
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProteomic analysis reveals mechanisms underlying increased efficacy of bleomycin by photochemical internalization in bladder cancer cells†
Odrun A.
Gederaas
 *ab,
Animesh
Sharma
ac,
Saide
Mbarak
a,
Bjørnar
Sporsheim
ae,
Anders
Høgset
f,
Vanya
Bogoeva
d,
Geir
Slupphaug
ac and
Lars
Hagen
*ac
*ab,
Animesh
Sharma
ac,
Saide
Mbarak
a,
Bjørnar
Sporsheim
ae,
Anders
Høgset
f,
Vanya
Bogoeva
d,
Geir
Slupphaug
ac and
Lars
Hagen
*ac
aDepartment of Clinical and Molecular Medicine, NTNU, Norwegian University of Science and Technology, N-7489 Trondheim, Norway
bDepartment of Natural Sciences, UiA, University of Agder, N-4630, Kristiansand, Norway. E-mail: odrun.a.gederaas@uia.no
cProteomics and Modomics Experimental Core, PROMEC, at NTNU and the Central Norway Regional Health Authority, Trondheim, Norway
dDepartment of Molecular Biology and Cell Cycle, Institute of Molecular Biology “Roumen Tsanev”, Bulgarian Academy of Sciences, 1113 Sofia, Bulgaria
eCMIC Cellular & Molecular Imaging Core Facility, Norwegian University of Science and Technology, NTNU, and the Central Norway Regional Health Authority Norway, Trondheim, Norway
fPCI Biotech AS, Ullernchaussen 64, 0379 Oslo, Norway
First published on 22nd June 2023
Abstract
Photochemical internalization (PCI) is a promising new technology for site-specific drug delivery, developed from photodynamic therapy (PDT). In PCI, light-induced activation of a photosensitizer trapped inside endosomes together with e.g. chemotherapeutics, nucleic acids or immunotoxins, allows cytosolic delivery and enhanced local therapeutic effect. Here we have evaluated the photosensitizer meso-tetraphenyl chlorine disulphonate (TPCS2a/fimaporfin) in a proteome analysis of AY-27 rat bladder cancer cells in combination with the chemotherapeutic drug bleomycin (BML). We find that BLMPCI attenuates oxidative stress responses induced by BLM alone, while concomitantly increasing transcriptional repression and DNA damage responses. BLMPCI also mediates downregulation of bleomycin hydrolase (Blmh), which is responsible for cellular degradation of BLM, as well as several factors known to be involved in fibrotic responses. PCI-mediated delivery might thus allow reduced dosage of BLM and alleviate unwanted side effects from treatment, including pulmonary fibrosis.
Introduction
Urinary bladder cancer is the fourth most common non-cutaneous malignancy and has a strong predominance in men.1 Transurethral resection of bladder tumours is the primary treatment,2 but is associated with a recurrence rate of approximately 60% after two years3 and disease progression to invasive cancer is observed in 25% of cases. Moreover, due to the requirement for costly follow-ups, bladder cancer has the highest life-time cost of all cancers in the US.4 Development of new non-invasive therapies, including combination treatment regimes, is thus of major importance.Bleomycin (BLM) is a water-soluble antibiotic of approximately 1500 Da that it is part of a specific group of glycopeptide-derived natural products isolated from the bacterium Streptomyces verticillus.5 BLM is used in combination with other antineoplastic agents to effectively treat lymphomas, testicular carcinomas, and squamous cell carcinomas of cervix, head, and neck.6,7 However, the therapeutic efficacy is limited by acquired drug resistance, renal and lung toxicity and development of lung fibrosis.8,9 The latter is attributed to low levels of the bleomycin-inactivating enzyme bleomycin hydrolase (BLMH) in the lung.10,11 The cytotoxic effects of BLM are believed to be mediated primarily via oxidative cleavage of DNA in the presence of redox-active metal ions such as Fe2+, thus creating both single- (SSB) and double- (DSB) strand breaks. However, considerable damage to RNA as well as lipid peroxidation has also been reported.12,13 This leads to G2/M-arrest and induction of apoptosis in BLM sensitive cells, apparently triggered by induction of ATM/ATR.14 Nevertheless, the molecular mechanisms underlying cytotoxicity as well as acquired BLM resistance remain inadequately understood. In contrast to many other chemotherapeutic drugs, BLM is rather large and hydrophilic, and does not readily pass the plasma membrane by passive diffusion. Instead, it binds to a surface receptor, potentially a glucose transporter,15 and is taken up by endocytosis. BLM thus accumulates in endosomes and lysosomes, which restricts its cytotoxicity.16 This has been confirmed by electro-permeabilization of cells in the presence of BLM, in which as few as 500 molecules of BLM introduced into the cytosol was sufficient to kill the cells.17
Photochemical internalization (PCI) is a relatively novel technology for cytosolic delivery of therapeutic agents based on principles of photodynamic therapy (PDT).18 A common mechanism of mammalian cells to take up extracellular substances that cannot directly pass the plasma membrane, is to invaginate the membrane and form an endocytic vesicle containing the substance. A therapeutic molecule taken up by endocytosis must then be released from endosomes to reach their specific intracellular target. This can be achieved by concomitant administration of e.g. an amphiphilic photosensitizer that incorporates in the outer leaflet of the plasma membrane and is subsequently transferred to the inner endosomal membrane.18 Upon light activation at appropriate wavelengths, the photosensitizer generates reactive oxygen species (ROS) that rupture the endosomal membrane, thereby releasing the entrapped drug. Rather than being degraded by lysosomal hydrolases, the drug is released to cytosol to act on its intracellular target.19 Such a delivery strategy would be attractive for several therapeutic agents, including proteins, nucleic acids, synthetic polymers, and other agents that that do not readily pass through the plasma membrane, such as BLM. In a mouse xenograft model, photochemical delivery of BLM aided by the photosensitizer aluminium phthalocyanine disulfonate (AlPcS2a) resulted in delayed tumour growth and mediated 60% complete response in two tumour models, whereas no complete response was observed with BLM alone.20 Promising effects were also observed in a phase I clinical study, including mainly head and neck cancer patients.21 Very recently, the photosensitizer meso-tetraphenylchlorin disulfonate (TPCS2a/fimaporfin, Amphinex®) was demonstrated to enhance the efficacy of gemcitabine in the treatment of inoperable periphilar cholangiocarcinoma (bile duct cancer).22 Fimaporfin-based PCI has also been shown to enhance the cytotoxic effect of BLM in rat bladder cancer cells23 and in clinically relevant animal model studies.24,25 PCI is currently also investigated for use as a vaccine therapy for cancer indications.26–28
In the present study we aimed to elucidate the molecular mechanisms underlying fimaporfin-enhanced BLM toxicity in the AY-27 rat bladder cancer cell line in more detail by employing stable isotope labelling by amino acids in cell culture (SILAC)-based quantitative proteomics. Deciphering cellular pathways affected by either single treatment or combined fimaporfin/BLM (BLMPCI), could aid future decisions in the treatment of bladder cancer.
Results and discussion
Cell viability analyses
The enhanced efficacy and reduced side effects associated with PCI-mediated delivery of BLM compared to standalone BLM treatment is well established, but the molecular mechanisms underlying this remain poorly defined. To this end, we undertook unbiased SILAC-based proteome profiling of AY-27 rat bladder carcinoma cells to monitor cellular responses after PCI-based delivery of low dose BLM, compared to BLM alone. To identify physiologically relevant responses, we aimed at employing a PCI treatment regimen that induced significant cytotoxicity, but still rendered most cells viable. As illustrated in Fig. 1, neither of the single (photosensitizer, BLM, light) nor the double photosensitizer/BLM or BLM/light treatments, mediated significantly reduced viability. Conversely, both PDT and BLMPCI mediated significant loss of viability. Whereas increasing light exposure time from 1 to 2 min did not increase the toxicity of PDT, only ∼20% viable cells remained after BLMPCI. We thus decided to progress with 1 min illumination for the SILAC experiments, which rendered 70–75% of the cells viable in both the PDT and the BLMPCI series. | ||
| Fig. 1 Viability of AY-27 cells after standalone or combined treatments with fimaporfin (TPCS2a, 0.2 μg mL−1), BLM (0.1 μM) and light (435 nm, 13 mW cm−2). After treatment, cells were incubated in regular culture medium for 24 h prior to viability analysis by MTT assay. Each bar represents mean values of three independent experiments, each with three technical replicates ± SD. *; p < 0.05, **; p < 0.01. | ||
SILAC analyses
Three independent biological experiments in each of the BLM and BLMPCI series were undertaken. Although not the primary focus of our study, we also included two independent experiments in which the cells were treated with the photosensitizer fimaporfin and illumination (denoted PDT) (Fig. S1, ESI†). MaxQuant analysis quantified 4443, 4428 and 4508 proteins in PDT, BLM and BLMPCI, respectively (FDR < 0.01, Table S1, ESI†). The total distributions of expressed proteins in PDT, BLM and BLMPCIversus control are illustrated in Fig. 2A. Among these, 51, 80 and 117 proteins (PDT, BLM and BLMPCI) displayed significant differential expression (p < 0.05 and >1.5-fold up- or downregulated) relative to controls and were considered further. The distribution of significantly up- and downregulated proteins in the three series are given in Fig. 2B and showed a striking difference between the treatments. Whereas 57.5% of the differentially expressed proteins (DEPs) in the BLM-treated cells were upregulated, additional PCI-treatment mediated a marked relative increase of upregulated proteins to 83.9%. Somewhat surprisingly, there was no overlap between the DEPs in the BLM and the BLMPCI series (Fig. 2C), suggesting that the treatments largely triggered different cellular responses. 11 DEPs overlapped between the PCIBLM and the PDT series, and in each case their direction of change was identical (Table S1, ESI†). Thus, these DEPs likely represent proteins primarily affected by fimaporfin and light treatment. Unsupervised hierarchical clustering based on the SILAC ratios resulted in separate clustering of the proteomes after each of the three treatments, in which BLMPCI clustered between PDT and BLM and most closely to PDT (Fig. 2D). This further supports that the PDT component excerpts an important effect upon the proteome after BLMPCI treatment. Gene Ontology (GO) analysis of cellular component enrichment revealed a striking difference in subcellular localization of the affected proteins after the different treatments (Fig. 2E). Most notably, BLMPCI mediated marked upregulation of nuclear proteins and proteins associated with ribonucleoprotein complexes, compared to BLM alone (left panel). A likely explanation to this is that PCI facilitates increased targeting of BLM to DNA and RNA, thereby introducing lesions that trigger DNA/RNA damage responses. This is also supported by previous results showing that PCI treatment elevated the levels of DNA damage in three different bladder cancer cell lines.23 Conversely, BLMPCI resulted in marked downregulation of mitochondrial and lysosomal proteins compared to the non-treated controls, suggesting that the BLMPCI treatment inflicted extensive mitochondrial and lysosomal/endosomal damage compared with BLM alone. In addition, peptide mass changes mediated by oxidative damage to lysosomal and mitochondrial proteins could contribute to this, by hampering their identification in the MS step.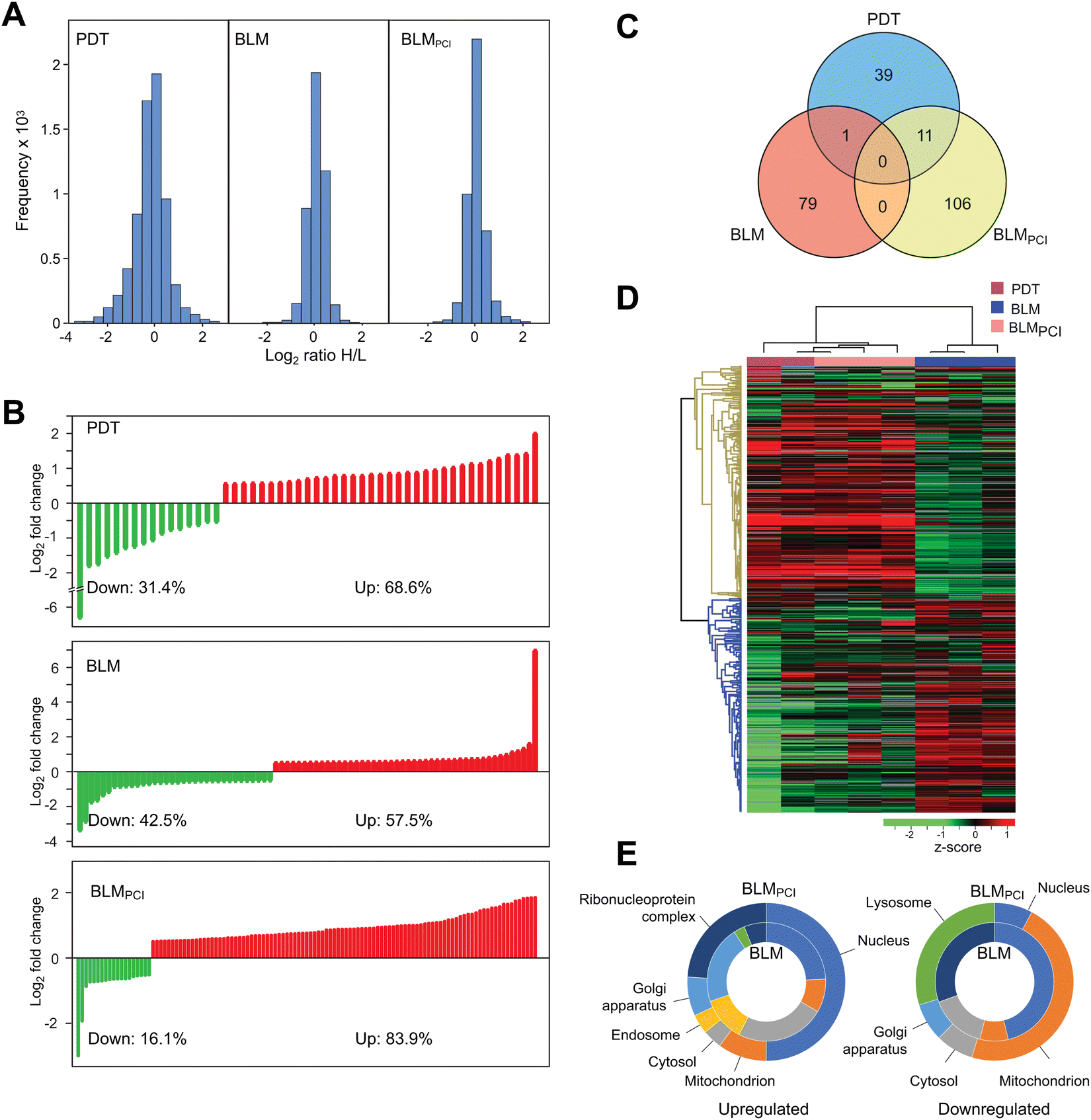 | ||
| Fig. 2 (A) Raw distribution of SILAC quantifications given as log2 (HEAVY (control)/LIGHT (treated)) ratios. (B) Distribution of DEPs (p < 0.05), fold change >|1.5| after BLM (upper panel) and BLMPCI (lower panel) treatment. (C) Venn diagram showing the number of unique and overlapping DEPs after the three treatments. (D) Two-way unsupervised hierarchical clustering of the 237 proteins that were differentially expressed after the different treatments. (E) GO annotation of the DEPs in (B) according to relative subcellular distribution within the most affected compartments. Inner circles represent up/downregulated proteins after BLM, while outer circles represent up/downregulated proteins after BLMPCI. | ||
BLMPCI attenuates upregulation of stress response proteins mediated by BLM alone
To search for modified biological pathways after different treatments, gene identifiers for the significant DEPs in the SILAC dataset were mapped in the ingenuity knowledge base, entered Ingenuity Pathway Analysis® (IPA) and plotted onto canonical pathways (Fig. 3, left panels). Here, BLM treatment alone mediated upregulation of energy-metabolic pathways; pentose phosphate pathway (PPP), glutaryl-CoA degradation and fatty acid β-oxidation. PPP has an important role in the response to oxidative stress by generating NADPH for the antioxidant machinery and by producing building blocks to repair DNA damage.29 Manual inspection of the data (Table S1, ESI†) also revealed that several other responses to oxidative stress were selectively upregulated after BLM treatment alone, including Glrx, Park7, Ppia, Psap, Gclm, Nqo1 and Txn. This also holds true for the four proteasomal subunits quantified, Psmb5, Psmb9, Psme1 and Psme4, and that are markers for cytosolic Fenton reactions.30 Potentially, the selective upregulation of fatty acid β-oxidation in the BLM arm could compensate for loss of glycolytic substrates by upregulated PPP. These results indicate that BLM alone induces several protective oxidative stress responses of the bladder cancer cells, in agreement with its proposed mechanism of action. Many of these responses appear to be attenuated by PCI delivery of BLM and may contribute to the increased cytotoxicity of the dual treatment. In support of this, IPA reported NRF2-mediated oxidative stress response as downregulated in BLMPCI (z-score −1.34, p = 3.4 × 10−3).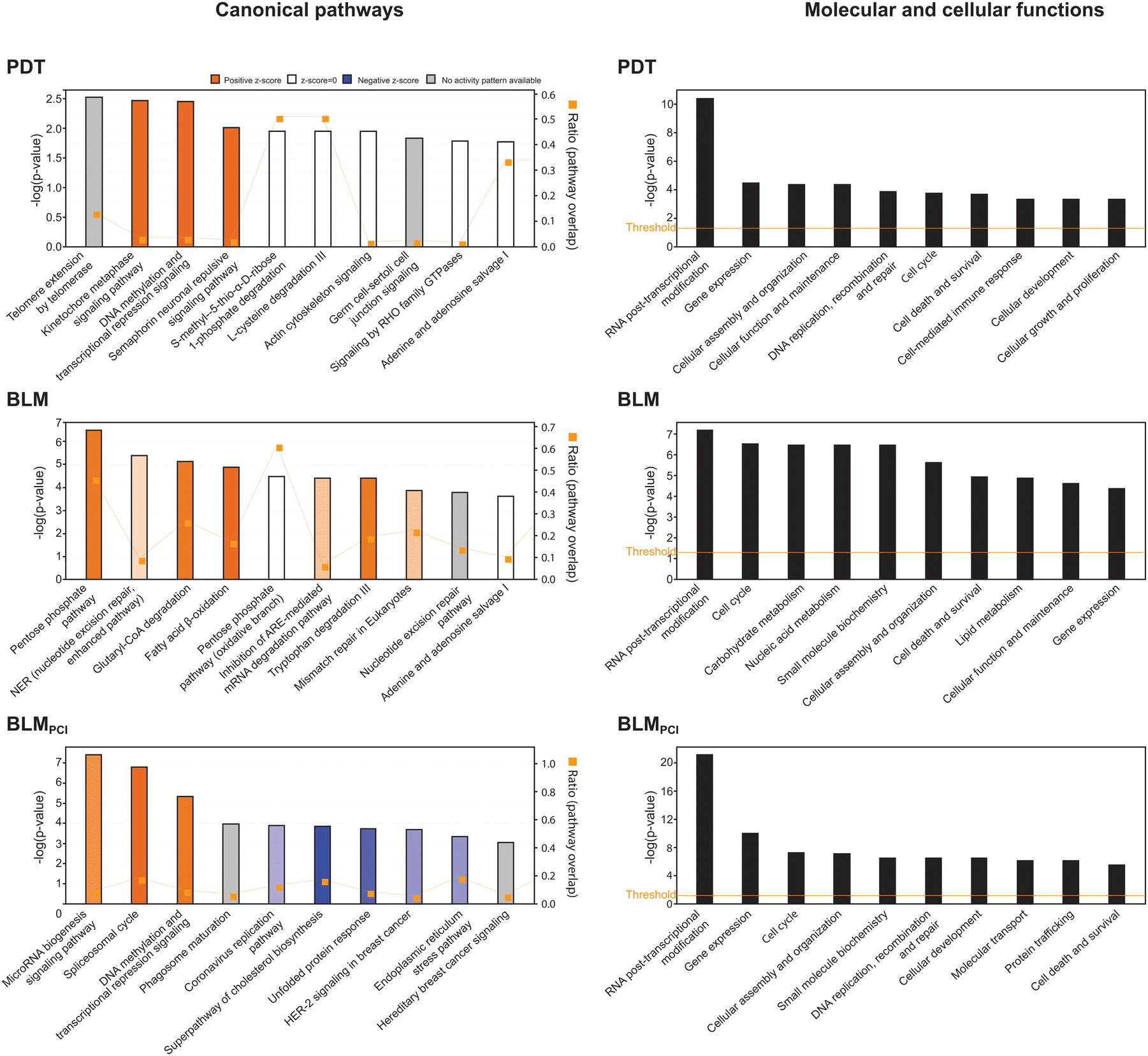 | ||
| Fig. 3 Results from IPA analysis showing the ten most significantly affected biological pathways (left panels) and molecular and cellular functions (right panels) after PDT, BLM and BLMPCI treatment. | ||
Attenuated stress responses also became evident when focusing on the most upregulated proteins after each treatment (Table 1). Neither of the ten most upregulated proteins after BLM alone were significantly affected by the PDT or BLMPCI treatments. Most upregulated was Kif21b (132-fold), a molecular motor that mediates pausing of microtubule growth31 and thus affects cellular structure and dynamics. Microtubule alterations were recently associated with BC aggressiveness and TUBB6 was identified as a biomarker of muscle invasion and poor prognosis.32 Kif21b was also found to be upregulated in a study of BLM-induced pulmonary fibrosis in mice.33 Thus, by employing PCI-mediated delivery and a reduced dose BLM, this serious adverse effect could potentially be alleviated. Vps26b (3.2-fold upregulated in BLM) is part of the retromer complex that recycles transmembrane receptors such as mannose 6-phosphate receptor (M6pr) from endosomes to the trans-Golgi network. Recent findings have, however, also associated Vps26B with microtubule structures by regulating mammalian ciliogenesis.34 A direct role of Vps26b in bladder cancer pathogenesis remains, however, to be investigated. Map3k20 (Zak, 2.3-fold upregulated in BLM) is a stress-activated, pro-apoptotic kinase involved in DNA damage checkpoint signalling and S and G2 cell cycle arrest.35 Recent findings demonstrate that it has a pivotal role in sensing ribosomal collisions that result when translation is arrested at mRNA lesions induced by e.g. UV-light, thereby inducing a ribotoxic stress response.36,37 BLM effectively induces oxidised bases such as 8-oxoG and FaPyG in DNA38 and such lesions are induced even more efficiently in mRNA and may lead to ribosomal stalling and defective protein synthesis.39 Thus, upregulation of Map3k20 could be a significant factor in the cellular response to BLM. It is also noteworthy that a newly identified small molecule inhibitor of Map3k20 ameliorated renal fibrosis. Since Map3k20 was not induced by PCI-mediated delivery of BLM it is tempting to speculate that this treatment modality may reduce pulmonary fibrosis during BLM treatment. PCNA was found to be 1.8-fold upregulated by BLM in agreement with previous findings and its established role in several DNA repair processes,40,41 but was not affected by BLMPCI.
| Gene symbol | Protein name | PDT FC | PDT p-value | BLM FC | BLM p-value | PCI FC | PCI p-value | Previously associated with bladder cancer and/or BLM response |
|---|---|---|---|---|---|---|---|---|
| PDT upregulated | ||||||||
| RGD1561149 | Similar to mKIAA1522 protein | 4.1 | 0.038 | 1.1 | 0.411 | 3.8 | 0.029 | |
| Lcor | Ligand dependent nuclear receptor corepressor | 2.8 | 0.039 | 1.8 | 1 | 2.6 | 0.097 | Predicts poor prognosis in BC86 |
| Nolc1 | Nucleolar and coiled-body phosphoprotein 1 | 2.7 | 0.029 | −1.3 | 0.021 | 2.1 | 0.010 | |
| Ahnak | AHNAK nucleoprotein | 2.7 | 0.007 | 1.1 | 0.190 | 1.9 | 0.032 | Predicts poor prognosis in BC87 |
| Ylpm1 | YLP motif-containing protein 1 | 2.5 | 0.014 | −1.4 | 0.063 | −1.2 | 0.420 | |
| Fip1l1 | S-Adenosylhomocysteine hydrolase-like protein 1 | 2.4 | 0.034 | 1.1 | 0.416 | 2.0 | 0.022 | |
| Zcchc7 | Zinc finger CCHC domain-containing protein 7 | 2.3 | 0.028 | −1.1 | 0.386 | 1.6 | 0.032 | |
| Ice2 | Interactor of little elongation complex ELL subunit 2 | 2.2 | 0.015 | −1.1 | 0.637 | 2.0 | 1 | |
| Tns4 | Tensin-4 | 2.2 | 0.027 | 1.1 | 0.535 | 1.0 | 0.990 | |
| Mfap1a | Microfibrillar-associated protein 1A | 2.1 | 0.016 | −1.1 | 0.547 | 1.8 | 0.022 | |
| PDT downregulated | ||||||||
| Tubgcp3 | Gamma-tubulin complex component | −77.0 | 0.022 | −19.2 | 0.452 | −45.5 | 0.115 | |
| Gsn | Gelsolin | −3.4 | 0.041 | −1.2 | 0.511 | −1.6 | 0.040 | Predicts poor prognosis in BC88 |
| Aprt | Adenine phosphoribosyltransferase | −3.4 | 0.007 | 1.51 | 0.205 | −1.3 | 0.394 | |
| Got1 | Aspartate aminotransferase | −2.9 | 0.049 | 1.51 | 0.060 | −1.2 | 0.819 | |
| Ube2n | Ubiquitin-conjugating enzyme E2 N | −2.8 | 0.035 | 1.35 | 0.081 | −1.2 | 0.231 | Predicts poor prognosis in BC89 |
| Cmpk1 | UMP-CMP kinase | −2.5 | 0.010 | 1.24 | 0.134 | −1.8 | 0.078 | |
| Sars | Serine-tRNA ligase, cytoplasmic | −2.4 | 0.037 | 1.34 | 0.446 | −1.4 | 0.147 | |
| Pin4 | Peptidyl-prolyl cis–trans isomerase | −2.3 | 0.002 | 1.40 | 0.097 | −1.2 | 0.068 | |
| Abcb6 | ATP-binding cassette sub-family B member 6 | −2.2 | 0.032 | 1.87 | 1 | −1.6 | 0.088 | |
| Crot | Peroxisomal carnitine O-octanoyltransferase | −1.9 | 0.027 | 1.14 | 0.010 | −1.1 | 0.501 | |
| BLM upregulated | ||||||||
| Kif21b | Kinesin-like protein KIF21B | 1.4 | 0.094 | 132 | 0.048 | 1.24 | 0.161 | Upregulated by BLM in mice90 |
| Vps26b | Similar to Vacuolar protein sorting 26 homolog | 1.3 | 0.057 | 3.2 | 0.047 | −1.2 | 0.538 | |
| Fam25A | Family with sequence similarity 25, member A | ND | 1 | 2.6 | 0.002 | ND | 1 | |
| Mrpl20 | Mitochondrial ribosomal protein L20 | 1.2 | 0.850 | 2.4 | 0.029 | 1.2 | 0.520 | |
| Map3k20 | Mitogen-activated protein kinase kinase kinase 20 | 1.1 | 1 | 2.3 | 0.003 | 1.0 | 0.203 | |
| Clcn2 | Chloride channel protein | ND | 1 | 2.1 | 0.037 | 1.2 | 0.377 | |
| Lrrc58 | Leucine-rich repeat-containing 58 | −1.3 | 1 | 2.0 | 0.037 | ND | 1 | |
| Elp6 | Elongator complex protein 6 | ND | 1 | 1.9 | 0.027 | ND | 1 | |
| Mat2b | Methionine adenosyltransferase 2 subunit beta | −2.0 | 0.453 | 1.9 | 0.024 | 1.1 | 0.322 | |
| Pcna | Proliferating cell nuclear antigen | −1.7 | 0.309 | 1.8 | 0.024 | 1.1 | 0.842 | Overexpressed in MBIC.91 Suggested drug target in BC.92 |
| BLM downregulated | ||||||||
| Ndel1 | Nuclear distribution protein nude homolog 1 | 1.2 | 1 | −11.0 | 0.020 | ND | 1 | Downregulated by BLM in mice90 |
| Fam185A | Protein FAM185A | ND | 1 | −7.8 | 0.017 | ND | 1 | |
| Keap1 | Kelch-like ECH-associated protein 1 | 1.3 | 1 | −3.6 | 0.049 | −1.1 | 0.489 | |
| Zyx | Zyxin | 1.5 | 1 | −3.5 | 0.024 | 1.3 | 1 | |
| Htra1 | Serine protease HTRA1 | −1.7 | 0.291 | −2.7 | 0.044 | −1.2 | 0.002 | Suggested early and sensitive urine biomarker in BC93 |
| Dbn1 | Drebrin | 1.1 | 0.604 | −2.4 | 0.003 | 1.3 | 0.045 | |
| Cuta | Divalent cation tolerant protein CUTA | ND | 1 | −2.0 | 0.001 | −1.7 | 1 | |
| Ctdnep1 | CTD nuclear envelope phosphatase 1 | 1.0 | 1 | −1.9 | 0.049 | −4.2 | 0.243 | |
| Arhgap11a | Rho GTPase-activating protein 11A | 1.6 | 0.064 | −1.9 | 0.029 | 1.5 | 0.279 | |
| Magt1 | Magnesium transporter protein 1 | −1.2 | 0.183 | −1.9 | 0.022 | −1.1 | 0.344 | |
| BLMPCI upregulated | ||||||||
| RGD1561149 | Similar to mKIAA1522 protein | 4.1 | 0.03 | 1.1 | 0.41 | 3.8 | 0.028 | Predicts favourable prognosis in renal cancer (proteinatlas.org) |
| Rpl38 | 60S ribosomal protein L38 | 4.6 | 0.09 | 1.2 | 0.32 | 3.8 | 0.014 | |
| Ccdc137 | Coiled-coil domain-containing 137 | 3.6 | 0.08 | −1.5 | 0.15 | 3.8 | 0.024 | |
| Mnt | MAX network transcriptional repressor | 2.9 | 1 | 1.5 | 0.22 | 3.7 | 0.027 | Myc antagonist |
| Sp1 | Transcription factor Sp1 | 3.5 | 0.18 | 1.1 | 0.43 | 3.7 | 0.035 | Predicts poor prognosis in BC94 |
| Gatad2a | GATA zinc finger domain-containing 2A | 3.6 | 0.31 | 1.2 | 0.43 | 3.5 | 0.023 | Transcription repressor |
| Maff | MAF bZIP transcription factor F | 6.0 | 0.23 | −1.1 | 0.86 | 3.3 | 0.023 | Transcription factor. Predicts increased survival in BC95 |
| Dhx38 | DEAH-box helicase 38 | −1.1 | 0.90 | 1.0 | 0.42 | 3.3 | 0.047 | Pre-mRNA-splicing factor |
| Zc3h4 | Zinc finger CCCH-type-containing 4 | 3.7 | 0.17 | 1.2 | 0.012 | 3.3 | 0.044 | |
| Hmga1 | High mobility group protein HMG-I/HMG-Y | 2.4 | 0.11 | −1.5 | 0.1 | 3.1 | 0.035 | High mRNA expression associated with poor prognosis in BC96 |
| BLMPCI downregulated | ||||||||
| Apob | Apolipoprotein B-100 | −3.9 | 0.135 | −5.3 | 0.073 | −8.4 | 0.013 | Suggested non-invasive biomarker for BC97 |
| Sprr2d | Small proline-rich protein 2D | −1.7 | 1 | −4.4 | 1 | −4.0 | 0.037 | |
| Gdpd3 | Glycerophosphodiester phosphodiesterase domain-containing 3 | −1.6 | 0.070 | 1.1 | 0.795 | −1.9 | 0.015 | Positive IHC biomarker for neoadjuvant response in BC98 |
| Ctsb | Cathepsin B | −2.9 | 0.085 | 1.1 | 0.167 | −1.8 | 0.030 | |
| Galk1 | Galactokinase 1 | −1.9 | 0.166 | 1.2 | 0.847 | −1.7 | 0.031 | Predicts poor prognosis in BC99 |
| Elovl5 | Elongation of very long chain fatty acids protein 5 | −4.2 | 0.217 | 1.7 | 0.224 | −1.7 | 0.037 | Predicts poor prognosis in renal cell carcinoma100 |
| Ctsl | Procathepsin L | −1.4 | 0.086 | 1.1 | 0.048 | −1.7 | 0.030 | Predicts poor prognosis in BC101 |
| Lipt2 | Putative lipoyltransferase 2, mitochondrial | 1.1 | 1 | 1.1 | 0.461 | −1.7 | 0.021 | |
| Mpv17 | Protein Mpv17 | ND | 1 | 0.9 | 0.482 | −1.7 | 0.046 | |
| Cnih4 | Cornichon family AMPA receptor auxiliary protein 4 | −3.4 | 0.334 | 1.0 | 0.391 | −1.7 | 0.006 | |
Three of the most upregulated proteins after BLM treatment, Fam25A, Lrrc58 and Elp6, were not detected in any of the BLMPCI samples, suggesting strong downregulation. In humans, FAM25A mRNA is highly expressed in cervical and uterine tissues, but any function of the protein remains elusive. The function of Lrrc58 is also not known, but the methylation status of human LRRC58 has been proposed part of a 16-gene methylation panel for early and differential diagnosis of prostate cancer.42 Elp6 is one of the six subunits of the Elongator complex. Elongator catalyses the primary modification of several tRNAs carrying a uridine at the anticodon wobble position (U34), by introducing either 5-carbamoylmethyluridine (ncm5U), 5-methoxycarbonyl-methyluridine (mcm5U) or 5-methoxycarbonylmethyl-2-thiouridine (mcm5s2U) at the C5 position.43,44 These modifications are specifically required for efficient decoding of AA-ending codons under stress conditions and mediate efficient translation of proteins involved in the response to oxidative stress and DNA damage.45,46 Although we did not detect significant changes in any of the other Elongator subunits, the potential involvement of Elp6 in response to BLM treatment and its potential downregulation by PCI-mediated BLM delivery, warrants further investigation.
Among the 46 upregulated proteins after BLM that met our statistical criteria, only two were common to a set of 88 upregulated proteins identified in a rat model with BLM-induced pulmonary fibrosis.47 This likely reflects different proteome responses in different cell types and that proteome data from tissue specimens are averaged over a complex mixture of cell types and extracellular proteins, which renders direct comparison of data complicated. The two commonly upregulated proteins in the data sets were Sfn (14-3-3 protein sigma) and aldehyde dehydrogenase, cytosolic 1 (Aldh1a7). Both 14-3-3 proteins and aldehyde dehydrogenases are often upregulated under conditions promoting oxidative stress and 14-3-3 proteins are involved in the regulation of several DNA repair pathways,48,49 in agreement with the proposed mechanisms underlying BLM toxicity.
PCI-mediated delivery of BLM significantly downregulates bleomycin hydrolase (Blmh)
Bleomycin hydrolase is a neutral cysteine protease of the papain superfamily. It is ubiquitously expressed across kingdoms and in mammals it has important roles in neonatal survival and epidermal integrity. It is also the sole enzyme required for BLM deamination and inactivation.8 Importantly, we found that Blmh was significantly downregulated (1.4-fold, p = 0.016) after BLMPCI (Table S1, ESI†). Conversely, it was found to be upregulated in all samples after BLM, although reported non-significantly (p = 0.18) due to large variation in the MS intensities. This observation is highly significant, since PCI-mediated delivery, in addition to allowing increased nuclear targeting, may prolong the cellular lifetime of BLM. Also, any modality that could allow reduced dose of BLM while maintaining cytotoxic effect could alleviate the renal and lung toxicity associated with BLM treatment.BLMPCI mediates enhanced transcriptional repression and DNA damage responses, compared to BLM alone
The most upregulated biological processes after BLMPCI were MicroRNA biogenesis signalling, spliceosomal cycle, and DNA methylation and transcriptional repression signalling (Fig. 3). DNA metabolic pathways were also the most affected after PDT-treatment (kinetochore metaphase signalling and DNA methylation and transcriptional repression signalling), although with lower p-values. This conforms to the co-segregation of PDT with BLMPCI in the hierarchical clustering analysis (Fig. 2D) and supports that PCI-mediated delivery of BLM allows enhanced nuclear targeting. Considerable overlap between pathways was also observed when the SILAC-results were plotted onto molecular functions. Here, RNA post-transcriptional modification was the most affected function after all three treatments, and by far most significant for BLMPCI (Fig. 3, right panels). The SILAC data were also analysed for enriched GO terms by employing the GOrilla single ranked list method50 (Table S2, ESI†). Here the most significantly upregulated biological process in and BLMPCI was (negative) regulation of transcription by RNA Polymerase II, while nucleobase-containing small molecule biosynthetic process was most downregulated. This is in agreement with previous studies demonstrating that a common cellular response to genotoxic agents is to downregulate overall transcription, while ensuring that a subset of transcripts is modulated to mediate a targeted DNA damage response (DDR) e.g., via altered transcriptional rate, splicing and 3′-processing.51,52Within microRNA biogenesis signalling, several components of the nuclear pore complex (Nup35, 37, 62, 98, 153, 160 and 188, Ahctf1 and Pom121) were uniquely upregulated after BLMPCI, suggesting increased nucleocytoplasmic shuttling. This is in accordance with increased spliceosomal cycle, in which snRNAs are exported to the cytoplasm prior to assembly of snRNPs and re-entry into the nucleus. Several spliceosomal proteins were upregulated, including Dhx16, Dhx38, Sf3b2 and Isy1. In addition to its role in splicing, human ISY1 was recently shown to be induced by oxidative stress and to enhance 5′-3-endonuclease activity of APE1, thereby increasing base excision repair (BER) of oxidised DNA bases.53 Several important effectors of epigenetic reprogramming were also uniquely upregulated in the BLMPCI arm, including the polycomb repressive complex 1 (PRC1) ubiquitin E3 ligases Ring1 and Rnf2 (Ring1B), the PRC2 histone lysine methyltransferases EZH1 and EED, and the PRC2 recruiter MTF2, which all mediate transcriptional repression.54
Two transcriptional repressors, Mnt and Gatad2a, and two transcription factors, Sp1 and Maff were also among the most upregulated proteins after BLMPCI (Table 1). Max-binding protein Mnt (3.7-fold up) acts as a heterodimer repressor in complex with Max. Since Max also is a transcription activator when heteroduplexed with Myc, Mnt acts as a tumour suppressor by antagonizing Myc-induced transcription. Upregulation of Mnt might thus be highly significant in the antitumor activity of BLMPCI, given that Myc overexpression is commonly observed in bladder cancer and is associated with tumour initiation and progression.55 Gatad2a is part of the nucleosome remodelling and deacetylase complex (NuRD), which comprises at least six subunits and has an important function in the DNA damage response.56 Recruitment of NuRD to sites of DNA damage creates a repressive environment that prevents transcription of damaged genes and facilitates DNA repair.57 Recent research has shown that NuRD exists in different forms with distinct functions, largely determined by variant composition of the subunits. Thus, NuRD containing Gatad2a, but not Gatad2b, is rapidly recruited to sites of DNA damage and facilitates repair of DSBs by homologous recombination.58 Two other subunits of the complex, Chd4 and Rbbp4, were also significantly upregulated after BLMPCI (Table S1, ESI†). CHD4 is a target for phosphorylation by ATM and acts in DNA repair by guiding assembly of DNA repair factors such as RNF168 and BRCA1 to promote homologous recombination repair of DSBs.59 The transcription factor SP1 is required for expression of numerous genes important for cell proliferation, apoptosis and DNA damage responses and is often overexpressed in human cancers and associated with poor prognosis.60 The tumour suppressor Bclaf1 was 2.9-fold upregulated in BLMPCI. Recent research indicates that BCLAF1 has a critical function in determining cellular fate after DNA-DSB induction. On the one hand, association of BCLAF1 with γH2AX-bound DSBs stabilizes Ku70/DNA-PK association and DSB repair by non-homologous end-joining. On the other hand, BCLAF1 promotes caspase-dependent apoptosis.61 Tp53bp1 and Rif1 were also upregulated after BLMPCI (1.6 and 1.7-fold, respectively). Both proteins were also upregulated after PDT, although sub-significantly (p = 0.09). These two proteins constitute a functional module that stabilizes the chromatin topology at DSBs to protect DNA ends against aberrant processing.62 Fbxo6, which is also involved in DDR constitutes a part of the SCF-type E3 ubiquitin ligase complex. Notably, Fbxo6, which is also involved in the DNA damage response, was not detectable after BLMPCI, whereas it was readily detectable in all other samples. Fbxo6 promotes degradation of activated checkpoint kinase Chek1 and could thus constitute part of the response to BLMPCI by promoting G2-arrest and activation of DNA DSB-repair by homologous recombination. In ovarian clear cells, CHEK1 inhibitors have been shown to selectively kill cells with sustained CHEK1 activation after BLM treatment63 and could constitute potential adjuvants in BLMPCI treatment.
Among the most upregulated proteins after BLMPCI, some are poorly characterized and not assigned to any of the pathways discussed above. RGD1561149 (3.8-fold) is an uncharacterized protein that is 81% identical to human KIAA1522, which is localized to the plasma membrane, cell junctions and nucleoplasm. In renal and pancreatic cancers KIAA1522 is an unfavourable prognostic marker, whereas in renal cancer it is a favourable prognostic marker (https://www.proteinatlas.org). The 60S ribosomal protein Rpl38 was 3.8-fold upregulated. Rpl38 promotes selective translation of a subset of Hox genes via IRES (internal ribosomal entry sites) present in their 5′-UTRs.64 Very recently, RPL38 was shown to bind the methyltransferase METTL3, thereby inducing m6A modification of SOCS2 mRNA and downregulation in human cartilage cells.65 To what extent RPL38 is involved in stress-regulated gene expression via m6A in bladder cancer remains to be investigated. Noteworthy, however, the m6A reader Igf2bp2, which increases stability of m6A-modified mRNAs, was significantly upregulated only after BLMPCI.66
Zc3h4 (3.3-fold upregulated after BLMPCI) is a nucleotide-binding CCCH-type zinc-finger protein, which often targets mRNAs encoding cytokines and inflammatory factors. Very recently, a role of Zc3h4 was identified in silica-induced epithelial to mesenchymal transition (EMT) leading to pulmonary fibrosis. Zfp36l2 (2.4-fold upregulated after BLMPCI) binds AU-rich elements (ARE) in the 3′-UTR of certain mRNAs, mediates signalling to the mRNA decay machinery and plays a key role in controlling S-phase progression in the case of genomic insult.67 It is also part of a nine-gene prognostic indicator panel for recurrence with muscle-invasive bladder cancer.68
While several DDR factors were found to be significantly upregulated after BLMPCI compared to BLM alone, some displayed an opposite trend. Pcna, Rpa1 and Rpa2 were 1.8-, 1.6- and 1.3-fold upregulated, respectively, after BLM alone but remained unaffected after BLMPCI. These proteins are also core factors of the chromosomal replisome and could thus reflect increased proliferation after BLM treatment. However, we found that Mki67 was threefold upregulated after BLMPCI (and fourfold, but sub-significantly after PDT) and remained unaffected after BLM alone. Mki67 is widely used as a proliferation marker in cancer histopathology and high expression correlates with poor survival of bladder cancer across several sub-groups.69 Generally, reduced proliferation confers increased resistance of cancer cells to chemotherapeutic agents. This has also been shown for BLM in several cell lines harbouring acquired BLM resistance.9 Conversely, cells in which increased resistance to fimaporfin-PDT has been induced by repeated treatments, demonstrate increased proliferation capacity.70 In agreement with this, we also observed a weak, but consistent increase in proliferation in AY-27 cells treated with the fimaporfin alone (Fig. 1). Given the significantly decreased cell viability observed after 1 min illumination in both the PDT and the BLMPCI protocols (Fig. 1), the increased Mki67 expression might seem paradoxical. However, recent research has shown that the Mki67 protein level is cell cycle dependent, peaking in late G2/M phase. Moreover, when cells are arrested by DNA damage, significant amounts of Mki67 are still detectable in the cells after 24 h.71 Thus, the increased Mki67 levels could result from increased number of cells arrested in G2/M after PDT and BLMPCI, and with elevated levels remaining at harvest 1 h post-treatment. To this end we undertook flow cytometric analysis of cells subjected to the different treatments. Here, BLM alone mediated a shift from G1- to S-phase whereas BLMPCI mediated a shift from G1 to G2/M-phase (Fig. 4, left panels). Although the changes in cell cycle distribution were modest, they are entirely in agreement with the observed upregulation of Pcna, Rpa1 and Rpa2 after BLM- and of Mki67 after BLMPCI. Interestingly, applying PCI-mediated delivery of BLM also resulted in a near doubling of necrotic cells compared with BLM alone. This was also observed after PDT. Conversely, the fraction of apoptotic cells remained less affected across the treatments (Fig. 4, right panels). A previous study in which AY-27 cell were treated with a different photosensitizer, ruthenium porphyrin, also demonstrated a similar outcome.72 This might seem somewhat paradoxical, since the AY-27 cells expressed ample amounts of Tp53 (Table S1 and Fig. S2, ESI†). However, we were not able to identify p21 (Cdkn1a), in any of the samples by MS (Table S1, ESI†) or by western analysis (data not shown), suggesting that Tp53 is functionally inactive in the cells. These results strongly support that BLMPCI induces cell death by necrosis in the AY-27 cells, and that the PDT component is a major contributor to this, in agreement with the general notion that necrosis is passive, accidental cell death resulting from environmental perturbations with uncontrolled release of inflammatory cellular contents.73
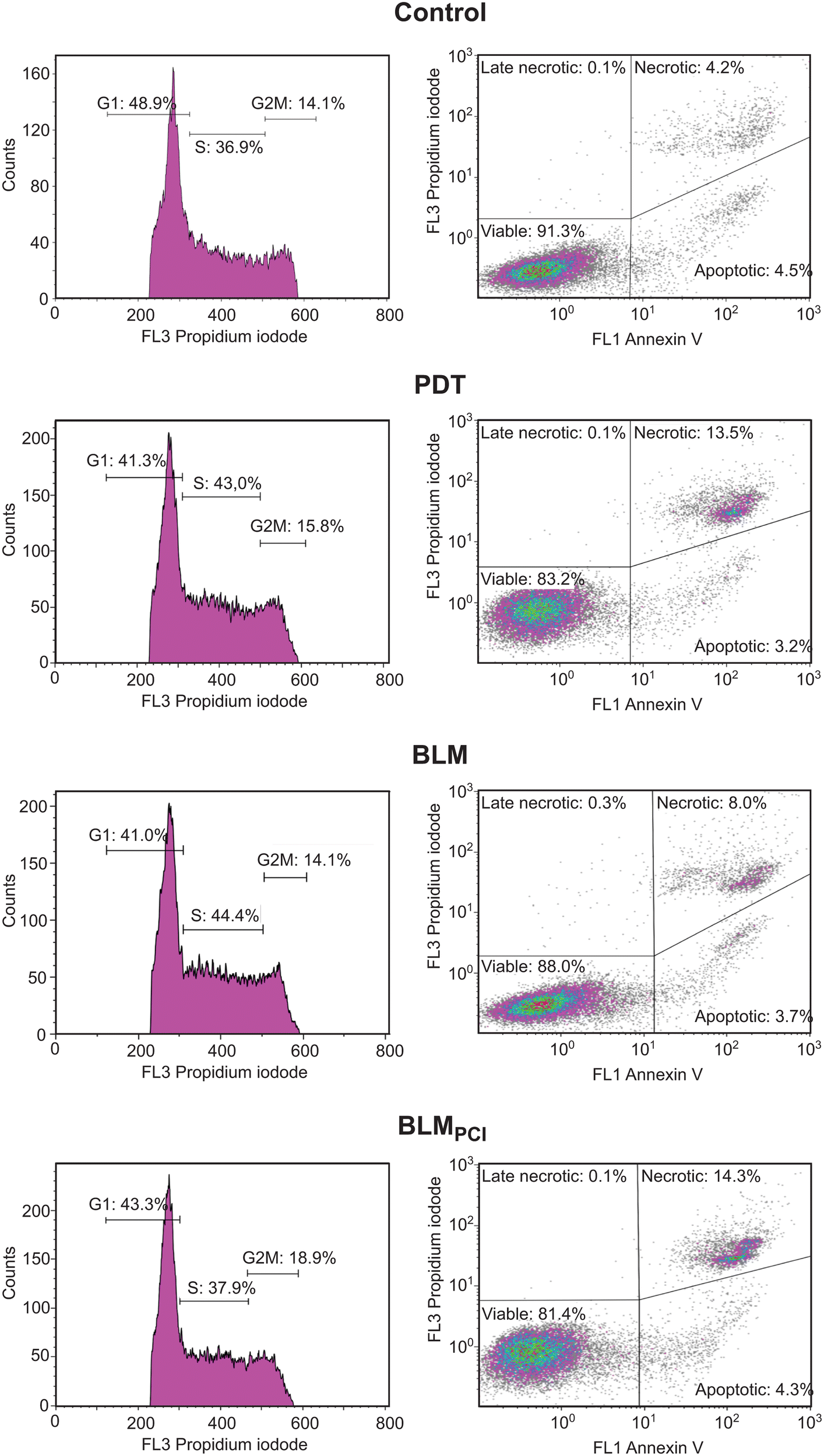 | ||
| Fig. 4 Cell cycle distribution (left panels) and fraction of apoptotic/necrotic cells (right panels) after the different treatments. | ||
BLMPCI attenuates downregulation of tumour suppressor proteins mediated by BLM alone
The tumour suppressor Keap1 was 3.6-fold downregulated in the BLM-treated cells, whereas it was not significantly changed after BLMPCI (Table 1). Keap1 is the inhibitory partner of the antioxidant transcription factor Nrf2, and its downregulation would increase Nrf2-mediated expression of antioxidant enzymes and promote resistance to chemotherapeutic agents and oxidative stress. PCI-mediated delivery may thus contribute to enhanced cytotoxicity of BLM. Zyx (zyxin) was 3.5-fold downregulated in BLM whereas no significant difference was observed in BLMPCI. It has actin polymerization activity, modulates cell adhesion and expression of integrins and may act as an oncogene or a tumour suppressor depending on the tumour in question. In a study encompassing 173 patients with bladder transitional cell carcinoma, low levels of zyxin were significantly associated with higher tumour grade and stage.74 The serine protease Htra1 was 2.7-fold downregulated in BLM and remained unchanged in BLMPCI. Htra1 functions as a tumour suppressor in various solid tumours and has been suggested as an early and highly sensitive biomarker for bladder cancer.75 Another actin remodeler, Dbn1 (drebrin) was 2.4-fold downregulated in BLM, whereas a weak, but significant upregulation was observed in BLMPCI. Drebrin is upregulated in bladder cancers compared to normal bladder tissue and mediates progranulin-dependent activation of AKT and MAPK and stimulation of proliferation and invasion.76 However, it has also been suggested to suppress dynamin-mediated endocytosis,77 which potentially could enhance the cellular uptake of BLM and contribute to its increased efficacy. Finally, we found that the Sirt1 regulator Rps19bp1 (Aros) was significantly upregulated (1.7-fold) after BLM, but unchanged after BLMPCI. SIRT1 may act as a tumour promoter or suppressor through its deacetylase activity towards e.g., p53, HSF1, STAT3 and mTOR.78 Likewise, AROS have been reported both as an activator and inhibitor of SIRT1.79 Any significance of AROS after BLM or BLMPCI treatment thus warrants further investigation.Conclusion
In the present study, we demonstrate that photochemical internalization of a low-dose BLM mediates a synergistic cytotoxic effect in the rat bladder cancer cell line AY-27. Whereas the PDT treatment excerpts a dominant effect on the cellular response, it concomitantly allows increased nuclear targeting of BLM and apparently attenuates several stress responses that previously have been associated with BLM treatment. The BLM-PCI combinatorial treatment yielded several significant findings compared to BLM treatment alone. Notably, it reduced the decrease in tumor suppressor proteins, led to upregulation of spliceosomal proteins and transcriptional repressors, caused a greater degree of DNA damage response, reduced the increase in stress response proteins and downregulated bleomycin hydrolyase. Among the proteins differentially affected by BLMPCI compared to BLM alone, several have also been associated with development of fibrosis. Thus, BLMPCI should be further evaluated in the treatment of bladder cancer as a potential means to reduce the effective BLM dose and thus alleviate adverse effects, including pulmonary fibrosis.Experimental
Chemicals
RPMI-1640 medium, L-glutamine, foetal bovine serum (FBS), sodium pyruvate, nonessential amino acids, trypsin, accutase and phosphate buffered saline (PBS) were from Gibco BRL, Life Technologies (Inchinnan, Scotland). Gentamicin sulphate was from Schering Corp. (Kenilworth, NJ) and absolute ethanol from Arcus A/S (Oslo, Norway), the MTT solution; (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) was from Sigma-Aldrich (St. Louis, MO) and bleomycin 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 IE/KY from Baxter (Deerfield, IL), USA. Cell apoptosis kit, comprising Alexa FluorTM 488 Annexin V and propidium iodide (PI) was from molecular probes (AA Leiden, The Netherlands). The photosensitizer meso-tetraphenylchlorine disulphonate, (TPCS2a/fimaporfin) in the Amphinex® formulation was provided by PCI Biotech (Oslo, Norway). The Amphinex® formulation contains 30 mg mL−1 fimaporfin in 3% polysorbate 80, 2.8% mannitol, 50 mM Tris–HCl pH 8.5.
000 IE/KY from Baxter (Deerfield, IL), USA. Cell apoptosis kit, comprising Alexa FluorTM 488 Annexin V and propidium iodide (PI) was from molecular probes (AA Leiden, The Netherlands). The photosensitizer meso-tetraphenylchlorine disulphonate, (TPCS2a/fimaporfin) in the Amphinex® formulation was provided by PCI Biotech (Oslo, Norway). The Amphinex® formulation contains 30 mg mL−1 fimaporfin in 3% polysorbate 80, 2.8% mannitol, 50 mM Tris–HCl pH 8.5.
Cell culture
The syngeneic rat bladder cancer cell line AY-27 was cultured in RPMI-1640 medium containing 10% FBS, L-glutamine (80 mg L−1), penicillin (100 U mL−1), streptomycin (100 U mL−1) and fungizone (0.25 mg mL−1), in an atmosphere of 95% air, 5% CO2 at 37 °C and subcultured twice a week. The cell line was kindly provided by Dr S. H. Selman, University of Toledo, USA. For SILAC experiments the labelled RPMI 1640 medium is described below.Blue light source for in vitro cell experiments
Culture dishes were illuminated (from below, at room temperature) by using a LumiSource® blue light box (PCI Biotech, Norway), consisting of 4 Osram tubes (18 W, peak wavelength 435 nm, irradiance of 13 mW cm−2). The light intensity at the level of the cells was 13 mW cm−2, measured with an Optometer UDT model 161, radiometer-photometer (United Detector Technology, Culver City, CA, USA), giving a total light dose of 7.8 J cm−2 on the cell level during a 10 min illumination period. The light was detected near the bottom of the cell dish and before passing the dishes. Both light sensitive solutions and cells were covered with aluminum foil during all experiments.Photodynamic experiments and viability assay
AY-27 cells were seeded in Petri dishes (∅ = 6 cm, Nunc Denmark) at a density of 0.5 × 106 cells per dish the day before adding of growth medium containing fimaporfin (0.2 μg mL−1, 18 h, 37 °C) in the dark (Fig. S1, ESI†). After incubation, the dishes were washed three times in PBS before adding of BLM (0.1 μM, 4 h, 37 °C) according to our established protocol for BLM-PCI.23 The blue light illumination was performed 1 h before the end of (BLM incubation). Then the cells were grown overnight (24 h) in regular culture medium prior to viability assays (MTT). Samples that were not incubated with photosensitizer or exposed to light were used as controls. Dishes containing only fimaporfin were considered as “dark toxicity” controls. The MTT solution (0.5 mg mL−1) was freshly made from MTT stock solution (5 mg mL−1) in culture medium and the adherent cultures were washed (PBS, 2 mL) before adding MTT (3 mL) followed by incubation for 1 h at 37 °C, 5% CO2. After removing the MTT solution, cells were detached by isopropanol (2 mL) and orbital shaking (80 rpm, 30 min). Then 0.1 mL of each supernatant was transferred to a cuvette containing isopropanol (0.9 mL), mixed, and absorbance measured by a double beam Shimadzu UV-1700 spectrophotometer. Isopropanol was used as a reference at excitation wavelength 595 nm.Flow cytometry
For each sample ∼106 cells were detached by trypsination (3–5 min, 1 mL/6 cm dish), diluted in 3 mL culture medium, counted in a Bürker chamber and centrifuged at 450 × g for 5 min. After 2 × washes in PBS and centrifugation as above, cells were resuspended in 1 × annexin-binding buffer to 1 × 106 cells mL−1. 100 μL of each sample was added to 5 μL Alexa FluorTM 488 annexin V 1 μL PI working solution (100 μg per mL 1 × annexin-binding buffer) and incubated at room temperature for 15 minutes. After addition of 400 μL 1 × annexin-binding buffer, each sample was immediately cooled on ice and then analyzed in a Beckman Coulter Gallios Flow Cytometer. Fluorescence was measured at 525 nm (FL1) and 620 nm (FL2) using 488 nm excitation wavelength. 20000 cells were counted per sample and Kaluza 1.2 software (Beckman Coulter) was used for data analysis.
SILAC LC-MS/MS analysis
AY-27 cells were grown in SILAC-RPMI 1640 medium with 10% heat inactivated and dialyzed FBS (Thermo Scientific), 2 mM L-glutamine, 2.5 μg mL−1 amphotericin B, 1% PenStrep, as either LIGHT (L-lysine-12C6 and L-arginine-12C6) or HEAVY (L-lysine-13C6,15N2 and L-arginine-13C6,15N4) and underwent six doublings before incorporation efficiency was evaluated by mass spectrometry. Prior to treatment, cells were seeded in 15 cm culture dishes (8 × 106 cells per dish, 25 mL) and incubated further for 24 h before replacement with either fresh medium (control and BLM series) or medium containing 0.2 μg mL−1 fimaporfin (PCI series). After 18 h incubation, cells were washed in PBS and medium with 0.1 μM BLM (BLM and BLMPCI series) or without BLM (controls) was added and the cells incubated for 4 h. The PCI series was irradiated with blue light (1 min, 435 nm, 13.0 mW cm−2) after 3 h and further incubated for 1 h. The experimental setup for the SILAC experiments is outlined in Fig. S1 (ESI†).Cells were lysed in 10 mM Tris–HCl pH 8, 4% SDS, 0.1 M DTT, cOmplete protease inhibitor (Roche) and phosphatase inhibitor I and II (Sigma-Aldrich) by sonication for 30 s using Branson Sonifier 450 (Branson, St. Louis, MO) with output control 2.5 and duty cycle 20%. Cell debris was pelleted by centrifugation at 13200 × g for 10 min and the supernatant harvested as protein extract. Protein concentration was measured using the MilliPore Direct Detect IR spectrometer. 50 μg (protein) each of HEAVY and LIGHT extract was mixed and proteins precipitated using chloroform/methanol.80 The protein pellet was dissolved in 150 μL 50 mM NH4HCO3, reduced with 10 mM DTT for 30 min at 55 °C and further alkylated using 20 mM iodoacetamide for 30 min at room temperature in the dark. Proteins were digested using 1.5 μg trypsin (Promega Corporation, Madison, WI) at 37 °C overnight. Peptides were desalted using homemade C18 Stagetips.81 Peptides were analyzed on a LC-MS/MS platform consisting of an Easy-nLC 1000 UHPLC system (Thermo Scientific) and QExactive Orbitrap mass spectrometer working in data dependent acquisition (DDA) mode using the following parameters: electrospray voltage 1.9 kV, HCD fragmentation with normalized collision energy 30, automatic gain control (AGC) target value of 3E6 for Orbitrap MS and 1E5 for MS/MS scans. Each MS scan (m/z 400–1600) was acquired at a resolution of 70000 FWHM, followed by 10 MS/MS scans triggered for intensities above 1.4 × 104, at a maximum ion injection time of 100 ms for MS and 60 ms for MS/MS scans. Peptides were injected onto a C-18 trap column (Acclaim PepMap100) (75 μm i.d. × 2 cm, C18, 3 μm, 100 Å, Thermo Scientific) and further separated on a C-18 analytical column (Acclaim PepMap100) (75 μm i.d. × 50 cm, C18, 2 μm, 100 Å, Thermo Scientific) using a gradient from 0.1% formic acid to 40% CH3CN, 0.1% formic acid at 250 nL min−1.
Bioinformatic and statistical analyses
Raw MS data were analyzed using Max Quant82 v.1.5.5.1, mapping the spectra over rat canonical proteome including isoforms downloaded from Uniprot in January 2018 with 31571 entries. FDR threshold of 0.01 is set at all the levels, i.e. PSM, sites, peptides, dependent-peptides and protein grouping levels.83 SILAC ratios were log2 transformed and subjected to Student's t-test to identify differentially expressed proteins (DEPs) using perseus platform. We have used Benjamini–Hochberg procedure as implemented in perseus for correction. Those as presented as respective q-values in the Table S1 (ESI†).84 Student's t-test was conducted over these values and relative up/down-regulated proteins (absolute fold-change >1.5 and p-value <0.05) were presented to Ingenuity Pathways Analysis tool (Ingenuity® Systems, https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis/). The data were also subjected to GO enrichment analysis using GOrilla50 (GO Ontology database released 2022-07-01) to identify significantly affected pathways.
Data availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium85via the PRIDE partner repository with the dataset identifier PXD006915.Author contributions
O. A. G., S. M., V. B., B. S. and L. H. carried out the investigation. A. S. and G. S. did the formal analysis of MS data. G. S. wrote the manuscript with support from O. A. G., A. S., B. S., A. H., V. B. and L. H. O. A. G. did the project administration.Conflicts of interest
The authors declare that they have no conflict of interest. Dr Anders Høgset is a co-owner of PCI Biotech ASA, Oslo, Norway.Acknowledgements
PROMEC and CMIC are funded by NTNU and the Liaison Committee for Education, Research and Innovation in Central Norway. PROMEC is a member of the National Network of Advanced Proteomics Infrastructure (NAPI), which is funded by the RCN INFRASTRUKTUR-program (295910). Data storage and handling is supported under the PRIDE and Norstore/Notur projects NN9036K/NS9036K, respectively. Additional funding was from the Cancer fund at St Olav's Hospital, Trondheim. V. B. acknowledges to Bulgarian National Science Fund for the grant KP-06-H41-9/2021. We thank Dr Steven H. Selman at the University of Toledo, Ohio, for kindly providing the AY-27 cell line.References
- R. L. Siegel, K. D. Miller and A. Jemal, CA-Cancer J. Clin., 2019, 69, 7–34 CrossRef PubMed.
- H. Yuan, J. Qiu, L. Liu and S. Zheng, et al. , PLoS One, 2013, 8, e74142 CrossRef CAS PubMed.
- K. Chamie, M. S. Litwin, J. C. Bassett and T. J. Daskivich, et al. , Cancer, 2013, 119, 3219–3227 CrossRef PubMed.
- B. Tomlinson, T. Y. Lin, M. Dall'Era and C. X. Pan, Nanomedicine, 2015, 10, 1189–1201 CrossRef CAS PubMed.
- C. A. Claussen and E. C. Long, Chem. Rev., 1999, 99, 2797–2816 CrossRef CAS PubMed.
- J. Chen and J. Stubbe, Nat. Rev. Cancer, 2005, 5, 102–112 CrossRef CAS PubMed.
- U. Galm, M. H. Hager, S. G. Van Lanen and J. Ju, et al. , Chem. Rev., 2005, 105, 739–758 CrossRef CAS PubMed.
- D. R. Schwartz, G. E. Homanics, D. G. Hoyt and E. Klein, et al. , Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 4680–4685 CrossRef CAS PubMed.
- R. M. Wang, Q. G. Zhang and G. Y. Zhang, Neurosci. Lett., 2004, 357, 13–16 CrossRef CAS PubMed.
- A. Jona, Z. Miltenyi, S. Poliska and B. L. Balint, et al. , PLoS One, 2016, 11, e0157651 CrossRef PubMed.
- T. Onuma, J. F. Holland, H. Masuda and J. A. Waligunda, et al. , Cancer, 1974, 33, 1230–1238 CrossRef CAS.
- S. M. Hecht, J. Nat. Prod., 2000, 63, 158–168 CrossRef CAS PubMed.
- L. F. Povirk, Mutat. Res., 1996, 355, 71–89 CrossRef PubMed.
- J. Cloos, O. Temmink, M. Ceelen and M. H. Snel, et al. , Environ. Mol. Mutagen., 2002, 40, 79–84 CrossRef CAS PubMed.
- B. R. Schroeder, M. I. Ghare, C. Bhattacharya and R. Paul, et al. , J. Am. Chem. Soc., 2014, 136, 13641–13656 CrossRef CAS PubMed.
- G. Pron, N. Mahrour, S. Orlowski and O. Tounekti, et al. , Biochem. Pharmacol., 1999, 57, 45–56 CrossRef CAS PubMed.
- B. Poddevin, S. Orlowski, J. Belehradek, Jr. and L. M. Mir, Biochem. Pharmacol., 1991, 42, S67–S75 CrossRef CAS PubMed.
- K. Berg, P. K. Selbo, L. Prasmickaite and T. E. Tjelle, et al. , Cancer Res., 1999, 59, 1180–1183 CAS.
- O. J. Norum, J. V. Gaustad, E. Angell-Petersen and E. K. Rofstad, et al. , Photochem. Photobiol., 2009, 85, 740–749 CrossRef CAS PubMed.
- K. Berg, A. Dietze, O. Kaalhus and A. Hogset, Clin. Cancer Res., 2005, 11, 8476–8485 CrossRef CAS PubMed.
- A. A. Sultan, W. Jerjes, K. Berg and A. Hogset, et al. , Lancet Oncol., 2016, 17, 1217–1229 CrossRef CAS PubMed.
- J. Trojan, A. Hoffmeister, B. Neu and S. Kasper, et al. , Oncologist, 2022, 27, e430–e433 CrossRef PubMed.
- Y. Baglo, L. Hagen, A. Hogset and F. Drablos, et al. , BioMed Res. Int., 2014, 2014, 921296 Search PubMed.
- O. J. Norum, A. S. V. Fremstedal, A. Weyergang and J. Golab, et al. , J. Controlled Release, 2017, 268, 120–127 CrossRef CAS PubMed.
- S. Sellevold, Q. Peng, A. S. V. Fremstedal and K. Berg, Photodiagnosis Photodyn. Ther., 2017, 20, 95–106 CrossRef CAS PubMed.
- M. Håkerud, P. K. Selbo, Y. Waeckerle-Men and E. Contassot, et al. , J. Controlled Release, 2015, 198, 10–17 CrossRef PubMed.
- M. Haug, G. Brede, M. Hakerud and A. G. Nedberg, et al. , Front. Immunol., 2018, 9, 650 CrossRef PubMed.
- T. Otterhaug, M. Haug, G. Brede and M. Håkerud, et al. , Cancer Immunol. Res., 2016, 4, A008 CrossRef.
- A. Kuehne, H. Emmert, J. Soehle and M. Winnefeld, et al. , Mol. Cell, 2015, 59, 359–371 CrossRef CAS PubMed.
- H. Sun, Y. Zhou, M. F. Skaro and Y. Wu, et al. , Cancer Res., 2020, 80, 1143–1155 CrossRef CAS PubMed.
- W. E. van Riel, A. Rai, S. Bianchi and E. A. Katrukha, et al. , eLife, 2017, 6, e24746 CrossRef PubMed.
- B. Kim, M. Jung, K. C. Moon and D. Han, et al. , Int. J. Cancer, 2023, 152, 320–330 CrossRef CAS PubMed.
- Y. M. Kulkarni, S. Dutta, A. K. Iyer and R. Venkatadri, et al. , Proteomics, 2016, 16, 33–46 CrossRef CAS PubMed.
- S. Xie, C. Dierlam, E. Smith and R. Duran, et al. , J. Cell Sci., 2022, 135(10) DOI:10.1242/jcs.259396.
- E. Tosti, L. Waldbaum, G. Warshaw and E. A. Gross, et al. , J. Biol. Chem., 2004, 279, 47652–47660 CrossRef CAS PubMed.
- C. C. Wu, A. Peterson, B. Zinshteyn and S. Regot, et al. , Cell, 2020, 182, 404–416e414 CrossRef CAS PubMed.
- A. C. Vind, G. Snieckute, M. Blasius and C. Tiedje, et al. , Mol. Cell, 2020, 78, 700–713e707 CrossRef CAS PubMed.
- D. H. Atha, E. Coskun, O. Erdem and A. Tona, et al. , J. Nucleic Acids, 2020, 2020, 8810105 Search PubMed.
- Q. Kong and C. L. Lin, Cell. Mol. Life Sci., 2010, 67, 1817–1829 CrossRef CAS PubMed.
- N. V. Tomilin, L. V. Solovjeva, M. P. Svetlova and N. M. Pleskach, et al. , Radiat. Res., 2001, 156, 347–354 CrossRef CAS PubMed.
- T. Lu, Y. Zhang, Y. Kidane and A. Feiveson, et al. , PLoS One, 2017, 12, e0170358 CrossRef PubMed.
- S. O. Vozianov, V. I. Kashuba, V. M. Grygorenko and V. V. Gordiyuk, et al. , Klin. Khir., 2016, 54–57 CAS.
- M. J. Johansson, A. Esberg, B. Huang and G. R. Bjork, et al. , Mol. Cell. Biol., 2008, 28, 3301–3312 CrossRef CAS PubMed.
- M. J. O. Johansson, F. Xu and A. S. Bystrom, Biochim. Biophys. Acta, Gene Regul. Mech., 2018, 1861, 401–408 CrossRef CAS PubMed.
- J. Fernandez-Vazquez, I. Vargas-Perez, M. Sanso and K. Buhne, et al. , PLoS Genet., 2013, 9, e1003647 CrossRef CAS PubMed.
- M. Tigano, R. Ruotolo, C. Dallabona and F. Fontanesi, et al. , Nucleic Acids Res., 2015, 43, 8368–8380 CrossRef PubMed.
- T. Yang, Y. Jia, Y. Ma and L. Cao, et al. , Am. J. Med. Sci., 2017, 353, 49–58 CrossRef PubMed.
- K. L. Pennington, T. Y. Chan, M. P. Torres and J. L. Andersen, Oncogene, 2018, 37, 5587–5604 CrossRef CAS PubMed.
- S. Singh, C. Brocker, V. Koppaka and Y. Chen, et al. , Free Radical Biol. Med., 2013, 56, 89–101 CrossRef CAS PubMed.
- E. Eden, R. Navon, I. Steinfeld and D. Lipson, et al. , BMC Bioinf., 2009, 10, 48 CrossRef PubMed.
- M. R. Murphy and F. E. Kleiman, Wiley Interdiscip. Rev.: RNA, 2020, 11, e1571 Search PubMed.
- C. B. Vagbo and G. Slupphaug, DNA Repair, 2020, 95, 102927 CrossRef PubMed.
- A. S. Jaiswal, E. A. Williamson, G. Srinivasan and K. Kong, et al. , DNA Repair, 2020, 86, 102769 CrossRef CAS PubMed.
- B. German and L. Ellis, Epigenomes, 2022, 6, 28 CrossRef CAS PubMed.
- M. Duenas, A. Perez-Figueroa, C. Oliveira and C. Suarez-Cabrera, et al. , Sci. Rep., 2019, 9, 10362 CrossRef PubMed.
- M. Sharifi Tabar, J. P. Mackay and J. K. K. Low, FEBS J., 2019, 286, 2043–2061 CrossRef CAS PubMed.
- D. M. Chou, B. Adamson, N. E. Dephoure and X. Tan, et al. , Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 18475–18480 CrossRef CAS PubMed.
- C. G. Spruijt, M. S. Luijsterburg, R. Menafra and R. G. Lindeboom, et al. , Cell Rep., 2016, 17, 783–798 CrossRef CAS PubMed.
- D. H. Larsen, C. Poinsignon, T. Gudjonsson and C. Dinant, et al. , J. Cell Biol., 2010, 190, 731–740 CrossRef CAS PubMed.
- K. Beishline and J. Azizkhan-Clifford, FEBS J., 2015, 282, 224–258 CrossRef CAS PubMed.
- Y. Y. Lee, Y. B. Yu, H. P. Gunawardena and L. Xie, et al. , Cell Death Dis., 2012, 3, e359 CrossRef CAS PubMed.
- F. Ochs, G. Karemore, E. Miron and J. Brown, et al. , Nature, 2019, 574, 571–574 CrossRef CAS PubMed.
- H. Shigetomi, T. Sudo, K. Shimada and C. Uekuri, et al. , Int. J. Gynecol. Cancer, 2014, 24, 838–843 CrossRef PubMed.
- S. Xue, S. Tian, K. Fujii and W. Kladwang, et al. , Nature, 2015, 517, 33–38 CrossRef CAS PubMed.
- L. Shi, H. Hu, P. Sun and Z. Li, et al. , Inflamm. Res., 2022, 71, 977–989 CrossRef CAS PubMed.
- H. Huang, H. Weng, W. Sun and X. Qin, et al. , Nat. Cell Biol., 2018, 20, 285–295 CrossRef CAS PubMed.
- A. Noguchi, S. Adachi, N. Yokota and T. Hatta, et al. , Biol. Open, 2018, 7, bio031575 CrossRef PubMed.
- Y. Han, Q. Zheng, Y. Tian and Z. Ji, et al. , J. Surg. Oncol., 2019, 119, 1145–1154 CrossRef CAS PubMed.
- Y. Tian, Z. Ma, Z. Chen and M. Li, et al. , PLoS One, 2016, 11, e0158891 CrossRef PubMed.
- C. E. Olsen, A. Weyergang, V. T. Edwards and K. Berg, et al. , Biochem. Pharmacol., 2017, 144, 63–77 CrossRef CAS PubMed.
- M. Sobecki, K. Mrouj, J. Colinge and F. Gerbe, et al. , Cancer Res., 2017, 77, 2722–2734 CrossRef CAS PubMed.
- V. Bogoeva, M. Siksjo, K. G. Saeterbo and T. B. Melo, et al. , Photodiagnosis Photodyn. Ther., 2016, 14, 9–17 CrossRef CAS PubMed.
- S. L. Fink and B. T. Cookson, Infect. Immun., 2005, 73, 1907–1916 CrossRef CAS PubMed.
- M. Sanchez-Carbayo, N. D. Socci, E. Charytonowicz and M. Lu, et al. , Cancer Res., 2002, 62, 6973–6980 CAS.
- T. Lorenzi, M. Lorenzi, E. Altobelli and D. Marzioni, et al. , Int. J. Cancer, 2013, 133, 2650–2661 CAS.
- S. Q. Xu, S. Buraschi, A. Morcavallo and M. Genua, et al. , Oncotarget, 2015, 6, 10825–10839 CrossRef PubMed.
- B. Li, S. Ding, N. Feng and N. Mooney, et al. , Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E3642–E3651 CrossRef CAS PubMed.
- N. Y. Song and Y. J. Surh, Ann. N. Y. Acad. Sci., 2012, 1271, 10–19 CrossRef CAS PubMed.
- T. Kokkola, T. Suuronen, F. Molnar and J. Maatta, et al. , FEBS Lett., 2014, 588, 1523–1528 CrossRef CAS PubMed.
- D. Wessel and U. I. Flugge, Anal. Biochem., 1984, 138, 141–143 CrossRef CAS PubMed.
- J. Rappsilber, Y. Ishihama and M. Mann, Anal. Chem., 2003, 75, 663–670 CrossRef CAS PubMed.
- J. Cox and M. Mann, Nat. Biotechnol., 2008, 26, 1367–1372 CrossRef CAS PubMed.
- E. Boutet, D. Lieberherr, M. Tognolli and M. Schneider, et al. , Methods Mol. Biol., 2016, 1374, 23–54 CrossRef CAS PubMed.
- S. Tyanova, T. Temu, P. Sinitcyn and A. Carlson, et al. , Nat. Methods, 2016, 13, 731–740 CrossRef CAS PubMed.
- J. A. Vizcaino, E. W. Deutsch, R. Wang and A. Csordas, et al. , Nat. Biotechnol., 2014, 32, 223–226 CrossRef CAS PubMed.
- R. Carrasco, L. Izquierdo, A. G. van der Heijden and J. J. Lozano, et al. , Sci. Rep., 2021, 11, 6132 CrossRef CAS PubMed.
- Z. Yao, H. Zhang, X. Zhang and Z. Zhang, et al. , Front. Genet., 2022, 13, 923768 CrossRef CAS PubMed.
- J. L. Yang, C. C. N. Wang, J. H. Cai and C. Y. Chou, et al. , Cancers, 2020, 12 CAS.
- H. Cai, H. Chen, Q. Huang and J. M. Zhu, et al. , Pathol. Oncol. Res., 2021, 27, 1609941 CrossRef PubMed.
- Y. M. Kulkarni, S. Dutta, A. K. Iyer and R. Venkatadri, et al. , Proteomics, 2016, 16, 33–46 CrossRef CAS PubMed.
- R. Stroggilos, M. Mokou, A. Latosinska and M. Makridakis, et al. , Int. J. Cancer, 2020, 146(1), 281–294 CrossRef CAS PubMed.
- C. K. Sogaard, A. Blindheim, L. M. Rost and V. Petrovic, et al. , Oncotarget, 2018, 9, 32448–32465 CrossRef PubMed.
- T. Lorenzi, M. Lorenzi, E. Altobelli and D. Marzioni, et al. , Int. J. Cancer, 2013, 133, 2650–2661 CAS.
- J. Zhu, Z. Lu, M. Ke and X. Cai, Int. Urol. Nephrol., 2022, 54, 1505–1512 CrossRef CAS PubMed.
- Z. Guo, H. Zhu, W. Xu and X. Wang, et al. , Mol. Carcinog., 2020, 59, 923–929 CrossRef CAS PubMed.
- X. Liu, Z. Zhou, Y. Wang and K. Zhu, et al. , Front. Oncol., 2020, 10, 589 CrossRef PubMed.
- Z. Andreu, R. Otta Oshiro, A. Redruello and S. Lopez-Martin, et al. , Eur. J. Pharm. Sci., 2017, 98, 70–79 CrossRef CAS PubMed.
- A. S. Baras, N. Gandhi, E. Munari and S. Faraj, et al. , PLoS One, 2015, 10, e0131245 CrossRef PubMed.
- M. Jiang, L. Ren, Y. Chen and H. Wang, et al. , Front. Mol. Biosci., 2021, 8, 613359 CrossRef CAS PubMed.
- S. Nitta, S. Kandori, K. Tanaka and S. Sakka, et al. , Cancer Sci., 2022, 113, 2738–2752 CrossRef CAS PubMed.
- J. A. Yan, H. Xiao, H. X. Ji and W. H. Shen, et al. , J. Int. Med. Res., 2010, 38, 1913–1922 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium85via the PRIDE partner repository with the dataset identifier PXD006915. See DOI: https://doi.org/10.1039/d2mo00337f |
| This journal is © The Royal Society of Chemistry 2023 |