
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrganosilica nanoparticles containing sodium borocaptate (BSH) provide new prospects for boron neutron capture therapy (BNCT): efficient cellular uptake and enhanced BNCT efficacy†
Mathilde
Laird
a,
Kotaro
Matsumoto
a,
Yuya
Higashi
a,
Aoi
Komatsu
a,
Art
Raitano
b,
Kendall
Morrison
b,
Minoru
Suzuki
 c and
Fuyuhiko
Tamanoi
*ad
c and
Fuyuhiko
Tamanoi
*ad
aInstitute for Integrated Cell-Material Sciences, Institute for Advanced Study, Kyoto University, Kyoto 606-8501, Japan. E-mail: tamanoi.fuyuhiko.2c@kyoto-u.ac.jp
bTAE Life Sciences, Drug Development Division, Santa Monica, CA 90404, USA
cInstitute for Integrated Radiation and Nuclear Science, Kyoto University, Kumatori 590-0494, Japan
dDepartment of Microbiology, Immunology and Molecular Genetics, University of California, Los Angeles, USA
First published on 3rd April 2023
Abstract
Boron neutron capture therapy (BNCT), a method based on the fission of boron-10 upon neutron irradiation, has emerged as an attractive option for radiation therapy. To date, the main drugs used in BNCT are 4-boronophenylalanine (BPA) and sodium borocaptate (BSH). While BPA has been extensively tested in clinical trials, the use of BSH has been limited, mainly due to its poor cellular uptake. Here, we describe a novel type of mesoporous silica-based nanoparticle containing BSH covalently attached to a nanocarrier. Synthesis and characterization of these nanoparticles (BSH-BPMO) are presented. The synthetic strategy involves a click thiol–ene reaction with the boron cluster, providing hydrolytically stable linkage with the BSH in four steps. The BSH-BPMO nanoparticles were efficiently taken up into cancer cells and accumulated in the perinuclear region. Inductively coupled plasma (ICP) measurements of boron uptake in cells highlight the important role of the nanocarrier in the enhancement of boron internalization. BSH-BPMO nanoparticles were also taken up and distributed throughout tumour spheroids. BNCT efficacy was examined by the neutron exposure of the tumour spheroids. BSH-BPMO loaded spheroids were completely destroyed upon neutron irradiation. In contrast, neutron irradiation of tumour spheroids loaded with BSH or BPA resulted in significantly less spheroid shrinkage. The significant difference in BNCT efficacy of the BSH-BPMO was correlated with the improved boron uptake via the nanocarrier. Overall, these results demonstrate the critical role of the nanocarrier in BSH internalization and the enhanced BNCT efficacy of the BSH-BPMO compared with BSH and BPA, two drugs used in BNCT clinical trials.
Introduction
Over the past few decades, Boron Neutron Capture Therapy, also called BNCT, has attracted increasing attention as a new type of radiotherapy aiming at lowering the side effect linked with common radiotherapy (i.e. damage to healthy tissues). As shown in Fig. 1, BNCT relies on the use of a neutron beam to trigger the nuclear fission of boron-10 atoms to lithium-7 and α-particles (helium-4).1 α-particles only travel short distances (typically cell distances) but can cause DNA double-strands to break, leading to cell death.1 The current objective of BNCT is to make radiation therapies safer by using a thermal neutron beam, which causes limited tissue damage in the absence of 10B, and by delivering 10B-enriched compounds to the cancer cells for local treatment of the undesirable cells by α-particles. BNCT using neutron beams generated in nuclear reactors has been used in clinical settings with encouraging results.2–6 Recent advances in compact neutron generators have stimulated the research on BNCT7 and are translating this therapy to hospital settings.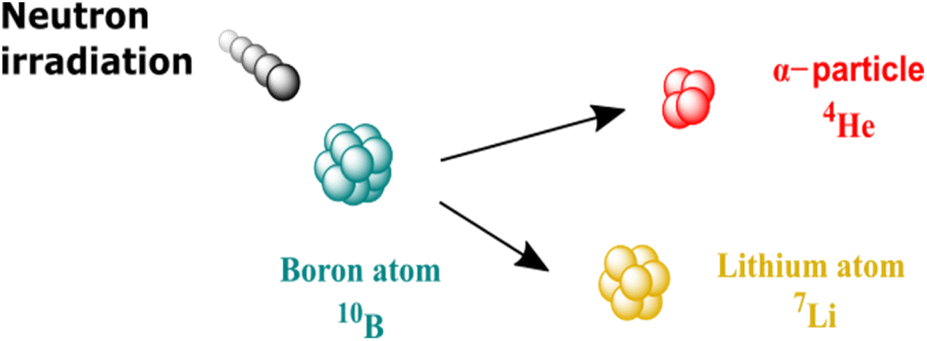 | ||
| Fig. 1 BNCT principle. | ||
Currently, two drugs, 4-boronophenylalanine (BPA), a boronic acid-modified amino acid, and sodium borocaptate (BSH), a cluster of twelve boron atoms, are used in clinical trials.2,8 In 2020, the Japanese Ministry of Health, Labour and Welfare approved BPA under the name of borofalan (10B),9 however, BSH is still only used in clinical trials. BPA is easily taken up into cells via the LAT1 amino acid transporter.10 BSH has other advantages including a high concentration of boron and high solubility in water. It has been used successfully to treat brain tumour,2,11 as BSH can be localized to a region of the disturbed blood–brain barrier (BBB).12 However, the use of BSH has been limited, mainly due to poor uptake into cells. Studies using ion microscopy showed that BSH can be taken up but was not strongly localized within the cells.13 Similar results for BSH localization were reported by using specific BSH monoclonal antibodies coupled with a dye and confocal microscopy.14 Kageji et al. also described the binding of BSH to the cell membrane.15 Therefore, we envision that one of the key issues surrounding the use of BSH is its inability to efficiently enter cells and that overcoming this problem may release the potential of this drug.
Nanoparticles provide an attractive solution to achieve efficient cellular uptake of various drugs. They can often take advantage of endocytosis mechanisms and are efficiently taken up into cells.16 Furthermore, they can be used to achieve intracellular localization of cargo molecules.16 They can be exploited to achieve tumour accumulation of cargo molecules.16 While liposomes and polymers have been extensively used as nanocarriers to date,17,18 mesoporous silica-based nanoparticles are considered an interesting platform for drug delivery due to their low toxicity and high stability in the bloodstream compared to the previously cited nanocarriers.19–21 Indeed, silica nanoparticles do not degrade easily in the bloodstream. Furthermore, silica nanoparticles can penetrate solid tumours and can thus promote efficient tumour treatment.22 From a chemical point of view, their high specific surface area provides an ideal substrate for attaching drugs and makes this material an appropriate choice as a drug carrier. Some 10B-containing silica nanoparticles have been described including mesoporous silica nanoparticles (MSN) with o-carborane impregnated into the pores,23 silica supports over which polymers modified with carborane were grafted,24,25 silica nanoparticles on which a silylated boron cluster was post-grafted26,27 and organically doped MSN on which BSH was post-grafted.28 Nevertheless, most of them have not been tested in BNCT assay.
In this paper, we design a new type of 10B-containing silica nanoparticle on which BSH is covalently grafted onto the organosilica network, thus avoiding premature release. The BSH boron cluster was grafted onto the silica network using a click thiol–ene reaction. To the best of our knowledge, it has not been used to date with B-SH compounds, thus providing a novel and effective platform for BSH grafting. BSH-BPMO has been characterized by various techniques including TEM, SEM, N2 physisorption, FTIR, and 13C and 29Si solid-state NMR. The uptake of the nanoparticles into cancer cells and tumour spheroids has been demonstrated. We developed a convenient BNCT assay to evaluate the efficacy of BSH-BPMO in BNCT and demonstrated that BSH-BPMO exhibits BNCT efficacy that is significantly improved compared to those of the two common BNCT drugs (BSH and BPA).
Experimental
Chemicals
1,2-Bis(triethoxysilyl)ethane (BTSE – Fluorochem), bis[3-(triethoxysilyl)propyl]tetrasulfide (BTSPS4 – Fluorochem), (3-aminopropyl)triethoxysilane (APTES – TCI), triethoxyvinylsilane (TCI), 2,2′-azobis[2-(2-imidazolin-2-yl)propane] dihydrochloride (AIBI – TCI), 3-(trihydroxysilyl)propyl methylphosphonate monosodium salt solution (MPTHS – Sigma Aldrich), cetyltrimethylammonium bromide (CTAB – Sigma Aldrich), Rhodamine B isothiocyanate (Funakoshi) and 10B-enriched sodium borocaptate (10BSH – KatChem) were used. 8 M sodium hydroxide solution and ethanol (99.5%) were obtained from Nacalai Tesque, ammonium nitrate was from Wako and anhydrous toluene was from Sigma Aldrich. Milli-Q water was used for the different synthesis steps.BSH-BPMO synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 20 min and washed twice with ethanol with centrifugation/sonication cycles. CTAB was removed from the pores by refluxing the particles overnight in a solution of ammonium nitrate (0.3 g) in ethanol (50 mL). The material was washed with ethanol three times (14000 rpm for 20 min), and dried completely. 120 to 150 mg of BPMO pink nanoparticles were recovered. 13C NMR (151 MHz, CP-MAS, δ, ppm): 4.7 (Si–CH2, BTSE), 10.4 (Si–CH2, BTSPS4), 15.8 (residual ethoxy), 22.4 (Si–CH2–CH2, BTSPS4), 41.8 (CH2–CH2–S, BTSPS4), 57.9 (residual ethoxy). IR (ν, cm−1): 1031 (Si–O–Si), 2897 (residual CH3, s), 2926 (CH2, as), 2977 (residual CH3, as). Zeta potential (BPMO, 25 °C, pH = 7.5): −38.0 mV.
000 rpm, 20 min) and washed three times with ethanol by centrifugation/sonication cycles. The powder was completely dried. Around 120 mg of BPMO-vinyl were recovered. 13C NMR (151 MHz, CP-MAS, δ, ppm): 4.7 (Si–CH2, BTSE), 11.1 (Si–CH2, BTSPS4), 15.8 (residual ethoxy), 22.5 (Si–CH2–CH2, BTSPS4), 41.2 (CH2–CH2–S, BTSPS4), 57.7 (residual ethoxy), 130.0 & 134.4 (CH2
000 rpm for 20 min and washed twice with ethanol with centrifugation/sonication cycles. CTAB was removed from the pores by refluxing the particles overnight in a solution of ammonium nitrate (0.3 g) in ethanol (50 mL). The material was washed with ethanol three times (14000 rpm for 20 min), and dried completely. 120 to 150 mg of BPMO pink nanoparticles were recovered. 13C NMR (151 MHz, CP-MAS, δ, ppm): 4.7 (Si–CH2, BTSE), 10.4 (Si–CH2, BTSPS4), 15.8 (residual ethoxy), 22.4 (Si–CH2–CH2, BTSPS4), 41.8 (CH2–CH2–S, BTSPS4), 57.9 (residual ethoxy). IR (ν, cm−1): 1031 (Si–O–Si), 2897 (residual CH3, s), 2926 (CH2, as), 2977 (residual CH3, as). Zeta potential (BPMO, 25 °C, pH = 7.5): −38.0 mV.
000 rpm, 20 min) and washed three times with ethanol by centrifugation/sonication cycles. The powder was completely dried. Around 120 mg of BPMO-vinyl were recovered. 13C NMR (151 MHz, CP-MAS, δ, ppm): 4.7 (Si–CH2, BTSE), 11.1 (Si–CH2, BTSPS4), 15.8 (residual ethoxy), 22.5 (Si–CH2–CH2, BTSPS4), 41.2 (CH2–CH2–S, BTSPS4), 57.7 (residual ethoxy), 130.0 & 134.4 (CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–Si & CH2CH–Si). IR (ν, cm−1): 1031 (Si–O–Si), 2896 (CH2, s), 2923 (CH2, as). Zeta potential (BPMO-vinyl, 25 °C, pH = 7.5): −26.5 mV.
000 rpm, 20 min), washed three times with ethanol with centrifugation/sonication cycles, completely dried. 100 to 110 mg of BPMO-vinyl-phos was recovered. 13C NMR (151 MHz, CP-MAS, δ, ppm): 5.1–12.2 (Si–CH2, BTSE & Si–CH2, MPTHS & Si–CH2–CH2, BTSPS4), 22.3 (Si–CH2–CH2, BTSPS4), 25.8 (Phos–CH2–CH2–), 41.2 (CH2–CH2–S, BTSPS4), 57.9 (residual ethoxy), 63.8 (Phos–CH2–), 130.8 & 134.7 (CH2CH–Si & CH2CH–Si). IR (ν, cm−1): 1032 (Si–O–Si), 2896 (CH2, s), 2924 (CH2, as). Zeta potential (BPMO-vinyl-phos, 25 °C, pH = 7.5): −50.1 mV. Phosphonated BPMO (BPMO w/o BSH) was prepared following the same protocol as for BPMO-vinyl-phos, described above. 77 mg of BPMO w/o BSH were recovered out of the initial 70 mg of BPMO. IR (ν, cm−1): 1027 (Si–O–Si), 2893 (CH2, s), 2926 (CH2, as). Zeta potential (BPMO w/o BSH, 25 °C, pH = 7.5): −47.8 mV.
000 rpm, 20 min), and washed twice with Milli-Q water and twice with ethanol. The powder was then completely dried. 36 mg of pink BSH-BPMO was recovered. 13C NMR (151 MHz, CP-MAS, δ, ppm): 5.0–11.4 (Si–CH2, BTSE & Si–CH2, MPTHS & Si–CH2–CH2, BTSPS4), 22.3 (Si–CH2–CH2, BTSPS4), 25.8 (Phos–CH2–CH2), 42.1 (CH2–CH2–S, BTSPS4 & CH2–SBSH), 58.1 (residual ethoxy), 64.1 (Phos–CH2), 130.0 & 134.5 (residual vinyl). 29Si NMR (119 MHz, δ, ppm): −57.6 (T2), −66.2 (T3). IR (ν, cm−1): 1031 (Si–O–Si), 2503 (B–H), 2896 (CH2, s), 2923 (CH2, as). Zeta potential (BSH-BPMO, 25 °C, pH = 7.5): −51.5 mV.
CH–Si & CH2CH–Si). IR (ν, cm−1): 1031 (Si–O–Si), 2896 (CH2, s), 2923 (CH2, as). Zeta potential (BPMO-vinyl, 25 °C, pH = 7.5): −26.5 mV.
000 rpm, 20 min), washed three times with ethanol with centrifugation/sonication cycles, completely dried. 100 to 110 mg of BPMO-vinyl-phos was recovered. 13C NMR (151 MHz, CP-MAS, δ, ppm): 5.1–12.2 (Si–CH2, BTSE & Si–CH2, MPTHS & Si–CH2–CH2, BTSPS4), 22.3 (Si–CH2–CH2, BTSPS4), 25.8 (Phos–CH2–CH2–), 41.2 (CH2–CH2–S, BTSPS4), 57.9 (residual ethoxy), 63.8 (Phos–CH2–), 130.8 & 134.7 (CH2CH–Si & CH2CH–Si). IR (ν, cm−1): 1032 (Si–O–Si), 2896 (CH2, s), 2924 (CH2, as). Zeta potential (BPMO-vinyl-phos, 25 °C, pH = 7.5): −50.1 mV. Phosphonated BPMO (BPMO w/o BSH) was prepared following the same protocol as for BPMO-vinyl-phos, described above. 77 mg of BPMO w/o BSH were recovered out of the initial 70 mg of BPMO. IR (ν, cm−1): 1027 (Si–O–Si), 2893 (CH2, s), 2926 (CH2, as). Zeta potential (BPMO w/o BSH, 25 °C, pH = 7.5): −47.8 mV.
000 rpm, 20 min), and washed twice with Milli-Q water and twice with ethanol. The powder was then completely dried. 36 mg of pink BSH-BPMO was recovered. 13C NMR (151 MHz, CP-MAS, δ, ppm): 5.0–11.4 (Si–CH2, BTSE & Si–CH2, MPTHS & Si–CH2–CH2, BTSPS4), 22.3 (Si–CH2–CH2, BTSPS4), 25.8 (Phos–CH2–CH2), 42.1 (CH2–CH2–S, BTSPS4 & CH2–SBSH), 58.1 (residual ethoxy), 64.1 (Phos–CH2), 130.0 & 134.5 (residual vinyl). 29Si NMR (119 MHz, δ, ppm): −57.6 (T2), −66.2 (T3). IR (ν, cm−1): 1031 (Si–O–Si), 2503 (B–H), 2896 (CH2, s), 2923 (CH2, as). Zeta potential (BSH-BPMO, 25 °C, pH = 7.5): −51.5 mV.
Characterization
Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images were taken on a JEOL JSM-75FCT and a JEOL JEM-2200FS microscope, respectively. Nitrogen adsorption isotherms were obtained at 77 K on a Belsorp mini II apparatus. Samples were dried at 80 °C for 17 h under vacuum. The specific surface areas were determined using the BET model in the relative pressure range between 0.05 and 0.25, taking 0.162 nm2 as the cross-sectional area. Pore size diameter was estimated by the BJH (Barrett–Joyner–Halenda) method on the desorption branch. Powder X-ray diffraction (XRD) patterns were collected on an X-ray Rigaku SmartLab X-ray diffractometer with Cu Kα radiation from 1.2 to 15°. 13C and 29Si CP-MAS NMR spectra were recorded on a 600 MHz solid-state NMR spectrometer (JNM-ECZ600R, JEOL) at a 20 kHz spinning rate. Fourier-transformed infrared spectra (FTIR) were obtained on an attenuated total reflectance (ATR) equipped Thermo Scientific Nicolet 5700 apparatus. Spectra were corrected with a classical “Extended ATR correction” function to correct the wavelength dependency of penetration depth in the sample. The residual CO2 band was set to zero. Raman spectra were obtained on a Horiba HR800 apparatus. The powders were analysed at a magnification of ×50, using 785 nm laser excitation and an exposure time of 30 s. Materials prepared without Rhodamine B were used for Raman experiments to avoid the saturation from the luminescent background generated by the rhodamine upon exposure to the 785 nm excitation source. The hydrodynamic radius and zeta potential of the particles were determined on a Malvern Zetasizer μV and a Malvern Zetasizer Nano-ZS at 25 °C. DLS sizes were retained using the mean value in number. An ICPE-9000 Shimadzu machine was used for the inductively coupled plasma atomic emission spectroscopy (ICP-AES) analysis. Nanoparticles (2 mg) were degraded in a mixture of nitric acid (1.9 mL) in water (8.1 mL) for 3 days prior to boron content determination by ICP.Cytotoxicity
OVCAR8 cells were seeded in 96-well plates with 2 × 104 cells in 190 μL of RPMI 1640 medium (Nacalai) per well. They were incubated at 37 °C in humidified 5% CO2 for 24 h, then 10 μL of BSH-BPMO suspension in sterilized water (0, 25, 50, 100, 200 μg mL−1) was added to the medium. After 24 h of incubation, lactate dehydrogenase (LDH, Dojindo Cytotoxicity LDH Assay Kit-WST) assay was performed and the absorbance at 490 nm was measured and compared to that of a lysed-cells control, according to the standard procedure.Tumour spheroid preparation
Tumour spheroids were prepared using human ovarian cancer cells OVCAR8 expressing green fluorescent protein (GFP). Cells were grown in RPMI 1640 medium (Nacalai) supplemented with 10% heat-inactivated FBS and 1% penicillin/streptomycin. 5.0 × 103 cells were inoculated in each well of a PrimeSurface 96U culture plate (MS-9096U, Sumitomo Bakelite Co., LTD, Japan). Cells were cultured at 37 °C in humidified CO2 for seven days with the medium being exchanged every two days. The spheroid diameter was around 200 μm.Cellular and spheroid uptake of BSH-BPMO for imaging
5.0 × 103 OVCAR8 cells were seeded in a 35 mmφ glass-bottom culture dish with 200 μL medium and incubated overnight at 37 °C in humidified 5% CO2. After 24 h, 10 μL of BSH-BPMO suspension in sterilized water (1 and 0.5 mg mL−1) was added to each dish, corresponding to 10 and 5 μg of BSH-BPMO. The cells were incubated for another 24 h, then washed three times with ice-cold PBS and fixed overnight with 4% paraformaldehyde (200 μL) at 4 °C. Hoechst 33258 dye (200 μL of a 1:1000 diluted solution) was used to stain the cell nucleus for 30 min in the dark and at room temperature prior to two other washes in PBS. After washing, the cells were kept in 200 μL of PBS and observed by confocal microscopy.
For the spheroid uptake, spheroids were prepared according to the protocol previously described. Briefly, 10 μL of BSH-BPMO suspension in sterilized water (2.5, 0.25 mg mL−1), corresponding to 25 and 2.5 μg of BSH-BPMO per spheroid, was added and the spheroids were incubated for 24 h at 37 °C in humidified 5% CO2. After incubation, spheroids were washed three times with ice-cold PBS and fixed overnight with 4% paraformaldehyde at 4 °C. Spheroids were then treated with 99.8% methanol to increase the permeability for 30 min at −80 °C. After three cycles of washing with ice-cold PBS, Hoechst staining was performed by treating with Hoechst 33258 solution dye (200 μL of a 1:1000 diluted solution) for 30 min in the dark prior to confocal microscopy. Confocal laser microscopy images were taken on a Nikon A1R confocal laser microscope.
Cellular uptake test by ICP
OVCAR8 cells (5.0 × 105) were seeded in a 10 cm diameter culture dish with 10 mL RPMI 1640 medium (Nacalai) and incubated at 37 °C in humidified 5% CO2 for five days. The medium was exchanged every two days for optimum cell growth. After five days, the medium was removed and replaced by 9.5 mL of fresh medium and 500 μL of boron-containing solution. The conditions were set to maintain a constant quantity of boron (64 μg) per dish. The solutions used were 6.4 mg mL−1 BSH-BPMO, 224 μg mL−1 free BSH and 2.67 mg mL−1 free BPA (BPA was prepared with 2.76 mg mL−1 fructose, corresponding to 1.2 eq. of BPA). The cells were incubated for 2 h or 24 h prior to removal of the medium. Cells were detached from the dish with trypsin and washed three times with fresh medium using centrifugation cycles of 5 min at 1500 rpm. Cell counting showed the presence of 1.0 × 107 cells per dish. For ICP analyses, cells were collected and immediately suspended using 2 mL of a mixture of 60% perchloric acid:30% hydrogen peroxide in a 1:2 proportion at 75 °C for 12 h. The mix was filtered with a 220 nm cut-off membrane and analysed by ICP-AES.
Neutron exposure experiments (BNCT)
Spheroids were prepared according to the protocol previously described and incubated with 10 μL of the following solutions: BSH-BPMO (500 μg mL−1), BPMO w/o BSH (500 μg mL−1), free BSH (15.6 μg mL−1), and free BPA (186 μg mL−1, prepared with 1.2 eq. of fructose i.e. 192 μg mL−1) corresponding to a constant 10B quantity of 89 ng per spheroid. For each condition, six spheroids were assigned (three for irradiation, and three for no irradiation). All tumour spheroids were incubated for 24 h at 37 °C in humidified 5% CO2 and washed three times with culture medium to remove the free BSH, BPA and nanoparticles that were not taken up. Spheroids were neutron-irradiated for 1 h at 1 MW with a thermal neutron beam at the Kyoto University Research Reactor (KUR), Heavy Water Neutron Irradiation Facility.30 Each spheroid received 4 × 1012 neutrons cm−2. After the irradiation, the spheroids were incubated for another three days at 37 °C in humidified 5% CO2. Samples were fixed overnight with 4% paraformaldehyde at 4 °C and stained with Hoechst 33258 solution dye (1:1000 diluted solution) for 30 min in the dark. Finally, the samples were observed by confocal microscopy on a Nikon A1R confocal laser microscope. The spheroid size was calculated with Image-J software (NIH) for the three spheroids of each condition set. The percentages were calculated by dividing the surface of the spheroid by the average size of the corresponding non-irradiated spheroids. Statistical data are presented in Table S2.† Error bars indicate standard errors. P-values were calculated for each condition in comparison to the no-loading condition.
Results and discussion
BSH-BPMO synthesis and characterization
BSH-BPMO nanoparticles were synthesized in a four-step reaction (Fig. 2). First, periodic mesoporous organosilica nanoparticles exhibiting high specific surface area were prepared. The surface of these particles was modified with vinyl groups to introduce reactive moieties for boron compound grafting. The dispersion of the particles in aqueous solutions was ensured by modification with phosphonate groups. Finally, BSH was grafted using a thiol–ene reaction between the vinyl moiety and the BSH in water. In order to carry out comparative studies, a phosphonated BPMO (BPMO w/o BSH) control was prepared by post-grafting the phosphonate silylated precursor on the BPMO (Fig. S1†).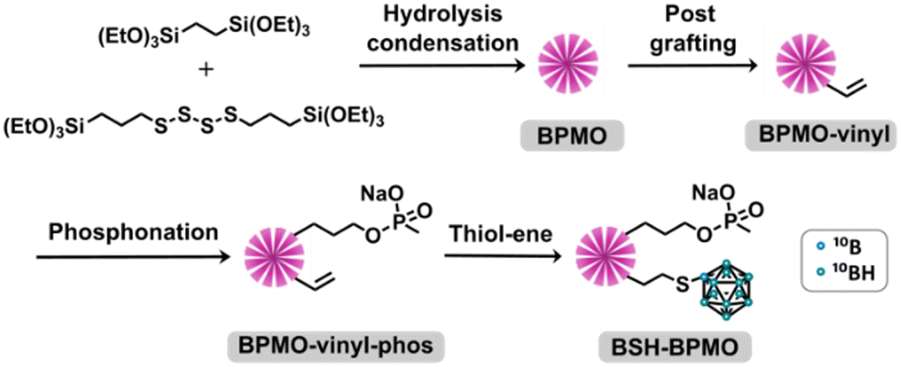 | ||
| Fig. 2 BSH-BPMO synthetic pathway. | ||
The periodic mesoporous organosilica nanoparticles used in the article (BPMO) were first synthesized by adapting the protocol previously described by our group,29 using 1,2-bis(triethoxysilyl)ethane (BTSE) and bis[3-(triethoxysilyl)propyl] tetrasulfide (BTSPS4) silica precursors. Rhodamine B isothiocyanate was reacted with (3-aminopropyl) triethoxysilane (APTES). The resulting Rhodamine B silylated derivative was incorporated into the BPMO synthesis, thus allowing easy observation of the nanoparticles by fluorescence microscopy at 561 nm (red). Images obtained by SEM and TEM show the formation of nanoparticles with an average diameter of 300 nm (Fig. 3a and b). The average hydrodynamic diameter of 357 nm, shown by DLS (Table S1†), is consistent with the microscopy observations. The nanoparticles were synthesized with a diameter of 300 nm to obtain an optimal specific surface area and pore organisation. Indeed, the N2-physisorption isotherm obtained (Fig. 3c) is a typical type I/IV isotherm. The low range of absorption at intermediate relative pressure, and parallel adsorption and desorption branches indicate a well-organised mesoporous network with low pore size dispersity. The BET specific surface area was calculated at 1200 m2 g−1 and the pore size diameter was estimated to be 2.5 nm (Fig. 3d). The organised pore network can also be observed in TEM (Fig. 3b) with a periodicity estimated to be around 4 nm. In Fig. S2,† X-ray diffraction (XRD) measurements confirm an organised network (with a distorted mesophase) as demonstrated by the two peaks observed for 2θ = 1.99 and 3.04°.31 The periodicity calculated from XRD data is 5.1 nm. Smaller nanoparticles (50, 100 and 250 nm) can be obtained at the expense of pore organisation.32 The incorporation of both BTSE and BTSPS4 into the network was evidenced by 13C solid-state NMR and Raman spectroscopy. 13C solid-state NMR (Fig. 4a) exhibits a very strong signal at 4.7 ppm from the CH2 of the BTSE. Signals at 10.4, 22.4 and 41.8 ppm are attributed to the CH2 moieties of the condensed BTSPS4, thus proving the successful incorporation of both precursors into the silsesquioxane network. The presence of the tetrasulfide is also confirmed by Raman spectroscopy (Fig. S3†) with the C–S stretching vibration observed at 632 cm−1.33
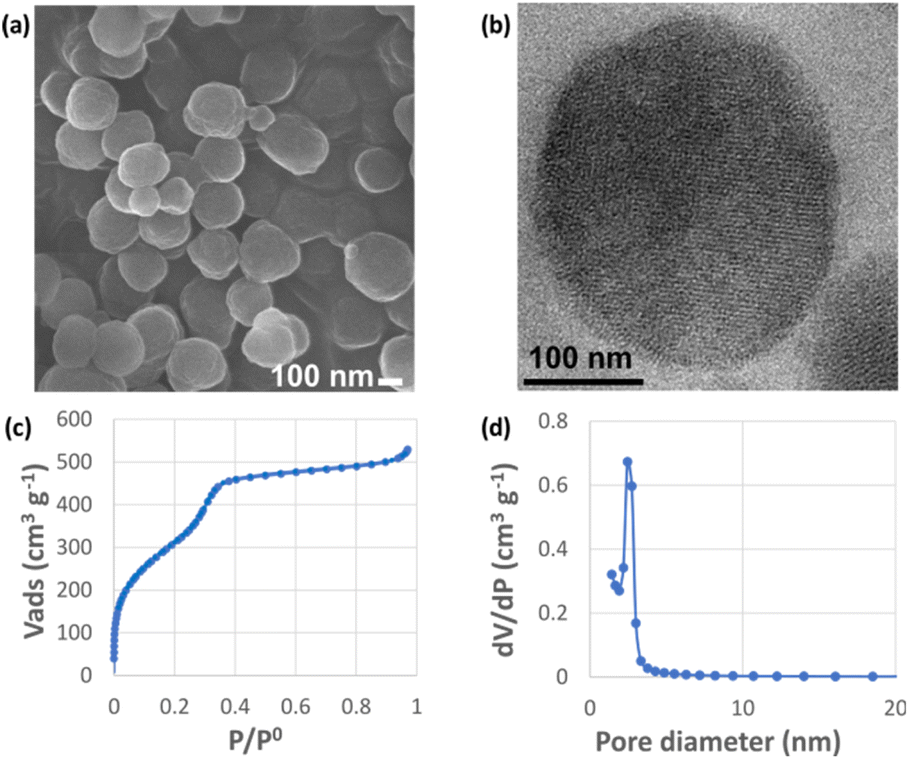 | ||
| Fig. 3 BPMO textural characterization: (a) SEM and (b) TEM images; (c) N2 physisorption isotherm; (d) BJH pore size distribution. | ||
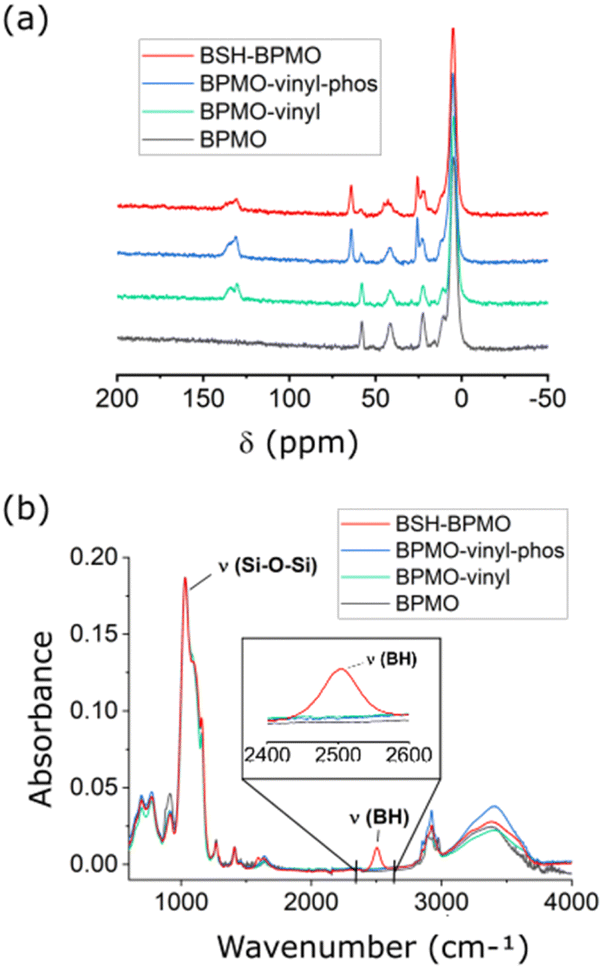 | ||
| Fig. 4 Chemical characterization of BPMO nanoparticles: (a) 13C solid-state NMR spectra of BPMO at different synthetic steps; (b) FTIR spectra. Note: The 13C NMR signal around 57 ppm arises from the residual ethoxy group. | ||
The BPMO nanoparticles were then post-grafted with triethoxyvinylsilane and characterized. The 13C solid-state NMR spectrum shows new signals at 130.0 and 134.4 ppm (in green in Fig. 4a). These signals are typical of the chemical shift expected for vinyl derivatives. Since the step described above tends to make the nanoparticles hydrophobic and lower their capacity to disperse in water, 3-(trihydroxysilyl)propyl methylphosphonate monosodium salt solution (MPTHS) was grafted (BPMO-vinyl-phos, Fig. 2). The 13C solid-state NMR exhibits two signals arising from the phosphonate moiety at 63.8 and 25.8 ppm. These signals can be attributed to the CH2 in the α- and β-position of the phosphonate while the CH2 near the silicon atom can be encountered in the cluster between 5.1 and 12.2 ppm. The significant decrease of the zeta potential value following phosphonation for BPMO-vinyl-phos and BPMO w/o BSH is presented in Table S1†.
The final synthetic step, a click thiol–ene reaction, was selected due to the high reaction yield (close to being quantitative in solution), the stability of the resulting covalent bond and the availability of vinyl derivatives.34 The reaction has been used with C–SH compounds,35 however, thiol–ene reactions involving B–SH compounds have not been reported. Hence, the most common reactions of BSH grafting rely on nucleophilic substitution over halogenated compounds, disulfide–thiol exchange or addition over maleimide moieties.28,36–41 The thiol–ene reaction is usually triggered by azobisisobutyronitrile (AIBN), which is soluble in organic solvents (toluene, chloroform, etc.). BSH is insoluble in these solvents and thus, one of the limitations of the use of this reaction with BSH is the necessity of carrying it out in solvents in which BSH is soluble. As the thiol–ene reaction can also occur in water,42 the reaction was thus performed with a water-soluble radical initiator: 2,2′-azobis[2-(2-imidazolin-2-yl)propane] dihydrochloride (AIBI). The SEM images (Fig. S4(a)†) and the DLS mean size of 336 nm (Table S1†) confirmed the presence of nanoparticles. 29Si solid-state NMR exhibited the typical features of the organosilica network with the T2 and T3 sites visible at respectively −57.6 and −66.2 ppm (Fig. S5†). In addition, the significant decrease of the vinylic signal intensity upon thiol–ene reaction (in red, Fig. 4a) is consistent with the successful grafting of BSH. The CH2 groups generated by the hydrothiolation reaction can be encountered as a cluster, together with the aliphatic signals of BTSE, BTSPS4 and MPTHS. BSH grafting was confirmed by FTIR which exhibits a band at 2503 cm−1, characteristic of the B–H stretching vibration (Fig. 4b).43,44 Boron content of 1.4 to 2% was measured by ICP. Similar to the other phosphonated nanoparticles that exhibit negative zeta potential values, the BSH-BPMO displays a zeta potential of −51.5 eV (Table S1 and Fig. S4(b)†). Several cycles of washing and dialysis over 72 h did not show any drug release by ICP or FTIR (ESI Part I and Fig. S6†). The boron content of BSH-BPMO is similar to that of other comparable materials.26,28,29 Compared with boron nitride45 or boron carbides,46,47 the boron content of BSH-BPMO is lower. However, boron nitride and boron carbides are fully inorganic compounds with restricted elemental composition. In contrast, our nanoparticle approach enables organic modification of the network or the surface to tune the properties. Indeed, Rhodamine B has been integrated into the network to make imaging possible and the surface was modified with phosphonate to enhance aqueous dispersion and cellular uptake. Due to silica particles being hybrid materials, they endow the nanocarrier with high blood stream stability, low toxicity and easy functionalization.19–21
Efficient uptake and perinuclear localization of BSH-BPMO in cancer cells
In vitro experiments were carried out using ovarian cancer cells (OVCAR8). Ovarian tumours express LAT1 at a high level,48 and thus ovarian cancer is a potential application of BNCT using BPA. Other studies using carborane derivatives in ovarian cancer were reported,49,50 demonstrating the interest in BNCT for ovarian cancers. Cytotoxicity of the BSH-BPMO nanoparticles was thus tested by lactate dehydrogenase (LDH) assay which is used as a cell membrane damage marker in ovarian cancer cells (OVCAR8). The BSH-BPMO nanoparticles were incubated with varying concentrations for one day at 37 °C prior to analysis. As a control, lysed cells were used. As can be seen in Fig. S7,† the LDH value was negligible at concentrations below 100 μg mL−1. Thus, BSH-BPMO is not toxic at the concentrations used for all the experiments described in this work.Cellular uptake of BSH-BPMO was examined by incubating ovarian cancer OVCAR8 cells with BSH-BPMO for 24 h, washing three times with PBS and then examining by confocal microscopy. The results shown in Fig. 5 were obtained by observing the nanoparticles at 561 nm (red) and the GFP-expressed OVCAR8 cells at 488 nm (green). Hoechst dye was used to stain the cell nucleus observed at 405 nm (blue). Importantly, the nanoparticles are taken up into OVCAR8 cells and localized in the perinuclear region, just outside the nucleus. Fig. S8† shows confocal microscopy images which demonstrate efficient uptake of the BSH-BPMO into the OVCAR8 cells in all focal planes and demonstrated the overlap of the Rhodamine B signal of the nanoparticles with the green signal of the GFP-marked cell. The perinuclear localization of the BSH-BPMO also highlights the importance of the nanocarriers, taken up by endocytosis,51,52 compared with free BSH which is taken up in limited quantity and accumulates on the cell membrane.15,53
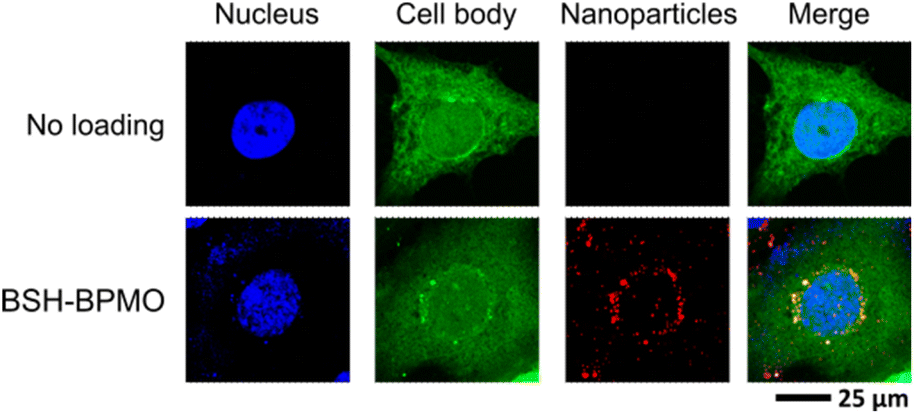 | ||
| Fig. 5 Cellular uptake image of BSH-BPMO in OVCAR8 cancer cells: Hoechst-dyed nuclei observed at 405 nm, GFP-expressed OVCAR8 cell at 488 nm and Rhodamine B containing nanoparticles at 561 nm. | ||
Boron uptake measurements in cancer cells
To quantify the boron uptake of the different drugs in OVCAR8 cancer cells, dishes of 1.0 × 107 cells were incubated with the same quantity of boron (64 μg of boron for BSH-BPMO, BSH or BPA prepared with fructose). Cells were washed at 2 and 24 h after incubation and prepared for ICP measurement. The results presented in Table 1 show that boron uptake was significantly higher for BSH-BPMO than with BSH or BPA. Indeed, boron uptake was undetected with BSH suggesting that the boron uptake was below the detection threshold. This observation is likely due to low BSH uptake and the washing step removing the extracellular BSH.15,53 In contrast, BSH-BPMO incubated cells exhibit significant quantities of boron at both incubation times. Furthermore, measurements evidence that the quantity of boron taken up via BSH-BPMO increases with the incubation time: 3.62 ± 0.09% and 19.15 ± 0.39% after 2 and 24 h respectively. Finally, with BPA, the proportion of boron taken up was measured to be 0.33 ± 0.02% after 24 h of incubation. This value is here again significantly lower than those obtained from BSH-BPMO and can be explained by a lower uptake or rapid exchange via the LAT1 amino acid transporter,10,54 thus preventing durable accumulation of BPA in the cell during the incubation (2 or 24 h) and washing period (30–40 min). The uptake of BSH is greatly improved by the use of the BPMO nanocarrier which enables the BSH contained in the nanoparticles to pass the cell barrier via endocytosis,51,52 thus resulting in BSH-BPMO exhibiting a significantly larger boron uptake than both BSH and BPA.Uptake and uniform distribution of BSH-BPMO in tumour spheroids
Due to their 3D structure, tumour spheroids are representative of tumours growing in vivo, especially in terms of diffusion of the particles.55,56 A further advantage of using spheroids is that the model can be extended in the future to tumour organoids that contain not only cancer cells but also fibroblasts and macrophages. Spheroid uptake was tested in spheroids prepared by growing OVCAR8 cells expressing GFP on a 96-well round bottom plate with low attachment coating. The spheroids prepared had a diameter of around 0.2 mm and contained 105 cells on average. Spheroids were incubated with BSH-BPMO for 24 h, washed three times with PBS and examined by confocal microscopy. The overlap of the signals from the nanoparticles (red) with the GFP signal from the cell (green) at each focal plane (Fig. 6 and S9†) proves that BSH-BPMO is efficiently taken up in the spheroid. Indeed, the nanoparticle signal is detected throughout the spheroid confirming the uniform distribution of the BSH-BPMO including the core (Fig. S9†). The uptake of BSH-BPMO was concentration-dependent (Fig. 6 and S10†). The distribution of silica nanoparticles in spheroids has been studied by Pratiwi et al.22 who demonstrated that it depends on several parameters including the surface charge, the size, the stability, etc. BSH-BPMO presents a structure which seems favourable for its diffusion in the spheroids.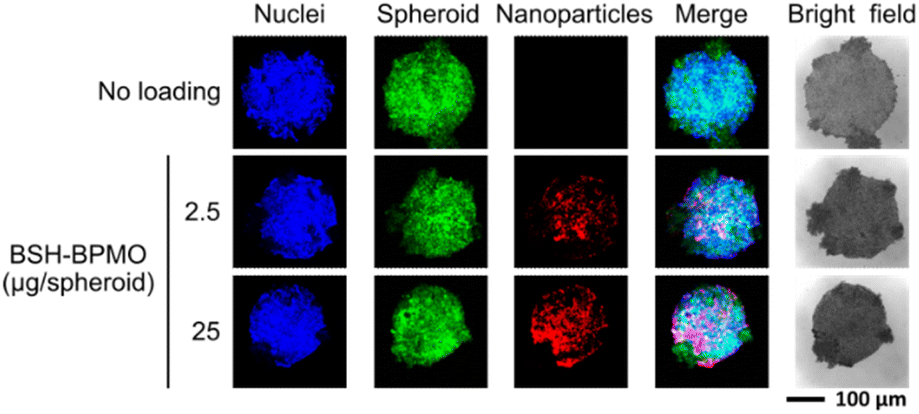 | ||
| Fig. 6 BSH-BPMO uptake in OVCAR8 spheroids with 104 cells: Hoechst-dyed nuclei observed at 405 nm, GFP-expressed OVCAR8 cell at 488 nm and Rhodamine B containing nanoparticles at 561 nm. | ||
Complete destruction of the tumour spheroids loaded with BSH-BPMO upon neutron exposure
Three-dimensional tumour spheroids of around 0.2 mm diameter were incubated with BSH-BPMO. For comparison, spheroids were also incubated with free BSH or free BPA (prepared with fructose), two drugs commonly used for BNCT.2,4,10 Additional spheroids were incubated with either no compound or BPMO w/o BSH (no boron) to represent boron-free controls. After 24 h, all spheroids were washed three times in PBS. Two sets of spheroids were prepared, of which one was submitted to neutron irradiation of 1 MW for 1 h, while the second set received no irradiation. The irradiation was done with neutrons whose energy was reduced to “thermal” neutrons at the Kyoto University Research Reactor in Kumatori, Japan (Fig. 7). Each spheroid of the irradiation set received 4 × 1012 neutrons cm−2. The spheroids were then incubated at 37 °C for three days after irradiation. The confocal microscopy images of the irradiated spheroids are shown in Fig. 8a. For comparison, results without irradiation are presented in Fig. S11.† The size of spheroids as a percentage of the non-irradiated spheroid size is presented in Fig. 8b.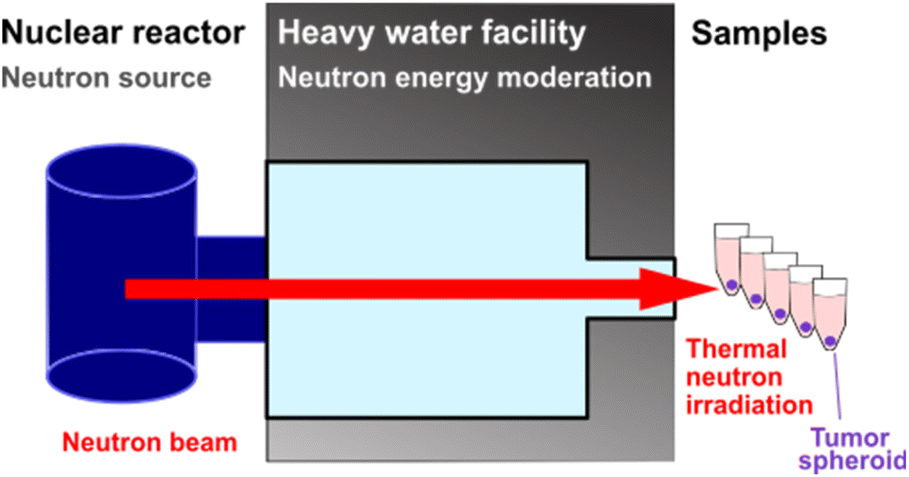 | ||
| Fig. 7 Thermal neutron irradiation setup. | ||
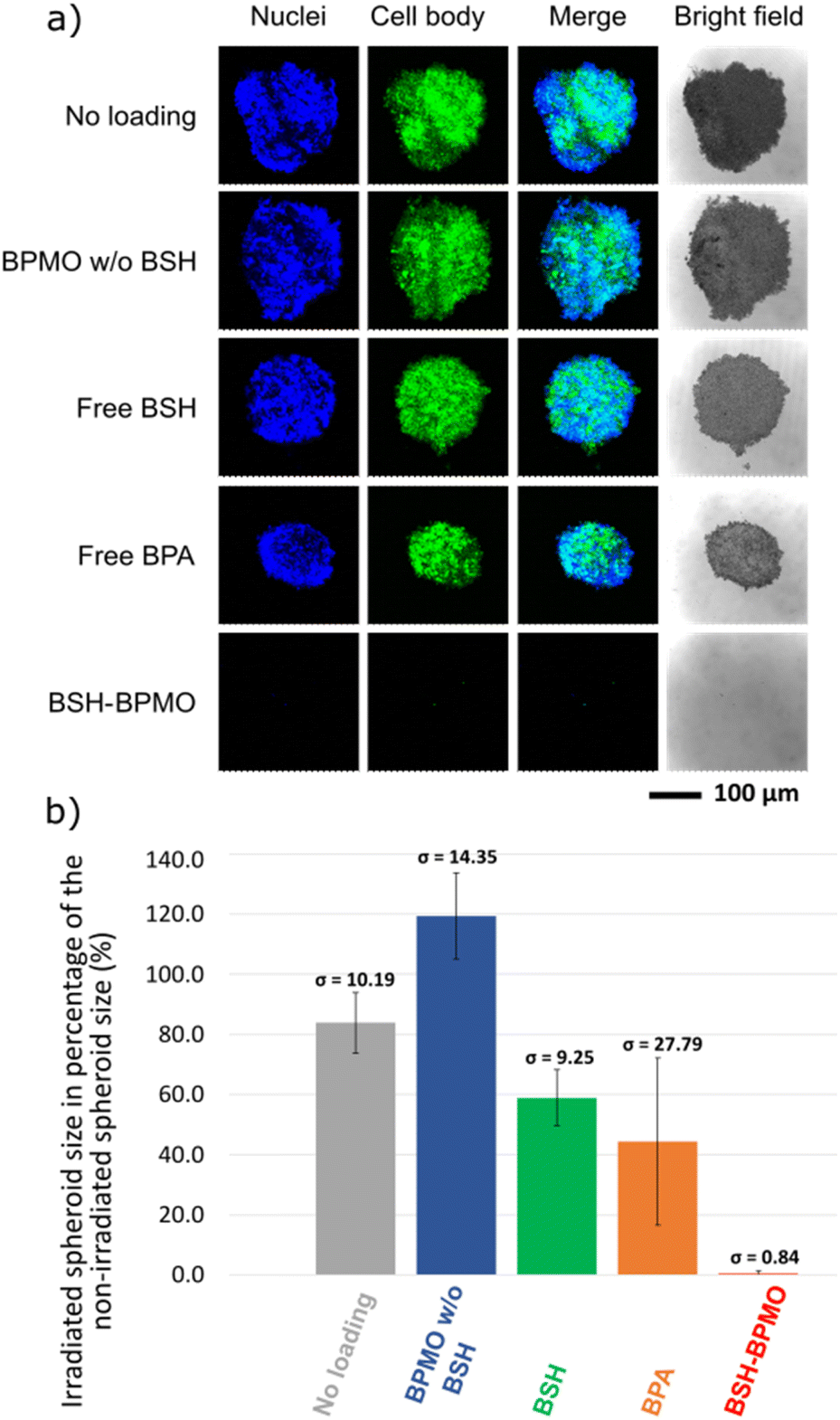 | ||
| Fig. 8 Neutron exposure results of (a) OVCAR8 spheroids observed by confocal microscopy after 1 h of neutron irradiation and three days of incubation: Hoechst-dyed nuclei observed at 405 nm and GFP modified OVCAR8 cell at 488 nm. (b) Spheroid shrinkage as a percentage of the non-irradiated spheroids control; error bars indicate standard deviation. All p-values were determined to be lower than 0.05 except for the BPA set where the p-value was calculated to be 0.052. Despite the value for BPA being slightly above the statistical threshold, the tumour loaded with BPA is visibly shrunk upon irradiation. | ||
As can be seen in Fig. 8, irradiation in the presence of BSH-BPMO shows a dramatic effect on spheroid survival with the complete destruction of the spheroids upon irradiation. Indeed, only residual GFP signals can be observed by confocal microscopy, on average 0.5% of the non-irradiated tumour size. In comparison, after irradiation, spheroids incubated with free BSH show minor shrinkage (corresponding to a spheroid size after irradiation of 59% that of the spheroid size without irradiation). The spheroids incubated with free BPA exhibit a size after irradiation corresponding to 44% of the reference non-irradiated spheroid. As already demonstrated by previous studies,2,4,10 BPA efficiently leads to tumour shrinkage and can give better outcomes than free BSH due to its greater uptake compared to BSH. Nevertheless, BSH-BPMO is significantly more efficient than both free BSH and free BPA. Indeed, it is the only drug leading to the complete degradation of the spheroids. In contrast, the examination of the “no loading” conditions with (Fig. 8) and without irradiation (Fig. S11†) evidenced a minor effect of the irradiation in the absence of boron. Similarly, BPMO w/o BSH, which does not contain boron, does not cause a size decrease of the tumour upon irradiation. The striking difference between the two commonly used drugs and BSH-BPMO may reflect the differences in drug uptake. According to the ICP uptake data mentioned above, at 24 h (incubation time used for the spheroid exposure experiment), the quantity of boron taken up via the BSH-BPMO is 60-fold higher than the quantity taken up via the free BPA at the concentration tested. These ICP uptake results (Table 1) are consistent with the complete destruction effect of the BSH-BPMO and enhanced efficacy compared to the two traditional drugs (BSH and BPA). As demonstrated previously, free BSH suffers from low cellular uptake and BPA exhibits poor cell retention due to the exchange via LAT1 transporters.10,54 In contrast, BSH-BPMO accumulates in the perinuclear region once taken up. Indeed, during the uptake process, the nanoparticles are taken up by endocytosis51,52 and localize near the nucleus (Fig. 5). This results in a facilitated uptake and high retention of the BSH in the cells.
Overall, BSH-BPMO appeared to be a better platform than the two most common drugs (BSH and BPA) currently used in BNCT clinical trials. Its capacity to favour cellular uptake and to be retained in the cell, thus preventing the rapid clearance phenomenon observed for molecular compounds, indicates that it is a potential platform for BNCT. However, further synthetic optimization studies are also being carried out to improve the boron content in BSH-BPMO and reduce the quantity of nanoparticles injected in mice. Indeed, 20–30 μg of boron per gram tumour are required to bring about the BNCT effect,57 which could require a significant concentration of nanoparticles and may lead to possible toxicity. The appropriate formulation of BSH-BPMO is also being investigated (size, surface modification, etc.).58 The results of these studies will be presented in future articles.
Conclusions
Periodic mesoporous organosilica nanoparticles containing BSH as a boronated agent have been synthesized. The synthetic procedure involves post-functionalization of the silica nanoparticles by vinyl groups followed by a thiol–ene reaction. This strategy offers a quick, cost-effective and efficient platform for covalently grafting BSH into nanocarriers. By this means, BSH-BPMO nanoparticles containing 1.4 to 2% boron have been obtained with no premature release observed over three days. The nanoparticles were efficiently taken up by OVCAR8 cancer cells and localized in the perinuclear region. ICP measurements of the boron uptake show enhanced uptake of boron via BSH-BPMO compared to BPA and BSH. The use of the BPMO nanocarrier efficiently overcomes one of the main issues of BSH by significantly enhancing the BSH uptake. Nanoparticles were also proved to be taken up into tumour spheroids and diffused throughout the spheroids. A comparative BNCT experiment between BSH-BPMO, BSH and BPA evidenced a significant improvement in the BNCT efficacy of BSH-BPMO compared to BSH and BPA. This correlates with the significantly enhanced uptake of BSH via the nanocarrier.BSH-BPMO gave encouraging results for the in vitro study in OVCAR8 cancer cells. The significant enhancement of BSH cellular uptake mediated by the nanocarrier opens a door for the use of BSH-derived drugs (until now often focused on head and neck cancers) to treat a variety of cancer types including those with low LAT1 receptor expression. The appropriate formulation of BSH-BPMO is currently being tested in mouse models.
Author contributions
ML, FT and KMa conceived the project. FT and KMa supervised the project. ML designed, synthesized, and characterized the nanoparticles and wrote the first draft of the manuscript. AK, KMa and ML carried out the cell and spheroid experiments. KMa, YH, MS and ML performed the neutron irradiation. All authors discussed the results and contributed to the final manuscript.Conflicts of interest
This work was partially supported by the Kyoto University–TAE Life Sciences collaboration program.Acknowledgements
This work was supported by JSPS KAKENHI under grant number JP15K21764. We would like to thank Satoshi Horike and Nao Horike (Kyoto University) for help with the solid-state NMR characterization. The Kyoto University iCeMS Analysis Center is gratefully acknowledged for its support in various physical characterizations of nanoparticles. We are grateful to Tomoko Inose for her help with Raman measurements.Notes and references
- R. F. Barth, J. A. Coderre, M. G. H. Vicente and T. E. Blue, Clin. Cancer Res., 2005, 11, 3987–4002 CrossRef CAS PubMed.
- T. D. Malouff, D. S. Seneviratne, D. K. Ebner, W. C. Stross, M. R. Waddle, D. M. Trifiletti and S. Krishnan, Front. Oncol., 2021, 11, 601820 CrossRef PubMed.
- A. Monti Hughes, Expert Rev. Mol. Med., 2022, 24, 1–11 CrossRef PubMed.
- H. He, J. Li, P. Jiang, S. Tian, H. Wang, R. Fan, J. Liu, Y. Yang, Z. Liu and J. Wang, Radiat. Oncol., 2021, 16, 1–8 CrossRef PubMed.
- M. A. Dymova, S. Y. Taskaev, V. A. Richter and E. V. Kuligina, Cancer Commun., 2020, 40, 406–421 CrossRef PubMed.
- M. Suzuki, Int. J. Clin. Oncol., 2020, 25, 43–50 CrossRef PubMed.
- R. F. Barth and J. C. Grecula, Appl. Radiat. Isot., 2020, 160, 109029 CrossRef CAS PubMed.
- S. Wang, Z. Zhang, L. Miao and Y. Li, Front. Oncol., 2022, 12, 1198 Search PubMed.
- H. Kanno, H. Nagata, A. Ishiguro, S. Tsuzuranuki, S. Nakano, T. Nonaka, K. Kiyohara, T. Kimura, A. Sugawara, Y. Okazaki, S. Takae, T. Nakabayashi, H. Arai and H. Suzuki, Oncologist, 2021, 26, e1250–e1255 CrossRef CAS PubMed.
- M. Lamba, A. Goswami and A. Bandyopadhyay, Chem. Commun., 2021, 57, 827–839 RSC.
- K. Hu, Z. Yang, L. Zhang, L. Xie, L. Wang, H. Xu, L. Josephson, S. H. Liang and M.-R. Zhang, Coord. Chem. Rev., 2020, 405, 213139 CrossRef CAS.
- H. Fukuda, Cells, 2021, 10, 2883 CrossRef CAS PubMed.
- X. Zha, W. A. Ausserer and G. H. Morrison, Cancer Res., 1992, 52, 5219–5222 CAS.
- A. Doi, S. Kawabata, K. Iida, K. Yokoyama, Y. Kajimoto, T. Kuroiwa, T. Shirakawa, M. Kirihata, S. Kasaoka, K. Maruyama, H. Kumada, Y. Sakurai, S. I. Masunaga, K. Ono and S. I. Miyatake, J. Neuro-Oncol., 2008, 87, 287–294 CrossRef CAS PubMed.
- T. Kageji, S. Nagahiro, B. Otersen, D. Gabel, M. Nakaichi and Y. Nakagawa, J. Neuro-Oncol., 2002, 59, 135–142 CrossRef PubMed.
- M. Sousa De Almeida, E. Susnik, B. Drasler, P. Taladriz-Blanco, A. Petri-Fink and B. Rothen-Rutishauser, Chem. Soc. Rev., 2021, 50, 5397–5434 RSC.
- J. Shi, P. W. Kantoff, R. Wooster and O. C. Farokhzad, Nat. Rev. Cancer, 2017, 17, 20–37 CrossRef CAS PubMed.
- D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751–760 CrossRef CAS PubMed.
- S. S. Park, M. Santha Moorthy and C.-S. Ha, NPG Asia Mater., 2014, 6, 1–21 Search PubMed.
- J. G. Croissant, X. Cattoën, M. Wong Chi Man, J.-O. Durand and N. M. Khashab, Nanoscale, 2015, 7, 20318–20334 RSC.
- S. Chinnathambi and F. Tamanoi, Pharmaceutics, 2020, 12, 890 CrossRef CAS PubMed.
- F. W. Pratiwi, C. C. Peng, S. H. Wu, C. W. Kuo, C. Y. Mou, Y. C. Tung and P. Chen, Biomedicines, 2021, 9, 1–14 Search PubMed.
- C.-H. Lai, N.-C. Lai, Y.-J. Chuang, F.-I. Chou, C.-M. Yang and C.-C. Lin, Nanoscale, 2013, 5, 9412–9418 RSC.
- I. V. Korolkov, K. Ludzik, A. L. Kozlovskiy, M. S. Fadeev, A. E. Shumskaya, Y. G. Gorin, M. Jazdzewska, M. Anisovich, V. S. Rusakov and M. V. Zdorovets, Colloids Surf., A, 2020, 601, 125035 CrossRef CAS.
- E. M. Brozek, A. H. Mollard and I. Zharov, J. Nanopart. Res., 2014, 16, 2407 CrossRef.
- F. Abi-Ghaida, S. Clément, A. Safa, D. Naoufal and A. Mehdi, J. Nanomater., 2015, 2015, 1–8 CrossRef.
- Y. Wang, Y. Xu, J. Yang, X. Qiu, N. Li, Y. Zhu, L. Yan, W. Li, X. Huang, K. Liang, R. Guo, H. Tan and H. Yu, Mater. Chem. Front., 2021, 5, 2771–2776 RSC.
- G. Vares, V. Jallet, Y. Matsumoto, C. Rentier, K. Takayama, T. Sasaki, Y. Hayashi, H. Kumada and H. Sugawara, Nanomed. Nanotechnol. Biol. Med., 2020, 27, 102195 CrossRef CAS PubMed.
- F. Tamanoi, S. Chinnathambi, M. Laird, A. Komatsu, A. Birault, T. Takata, T. L.-H. Doan, N. X. D. Mai, A. Raitano, K. Morrison, M. Suzuki and K. Matsumoto, Int. J. Mol. Sci., 2021, 22, 2251 CrossRef CAS PubMed.
- Y. Sakurai and T. Kobayashi, Nucl. Instrum. Methods Phys. Res., Sect. A, 2000, 453, 569–596 CrossRef CAS.
- J. M. Kim, Y. Sakamoto, Y. K. Hwang, Y.-U. Kwon, O. Terasaki, S.-E. Park and G. D. Stucky, J. Phys. Chem. B, 2002, 106, 2552–2558 CrossRef CAS.
- N. X. D. Mai, Y. T. Dang, H. K. T. Ta, J.-S. Bae, S. Park, B. T. Phan, F. Tamanoi and T. L. H. Doan, J. Mater. Sci., 2021, 56, 3713–3722 CrossRef CAS.
- Y. Minoura and T. Moriyoshi, Nippon Kagaku Zassi, 1962, 83, 909–912 CrossRef CAS.
- S. Shenoi-Perdoor, A. Noureddine, F. Dubois, M. Wong Chi Man and X. Cattoën, in Handbook of Sol-Gel Science and Technology, Springer International Publishing, Cham, 2018, pp. 3001–3040 Search PubMed.
- A. B. Lowe, Polym. Chem., 2010, 1, 17–36 RSC.
- Z. Gao, Y. Horiguchi, K. Nakai, A. Matsumura, M. Suzuki, K. Ono and Y. Nagasaki, Biomaterials, 2016, 104, 201–212 CrossRef CAS PubMed.
- S. Kusaka, Y. Hattori, K. Uehara, T. Asano, S. Tanimori and M. Kirihata, Appl. Radiat. Isot., 2011, 69, 1768–1770 CrossRef CAS PubMed.
- P. Mi, H. Yanagie, N. Dewi, H. C. Yen, X. Liu, M. Suzuki, Y. Sakurai, K. Ono, H. Takahashi, H. Cabral, K. Kataoka and N. Nishiyama, J. Controlled Release, 2017, 254, 1–9 CrossRef CAS PubMed.
- H. Michiue, Y. Sakurai, N. Kondo, M. Kitamatsu, F. Bin, K. Nakajima, Y. Hirota, S. Kawabata, T. I. Nishiki, I. Ohmori, K. Tomizawa, S. ichi Miyatake, K. Ono and H. Matsui, Biomaterials, 2014, 35, 3396–3405 CrossRef CAS PubMed.
- M. Umano, K. Uechi, T. Uriuda, S. Murayama, H. Azuma, A. Shinohara, Y. Liu, K. Ono, M. Kirihata, H. Yanagie and T. Nagasaki, Appl. Radiat. Isot., 2011, 69, 1765–1767 CrossRef CAS PubMed.
- S. Kimura, S. I. Masunaga, T. Harada, Y. Kawamura, S. Ueda, K. Okuda and H. Nagasawa, Bioorg. Med. Chem., 2011, 19, 1721–1728 CrossRef CAS PubMed.
- I. Munar, V. Flndlk, I. Degirmenci and V. Aviyente, J. Phys. Chem. A, 2020, 124, 2580–2590 CrossRef CAS PubMed.
- A. Starobrat, T. Jaroń and W. Grochala, Dalton Trans., 2018, 47, 4442–4448 RSC.
- V. D'Anna, A. Spyratou, M. Sharma and H. Hagemann, Spectrochim. Acta, Part A, 2014, 128, 902–906 CrossRef PubMed.
- M. Kaur, P. Singh, K. Singh, U. S. Gaharwar, R. Meena, M. Kumar, F. Nakagawa, S. Wu, M. Suzuki, H. Nakamura and A. Kumar, Mater. Lett., 2020, 259, 126832 CrossRef CAS.
- P. Singh, M. Kaur, K. Singh, R. Meena, M. Kumar, J. H. Yun, A. Thakur, F. Nakagawa, M. Suzuki, H. Nakamura and A. Kumar, Phys. E, 2021, 132, 114766 CrossRef CAS.
- M. Kaur, P. Singh, R. Meena, F. Nakagawa, M. Suzuki, H. Nakamura and A. Kumar, J. Cluster Sci., 2021, 32, 221–225 CrossRef CAS.
- K. Kaira, K. Nakamura, T. Hirakawa, H. Imai, H. Tominaga, N. Oriuchi, S. Nagamori, Y. Kanai, N. Tsukamoto, T. Oyama, T. Asao and T. Minegishi, Am. J. Transl. Res., 2015, 7, 1161–1171 Search PubMed.
- I. Romero-Canelón, B. Phoenix, A. Pitto-Barry, J. Tran, J. J. Soldevila-Barreda, N. Kirby, S. Green, P. J. Sadler and N. P. E. Barry, J. Organomet. Chem., 2015, 796, 17–25 CrossRef.
- D. Alberti, A. Toppino, S. Geninatti Crich, C. Meraldi, C. Prandi, N. Protti, S. Bortolussi, S. Altieri, S. Aime and A. Deagostino, Org. Biomol. Chem., 2014, 12, 2457–2467 RSC.
- M. Ekkapongpisit, A. Giovia, C. Follo, G. Caputo and C. Isidoro, Int. J. Nanomed., 2012, 7, 4147–4158 CAS.
- J. Lu, M. Liong, S. Sherman, T. Xia, M. Kovochich, A. E. Nel, J. I. Zink and F. Tamanoi, Nanobiotechnology, 2007, 3, 89–95 CrossRef CAS PubMed.
- V. Panov, Y. Salomon, G. W. Kabalka and P. Bendel, Radiat. Res., 2000, 154, 104–112 CrossRef CAS PubMed.
- H. Fukuda and J. Hiratsuka, Appl. Radiat. Isot., 2020, 166, 109308 CrossRef CAS PubMed.
- G. Mehta, A. Y. Hsiao, M. Ingram, G. D. Luker and S. Takayama, J. Controlled Release, 2012, 164, 192–204 CrossRef CAS PubMed.
- F. Hirschhaeuser, H. Menne, C. Dittfeld, J. West, W. Mueller-Klieser and L. A. Kunz-Schughart, J. Biotechnol., 2010, 148, 3–15 CrossRef CAS PubMed.
- R. F. Barth, P. Mi and W. Yang, Cancer Commun., 2018, 38, 1–15 CrossRef PubMed.
- S. Jafari, H. Derakhshankhah, L. Alaei, A. Fattahi, B. S. Varnamkhasti and A. A. Saboury, Biomed. Pharmacother., 2019, 109, 1100–1111 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2na00839d |
| This journal is © The Royal Society of Chemistry 2023 |