermal properties and some applications in imaging, sensing, biology, and medicine, Acc. Chem. Res., 2008, 41(12), 1578e86 CrossRef PubMed.
- C. J. Murphy, L. B. Thompson, A. M. Alkilany, P. N. Sisco, S. P. Boulos and S. T. Sivapalan, et al., The many faces of gold nanorods, J. Phys. Chem. Lett., 2010, 1(19), 2867e75 Search PubMed.
- N. R. Jana, L. Gearheart and C. J. Murphy, J. Phys. Chem. B, 2001, 105, 4065–4067 CrossRef CAS.
- D. K. Smith and B. A. Korgel, The importance of the CTAB ligand on the colloidal seed-mediated synthesis of gold nanorods, Langmuir, 2008, 24(3), 644–649 CrossRef CAS PubMed.
- N. Garg, C. Scholl, A. Mohanty and R. Jin, The role of bromide ions in seeding growth of Au nanorods, Langmuir, 2010, 26(12), 10271–10276 CrossRef CAS PubMed.
- Y. Xu, L. Chen, X. Ye, X. Wang, J. Yu, Y. Zhao, M. Cao, Z. Xia, B. Sun and Q. Zhang, Cooperative interactions among CTA+, Br− and Ag+ during seeded growth of gold nanorods, Nano Res., 2017, 10(6), 2146–2155 CrossRef CAS.
- F. Hubert, F. Testard and O. Spalla, Cetyltrimethylammonium bromide silver bromide complex as the capping agent of gold nanorods, Langmuir, 2008, 24, 9219–9222 CrossRef CAS PubMed.
- S. R. Jackson, J. R. McBride, S. J. Rosenthal and D. W. Wright, Where's the silver? Imaging trace silver coverage on the surface of gold nanorods, J. Am. Chem. Soc., 2014, 136, 5261–5263 CrossRef CAS PubMed.
- C. Oliveira, C. Ribeiro Chaves, P. Bargiela, M. da Grac, C. da Rocha, A. Ferreira da Silva Jos, F. Diniz Chubaci, M. Boström, C. Persson and M. Malta, Surface studies of the chemical environment in gold nanorods supported by X-ray photoelectron spectroscopy (XPS) and ab initio calculations, J. Mater. Res. Technol., 2021, 15, 768–776 CrossRef CAS.
- S. E. Lohse and C. J. Murphy, The quest for shape control: a history of gold nanorod synthesis, Chem. Mater., 2013, 25(8), 1250–1261 CrossRef CAS.
- L. Smykalla, P. Shukrynau, M. Korb, H. Lang and M. Hietschold, Surface-confined 2D polymerization of a brominated copper-tetraphenylporphyrin on Au(111), Nanoscale, 2015, 7, 4234–4241 RSC.
- A. Di Cicco, G. Aquilanti, M. Minicucci, E. Principi, N. Novello, A. Cognigni and L. Olivi, Novel XAFS capabilities at Elettra synchrotron light source, J. Phys.: Conf. Ser., 2009, 190, 012043 CrossRef.
- F. d'Acapito, G. O. Lepore, A. Puri, A. Laloni, F. La Manna, E. Dettona, A. De Luisa and A. Martin, J. Synchrotron Radiat., 2019, 26, 551–558 CrossRef PubMed.
- C. Meneghini, F. Bardelli and S. Mobilio, ESTRA-fitEXA: a software package for EXAFS data analysis, Nucl. Instrum. Methods Phys. Res., Sect. B, 2012, 285, 153–157 CrossRef CAS.
- G. Bunker, Introduction to XAFS: A Practical Guide to X-Ray Absorption Fine Structure Spectroscopy, Cambridge University Press, Cambridge, 2010 Search PubMed.
- D. E. Sayers, E. A. Stern and F. W. Lytle, New technique for investigating noncrystalline structures: fourier analysis of the extended X-ray absorption fine structure, Phys. Rev. Lett., 1971, 27, 1204–1207 CrossRef CAS.
- A. L. Ankudinov, B. Ravel, J. J. Rehr and S. D. Conradson, Real space multiple scattering calculation of xanes, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 7565 CrossRef CAS.
- V. Secchi, S. Franchi, D. Ciccarelli, M. Dettin, A. Zamuner, A. Serio, G. Iucci and C. Battocchio, Biofunctionalization of TiO2 surfaces with self-assembling layers of oligopeptides covalently grafted to chitosan, ACS Biomater. Sci. Eng., 2019, 5, 2190–2199 CrossRef CAS PubMed.
- S. D. Techane, L. J. Gamble and D. G. Castner, X-ray photoelectron spectroscopy characterization of gold nanoparticles functionalized with amine-terminated alkanethiols, Biointerphases, 2011, 6, 98 CrossRef CAS PubMed.
- D. Kwaśniewska and J. Kiewlicz, Study of interaction between cationic ligand (CTAB) and ascorbic acid/ascorbic acids derivatives by tensiometric and spectroscopic methods, J. Mol. Liq., 2022, 354, 118917 CrossRef.
- I. Haidar, A. Day, P. Decorse, S. Lau-Truong, A. Chevillot-Biraud, J. Aubard, N. Felidj and L. Boubekeur-Lecaque, Tailoring the shape of anisotropic core–shell Au–Ag nanoparticles in dimethyl sulfoxide, Chem. Mater., 2019, 31(8), 2741–2749 CrossRef CAS.
- S. Jin Heo, S. Yoon, S. H. Oh and H. J. Kim, Modification of hybrid active bilayer for enhanced efficiency and stability in planar heterojunction colloidal quantum dot photovoltaics, Nanoscale Res. Lett., 2013, 8, 488 CrossRef PubMed.
- A. G. Shard, Detection limits in XPS for more than 6000 binary systems using Al and Mg KαX-rays, Surf. Interface Anal., 2014, 46, 175 CrossRef CAS.
- I. Berlanga, Synthesis of non-uniform functionalized amphiphilic block copolymers and giant vesicles in the presence of the Belousov–Zhabotinsky reaction, Biomolecules, 2019, 9, 352 CrossRef CAS PubMed.
- F. C. Loh, K. L. Tan and E. T. Kang, XPS studies of charge transfer interactions in some poly(N-vinylcarbazole)/acceptor complexes, Eur. Polym. J., 1991, 27(10), 1055 CrossRef CAS.
- I. Venditti, G. Iucci, I. Fratoddi, M. Cipolletti, E. Montalesi, M. Marino, V. Secchi and C. Battocchio, Direct conjugation of resveratrol on hydrophilic gold nanoparticles: structural and cytotoxic studies for biomedical applications, Nanomaterials, 2020, 10, 1898 CrossRef CAS PubMed.
- G. Beamson and D. Briggs, High Resolution XPS of Organic Polymers: The Scienta ESCA 300 Database, John Wiley & Sons, Chichester, U.K., 1992 Search PubMed.
- M. Benfatto and C. Meneghini, A close look into the low energy region of the XAS spectra: the XANES region, in Synchrotron Radiation, ed. S. Mobilio, F. Boscherini and C. Meneghini, Springer, Berlin, Heidelberg, 2015 Search PubMed.
- C. Nayak, D. Bhattacharyya, K. Bhattacharyya, A. K. Tripathi, R. D. Bapat, S. N. Jha and N. K. Sahoo, Insight into growth of Au-Pt bimetallic nanoparticles: an in situ XAS study, J. Synchrotron Radiat., 2017, 24(Pt 4), 825–835 CrossRef CAS PubMed.
- B. A. Kyffin, D. M. Pickup, G. Mountjoy, F. Foroutan, I. Abrahams and D. Carta, Atomic-scale structural characterization of silver-doped phosphate-based glasses prepared by coacervation, J. Phys. Chem. C, 2021, 125(22), 12256–12268 CrossRef CAS.
- E. A. Pradas del Real and G. Sarret, Ag K Edge XAS Fluorescence of Frozen AgNO3 Solution, SSHADE/FAME (OSUG Data Center), 2013, Dataset/Spectral Data, DOI:10.26302/SSHADE/EXPERIMENT_GS_20170713_007.
- E. A. Pradas del Real and G. Sarret, Ag K Edge XAS Trasmission of AgCl, SSHADE/FAME (OSUG Data Center), 2014, Dataset/Spectral Data, DOI:10.26302/SSHADE/EXPERIMENT_GS_20170713_002.
- E. A. Pradas del Real and G. Sarret, Ag K Edge XAS Trasmission of AgO, SSHADE/FAME (OSUG Data Center), 2013, Dataset/Spectral Data, DOI:10.26302/SSHADE/EXPERIMENT_GS_20170712_002.
- R. W. G. Wyckoff, Second edition. Interscience Publishers, New York, Cubic closest packed, ccp, structure, Cryst. Struct., 1963, 7–83 Search PubMed.
- C. Battocchio, C. Meneghini, I. Fratoddi, I. Venditti, M. V. Russo, G. Aquilanti, C. Maurizio, F. Bondino, R. Matassa, M. Rossi, S. Mobilio and G. Polzonetti, Silver nanoparticles stabilized with thiols: a close look at the local chemistry and chemical structure, J. Phys. Chem. C, 2012, 116(36), 19571–19578 CrossRef CAS.
- J. Spreadborough and J. W. Christian, High-temperature X-ray diffractometer, J. Sci. Instrum., 1959, 36, 116–118 CrossRef.
- H. Takahashi, S. Tamaki and S. Sato, Electron density distribution in Ag Br, J. Phys. Soc. Jpn., 1987, 56, 3593–3597 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Table S1: experimental conditions and plasmonic peaks for the synthesised AuNRs. Table S2: XPS data analysis results; Fig. S1: EDX data; Fig. S2: C 1s XPS spectra, also reporting the in-depth description of each spectral component. See DOI: https://doi.org/10.1039/d3na00356f |
| This journal is © The Royal Society of Chemistry 2023 |