
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanotechnology-based diagnostics and therapeutics in acute lymphoblastic leukemia: a systematic review of preclinical studies†
Reyhane
Khademi‡
 *abc,
Zahra
Mohammadi
de,
Rahele
Khademi
ab,
Amene
Saghazadeh
fg and
Nima
Rezaei
*fgh
*abc,
Zahra
Mohammadi
de,
Rahele
Khademi
ab,
Amene
Saghazadeh
fg and
Nima
Rezaei
*fgh
aSystematic Review and Meta-Analysis Expert Group (SRMEG), Universal Scientific Education and Research Network (USERN), Tehran, Iran. E-mail: khademi.1358@yahoo.com
bImmunology Board for Transplantation and Cell-Based Therapeutics (Immuno_TACT), Universal Scientific Education and Research Network (USERN), Tehran, Iran
cDepartment of Medical Laboratory Sciences, School of Para-medicine, Ahvaz Jundishapour University of Medical Sciences, Ahvaz, Iran
dRadiological Technology Department of Actually Paramedical Sciences, Babol University of Medical Sciences, Babol, Iran
eSystematic Review and Meta-Analysis Expert Group (SRMEG), Universal Scientific Education and Research Network (USERN), Babol, Iran
fResearch Center for Immunodeficiencies, Children's Medical Center, Tehran University of Medical Sciences, Dr Qarib St, Keshavarz Blvd, Tehran 14194, Iran. E-mail: rezaei_nima@tums.ac.ir; Rezaei_nima@yahoo.com; Fax: +98-21-6692-9235; Tel: +98-21-6692-9234
gIntegrated Science Association (ISA), Universal Scientific Education and Research Network (USERN), Tehran, Iran
hDepartment of Immunology, School of Medicine, Tehran University of Medical Sciences, Tehran, Iran
First published on 11th January 2023
Abstract
Background: Leukemia is a malignant disease that threatens human health and life. Nano-delivery systems improve drug solubility, bioavailability, and blood circulation time, and release drugs selectively at desired sites using targeting or sensing strategies. As drug carriers, they could improve therapeutic outcomes while reducing systemic toxicity. They have also shown promise in improving leukemia detection and diagnosis. The study aimed to assess the potential of nanotechnology-based diagnostics and therapeutics in preclinical human acute lymphoblastic leukemia (h-ALL). Methods: We performed a systematic search through April 2022. Articles written in English reporting the toxicity, efficacy, and safety of nanotechnology-based drugs (in the aspect of treatment) and specificity, limit of detection (LOD), or sensitivity (in the aspect of the detection field) in preclinical h-ALL were included. The study was performed according to PRISMA instructions. The methodological quality was assessed using the QualSyst tool. Results: A total of 63 original articles evaluating nanotechnology-based therapeutics and 35 original studies evaluating nanotechnology-based diagnostics were included in this review. As therapeutics in ALL, nanomaterials offer controlled release, targeting or sensing ligands, targeted gene therapy, photodynamic therapy and photothermic therapy, and reversal of multidrug-resistant ALL. A narrative synthesis of studies revealed that nanoparticles improve the ratio of efficacy to the toxicity of anti-leukemic drugs. They have also been developed as a vehicle for biomolecules (such as antibodies) that can help detect and monitor leukemic biomarkers. Therefore, nanomaterials can help with early diagnostics and personalized treatment of ALL. Conclusion: This review discussed nanotechnology-based preclinical strategies to achieve ALL diagnosis and therapy advancement. This involves modern drug delivery apparatuses and detection devices for prompt and targeted disease diagnostics. Nonetheless, we are yet in the experimental phase and investigational stage in the field of nanomedicine, with many features remained to be discovered as well as numerous problems to be solved.
1 Introduction
Acute lymphocytic leukemia (ALL) is a kind of leukemia that develops when lymphocytes in bone marrow (BM) proliferate abnormally.1–8 It is divided into two types based on the cell of origin: B-ALL and T-ALL. There are four stages of the treatment for ALL, which usually take two to three years: induction, consolidation, intensification, and long-term maintenance. Besides, routine CNS prophylaxis is proposed, and allogeneic hematopoietic stem cell transplantation (Allo-HSCT) stands as the standard consolidation treatment in high-risk patients with an available donor.7–11 If not treated, the leukemic cells are distributed to the blood circulation system and subsequently to vital body organs. Moreover, liquid tumors, such as leukemia, cannot be surgically removed, unlike solid tumors.4 Current treatment regimens face significant obstacles, including (i) the need to use a high amount of agents to guarantee their delivery to target cells, (ii) the requirement to combine numerous agents to boost therapeutic efficiency and diminish the multidrug resistance (MDR) process, and (iii) the destruction of healthy tissues and drastic toxicity.2–4,12–14 It has been demonstrated that while standard therapy can eliminate the bulk of the disease cell population, several resistant leukemic stem cells (LSCs) remain alive, leading to chemoresistance and unfavorable side effects.1–4,15,16 There is a high societal cost in terms of healthcare and the quality of life for patients from the start of treatment until unending relapse-dependent therapy.6 As a result, finding and developing smart delivery methods that efficiently preserve and distribute therapeutic medications, improve targeting capabilities, and accomplish controlled release are critical to healing leukemia.2–4,17–19Conventional detection methods show some restrictions, such as being costly, laborious, and time-consuming.20 They require a set of elaborated devices while suffering from low sensitivity and need several processing phases, which makes them unfavourable for easy and fast medical monitoring or analysis.21 Hence, due to the demand for the discovery of better diagnostics and treatment approaches with the highest possible specificity and efficiency and minor toxicity, researchers are trying to develop modern strategies.13
Nanotechnology has recently been developed as a delivery system. A system with a size of 1–1000 nm is referred to as a nanosystem. It first appeared in the medical area in the 1990s, giving rise to nanomedicine. Nanotechnologies can encapsulate and distribute hydrophobic compounds that are difficult to freely administrate and enhance their solubility and biocompatibility.2,19,22 The materials used for nanosystems must be biocompatible, non-toxic, biodegradable, and sufficiently stable for in vivo administration.2,9,10,12,15,19,23,24 Nanomaterials also become great micro-spectroscopic contrasting agents or labels for imaging,9,15,19,25,26 which helps in fast, specific, and sensitive diagnostics and is beneficial in the discovery of minimal residual diseases (MRDs) after treatment.27 Their extremely large surface areas give them multifunctional capability to load several therapeutic drugs accompanied by other factors like stealth agents, targeting elements, triggering strategies, magnetic features, or imaging traits. The multifunctional trait of nano-delivery complexes has been more studied in developing theranostic (therapeutic + diagnostic) strategies for attacking cancer through encompassing a multitude of drug agents with imaging probes to monitor and scan the therapeutic agents distributed in the body.12,16,28–30 Theranostics provides real-time evaluation of the growth or spoiling of cancer cells.28,29 However, non-viral nanoparticle delivery methods are still in the experimental stage. Several issues are to be addressed, including side effects, controlled release, targeted therapy, and the possibility of combinatorial therapy.2–4,10,31
This study systematically evaluated preclinical human-ALL research to explain the potential benefits and limitations of nanomaterial application and provide an overview of their diagnostic and therapeutic efficacy and possible toxicity in vitro and/or in vivo. To our knowledge, this is the first in-depth systematic review of the literature on the function of nanosystems in preclinical h-ALL.
2 Methods
This study followed PRISMA instructions.322.1. Data sources
Electronic databases were used to conduct the literature search (PubMed, Scopus, and ISI Web of Science). The search was conducted using a set of keywords relevant to nano-based materials and their use in treatment or detection applications. The database search had no time limit and occurred on April 15, 2022. ESI File 1† includes database search terms for each database.2.2. Data sources
During the study selection, a two-stage method was followed. The review writers assessed the title, abstract, and keywords against the qualifying criteria in the first step. If the studies were eligible or doubted to be included, they were selected for the second stage of the review, i.e., detailed review. In the second step, the full text of the articles was reviewed, and the review authors evaluated the studies if they satisfied the eligibility criteria. Any discrepancies between reviewers were handled following discussion and agreement.2.3. Eligibility criteria
Studies were included if they met the following criteria: (1) were written in English with the full text available; (2) used human-ALL as the objective disease; (3) were studied in vitro by using cellular lines and/or in vivo by using animal models; (4) research papers offered at least one treatment plan and/or one detection plan in comparison to routine approaches. Review articles were excluded. Interventions directing therapeutic substances coupled with nanostructures were of interest.2.4. Data items and quality assessment
We created a data extraction sheet that was pilot tested and optimized for gathering data for this review. The information obtained from each selected paper included: (1) in vitro and/or in vivo patterns used, comparing the free drug effects against their nano-conjugations; (2) theranostics results depicted in words of diagnostic/therapeutic plans used, and therapeutic efficiency gained (determined by cellular viability, tumor size/volume diminution or histological examinations); (3) toxicity effects (also determined by cellular viability, animal survival rate or animal weight variations, or histological examinations); (4) nanomaterials as diagnostic tools in monitoring leukemic cells (also determined by specificity, sensitivity or LOD).The studies were selected by full-text evaluation. Then, two reviewers separately assessed them for methodological quality based on criteria taken from the QualSyst tool for quantitative/qualitative research33 to meet the prerequisites of the present systematic review (Table S1 ESI†). All selected studies covered the minimum threshold for inclusion.
2.5. Outcomes
The primary outcomes were those associated with therapeutic effects and diagnostic applications. Therapeutic efficacy in vitro is assessed using tumor cell IC50/viability or differentiation status and the extent of tumor growth regression. In vivo, the quality of mice life, survival rate, remission status, event-free survival (EFS), overall survival (OS), and relapse rate apply for treatment evaluation. Secondary outcomes included the toxicity, side effects, and safety of the proposed nanomaterial systems for therapeutic reasons. The LOD, sensitivity, and specificity were of interest for detection purposes.3 Results
The PRISMA flow diagram displays the study selection process (Fig. 1). Of 2804 studies found from PubMed, Web of Science, and Scopus, 242 were removed as duplicates, and 2562 were screened. According to defined exclusion criteria, 2410 studies were excluded through abstract/title screening. One hundred and fifty two studies for treatment and detection were considered for the full-text evaluation, and 54 studies were also excluded because of not satisfying the inclusion criteria, using other cell lines instead of humane ALL (n = 5),34–38 not being totally relevant to our objective, or lacking detailed and obvious data (n = 23 for treatment;39–61n = 14 for detection)20,62–74 and not having full-text publications available (n = 12).64,75–85 Finally, 63 articles matched the inclusion criteria for the treatment section (n = 63) and 35 articles for the detection section (n = 35). Tables 1–6 summarize the characteristics and findings of the studies included. | ||
| Fig. 1 PRISMA flow diagram of study selection. | ||
| Cell line (year of study) | Nanomaterial/size | Drug, bioactive, gene, photosensitizer | Nano-drug efficacy on leukemic cells | Nano-drug cytotoxic effect on non-target/healthy cells | Release status | Trigger |
|---|---|---|---|---|---|---|
| a ND: not determined. | ||||||
| CCRF-CEM (2009)106 | DNA aptamer sgc8c | Doxorubicin (DOX) | Similar to the free drug | Nontoxic | (Acid-labile linkages) | PH |
| Jurkat (2011)22 | PEG-hydrophobic poly(β-amino ester) (PbAE)/ND | Paclitaxel/paclitaxel oleate in poly(ε-caprolactone) (PCL) | Similar to the free drug | NDa | Controlled release (slow, sustained) | PH |
| CCRF-CEM (2017)108 | sgc8-polyrotaxane-based nanoconstruct (PRNC), PEG/400 nm | Doxorubicin (DOX) | Similar to the free drug | Nontoxic, Safer | Controlled release | PH |
| CCRF-CEM (2018)109 | Liposomal – PEG/110.37 nm | Vincristine (VCR) | Weaker cytotoxic than the free drug targeting form is similar to the free drug | Nontoxic, safer | Controlled release (sustained) | PH |
| 6T-CEM (T-ALL) (2018)105 | Chitosan (CS), CSNP/ND | Zinc ion (Zn) antioxidant | More cytotoxic than the free drug | ND | ND | PH |
| Jurkat (2021)111 | PLGA/ND | Mercaptopurine (6 MP) | Weaker cytotoxic than the free drug | Nontoxic, safer | Burst release initiation, and then slow, sustained release | PH |
| Molt-4 (2011)107 | Sgc8-single-walled carbon nanotubes (SWNTs)/6 nm | Daunorubicin (Dau) | Similar to free drugs | Nontoxic, safer | Controlled release | PH |
| Molt-4 (2015)103 | sgc8-AuNPs/15 nm | Daunorubicin (Dau) | More cytotoxic than the free drug | Nontoxic, safer | Controlled release | PH |
| Molt-4 (2016)104 | sgc8-AuNPs/15 nm | Daunorubicin (Dau) | More cytotoxic than the free drug | Nontoxic, safer | Controlled release | PH |
| CCRF-CEM (2019)112 | Sgc8-MSN/103.24 nm | Doxorubicin (DOX) | Weaker cytotoxic than the free drug targeting form is similar to the free drug | Nontoxic, safer | Controlled release (slow, sustained) | PH |
| Jurkat cells co-culture assay with Nalm-6 (2020)110 | Ionizable lipid nanoparticles (LNPs), C14-4/65.19 nm | CAR T cell: mRNA viral delivery, electroporation EP | similar potent cancer cell killing with C14-4 LNPs, EP, or lentivirus | Nontoxic, safer | Controlled release | PH |
| CEM (2015)121 | AgNPs-mesoporous silica nanospheres (MSNs) – PEG/90 nm | Paclitaxel (PTX) | Cytotoxic effect weaker than the free drug targeting form is similar to the free drug | Nontoxic, safer | Controlled release | GSH |
| CCRF-CEM (2019)120 | Polymeric micelles (PCL-ss-Sgc8-BSA (polycaprolactone)/165.1 ± 5.2 nm | Ara-C | Similar to the free drug targeting form is more cytotoxic than the free drug | Nontoxic | Controlled release | GSH |
| CCRF-CEM (2011)115 | sgc8-hp-Au NP/21.± 1.6 nm | Doxorubicin (DOX) | Similar to the free drug | Nontoxic, safer | Controlled release | Light, temperature |
| CCRF-CEM (2011)128 | sgc8-G-quadruplex/ND | Photosensitizer TMPyP4 (porphine: parent of porphyrin) | More effective than TMPyP4 alone | Nontoxic, safer | ND | Irradiation |
| Jurkat (2016)100 | poly(methyl methacrylate) (PMMA)/100 nm | Zinc(II) phthalocyanine (ZnPc), (photosensitizer: PS) | More effective than free ZnPc | Nontoxic | Controlled release (slow, sustained), diffusion | Irradiation |
| CCRF-CEM LCK high (2020)129 | DMAP-EDCI-DMF + zinc(II) phthalocyanine (ZnPc), (photosensitizer: PS): C5/ND | Dasatinib (small-molecule-tyrosine kinase inhibitor) | More cytotoxic than the free drug | Nontoxic | ND | Irradiation |
| Daudi cell (2002)114 | PEG-dipeptidyl linker-mAb/ND | Adriamycin (ADR) | Weaker cytotoxic than the free drug | Nontoxic | Controlled release (slow, sustained) | Enzyme |
4 Discussion
Although the study designs, interventions, and duplicated results differed substantially, our team tried to descript systematically the results of the included studies (organized in Tables 1–6). We also tried to discuss their correlations, and limitations, in a feasible qualitative evaluation as an alternative of a meta-analysis. The intention of this systematic review was to study the therapeutic, diagnosis and theranostic effects of various nanomaterials on human Acute Lymphoblastic Leukemia (h-ALL) pre-clinically. This systematic study was conducted since the NP system has not been analyzed in the aspect of either the therapy effect or diagnostics, and without this conception, the NP role in both the features is compromised.4.1. A brief overview of nanotechnology tools
It is worth to have a brief introduction of nanomaterials with their division in two main categories of organic and inorganic groups.Organic nanoparticles (including PEG, liposomes, micelles, polysaccharides, proteins, and dendrimers) are usually biocompatible and biodegradable. Their high surface-to-volume ratio allows them to load many pharmaceuticals, while their surface chemistry and chemical features enable them to release the loaded molecule in a regulated control manner.15,86 Pegylated nanoparticles (polyethylene glycol, PEG) may circulate in the bloodstream for a long time, namely as “stealth particles,” since they can elude immune system detection and clearance.87,88
Liposomes are used as a drug delivery vehicle because of their unique ability to solubilize water-insoluble organic compounds, preserve pharmaceuticals from degradation, are straightforward to transport to the target location, and have low nonspecific toxicity. The liposome is kept in the circulation in typical healthy tissues because endothelial cells' tight connections prevent particles from leaking out of the channel.10,89 Generally, with a hydrophilic PEG shell, polymeric micelles are an excellent drug delivery strategy for weakly water-soluble anticancer medicines. The polymeric micelles circulate in the blood for a long period and aggregate more at the tumor site, resulting in a more consistent drug release profile.10,89
Natural biopolymers include polysaccharides (chitosan) and protein nanoparticles like transferrin (TF) and albumin.90–92 Human serum albumin (HSA) nanoparticles with free functional groups on their surfaces can be modified to increase complex stability or targeting ability.74,93 Because tumor tissues have a faster metabolic rate and utilize HSA as a nutrient, significant amounts of HSA accumulation occur in cancer sites, making it an ideal carrier for anti-cancer medication delivery.12,94 HSA's intrinsic autofluorescence is accompanied by several drug binding sites as a natural carrier of therapeutic molecules, making it a suitable biological theranostic agent.74
A dendrimer is a polymer nanocarrier with a spherical center and regular branches around it. Various generations of dendrimers were created by modifying the chemical groups on their surfaces (e.g., charge, basicity, and hydrogen bonding capability). Dendrimer–drug conjugates are also made by covalently connecting an antineoplastic agent to dendrimer's peripheral groups.89,91 As a result, many drug molecules may be attached to each dendrimer molecule (multivalent), and the type of the connection bonds aids in the control of therapeutic molecule release.89 Multivalent interactions can overcome monovalent ones that are innately weak.95 Another advantage of the dendrimer is attachment and assembling with DNA clusters (i.e., DNA PAMAM).16,89
Inorganic nanoparticles have a wider range of size and composition-dependent physical characteristics, making them ideal for biological applications, including cell imaging and molecular detection. However, because they are less biocompatible, they are frequently mixed with organic materials for nano-safety. Many forms of inorganic nanoparticles, such as carbon nanotubes (CNTs), quantum dots, magnetic nanoparticles, silica-based, and noble metal nanoparticles (gold and silver, in particular), are employed in anti-cancer applications. These plasmonic-active nanoparticles with strong near-infrared (NIR) resonances can be effective light-to-heat converters for initiating hyperthermia inside living cells. Due to their high surface-to-volume ratio, they can also transport many different chemicals and pharmaceuticals for delivery applications.15,16,96,97
The construction, superficial area, mechanical rigidity, metallic properties, conductivity (both electrical and thermal), and ultra-light weight are some unique physical and chemical characteristics of CNTs. They display higher drug loading than standard liposomes and dendrimer drug carriers based on their large specific surface area. CNT-based medication delivery improves the solubility, blood circulation, and effective regulation of therapeutic release, resulting in lower doses and higher medicinal efficacy. They can be excreted from the body by the kidneys.15,89
Quantum dots (QDs), 2–10 nm semiconducting materials, were firstly introduced in 1980. Compared to traditional fluorescent immunolabelling, Ruan et al. found that QD-based immunolabelling had a more constant photo-intensity leading to significant time and cost savings. Moreover, QD signals have a more selective and brighter photostability than typical organic dyes. Multiplexed cancer biomarker imaging in situ on intact tumor tissue specimens for tumor pathology analysis at the histological and molecular levels is possible with QD-based nanotechnology. Compared to typical immunochemistry experiments, QD immunostaining has been more accurate in detecting low protein expression levels accompanied by a reduced background. QDs have the potential to replace untargeted drug delivery and thereby reduce chemotherapy's undesired effects.16,89,91
4.2. Nanomaterials as therapeutics in ALL
Although the study designs differed substantially, we tried to systematically descript the included therapy related studies' results (Tables 1–3) and discuss their correlations and limitations in a qualitative manner.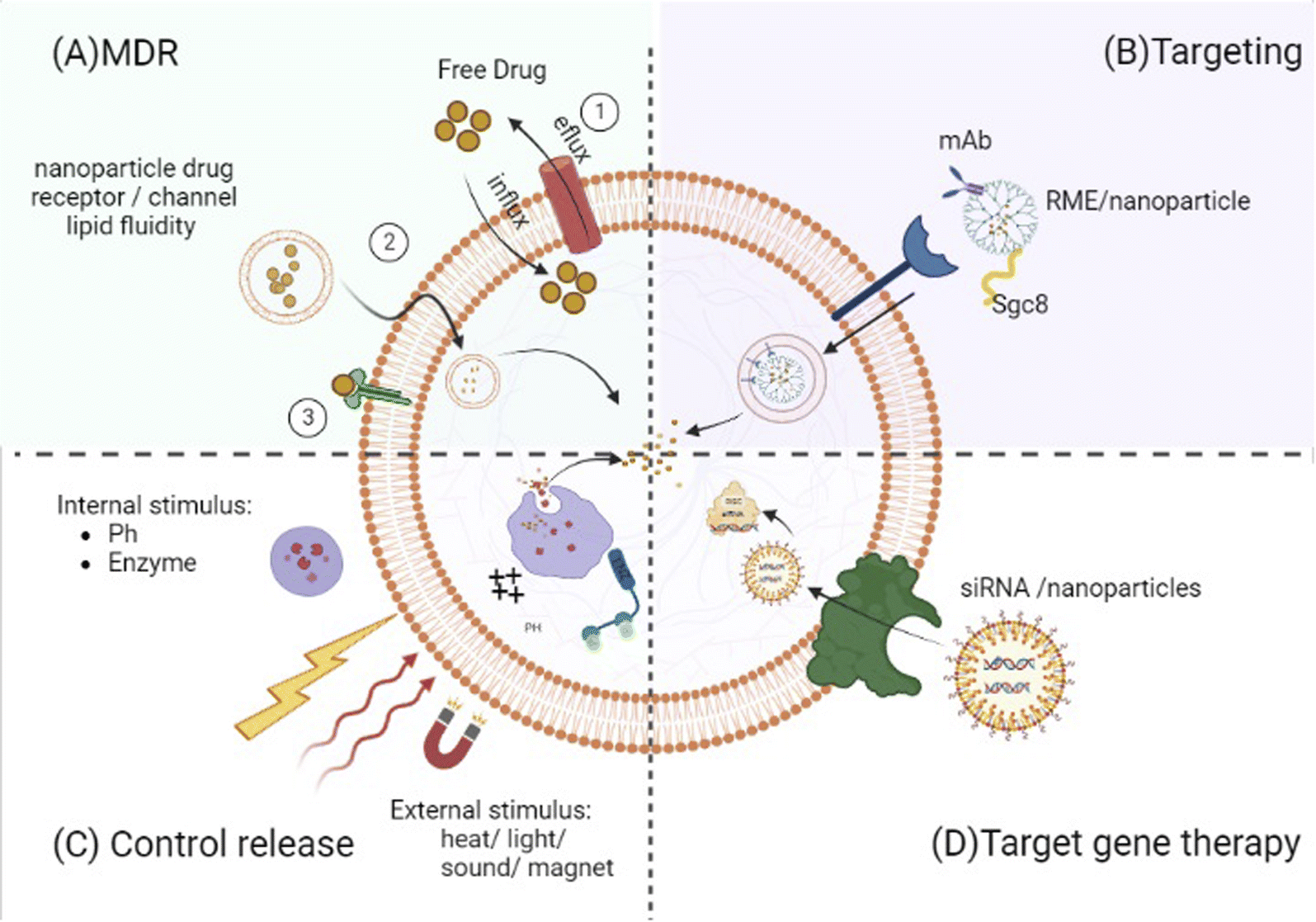 | ||
| Fig. 2 Control release: drug content liberation from nanocarriers can be stimulated by certain microenvironmental parameters at the target location (e.g., pH change and enzymatic activities) or by external stimuli (e.g., heat, light, electric and magnetic field or ultrasound). Targeting: targeting ligands can specifically attach to the malignant cell (e.g., monoclonal antibodies (mAbs) or sgc8 aptamer). Targeted gene therapy: nanoparticle formation of targeted gene therapy agents (e.g., siRNA and TKIS) increased their anti-leukemic potency by enhancement of their stability and/or concentration. MDR (multi-drug resistance). Free small drugs move into malignant cells through passive transmission across the cell membrane, and they come into contact with membrane proteins such as drug efflux pumps leading to less than optimum amounts in the target cell. Lipid packing density and velocity can affect diffusion through the membrane. Increased membrane fluidity implies considerable drug permeability by delivering therapeutic agents into cancer cells without relying on specific receptors or channels, and nanoparticles can circumvent this resistance mechanism. | ||
It has been observed that many tumor cells overexpress certain proteases such as cathepsin and matrix metalloproteases. CALLA is an endoprotease known as NEP (E.C.3.4.24.11.) related to refractory ALL. Accordingly, a dipeptide linker was employed to conjugate adriamycin (ADM) to be cleaved selectively with the CALLA tumor-specific enzyme.114 Chemically induced drug liberation, such as glutathione (GSH)-mediated release, is an ordinary strategy in drug delivery because of significant intracellular and extracellular variation of GSH amounts in some tumor cells. GSH can break disulfide bonds between drugs and nanocarriers and permit them to be released.15,120,121
Release methods dependent on particular changes in the surrounding media, such as pH, may, on the other hand, result in unpredicted distribution of payloads to different cellular sites and cell types. Developing a finely responsive system that is sensitive to minor environmental changes remains a key issue. As a result, far-away triggers that can be regulated exogenously come into the scene.115 For example, a heat-dependent drug liberation can be designed in the nanosystem coupled with a temperature-sensitive polymer.15 The temperature-dependent laser light causes the slow release of Dox molecules from the nanocarrier hp-AuNP by the input photon energy leading to a local photothermal heating reaction (hyperthermia).115
Several nanoparticles that selectively and serially react to two or more stimuli have been created to increase controlled drug release. Various combinations of stimuli were tested with activation by synchronic triggers in one cellular area or in a serial manner as the particle moves through different biological sections. These strategies resulted in greater drug release, which promised curative efficiency improvement.15,59,115
Last but not least, it is worth mentioning that drug diffusion,100,102,122 solvent,122,123 degradation, erosion,99 swelling, and selective burst,74,111,120 in addition to stimuli-controlled release are some strategies of drug release from nanocarriers. As more water enters the nanocapsules, water–polymer interactions become more likely than polymer–polymer interactions, causing the separation of polymer chains and disassembly of the nanoparticle in the erosion release profile.99 The slow and sustained release of the drug encapsulated within the stable core of serum albumin nanoparticles may be preceded by a fast initial burst release or erosion due to the breaking of the surface-adsorbed drug.74 The PEG section also plays a key role in controlling the drug release rate that ensures prolonged release, which may cause drug diffusion and dissolution from the reservoir to be slow and sustained.24,109 Generally, the quick release is thought to aid in achieving effective blood concentrations and promoting immediate illness alleviation, while the sustained release aids in maintaining a stable blood concentration. As a result, a lengthy exposure duration maintains significantly larger agent concentrations in the cells, resulting in more damage and increased cell death.111
| Ligand/nano-drug target | Function | Internalization mechanism |
|---|---|---|
| a ND: not determined. | ||
| Anisamide (AA)126 | Targets overexpressed sigma receptors | ND |
| Sgc8 (94, 103, 104, 106–109, 112, 113, 115, 118–121, 128, 141 and 194) | Targets PTK7 (protein tyrosine kinase-7) overexpressed in T-ALL cells | Internalized via endocytosis |
| Anti-CD19(Ab)131,133,195 | Targets CD19 overexpressed in B-ALL cells | Internalized via endocytosis |
| NL-1 antibody114 | Targets overexpressed CALLA (CD10) in B-ALL cells | Internalized via endocytosis |
| Anti-CD3e f(ab')2 fragments132 | Targets CD3 on T-cells | Internalized via endocytosis |
| Heptapeptide DT7, transferrin90,136 | Targets the transferrin receptor (TfR, also named CD71) overexpressed in T-ALL cells | Internalized via endocytosis |
| Peptidomimetic LLP2A125 | Targets activated α4β1 integrin expressed on childhood ALL cells | Internalized via endocytosis |
| Nonapeptide CD21 (CR2)135 | Targets CD21 (CR2) | Internalized via endocytosis, pinocytosis |
| CD22ΔE12 SiRNA50,146 | Targets CD22ΔE12 in aggressive B-ALL cells | None |
| WHI-P131 (ref. 87) | Targets tyrosine kinase JAK3 that regulates the activation of some important oncogenic proteins | None |
| C61 (ref. 145 and 146) | Targets the SYK inhibitor in the resistance B-ALL cells | None |
| Dasatinib129 | Targets LCK tyrosine kinase | None |
| Amino acid L-Phe150 | Targets Pyruvate kinase, PK, to reduce the ROS level | None |
Several chemical techniques for targeting the functionalization of nanomaterials with particular monoclonal antibodies (mAbs) for specific recognition of cell-surface proteins have been discussed.114,131–134 Pendant-type immunoliposomes covered with a mAb demonstrate a greater internalization efficiency than immunoliposomes with antibodies covalently attached to the nanosystems.131 Small benzamides, such as anisamide (AA), can also target overexpressed sigma receptors in various human cancer cell types.126 Due to the upregulation of receptors on the surface of cancer cells, targeted nanoparticles can discriminate between cancer and normal cells.95 In this review, we can point to some peptidomimetic ligands with high affinity and high specificity like LLP2A against activated α4β1 integrin,125 a nonapeptide for CD21 (CR2),135 and heptapeptide DT7, as a new target for the transferrin receptor (TFR or CD71 which is highly expressed in T-ALL cell lines).136
Because ALL is a liquid tumor, passive targeting through the increased permeability and retention (EPR) effect is not applicable. The gathering of non-targeted nanoparticles in the liver and spleen can be beneficial in the treatment of ALL, which are the main organs for leukemic blast accumulation and proliferation. In contrast, nanoparticle accumulation reduces its active filtration or clearance by organs like lungs and kidneys.17 The biology and cytotoxicity effects of nanoparticles are governed by their physicochemical qualities. For example, the charge of the nanostructure has been linked to the interactions with various cellular characteristics or receptors and causes distinct results in target cells. Thus, nanostructures prefer cancer cells to PBMCs (despite sharing the same origin with leukemic cells).105,137,138 However, the absence of targeting ligands restricts the uptake rate of leukemic cells, mostly in vivo, regardless of their existence in the systemic circulation flow.17 The IC50 values of some drug-conjugated nanoparticles of therapeutic agents were similar to or higher than those of the free drug in some studies.17,94,102,108,109,111,119,120,122,123,128,131,134 A possible explanation is that the special receptor for conventional drugs (e.g., multidrug and toxin extrusion protein 1, MATE1, accepting imatinib as a substrate) is not available within the nanosystem formulation.139 Furthermore, the drug–nanoparticle complex may represent a negatively charged nanomaterial similar to the charge of the cell membrane, and consequently, it is impossible to be taken up via passive processes. Thus, without targeting the nanoparticles, the drug uptake and efficacy may be diminished.119 Finally, even in targeted-nanoparticle ones, the nanoparticle must first be internalized via endocytosis or phagocytosis and then released into the space.102,111,115,140 In contrast to the time delay for cellular uptake and release of drugs from nanosystems, free drugs diffuse immediately. IC50 values are influenced by the drug toxicity and the cell viability, as the termination point. A time delay for cytotoxicity induction is an important item in accepting the absence of significant differences in the IC50 values between free and encapsulated forms.17,74 The internalization mechanisms of nanoparticles include macropinocytosis,135 caveolin,119 and clathrin-dependent15 endocytosis.133 Targeting agents usually enter cells via receptor-mediated endocytosis (RME).107,118,136
It has been shown that targeted drug delivery may not significantly change the cell viability or drug's intracellular concentration compared to the drug alone. In contrast, high toxicity and intracellular drug concentration were seen in the drug–targeted-nanoparticle complex.107 It can be explained by the fact that the large surface area of nanoparticles offers the possibility of the presence of multiple building blocks of the targeting ligand.104
The Sgc8 aptamer is the most commonly used targeting ligand in the studies evaluated in this review. It binds specifically to PTK7, a transmembrane receptor protein tyrosine kinase and a potential biomarker for T-ALL.94,103,104,106–109,112,113,115,118–121,128,141,142 Moreover, PTK7 knockdown suppresses the Wnt signaling pathway, affecting cell proliferation and tumorigenesis.142 The more stability of aptamers rather than antibodies makes them suitable for difficult conditions such as high temperatures.143 Moreover, aptamers have a smaller size relative to antibodies, leading to more and faster penetration into cancer tissues.94 Some studies used aptamers as both drug delivery carriers and targeting ligands.103,104,106,141 However, besides the high cost, it is necessary to point to its renal filtration and nuclease degradation in the biological environment. Researchers are trying to make modifications to further improve aptamers' properties, like conjugating them to nanoparticles.1,103,104,142 Besides, a high salt concentration in biological liquids could aggregate nanoparticles (e.g., AuNPs or SWNTs). Conjugation with an aptamer could enhance the solubility of nanoparticles and prevent them from aggregation.103,104 A poly-aptamer–drug complex as a polyvalent aptamer complex displayed noticeably more efficiency than its monovalent analog in both cytotoxicity and selectivity as opposed to leukemia cells because of the amplified binding affinity of multivalence.94,103,118,142 The feasibility of incorporation of multiple drug molecules within one aptamer was assumed. Although this operation is relatively impossible in the usual mAb-based immunoconjugate methodology, the IC50 of sgc8c–3Dox compared to that of sgc8c–Dox with target cells showed no significant difference, which was less than expectations. This phenomenon was attributed to the reduced binding space and, as a result, the ability of the aptamer in the form of sgc8c–3Dox to bind to target cells.106
Most nanoparticle formulations decorated with target agents enhanced the anti-leukemic effect in vitro and in vivo more than typical drug solutions. Leukemic mice that underwent the treatment of drug–targeted-nanoparticles lived longer and exhibited lower obvious systemic toxicity than mice treated with traditional free therapeutic agents.108,109,120,121,133
An ideal and most favorable nanoparticle system would put together the high stability level of drugs with high loading proportions to reduce the amount of the drug content essential for administration and improve the selectivity for target tissue sites.124 Some studies showed that drugs nanoencapsulated without targeting ligands have more potent anti-leukemic activities than free ones. The nanomaterials utilized included dendrimers,100,138 chitosan (CS),99,105 human serum albumin nanoparticles (HSA),94 liposomes,126 and polymer PLGA-PEG.144 The following factors can explain the improved efficacy of nanoencapsulation: (i) a high rate of metabolism in tumor cells and, as a result, higher consumption of nanomaterials (e.g., HSA) as a source of energy,94 (ii) protection of agents attached from enzymatic degradation in biological media,92,126,138 and (iii) improved uptake, better internalization properties, control, and sustained release mediated by nanoencapsulation.99,100,105,122,126,144 Poor aqueous solubility limits the hydrophobic chemotherapeutic drugs' utility, leading to accumulation and aggregation in biologic environments. Biomaterial-based strategies can address this problem and help as drug delivery apparatus by increasing drug uptake.74,99,100,122,138,144 Cationic nanoformulations interact strongly with negatively charged cell membrane surfaces for immediate internalization.105,126,138
Albumin-bound molecules aggregate preferentially in tumors due to the secretion of the albumin-binding SPARC (secreted protein, acidic, and rich in cysteine) protein. The reduced apoptosis of cells might, on the other hand, be due to albumin's well-known anti-apoptotic actions.74
Instead of passive diffusion of the medication across the membrane, encapsulating the drug in liposomes permits it to be delivered into the cells' interior by vascular fusion with the membrane.90 The trafficking channels and amounts of enzymes for cleaving drugs from their carrier differ in leukemic and normal cells. The conjugate's cellular accumulation depends on the drug influx and efflux activities. Free drugs were usually transported quicker to leukemic cells than their conjugate. Additionally, the rate of drug-conjugated efflux in leukemia cells was lower than in free ones, while they were equivalent in normal PBMC.90
Although high-dose imatinib, a BCR-ABL tyrosine kinase inhibitor, was recommended to overcome imatinib resistance, it had to be stopped due to toxicity. The IC50 of imatinib-PEG-liposomes (liposomal nanoparticles (LNPs)) was greater than that of free imatinib, which can be explained by the nanomaterial covering the particular receptor, MATE1. Imatinib-CD19-PEG-liposomes had stronger anti-leukemic effects, with less damage to normal hematopoietic progenitor cells than free imatinib, and prevented the development of cobblestone areas (CAs) of leukemia cells from patients with imatinib resistance.131
WHI-P131-NP, a nanoencapsulated tyrosine kinase JAK3 inhibitor, outperformed WHI-P131 and vincristine in vivo, regardless of chemosensitivity or resistance profile.87
Besides the damage to non-hematopoietic organs as a key restriction,127 TBI-based therapy regimens cannot prevent leukemic relapses after HSCT, especially in high-risk cases with radiation-resistant BPL cells. The invention of novel drugs that can dominate the radiation resistance BPL cells would be a great step in attempts to improve post-HSCT results.146 Spleen tyrosine kinase (SYK) is a chief regulator of anti-apoptotic signaling pathways in B-ALL cells. Compound 61 (C61), as a highly selective SYK inhibitor, induces apoptosis in the resistance malignant B-ALL cells. C61's low water solubility and life-threatening off-target adverse effects, on the other hand, have proven to be substantial roadblocks to its continued development as an anti-leukemic therapeutic candidate. The anti-leukemic potency of C61-LNP 25A, a safe liposomal nanoparticle formulation, was much higher than irradiation against aggressive radiation-resistant B-precursor ALL. Because irradiation/chemotherapeutic-resistant leukemic cells do not have cross-resistance to C61, novel combination strategies with multiple chemotherapy agents can be explored.145 Furthermore, the C61-LNP + TBI combination was outstandingly more efficient than either TBI alone or C61-LNP alone without causing significant adverse effects.146
Dysfunctional CD22 following deletion of exon 12 (CD22ΔE12) represents aggressive B-ALL cells. The mutant protein is deficient in most intracellular domains considered the key regulatory signal transduction items.4,6 The expression of genes correlated with the MAPK, PI3-K/mTOR, and WNT pathways was elevated differently in response to CD22ΔE12.51 Leukemia gene therapy by small interfering RNA (siRNA) (an effector of the RNA interfering pathway that may down-regulate specific genes linked to disease pathology) has shown considerable promise in leukemia treatment.4,6 Due to fast enzymatic breakdown in the blood, fast clearance rate, and poor entrance rate into target cells, systemically given unformulated siRNA has little RNAi action in vivo.6,50 Genes and short RNAs can be electrostatically linked to nanoparticles or conjugated onto their surfaces. Natural organic polymeric nanoparticles, synthetic polymers, and inorganic nanoparticles (including carbon nanotubes, gold nanoparticles, and quantum dots44) have all been extensively exploited as cancer gene carriers in numerous cancer treatment studies.89 Nanocarriers in gene therapy should have three critical characteristics: high loading efficiency, the ability to be carried from endosomes to the cytosol, and the ability to release the genetic material.110,147 Using lipid nanoformulations (LNFs) of CD22ΔE12-siRNA, both CD22ΔE12-radiation and -chemotherapy-resistant leukemic clones were killed. A CD22ΔE12-siRNA LNF, alone or combined with chemotherapeutic medicines, was much more effective than those treatments alone in preventing leukemic clonogenicity in vitro and in vivo.50
Current high-cost clinical-scale T lymphocyte production (for chimeric antigen receptors, CAR-T cell) necessitates a series of complex methods, including extracting, genetically altering, and selectively expanding the genetically engineered T cells before re-injecting them into the patient.6,110,132 These present CAR T cell engineering strategies rely on viral delivery, which results in persistent CAR expression and the danger of severe consequences associated with viral transduction. Although mRNA has been introduced as a possible technique for producing transitory CAR expression in T cells without causing genomic changes and mitigating the negative consequences of viral expression, it is most typically used in electroporation (EP) for T cell transfection, which can be cytotoxic. Using C14-4 ionizable lipid nanoparticles (LNPs), Billingsley et al. effectively transferred CD19-targeted CAR mRNA to primary human T cells with the same powerful cancer cell killing of EP or lentivirus CAR-T cells generated while reducing cytotoxicity related to EP.110 In another study, nanoparticles containing CD19-specific CAR genes were shown to alter T-cell specificity in vivo as they circulated, resulting in leukemia regression with success comparable to standard adoptive transfer of laboratory-made CAR T-cells (via lentivirus) while avoiding its complications.132
The overexpression of nucleophosmin (NPM) has also been linked to the development of multidrug resistance (MDR) in ALL, prompting the creation of a synthetically produced recombinant NPM binding protein (NPMBP).148,149 Despite having degradable free NPMBP, the DOX-PMs-NPMBP nanoparticles dramatically reduce leukemia cell proliferation, cause apoptosis, and increase the anti-leukemia effect in resistant ALL cells in vitro and in vivo.148
A series of L-phenylalanine-based polymers (ester amide) (Phe-PEA), particularly Metabolic Reprogramming Immunosurveillance Activation Nanomedicine (MRIAN), showed specificity-enhanced BM accumulation and crossing of the brain–blood barrier. It could enhance uptake by T-ALL and myeloid-derived suppressor cells (MDSCs) as an efficient delivery approach while sparing non-leukemic normal cells and reducing tissue toxicity. Accordingly, free Dox and even liposome-encapsulated Dox demonstrated lower cellular absorption rates than MRIAN-Dox-PLGA NP (Doxil). Besides the role of a drug carrier, it disturbed the immunosuppressive function of MDSCs by degradation to L-Phe amino acid.150
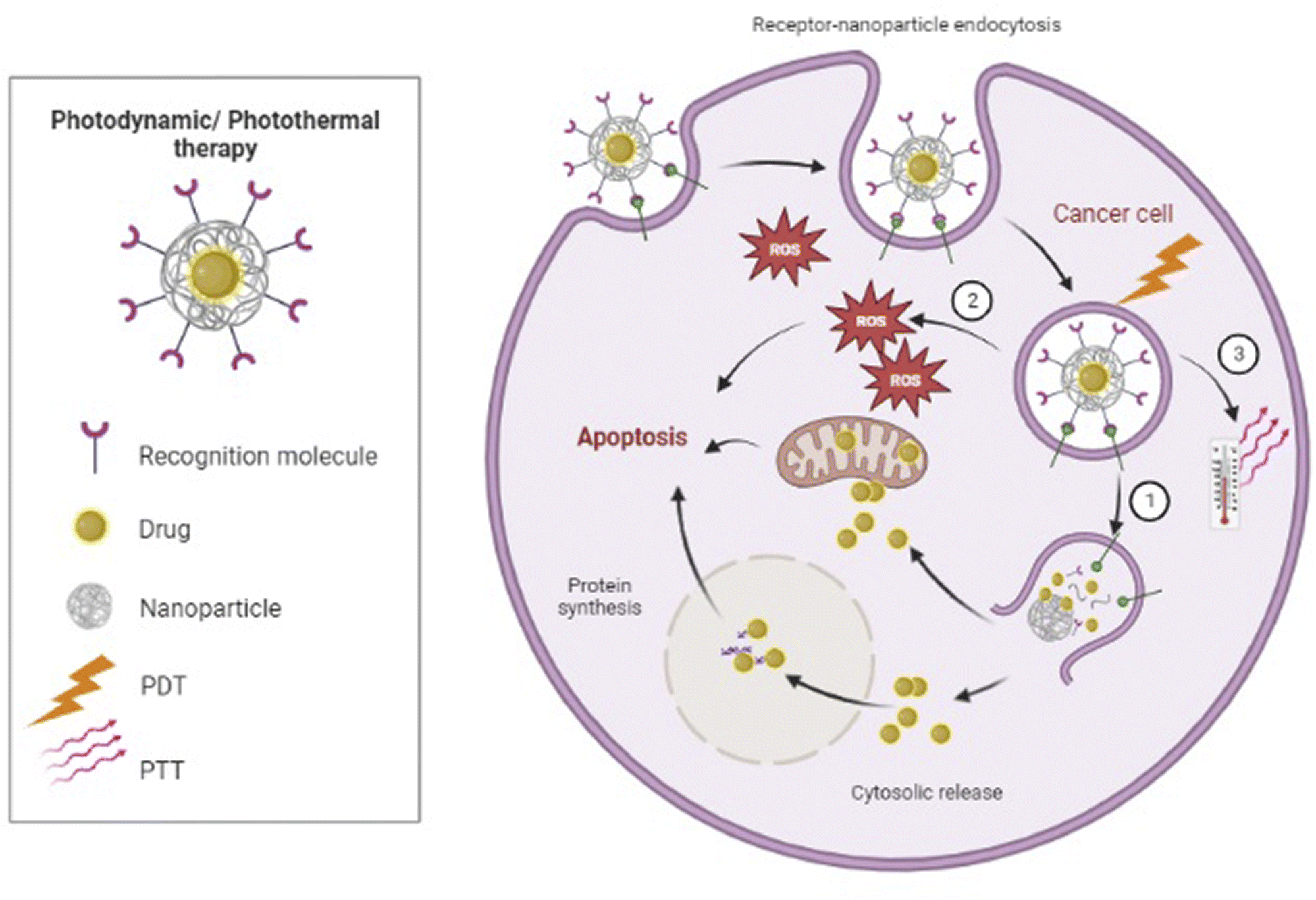 | ||
| Fig. 3 Photodynamic therapy (PDT): ① the irradiation light would be utilized for the release of the drug encapsulated. ② The irradiation light excite the photosensitizer (PS) in the tumor tissue, causing the PS to form a ROS that kills tumor cells. Photothermic therapy (PTT): ③ upon radiation absorption, CNTs or metal nanoparticles can transform photon energy to thermal energy, which results in a temperature rise and subsequently makes cellular destruction. | ||
Non-invasive photothermic therapy (PTT) is another nanoparticle-based therapeutic strategy. Upon radiation absorption, CNTs or metal nanoparticles such as Au-based nanomaterials can transform photon energy to thermal energy, which results in a temperature rise and subsequently causes cellular destruction or death due to hyperthermia155,156 (Fig. 3). Nonetheless, PTT as a single therapy is generally not enough for complete tumor ablation.155 Wang et al. designed a model of multimodal therapy, PDT accompanied by PTT, for attacking and destroying T-ALL, but their study did not fit our inclusion criteria. However, they demonstrated that PTT/PDT had a synergistic effect and could kill more tumor cells than either therapeutic strategy alone.156
By altering the pharmacokinetic features of therapeutic agents, nanomedicines will raise the probability of active chemicals showing increased circulatory retention and local concentrations at the place of interest but, at minimum, exposed to healthy organs.10,50,87,145,146,148,157,158 Accordingly, nanomedicines may somewhat, if not fully, overcome MDR. Because free small drugs almost entirely move into malignant cells through passive transmission across the cell membrane, they contact membrane proteins such as drug efflux pumps leading to less than optimum amounts in the target cell, resulting in MDR10 (Fig. 2). Nanomedicines are still frequently taken up through endocytosis,10,133 so receptor-mediated endocytosis is considered for 100–200 nm nanoparticles, whereas phagocytosis is used for bigger (500 nm) and smaller (25 and 50 nm) ones.101 As a result, they skip the drug efflux mechanism, resulting in a significantly higher concentration within the cell than with a free drug.10 Some anti-leukemic drugs, such as cisplatin and doxorubicin, have induced apoptosis in tumor cells via increasing cell membrane fluidity and clustering of lipid rafts.52 The lipid packing density and velocity can affect diffusion through the membrane. Increased membrane fluidity implies considerable drug permeability. Cholesterol, which is known to diminish the fluidity of membranes, limits the drug uptake and accumulation in both drug-sensitive and drug-resistant cells. As a result, liposomes with no encapsulated therapeutic agents could enhance the cytotoxicity of vinblastine (VLB) in CEM/VLB100 cells (vinblastine resistant) by around ten-fold and were even significantly more efficient than the efflux pump inhibitor, verapamil, alone. On the other hand, parent-sensitive CEM cells were unaffected by this liposome treatment. This led to the hypothesis that liposomal lipids inserted into the plasma membranes of resistant cells modified their characteristics and fluidity, allowing for a considerable restoration of drug accumulation with no effect on the rate of drug efflux from those cells.159 The activation of apoptosis, the reduction of tumor volume, a decrease of ascites side effects, and longer life were also validated in mice treated intravenously with hybrid liposomes (HL-25) without any medications. These inhibitory effects on tumor cell proliferation should be linked to membrane fluidity.41
Some therapeutic medications require specific proteins to traverse plasma membranes or move across intracellular compartments, and drug resistance develops when their expression is reduced. By delivering therapeutic agents into cancer cells without relying on those particular proteins, nanoparticles can circumvent this resistance mechanism (Fig. 2). For example, because of various endocytic routes between cell lines,90,138 the entrance mechanism or process of the (cytarabine) Ara–CTP-dendrimer (nanoparticle) complex differs in different cells. Such behavior points to the dendrimer's role as a drug importation mechanism. Interestingly, this additional process does not exist in normal PBMCs, where the presence of the dendrimer hinders Ara–CTP activation.138
BCL2, an anti-apoptotic factor, has an important role in T-ALL cell survival. The combined treatment of DEX and GSI encapsulated in nanomaterial inhibited BCL2 expression more synergistically than the free combination model.136
Most in vivo studies (14/18) included were prosperous in attacking and eliminating cancer cells in vitro (Table 3).17,108,109,111,120,121,129,132,133,136,144,146,148,160 By analyzing therapeutic outcomes in vivo, researchers discovered that nano-drug encapsulations boosted anti-tumor activity compared to free chemotherapeutic drugs. Importantly, nanoparticles have been shown to pass the blood–brain barrier and circulate to sanctuary areas, such as the CNS, where higher medication exposure improves overall therapeutic effectiveness.93,150,160 One immediate conclusion is that the high level of tumor uptake might clarify the suitable achievements of these formulations in lowering the tumor burden and increasing the survival rate.17,108,109,111,121,133,136,146,148,161,162 This higher uptake by tumor, as previously referred to, can be attributed to the targeting moiety (sgc8, CD19, targeted tyrosine kinase, and transferrin receptor (TfR, also named CD71)) or stimuli-responsive behavior (pH and GSH-triggered) in some studies.108,120,121,129,133,134,136,144
| Nanomaterial | Drug, bioactive, photosensitizer, or gene | Therapeutic strategy | Animal model/cell line | Bio-distribution | Tumor growth inhibition (tumor ablation, recurrence) | Survival rate | Antileukemic efficacy of the nano-drug | Side effects (systematic toxicity, body weight, and histologic assessment) |
|---|---|---|---|---|---|---|---|---|
| a ND: not determined. | ||||||||
| Liposome163 | Vincristine (VCR) | Liposomal vincristine 50–60 nm, 1 mg kg−1 intraperitoneal | Xenograft NOD/SCID mice, B-ALL | ND | 20% delay in tumor progression | Increased lifespan | Enhanced Antileukemic efficacy compared to the free drug | Well tolerated, safer |
| Liposome161 | cytarabine![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :daunorubicin cocktail (300:4.5 mg kg−1) :daunorubicin cocktail (300:4.5 mg kg−1) |
CPX-351: Cyt:Daun 100 ± 20 nm, 10:4.4 mg kg−1 i.v. |
SCID/Rag2M mice, CCRF-CEM | Considerable and constant tumor uptake (BM) | ND | Increased lifespan | Enhanced Antileukemic efficacy compared to free-drug cocktail | ND |
| Liposome162 | cytarabine:daunorubicin cocktail (300:4.5 mg kg−1) |
CPX-351: Cyt:Daun 10:4.4 mg kg−1 i.v. |
Xenograft Rag2-M mice, CCRF-CEM | Considerable and constant tumor uptake (BM) | Long-term remission in 33% of mice | Increases lifespan | Enhanced Antileukemic efficacy with consolidation compared to induction therapy alone, enhanced Antileukemic efficacy compared to free-drug cocktail | Severe and prolonged cytopenia, slow BM recovery |
| Liposome-PEG87 | WHI-P131 (CAS 202475-60-3) JAK3 tyrosine kinase inhibitor/vincristine (VCR) 0.050 mg kg−1 | WHI-P131-NP 100 nm, 100 mg kg−1 day−1 × 2 days i.p. | Xenograft SCID mouse, resistant B-ALL RS4; 11 | ND | No signs of overt leukemia | Increased lifespan | Enhanced Antileukemic efficacy compared to the free drug and vincristine (VCR) | ND |
| Liposomal Nanoformulation (LNF)-PEG146 | TBI treatment (200y γ-rays) | C61-LNP 136.3 ± 1.2 nm, 80 mg kg−1 i.v./C61-LNP + 2 Gy γ-rays | Xenograft NOD/SCID mouse, relapsed BPL xenograft NOD/SCID | ND: favorable pharmacokinetic of 25A145 | ND | Increased lifespan | Enhanced Antileukemic efficacy compared to the free drug | Well tolerated, safer |
| TBI treatment (400y γ-rays) | C61-LNP 136.3 ± 1.2 nm, 80 mg kg−1 i.v./C61-LNP + 4 Gy γ-rays | CD22ΔE12×BCR-ABL double-Tg mice, radiation-resistant BPL | Lower tumor growth/burden rate remission C61-LNP + TBI mice: 100%, TBI: 50%, C61-LNP: (0) | Increased lifespan | Enhanced Antileukemic efficacy compared to the free drug | |||
| Liposome-PEG109 | Vincristine (VCR) | Sgc8-VCR-Lipo/VCR-Lipo 100 nm i.v. | Female BALB/c nude mice, CCRF-CEM | Liver, tumor accumulation | Lower tumor growth/burden rate | Increased lifespan | Enhanced Antileukemic efficacy compared to the free drug | Well tolerated, safer |
| PEG-PCL-ECT2 (amphiphilic block copolymers)17 | Dexamethasone (Dex) | Dex-NP-PEG 110 nm, 5 mg kg−1 i.v. | Xenograft NSG-B2m mice, human ALL | Spleen, liver accumulate, sustained plasma circulation, lower levels in the kidney and lung (clearance organs) | ND | Increase lifespan | Enhanced Antileukemic efficacy compared to the free drug | Well tolerated, safer |
| Amphiphilic block copolymers-PEG-PCL133 | DOX (doxorubicin) | Anti-CD19-DOX-NPs 75 nm, 2.5 mg kg−1 i.v. | Xenograft NSG-B2m mice, RS4; 11 B-ALL | BALB/c mice: liver, spleen accumulation (targeted NPs) with lower levels in clearance organs | ND | Increased lifespan | Enhanced Antileukemic efficacy compared to the free drug | Well tolerated, safer |
| PEG poly(lactic-co-glycolic acid)144 | IRAK1/4 and ABT-737 cocktail | IRAK/ABT-PEG-PLGA polymer NP 100 nm, 10 mg kg−1 IRAK inhibitor and 40 mg kg−1 ABT-737 | Xenograft female NPG mice, Jurkat | ND | Lower tumor growth/burden rate | Increased lifespan | Enhanced Antileukemic efficacy compared to the free drug | ND |
| Polyrotaxane-based nanoconstruct (PRNC), PEG108 | DOX (doxorubicin) | Sgc8-DOX-PRNCs-PEG i.v. | Balb/c nude mice, CCRF–CEM | ND: rapid clearance of NPs | Lower tumor growth/burden rate | ND | Enhanced Antileukemic efficacy compared to the free drug | Well tolerated, safer |
| Silver (AgNPs)-mesoporous silica nanospheres (MSNs)-PEG121 | Paclitaxel (PTX) | Sgc8-PTX-MSNs-AgNPs-PEG 90 nm, i.v. 1 mg kg−1 | BALB/c nude mice, CEM | Tumor accumulation | Lower tumor growth/burden rate | ND | Enhanced Antileukemic efficacy compared to the free drug | Well tolerated, safer |
| Polymeric micelles (PCL-ss-Sgc8-BSA (polycaprolactone)120 | Ara-C 20 mg kg−1 | Sgc8-PCL-ss-Ara-BSA 165.1 ± 5.2 nm, 20 mg kg−1 i.v. | Tumor-bearing mice, CCRF-CEM | ND | Lower tumor growth/burden rate | Increased lifespan | Enhanced Antileukemic efficacy compared to the free drug | Well tolerated, safer |
| Poly(beta-amino ester) (PBAE) polymer-(microtubule132 | Plasmid DNA encoding the leukemia-specific 194-1BBz CAR (+iPB7 transposase). Infusion of 194-1BBz CAR-T cells transduced ex vivo with 194-1BBz-encoding lentivirus vectors | anti CD3-antiCD19-194-1BBz CAR-(PBAE)-(MTAS)-(NLS) i.v. | Albino C57BL/6 mice, B-ALL (Eμ-ALL01) | Liver accumulation (non-targeted NPs). Spleen, lymph nodes, bone marrow accumulation (targeted NPs) | Lower tumor growth/burden rate | Similar to the conventional group | Similar to the conventional CAR T-cell group | Well tolerated, safer |
| DMAP-EDCI-DMF + zinc(II) phthalocyanine (ZnPc), (photosensitizer: PS): C5 (ref. 129) | Dasatinib (small-molecule-tyrosine kinase inhibitor) | DMAP-EDCI-DMF + (ZnPc), (photosensitizer: PS)–dasatinib: C4 (PDT), tail vein | Nude mice, CCRF-CEM | Tumor accumulation. Rapid clearance of C4 | Lower tumor growth/burden rate | ND | Enhanced Antileukemic efficacy compared to the free drug under light exposure | Well tolerated, safer |
| Copolymer mPEG-PTMC160 | Dexamethasone (Dex) 2.5–5 mg kg−1 i.p. | mPEG-PTMC-Dex 41 nm, 5 mg kg−1 i.v. | NSG mice, primary T-ALL | Broad distribution. Accumulation in the major organs. Circulate to sanctuary sites (BM and BBB) | ND | ND | Enhanced Antileukemic efficacy compared to the free drug | Well tolerated, safer |
| PEG-PLA (polymeric micelles (PMs))148 | DOX (doxorubicin) 3 mg kg−1 i.p. | DOX-PMs-NPMBP 3 mg kg−1 i.p. | BALB/C-nude mice, Nalm6, resistance Nalm6 (B-ALL cell line) | ND | Lower tumor growth/burden rate | ND | Enhanced Antileukemic efficacy compared to the free drug | Well tolerated, safer |
| DOX-PMs-NPMBP 1.5 mg kg−1 i.p. | NCG mice, Nalm6, resistance Nalm6 (B-ALL cell line) | ND | Lower tumor growth/burden rate | Increased lifespan | ||||
| PLGA111 | 6-Mercaptopurine (6 MP) 15.75 mg kg−1 oral | PLGA-6-MP 138.01 ± 0.39 nm 15.75 mg kg−1 oral | Female NPG, Jurkat T-ALL | ND | Lower tumor growth/burden rate | Increased lifespan | Enhanced Antileukemic efficacy compared to the free drug | Well tolerated, safer |
| DSPE-PEG-DT7 (peptide-modified lecithin nanoparticles): TLnp136 | Gamma-secretase inhibitors (GSIs) with dexamethasone (DEX): D&G-sol | DT7-DEX + GSI co-loaded lecithin nanoparticles (TLnp/D&G) 32.36 ± 2.15 nm DEX 30 mg kg−1, GSI 30 mg kg−1 i.v. | NSG, CEM | Tumor accumulation | Lower tumor growth/burden rate | Increased lifespan | Enhanced Antileukemic efficacy compared to the free drug | Well tolerated, safer |
In studies in which there are no bio-distribution details to realize tumor uptake or accumulation,87,120,144,163 some properties of the formulizations might indicate their treatment success. The pharmacokinetic index has clearly improved, with a longer circulation time and a small distribution mass restricted to plasma volume, decreased clearance, and better efficacy/toxicity profiles than free drugs.10 To improve effectiveness, a threshold concentration in bone marrow must be reached and maintained for a long period.162 The nanoparticles' distribution and accumulation level on different organs are strongly linked to their size or scale, the form of shape, and surface charge. Particles of 50–250 nm size mostly accumulated in the liver and spleen for several months, while only particles smaller than 10 nm accumulated and settled in other organs such as the kidneys, lungs, brain, and so on.15,160 A long-time circulation would increase the chances of nanoparticles coming into contact with malign cells in the peripheral blood, a common target location in hematological malignancies. Nanomedicines tend to collect in the liver, spleen, and bone marrow as the mononuclear phagocytic system (MPS) organs annex to tumor sites.10 These organs are also main and/or secondary target areas in most hematological malignancies, which can boost therapy effectiveness even more.10,17,109,133,160 Furthermore, because nanoparticles accumulate less in active filtration organs such as the lungs and kidneys, encapsulated drugs may have a longer half-life than free drugs. Although the dosage employed in the mice did not result in a definite cure, it is notable that nanoparticle encapsulation considerably improved medication effectiveness at lower doses.17,133 Although smaller particles have superior plasma profiles,133 the drug encapsulation amount is limited by nanoparticles smaller than 50 nm.10
There is a long-term circulation of neutral nanocarriers in the blood. Nanocarriers with a negative charge are eliminated from Kupffer cells, whereas positively charged nanocarriers are removed via opsonization.89 Scavenger receptors162 facilitated the absorption of negatively charged nanoparticles (e.g., liposomes)87,163 by bone marrow macrophages. Polymeric micelles (PMs) composed of block copolymers with the hydrophilic shell ensure colloidal stableness and an elongated circulation in vivo.10,120 The degree of PEGylation also influences bone marrow uptake. Slow liberation from long-flowing PEGylated liposomes, on the other hand, might reduce drug concentrations at the (main) desired site.10
Nanomedicines taken orally can be absorbed by microfold cells in Peyer's patches. This leads to improved systemic medication distribution and passive lymphatic targeting. Despite the higher therapeutic efficacy, Zou et al. found that nano-encapsulated 6-mercaptopurine (6-MP) had no sustained releasing impact in vivo. This phenomenon was described by the presence of esterase, which might breakdown the PLGA skeleton via the hydrolysis of ester bonds.111 However, the methylated metabolite of 6-MP (6-MMP) may be harmful to the liver and BM. This study implied that hepatotoxicity and myelotoxicity were lower in the drug-nanoencapsulated group than in the free drug suspension group, owing to the avoidance of direct drug metabolism by liver microsomal enzymes after encapsulation.111
A combination of multiple drugs is frequently employed to attack the tumor cells by the restriction of several oncogenic pathways simultaneously. This often eventuates better anti-leukemic strategies with the drug's effective concentration in leukemia cells, resulting in their long-term death.10,136,144,161,162 A 5:1 cytarabine:daunorubicin molar ratio revealed the reliable modulation of synergy and the lowest possibility of antagonism. In the combination model of the free cytarabine: daunorubicin cocktail, the administered drug ratios depicted an antagonistic couple in vitro after about two hours. The ratios could be kept for an extended period after administration in vivo by the encapsulated form of liposomes and retained the two combined drugs in the desired ratio pattern of their pharmacokinetics. CPX-351 illustrated a notable advancement in the therapeutic profile compared to the free-drug combination form. This strategy keeps the drug ratios away from antagonistic forms in the plasma and locations of growing tumors. When the amounts of drugs in individual liposomal cytarabine and individual liposomal daunorubicin were similar to those related to CPX-351 and tested in tumor-bearing mice, the efficacy obtained from CPX-351 was much better than in two separate liposomal drug agents.161,162 Similarly, the co-delivery of DEX and GSI via nanocarriers reduced AKT (Ser473) phosphorylation and downregulated the anti-apoptotic protein BCL2, resulting in a considerable improvement in anti-leukemia efficacy.136
Metformin inhibits angiogenesis in certain cancers by acting synergistically with cisplatin. Specially formulated lipid-based cubosomal nanoformulations were utilized as drug carriers to promote chemical entrance in low dosages. Combining the medications in a carrier, on the other hand, had an antagonistic impact, indicating that metformin is not a viable alternative for sensitizing leukemia cells to cisplatin.164
Most therapeutic agents are also poorly soluble in water, wherein solubility limits absorption and nanoencapsulations offer them high solubility, which results in higher uptake and sustained release in tumor cells.111,144,145 C61-LNP formulation (alone or in combination with irradiation) intensified the anti-leukemic power of low-dose TBI both in vitro and in vivo. It elevated the event-free survival time and the survival outcome in NOD/SCID mouse models of relapsed BPL more than in untreated control NOD/SCID mice or mice handled with two Gy single-dose TBI alone.145,146 It is worth saying that the treatment protocol of C61-LNP formulation alone against CD22ΔE12×BCR–ABL double-Tg mice (spontaneously expand radiation-resistant lethal BPL) was similar to the untreated ones.146 The study did not notice any explanation for it. Additionally, the report was without biodistribution information, and thus it is arduous to describe the possible proofs why this formulation is not efficient. On the other hand, the in vivo NOD/SCID mouse model showed the anti-tumor efficacy of this agent.146 In another previous study by the same authors, although there were no BM accumulation data, C61-LNP illustrated good pharmacokinetics and a harmless profile in mice.145 Hence, it is believed that this cannot be relevant to the absence of tumor uptake. We can at least hypothesize some possible factors like a larger tumor burden or the type of animal model in which a special BM microenvironment protects leukemic cells from nanoparticle encapsulation of targeted therapies. This protecting model might be an inducing factor of the malignant cell refractory.
In vitro toxicity assessment needs a certain critical look. Several studies applied a cell line representative of healthful tissues to assess the cytotoxicity, and a cancer cell line was regarded for therapeutic assessments.50,94,103,104,107,108,115,118,119,121,128,133,134,141 On the other hand, cancer cell lines might be more resistant or sensitive depending on their genotype. As a result, primary cell lines would be better for assessing cytotoxicity.12
In vivo toxicity evaluations were mostly based on histological tests of healthful tissues or monitoring of animal weight changes after treatment. No toxicity issues were noticed in any of the papers in which these evaluations were made (Table 3). In comparison to control animals, no significant changes were found in any of the cases, and all parameters were within the reference range. Some tests confirmed the compound's in vivo biosafety as well as the H&E staining findings of normal organ sections, which showed no clear symptoms of organ damage or inflammation.111,129,136,148 Although some studies have shown that nanoparticles can accumulate in the liver and kidneys,17,109,132,133,145,160 the studies did not analyze important liver biochemical profiles such as serum measurements of alkaline phosphatase (ALP), alanine aminotransferase (ALT), aspartate transaminase (AST), or serum evaluation of blood urea nitrogen (BUN) and creatinine (CR) for the kidney to evaluate toxicity further. It is also critical to perform a hematological examination. Once more, just five of the 18 in vivo studies considered in this review had a hematological assessment.120,132,136,150,162 One of them was in vitro,120 in which the parameters were within the reference values. Lim et al.162 assessed circulating platelets and bone marrow hematopoiesis and discovered toxicity related to the administration dose and schedule time. Elevated cytokines, such as tumor necrosis factor-α (TNF-α), indicate toxicity as part of the inflammatory response after intravenous delivery.12 By contrast, preclinical papers have primarily planned xenogeneic models of immunodeficient mice that do not precisely signify interactions between nanomedicines utilized with the immune system. This consideration was only seen in one among the 63 studies included in this review, and treatments based on nanoparticles aroused only modest expression levels of inflammatory cytokines (e.g., interferon-gamma, interleukin-12, and interleukin-6). Cell counts and blood chemistry parameters also illustrated no abnormalities, indicating that general toxicities did not occur.132
4.3. Nanomaterials as diagnostics in ALL
4.3.2.1. Biosensor classification based on the type of transducer. Hematological cancers, such as acute leukemia, may now be better diagnosed and treated because of the development of biosensors. Biosensors can be classified into optical, mass-based, calorimetric, and electrochemical types based on the type of transducer (Table 4). Enzymes, antibodies, cells, nucleic acids, and aptamers may be utilized as biosensor biocomponents for analyte identification. Nanoparticle-based biosensors provide great sensitivity and specificity for developing any type of biosensor. Polyaniline nanofibers (PANI-NFs) are more attractive biosensors due to higher conductivity, lower cytotoxicity, and better biocompatibility. Stability in immobilized live cells and strong electrical characteristics are further advantages of gold nanoparticles (AuNPs). AuNPs/PANI-NF nanocomposites are excellent in the immobilization of cells. Biosensors based on a polyaniline–gold composite can detect biomarkers in the range of 10−18 M.166–172 Moreover, these biosensors are used in the specific and quantitative detection of many kinds of cancers by targeting molecular and cellular diagnosis levels. According to prior investigations, a biosensor including folic acid (FA) and AuNPs was able to identify 104 cancer cells at a level equivalent to flow cytometry (FCM).
| Cancer | Biosensor | Method | Sensitivity, quality and quantity | Advantage |
|---|---|---|---|---|
| ALL166 | Electro-synthesized poly(catechol), graphene sheets, biosynthesized gold nanoparticles | Using DPV, EIS, CV, and chronoamperometry | Detection limits of 1.0 pM for the DNA strand | Continuous monitoring, molecular diagnosis of the BCR/ABL oncogene, and simple, rapid, and quantitative detection of many kinds of cancers |
| CML and ALL167 | Polyaniline–gold composite | Molecular assay | A detection limit as low as 69.4 × 10−18 M (41 DNA copies per μL) | High specificity and selectivity |
| T-ALL119 | Poly-HRP complex and CDNA-modified MNPs (CDN-MNPs) for miRNA detection | Conventional methods for detecting miRNA | Down to 100 fM in human serum | Process completion in a short time (about one hour) |
| Clinical cancer screening and early diagnosis170 | Gold nanoparticles (AuNPs) | Scanning electrochemical microscopy (SECM) | The detection limit of 4.38 × 10−12 M | Useful in the development for the detection of other antigens |
| Breast cancer, MCF-7 cell line (cell detection)169 | Surface-enhanced Raman spectroscopic tagging material (SERS dots) | Raman signal | Specific for the targets | Molecular diagnosis of the BCR/ABL oncogene. Simple, rapid, and quantitative detection of many kinds of cancers |
| HeLa cells (human epithelial cervical cancer) and CERF-CEM cells (T cell)196 | The FITC-FA-AuNPs (FFANPs) | Diphenyl alaninamide nanoparticles (FFANPs) | Sensitive detection of 10000 Hela cells by FCM assay of FFANPs |
Simple and cost-effective |
| Human T-lymphoblast cell line MOLT-4 derived from patients with acute lymphoblastic leukemia197 | Monoclonal antibody-targeted ZnO NRs | Flow cytometry | 3 to 128 cells per 1.0 mm2 | Suitability for the detection of other relevant analytes |
DNA-based aptamers may be used as biosensors in the same way as antibodies but with greater selectivity and affinity for certain biomarkers (Table 5).173 In clinical applications, to ensure the reliability of the positive result of a DNA test, an aptamer test will be used. Aptamer-modified nanomaterials such as aptamer modified Quantum Dots (QDs), AuNPs, dye-doped silica NPs, and magnetic nanoparticles (MNPs) are extremely useful diagnostic tools for specific targets with more efficient function than aptamer free modification with nanomaterials. Aptamers' optical, electrochemical, magnetic, and mechanical capabilities, as well as the ease with which they may be synthesized, modified, and tailored to work with different detection modalities, all contribute to their great efficiency in recognizing and delivering drugs to target cells.174
| Cancer | Biosensor | Method | Sensitivity, quality and quantity | Advantage |
|---|---|---|---|---|
| Molt-4 cells198 | Dual-aptamer (Sgc8c and ATP aptamers)-functionalized graphene oxide (DAFGO) complex | Flow cytometry analysis, fluorescence imaging | High targeting | Sensitive and selective detection of Molt-4 cells |
| ALL cells199 | Specific DNA aptamer sgc8c | Topographic and recognition imaging (TREC) | High density and homogeneous lateral distribution in ALL-cells | Sensitive and selective detection of Molt-4 cells |
| CCl-119 cells180 | Biotinylated aptamers in conjunction with metal-labeled neutravidin | Mass cytometry, CyTOF experiments | Differentiating positive, CCL-119, and negative, Ramos cell lines | Successfully utilized for mass cytometry experiments on par with commercially available antibodies |
| Ramos cells179 | Aptamer-nanoparticle strip biosensor (ANSB) | SELEX (systematic evolution of ligands by exponential enrichment) | Detecting a minimum of 4000 Ramos cells without instrumentation and 800 Ramos cells with a portable strip reader | A simple, rapid, and low-cost tool for both the qualitative and quantitative detection of cancer |
| T-ALL cells185 | Aptamer-conjugated magnetic beads (apt-MBs) | Magnet–QCM system | The detection limit of 8 × 103 cells mL−1 for human acute leukemia cells | Required no further labeling of cells, the potential for specific detection of various kinds of cancer cells |
| Ramos cells184 | Self-assembled aptamer-micelle nanostructure (TDO5-micelle) | Fluorescence shift | About 0.005 nM or 5 nM, based on DNA-lipid concentration | Rapid recognition ability with enhanced sensitivity and low critical micelle concentration |
| CCRF-CEM cells183 | Single-stranded DNA aptamer | Gold nanoparticle (Au NP) labeling with backscattered electron (BE) imaging of field emission scanning electron microscopy (FESEM) | Sensitive and reversible probes to label target biomolecules on cells. | The high detection sensitivity of the colloidal probe method |
| HL-60 and CEM as AML and ALL cells182 | Hierarchical assembly of dual aptamer functionalized, multilayered graphene–Au nanoparticle | Electrodeposition | The detection limit as low as 350 cells per mL, and a wide linear range | Diagnostic tool for early detection and classification of human acute leukemia |
| CEM and Ramos cells200 | QDs-bsb-apt | Quantifying the fluorescence signal of the QDs | CEM cells (71.6%) could be detected by employing the QDs-bsb-Sgc8 complex, while FITC-Sgc8 detected 57.5% of cells | The detection efficiency of QDs-bsb-Sgc8 was 1.2-fold higher than that of the traditional organic dye modified aptamer FITC |
| CCRF-CEM cells201 | Silver decahedral nanoparticle (Ag10NP)-based FRET (fluorescence resonance energy transfer) sensor (Ag10NP-based FRET sensor (Ag10-Sgc8-F/Q). | FREF-based methods | Highly sensitive and specific for CCRF-CEM cell imaging | Simple, inexpensive, and convenient for target cell imaging. |
| CCRF-CEM cells21 | Terbium(III)-aptamer (Tb3+-apt) | Fluorescence spectrophotometer | The detection limit of 5 cells per ml of the binding buffer | Rapid, sensitive, and economical diagnosis of various types of leukemia at the early stage |
| CCRF-CEM cells202 | ZnO nanodisks(NDs)@g-C3N4 quantum dot conjugation to the Sgc8c aptamer | Photoelectrochemical (PEC) | The detection limit down to 20 cell per mL | Wide detection range, low detection limit, excellent selectivity, and reproducibility |
| ALL186 | Aptamer-based electrochemical nano-biosensor (graphitic carbon nitride (Au/g-C3N4 /aptamer nanocomposite) | Gene detection (miRNA-128) | The limit of detection of 0.0034 fM concentration of miRNA-128 detection | Needed a short time (about 45 minutes) to detect miRNA-128 as a symptom of the disease |
| CEM cells and Ramos cells203 | Aptamer-functionalized copolymer/TPdye fluorescent organic dots | Two-photon imaging | Tissue imaging up to 210 mm | A powerful tool for cancer cell-targeted imaging |
Biosensor-based nanoparticles have superior properties, including electron transfer facilitation, unique optical and plasmonic effects, biocompatibility, and an excellent affinity for biomolecules, facilitating the immobilization of antibodies, enzymes, nucleic acids, and proteins, compared with their bulk counterparts.175,176
4.3.2.2. Biosensor classification based on the identification purposes. Biosensors are divided into three categories based on their identification purposes. The first cellular target identifies cancer stem cells, whose sensitivity and specificity are expressed by identifying the number of cells per milliliter. For this purpose, many studies have been done. Using an aptamer-based electrochemical biosensor (sgc8c aptamer-gold nanoparticle-coated magnetic Fe3O4 nanoparticles (Apt-GMNPs)), a highly sensitive, selective, and simple method for early-stage detection of leukemia cancer was introduced. So, specifically targeting the cells of interest by using modified nanoparticles improved sensitivity detection by limiting detection as low as ten cells per ml.177 The second identified cancer cells based on peptide or protein targets and even related chemical metabolites, in which sensitivity and accuracy are expressed as a unit of concentration. In this regard, an aptasensor with a self-assembled aptamer–micelle nanostructure (TDO5-micelle) can detect DNA-lipid concentrations as low as 0.005 nM.21,65,178–185 The third one is nucleic acid targets such as biomarker microRNAs or other non-coding DNA/RNAs. Depending on the method used, the sensitivity and specificity may be expressed in terms of the copy number or something similar. Nanoparticles may be enhanced by employing carriers for targeting, such as integrating nano-designs with organ-specific response receptors.119 An aptamer-based electrochemical nano-biosensor demonstrates efficacy in the early identification of ALL by gene detection (microRNA-128). In this work, Graphitic Carbon Nitride (G-C3N4) has been used in the square wave voltammetry (SWV) technique. The LOD of this nano-complex was obtained to be about 0.0034 fM. So, electrochemical biosensors show suitable practical application prospects for analyzing miRNAs with high sensitivity.186,187 Until now, various types of biosensors such as electrochemical (EC) methods, chemiluminescence (CL) high-performance liquid chromatography (HPLC), UV-vis spectrophotometry, surface-enhanced Raman scattering spectroscopy and other methods were used for 6-mercaptopurine detection in order to monitor the concentrations of 6-mercaptopurine in human serum (Table 6). It is remarkable that fluorescent nanosensors were developed for this purpose with an acceptable detection limit (0.198 nM).188–190
| Cancer | Biosensor | Method | Sensitivity, quality and quantity | Advantage |
|---|---|---|---|---|
| ALL168 | 6-Thiouric acid | Electrochemical surface-enhanced Raman spectroscopy (EC-SERS) | Excellent signal for 6-TUA down to μM concentrations in synthetic urine | Detected rapidly at clinically relevant concentrations, without the need for sample pre-treatment (less than 1 minute) |
| ALL204 | Chip-based capillary electrophoresis (CE) (CE-EC detection) | PMMA-based microfluidic chip | The detection limit of 100 nM | High detection speed, negligible sample consumption, low power requirement, easy integration |
| ALL171 | Gold nanoparticles (AuNPs) | SPR | More sensitive for detecting different purines at a concentration of 10−7 M for 6-thioguanine and 6-mercaptopurine | High detection speed, negligible sample consumption, low power requirement, and easy integration |
Many hurdles, including reliability, repeatability, quantitative detection findings, and biosafety, must be overcome to expedite the translation of nanotechnology as a diagnostic tool into clinical applications. Nonspecific nanoparticle probe binding, aggregation, and unsuitable detection circumstances are only a few issues that might impact nanoparticle-based detection results. Signal fluctuations can also be due to the complex makeup of bodily fluids.165
5. Conclusion
This study examined nanomedicines' recent benefits and drawbacks, advances in drug delivery, and modern diagnostic approaches.24,191 Originally, nanotechnology usage greatly relied on solubility and absorption improvement, bioavailability, extended blood circulation time, targeting strategy, and sensitive or controlled-release drug agents.91 The aim is to enhance treatment efficacy and alleviate the general toxicity. The co-loading of several drug agents may help abolish the restrictions in which drugs are impossible to administer at the same time and reduce the refractory to the therapeutic drug. The real-time following of this system is feasible via processing the biological probing agents.4 Nanoparticles can be effective in overcoming the MDR problem.10 Progressions in nanotechnology development enable the loading of higher intracellular concentrations of drug agents and, consequently, improve their effectiveness. Finally, even by reducing drug consumption, this strategy helps achieve a constant and longer remission duration in patients.192A variety of nanoparticle-based assays improved diagnostic selectivity and sensitivity or added whole new capabilities, such as easy analysis, that were not possible with older methods. These advancements will enhance cancer patient survival rates by allowing early identification, leading to improved cancer treatment options.86,165 Even integrating diagnosis with treatment strategies is also coming to the real world.91 Nanotechnology can provide the possibility of diagnostics accompanied by the treatment scanning of hematologic malignancies in real time.4,86
Both in vitro and animal studies have presented a valuable picture for realizing the illness behavior and nanomaterial interactions with alive tissues or efficacy on malignant cells.193 Nonetheless, we are yet in the experimental phase and investigational stage in the field of nanomedicine or nanotechnology. We must evaluate some features and overcome numerous problems, such as biocompatibility, long-term toxicity, immunogenicity, targeting, pharmacokinetics, biodistribution and clearance pathways of the entire nanoparticles, and inadequate drug release response before entering clinical applications.4 Generally, the experimental conditions of nanoparticle efficacy assessment against leukemia vary considerably between different preclinical studies in addition to shortage evaluation of in vivo stability, toxicity, safety, and biodistribution, which results in their lower and variable clinical impact.86 There may also be some issues with the drug-responsive liberation process, like an irreversible stimulus-response and low targeting. The scarcity of exact animal models to mimic leukemia more closely affects the accurate evaluation of the leukemia mechanism [4], making it difficult to anticipate their successful results for clinical trials.10
Author contributions
Re. K. conceptualized the study, searched the databases, screened the literature, reviewed the studies, extracted the data, assessed the included studies' quality, and prepared the original draft of the treatment section. Z. M. extracted the data, assessed the included studies' quality, and prepared the original draft of the diagnosis section. Ra. K. conceptualized the study, searched the databases, screened the literature, extracted the data, and designed the graphical abstract. A. S. conceptualized the study and edited the manuscript. N. R. conceptualized the study, appraised the manuscript, and supervised the project.Conflicts of interest
The authors have no conflicts of interest to disclose.Acknowledgements
The authors would like to thank Dr Malakeh Malekzadeh for her assistance with research in the diagnosis section.References
- R. Yazdian-Robati, A. Arab, M. Ramezani, K. Abnous and S. M. Taghdisi, Int. J. Pharm., 2017, 529, 44–54 CrossRef CAS PubMed.
- M. Houshmand, F. Garello, P. Circosta, R. Stefania, S. Aime, G. Saglio and C. Giachino, Nanomaterials, 2020, 10 Search PubMed.
- V. Cordo, J. C. G. van der Zwet, K. Canté-Barrett, R. Pieters and J. P. P. Meijerink, Blood Cancer Discov., 2021, 2, 19–31 CrossRef CAS PubMed.
- J. Shen, Z. Lu, J. Wang, T. Zhang, J. Yang, Y. Li, G. Liu and X. Zhang, ACS Biomater. Sci. Eng., 2020, 6, 6478–6489 CrossRef CAS PubMed.
- Z. Hong, Z. Wei, T. Xie, L. Fu, J. Sun, F. Zhou, M. Jamal, Q. Zhang and L. Shao, J. Hematol. Oncol., 2021, 14, 48 CrossRef CAS PubMed.
- R. Stephenson and A. Singh, Adv. Drug Delivery Rev., 2017, 114, 285–300 CrossRef CAS.
- L. V. Cappelli, D. Fiore, J. M. Phillip, L. Yoffe, F. Di Giacomo, W. Chiu, Y. Hu, C. Kayembe, M. Ginsberg, L. Consolino, J. G. Barcia Durán, N. Zamponi, A. M. Melnick, F. Boccalatte, W. Tam, O. Elemento, S. Chiaretti, A. R. Guarini, R. Foà, L. Cerchietti, S. Rafii and G. G. Inghirami, Blood, 2022 DOI:10.1182/blood.2022015414.
- C. Thakur, P. Nayak, V. Mishra, M. Sharma and G. K. Saraogi, in Nano Drug Delivery Strategies for the Treatment of Cancers, ed. A. K. Yadav, U. Gupta and R. Sharma, Academic Press, 2021, pp. 225–243, DOI:10.1016/B978-0-12-819793-6.00010-2.
- R. Basha, N. Sabnis, K. Heym, W. P. Bowman and A. G. Lacko, Front. Oncol., 2014, 4, 101 Search PubMed.
- A. K. Deshantri, A. V. Moreira, V. Ecker, S. N. Mandhane, R. M. Schiffelers, M. Buchner and M. Fens, J. Controlled Release, 2018, 287, 194–215 CrossRef CAS PubMed.
- G. Visani, F. Loscocco and A. Isidori, Nanomedicine, 2014, 9, 2415–2428 CrossRef CAS PubMed.
- T. Viseu, C. M. Lopes, E. Fernandes, M. E. C. D. R. Oliveira and M. Lúcio, Pharmaceutics, 2018, 10(4), 282 CrossRef CAS PubMed.
- A. Andleeb, A. Andleeb, S. Asghar, G. Zaman, M. Tariq, A. Mehmood, M. Nadeem, C. Hano, J. M. Lorenzo and B. H. Abbasi, Cancers, 2021, 13, 2818 CrossRef CAS.
- C. Wang, W. Zhang, Y. He, Z. Gao, L. Liu, S. Yu, Y. Hu, S. Wang, C. Zhao, H. Li, J. Shi, W. Zhou, F. Li, H. Yue, Y. Li, W. Wei, G. Ma and D. Ma, Nat. Nanotechnol., 2021, 16, 1413–1423 CrossRef CAS PubMed.
- A. S. Tatar, T. Nagy-Simon, C. Tomuleasa, S. Boca and S. Astilean, J. Contr. Release, 2016, 238, 123–138 CrossRef CAS PubMed.
- F. Asghari, R. Khademi, F. Esmaeili Ranjbar, Z. Veisi Malekshahi and R. Faridi Majidi, Int. J. Stem Cells, 2019, 12, 227–239 CrossRef CAS PubMed.
- V. Krishnan, X. Xu, S. P. Barwe, X. Yang, K. Czymmek, S. A. Waldman, R. W. Mason, X. Jia and A. K. Rajasekaran, Mol. Pharm., 2013, 10, 2199–2210 CrossRef CAS PubMed.
- C. Cao, N. Yang, H. Dai, H. Huang, X. Song, Q. Zhang and X. Dong, Nanoscale Adv., 2021, 3, 106–122 RSC.
- T. Limongi, F. Susa and V. Cauda, Hematol. Med. Oncol., 2019, 4, 1000183 CrossRef PubMed.
- Y. Zhong, K. Beimnet, Z. Alli, A. Arnoldo, P. E. Kowalski, G. R. Somers, C. Hawkins and M. Abdelhaleem, J. Mol. Diagn., 2020, 22, 72–80 CrossRef CAS.
- S. Wu, N. Yang, L. Zhong, Y. Luo, H. Wang, W. Gong, S. Zhou, Y. Li, J. He and H. Cao, Analyst, 2019, 144, 3843–3852 RSC.
- B. B. Lundberg, Int. J. Pharm., 2011, 408, 208–212 CrossRef CAS PubMed.
- A. Wicki, D. Witzigmann, V. Balasubramanian and J. Huwyler, J. Contr. Release, 2015, 200, 138–157 CrossRef CAS.
- G. Soni and K. S. Yadav, Mater. Sci. Eng. C, 2015, 47, 156–164 CrossRef CAS PubMed.
- Y. Ye, J. Wang, Q. Hu, G. M. Hochu, H. Xin, C. Wang and Z. Gu, ACS Nano, 2016, 10, 8956–8963 CrossRef CAS PubMed.
- R. S. Riley, C. H. June, R. Langer and M. J. Mitchell, Nat. Rev. Drug Discovery, 2019, 18, 175–196 CrossRef CAS PubMed.
- J. J. Lee, L. Saiful Yazan and C. A. Che Abdullah, Int. J. Nanomed., 2017, 12, 2373–2384 CrossRef CAS PubMed.
- R. Vinhas, M. Cordeiro, F. F. Carlos, S. Mendo, A. R. Fernandes, S. Figueiredo and P. V. Baptista, Nanobiosensors Dis. Diagnosis, 2015, 4, 11 Search PubMed.
- P. Pedrosa, R. Vinhas, A. Fernandes and P. V. Baptista, Nanomaterials, 2015, 5, 1853–1879 CrossRef CAS PubMed.
- W. Mekseriwattana, P. Guardia, B. T. Herrero, J. M. de la Fuente, C. Kuhakarn, A. Roig and K. P. Katewongsa, Nanoscale Adv., 2022, 4, 1988–1998 RSC.
- B. Ramdass, A. Chowdhary and P. S. Koka, J. Stem Cell., 2013, 8, 151–187 Search PubMed.
- D. Moher, A. Liberati, J. Tetzlaff and D. G. Altman, PLoS Med., 2009, 6, e1000097 CrossRef PubMed.
- K. F. Schulz, D. G. Altman and D. Moher, BMC Med., 2010, 8, 18 CrossRef PubMed.
- T. Ouchi, H. Kobayashi and T. Banba, Br. Polym. J., 1990, 23, 221–228 CAS.
- Y. Ohya, K. Nonomura and T. Ouchi, J. Bioact. Compat Polym., 1995, 10, 223–234 CrossRef CAS.
- M. S. Webb, A. H. Sarris, F. Cabanillas, L. D. Mayer, M. B. Bally, C. Burge and P. R. Cullis, J. Liposome Res., 2000, 10, 501–512 CrossRef CAS.
- A. K. Sharma, L. Zhang, S. Li, D. L. Kelly, V. Y. Alakhov, E. V. Batrakova and A. V. Kabanov, J. Controlled Release, 2008, 131, 220–227 CrossRef CAS PubMed.
- N. Eslahi, A. Shakeri-Zadeh, K. Ashtari, V. Pirhajati-Mahabadi, T. T. Moghadam, R. Shabani, K. Kamrava, Z. Madjd, C. Maki, H. R. Asgari and M. Koruji, Cell J., 2019, 21, 14–26 Search PubMed.
- J. Koechling, Y. Rott, S. Anidt, J. Green, D. Anderson, R. Langer, R. Stripecke, B. Wittig and M. Schmidt, Blood, 2007, 110, 366B CrossRef.
- Y. Tong, C. Li, F. Liang, J. Chen, H. Zhang, G. Liu, H. Sun and J. H. T. Luong, Nucl. Instrum. Methods Phys. Res., Sect. B, 2008, 266, 5041–5046 CrossRef CAS.
- H. Ichihara, J. Ueno, M. Umebayashi, Y. Matsumoto and R. Ueoka, Int. J. Pharm., 2011, 406, 173–178 CrossRef CAS PubMed.
- F. A. Fakhr, A. Khanafari, M. Baserisalehi, R. Yaghoobi and S. Shahghasempour, Afr. J. Microbiol. Res., 2012, 6, 6235–6242 CAS.
- D. Pramanik, N. R. Campbell, S. Das, S. Gupta, V. Chenna, S. Bisht, P. Sysa-Shah, D. Bedja, C. Karikari, C. Steenbergen, K. L. Gabrielson, A. Maitra and A. Maitra, Oncotarget, 2012, 3, 640–650 CrossRef PubMed.
- N. Satake, G. Barisone, E. Diaz, N. Nitin, J. Nolta and K. Lam, Proceedings of SPIE - the International Society for Optical Engineering, Baltimore, MD, 2012 Search PubMed.
- M. A. Orlova, E. Y. Osipova, S. A. Rumiantsev and D. A. Zaitsev, Russ. Chem. Bull., 2013, 62, 1120–1124 CrossRef CAS.
- R. Raghunathan, S. Mahesula, K. Kancharla, P. Janardhanan, Y. L. Jadhav, R. Nadeau, G. P. Villa, R. L. Cook, C. M. Witt, J. A. Gelfond, T. G. Forsthuber and W. E. Haskins, Particle & particle systems characterization : measurement and description of particle properties and behavior in powders and other disperse systems, 2013, vol. 30, pp. 355–364 Search PubMed.
- M. A. Orlova, A. A. Poloznikov and A. P. Orlov, Moscow Univ. Chem. Bull., 2014, 69, 142–147 CrossRef.
- M. G. Kim, J. Y. Park, W. Miao, J. Lee and Y. K. Oh, Biomaterials, 2015, 48, 129–136 CrossRef CAS PubMed.
- T. Pivetta, V. Lallai, E. Valletta, F. Trudu, F. Isaia, D. Perra, E. Pinna and A. Pani, J. Inorg. Biochem., 2015, 151, 107–114 CrossRef CAS PubMed.
- F. M. Uckun, H. Ma, J. Cheng, D. E. Myers and S. Qazi, Br. J. Haematol., 2015, 169, 401–414 CrossRef CAS PubMed.
- F. M. Uckun, L. G. Mitchell, S. Qazi, Y. Liu, N. Zheng, D. E. Myers, Z. Song, H. Ma and J. Cheng, EBioMedicine, 2015, 2, 649–659 CrossRef PubMed.
- Y. Matsumoto, K. Kuwabara, H. Ichihara and M. Kuwano, Bioorg. Med. Chem. Lett., 2016, 26, 301–305 CrossRef CAS PubMed.
- Z. Hasanzade and H. Raissi, Appl. Surf. Sci., 2017, 422, 1030–1041 CrossRef CAS.
- J. H. Kang, K. R. Kim, H. Lee, D. R. Ahn and Y. T. Ko, Colloids Surf. B Biointerfaces, 2017, 157, 424–431 CrossRef CAS PubMed.
- S. Chen, J. Zaifman, J. A. Kulkarni, I. V. Zhigaltsev, Y. K. Tam, M. A. Ciufolini, Y. Y. C. Tam and P. R. Cullis, J. Contr. Release, 2018, 286, 46–54 CrossRef CAS PubMed.
- S. Kallus, B. Englinger, J. Senkiv, A. Laemmerer, P. Heffeter, W. Berger, C. R. Kowol and B. K. Keppler, Nanomed. Nanotechnol. Biol. Med., 2018, 14, 2632–2643 CrossRef CAS PubMed.
- R. R. Nair, D. Piktel, W. J. Geldenhuys and L. F. Gibson, Leuk. Res., 2018, 72, 59–66 CrossRef CAS PubMed.
- A. C. Uscanga-Palomeque, K. M. Calvillo-Rodriguez, L. Gomez-Morales, E. Larde, T. Denefle, D. Caballero-Hernandez, H. Merle-Beral, S. A. Susin, P. Karoyan, A. C. Martinez-Torres and C. Rodriguez-Padilla, Cancer Sci., 2019, 110, 256–268 CrossRef CAS PubMed.
- S. Zong, L. Wang, Z. Yang, H. Wang and Z. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 5896–5902 CrossRef CAS PubMed.
- H. Al Faruque, E. S. Choi, H. R. Lee, J. H. Kim, S. Park and E. Kim, Nanoscale, 2020, 12, 2773–2786 RSC.
- M. Mohseni, C. Kucharski, K. C. R. Bahadur, M. Nasrullah, X. Y. Jiang, H. Uludag and J. Brandwein, PLoS One, 2021, 16 Search PubMed.
- Y. A. Grechkin, S. L. Grechkina, E. A. Zaripov, S. V. Fedorenko, A. R. Mustafina and M. V. Berezovski, Biomedicines, 2020, 8, 14 CrossRef CAS PubMed.
- N. F. Ramandi, I. S. Mashhadi, A. Sharif, N. Saeedi, M. A. Ashabi, M. Faranoush, A. Ghassempour and H. Y. Aboul-Enein, J. Chromatogr., B, 2022, 1190, 123091 CrossRef PubMed.
- X. Hu, Y. Wang, X. Zuping, P. Song, A. J. Wang, Z. Qian, P. X. Yuan, T. Zhao and J. J. Feng, Anal. Chem., 2022, 94, 3708–3717 CrossRef CAS PubMed.
- D. Woods, K. Winchester, A. Towerman, K. Gettinger, C. Carey, K. Timmermann, R. Langley and E. Browne, J. Pediatr. Oncol. Nurs., 2017, 34, 387–396 CrossRef PubMed.
- M. Ramachandran, A. Dimberg and M. Essand, The cancer-immunity cycle as rational design for synthetic cancer drugs: Novel DC vaccines and CAR T-cells, in Seminars in Cancer Biology, Elsevier, 2017, pp. 23–35 Search PubMed.
- S. Sarangapani, R. E. Mohan, A. Patil, M. J. Lang and A. Asundi, presented in 5th International Conference on Optical and Photonics Engineering, International Society for Optics and Photonics, 2017, p. 1044936 Search PubMed.
- X. Li, B. Zhou, Z. Zhao, Z. Hu, S. Zhou, N. Yang, Y. Huang, Z. Zhang, J. Su and D. Lan, J. Biomed. Nanotechnol., 2016, 12, 2151–2160 CrossRef CAS PubMed.
- P. S. Kedar, V. Gupta, P. Warang, A. Chiddarwar and M. Madkaikar, Hematology, 2018, 23, 567–573 CrossRef CAS PubMed.
- E. A. Motea, I. Lee and A. J. Berdis, ACS Chem. Biol., 2012, 7, 988–998 CrossRef CAS PubMed.
- D. Shangguan, Z. Cao, L. Meng, P. Mallikaratchy, K. Sefah, H. Wang, Y. Li and W. Tan, J. Proteome Res., 2008, 7, 2133–2139 CrossRef CAS PubMed.
- J. Skulan, T. Bullen, A. D. Anbar, J. E. Puzas, L. Shackelford, A. LeBlanc and S. M. Smith, Clin. Chem., 2007, 53, 1155–1158 CrossRef CAS PubMed.
- H. Wang, H. Zeng, G. Shen and R. Yu, Anal. Chem., 2006, 78, 2571–2578 CrossRef CAS PubMed.
- B. Patra, A. K. Mishra and R. S. Verma, J. Sci. Adv. Mater. Dev., 2022, 7 CAS.
- J. D. Cohen and H. I. Robins, Cancer Res., 1987, 47, 4335–4337 CAS.
- P. Jungmayr and T. Müller-Bohn, Dtsch. Apoth. Ztg., 2007, 147, 66–73 Search PubMed.
- Y. Godfrin, Innov. Pharm. Technol., 2009, 60–62 CAS.
- K. I. Batarseh, Curr. Med. Chem., 2013, 20, 2363–2373 CrossRef CAS PubMed.
- A. A. Philchenkov, E. D. Shishko, M. P. Zavelevich, L. M. Kuiava, K. Miura, D. Y. Blokhin, I. O. Shton and N. F. Gamaleia, Exp. Oncol., 2014, 36, 241–245 CAS.
- H. Pan, S. Li, M. Li, Q. Tao, J. Jia, W. Li, L. Wang, Z. Guo, K. Ma, Y. Liu and C. Cui, Pharmazie, 2020, 75, 318–323 CAS.
- J. Varshosaz, S. Fardshouraki, M. Mirian, L. Safaeian, S. Jandaghian and S. Taymouri, Anti Cancer Agents Med. Chem., 2020, 20, 1966–1980 CrossRef CAS.
- K. Sumi, K. Tago, Y. Nakazawa, K. Takahashi, T. Ohe, T. Mashino and M. Funakoshi-Tago, Eur. J. Pharmacol., 2022, 916 Search PubMed.
- Y. F. Zhang, J. N. An, Y. Shao, N. Yu, S. J. Yue, H. L. Sun, J. B. Zhang, W. X. Gu, Y. F. Xia, J. P. Zhang, Y. Xu and Z. Y. Zhong, Biomacromolecules, 2022, 23, 377–387 CrossRef CAS PubMed.
- P. Nandy, A. P. Periclou and V. I. Avramis, Anticancer Res., 1998, 18, 727–737 CAS.
- L. Mayer, H. Carol, C. L. Morton, T. Harasym, M. A. Smith and R. B. Lock, Mol. Cancer Therapeut., 2009, 8 Search PubMed.
- R. Vinhas, R. Mendes, A. R. Fernandes and P. V. Baptista, Front. Bioeng. Biotechnol., 2017, 5, 79 CrossRef PubMed.
- F. M. Uckun, I. Dibirdik, S. Qazi and S. Yiv, Arzneim.-Forsch., 2010, 60, 210–217 CAS.
- W. M. Becicka, P. A. Bielecki, M. E. Lorkowski, T. J. Moon, Y. Zhang, P. U. Atukorale, G. Covarrubias and E. Karathanasis, Nanoscale Adv., 2021, 3, 4961–4972 RSC.
- V. K. Chaturvedi, A. Singh, V. K. Singh and M. P. Singh, Curr. Drug Metab., 2019, 20, 416–429 CrossRef CAS PubMed.
- M. Szwed, A. Matusiak, A. Laroche-Clary, J. Robert, I. Marszalek and Z. Jozwiak, Toxicol. Vitro, 2014, 28, 187–197 CrossRef CAS.
- J. K. Patra, G. Das, L. F. Fraceto, E. V. R. Campos, M. d. P. Rodriguez-Torres, L. S. Acosta-Torres, L. A. Diaz-Torres, R. Grillo, M. K. Swamy, S. Sharma, S. Habtemariam and H.-S. Shin, J. Nanobiotechnol., 2018, 16, 71 CrossRef PubMed.
- B. V. Lima, M. J. Oliveira, M. A. Barbosa, R. M. Gonçalves and F. Castro, Biomater. Sci., 2021, 9, 3209–3227 RSC.
- H. Nekounam, R. Dinarvand, R. Khademi, R. Karimi, H. Arzani, N. Mahmoodi, E. Hasanzadeh, M. Kamali, M. Khosravani, bioRxiv, 2021, bioRxiv:2021.2005.2015.444280, DOI:10.1101/2021.05.15.444280.
- S. M. Taghdisi, N. M. Danesh, M. Ramezani and K. Abnous, RSC Adv., 2016, 6, 46366–46371 RSC.
- C. Chittasupho, C. Aonsri and W. Imaram, Bioorg. Chem., 2021, 107 Search PubMed.
- H. Zhao, H. Wang, H. Li, T. Zhang, J. Zhang, W. Guo, K. Fu and G. Du, Nanoscale Adv., 2022, 4, 1815–1826 RSC.
- A. G. Leonel, A. A. P. Mansur, S. M. Carvalho, L. E. F. Outon, J. D. Ardisson, K. Krambrock and H. S. Mansur, Nanoscale Adv., 2021, 3, 1029–1046 RSC.
- L. Zhou, Y. Zhang and Y. Ma, Nanoscale Adv., 2022, 4, 4059–4065 RSC.
- A. F. Chamorro Rengifo, N. Stefanes, J. Toigo, C. Mendes, M. C. Santos-Silva, R. J. Nunes, A. L. Parize and E. Minatti, Mater. Sci. Eng. C, 2019, 105, 110051 CrossRef CAS PubMed.
- P. E. Feuser, P. C. Gaspar, A. V. Jacques, A. C. Tedesco, M. C. D. Santos Silva, E. Ricci-Junior, C. Sayer and P. H. H. de Araujo, Mater. Sci. Eng. C, 2016, 60, 458–466 CrossRef CAS PubMed.
- P. E. Feuser, P. C. Matos dos Santos, A. P. Cordeiro, N. M. Stefanes, L. O. Walter, M. F. Maioral, M. C. Santos-Silva, P. H. Hermes de Araújo and C. Sayer, J. Drug Deliv. Sci. Technol., 2022, 67 CAS.
- H. S. Rahman, A. Rasedee, C. W. How, A. B. Abdul, N. A. Zeenathul, H. H. Othman, M. I. Saeed and S. K. Yeap, Int. J. Nanomed., 2013, 8, 2769–2781 CrossRef PubMed.
- N. M. Danesh, P. Lavaee, M. Ramezani, K. Abnous and S. M. Taghdisi, Int. J. Pharm., 2015, 489, 311–317 CrossRef CAS PubMed.
- S. M. Taghdisi, N. M. Danesh, P. Lavaee, A. S. Emrani, K. Y. Hassanabad, M. Ramezani and K. Abnous, Mater. Sci. Eng. C, 2016, 61, 753–761 CrossRef CAS PubMed.
- K. Saravanakumar, E. Jeevithan, R. Chelliah, K. Kathiresan, W. Wen-Hui, D. H. Oh and M. H. Wang, Int. J. Biol. Macromol., 2018, 119, 1144–1153 CrossRef CAS PubMed.
- Y. F. Huang, D. Shangguan, H. Liu, J. A. Phillips, X. Zhang, Y. Chen and W. Tan, Chembiochem, 2009, 10, 862–868 CrossRef CAS PubMed.
- S. M. Taghdisi, P. Lavaee, M. Ramezani and K. Abnous, Eur. J. Pharm. Biopharm., 2011, 77, 200–206 CrossRef CAS PubMed.
- D. Jang, Y. M. Lee, J. Lee, J. Doh and W. J. Kim, Sci. Rep., 2017, 7, 40739 CrossRef CAS PubMed.
- S. Duan, Y. Yu, C. Lai, D. Wang, Y. Wang, D. Xue, Z. Hu and X. Lu, J. Biomed. Nanotechnol., 2018, 14, 910–921 CrossRef CAS PubMed.
- M. M. Billingsley, N. Singh, P. Ravikumar, R. Zhang, C. H. June and M. J. Mitchell, Nano Lett., 2020, 20, 1578–1589 CrossRef CAS PubMed.
- Y. Zou, D. Mei, J. Yuan, J. Han, J. Xu, N. Sun, H. He, C. Yang and L. Zhao, Int. J. Nanomed., 2021, 16, 1127–1141 CrossRef PubMed.
- Y. Yang, W. H. Zhao, W. W. Tan, Z. Q. Lai, D. Fang, L. Jiang, C. T. Zuo, N. Yang and Y. R. Lai, Nanoscale Res. Lett., 2019, 14 Search PubMed.
- S. Bi, S. Yue, W. Song and S. Zhang, Chem. Commun., 2016, 52, 12841–12844 RSC.
- T. Suzawa, S. Nagamura, H. Saito, S. Ohta, N. Hanai, J. Kanazawa, M. Okabe and M. Yamasaki, J. Contr. Release, 2002, 79, 229–242 CrossRef CAS PubMed.
- Y. L. Luo, Y. S. Shiao and Y. F. Huang, ACS Nano, 2011, 5, 7796–7804 CrossRef CAS PubMed.
- Y. S. Youn and Y. H. Bae, Adv. Drug Delivery Rev., 2018, 130, 3–11 CrossRef CAS PubMed.
- S. Mondal, M. Saha, M. Ghosh, S. Santra, M. A. Khan, K. Das Saha and M. R. Molla, Nanoscale Adv., 2019, 1, 1571–1580 RSC.
- H. Zhang, Y. Ma, Y. Xie, Y. An, Y. Huang, Z. Zhu and C. J. Yang, Sci. Rep., 2015, 5, 10099 CrossRef CAS PubMed.
- M. Liu, W. Ma, Q. Li, D. Zhao, X. Shao, Q. Huang, L. Hao and Y. Lin, Cell Prolif., 2019, 52 Search PubMed.
- Z. Fang, X. Wang, Y. Sun, R. Fan, Z. Liu, R. Guo and D. Xie, Nanoscale, 2019, 11, 23000–23012 RSC.
- C. Liu, J. Zheng, L. Deng, C. Ma, J. Li, Y. Li, S. Yang, J. Yang, J. Wang and R. Yang, ACS Appl. Mater. Interfaces, 2015, 7, 11930–11938 CrossRef CAS PubMed.
- R. Ridolfo, B. C. Ede, P. Diamanti, P. B. White, A. W. Perriman, J. C. M. van Hest, A. Blair and D. S. Williams, Small, 2018, 14, e1703774 CrossRef PubMed.
- R. C. Deller, P. Diamanti, G. Morrison, J. Reilly, B. C. Ede, R. Richardson, K. Le Vay, A. M. Collins, A. Blair and A. W. Perriman, Mol. Pharm., 2017, 14, 722–732 CrossRef CAS PubMed.
- N. Dinauer, S. Balthasar, C. Weber, J. Kreuter, K. Langer and H. von Briesen, Biomaterials, 2005, 26, 5898–5906 CrossRef CAS PubMed.
- N. Satake, J. Lee, K. Xiao, J. Luo, S. Sarangi, A. Chang, B. McLaughlin, P. Zhou, E. Kenney, L. Kraynov, S. Arnott, J. McGee, J. Nolta and K. Lam, Proceedings of SPIE - the International Society for Optical Engineering, Orlando, FL, 2011 Search PubMed.
- R. C. Huxford, K. E. Dekrafft, W. S. Boyle, D. Liu and W. Lin, Chem. Sci., 2012, 3, 198–204 RSC.
- S. Habringer, C. Lapa, P. Herhaus, M. Schottelius, R. Istvanffy, K. Steiger, J. Slotta-Huspenina, A. Schirbel, H. Hanscheid, S. Kircher, A. K. Buck, K. Gotze, B. Vick, I. Jeremias, M. Schwaiger, C. Peschel, R. Oostendorp, H. J. Wester, G. U. Grigoleit and U. Keller, Theranostics, 2018, 8, 369–383 CrossRef CAS PubMed.
- K. Wang, M. You, Y. Chen, D. Han, Z. Zhu, J. Huang, K. Williams, C. J. Yang and W. Tan, Angew. Chem., Int. Ed., 2011, 50, 6098–6101 CrossRef CAS PubMed.
- G. Yuan, M. Yao, H. Lv, X. Jia, J. Chen and J. Xue, J. Med. Chem., 2020, 63, 15655–15667 CrossRef CAS PubMed.
- H. Lu, W. Li, P. Qiu, X. Zhang, J. Qin, Y. Cai and X. Lu, Nanoscale Adv., 2022, 4, 4304–4313 RSC.
- M. Harata, Y. Soda, K. Tani, J. Ooi, T. Takizawa, M. Chen, Y. Bai, K. Izawa, S. Kobayashi, A. Tomonari, F. Nagamura, S. Takahashi, K. Uchimaru, T. Iseki, T. Tsuji, T. A. Takahashi, K. Sugita, S. Nakazawa, A. Tojo, K. Maruyama and S. Asano, Blood, 2004, 104, 1442–1449 CrossRef CAS PubMed.
- T. T. Smith, S. B. Stephan, H. F. Moffett, L. E. McKnight, W. Ji, D. Reiman, E. Bonagofski, M. E. Wohlfahrt, S. P. S. Pillai and M. T. Stephan, Nat. Nanotechnol., 2017, 12, 813–822 CrossRef CAS PubMed.
- V. Krishnan, X. Xu, D. Kelly, A. Snook, S. A. Waldman, R. W. Mason, X. Jia and A. K. Rajasekaran, Mol. Pharm., 2015, 12, 2101–2111 CrossRef CAS PubMed.
- Z. Lin, R. Peng, Y. Sun, L. Zhang and Z. Zhang, Biosci. Rep., 2021, 41 CAS.
- V. Omelyanenko, P. Kopeckova, R. K. Prakash, C. D. Ebert and J. Kopecek, Pharmaceut. Res., 1999, 16, 1010–1019 CrossRef CAS PubMed.
- Y. Zhou, L. Guan, W. Li, R. Jia, L. Jia, Y. Zhang, X. Wen, S. Meng, D. Ma, N. Zhang, M. Ji, Y. Liu and C. Ji, Cancer Lett., 2022, 533 Search PubMed.
- A. C. Martínez-Torres, D. G. Zarate-Triviño, H. Y. Lorenzo-Anota, A. Ávila-Ávila, C. Rodríguez-Abrego and C. Rodríguez-Padilla, Int. J. Nanomed., 2018, 13, 3235–3250 CrossRef PubMed.
- A. Szulc, L. Pulaski, D. Appelhans, B. Voit and B. Klajnert-Maculewicz, Chem. Commun., 2016, 513, 572–583 CAS.
- S. Harrach, C. Schmidt-Lauber, T. Pap, H. Pavenstädt, E. Schlatter, E. Schmidt, W. E. Berdel, U. Schulze, B. Edemir, S. Jeromin, T. Haferlach, G. Ciarimboli and J. Bertrand, Blood Cancer J., 2016, 6, e470 CrossRef CAS PubMed.
- H. S. Rahman, A. Rasedee, A. B. Abdul, N. Allaudin, Zeenathul, H. H. Othman, S. K. Yeap, C. W. How, W. A. G. Wan and N. Hafiza, Int. J. Nanomed., 2014, 9, 527–538 Search PubMed.
- S. M. Taghdisi, K. Abnous, F. Mosaffa and J. Behravan, J. Drug Target., 2010, 18, 277–281 CrossRef CAS PubMed.
- Y. Liu, M. Lv, P. Bai, L. Wang, Y. Tan, H. Jian, R. Zhang, J. Zhu, S. Qu, S. Luo, L. Jiang, H. Nie, D. Guo, Z. Yu, Y. Li and W. Liang, Int. J. Leg. Med., 2021, 135, 23–41 CrossRef PubMed.
- H. Abbasi, N. Rahbar, M. Kouchak, P. Khalil Dezfuli and S. Handali, J. Liposome Res., 2021, 1–16, DOI:10.1080/08982104.2021.1903035.
- X. Wu, L. Wang, Y. Qiu, B. Zhang, Z. Hu and R. Jin, Int. J. Nanomed., 2017, 12, 8025–8034 CrossRef CAS PubMed.
- F. M. Uckun, S. Qazi, I. Cely, K. Sahin, A. Shahidzadeh, I. Ozercan, Q. Yin, P. Gaynon, A. Termuhlen, J. Cheng and S. Yiv, Blood, 2013, 121, 4348–4354 CrossRef CAS PubMed.
- F. M. Uckun, D. E. Myers, J. Cheng and S. Qazi, EBioMedicine, 2015, 2, 554–562 CrossRef PubMed.
- M. H. Amer, Mol. Cell Ther., 2014, 2, 27 CrossRef PubMed.
- D. Gan, Y. Chen, Z. Wu, L. Luo, S. K. Yirga, N. Zhang, F. Ye, H. Chen, J. Hu and Y. Chen, Front. Pharmacol., 2021, 12 Search PubMed.
- Y. Y. Chen, D. H. Gan, L. P. Luo, Z. J. Wu, Y. W. Chen, H. J. Chen and J. D. Hu, Blood, 2020, 136 CAS.
- C. Li, X. You, X. Xu, B. Wu, Y. Liu, T. Tong, J. Chen, Y. Li, C. Dai, Z. Ye, X. Tian, Y. Wei, Z. Hao, L. Jiang, J. Wu and M. Zhao, Adv. Sci., 2022, e2104134 CrossRef PubMed.
- T. A. Tabish, S. Zhang and P. G. Winyard, Redox Biol., 2018, 15, 34–40 CrossRef CAS PubMed.
- B. Zhang, Y. Wang, J. Liu and G. Zhai, Curr. Med. Chem., 2017, 24, 268–291 CrossRef CAS PubMed.
- B. P. Coughlin, P. T. Lawrence, I. R. Lui, C. J. Luby, D. J. Spencer, E. C. H. Sykes and C. R. Mace, J. Nanopart. Res., 2020, 22(2) DOI:10.1007/s11051-020-4765-1.
- Satrialdi, Y. Takano, E. Hirata, N. Ushijima, H. Harashima and Y. Yamada, Nanoscale Adv., 2021, 3(20), 5919–5927 RSC.
- L. Zou, H. Wang, B. He, L. Zeng, T. Tan, H. Cao, X. He, Z. Zhang, S. Guo and Y. Li, Theranostics, 2016, 6, 762–772 CrossRef CAS PubMed.
- J. Wang, M. You, G. Zhu, M. I. Shukoor, Z. Chen, Z. Zhao, M. B. Altman, Q. Yuan, Z. Zhu, Y. Chen, C. Z. Huang and W. Tan, Small, 2013, 9, 3678–3684 CrossRef CAS PubMed.
- L. F. Verdonck, H. M. Lokhorst, D. J. Roovers and H. G. van Heugten, Leuk. Res., 1998, 22, 249–256 CrossRef CAS PubMed.
- R. R. Nair, D. Piktel, Q. A. Hathaway, S. L. Rellick, P. Thomas, P. Saralkar, K. H. Martin, W. J. Geldenhuys, J. M. Hollander and L. F. Gibson, Pharm. Res., 2020, 37 CAS.
- L. Warren, J. C. Jardillier, A. Malarska and M. G. Akeli, Cancer Res., 1992, 52, 3241–3245 CAS.
- B. C. Ede, P. Diamanti, D. S. Williams and A. Blair, Sci. Rep., 2021, 11 Search PubMed.
- P. Tardi, S. Johnstone, N. Harasyrn, S. W. Xie, T. Harasyrn, N. Zisman, P. Harvie, D. Bermudes and L. Mayer, Leuk. Res., 2009, 33, 129–139 CrossRef CAS PubMed.
- W. S. Lim, P. G. Tardi, X. Xie, M. Fan, R. Huang, T. Ciofani, T. O. Harasym and L. D. Mayer, Leuk. Lymphoma, 2010, 51, 1536–1542 CrossRef CAS PubMed.
- J. L. Millar, B. C. Millar, R. L. Powles, J. P. C. Steele, R. D. Clutterbuck, P. L. R. Mitchell, G. Cox, E. Forssen and D. Catovsky, Br. J. Haematol., 1998, 102, 718–721 CrossRef CAS PubMed.
- M. M. Saber, A. M. Al-mahallawi and B. Stork, Biomed. Pharmacother., 2021, 143 Search PubMed.
- Y. Zhang, M. Li, X. Gao, Y. Chen and T. Liu, J. Hematol. Oncol., 2019, 12, 137 CrossRef PubMed.
- M. Mazloum-Ardakani, B. Barazesh, A. Khoshroo, M. Moshtaghiun and M. H. Sheikhha, Bioelectrochemistry, 2018, 121, 38–45 CrossRef CAS PubMed.
- K. Y. Avelino, I. A. Frias, N. Lucena-Silva, R. G. Gomes, C. P. de Melo, M. D. Oliveira and C. A. Andrade, Colloids Surf. B Biointerfaces, 2016, 148, 576–584 CrossRef CAS PubMed.
- B. Greene, D. Alhatab, C. Pye and C. Brosseau, J. Phys. Chem. C, 2017, 121, 8084–8090 CrossRef CAS.
- J.-H. Kim, J.-S. Kim, H. Choi, S.-M. Lee, B.-H. Jun, K.-N. Yu, E. Kuk, Y.-K. Kim, D. H. Jeong and M.-H. Cho, Anal. Chem., 2006, 78, 6967–6973 CrossRef CAS PubMed.
- W. Song, Z. Yan and K. Hu, Biosens. Bioelectron., 2012, 38, 425–429 CrossRef CAS PubMed.
- S. Kainth and S. Basu, Plasmonics, 2018, 13, 1785–1793 CrossRef CAS.
- S. Zhang, L. Zhang, X. Zhang, P. Yang and J. Cai, Analyst, 2014, 139, 3629–3635 RSC.
- N. S. Fracchiolla, S. Artuso and A. Cortelezzi, Sensors, 2013, 13, 6423–6447 CrossRef CAS PubMed.
- Q. Liu, C. Jin, Y. Wang, X. Fang, X. Zhang, Z. Chen and W. Tan, NPG Asia Mater., 2014, 6, e95 CrossRef CAS PubMed.
- M. Pirzada and Z. Altintas, Sensors, 2019, 19, 5311 CrossRef PubMed.
- J. Yoon, M. Shin, T. Lee and J.-W. Choi, Materials, 2020, 13, 299 CrossRef CAS PubMed.
- S. M. Khoshfetrat and M. A. Mehrgardi, Bioelectrochemistry, 2017, 114, 24–32 CrossRef CAS PubMed.
- D. Shangguan, Z. C. Cao, Y. Li and W. Tan, Clin. Chem., 2007, 53, 1153–1155 CrossRef CAS PubMed.
- G. Liu, X. Mao, J. A. Phillips, H. Xu, W. Tan and L. Zeng, Anal. Chem., 2009, 81, 10013–10018 CrossRef CAS PubMed.
- G. G. Mironov, A. Bouzekri, J. Watson, O. Loboda, O. Ornatsky and M. V. Berezovski, Anal. Bioanal. Chem., 2018, 410, 3047–3051 CrossRef CAS PubMed.
- Z. Yan, M. Yang, Z. Wang, F. Zhang, J. Xia, G. Shi, L. Xia, Y. Li, Y. Xia and L. Xia, Sensor. Actuator. B Chem., 2015, 210, 248–253 CrossRef CAS.
- T. Zheng, T. Tan, Q. Zhang, J.-J. Fu, J.-J. Wu, K. Zhang, J.-J. Zhu and H. Wang, Nanoscale, 2013, 5, 10360–10368 RSC.
- H. Kim, H. Terazono, M. Hayashi, H. Takei and K. Yasuda, Jpn. J. Appl. Phys., 2012, 51, 06FH01 CrossRef.
- Y. Wu, K. Sefah, H. Liu, R. Wang and W. Tan, Proc. Natl. Acad. Sci. U.S.A., 2010, 107, 5–10 CrossRef CAS PubMed.
- Y. Pan, M. Guo, Z. Nie, Y. Huang, C. Pan, K. Zeng, Y. Zhang and S. Yao, Biosens. Bioelectron., 2010, 25, 1609–1614 CrossRef CAS PubMed.
- N. Naderian, M. Pourmadadi, H. Rashedi and F. Yazdian, Design of a Novel Nanobiosensor for the Diagnosis of Acute Lymphoid Leukemia (ALL) by Measurement of miRNA-128, in 2020 27th National and 5th International Iranian Conference on Biomedical Engineering (ICBME), 2020, IEEE, pp. 41–46 Search PubMed.
- M. El Aamri, G. Yammouri, H. Mohammadi, A. Amine and H. Korri-Youssoufi, Biosensors, 2020, 10, 186 CrossRef CAS PubMed.
- Y. Yuan, Y. Wang, S. Liu, Y. Li, R. Duan, H. Zhang and X. Hu, RSC Adv., 2016, 6, 52255–52263 RSC.
- W. Tang, W. Li, Y. Li, M. Zhang and X. Zeng, New J. Chem., 2015, 39, 8454–8460 RSC.
- Z. Chen, G. Zhang, X. Chen, J. Chen, J. Liu and H. Yuan, Biosens. Bioelectron., 2013, 41, 844–847 CrossRef CAS PubMed.
- C. Maksoudian, N. Saffarzadeh, E. Hesemans, N. Dekoning, K. Buttiens and S. J. Soenen, Nanoscale Adv., 2020, 2, 3734–3763 RSC.
- Y. D. Taghipour, R. Bahramsoltani, A. M. Marques, R. Naseri, R. Rahimi, P. Haratipour, A. Iranpanah, A. I. Panah, M. H. Farzaei and M. Abdollahi, Daru, 2018, 26, 229–239 CrossRef PubMed.
- G. Brakmane, M. Winslet and A. M. Seifalian, Aliment. Pharmacol. Ther., 2012, 36, 213–221 CrossRef CAS PubMed.
- Z. Zhang, C. Tang, R. Hammink, F. H. T. Nelissen, H. A. Heus and P. H. J. Kouwer, Chem. Commun., 2021, 57, 2744–2747 RSC.
- S. Li, L. Zhang, M. Li, J. Huang, B. Cui, J. Jia, Z. Guo, K. Ma and C. Cui, J. Pharm. Sci., 2021, 110, 2733–2742 CrossRef CAS PubMed.
- J. Ai, Y. Xu, D. Li, Z. Liu and E. Wang, Talanta, 2012, 101, 32–37 CrossRef CAS PubMed.
- A. Tamashevski, Y. Harmaza, E. Slobozhanina, R. Viter and I. Iatsunskyi, Molecules, 2020, 25, 3168 CrossRef CAS PubMed.
- A. Bahreyni, R. Yazdian-Robati, M. Ramezani, M. Rasouli, M. Alinezhad Nameghi, M. Alibolandi, K. Abnous and S. M. Taghdisi, J. Biomater. Appl., 2017, 32, 74–81 CrossRef CAS PubMed.
- M. Leitner, A. Poturnayova, C. Lamprecht, S. Weich, M. Snejdarkova, I. Karpisova, T. Hianik and A. Ebner, Anal. Bioanal. Chem., 2017, 409, 2767–2776 CrossRef CAS PubMed.
- Y. Yu, S. Duan, J. He, W. Liang, J. Su, J. Zhu, N. Hu, Y. Zhao and X. Lu, Oncol. Rep., 2016, 36, 886–892 CrossRef CAS PubMed.
- H. Li, H. Hu and D. Xu, Anal. Chem., 2015, 87, 3826–3833 CrossRef CAS PubMed.
- X. Pang, C. Cui, M. Su, Y. Wang, Q. Wei and W. Tan, Nano Energy, 2018, 46, 101–109 CrossRef CAS PubMed.
- H. Yan, W. Ren, S. Liu and Y. Yu, Anal. Chim. Acta, 2020, 1106, 199–206 CrossRef CAS PubMed.
- C. Chen, Y. Ho, S. Wu, G.-L. Chang and C. Lin, J. Nanosci. Nanotechnol., 2009, 9, 718–722 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2na00483f |
| ‡ These authors equally contributed to the manuscript. |
| This journal is © The Royal Society of Chemistry 2023 |