
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Planar aromatic anchors control the electrical conductance of gold|molecule|graphene junctions†
Luke J.
O'Driscoll
 a,
Michael
Jay
b,
Benjamin J.
Robinson
b,
Hatef
Sadeghi
c,
Xintai
Wang
d,
Becky
Penhale-Jones
b,
Martin R.
Bryce
*a and
Colin J.
Lambert
*b
a,
Michael
Jay
b,
Benjamin J.
Robinson
b,
Hatef
Sadeghi
c,
Xintai
Wang
d,
Becky
Penhale-Jones
b,
Martin R.
Bryce
*a and
Colin J.
Lambert
*b
aDept. of Chemistry, Durham University, Durham, DH1 3LE, UK. E-mail: m.r.bryce@durham.ac.uk
bDept. of Physics, Lancaster University, Lancaster, LA1 4YB, UK. E-mail: c.lambert@lancaster.ac.uk
cDept. of Engineering, Warwick University, Coventry CV4 7AL, UK
dSchool of Information Science and Technology, Dalian Maritime University, Dalian, China
First published on 27th March 2023
Abstract
The synthesis of a family of alkanethiol molecules with planar aromatic head groups, designed to anchor molecules effectively to graphene electrodes, is reported. Characterisation of self-assembled monolayers of these molecules on a gold surface via conductive atomic force microscopy shows that when an aromatic head group is present, the conductance Ggraphene obtained using a graphene coated probe is higher than the conductance GPt obtained using a platinum (Pt) probe. For Pt probe and graphene probe junctions, the tunnelling decay constant of benzyl ether derivatives with an alkanethiol molecular backbone is determined as β = 5.6 nm−1 and 3.5 nm−1, respectively. The conductance ratio Ggraphene/GPt increases as the number of rings present in the aromatic head unit, n, increases. However, as the number of rings increases, the conductance path length increases because the planar head groups lie at an angle to the plane of the electrodes. This means that overall conductance decreases as n increases. Density functional theory-based charge transport calculations support these experimental findings. This study confirms that planar aromatic head groups can function as effective anchoring units for graphene electrodes in large area molecular junctions. However, the results also indicate that the size and geometry of these head groups must be considered in order to produce effective molecular designs.
Introduction
Molecular junctions, in which one or more molecules bridge the gap between a source and drain electrode, have been intensely studied during the past two decades.1–17 A variety of methods for engineering the transport properties of molecules have been developed, based on manipulating conformation,18–22 anchor groups,8,23–27 electrodes,28–31 quantum interference,32–42 and heteroatom substitution,43–48 both in single-molecule junctions and large-area junctions comprising self-assembled monolayers (SAMs) of organic molecules.29,49–51 Typically in single-molecule experiments using mechanically controllable break junction (MCBJ)1 or scanning tunnelling microscope (STM)2 contacting methodologies, gold is the electrode material of choice. However, there is a growing interest in the study of molecular junctions formed using graphene electrodes. For this purpose, planar aromatic moieties are desirable terminal groups for anchoring molecules to graphene electrodes owing to the strong π–π interactions between their planar π systems and graphitic surfaces.25,28,30,52 Methods used to determine the conductance of larger area junctions, such as conductive atomic force microscopy (cAFM), are well suited to investigations using different electrode materials.53,54A major goal of molecular-scale electronics is the control of transport properties by systematically varying structural features of the molecule.55 Studies of polycyclic aromatic hydrocarbons on graphene surfaces have shown an increase in the binding energy52 as the number of fused rings increases. Additionally, in molecular junctions where a polycyclic aromatic hydrocarbon lies parallel to both electrodes, the “cross-plane conductance”56 (i.e. the conductance perpendicular to the plane of the electrodes and substrate) increases with the number of fused rings. This present study investigates whether these trends persist when planar aromatics are used as the top contact groups in Au|SAM|Pt and Au|SAM|graphene junctions. Firstly, the synthesis of a family of alkanethiol molecules with planar aromatic head groups of differing sizes is presented. The preparation and characterisation of SAMs of these molecules on Au substrates is reported, followed by cAFM studies of the SAMs in which the effect on conductance of using either a Pt tip or graphene-coated Pt tip was investigated. The experimental results are then compared with charge transport calculations, based on density functional theory (DFT) and molecular dynamics (MD) simulations.
Results and discussion
Molecular design and synthesis
The investigated molecules are shown in Fig. 1. With the exception of commercial octanethiol (C8SH), each molecule is based on an α,ω-disubstituted linear alkyl chain, bearing an ether functionality at one end and an acetyl-protected thiol at the other. The following naming convention is used: the nature of the ether-linked head group (if present), is stated (using standard chemical nomenclature where possible; AM and PyrM refer to anthracenemethylene and pyrenemethylene groups, respectively), followed by the length of the alkyl chain as number of carbons (CX) then the terminal functionality (SAc for protected species, S for assembled molecules, presumed to be thiolates). For example, BnOC8SAc is an octyl chain substituted at each terminus; at one with a benzyl ether and at the other with a thioacetate functionality.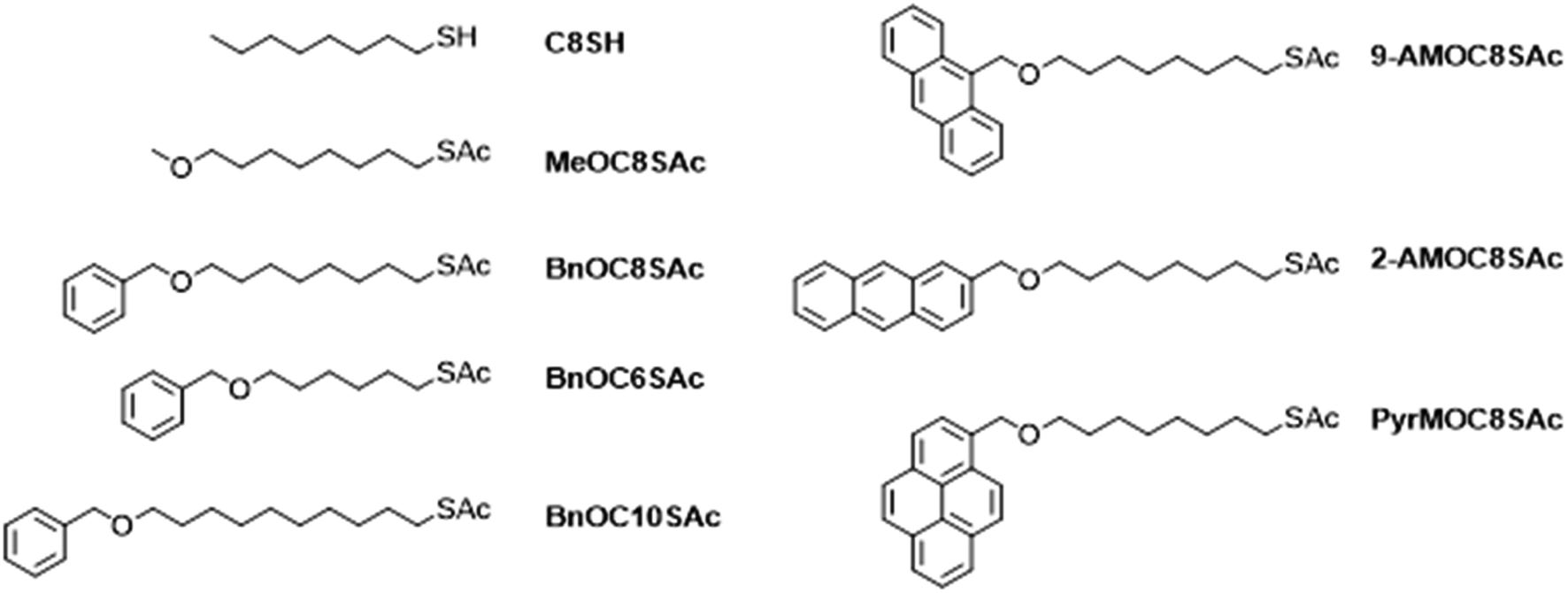 | ||
| Fig. 1 Structures of the molecules investigated in this study. The acetyl protecting group cleaves in situ during assembly on Au surfaces. | ||
The (protected) thiol is designed to assemble on Au surfaces through S–Au bonds (after in situ loss of the acetyl protecting group), forming a SAM in which the alkyl or aryl head group is available to interact with an AFM tip. Alkyl chains are known to be less conductive than conjugated backbones and are uncommon in molecules designed for modern single-molecule conductance studies,7 although they are still useful benchmark compounds57 and were widely used in earlier studies.3,58–60 However, low molecular conductance is less problematic when studying large area molecular junctions as many molecules are measured in parallel. Alkyl backbones are synthetically convenient and were anticipated to result in dense, reasonably ordered SAMs as observed for simple alkanethiols and structurally similar species.29,61 Furthermore, it was anticipated that the increased flexibility of alkyl chains compared to widely-used, rigid, conjugated backbones such as oligo(phenylene ethynylene)s16 would afford more conformational freedom to the planar head groups, potentially permitting more efficient contact between the SAMs and the top electrode.
The studied compounds (Fig. 1) comprise two homologous series. In the first, the alkyl chain has a constant length (C8, i.e., 1,8-disubstituted octanes) and the nature of the head group varies through the series: none, MeO, BnO, 9-AMO, 2-AMO or PyrMO. In the second series the alkyl chain length is varied (C6, C8, C10), while the head group remains the same (BnO, i.e., benzyl ether). The different head groups were selected to investigate the effect of the presence, size and geometry of an aromatic anchor group on molecular conductance. The species bearing aryl ethers were prepared by statistical desymmetrisation of α,ω-alkanediols62 by reaction with the appropriate benzylic bromide,63 activation of the remaining alcohol as a mesylate and finally a nucleophilic substitution using potassium thioacetate. MeOC8SAc was prepared instead by desymmetrisation of 1,8-dibromooctane. Full details of the synthesis and characterisation of these molecules and their precursors are given in the Section S1 of the ESI.†
SAM preparation and characterisation
The molecules were deposited onto a template stripped Au substrate (AuTS) by self-assembly as described in Section S2.1 of the ESI.† SAM characterisation is also described in Section S2.1 of the ESI,† together with representative images. In brief, the deposition of the thin film was monitored by a quartz crystal microbalance (QCM) to confirm SAM formation by determining the density of molecules adsorbed on the AuTS surface. Atomic force microscopy (AFM) was used to determine the thickness of the molecular film via a nano-scratching method.64,65 This provided an indicator of the binding geometry of the molecule on the surface, as detailed in Table S1 in the ESI.† All the measured molecules were determined to have a tilt angle of 35°–55° with respect to the normal to the Au substrate. This is consistent with previous reports; the typical value for alkanethiols is 30°–35°,61 and the coexistence of regions with a tilt angle of around 50° has also been reported.66 SAM quality was further investigated by AFM topography. The roughness of the measured samples was in the range of 1.2 to 2.1 Å, which is comparable with the roughness of a clean AuTS substrate (ca. 1.5 Å), indicating that the contour of the molecular layer followed the underlying gold surface.Electrical measurements
The electron transport properties of the SAMs were characterised using cAFM (see Section S2.2 of the ESI†). The AuTS and an AFM probe coated with a conductive layer were used as source and drain respectively. dI/dV curves were obtained via mathematic differentiation of IV curves collected at a constant normal force of 2 nN shown to provide good electrical contact but negligible compression of the molecular film.67 Histograms shown in Fig. 2 represent dI/dV at near 0 bias (±10 mV), a region which has been shown to exhibit the strongest dependence on the quantum transport properties of the molecular junction.68 Previous reports have shown that electrical transport in SAMs is strongly dependent on the mechanically induced molecular geometry of the probe-sample junction69 arising from either a change in the number of molecules in the junction or a change in the tilt angle of the molecules relative to the substrate.70–72 The precise nature of possible mechanical deformation of the SAMs reported here is beyond the scope of this study. However, we have ensured consistent compression of the SAMs by performing all measurements at the same low applied force, as determined by equipment integrated thermal calibration of the AFM probe and force curve calibrations of the junction. | ||
| Fig. 2 Experimental results for the electrical conductances of different SAMs. Conductance distribution of SAMs obtained using (a) a Pt coated probe and (b) a graphene coated probe. The distribution of conductances versus the number of alkyl units, obtained using (c) a Pt coated probe and (d) a graphene coated probe for molecules with a benzyl head group (average conductance is indicated by the yellow squares). | ||
Both Pt and graphene54 were used as the conductive coating layer for the AFM probe. To estimate the number of molecules present in each junction (Table S2†), and hence the average conductance per molecule in a given SAM, the contact area between the AuTS and probe was estimated by the JRK72–74 model (as detailed in Section S2.2 of the ESI†).
The resulting histograms of molecular conductance are shown in Fig. 2a and b. The statistically most probable conductance of each species is listed in Table 1. Fig. 2c shows the conductance decay of SAMs with benzyl head groups as a function of the number of alkyl units in the molecular backbone in Au|SAM|Pt junctions. This follows the characteristic relationship for conductance decay in molecular junctions: G ∝ e−βL where G is molecular conductance, L is molecular length obtained from density functional theory (DFT) calculations, and β is the tunnelling decay constant of the molecular backbone. For the Pt probe junctions, we measure β = 5.6 nm−1, slightly below the typical range for alkanethiols and alkanedithiols in metallic junctions (ca. 8–10 nm−1).75,76 In comparison, for the graphene probe junctions we measure β = 3.5 nm−1 (Fig. 2d) which is consistent with recent studies that have shown that metal|molecule|graphene junction architectures afford lower β values than metal|molecule|metal junction architectures for alkane derivatives including alkanedithiols.76,77
| Molecule | log(GPt(S)) | log(Ggraphene(S)) | G graphene/GPt |
|---|---|---|---|
| C8S | −6.58 | −6.31 | 1.86 |
| MeOC8S | −7.81 | −7.82 | 0.98 |
| BnOC8S | −8.94 | −8.62 | 2.09 |
| 9-AMOC8S | −8.89 | −8.28 | 4.07 |
| 2-AMOC8S | −9.25 | −9.13 | 1.32 |
| PyrMOC8S | −10.29 | −9.58 | 5.13 |
Transport calculations
Charge transport simulations of molecular junctions were carried out using the SIESTA78 density functional theory (DFT) code, combined with the Green's function transport code Gollum79 (see Section S3 of ESI†). In the simulations, the thiol terminal group is bound to a Au bottom electrode and the other end of the molecule (usually an ether-linked head group) is in contact with a top Au electrode. Further details are provided in Section S3.1 of the ESI.† Where head groups are present, the angle θ between the plane of the aromatic head group and the plane of the top electrode must be considered. The following discussion will use the formalism that θ = 0° when these planes are parallel and θ = 90° when these planes are perpendicular.Molecular dynamics simulations modelling a SAM of BnOC8S showed that in the absence of a top graphene electrode, the headgroups tend to be positioned in an upright manner, with θ close to 90° (Fig. S46†). This is unsurprising as this conformation would be expected to minimise steric clash between neighbouring molecules. However, in the presence of a graphene top contact, the distribution of angles is centred at θ ≈ 55° (Fig. S46†). This rearrangement implies a shorter conductance pathway than would be expected for the fully extended molecule, which is likely to result in increased conductance. A more detailed discussion can be found in Section S3.2 of the ESI.†
The MD simulations do not represent the distribution of angles in the cAFM measurements described above, which will depend upon additional factors, such as surface roughness and molecular packing in the SAM. Due to the computational expense of carrying out DFT simulations on large distributions of angles, the two extremes of angle θ were studied for each molecule. Fig. 3a shows the simulated transmission functions T(E) for the molecular junctions in which the aromatic head groups are approximately perpendicular to the top electrode (θ ≈ 90°), while Fig. 3b shows the equivalent data for approximately parallel head groups (θ ≈ 0°). The electrical conductance of such junctions is approximately G = G0T(EF), where EF is the Fermi energy and G0 ≈ 77 μS is the conductance quantum.
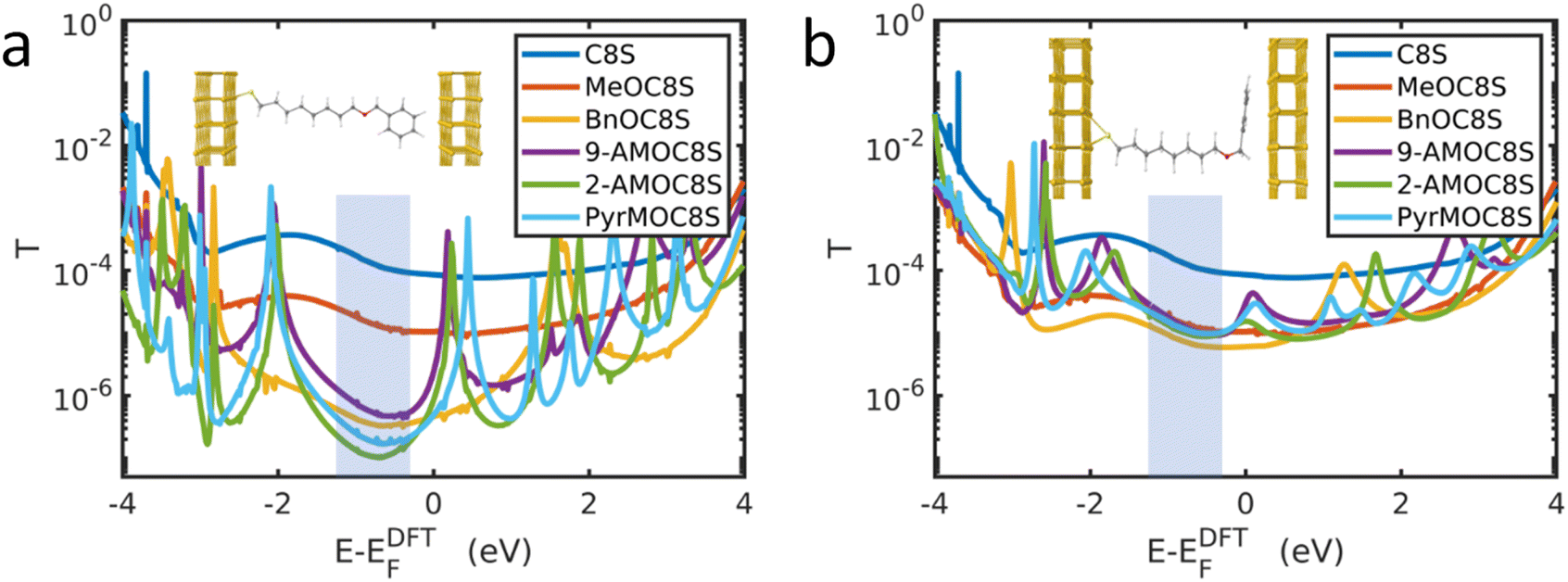 | ||
| Fig. 3 DFT-based transmission functions for Au|molecule|Au junctions. Where aromatic head groups are present (a) shows aromatic head groups approximately perpendicular to the top Au lead (i.e., θ ≈ 90°, see inset) and (b) shows aromatic head groups approximately parallel to the top Au lead (i.e., θ ≈ 0°, see inset). EDFTF is the DFT-predicted Fermi energy. In practice, EDFTF may not coincide with the experimental value EF, which typically lies near the middle of the HOMO–LUMO gap, indicated by the shaded regions. | ||
Past comparisons between experiment and theory suggest that EF typically lies near the middle of the HOMO–LUMO gap,44,80,81 indicated by the shaded regions in Fig. 3. The trends in T(EF) observed in Fig. 3a are in qualitative agreement with the experimentally measured conductances. Starting from the relatively high T(EF) of the alkanethiol C8S, the addition of the methoxy group causes the transmission function in the shaded region to fall by around an order of magnitude. Fig. 3a shows that for fully extended, near-perpendicular conformations (θ ≈ 90°), T(EF) is lowered further by the addition of the aromatic head groups, with the head groups which increase the molecular length the most (PyrM, 2-AM) having the lowest transmission. Fig. 3b shows that when an aromatic head group is present, near-parallel configurations (θ ≈ 0°) are more conductive than those in which the head group is perpendicular to the Au lead. In the parallel configuration, the transmission of all ether-functionalised species is similar in the HOMO–LUMO gap. The implication is that in the experimental system it is unlikely that the aromatic head groups lie perpendicular to the top electrode surface. Rather, as supported by the MD simulations discussed above, the head groups present in the experimental junctions may display a distribution of angles between the limits of parallel and perpendicular configurations. Realistic transmission curves are therefore expected to lie between those shown in Fig. 3a and b. However, the intensive calculations required to afford such data are beyond the scope of the present study.
Discussion
When considering the C8 alkyl chain series, two trends are apparent in the experimental data as the number n of fused rings in the aromatic head group changes. The first trend is that the conductance tends to decrease as the size of the aromatic head group increases. This result is seemingly in contrast to previous studies. It has been reported that the electrical conductance of planar aromatic molecules adsorbed on planar electrodes increases with their size, when current flows perpendicular to the plane of the molecules.56 Additionally, the binding energy between an aromatic head group and a planar electrode increases with the size of the head group.52,82 However, these studies are concerned with aromatic systems that are coplanar to the electrode surface. The MD simulations, the agreement between experimental data and trends observed in Fig. 3a, and SAM thickness analysis (Fig. S41 and S42†) indicate that in the present work the aromatic head groups are tilted with respect to the probe. A consequence of this tilting is that the current pathway is longer for species with larger head groups. The observation that conductance falls with increasing head group size (and therefore molecular length) supports the presence of tilted aromatic units. In contrast, 9-AMOC8S has a similar (Pt probe) or lower (graphene probe) conductance than BnOC8S, despite its larger head group. In this case, the position of the additional fused rings in the former system is such that the effective length of the molecule remains similar to the latter. The head group of 9-AMOC8S effectively becomes wider rather than longer and a reduction in conductance is not observed.The second trend is the increase in the ratio between the conductance of Au|SAM|graphene junctions and the conductance of Au|SAM|Pt junctions (Table 1) as the number of fused rings in the ether-linked head group, n, increases. For MeOC8S (n = 0), BnOC8S (n = 1), 9-AMOC8S (n = 3) and PyrMOC8S (n = 4), the ratio Ggraphene/GPt is approximately equal to n + 1, indicating that the larger aromatics are more optimised to graphene contacts than Pt contacts. This observation can be attributed to π–π interactions,52,82 which are expected between aromatic species and graphene but not Pt. The presence of π–π interactions when a graphene electrode is used would favour a smaller average value of θ than for Pt, which the computational studies above indicate would result in increased conductance. For unsubstituted polyacenes adsorbed directly on graphene, the calculated binding energy increases linearly with the number of fused rings.82 Therefore, in the present systems, the strength of π–π interactions, and the concomitant reduction of θ, would be expected to increase as n increases. This accounts for the observed trend in Ggraphene/GPt. Additionally, the atomically smooth graphene surface may accommodate conformational changes in the SAM more readily than the sharper Pt probe.
Two of the studied molecules do not follow the observed Ggraphene/GPt = n + 1 pattern. C8S is shorter than the other species and does not contain an ether linker, limiting its possible conformations in a SAM. Therefore, it is not unreasonable that C8S behaves differently to the other molecules. 2-AMOC8S shows a much smaller Ggraphene/GPt ratio (1.32) than would be expected based on observations for the remainder of the ether-linked series (Table 1). This may relate to the connectivity of the anthracene unit in 2-AMOC8S where in certain conformations the additional rings have a much larger effect on molecular length versusBnOC8S in comparison to 9-AMOC8S or PyrMOC8S. This geometric difference could impact SAM packing and the energy barrier associated with deviating from θ ≈ 90° in the presence of a top electrode. It is possible that related conformational effects are responsible for the relatively poor fit of the conductance data for 2-AMOC8S to a Gaussian curve in comparison to that of the other molecules in Fig. 2a.
The above observations indicate that polycyclic aromatic anchoring units have potential as graphene contacts in Au|SAM|graphene molecular junctions. The model systems studied in this work serve as a proof of concept and will inform design strategies for future SAM based devices. In order to best utilise the favourable interactions between aromatic species and graphene, more complex molecular designs are necessary. Achieving high molecular conductance will require consideration of factors such as: (i) how best to include large aromatic systems without significantly increasing molecular length; (ii) how to minimise θ while accounting for geometric requirements at the Au electrode and the desirability of conjugated molecular backbones. To minimise θ, it may prove important to reduce steric clash between neighbouring aromatic head groups. Methods to achieve this could include the use of large-footprint anchoring units54,83 on the gold surface to increase head group spacing, or the use of mixed monolayers containing functional species bearing aromatic contact units alongside simple “spacer” molecules.84
Conclusions
The self-assembly and conductance properties of a series of alkanethiol derivatives bearing ether-linked aromatic head groups, and reference compounds, were investigated using AFM imaging, QCM studies and cAFM experiments. DFT and MD studies support the experimental observations. A sub-series of species with increasing alkyl linker length were observed to show an expected exponential conductance decay with increasing molecular length, with a tunnelling decay constant of β = 5.6 nm−1 in Au|SAM|Pt junctions and β = 3.5 nm−1 in Au|SAM|graphene junctions. Using both Pt and graphene top electrodes, conductance was observed to decrease as the size of the aromatic head group increased. This was attributed to an increasing junction length caused by the head groups being tilted with respect to the planar electrode, as larger head groups have a greater impact on the length of an extended molecule. The ratio of the conductances measured using graphene and Pt top electrodes, Ggraphene/GPt, generally increased with the number of aromatic rings in the head group. As π–π interactions are known to increase in strength for progressively larger aromatic systems, this effect is attributed to an increasing tendency towards coplanarity with the graphene electrode as the size of the aromatic head group increases. These results indicate the effectiveness of aromatic head groups as contacts for graphene electrodes in asymmetric metal|SAM|graphene molecular junctions. Furthermore, they highlight some important considerations that must be made when designing optimised molecules for such junctions. The geometry of the head group is key; to maximise conductance it should lie near parallel to the graphene electrode in a molecular junction and have minimal impact on the length of the conductance pathway. These observations contribute to the design of the next generation of molecules for use in hybrid nanodevices.Author contributions
L. J. O. synthesised the molecules. X. W. and B. P.-J. performed device fabrication and measurements. M. J. carried out the calculations with assistance from H. S. The funding for this work was obtained by B. J. R., M. R. B. and C. J. L. who supervised the research and contributed to interpreting the results. L. J. O. and M. R. B. coordinated the writing of the manuscript, with contributions from all co-authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the UK EPSRC (grants EP/M014452/1, EP/N03337X/1 and EP/P027520/1) and by the European Commission FET Open projects 767187 – QuIET and 766853 – EFINED. H. S. acknowledges the UKRI for funding: MR/S015329/2 and MR/X015181/1.References
- M. A. Reed, C. Zhou, C. J. Muller, T. P. Burgin and J. M. Tour, Science, 1997, 278, 252–254 CrossRef CAS.
- B. Xu and N. J. Tao, Science, 2003, 301, 1221–1223 CrossRef CAS PubMed.
- X. Li, J. He, J. Hihath, B. Xu, S. M. Lindsay and N. Tao, J. Am. Chem. Soc., 2006, 128, 2135–2141 CrossRef CAS PubMed.
- S. H. Choi, B. Kim and C. D. Frisbie, Science, 2008, 320, 1482–1486 CrossRef CAS PubMed.
- L. Sun, Y. A. Diaz-Fernandez, T. A. Gschneidtner, F. Westerlund, S. Lara-Avila and K. Moth-Poulsen, Chem. Soc. Rev., 2014, 43, 7378–7411 RSC.
- C. J. Lambert, Chem. Soc. Rev., 2015, 44, 875–888 RSC.
- T. A. Su, M. Neupane, M. L. Steigerwald, L. Venkataraman and C. Nuckolls, Nat. Rev. Mater., 2016, 1, 16002 CrossRef CAS.
- E. Leary, A. La Rosa, M. T. González, G. Rubio-Bollinger, N. Agraït and N. Martín, Chem. Soc. Rev., 2015, 44, 920–942 RSC.
- D. Xiang, X. Wang, C. Jia, T. Lee and X. Guo, Chem. Rev., 2016, 116, 4318–4440 CrossRef CAS PubMed.
- L. Rincón-García, C. Evangeli, G. Rubio-Bollinger and N. Agraït, Chem. Soc. Rev., 2016, 45, 4285–4306 RSC.
- K. Wang and B. Xu, Top. Curr. Chem., 2017, 375, 17 CrossRef PubMed.
- L. Cui, R. Miao, C. Jiang, E. Meyhofer and P. Reddy, J. Chem. Phys., 2017, 146, 092201 CrossRef.
- P. Gehring, J. M. Thijssen and H. S. J. van der Zant, Nat. Rev. Phys., 2019, 1, 381–396 CrossRef.
- N. Xin, J. Guan, C. Zhou, X. Chen, C. Gu, Y. Li, M. A. Ratner, A. Nitzan, J. F. Stoddart and X. Guo, Nat. Rev. Phys., 2019, 1, 211–230 CrossRef.
- F. Evers, R. Korytár, S. Tewari and J. M. van Ruitenbeek, Rev. Mod. Phys., 2020, 92, 035001 CrossRef CAS.
- L. J. O'Driscoll and M. R. Bryce, Nanoscale, 2021, 13, 10668–10711 RSC.
- C. J. Lambert, Quantum Transport in Nanostructures and Molecules, IOP Publishing, 2021 Search PubMed.
- L. Venkataraman, J. E. Klare, C. Nuckolls, M. S. Hybertsen and M. L. Steigerwald, Nature, 2006, 442, 904–907 CrossRef CAS PubMed.
- W. Haiss, C. Wang, I. Grace, A. S. Batsanov, D. J. Schiffrin, S. J. Higgins, M. R. Bryce, C. J. Lambert and R. J. Nichols, Nat. Mater., 2006, 5, 995–1002 CrossRef CAS PubMed.
- D. Stefani, K. J. Weiland, M. Skripnik, C. Hsu, M. L. Perrin, M. Mayor, F. Pauly and H. S. J. van der Zant, Nano Lett., 2018, 18, 5981–5988 CrossRef CAS PubMed.
- F. Jiang, D. I. Trupp, N. Algethami, H. Zheng, W. He, A. Alqorashi, C. Zhu, C. Tang, R. Li, J. Liu, H. Sadeghi, J. Shi, R. Davidson, M. Korb, A. N. Sobolev, M. Naher, S. Sangtarash, P. J. Low, W. Hong and C. J. Lambert, Angew. Chem., Int. Ed., 2019, 58, 18987–18993 CrossRef CAS PubMed.
- C. Tang, Y. Tang, Y. Ye, Z. Yan, Z. Chen, L. Chen, L. Zhang, J. Liu, J. Shi, H. Xia and W. Hong, Chem, 2020, 6, 2770–2781 CAS.
- E. Gorenskaia, M. Naher, L. Daukiya, S. A. Moggach, D. Costa Milan, A. Vezzoli, C. J. Lambert, R. J. Nichols, T. Becker and P. J. Low, Aust. J. Chem., 2021, 74, 806–818 CrossRef CAS.
- W. Hong, D. Z. Manrique, P. Moreno-García, M. Gulcur, A. Mishchenko, C. J. Lambert, M. R. Bryce and T. Wandlowski, J. Am. Chem. Soc., 2012, 134, 2292–2304 CrossRef CAS PubMed.
- B. Limburg, J. O. Thomas, G. Holloway, H. Sadeghi, S. Sangtarash, I. C.-Y. Hou, J. Cremers, A. Narita, K. Müllen, C. J. Lambert, G. A. D. Briggs, J. A. Mol and H. L. Anderson, Adv. Funct. Mater., 2018, 28, 1803629 CrossRef.
- I. L. Herrer, A. K. Ismael, D. C. Milán, A. Vezzoli, S. Martín, A. González-Orive, I. Grace, C. Lambert, J. L. Serrano, R. J. Nichols and P. Cea, J. Phys. Chem. Lett., 2018, 9, 5364–5372 CrossRef CAS PubMed.
- C. Huang, S. Chen, K. Baruël Ørnsø, D. Reber, M. Baghernejad, Y. Fu, T. Wandlowski, S. Decurtins, W. Hong, K. S. Thygesen and S.-X. Liu, Angew. Chem., Int. Ed., 2015, 54, 14304–14307 CrossRef CAS PubMed.
- G. Wang, Y. Kim, M. Choe, T.-W. Kim and T. Lee, Adv. Mater., 2011, 23, 755–760 CrossRef CAS PubMed.
- C. A. Nijhuis, W. F. Reus and G. M. Whitesides, J. Am. Chem. Soc., 2009, 131, 17814–17827 CrossRef CAS PubMed.
- P. Gehring, J. K. Sowa, J. Cremers, Q. Wu, H. Sadeghi, Y. Sheng, J. H. Warner, C. J. Lambert, G. A. D. Briggs and J. A. Mol, ACS Nano, 2017, 11, 5325–5331 CrossRef CAS PubMed.
- I. J. Planje, R. J. Davidson, A. Vezzoli, A. Daaoub, S. Sangtarash, H. Sadeghi, S. Martín, P. Cea, C. J. Lambert, A. Beeby, S. J. Higgins and R. J. Nichols, ACS Sens., 2021, 6, 530–537 CrossRef CAS PubMed.
- G. C. Solomon, D. Q. Andrews, T. Hansen, R. H. Goldsmith, M. R. Wasielewski, R. P. Van Duyne and M. A. Ratner, J. Chem. Phys., 2008, 129, 054701 CrossRef PubMed.
- K. Yoshizawa, T. Tada and A. Staykov, J. Am. Chem. Soc., 2008, 130, 9406–9413 CrossRef CAS PubMed.
- T. Markussen, R. Stadler and K. S. Thygesen, Nano Lett., 2010, 10, 4260–4265 CrossRef CAS PubMed.
- C. M. Guédon, H. Valkenier, T. Markussen, K. S. Thygesen, J. C. Hummelen and S. J. van der Molen, Nat. Nanotechnol., 2012, 7, 305–309 CrossRef PubMed.
- H. Vazquez, R. Skouta, S. Schneebeli, M. Kamenetska, R. Breslow, L. Venkataraman and M. S. Hybertsen, Nat. Nanotechnol., 2012, 7, 663–667 CrossRef CAS PubMed.
- C. R. Arroyo, S. Tarkuc, R. Frisenda, J. S. Seldenthuis, C. H. M. Woerde, R. Eelkema, F. C. Grozema and H. S. J. van der Zant, Angew. Chem., Int. Ed., 2013, 52, 3152–3155 CrossRef CAS PubMed.
- D. Z. Manrique, C. Huang, M. Baghernejad, X. Zhao, O. A. Al-Owaedi, H. Sadeghi, V. Kaliginedi, W. Hong, M. Gulcur, T. Wandlowski, M. R. Bryce and C. J. Lambert, Nat. Commun., 2015, 6, 6389 CrossRef CAS PubMed.
- L. J. O'Driscoll and M. R. Bryce, Nanoscale, 2021, 13, 1103–1123 RSC.
- L. J. O'Driscoll, S. Sangtarash, W. Xu, A. Daaoub, W. Hong, H. Sadeghi and M. R. Bryce, J. Phys. Chem. C, 2021, 125, 17385–17391 CrossRef PubMed.
- J. E. Greenwald, J. Cameron, N. J. Findlay, T. Fu, S. Gunasekaran, P. J. Skabara and L. Venkataraman, Nat. Nanotechnol., 2021, 16, 313–317 CrossRef CAS PubMed.
- H. Chen, S. Hou, Q. Wu, F. Jiang, P. Zhou, L. Zhang, Y. Jiao, B. Song, Q.-H. Guo, X.-Y. Chen, W. Hong, C. J. Lambert and J. F. Stoddart, Matter, 2021, 4, 3662–3676 CrossRef CAS.
- S. Sangtarash, H. Sadeghi and C. J. Lambert, Phys. Chem. Chem. Phys., 2018, 20, 9630–9637 RSC.
- X. Liu, S. Sangtarash, D. Reber, D. Zhang, H. Sadeghi, J. Shi, Z.-Y. Xiao, W. Hong, C. J. Lambert and S.-X. Liu, Angew. Chem., Int. Ed., 2017, 56, 173–176 CrossRef CAS PubMed.
- Y. Yang, M. Gantenbein, A. Alqorashi, J. Wei, S. Sangtarash, D. Hu, H. Sadeghi, R. Zhang, J. Pi, L. Chen, X. Huang, R. Li, J. Liu, J. Shi, W. Hong, C. J. Lambert and M. R. Bryce, J. Phys. Chem. C, 2018, 122, 14965–14970 CrossRef CAS.
- M. Gantenbein, L. Wang, A. A. Al-jobory, A. K. Ismael, C. J. Lambert, W. Hong and M. R. Bryce, Sci. Rep., 2017, 7, 1794 CrossRef PubMed.
- L. Palomino-Ruiz, S. Rodríguez-González, J. G. Fallaque, I. R. Márquez, N. Agraït, C. Díaz, E. Leary, J. M. Cuerva, A. G. Campaña, F. Martín, A. Millán and M. T. González, Angew. Chem., Int. Ed., 2021, 60, 6609–6616 CrossRef CAS PubMed.
- I. M. Grace, G. Olsen, J. Hurtado-Gallego, L. Rincón-García, G. Rubio-Bollinger, M. R. Bryce, N. Agraït and C. J. Lambert, Nanoscale, 2020, 12, 14682–14688 RSC.
- C. Jia, M. Famili, M. Carlotti, Y. Liu, P. Wang, I. M. Grace, Z. Feng, Y. Wang, Z. Zhao, M. Ding, X. Xu, C. Wang, S.-J. Lee, Y. Huang, R. C. Chiechi, C. J. Lambert and X. Duan, Sci. Adv., 2018, 4, eaat8237 CrossRef CAS PubMed.
- M. Famili, C. Jia, X. Liu, P. Wang, I. M. Grace, J. Guo, Y. Liu, Z. Feng, Y. Wang, Z. Zhao, S. Decurtins, R. Häner, Y. Huang, S.-X. Liu, C. J. Lambert and X. Duan, Chem, 2019, 5, 474–484 CAS.
- X. Wang, T. L. R. Bennett, A. Ismael, L. A. Wilkinson, J. Hamill, A. J. P. White, I. M. Grace, O. V. Kolosov, T. Albrecht, B. J. Robinson, N. J. Long, L. F. Cohen and C. J. Lambert, J. Am. Chem. Soc., 2020, 142, 8555–8560 CrossRef CAS PubMed.
- S. Bailey, D. Visontai, C. J. Lambert, M. R. Bryce, H. Frampton and D. Chappell, J. Chem. Phys., 2014, 140, 054708 CrossRef PubMed.
- Z. Wei, T. Hansen, M. Santella, X. Wang, C. R. Parker, X. Jiang, T. Li, M. Glyvradal, K. Jennum, E. Glibstrup, N. Bovet, X. Wang, W. Hu, G. C. Solomon, M. B. Nielsen, X. Qiu, T. Bjørnholm, K. Nørgaard and B. W. Laursen, Adv. Funct. Mater., 2015, 25, 1700–1708 CrossRef CAS.
- L. J. O'Driscoll, X. Wang, M. Jay, A. S. Batsanov, H. Sadeghi, C. J. Lambert, B. J. Robinson and M. R. Bryce, Angew. Chem., Int. Ed., 2020, 59, 882–889 CrossRef PubMed.
- M. S. Hybertsen and L. Venkataraman, Acc. Chem. Res., 2016, 49, 452–460 CrossRef CAS PubMed.
- S. Zhao, Q. Wu, J. Pi, J. Liu, J. Zheng, S. Hou, J. Wei, R. Li, H. Sadeghi, Y. Yang, J. Shi, Z. Chen, Z. Xiao, C. Lambert and W. Hong, Sci. Adv., 2020, 6, eaba6714 CrossRef CAS PubMed.
- F. H. van Veen, L. Ornago, H. S. J. van der Zant and M. El Abbassi, J. Phys. Chem. C, 2022, 126, 8801–8806 CrossRef CAS.
- B. Mann and H. Kuhn, J. Appl. Phys., 1971, 42, 4398–4405 CrossRef CAS.
- A. Ulman, J. E. Eilers and N. Tillman, Langmuir, 1989, 5, 1147–1152 CrossRef CAS.
- R. Haag, M. A. Rampi, R. E. Holmlin and G. M. Whitesides, J. Am. Chem. Soc., 1999, 121, 7895–7906 CrossRef CAS.
- C. Vericat, M. E. Vela, G. Benitez, P. Carro and R. C. Salvarezza, Chem. Soc. Rev., 2010, 39, 1805–1834 RSC.
- B. V. Subba Reddy, B. Anusha, U. V. Subba Reddy, J. S. Yadav and C. Suresh Reddy, Helv. Chim. Acta, 2013, 96, 1983–1990 CrossRef CAS.
- L. J. O'Driscoll, D. J. Welsh, S. W. D. Bailey, D. Visontai, H. Frampton, M. R. Bryce and C. J. Lambert, Chem.–Eur. J., 2015, 21, 3891–3894 CrossRef PubMed.
- R. Garcia, R. V. Martinez and J. Martinez, Chem. Soc. Rev., 2006, 35, 29–38 RSC.
- N. A. Amro, S. Xu and G. Y. Liu, Langmuir, 2000, 16, 3006–3009 CrossRef CAS.
- E. Barrena, E. Palacios-Lidón, C. Munuera, X. Torrelles, S. Ferrer, U. Jonas, M. Salmeron and C. Ocal, J. Am. Chem. Soc., 2004, 126, 385–395 CrossRef CAS PubMed.
- Z. Wei, T. Li, K. Jennum, M. Santella, N. Bovet, W. Hu, M. B. Nielsen, T. Bjørnholm, G. C. Solomon, B. W. Laursen and K. Nørgaard, Langmuir, 2012, 28, 4016–4023 CrossRef CAS PubMed.
- J. Ning, R. Li, X. Shen, Z. Qian, S. Hou, A. R. Rocha and S. Sanvito, Nanotechnology, 2007, 18, 345203 CrossRef.
- X. Wang, A. Ismael, A. Almutlg, M. Alshammari, A. Al-Jobory, A. Alshehab, T. L. R. Bennett, L. A. Wilkinson, L. F. Cohen, N. J. Long, B. J. Robinson and C. Lambert, Chem. Sci., 2021, 12, 5230–5235 RSC.
- T. Frederiksen, C. Munuera, C. Ocal, M. Brandbyge, M. Paulsson, D. Sanchez-Portal and A. Arnau, ACS Nano, 2009, 3, 2073–2080 CrossRef CAS PubMed.
- Y. Qi, I. Ratera, J. Y. Park, P. D. Ashby, S. Y. Quek, J. B. Neaton and M. Salmeron, Langmuir, 2008, 24, 2219–2223 CrossRef CAS PubMed.
- H. Song, H. Lee and T. Lee, J. Am. Chem. Soc., 2007, 129, 3806–3807 CrossRef CAS PubMed.
- N. A. Burnham, R. J. Colton and H. M. Pollock, Phys. Rev. Lett., 1992, 69, 144–147 CrossRef CAS PubMed.
- K. L. Johnson, K. Kendall, A. D. Roberts and D. Tabor, Proc. R. Soc. London, Ser. A, 1971, 324, 301–313 CAS.
- V. B. Engelkes, J. M. Beebe and C. D. Frisbie, J. Am. Chem. Soc., 2004, 126, 14287–14296 CrossRef CAS PubMed.
- C. He, Q. Zhang, T. Gao, C. Liu, Z. Chen, C. Zhao, C. Zhao, R. J. Nichols, Y. J. Dappe and L. Yang, Phys. Chem. Chem. Phys., 2020, 22, 13498–13504 RSC.
- Q. Zhang, L. Liu, S. Tao, C. Wang, C. Zhao, C. González, Y. J. Dappe, R. J. Nichols and L. Yang, Nano Lett., 2016, 16, 6534–6540 CrossRef CAS PubMed.
- J. M. Soler, E. Artacho, J. D. Gale, A. García, J. Junquera, P. Ordejón and D. Sánchez-Portal, J. Phys.: Condens. Matter, 2002, 14, 2745 CrossRef CAS.
- J. Ferrer, C. J. Lambert, V. M. García-Suárez, D. Z. Manrique, D. Visontai, L. Oroszlany, R. Rodríguez-Ferradás, I. Grace, S. W. D. Bailey, K. Gillemot, H. Sadeghi and L. A. Algharagholy, New J. Phys., 2014, 16, 093029 CrossRef.
- S. Sangtarash, C. Huang, H. Sadeghi, G. Sorohhov, J. Hauser, T. Wandlowski, W. Hong, S. Decurtins, S.-X. Liu and C. J. Lambert, J. Am. Chem. Soc., 2015, 137, 11425–11431 CrossRef CAS PubMed.
- J. Bai, A. Daaoub, S. Sangtarash, X. Li, Y. Tang, Q. Zou, H. Sadeghi, S. Liu, X. Huang, Z. Tan, J. Liu, Y. Yang, J. Shi, G. Mészáros, W. Chen, C. Lambert and W. Hong, Nat. Mater., 2019, 18, 364–369 CrossRef CAS PubMed.
- Y. Li, X. Tu, H. Wang, S. Sanvito and S. Hou, J. Chem. Phys., 2015, 142, 164701 CrossRef PubMed.
- M. Valášek and M. Mayor, Chem.–Eur. J., 2017, 23, 13538–13548 CrossRef PubMed.
- S. Kumar, J. T. van Herpt, R. Y. N. Gengler, B. L. Feringa, P. Rudolf and R. C. Chiechi, J. Am. Chem. Soc., 2016, 138, 12519–12526 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of synthesis and characterisation of molecules; SAM formation and characterisation; computational studies. See DOI: https://doi.org/10.1039/d2na00873d |
| This journal is © The Royal Society of Chemistry 2023 |