
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTime-dependent exfoliation study of MoS2 for its use as a cathode material in high-performance hybrid supercapacitors†
Surbhi
Priya
 a,
Debabrata
Mandal
b,
Ananya
Chowdhury
c,
Sakshi
Kansal
a and
Amreesh
Chandra
*abc
a,
Debabrata
Mandal
b,
Ananya
Chowdhury
c,
Sakshi
Kansal
a and
Amreesh
Chandra
*abc
aDepartment of Energy Science and Engineering, Indian Institute of Technology Kharagpur, Kharagpur, India. E-mail: achandra@phy.iitkgp.ac.in
bSchool of Nanoscience and Technology, Indian Institute of Technology Kharagpur, Kharagpur, India
cDepartment of Physics, Indian Institute of Technology Kharagpur, Kharagpur, India
First published on 10th January 2023
Abstract
Quick and precise exfoliation of bulk molybdenum sulphide into few layers can bring a quantum leap in the electrochemical performance of this material. Such a cost-effective exfoliation route to obtain few layers of MoS2 nanosheets with a high mass yield of ∼75% is presented in this study. The electrochemical behaviours of three types of samples, namely pristine MoS2 and MoS2 exfoliated for 3 h and 5 h, were compared and the reasons leading to their performance modulation are explained. The performance could be tuned by changing the nature of the electrolytes, as shown using three different electrolytes, i.e. H2SO4, Na2SO4, and KOH. The electrochemical performance of a supercapacitor device fabricated using the 5 h-exfoliated sample showed many fold improvement. The strategy of combining with a 2D material-based anode is an interesting way forward for such devices. In addition, the anode material has to be carefully chosen so that high performance can be ensured. The usefulness of 2D flake-like WO3 as an anode was investigated first before establishing its worthiness in a hybrid device. The hybrid device was able to deliver an excellent energy density of 33.74 W h kg−1 with long-term cycling stability and coulombic efficiency, thus proving its applicability for high-performance energy-storage devices.
Introduction
The group of layered materials with the molecular formula MX2 (where M is a transition metal and X is a chalcogen atom) are known as layered transition metal dichalcogenides (TMDCs). They come under the broad spectrum of 2D materials having the space group P63/mmc.1,2 In these materials, the layers of the covalently bonded in-plane atoms are attached through weak van der Waals forces. Therefore, the application of an external force can lead to the exfoliation of these weakly bonded 2D layers. Amongst many TMDCs, molybdenum sulphide is attracting appreciable interest owing to its extraordinary physical, electrical, and optical properties.3–6 MoS2 is a typical layered transition metal dichalcogenide [LTMDC] with a three-atom thick layer, which comprises a plane of molybdenum sandwiched between two planes of sulphur atoms. The in-plane atoms are covalently bonded with each other, while the stacked layers are connected through van der Waals forces. The monolayer of MoS2 constitutes a S–Mo–S layer. When three or four stacked layers are held together by van der Waals forces, the materials are known as tri-layer structures, and finally, 3-D bulk molybdenum sulphide forms if more than 100 MoS2 monolayers are stacked together. The unit cell of MoS2 consists of a honeycomb lattice structure with lattice parameters a = 3.12 Å and c = 3.11 Å. Here, individual Mo atoms are arranged in triangular prismatic coordination with the P63/mmc space group and non-centro-symmetric crystal structure.7 Due to its unique layered structure (interlayer spacing = 0.62 nm), MoS2 exhibits various exciting properties, such as a tuneable bandgap, large surface area, and high electron mobility rate. The majority of the properties of monolayer or few-layered MoS2 differ from their bulk counterparts. Therefore, various preparation protocols, such as scotch tape exfoliation, hydrothermal, chemical vapour deposition, and liquid exfoliation, are being explored. However, obtaining samples through ion intercalation followed by sonication is quite interesting because large flakes can be produced with high yield.Coleman et al. recently suggested the feasible liquid-phase exfoliation of bulk MoS2 powders in a suitable organic solvent with the help of ultrasonication.8 Wan et al. used a CVD approach to create fractal-shaped single-layer MoS2. In comparison to a triangle-shaped structure, these MoS2, with a large number of exposed edge sites, also demonstrated greater HER characteristics.9 However the synthesis of MoS2 nanoflakes via ion intercalation followed by liquid-phase exfoliation necessitates a protracted intercalation stage (24 h or days), which limits its usage at the industrial level. Further, amongst all the methods discussed above, scalability remains a major issue. Therefore, an effective, scalable and fast process to exfoliate MoS2 is required. In the present work, a novel modification to the liquid-phase exfoliation technique is proposed, which can lead to a rapid and efficient route for exfoliating bulk into few-layered-thick MoS2. The whole process can be completed within a few hours, which makes the strategy practical, economical and industrially applicable. Along with the quick exfoliation, a ∼75% yield (exfoliated MoS2) can be easily achieved.
While exfoliation through LixMoS2 allows easy access to the MoS2 monolayers, the change in their physical properties through exfoliation is not completely understood yet. Therefore, time-dependent analysis of MoS2 exfoliation was conducted here, which led to a clear strategy being developed to tune the surface area and associated electrochemical response characteristics. The exfoliated material delivered excellent physiochemical performance, making its use in energy-storage devices worthwhile. However, a few challenges remain, such as the limited voltage window, due to the probability of the hydrogen and oxygen evolution processes, and the relatively poor electrical conductivity, that restrict the full use of MoS2 electrodes in energy-storage applications. To mitigate these issues and fabricate MoS2-based supercapacitors, a careful selection of the second electrode material is essential. In order to increase the voltage window, carbonaceous materials can be used as they exhibit a large voltage window of 0 to −1 V. This can increase the operating voltage window of the device to ∼2 V. However, carbon material stores charge according to an electric double-layer charge-storage mechanism, which significantly limits the specific capacitance value. Unlike carbon materials, metal oxides store charge through a pseudocapacitive mechanism, based on quick and reversible redox reactions. WO3 is fast emerging as an excellent anode material. It also has reasonable conductivity, a high theoretical capacitance, and multiple valence states, which lead to its large electrochemical performance.10–12
As explained earlier, the use of a 2D layered structure is leading to appreciable improvements in supercapacitor performance. Therefore, 2D WO3 was also synthesized during the work. The compatible structures of the two electrode materials, i.e. MoS2 and WO3, would ensure a reduced ion-diffusion path and resistance, leading to high electrochemical performance.13,14 The fabricated device achieved a maximum specific capacitance of 84 F g−1 at a current density of 1 A g−1, with an extended working potential window of 1.6 V. Moreover, the device also exhibited a very high energy and power density of 33.74 W h kg−1 and 875 W kg−1, respectively, at 1 A g−1 current density, with long term cycling stability, making it useful for several applications.
Experimental
Exfoliation of MoS2
First, 2.5 g of bulk MoS2 was mixed with a solution of 20 ml N-methyl-2-pyrrolidone (NMP), 0.5 M NaBH4, and 1 M lithium chloride. The mixed solution was stirred for 30 min. The pre-treatment of bulk MoS2 with NaBH4 generated H2 gas, which penetrated through the layers of MoS2 and expanded the interlayer spacing, leading to an easy exfoliation. Following pre-treatment, the complete solution was subjected to sonication using an ultrasonic bath (power = 200 W) for 5 h. In a water-cooled bath at 2.5 °C, the samples were sonicated continuously for different time durations of 3 h and 5 h with a probe sonic tip (5 mm, tapered microtip 630-0419 vibra cell) at a power output of 120 W. To avoid overheating, the tip was pulsed for 10 s ON and 5 s OFF cycles. A black dispersion was formed and collected on an hourly basis for time-dependent analysis. Wet sediments were collected and washed twice using dimethyl formamide (DMF), in order to collect solvated LixMoS2 ((Li–solvent)xMoS2) with no hexane. The collected supernatant was mildly sonicated in DMF at room temperature for 4 min. Finally, the yellowish-green precipitate was washed with ethanol and acetone twice and dried at 80 °C for 12 h. Generally, the lesser the difference in the surface energy between the layered material and solvent, the better the exfoliation.15–17 Herein, NMP was used as it has a surface tension of 40 mJ m−2, which is similar to the surface energy of several layered materials.18 The complete exfoliation process is schematically explained in Fig. 2(a).Synthesis of WO3 nanoparticles
A simple hydrothermal method was used to synthesize 2D flakes of WO3. In 20 ml of DI water, 11 mM Na2WO4·2H2O was added to form a homogenous solution. In order to maintain a pH value of 1.5, 6 M HCl was added drop-wise. Following 5 min of stirring, 11 mM H2C2O4 and 22 mM NaCl were added to the above mixture. Finally, this homogenous solution was transferred into a 50 ml Teflon-lined vessel and heated at 120 °C for 5 h. The precipitate was collected, centrifuged and dried at 60 °C. A schematic of the synthesis protocol is shown in the ESI (Scheme S1†).Physiochemical measurement method
The confirmation of the phase was carried out by evaluating the X-ray diffraction (XRD) patterns obtained using a Rigaku X-ray diffractometer with Cu Kα1 (λ = 1.5406 Å) as the incident radiation. A Jobin Yvon Horiba T64000 Raman spectrometer with an excitation wavelength of 514.5 nm was used for the Raman investigation. A Carl Zeiss SUPRA 40 field-emission scanning electron microscope (FESEM) and a high-resolution transmission electron microscope (HRTEM) were used to examine the morphology of the particles (model: JEM-2100 HRTEM, JEOL). The surface area analysis and porosity of the synthesized powders were determined using a Quantachrome Chem BET analyzer, and the thickness measurement was done through atomic force microscopy (model: Agilent Technology).Electrochemical measurement method
For the electrochemical measurements, three-electrode analysis was performed with Ag/AgCl as the reference electrode and platinum rod as the counter electrode. The ink of the working electrode was prepared by mixing 80 wt% of the active material, 10 wt% of carbon black, and 10 wt% of polyvinylidenefluoride (PVDF) in acetone. This slurry (ink) was then drop-cast on to a graphite sheet with 1 × 1 cm dimensions and dried at 80 °C for 15 h. For device fabrication, exfoliated MoS2 and WO3 nanoparticles based electrodes were used as the positive and negative electrode, respectively.Results and discussions
Structural analysis
The crystallinity of the bulk MoS2 and few-layer exfoliated MoS2 was determined using the XRD data. The XRD patterns of all the samples, shown in Fig. 1, could be indexed using the JCPDS card no. (JCPDF-06-0097).19,20 As the exfoliation time increased gradually, the normalized peak intensity showed a suppression and, for the material exfoliated for 5 h, a very low intensity peak was seen. The Raman spectra of the pristine MoS2 and exfoliated nanosheets with the increase in exfoliation time are shown in Fig. 2(b). In the case of MoS2, the mode caused by sulphur atoms vibrating perpendicular to the basal plane was A1g, and the one caused by molybdenum and sulphur atoms vibrating parallel to the basal plane was the E2g mode. The spectrum for the pristine sample clearly showed two distinct peaks at 403 and 376.88 cm−1, which corresponded to the out-of-plane A1g and in-plane E2g modes, respectively. The difference in frequency observed between the two phonon modes was 27 cm−1. The out-of-plane A1g and in-plane E2g modes in the 5 h-exfoliated sample were observed at 405 and 380 cm−1, respectively. The peak shift position in the exfoliated sample confirmed the formation of the few-layered MoS2.21–23 The SEM image (Fig. 2(c)) of the sample exfoliated for 3 h showed a flake-like morphology, but the crystals remained in the micron level, revealing an incomplete exfoliation. Following 5 h of sonication, the SEM image (Fig. 2(d)) of the sample clearly showed a flake-like morphology in the size range of 100 nm. In this sample, the flakes opening gradually increased and the appearances of the flakes were clear and prominent. The TEM image of the 5 h-exfoliated MoS2, shown in Fig. 2(d), reconfirmed the formation of a few layers of MoS2.24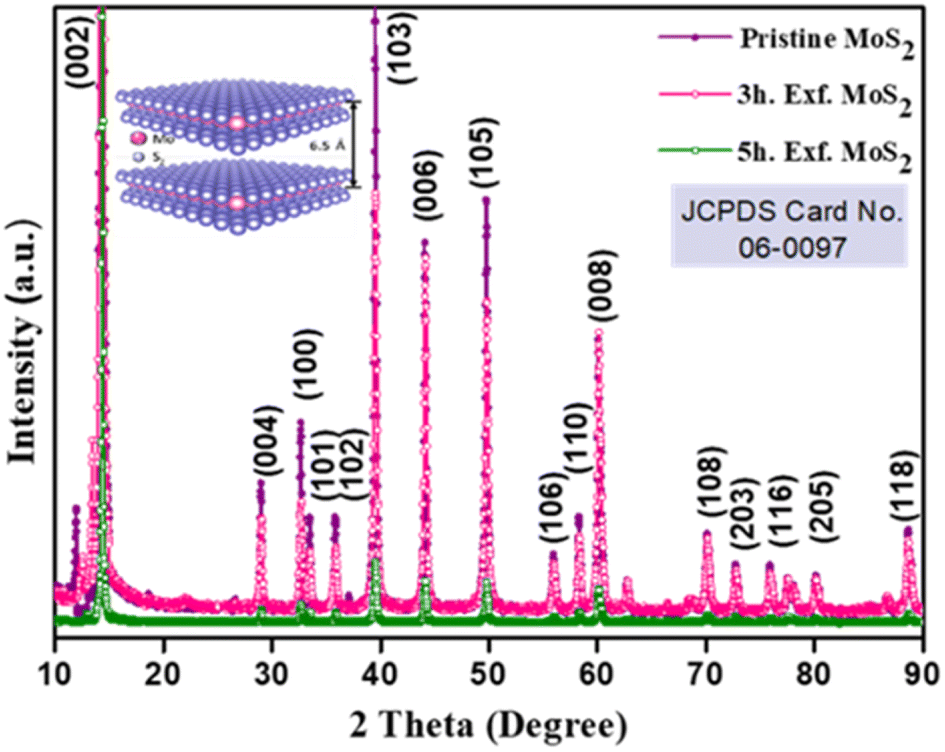 | ||
| Fig. 1 XRD profile of the pristine and exfoliated MoS2 samples. | ||
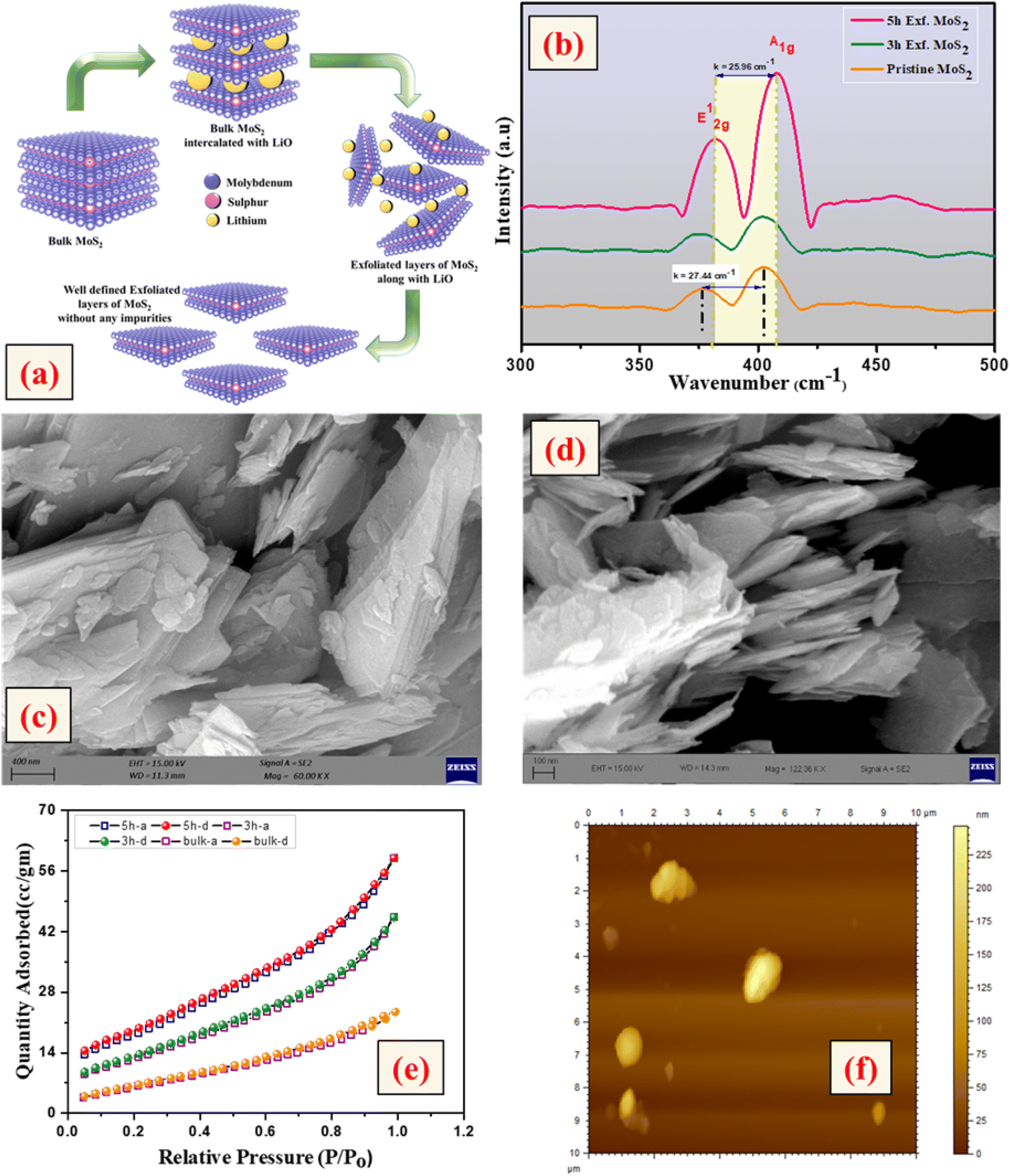 | ||
| Fig. 2 (a) Exfoliation process of MoS2, (b) Raman profile, (c) SEM image of the pristine sample, (d) SEM and TEM (inset) images of the 5 h-exfoliated sample, (e) BET profile and (f) AFM images of the pristine and exfoliated MoS2 samples. | ||
N2 adsorption–desorption curves were used to evaluate the surface area of the samples exfoliated for different time periods. As shown in Fig. 2(e), all the samples showed a type-IV isotherm indicating a mesoporous structure. The specific surface area was recorded to be 17, 44, and 62 m2 g−1 for the pristine, and 3 h- and 5 h-exfoliated MoS2, respectively. This result corroborated the inferences drawn from the microscopic characterizations. The thickness of the nanosheets present in the sample exfoliated for 5 h was found to be 11 nm, using the AFM data shown in Fig. 2(f). The EDAX analysis, given in the ESI (Fig. S1†), confirmed the presence of molybdenum and sulphur atoms. All the physiochemical characterizations of WO3 are also discussed in the ESI (Fig. S2†).
Electrochemical analysis
| MoS2 + 2H+ + e− ↔ MoS–SH | (1) |
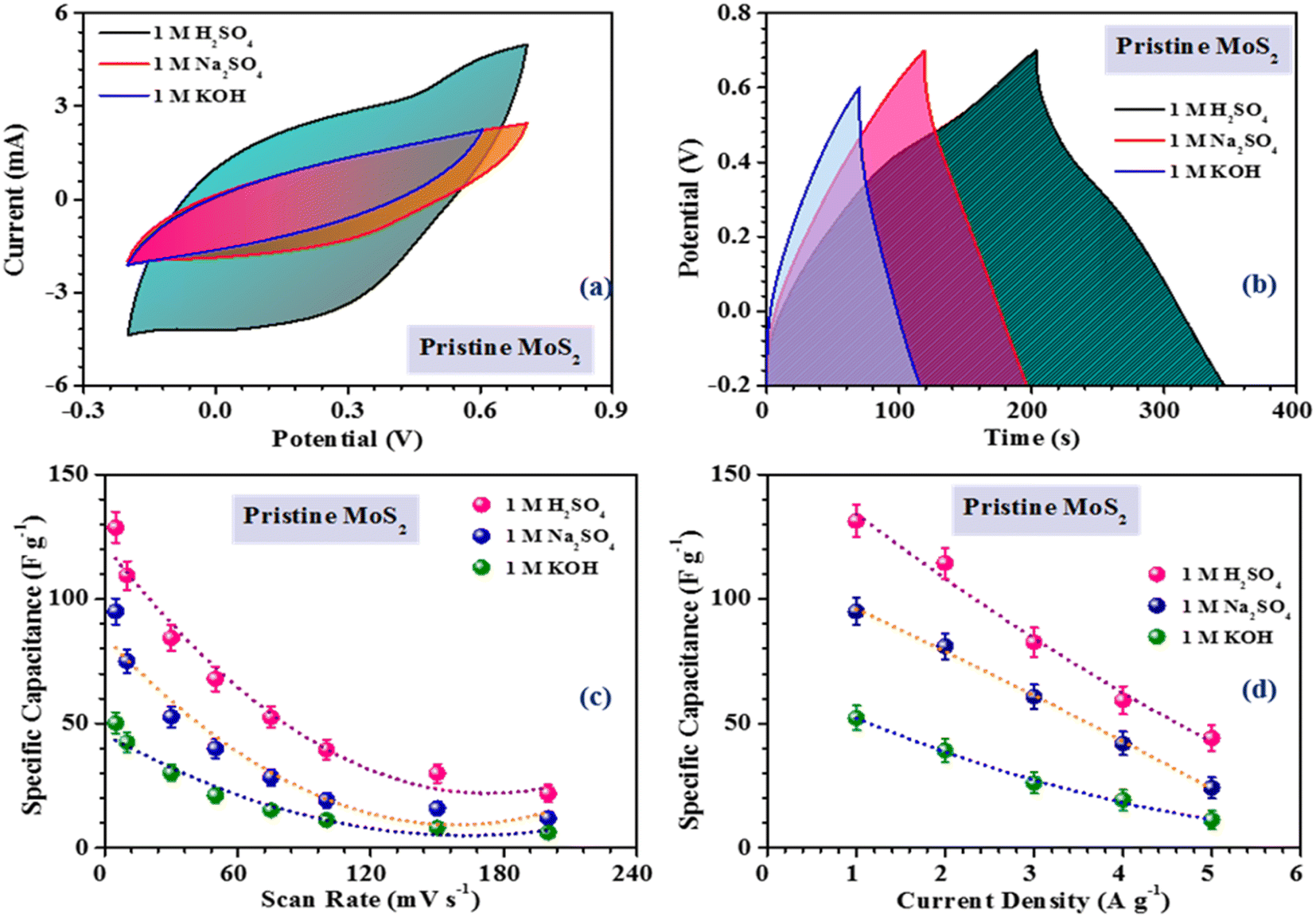 | ||
| Fig. 3 Comparison of the (a) CV profiles, (b) CD profiles. Variation of the specific capacitance with the (c) scan rate and (d) current density in pristine MoS2 combining with different electrolytes. | ||
During the electrochemical reaction, proton intercalation can lead to the formation of MO–SH species.25 In addition, there is a contribution from non-faradaic reactions, which involve the adsorption of H+ on the edge sites of MoS2 flakes, which can also contribute to the electrochemical activity, as indicated below :26
| (MoS2)surface + H+ + e− ⇔ (MoS2–H+)surface | (2) |
The inferences were further corroborated by the galvanostatic charge–discharge measurements. Fig. 3(b) shows the CD profiles of pristine MoS2 at 1 A g−1 current density in different electrolytes. As seen earlier, the material showed the best performance in an acidic electrolyte. The specific capacitance values from the CV and CD profiles were estimated using the following formulas:27
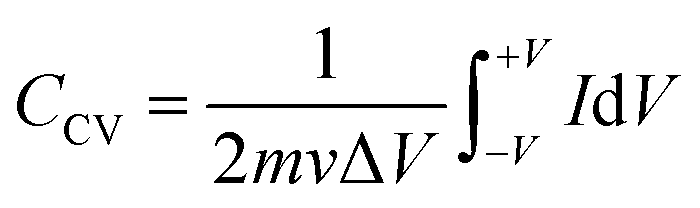 | (3) |
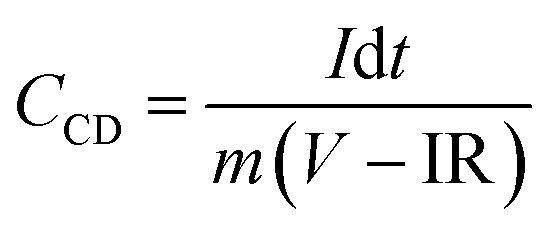 | (4) |
Fig. 4(a), (c) and (e) show the CV profiles of pristine MoS2 and the samples exfoliated for 3 h and 5 h. It was clear that, with increasing the exfoliation time, the area under the curve as well as the current value showed an increasing trend. This clearly indicated an overall increment in the electrochemical activity as well as the conductivity of the samples. Additionally, a few prominent redox peaks were observed in the CV profiles, confirming the pseudocapacitive nature of the samples. This can be clearly seen in the comparison of the CV profiles, as shown in Fig. 5(a). Fig. 5(b) depicts the variation of the specific capacitance in the MoS2 samples with increasing the scan rate. The pristine MoS2-based electrode delivered a maximum specific capacitance of 129 F g−1 at a 10 mV s−1 scan rate, whereas the samples exfoliated for 3 h and 5 h delivered specific capacitances of 232 and 322 F g−1, respectively. Hence, after 5 h of exfoliation, a ∼2.5-fold increment in the specific capacitance value of MoS2 was observed. For all the samples, the value of the specific capacitance was higher at low scan rates before showing gradual decrement at higher scan rates. This happened because, at lower scan rates, the ions have more time to diffuse and penetrate the electrode surface, leading to higher storage.
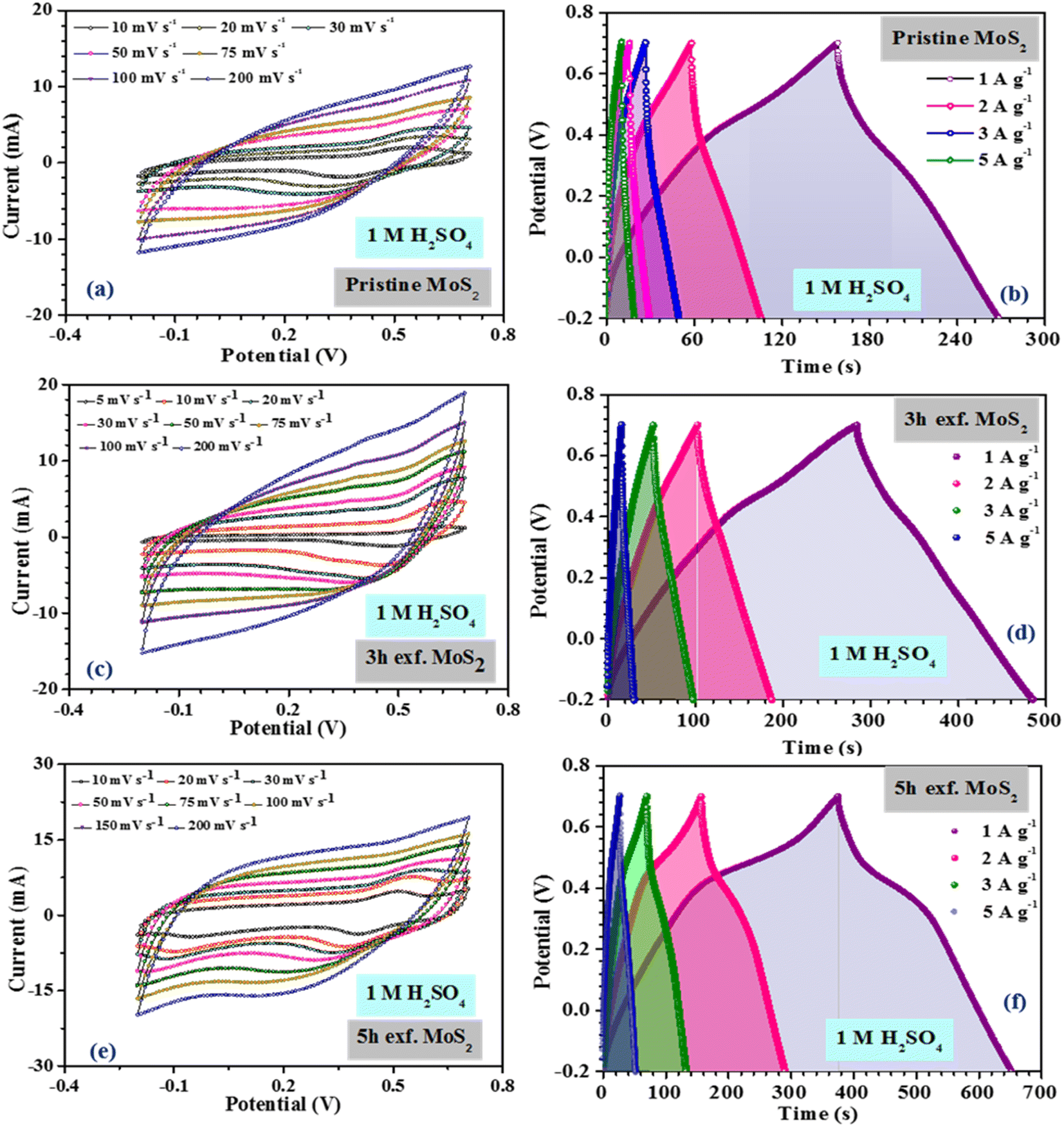 | ||
| Fig. 4 (a), (c), (e) CV, (b), (d) and (f) CD profiles of the pristine and exfoliated samples in 1 M H2SO4 electrolyte. | ||
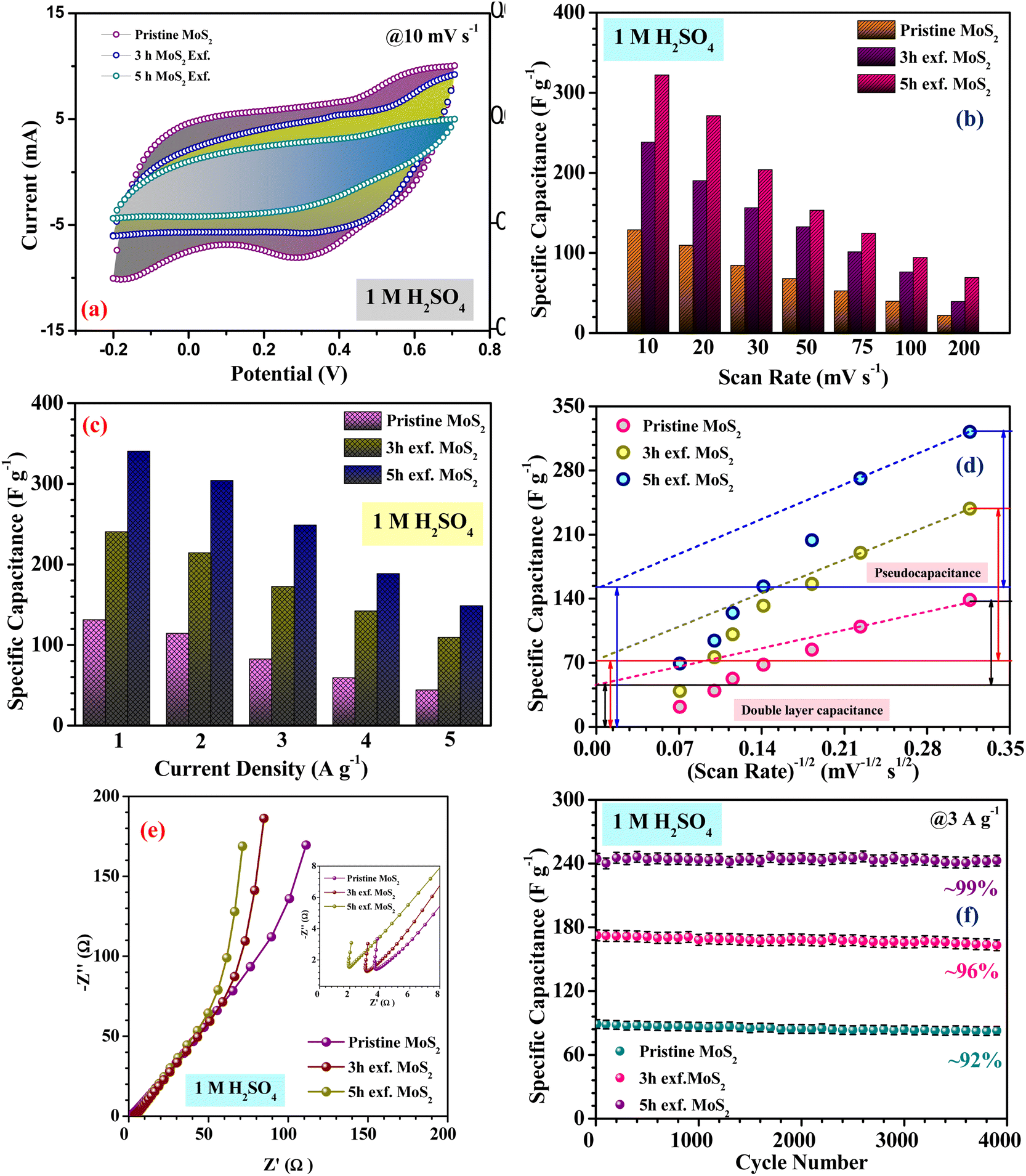 | ||
| Fig. 5 (a) Comparison of the CV profiles, (b) and (c) specific capacitance variation, (d) quantification graph, (e) Nyquist plots and (f) cycling stability of the pristine and exfoliated samples in 1 M H2SO4. | ||
In the galvanostatic charge–discharge curves, two different discharge behaviours, namely linear and non-linear, were observed. The linear region implied that the electrode was able to store charge owing to adsorption–desorption reactions at the electrode–electrolyte interface on the surface. The non-linear region, on the other hand, was evidence that the electrode could also store charge because of the redox or intercalation mechanisms associated with a pseudocapacitive nature. The galvanostatic charge–discharge curves for the MoS2 samples at current densities ranging from 1–5 A g−1 are presented in Fig. 4(b), (d) and (f). The material showed a nearly symmetric charge–discharge response with a coulombic efficiency of more than 80%. From the GCD profiles, it could be observed that the discharge time showed an increasing trend with increasing the exfoliation time, which reaffirmed the higher electrochemical activity. The specific capacitance values calculated from the CD profiles at a 1 A g−1 current density for the pristine, and 3 h- and 5 h-exfoliated samples were ∼131, 242, and 340 F g−1, respectively. Therefore, once again, a more than 2.5-fold increment in the specific capacitance value was obtained after exfoliation. In the GCD curve, a plateau region, corresponding to the redox reaction, was clearly visible. Further, the coulombic efficiency of the 5 h-exfoliated MoS2 sample was higher than that of the pristine sample. Fig. 5(c) depicts the specific capacitance variation in the MoS2 samples with the current density. With increasing the current density, the specific capacitance showed a decreasing trend. The 5 h-exfoliated MoS2 showed a ∼50% rate capability at a high current density of 5 A g−1, by delivering ∼167 F g−1 specific capacitance.
From the redox peaks and the plateau region of the CV and CD profiles of MoS2, it was clear that here the total specific capacitance was a manifestation of the combined contributions from the double-layer capacitance and pseudocapacitance. At lower scan rates, the pseudocapacitance dominated, as the electrolyte ions had sufficient time for intercalation and could utilize all the active sites of the material. In comparison, at higher scan rates, the redox processes were restricted because of the limited reaction time. To visualize this graphically, the specific capacitances of the pristine and exfoliated MoS2 were plotted with the scan rate−1/2. At a higher scan rate, the data points deviated from the linear fit. The extrapolation of the fitted line towards the zero point, i.e. infinity scan rate, denotes the expected value of EDLC, as the pseudocapacitance would be practically zero there. Finally, the pseudocapacitance was calculated by subtracting the EDLC value from the total specific capacitance. The quantification graphs of all the samples are shown in Fig. 5(d). The pseudocapacitance contribution in pristine MoS2 was found to be ∼42%. Following exfoliation, the pseudocapacitance contribution in the 3 h- and 5 h-exfoliated MoS2 samples were estimated to be ∼66 and 69%, respectively. The pseudocapacitive nature could be inferred as a redox pair peak was seen at the low scan rate of 10 mV s−1 in the sample exfoliated for 5 h. These redox peaks were due to the insertion/desertion of protons into the MoS2 interlayer. The sulphur atoms, present in the layers of MoS2, reversibly bind with protons in the acidic electrolyte, thus changing the oxidation state of the neighbouring Mo atoms from 4+ to (4 − δ)+ according to the proton attachment reaction.28 This reversible proton attachment distorted the CV curve, confirming the pseudocapacitive behaviour.
These findings were further supported by the galvanostatic charge–discharge measurements. There was a significant increase in the specific capacitance in the case of the sample exfoliated for 5 h. The enhancement of the electrochemical performance was attributed to the improved electrical conductivity, efficient ion transportation and, increase in electroactive sites. The exfoliation of MoS2 induced the weak van der Waals force between the layers to break, leading to the formation of layers. These layer made the sites on the edge plane available for reaction and ensured an increase in the electroactive sites. Along with the increment in the specific capacitance value, there was a significant enhancement in the rate capability of the material, even at higher current density. Further, the electrochemical measurement of the sample exfoliated for 6 h was also investigated. However, the area under the CV profile did not change significantly for 6 h exfoliation, as shown in the ESI.† From Fig. S7(a) and (b),† we confirmed that the sizes of the MoS2 flakes were almost same in 5 h exfoliation and 6 h exfoliation. H+ diffusion in the inter MoS2 flakes was almost similar for the 5 h exfoliation and 6 h exfoliation because the size of the dispersed particle did not change inside the solvent with the higher exfoliation time. However, from a thermodynamic point of view, the surface tension is the key parameter for this. The intermolecular attraction between a solid surface and solvent drop is the cause of surface tension and the differences in surface tension create Gibbs free energy change during exfoliation. Therefore, the surface tension for the 5 h- and 6 h-exfoliated samples was very similar.
Further, to estimate the equivalent series resistance of the samples, EIS spectra were collected for the MoS2 samples. The corresponding Nyquist plots are shown in Fig. 5(e). The fitted Nyquist plot and the equivalent circuit for each sample is shown in the ESI (Fig. S6†), with the corresponding charge transfer and series resistance values tabulated in Table S1.†
The intercept value of the plot in the high-frequency range corresponds to the ESR value of the material. The estimated ESR values of the pristine, and 3 h- and 5 h-exfoliated MoS2 were ∼4.1, 3.5, and 2.7 Ω, respectively. The low series resistance and improved slope in the low-frequency region proved the high electrochemical activity and superior capacitive nature of the 5 h-exfoliated MoS2 sample. Finally, the cycling stabilities of all the materials were also analyzed to test for their practical applicability. Repetitive charge–discharge measurements at 3 A g−1 current density were carried out for 4000 cycles. The pristine MoS2 successfully retained 92% of its initial capacitance value after 4000 cycles, while, after exfoliation, the 5 h-exfoliated MoS2 showed excellent cycling stability with ∼99% performance retention, as shown in Fig. 5(f). This further proved the importance of exfoliation and the usefulness of the obtained material for practical energy applications. As mentioned before, in this work, WO3 was used as the negative electrode material for asymmetric device fabrication. Therefore, electrochemical studies were carried out on the synthesized WO3 nanomaterials, to check their viability as a negative electrode. The corresponding electrochemical results are shown in the ESI (Fig. S4†). The CV profiles of WO3 revealed its capacitive nature within a stable potential window of 0.8 V (i.e. −0.8 to 0 V). The large area under the CV curve indicated the high electrochemical activity of the material. The specific capacitance values, at various scan rates were estimated using the formula mentioned earlier. The WO3-based electrode delivered a maximum specific capacitance of 50 F g−1 at a 10 mV s−1 scan rate. The results from the CV measurements were further verified by charge–discharge data analysis. The GCD profiles indicated a high coulombic efficiency and good capacitive nature of the material even at a high current density. The material delivered a maximum specific capacitance value of 48 F g−1 at a current density of 1 A g−1. The electrochemical performance of the exfoliated MoS2 was compared with some of the recently reported materials in the literature, as tabulated in Table 1.
| Material | Exfoliation method | Exfoliation time | Application | Capacitance | References |
|---|---|---|---|---|---|
| Exfoliated MoS2 | Liquid exfoliation | Continuously 5 days | Photoanode | — | 28 |
| Exfoliated MoS2 | Liquid exfoliation | 30 h | Photocatalysis | — | 29 |
| Exfoliated MoS2 | Liquid exfoliation | 10 h | Flexible supercapacitor | 1060 mF g−1 | 30 |
| MoS2 nanospheres | — | — | Supercapacitor | 106 F g−1 | 31 |
| MoS2 nanorods | — | — | Supercapacitor | 275 F g−1 | 32 |
| MoS2 microspheres | Biopolymer-assisted hydrothermal | — | Supercapacitor | 145 F g−1 | 33 |
| MoS2 nanosheets | Hydrothermal | — | Supercapacitor | 129.2 F g−1 | 34 |
| MoS2/C composite | Hydrothermal | — | — | 201.4 F g−1 | 35 |
| MoS2-titanium plate | Hydrothermal | — | — | 133 F g−1 | 36 |
| Exfoliated MoS 2 flakes | Liquid exfoliation | 5 h | Supercapacitor | 340 F g−1 | Present work |
Device performance
Following the three-electrode characterization, the device performance of the materials was investigated. For device fabrication, pristine and 5 h-exfoliated MoS2 were used as the positive electrode, while WO3 served as a negative electrode in both cases. Also, 1 M H2SO4 was used as the electrolyte. A Whatman glass microfiber filter, pre-soaked in H2SO4, was used as a separator. Both the devices were fabricated while ensuring a mass balance condition. The details of the device fabrication process, along with a schematic representation are given in the ESI (Scheme S2†).The electrochemical performance of the pristine MoS2//WO3 asymmetric device is shown in the supplementary information (Fig. S5†). The device showed an optimized working potential of 1.6 V (i.e. 0–1.6 V). Weak redox peaks, corresponding to the reduction and oxidation reactions, were observed in the CV profiles. These were more prominent at lower scan rates, where the slow scanning allowed the occurrence of redox reactions at the electrode–electrolyte interface. Galvanostatic charge–discharge measurements at different current densities for the pristine MoS2//WO3 asymmetric device were also performed. The device revealed a good capacitive performance with a high coulombic efficiency of ∼93%. The device delivered a maximum specific capacitance of ∼40 F g−1 at 1 A g−1 current density. The variation of the specific capacitance value with increasing the current density is given in the ESI (Fig. S5†).
Fig. 6(a) indicates the CV profiles of the 5 h-exfoliated MoS2//WO3 device at scan rates ranging from 10 to 200 mV s−1. The enhancement in the electrochemical activity of the material after exfoliation was clearly visible in the CV profiles with high current values. Additionally, the redox peaks were also quite prominent. The CD measurements of the device further confirmed these findings, as shown in Fig. 6(b), with a higher discharge time and improved coulombic efficiency. The specific capacitance value of the 5 h-exfoliated MoS2//WO3 asymmetric device delivered a maximum specific capacitance of ∼79 and 84 F g−1 at a scan rate and current density of 10 mV s−1 and 1 A g−1, respectively. Therefore, a more than two-fold increment in the specific capacitance value was observed compared to the previous case, which clearly supported the enhancement of the electrochemical activity after exfoliation. The variation of the specific capacitance for the 5 h-exfoliated MoS2//WO3 with the scan rate and current density is depicted in Fig. 6(c).
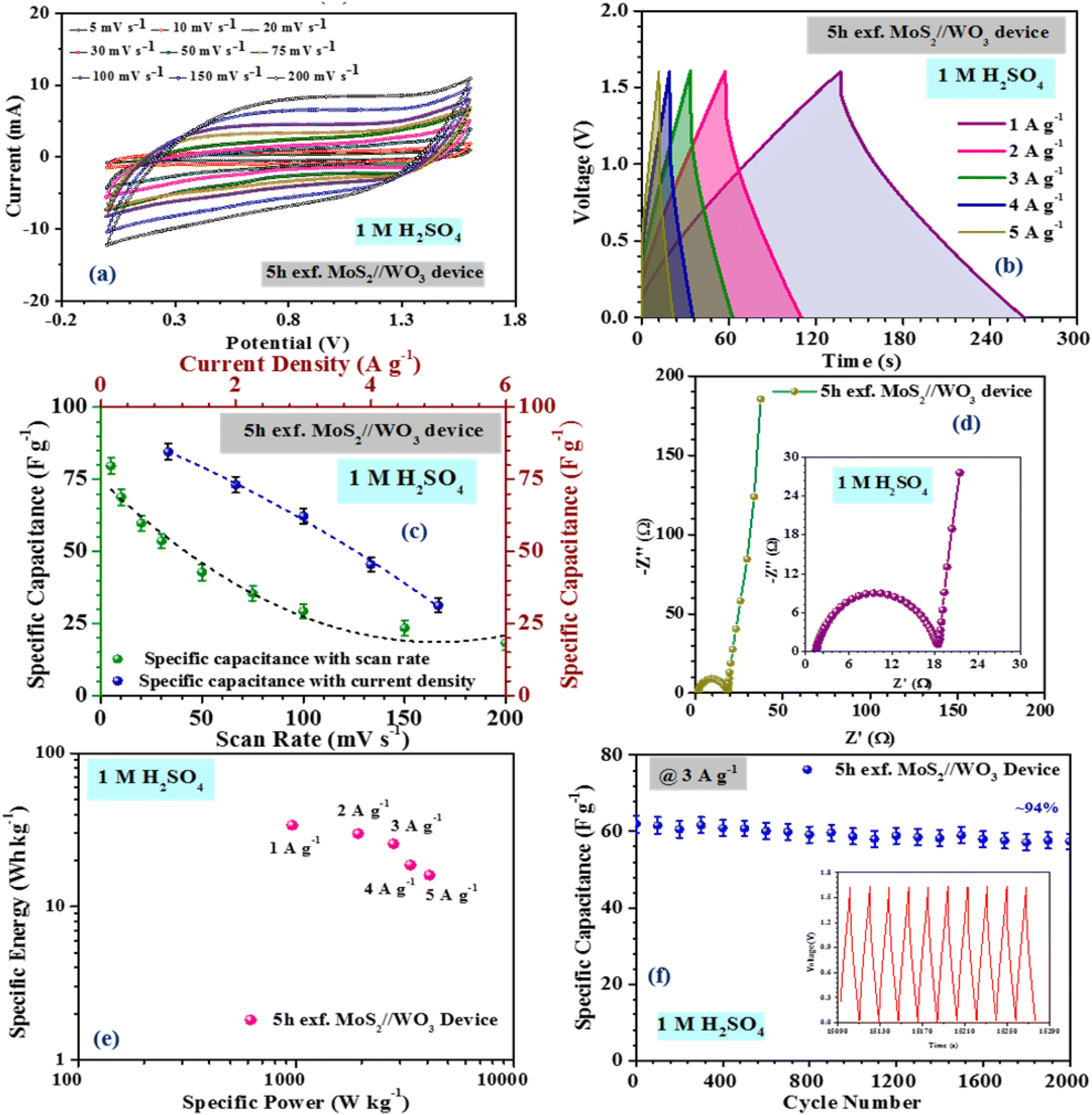 | ||
| Fig. 6 (a) CV, (b) CD profiles, (c) specific capacitance variation, (d) Nyquist plot, (e) Ragone plot and (f) cycling stability of the as-fabricated 5 h-exfoliated MoS2//WO3 device. | ||
Electrochemical impedance measurements of the 5 h-exfoliated MoS2//WO3 device were also carried out within the frequency range of 1 mHz–100 kHz, using an AC signal of 5 mV. The corresponding Nyquist plot is shown in Fig. 6(d). The vertical rise of the impedance value at the lower frequency region confirmed the good capacitive nature of the device. The inset of the figure shows a selected region of the Nyquist plot. The small semicircle indicates the charge-transfer resistance at the electrode–electrolyte interface, which was estimated to be ∼15.78 Ω for this device. Further, the equivalent series resistance of the device was ∼1.89 Ω, as estimated from the first intercept of the Nyquist plot. The fitted Nyquist plot along with the equivalent circuit and the values of the series resistance, Warburg resistance, and charge-transfer resistance for the 5 h-exfoliated MoS2//WO3 device are shown in the ESI (Fig. S6(d) and Table S2†). Compared to the pristine MoS2-based device, the lower series resistance and charge-transfer resistance in this case would ensure a performance enhancement.
The specific energy and power of the devices were calculated at various current densities using the following relations:37:
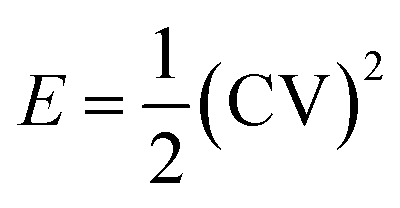 | (5) |
| P = E/t | (6) |
To investigate the cycling stability, both the devices were subjected to repetitive charging and discharging for 2000 cycles, at 3 A g−1 current density, and the corresponding findings are depicted in Fig. 6(f). The 5 h-exfoliated MoS2//WO3 device showed better electrochemical activity. It was also able to maintain its performance more efficiently, retaining ∼94% of the initial specific capacitance value after 2000 charge–discharge cycles. This clearly proved the usefulness of the exfoliated samples for various practical applications. For practical demonstration of the MoS2//WO3 device, we tried to light up an LED strap and a toy candle using the fabricated device. Since, the as-fabricated asymmetric device exhibited a potential window of 1.6 V, therefore to power an LED strap of 13 V, 9 MoS2//WO3 devices were stacked together and then connected to the source. Some applications of the fabricated device are shown in Fig. S8,† which prove the practical importance of the fabricated device.
Conclusion
In summary, a fast and efficient method for exfoliating bulk MoS2 into monolayers was established. Here, the excessively strong reduction ability of LiCl induced the decomposition of MoS2 into Mo and Li2S, resulting in a disruption of its layered structure. In addition, the complete lithium intercalation of MoS2 with Li could be accomplished within 5 h at room temperature, which was significantly faster than conventional methods reported in the literature. Following a simple exfoliation, the as-obtained MoS2 nanosheets demonstrated an ideal thickness of 12 nm. These MoS2 nanosheets were able to exhibit excellent electrochemical storage capability with a high reversible capacity of 340 and 256 F g−1 at 1 A g−1, and an outstanding cycling stability, with >95% capacity retention after 4000 cycles. The exfoliated samples also proved their usefulness in hybrid supercapacitors, where they could combine with a suitable negative electrode for delivering a high storage ability.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors acknowledge the funding received from Defence Research Development and Organisation (DRDO, India) for the project entitled, “Functional and flexible polymer nanocomposites using hierarchical nano metal oxides for defence applications” (Grant No. ERIP/ER/202202004/M/01/1796).References
- Q. Fu, J. Han, X. Wang, P. Xu, T. Yao, J. Zhong, W. Zhong, S. Liu, T. Gao, Z. Zhang, L. Xu and B. Song, Adv. Mater., 2021, 33, e1907818 CrossRef PubMed
.
- S. Manzeli, D. Ovchinnikov, D. Pasquier, O. V. Yazyev and A. Kis, Nat. Rev. Mater., 2017, 2, 17033 CrossRef CAS
- W. Zhao, Z. Ghorannevis, L. Chu, M. Toh, C. Kloc, P. H. Tan and G. Eda, ACS Nano, 2013, 7, 791 CrossRef CAS PubMed
- K. F. Mak, C. Lee, J. Hone, J. Shan and T. F. Heinz, Phys. Rev. Lett., 2010, 105, 136805 CrossRef PubMed
- P. B. James and M. T. Lavik, Acta Crystallogr., 1963, 16, 1183 CrossRef CAS
- D. Puotinen and R. E. Newnham, Acta Crystallogr., 1961, 14, 691 CrossRef CAS
- R. A. Bromley, The Philosophical Magazine: A Journal of Theoretical Experimental and Applied Physics, 2010, 23, 1417 CrossRef
- J. N. Coleman, M. Lotya, A. O'Neill, S. D. Bergin, P. J. King, U. Khan, K. Young, A. Gaucher, S. De, R. J. Smith, I. V. Shvets, S. K. Arora, G. Stanton, H. Y. Kim, K. Lee, G. T. Kim, G. S. Duesberg, T. Hallam, J. J. Boland, J. J. Wang, J. F. Donegan, J. C. Grunlan, G. Moriarty, A. Shmeliov, R. J. Nicholls, J. M. Perkins, E. M. Grieveson, K. Theuwissen, D. W. McComb, P. D. Nellist and V. Nicolosi, Science, 2011, 331, 568 CrossRef CAS PubMed
- Y. Wan, Z. Zhang, X. Xu, Z. Zhang, P. Li, X. Fang, K. Zhang, K. Yuan, K. Liu, G. Ran, Y. Li, Y. Ye and L. Dai, Nano Energy, 2018, 51, 786 CrossRef CAS
- Y. Zhuo, E. Prestat, I. A. Kinloch and M. A. Bissett, ACS Appl. Energy Mater., 2022, 5, 61 CrossRef CAS
- A. Rajapriya, S. Keerthana, C. Viswanathan and N. Ponpandian, J. Alloys Compd., 2021, 859 Search PubMed
- S. Yao, F. Qu, G. Wang and X. Wu, J. Alloys Compd., 2017, 724, 695 CrossRef CAS
- V. Lokhande, A. Lokhande, G. Namkoong, J. H. Kim and T. Ji, Results Phys., 2020, 12, 2012 CrossRef
- M. Ashraf, S. S. Shah, I. Khan, M. A. Aziz, N. Ullah, M. Khan, S. F. Adil, Z. Liaqat, M. Usman, W. Tremel and M. N. Tahir, Chemistry, 2021, 27, 6973 CrossRef CAS PubMed
- C. Backes, N. C. Berner, X. Chen, P. Lafargue, P. LaPlace, M. Freeley, G. S. Duesberg, J. N. Coleman and A. R. McDonald, Angew. Chem., Int. Ed., 2015, 54, 2638 CrossRef CAS PubMed
- D. Wang, F. Wu, Y. Song, C. Li and L. Zhou, J. Alloys Compd., 2017, 728, 1030 CrossRef CAS
- H. Liu, L. Xu, W. Liu, B. Zhou, Y. Zhu, L. Zhu and X. Jiang, J. Colloid Interface Sci., 2018, 515, 27 CrossRef CAS PubMed
- A. Gupta, V. Arunachalam and S. Vasudevan, J. Phys. Chem. Lett., 2016, 7, 4884 CrossRef CAS PubMed
- X. Zhou, B. Xu, Z. Lin, D. Shu and L. Ma, J. Nanosci. Nanotechnol., 2014, 14, 7250 CrossRef CAS PubMed
- K. M. Garadkar, A. A. Patil, P. P. Hankare, P. A. Chate, D. J. Sathe and S. D. Delekar, J. Alloys Compd., 2009, 487, 786 CrossRef CAS
- H. Li, Q. Zhang, C. C. R. Yap, B. K. Tay, T. H. T. Edwin, A. Olivier and D. Baillargeat, Adv. Funct. Mater., 2012, 22, 1385 CrossRef CAS
- S. A. Bhakhar, N. F. Patel, C. K. Zankat, M. Tannarana, G. K. Solanki, K. D. Patel, V. M. Pathak and P. Pataniya, Mater. Sci. Semicond. Process., 2019, 98, 13 CrossRef CAS
- A. Jawaid, D. Nepal, K. Park, M. Jespersen, A. Qualley, P. Mirau, L. F. Drummy and R. A. Vaia, Chem. Mater., 2015, 28, 337 CrossRef
- A. Chowdhury, S. Biswas, D. Mandal and A. Chandra, J. Alloys Compd., 2022, 902, 163733 CrossRef CAS
- Z. Zhang, L. Fan, W. Liao, F. Zhao, C. Tang, J. Zhang, M. Feng and J.-Q. Lu, J. Catal., 2022, 405, 333 CrossRef CAS
- J. M. Soon and K. P. Loh, Electrochem. Solid-State Lett., 2007, 10, A250 CrossRef CAS
- Y. Xie and P. Sun, J. Nanopart. Res., 2018, 20 CAS
- Z. Masoumi, M. Tayebi and B. K. Lee, Ultrason. Sonochem., 2021, 72, 105403 CrossRef CAS PubMed
- D. Sahoo, B. Kumar, J. Sinha, S. Ghosh, S. S. Roy and B. Kaviraj, Sci. rep., 2020, 10, 10759 CrossRef CAS PubMed
- A. Ghorai, S. K. Ray and A. Midya, ACS Appl. Nano Mater., 2019, 2, 1170 CrossRef CAS
- K. Krishnamoorthy, G. K. Veerasubramani, S. Radhakrishnan and S. J. Kim, Mater. Res. Bull., 2014, 50, 499 CrossRef CAS
- H. Xiao, S. Wang, S. Zhang, Y. Wang, Q. Xu, W. Hu, Y. Zhou, Z. Wang, C. An and J. Zhang, Mater. Chem. Phys., 2017, 192, 100 CrossRef CAS
- L. Ma, L.-M. Xu, X.-P. Zhou and X.-Y. Xu, Mater. Lett., 2014, 132, 291 CrossRef CAS
- K.-J. Huang, J.-Z. Zhang, G.-W. Shi and Y.-M. Liu, Electrochim. Acta, 2014, 132, 397 CrossRef CAS
- L.-Q. Fan, G.-J. Liu, C.-Y. Zhang, J.-H. Wu and Y.-L. Wei, Int. J. Hydrogen Energy, 2015, 40, 10150 CrossRef CAS
- L. Wang, Y. Ma, M. Yang and Y. Qi, Appl. Surf. Sci., 2017, 396, 1466 CrossRef CAS
- D. Mandal, J. Halder, P. De, A. Chowdhury, S. Biswas and A. Chandra, ACS Appl. Energy Mater., 2022, 5, 7735 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2na00807f |
| This journal is © The Royal Society of Chemistry 2023 |